
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA polymer deposition-mediated surface-charge reformation strategy: reversing the MOF biomineralization behavior†
Yanbin
Xu
a,
Huangsheng
Yang
 b,
Anlian
Huang
b,
Linjing
Tong
b,
Wei
Huang
c,
Guosheng
Chen
*b,
Wei
Yi
*a,
Siming
Huang
*a and
Gangfeng
Ouyang
bc
b,
Anlian
Huang
b,
Linjing
Tong
b,
Wei
Huang
c,
Guosheng
Chen
*b,
Wei
Yi
*a,
Siming
Huang
*a and
Gangfeng
Ouyang
bc
aGuangzhou Municipal and Guangdong Provincial Key Laboratory of Molecular Target & Clinical Pharmacology, The NMPA and State Key Laboratory of Respiratory Disease, School of Pharmaceutical Sciences and the Fifth Affiliated Hospital, Guangzhou Medical University, Guangzhou, 511436, China. E-mail: yiwei@gzhmu.edu.cn; huangsm@gzhmu.edu.cn
bMOE Key Laboratory of Bioinorganic and Synthetic Chemistry/KLGHEI of Environment and Energy Chemistry, School of Chemistry, Sun Yat-Sen University, Guangzhou, 510006, China. E-mail: chengsh39@mail.sysu.edu.cn
cSchool of Chemical Engineering and Technology, Sun Yat-Sen University, Zhuhai, 519082, China
First published on 30th October 2024
Abstract
Biomineralization of a porous metal–organic framework (MOF) shell onto biomacromolecule templates is a burgeoning strategy to construct robust biocatalysts. However, it strongly relies on the interfacial interaction between MOF precursors and enzyme surface, significantly limiting the generalization of this nanotechnology. Herein, we identify polymers that are well-suited for deposition onto target biomacromolecules via supramolecular interactions and introduce a polymer deposition-mediated surface-charge reformation strategy to facilitate the biomineralization of porous MOFs, including ZIF-8, ZIF-90, and ZIF-zni onto enzymes. We investigate nine commercially available polymers to find that those with dense –SO3H and –COOH groups effectively regulate the surface-charge properties of the enzymes that are unfavorable for biomineralization. The polymer–enzyme complex thus formed retains its original bioactivity and offers significantly elevated sites to accumulate metal precursors, triggering the in-place MOF biomineralization. We demonstrate that this approach allows access to diverse MOF biocatalysts independent of the enzyme surface chemistry, which are difficult to be synthesized by previous biomineralization methods. Given the highly specific bioactivity and structural stability of the MOF biocatalysts, a chemiluminiscence sensor platform is developed for the sensitive detection of hydrogen sulfide (H2S) biomarkers, with a low limit of detection of 0.09 nM that is superior to most of the reported methods. This study provides an effective and universal strategy for MOF biomineralization using fragile enzymes as biotemplates and offers new insights into accessing multifunctional MOF hybrid biocatalysts.
Introduction
Biomineralization is a mineral deposition process mediated by organisms and represents one of the most important biomanufacturing technologies that have been extensively discovered in intracellular and extracellular events.1,2 It is highly associated with interplays between mineral precursors and biointerfaces, facilitating the in-place growth of inorganic minerals onto varying organism templates from large-sized mussels to small-sized cells and proteins.3–6 Inspired by this bioprocess, several efforts have been devoted to synthesizing functional materials using biomacromolecules as templates, termed biomimetic mineralization.7–10 This biomimetic strategy offers new insights into accessing new hybrid nanoarchitecture that integrates material attributes and biomacromolecular functions, thus holding numerous potentials in biocatalysis, biosensing, environmental remediation, and nanomedicine, among others.11–14From a structural perspective, a biomimetic mineral with high porosity is essential for leveraging the functions of interior biomacromolecules because it can ensure mass transfer and render the biomacromolecules highly accessible. In this context, porous reticular frameworks, such as metal–organic frameworks (MOFs), are ideal materials to target this goal.15 In fact, in the past decade, increasing MOF approaches have been developed for biomimetic mineralization.16–19 In this regard, zeolite imidazole frameworks (ZIFs), such as ZIF-8, are prioritized due to their structural advantages of high porosity and water-stability, as well as their mild crystallization friendliness to biomacromolecules (water phase, at room temperature, etc.).8,20–23 The mechanism of this ZIF biomimetic mineralization is considered to be strongly associated with the interfacial chemistry of a biomacromolecular template.22,24–28 So far, apart from some proteins (including enzymes), other biomolecules, such as hyaluronic acid, heparin, chondroitin sulfate and dermatan sulfate, have been used as templates to initiate the ZIF biomimetic mineralization.28 In these examples, the electrostatic interactions between the negatively charged moieties on these biomolecule surfaces and the metal ion precursors (Zn2+) mediate the ZIF nucleation and dictate the biomineralization process. Owing to the interfacial interactions, it has become feasible to extend the in-place synthesis of porous ZIF minerals onto bacterium, cell, and tobacco mosaic virus, among others,26,29–33 exhibiting multifunctional properties at the intersection of chemistry and biological science.
In principle, the biomacromolecule with a positively charged surface will fail to induce the biomimetic mineralization of ZIF22 due to unfavorable electrostatic repulsion. To address this problem, the surface-charge modification of biomacromolecules via chemical coupling has been recently proposed, and the feasibility of facilitating the ZIF biomimetic mineralization onto positively charged biomacromolecules has been demonstrated.24,25 For instance, the pioneering work reported by Falcaro and Doonan proved that the chemical modification of the protein surface by succinylation or acetylation led to the zeta potential decrease of protein without enough negative charge to below −30 mV, which facilitated the ZIF-8 biomimetic mineralization.24 Our group further found that this biomineralization regulated by chemical modification could, in turn, affect the catalytic activity of the encapsulated enzymes.25 As the accessible functional sites on a biomacromolecule are limited, these chemical modification approaches may result in insufficient efficiency in reforming the surface charges, leading to the failure of biomineralization. In addition, the chemical coupling method poses a risk of perturbing the global conformation of a biomacromolecule, thereby compromising the biofunctionality of the resulting biocomposites.
It is plausible that reformatting the surface charge of a biomacromolecule through supramolecular assembly is a simple yet efficient alternative.22,34 Herein, we identify polymers suitable for deposition onto a target biomacromolecule by supramolecular interactions and describe a polymer deposition-mediated surface-charge reformation strategy to facilitate the biomineralization of porous ZIFs onto functional biomacromolecules, such as enzymes (Scheme 1). Nine commercially available polymers are investigated, among which the polymers with dense carboxyl (–COOH) and sulfonic acid (–SO3H) groups are sufficient enough to reform the surface-charge properties of a series of positively charged biomacromolecules. These polymer-deposited biomacromolecules offer dynamically elevated sites for accumulating ZIF precursors, contributing to a porous biomineral with high enzyme loading and well-preserved biofunctionality. Given the highly specific bioactivity and structural stability of the MOF biocatalysts, a chemiluminescence platform is developed for H2S biomarker detection based on a confined biocatalysis principle, with a limit of detection (LOD) as low as 0.09 nM. This work offers a facile yet efficient strategy to address the biomineralization dilemma of MOFs on vulnerable enzymes.
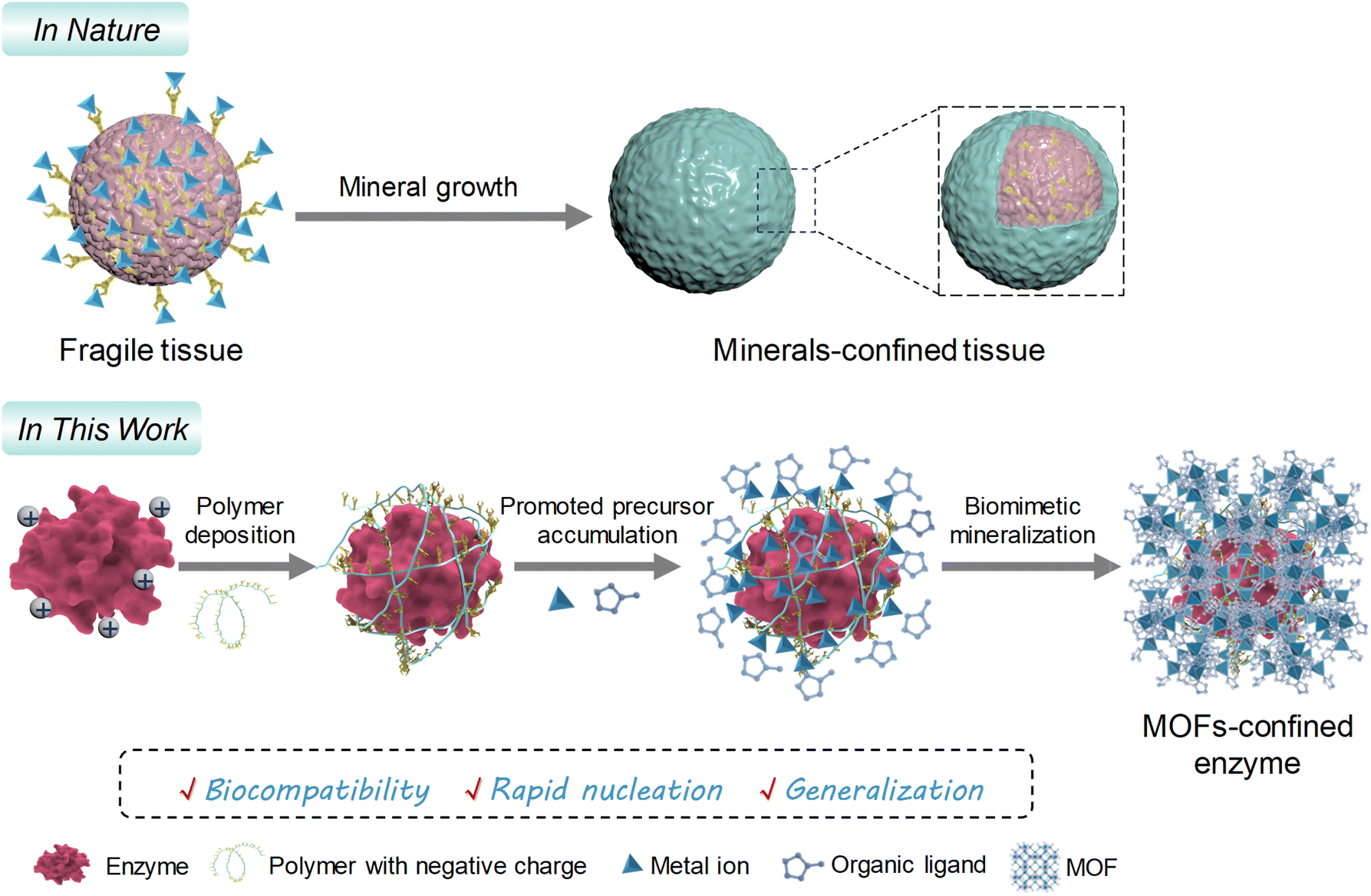 | ||
| Scheme 1 Schematic representation of the polymer deposition-mediated surface-charge reformation strategy for promoting biomineralization of MOFs on fragile enzymes with positive surface charges. | ||
Results and discussion
Polymer screening and polymer deposition-mediated biomineralization of ZIF
Natural biomineralization mechanisms indicate that mineral formation is closely associated with the interactions of the acidic groups of –COOH, hydroxyl (–OH), –SO3H, or phosphate (PO43−) on the biosurface with metal precursors.35–39 In addition, previous studies have confirmed the positive role of –COOH and –SO3H moieties in biomolecules for accelerating the ZIF-8 nucleation.27,28 Inspired by this, we chose a series of polymers involving common pendant groups, including polystyrolsulfon acid (PSSA), polyacrylic acid (PAA), hyaluronic acid (HA), sodium alginate (SA), polyphosphoric acid (PPA), polyvinyl alcohol (PVA), α-cyclodextrin (α-CD), β-cyclodextrin (β-CD) and γ-cyclodextrin (γ-CD), to reform the surface chemistry of an enzyme through electrostatic interactions. The structures, functional groups, molecular weights, and zeta potentials of these polymers are summarized in Fig. 1a and Table S1.† The zeta potentials were all detected to be negative, suggesting their potential for reversing the surface charge of an enzyme from positive to negative. As a proof-of-concept, we first chose horseradish peroxidase (HRP, from horseradish) possessing a pI value of 9.6 with a positive surface charge as a model enzyme, which would be unfavorable for biomimetic mineralization. As shown in Fig. 1b, the native HRP showed an average zeta potential of 4.07 mV. However, after interaction with these negative charge-rich polymers (the dosage of each polymer was kept at 0.5 mg) and removal of the free polymers in the supernatant (detailed procedures presented in the Experimental section of ESI†), the zeta potentials of the polymer–HRP complexes became negative, indicating the successful surface-charge reformation by a simple polymer deposition (Fig. 1b and c). Subsequently, the secondary structure of HRP in the polymer–HRP complexes was determined using a circular dichroism (CD) method. In the CD spectra, the characteristic bands at 208 nm and 222 nm attributing to the α-helix structure of HRP40 were detected in all the polymer–HRP complexes, demonstrating the negligible transformation of the secondary structure of the HRP (Fig. S1†). Furthermore, the catalytic activities of polymer–HRP complexes were estimated by tracing the oxidation of chromogenic reagent 3,3′,5,5′-tetramethylbenzidine (TMB) at 652 nm with the addition of H2O2 using ultraviolet-visible (UV-Vis) spectroscopy. All the polymer–HRP complexes showed comparable activity to the free HRP (Fig. S2†), indicating that the polymer deposition did not affect the accessibility of the catalytic center. Overall, these results demonstrated the biocompatibility of the polymer deposition-mediated surface-charge reformation protocol without disturbing the structure and activity of the enzyme template.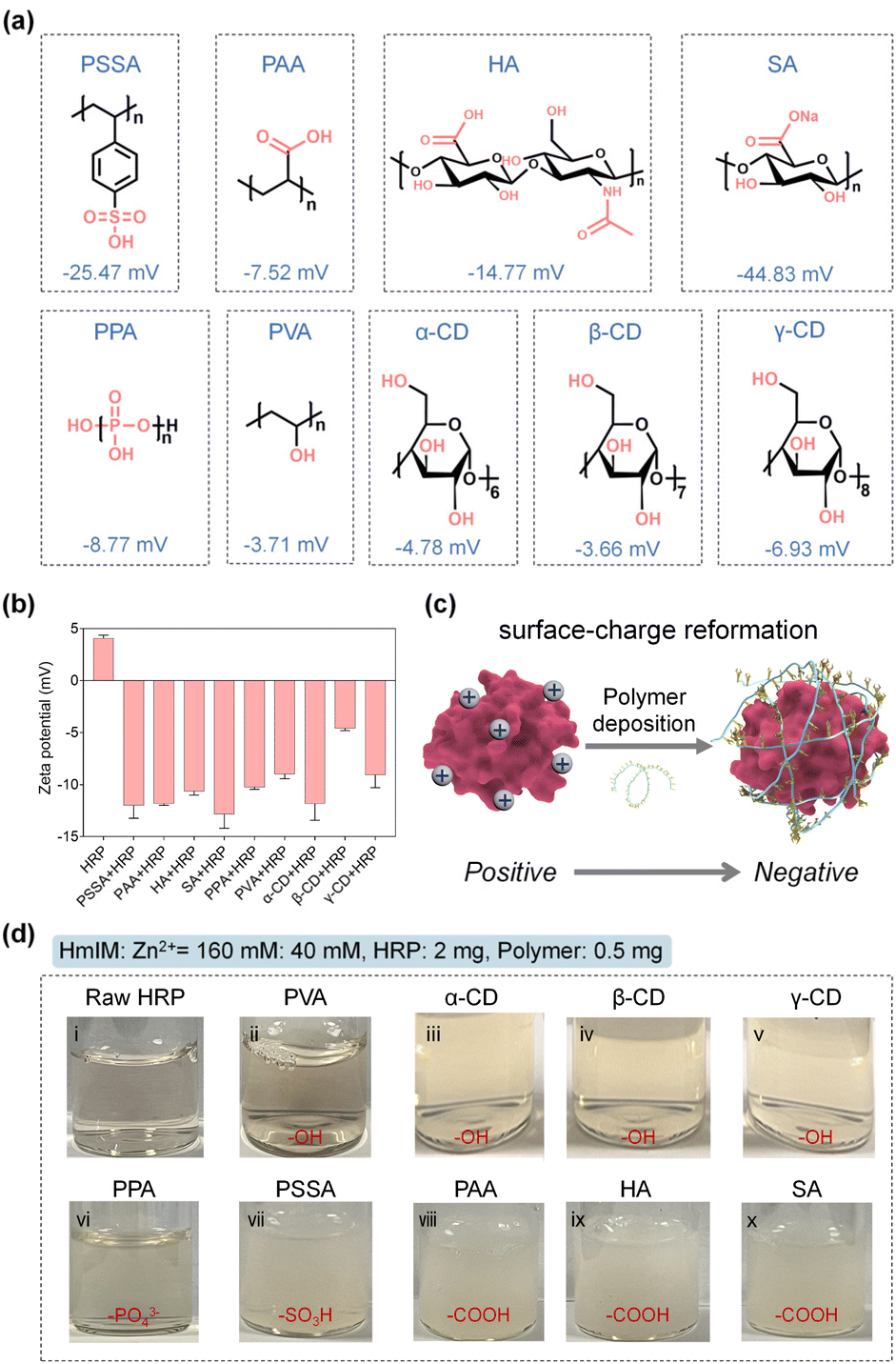 | ||
| Fig. 1 (a) Structures and average zeta potentials of the polymers selected for the enzyme modification. (b) The zeta potentials of HRP before and after the modification of negatively charged polymers. (c) A schematic representation of the polymer deposition-mediated surface-charge reformation. (d) The photographs recording the growth phenomenon of ZIF-8 minerals in the presence of raw HRP or the polymers–HRP complexes. | ||
After ascertaining the feasibility of reformatting the enzyme's surface charge, the ZIFs biomineralization behaviors of the resulting polymer–enzyme complexes were evaluated. ZIF-8, a kind of ZIF crystal formed by the coordination interaction between zinc ions (Zn2+) and 2-methylimidazole (HmIM), was selected as the model ZIF mineral due to its rapid and biocompatible crystallization conditions.8,20,21 As displayed in Fig. 1d, in the raw HRP solution containing HRP (2 mg), Zn2+ (40 mM) and HmIM (160 mM), no precipitate was produced (Fig. 1d(i)), suggesting that the native HRP could not trigger the mineralization enhancement of ZIF-8. These observations were also in line with a previous report.25 In addition, the PVA-HRP, α-CD-HRP, β-CD-HRP and γ-CD-HRP complexes with –OH modifications (Fig. 1d(ii)–(v)) and PPA-HRP with –PO43− modification (Fig. 1d(vi)) could not manage the rapid growth of ZIF-8 minerals as well. As a comparison, polymer–HRP complexes with –SO3H or –COOH modifications (i.e. PSSA, PAA, HA, and SA) could induce the generation of massive precipitates immediately (Fig. 1d(vii)–(x)). These results suggest that in addition to the surface-charge properties, the types of functional groups are decisive for the nucleation of ZIF-8 onto an enzyme. These findings imply that both the –COOH and –SO3H groups in the polymer play a positive role in ZIF-8 biomineralization.
To further demonstrate that stimulative biomineralization is, indeed, dominated by the deposited polymer, we conducted the following control experiments. We first validated that the raw PSSA, PAA, HA, or SA polymer were able to trigger the ZIF-8 biomineralization (Fig. S3 and S4†), highlighting the capability of these deposited polymers to function as biomimetic mineralization agents for directing ZIF-8 growth onto the enzyme. Amination of a protein can substantially improve its negative surface-charge, leading to a failure of biomimetic mineralization.24 Bovine serum albumin (BSA) has been extensively used as the protein for ZIF-8 biomineralization.8,41–43 Upon aminating BSA (the aminated BSA was termed as BSA-NH2), its zeta potential was changed from −18.23 ± 0.21 mV to 22.23 ± 0.33 mV, leading to the unsuccessful ZIF-8 biomineralization (Fig. S5†). We found that, when the BSA-NH2 undergoes PSSA deposition (termed as PSSA-BSA-NH2, zeta potential determined to be −0.79 ± 0.04 mV), the biomineralization ability is restored (Fig. S5–S7†), further ascertaining that the deposited polymers dictate ZIF-8 biomineralization.
Next, we leveraged powder X-ray diffraction (PXRD) to survey the nucleation kinetics of ZIF-8 under the optimized polymers of PSSA, PAA, HA, and SA, which could accelerate ZIF-8 biomineralization. The PXRD patterns in Fig. S8† showed that when using the polymers of PSSA, HA and SA, the characteristic peaks of ZIF-8 occurred in as short duration as 10 s, whereas when using the PAA polymer, the characteristic peaks of ZIF-8 occurred after a min. The results suggested that although all the polymers enabled the formation of ZIF-8 biominerals, the nucleation speeds of the ZIF biominerals were associated with the polymer types. The electrostatic potential (ESP) simulation (vide infra the section titled Mechanism of the polymer deposition-mediated biomineralization) suggested that all the PSSA, HA and SA molecules presented higher metal-binding affinity than PAA. It is plausible that the different nucleation rate is affected by the metal-binding affinity of the polymers.
Furthermore, the performance of ZIF-8 biomineralization using our polymer deposition method and the previously reported chemical coupling approach was compared. We exploited the chemical coupling approach to change the surface charge of HRP using acetic anhydride as the coupling agent (Fig. S9a†).24 The zeta potential of HRP after acetylation (termed acetylated HRP) changed to −15.97 ± 0.35 mV, which indicated the successful reformation of HRP surface charge (Fig. S9b†). However, under the same dosages of both enzyme and ZIF-8 precursors, the acetylated HRP could hardly trigger the growth of ZIF-8 biominerals (Fig. S9c†), which might have resulted from the inadequate functional sites to trigger the MOF nucleation. These results indicated that the simple surface deposition of –COOH and –SO3H-rich polymers is favorable for facilitating the biomimetic mineralization of ZIFs onto the enzymes.
Structural characterization of the as-obtained ZIF-8 biominerals
The accessible ZIF-8 biominerals, termed as PSSA-HRP@ZIF-8, PAA-HRP@ZIF-8, HA-HRP@ZIF-8, and SA-HRP@ZIF-8, were evaluated using techniques such as PXRD, scanning electron microscopy (SEM), Fourier Transform-infrared (FT-IR) spectroscopy, and confocal laser scanning microscopy (CLSM). The PXRD peaks of these ZIF-8 biominerals matched well with those of the standard ZIF-8 (Fig. 2a), and the SEM images resembled the dodecahedron morphology to ZIF-8 crystal, evidencing the successful biosynthesis of ZIF-8 crystals (Fig. 2b). Subsequently, to determine whether increasing the polymer dosage further facilitated the ZIF-8 mineralization process, the dosages of COOH- and SO3H-rich polymers were increased from 0.5 mg to 1.0 mg and to 4.0 mg, respectively, during the biomineralization procedure. In all cases with 1.0 and 4.0 mg dosages of each polymer, the ZIF-8 biominerals could be acquired, as seen in the PXRD patterns (Fig. S10†) and SEM images (Fig. S11†). Afterwards, the loadings of HRP in each biomineral were determined using a standard BCA assay (Table S2 and Fig. S12†). The results demonstrated that the encapsulation of HRP is polymer dosage-dependent, of which 0.5 mg of the polymer in each case affords the best loading efficiency. It is plausible that the increase in polymer dosage offers competitive nucleation sites for ZIF crystallization in the solution phase rather than onto polymer–enzyme complexes, leading to the decreased loading of HRP. We found that the highest enzyme loading as high as 14.02% (w/w) occurred in the PSSA–HRP@ZIF-8 biomineral; this was higher than most of the reported works for HRP immobilization using MOFs (Table S3†). Note that the loading content was very close to that of previous work (14.4%, w/w) in which the HRP was modified by polyvinylpyrrolidone/cysteine (PVP/Cys),22 underscoring the impressive efficiency of this polymer deposition strategy for encapsulating HRP into ZIF-8.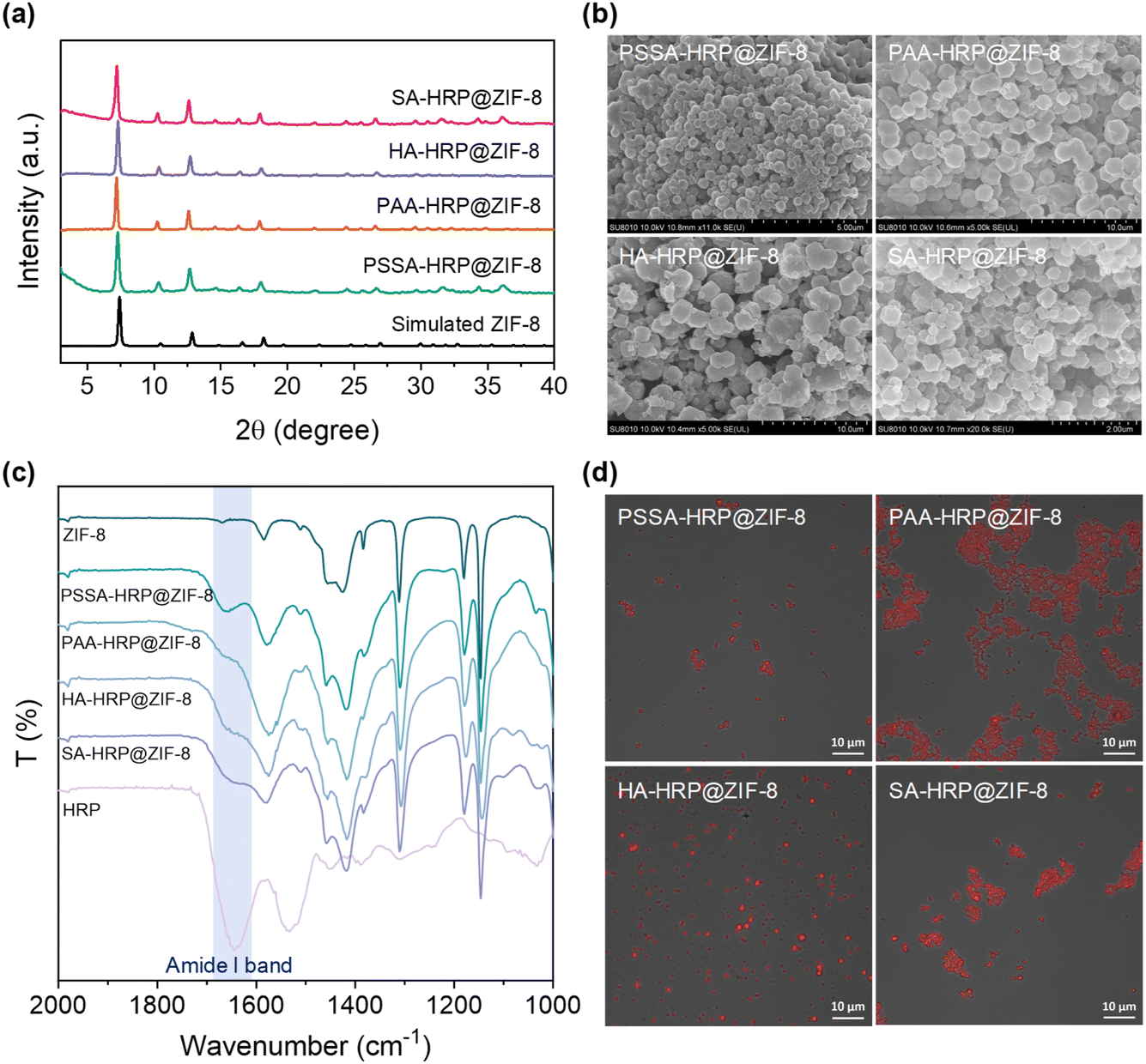 | ||
| Fig. 2 PXRD patterns (a), SEM images (b), FT-IR spectra (c) and the merged CLSM images (d) of the PSSA-HRP@ZIF-8, PAA-HRP@ZIF-8, HA-HRP@ZIF-8, and SA-HRP@ZIF-8, where the polymer dosage for synthesis was kept at 0.5 mg for all biominerals. | ||
The successful encapsulation of HRP within the ZIF-8 biominerals was further confirmed using FT-IR and CLSM. In the FT-IR spectra of PSSA-HRP@ZIF-8, PAA-HRP@ZIF-8, HA-HRP@ZIF-8, and SA-HRP@ZIF-8 samples, the characteristic bands emerged around 1650 cm−1 that were ascribed to the amide I band of proteins, indicating the incorporation of HRP in ZIF-8 biominerals (Fig. 2c). To witness the location of HRP, it was pre-labeled with red fluorescent dye rhodamine B (RhB) prior to biomineralization. With CLSM imaging (Fig. S13† and 2d), the red fluorescent-labelled HRP overlapped well with the ZIF-8 crystals in all biominerals, manifesting that the encased HRP molecules were uniformly distributed throughout the ZIF-8 biominerals.
Next, the bioactivities of all polymer-HRP@ZIF-8 biominerals were further estimated under the same HRP amount (Fig. S14†), and 0.5 mg polymer dosage resulted in the highest catalytic activity of the PSSA-HRP@ZIF-8 and SA-HRP@ZIF-8 biominerals. Although PAA-HRP@ZIF-8 and HA-HRP@ZIF-8 were synthesized by adding 0.5 mg of polymer, they did not exhibit the best catalytic activity; the activity variation between different samples remained limited. Therefore, 0.5 mg was chosen as the optimal polymer dosage for ZIF-8 biomineralization and employed for the following experiments.
Enzyme catalytic kinetics and stability in different ZIF-8 biominerals
The influence of ZIF-8 biomineralization mediated by different types of polymers on the catalytic kinetics of encapsulated enzymes was studied. The H2O2-dependent catalytic kinetics were monitored (Fig. S15†). Subsequently, the plots of the initial catalytic rate (V0) against H2O2 concentration were fitted by the Michaelis–Menten model to evaluate the kinetic parameters, which are displayed in Fig. 3a and b. It is observed that the Michaelis constant of Km, reflecting the substrate-binding capacity of the catalytic center,44 was different among four polymer-HRP@ZIF-8 biominerals. PSSA-HRP@ZIF-8 and PAA-HRP@ZIF-8 presented lower Km values, indicating the favorable binding of H2O2 with the catalytic center of HRP inside these two ZIF-8 biominerals. This behaviour might originate from the different defect formations (amount, geometric size, etc.) induced by different kinds of polymers in ZIF-8 that affected the mass transfer of H2O2 across the ZIF-8 shell. As a result, the catalytic efficiencies (kcat/Km)45 of PSSA-HRP@ZIF-8 and PAA-HRP@ZIF-8 were calculated to be 72.85 and 70.58 s−1 mM−1, respectively, superior to that of HA-HRP@ZIF-8 (56.83 s−1 mM−1) and SA-HRP@ZIF-8 (50.52 s−1 mM−1). These results suggested that the different polymers could affect the catalytic kinetics of the resulting ZIF biominerals; PSSA and PAA polymers were the priorities for ZIF-8 biomineralization. Notably, free HRP exhibited a 3.19–4.98 times lower Km value (0.0581 mM, Fig. S16†) than the polymer-HRP@ZIF-8 composites (from 0.1852 to 0.2894 mM, Fig. 3b), as the microporous ZIF-8 biominerals inevitably lowered the mass transfer rate of the catalytic substrate, which was widely discovered in the previous reports.34,46,47 | ||
| Fig. 3 (a) The plots of initial catalytic rate (V0) against substrate [H2O2] concentration for PSSA-HRP@ZIF-8, PAA-HRP@ZIF-8, HA-HRP@ZIF-8, and SA-HRP@ZIF-8 biominerals. (b) The calculated kinetic parameters of these four polymer-HRP@ZIF-8 biominerals. (c) The retained catalytic capability of free HRP, PSSA-HRP@ZIF-8, PAA-HRP@ZIF-8, HA-HRP@ZIF-8, and SA-HRP@ZIF-8 after different treatments. Conditions: 300 μg mL−1 TMB and 0.1 mM H2O2. (d) The retained catalytic capability of PSSA-HRP@ZIF-8, PAA-HRP@ZIF-8, HA-HRP@ZIF-8, and SA-HRP@ZIF-8 biohybrids after storage at ambient conditions. | ||
The ZIF-8 mineral sets up a suit of armor to shield the encapsulated enzyme against the exterior environment. Next, we investigated the protective effect of different ZIF-8 biominerals for HRP in different challenging environments, including a high-concentration urea solution (6 M), and several organic solvents, including dimethyl sulfoxide (DMSO), N,N′-dimethylformamide (DMF), methanol, acetone, and acetonitrile, for 30 min. As depicted in Fig. 3c, it was found that the activity of native free HRP decreased to less than 70% of its original activity after exposure to these environments. Of specific note, after the treatments with DMSO, DMF and methanol solvents, the free HRP was completely deactivated. In contrast, all the ZIF-8 minerals protected the enzymes from deactivation, which has also been verified in previous works.8,22,43,48 Especially, the HA-HRP@ZIF-8 and PSSA-HRP@ZIF-8 samples could preserve more than 78% of their original activity in all testing conditions. The results validated that the polymer-inducing ZIF biominerals still had an excellent protective effect on interior enzymes. Moreover, the storage stability of these ZIF biominerals at ambient conditions was also surveyed. As presented in Fig. 3d, higher than 74% of the catalytic capability could be preserved after exposing the materials to ambient conditions for 7 days, suggesting the functional stability of the ZIF biominerals.
Mechanism of the polymer deposition-mediated biomineralization
We calculated the ESP values of four polymers that could induce biomineralization and surveyed their potential binding sites for interaction with metal precursors. As described in Fig. 4a, the negative charges were mainly centered on the sulfonic acid moiety of PSSA, the carboxyl moiety of SA, the amide moiety of HA, and the carboxyl moiety of PAA, where the interactions of Zn2+ precursors would be energetically favorable. Of specific note, the lowest ESP value exhibited the order of PSSA (−6.39 eV) < SA (−5.75 eV) < HA (−2.72 eV) < PAA (−1.98 eV), suggesting that the PSSA and SA favored the accumulation of Zn2+ by electrostatic attraction, which was supported well by the higher enzyme loading contents observed in PSSA- and SA-mediated MOF biomineralization (Table S2†). Considering the lowest ESP of PSSA and highest catalytic activity of PSSA-HRP@ZIF-8 biominerals (Fig. 3a, b, and 4a), the mechanism of the PSSA-mediated biomineralization was investigated using different spectroscopic methods, including UV-Vis absorbance spectroscopy, fluorescence spectroscopy, inductively coupled plasma mass spectrometry (ICP-MS), FT-IR and X-ray photoelectron spectroscopy (XPS). As described in Fig. 1b, the zeta potential of HRP changed from 4.07 ± 0.30 mV to −21.6 ± 0.30 mV after interacting with the negatively charged PSSA, implying the formation of the PSSA–HRP complex. This intermolecular interaction was further validated using fluorescence and UV-Vis absorbance spectra. The PSSA presented strong fluorescence emission peaks at 355 nm and 330 nm, attributed to the extended π–π conjugation within the styrene sulfonic acid monomer.49 In the presence of HRP, blue shifts were observed with the peaks moving to 350 nm and 320 nm, respectively, along with an obviously decreased fluorescence intensity (Fig. 4b). This indicated that the electrostatic interaction between negatively charged PSSA and positively charged HRP led to an increase in the electron band gap in PSSA. In addition, the characteristic Soret absorbance band of native HRP at around 405 nm12 displayed a blue shift to 400 nm after introducing PSSA (Fig. 4c), suggesting the intermolecular electrostatic interaction. | ||
| Fig. 4 (a) ESP maps of PSSA, SA, HA and PAA using their corresponding monomer as the simulation fragment. The inserted values were the minimal ESP values. (b) The fluorescence spectra of PSSA, PSSA-HRP, and a mixture of PSSA-HRP and Zn2+ (i.e. PSSA-HRP-Zn2+). (c) The UV-Vis spectra of HRP, PSSA, PSSA-HRP complex, and PSSA-HRP-Zn2+. (d) The FT-IR spectra of HRP, PSSA, HRP-Zn2+, HRP-PSSA, and PSSA-HRP-Zn2+. (e) The high-resolution XPS spectra of Zn 2p of pure ZIF-8 and the PSSA-HRP@ZIF-8. (f) Schematic diagram of the mechanism of the polymer deposition-mediated biomineralization of ZIF-8 on positively charged enzymes. | ||
The above spectroscopic changes manifested the formation of the PSSA–HRP complex via electrostatic interactions. Afterwards, the accumulation ability of Zn2+ by raw HRP and PSSA-HRP complex was assessed using ICP-MS. It showed that the PSSA–HRP complex presented ∼3.6 times the accumulation of Zn2+ compared to the raw HRP (Fig. S17†). The insight into the fluorescence spectra found that the emission at 350 nm of the PSSA–HRP complex was almost dismissed (Fig. 4b), implying the electron transfer from PSSA to Zn2+ by the coordination interaction between –SO3− and Zn2+. At the same time, the new UV-Vis absorbance band of the PSSA–HRP complex around 500 nm appeared after adding Zn2+ (Fig. 4c). This characteristic peak was ascribed to the electron transfer peak from ligand to metal,50,51 which was also supported by the coordination interaction between –SO3− and Zn2+. In the FT-IR spectra (Fig. 4d), the peak located around 1171 cm−1 was assigned to the antisymmetric S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the SO3− groups,52 whereas this characteristic peak was shifted to 1163 cm−1 after the system was treated with Zn2+ ions, further suggesting the strong interaction of Zn2+ by SO3− groups in PSSA. This was in agreement with the ESP calculation (Fig. 4a), in which the SO3− moiety of PSSA was energetically favorable for the interaction with Zn2+. Collectively, these spectroscopic analyses adequately elucidated the enhanced accumulation of Zn2+ onto the PSSA–HRP complex, attributed to the strong coordination interaction between –SO3− and Zn2+.
O stretching vibration of the SO3− groups,52 whereas this characteristic peak was shifted to 1163 cm−1 after the system was treated with Zn2+ ions, further suggesting the strong interaction of Zn2+ by SO3− groups in PSSA. This was in agreement with the ESP calculation (Fig. 4a), in which the SO3− moiety of PSSA was energetically favorable for the interaction with Zn2+. Collectively, these spectroscopic analyses adequately elucidated the enhanced accumulation of Zn2+ onto the PSSA–HRP complex, attributed to the strong coordination interaction between –SO3− and Zn2+.
Furthermore, the as-synthesized PSSA-HRP@ZIF-8 biomineral was collected for XPS tests (Fig. S18†). We found that the high-resolution XPS peaks of Zn 2p in the PSSA-HRP@ZIF-8 sample shifted to higher binding energies as compared to those in standard ZIF-8 (Fig. 4e). This might have resulted from the additional Zn–O coordination interaction between Zn2+ and PSSA, wherein the higher electronegativity of O than that of N elements led to a more electron-deficient state of Zn2+. This indicated that the coordination linkage of ZIF-8 in PSSA-HRP@ZIF-8 biominerals consisted of Zn-HmIM and Zn-PSSA interactions (Fig. 4f). Overall, the biomineralization mechanism could be described as follows: the negatively charged polymers were spontaneously deposited onto positively charged enzymes via electrostatic interactions, reversing the surface charge that promoted Zn2+ accumulation, thereby enabling the in-place nucleation of ZIF-8.
Generalization for other enzymes and ZIF minerals
The feasibility of the polymer deposition-mediated biomineralization protocol for other positively charged enzymes, including cytochrome c (Cyt c, pI = 9.1), lysozyme (Lys, pI = 11), and trypsin (Try, pI = 10.7), was explored. As displayed in Fig. S19,† the introduction of PSSA, PAA, HA, or SA could indeed trigger the formation of Cyt c@ZIF-8, Lys@ZIF-8 and Try@ZIF-8 biominerals. All the biominerals exhibited a typical dodecahedron-shaped structure, with identical PXRD diffraction peaks as simulated ZIF-8 (Fig. 5a and S20–S22†). The amide I bands that appeared in the FT-IR spectra attested the successful encapsulation of enzymes within the ZIF biominerals (Fig. 5b), with loadings as high as 17.49% (w/w) (Fig. S23 and Table S4†). | ||
| Fig. 5 (a) PXRD patterns of different polymer–enzyme@ZIF-8. (b) FT-IR spectra of the polymer–enzyme@ZIF-8. (c) PXRD patterns and SEM images of PSSA-HRP@ZIF-90. (d) PXRD patterns and SEM images of PSSA-HRP@ZIF-zni. The ligand-to-metal ratio to synthesize the ZIF-90 and ZIF-zni biominerals were all 160 mM:40 mM, and the PSSA and enzyme usages were 0.5 mg and 2 mg, respectively. | ||
In addition, facile biomineralization was also extendable to other porous ZIFs, such as ZIF-90 (imidazole-2-formaldehyde (ICA) as ligand and Zn2+ as metal node) and ZIF-zni (imidazole, IM as ligand and Zn2+ as metal node). In this work, PSSA was taken as the polymer, and the PSSA-HRP complex rapidly triggered the nucleation and growth of ZIF-90 and ZIF-zni biominerals with high HRP loadings of 5.55% and 12.75%, respectively (Fig. S24, S25 and Table S5†). The PXRD patterns and SEM images verified the highly crystalline structures, in line with the simulated models (Fig. 5c and d). Likewise, the BSA and BSA-NH2 were used as the positive and negative controls to consolidate the mechanism of polymer deposition-mediated biomineralization (Fig. S26–S31†). The ZIF-90 and ZIF-zni biominerals were rapidly induced by the PSSA-BSA-NH2 complex but not by BSA-NH2, indicating the critical role of deposited PSSA in dictating biomineralization (Figs. S26–S31†). Taken together, the results suggested the generalization of this polymer deposition-mediated biomineralization protocol for different enzymes and ZIF minerals.
Chemiluminescence biosensor for sensitive detection of H2S
H2S, as an indispensable gas signal molecule, involves in many signal transduction pathways to modulate humans' normal physiological functions, such as relaxing smooth muscles, regulation of cardiac function, inflammation, neuromodulation, and activation of autophagy.53,54 More importantly, endogenous H2S is also closely associated with many pathological states, including Alzheimer's disease, Parkinson's disease, down syndrome, diabetes, and liver cirrhosis.55–58 Therefore, H2S is recognized as a vital disease biomarker, and the development of a sensitive H2S detection platform is of significant importance.Given the better stability of encapsulated enzymes than that of free enzymes in external environments, we herein developed a chemiluminescence H2S sensor based on the biocatalytic function of PSSA-HRP@ZIF-8 biomineral. Briefly, luminol could be activated to emit chemiluminescence through biocatalysis of PSSA-HRP@ZIF-8. In the presence of H2S, the chemiluminescence biocatalysis was inhibited (Fig. 6a), where the H2S concentration was proportional to the reduction in chemiluminescence. As a proof-of-concept, sodium bisulfide (NaHS) was used as the simulant of H2S. In the sensing procedures, PBS solution (10 mM, pH = 7.4) was used as the medium to avoid pH variation. It has been reported that ZIF-8 would be decomposed after incubating in a phosphate solution for a period of time.59 Thus, we conducted a series of tests, including ICP-MS, PXRD, SEM, and enzymatic activity measurements, to ensure that during 1 min of sensing, the ZIF-8 biocomposite architecture had not yet decomposed and, synchronously, the HRP could be retained within the ZIF-8 biomineral at the moment (details presented in Fig. S32 and Table S6†).
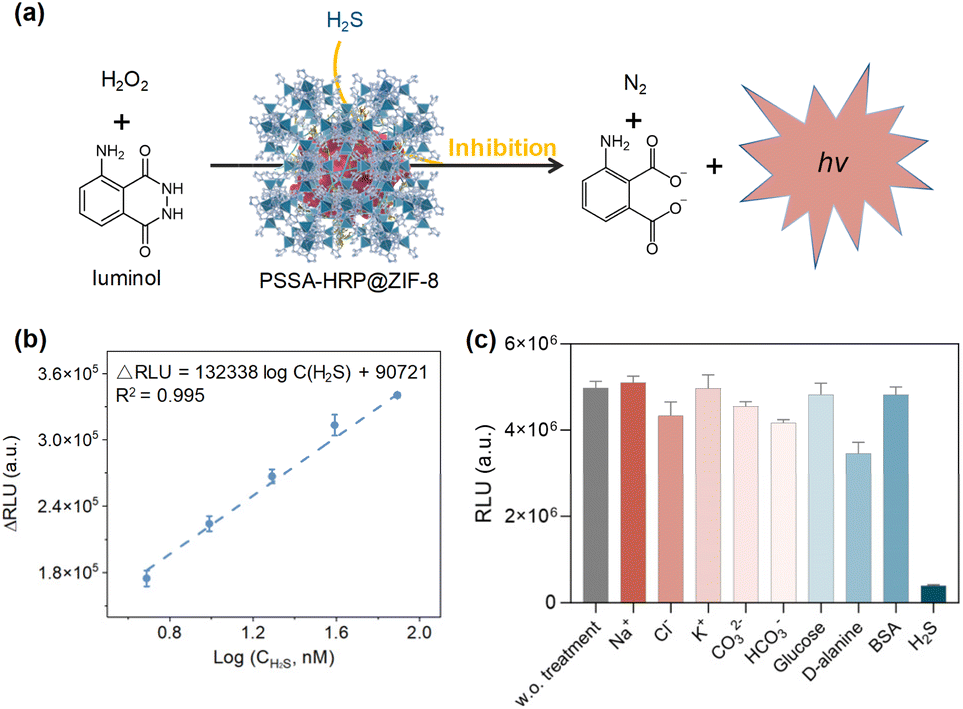 | ||
| Fig. 6 (a) Schematic illustration of H2S sensing based on the PSSA-HRP@ZIF-8 composite combined with the luminol-H2O2 system. (b) The linear range for H2S quantification. The concentration of HRP was set at 0.1 μg mL−1. (c) The selectivity of the proposed biosensor. The concentrations of H2S, BSA, and other interfering substances were 1 mM, 2 mM, and 10 mM, respectively. | ||
To acquire the best performance for H2S sensing, we optimized the concentrations of luminol and H2O2. As presented in Fig. S33,† the signal intensity increased with the increasing luminol concentration in the range of 0.3–1.5 mM and reached a plateau when the concentrations were higher than 1.5 mM. As for the optimization of H2O2, the signal intensity reached the highest at a concentration of 5 mM (Fig. S34†). Under the optimal conditions of luminol (1.5 mM) and H2O2 (5 mM), the decrease in chemiluminescence signal was highly related to the H2S concentration, with good linearity ranging from 4.88 nM to 78.13 nM (R2 = 0.995) and a low LOD of 0.09 nM based on 3δ/k method (Fig. 6b, and S35†), significantly outperforming the previously reported methods (Table S7†). In addition, the good selectivity and anti-interference capability showcased the great potential of this chemiluminescence sensor for the diagnosis of H2S-associated diseases (Fig. 6c, and S36†). Furthermore, after five successive repeat usages of the sensor, the signal intensities remained approximately the same, demonstrating the good recyclability of the proposed biosensor (Fig. S37†).
Conclusions
We have developed a facile and biocompatible polymer deposition-mediated strategy facilitating the rapid in-place growth of MOF minerals onto the enzyme surfaces. The deposition of negatively charged polymers can reverse the positive surface charge of an enzyme by electrostatic interactions, without compromising the enzymatic activity. Both the experimental results and simulation validate that the –SO3H and –COOH groups decorated on the polymers offer energetically favorable binding sites for accumulating metal precursors onto the enzyme surface, thus inducing different MOF growth using the enzyme as a biotemplate. This method allows the facile synthesis of MOF biocatalysts independent of the surface charge chemistry of an enzyme. This characteristic is rare in previously reported biomineralization approaches. In addition, the feasibility of the biocatalytic MOF for ultrasensitive sensing of H2S biomarker was verified, with a low LOD of 0.09 nM, superior to most of the reported detection methods. This work demonstrates the advantages of the polymer deposition approach for regulating the enzyme surface chemistry and governing the biomineralization behavior of MOFs onto biotemplates, which is beneficial for engineering versatile MOF biocatalysts that integrate reticular chemistry attributes and biological functions for multifunctional catalytic platforms.Data availability
The data supporting this article have been included in the main text and ESI.†Author contributions
S. H., G. C. and W. Y. conceived the idea and supervised the project. Y. X. carried out most of the experimental work and statistical analysis. H. Y., A. H. and W. H. provided assistance with PXRD, SEM, FT-IR and CLSM characterization. L. T. performed the ESP calculations. G. O. coordinated the entire project. All authors contributed to the discussion and manuscript writing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We acknowledge financial support from the projects of the National Natural Science Foundation of China (22104159, 22174164) and Guangdong Basic and Applied Basic Research Foundation (2023A1515011632, 2024B1515020070).References
- L. A. Estroff, Chem. Rev., 2008, 108, 4329–4331 CrossRef CAS PubMed.
- F. C. Meldrum and H. Cölfen, Chem. Rev., 2008, 108, 4332–4432 CrossRef CAS.
- J. H. Harding, D. M. Duffy, M. L. Sushko, P. Mark Rodger, D. Quigley and J. A. Elliott, Chem. Rev., 2008, 108, 4823–4854 CrossRef CAS.
- X. Zhang, Z. Fan, Q. Lu, Y. Huang, D. L. Kaplan and H. Zhu, Acta Biomater., 2013, 9, 6974–6980 CrossRef CAS.
- S. Akkineni, C. Zhu, J. Chen, M. Song, S. E. Hoff, J. Bonde, J. Tao, H. Heinz, S. Habelitz and J. J. De Yoreo, Proc. Natl. Acad. Sci. U.S.A., 2022, 119, e2106965119 CrossRef CAS PubMed.
- C. Shao, R. Zhao, S. Jiang, S. Yao, Z. Wu, B. Jin, Y. Yang, H. Pan and R. Tang, Adv. Mater., 2018, 30, 1704876 CrossRef PubMed.
- X. Liu, F. Zhang, X. Jing, M. Pan, P. Liu, W. Li, B. Zhu, J. Li, H. Chen, L. Wang, J. Lin, Y. Liu, D. Zhao, H. Yan and C. Fan, Nature, 2018, 559, 593–598 CrossRef CAS PubMed.
- K. Liang, R. Ricco, C. M. Doherty, M. J. Styles, S. Bell, N. Kirby, S. Mudie, D. Haylock, A. J. Hill, C. J. Doonan and P. Falcaro, Nat. Commun., 2015, 6, 7240 CrossRef CAS PubMed.
- W. Liang, F. Carraro, M. B. Solomon, S. G. Bell, H. Amenitsch, C. J. Sumby, N. G. White, P. Falcaro and C. J. Doonan, J. Am. Chem. Soc., 2019, 141, 14298–14305 CrossRef CAS.
- Y. Zheng, S. Zhang, J. Guo, R. Shi, J. Yu, K. Li, N. Li, Z. Zhang and Y. Chen, Angew. Chem., Int. Ed., 2022, 61, e202208744 CrossRef CAS PubMed.
- S. Yao, B. Jin, Z. Liu, C. Shao, R. Zhao, X. Wang and R. Tang, Adv. Mater., 2017, 29, 1605903 CrossRef.
- W. Huang, H. Yuan, H. Yang, X. Ma, S. Huang, H. Zhang, S. Huang, G. Chen and G. Ouyang, Nat. Commun., 2023, 14, 3644 CrossRef CAS.
- Y.-Z. Wang, S. B. Shah, J.-Y. Liu, H. Hu and Y.-C. Yong, Appl. Catal., B, 2024, 351, 124015 CrossRef CAS.
- J.-S. Lei, Y. Zheng, Y.-F. Meng, F. Wang, Y.-H.-Z. Feng, H.-C. Wang, L.-B. Mao, S.-H. Yu and Z.-L. Wang, Adv. Funct. Mater., 2022, 32, 2202928 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef.
- S. Huang, X. Kou, J. Shen, G. Chen and G. Ouyang, Angew. Chem., Int. Ed., 2020, 59, 8786–8798 CrossRef CAS.
- W. Liang, P. Wied, F. Carraro, C. J. Sumby, B. Nidetzky, C.-K. Tsung, P. Falcaro and C. J. Doonan, Chem. Rev., 2021, 121, 1077–1129 CrossRef CAS.
- K.-Y. Wang, J. Zhang, Y.-C. Hsu, H. Lin, Z. Han, J. Pang, Z. Yang, R.-R. Liang, W. Shi and H.-C. Zhou, Chem. Rev., 2023, 123, 5347–5420 CrossRef CAS PubMed.
- J. Cases Díaz, B. Lozano-Torres and M. Giménez-Marqués, Chem. Mater., 2022, 34, 7817–7827 CrossRef PubMed.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U.S.A., 2006, 103, 10186–10191 CrossRef CAS.
- X. Huang, Y. Lin, J. Zhang and X. Chen, Angew. Chem., Int. Ed., 2006, 45, 1557–1559 CrossRef CAS PubMed.
- G. Chen, S. Huang, X. Kou, S. Wei, S. Huang, S. Jiang, J. Shen, F. Zhu and G. Ouyang, Angew. Chem., Int. Ed., 2019, 58, 1463–1467 CrossRef CAS.
- L. Tong, S. Huang, Y. Shen, S. Liu, X. Ma, F. Zhu, G. Chen and G. Ouyang, Nat. Commun., 2022, 13, 951 CrossRef CAS PubMed.
- N. K. Maddigan, A. Tarzia, D. M. Huang, C. J. Sumby, S. G. Bell, P. Falcaro and C. J. Doonan, Chem. Sci., 2018, 9, 4217–4223 RSC.
- G. Chen, X. Kou, S. Huang, L. Tong, Y. Shen, W. Zhu, F. Zhu and G. Ouyang, Angew. Chem., Int. Ed., 2020, 59, 2867–2874 CrossRef CAS.
- S. Li, M. Dharmarwardana, R. P. Welch, C. E. Benjamin, A. M. Shamir, S. O. Nielsen and J. J. Gassensmith, ACS Appl. Mater. Interfaces, 2018, 10, 18161–18169 CrossRef CAS.
- E. Astria, M. Thonhofer, R. Ricco, W. Liang, A. Chemelli, A. Tarzia, K. Alt, C. E. Hagemeyer, J. Rattenberger, H. Schroettner, T. Wrodnigg, H. Amenitsch, D. M. Huang, C. J. Doonan and P. Falcaro, Mater. Horiz., 2019, 6, 969–977 RSC.
- M. J. Velásquez-Hernández, E. Astria, S. Winkler, W. Liang, H. Wiltsche, A. Poddar, R. Shukla, G. Prestwich, J. Paderi, P. Salcedo-Abraira, H. Amenitsch, P. Horcajada, C. J. Doonan and P. Falcaro, Chem. Sci., 2020, 11, 10835–10843 RSC.
- M. Liu, L. Zhang, R. Yang, H. Cui, Y. Li, X. Li and H. Huang, J. Hazard. Mater., 2024, 461, 132475 CrossRef CAS.
- K. Liang, J. J. Richardson, J. Cui, F. Caruso, C. J. Doonan and P. Falcaro, Adv. Mater., 2016, 28, 7910–7914 CrossRef CAS PubMed.
- T. Chen, J. Yi, Y. Zhao and X. Chu, J. Am. Chem. Soc., 2018, 140, 9912–9920 CrossRef CAS.
- F. Liao, W. Lo, Y. Hsu, C. Wu, S. Wang, F. Shieh, J. V. Morabito, L. Chou, K. C.-W. Wu and C. Tsung, J. Am. Chem. Soc., 2017, 139, 6530–6533 CrossRef CAS.
- K. Wei, M. He, J. Zhang, C. Zhao, C. Nie, T. Zhang, Y. Liu, T. Chen, J. Jiang and X. Chu, Angew. Chem., Int. Ed., 2023, 62, e202307025 CrossRef CAS.
- G. Chen, S. Huang, X. Kou, F. Zhu and G. Ouyang, Angew. Chem., Int. Ed., 2020, 59, 13947–13954 CrossRef CAS PubMed.
- D. Ren, Z. Li, Y. Gao and Q. Feng, Biomed. Mater., 2010, 5, 055009 CrossRef PubMed.
- M. Golda-Cepa, K. Riedlová, W. Kulig, L. Cwiklik and A. Kotarba, ACS Appl. Mater. Interfaces, 2020, 12, 12426–12435 CrossRef CAS.
- K. Wang, F. Luo, L. Wang, B. Zhang, Y. Fan, X. Wang, D. Xu and X. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 49519–49534 CrossRef CAS.
- K. Kahil, S. Weiner, L. Addadi and A. Gal, J. Am. Chem. Soc., 2021, 143, 21100–21112 CrossRef CAS PubMed.
- Y. Liu, Z. Jiang, S. Tong, Y. Sun, Y. Zhang, J. Zhang, D. Zhao, Y. Su, J. Ding and X. Chen, Adv. Mater., 2023, 35, 2203291 CrossRef CAS PubMed.
- F. Gui, F. Chen, J. Wu, Z. Wang, X. Liao and X. Hu, Food Chem., 2006, 97, 480–489 CrossRef CAS.
- W. Liang, R. Ricco, N. K. Maddigan, R. P. Dickinson, H. Xu, Q. Li, C. J. Sumby, S. G. Bell, P. Falcaro and C. J. Doonan, Chem. Mater., 2018, 30, 1069–1077 CrossRef CAS.
- D. Tocco, D. Chelazzi, R. Mastrangelo, A. Casini, A. Salis, E. Fratini and P. Baglioni, J. Colloid Interface Sci., 2023, 641, 685–694 CrossRef CAS PubMed.
- A. Huang, L. Tong, X. Kou, R. Gao, Z.-W. Li, S. Huang, F. Zhu, G. Chen and G. Ouyang, ACS Nano, 2023, 17, 24130–24140 CrossRef CAS PubMed.
- S. Huang, G. Chen and G. Ouyang, Chem. Soc. Rev., 2022, 51, 6824–6863 RSC.
- R. Eisenthal, M. J. Danson and D. W. Hough, Trends Biotechnol., 2007, 25, 247–249 CrossRef CAS PubMed.
- G. Chen, S. Huang, Y. Shen, X. Kou, X. Ma, S. Huang, Q. Tong, K. Ma, W. Chen, P. Wang, J. Shen, F. Zhu and G. Ouyang, Chem, 2021, 7, 2722–2742 CAS.
- Y. Feng, Q. Ma, Z. Wang, Q. Zhang, L. Zhao, J. Cui, Y. Du and S. Jia, Chin. J. Catal., 2024, 60, 386–398 CrossRef CAS.
- X. Wu, C. Yang and J. Ge, Bioresour. Bioprocess., 2017, 4, 24 CrossRef.
- Y. Gu, H. Lai, Z.-Y. Chen, Y. Zhu, Z. Sun, X. Lai, H. Wang, Z. Wei, L. Chen, L. Huang, Y. Zhang, F. He and L. Tian, Angew. Chem., Int. Ed., 2023, 62, e202303476 CrossRef CAS PubMed.
- M. Luísa Ramos, L. L. G. Justino, A. Branco, S. M. Fonseca and H. D. Burrows, Polyhedron, 2013, 52, 743–749 CrossRef.
- H. S. Elshafie, S. H. Sakr, S. A. Sadeek and I. Camele, Chem. Biodiversity, 2019, 16, e1800633 CrossRef.
- R. T. S. Muthu Lakshmi, M. K. Vyas, A. S. Brar and I. K. Varma, Eur. Polym. J., 2006, 42, 1423–1432 CrossRef.
- J. P. Collman, S. Ghosh, A. Dey and R. A. Decréau, Proc. Natl. Acad. Sci. U.S.A., 2009, 106, 22090–22095 CrossRef CAS.
- H. Ye, L. Sun, Z. Pang, X. Ji, Y. Jiao, X. Tu, H. Huang, X. Tang, Z. Xi and L. Yi, Anal. Chem., 2022, 94, 1733–1741 CrossRef CAS.
- K. Eto, T. Asada, K. Arima, T. Makifuchi and H. Kimura, Biochem. Biophys. Res. Commun., 2002, 293, 1485–1488 CrossRef CAS.
- K. Kida, M. Yamada, K. Tokuda, E. Marutani, M. Kakinohana, M. Kaneki and F. Ichinose, Antioxid. Redox Signaling, 2011, 15, 343–352 CrossRef CAS.
- B. Ke, W. Wu, W. Liu, H. Liang, D. Gong, X. Hu and M. Li, Anal. Chem., 2016, 88, 592–595 CrossRef CAS PubMed.
- S. Fiorucci, E. Antonelli, A. Mencarelli, S. Orlandi, B. Renga, G. Rizzo, E. Distrutti, V. Shah and A. Morelli, Hepatology, 2005, 42, 539–548 CrossRef CAS.
- M. de J. Velásquez-Hernández, R. Ricco, F. Carraro, F. T. Limpoco, M. Linares-Moreau, E. Leitner, H. Wiltsche, J. Rattenberger, H. Schröttner, P. Frühwirt, E. M. Stadler, G. Gescheidt, H. Amenitsch, C. J. Doonan and P. Falcaro, CrystEngComm, 2019, 21, 4538–4544 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05935b |
| This journal is © The Royal Society of Chemistry 2024 |