
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnified enantiospecific synthesis of drimane meroterpenoids enabled by enzyme catalysis and transition metal catalysis†
Yipeng
You‡
 a,
Xue-Jie
Zhang‡
b,
Wen
Xiao
b,
Thittaya
Kunthic
b,
Zheng
Xiang
*bc and
Chen
Xu
*a
a,
Xue-Jie
Zhang‡
b,
Wen
Xiao
b,
Thittaya
Kunthic
b,
Zheng
Xiang
*bc and
Chen
Xu
*a
aShenzhen Grubbs Institute, Department of Chemistry, Guangdong Provincial Key Laboratory of Catalysis, Southern University of Science and Technology, 1088 Xueyuan Avenue, Shenzhen, P. R. China. E-mail: xuc@sustech.edu.cn
bState Key Laboratory of Chemical Oncogenomics, Shenzhen Key Laboratory of Chemical Genomics, AI for Science (AI4S) Preferred Program, School of Chemical Biology and Biotechnology, Peking University Shenzhen Graduate School, University Town of Shenzhen, Nanshan District, Shenzhen 518055, P. R. China. E-mail: zxiang@pku.edu.cn
cInstitute of Chemical Biology, Shenzhen Bay Laboratory, Gaoke Innovation Center, Guangqiao Road, Guangming District, Shenzhen 518132, P. R. China
First published on 31st October 2024
Abstract
Merging the advantages of biocatalysis and chemocatalysis in retrosynthetic analysis can significantly improve the efficiency and selectivity of natural product synthesis. Here, we describe a unified approach for the synthesis of drimane meroterpenoids by combining heterologous biosynthesis, enzymatic hydroxylation, and transition metal catalysis. In phase one, drimenol was produced by engineering a biosynthetic pathway in Escherichia coli. Cytochrome P450BM3 from Bacillus megaterium was engineered to catalyze the C-3 hydroxylation of drimenol. By means of nickel-catalyzed reductive coupling, six drimane meroterpenoids (+)-hongoquercins A and B, (+)-ent-chromazonarol, 8-epi-puupehenol, (−)-pelorol, and (−)-mycoleptodiscin A were synthesized in a concise and enantiospecific manner. This strategy offers facile access to the congeners of the drimane meroterpenoid family and lays the foundation for activity optimization.
Introduction
The successful total synthesis of natural products depends on orchestrating an array of reactions, careful control of stereochemistry, and protection–deprotection tactics. Based on the principle of retrosynthetic analysis,1 various strategies have been developed to plan a synthetic route by taking a complex target molecule back to simple building blocks.2 Notably, chemical total synthesis has significantly benefited from the advancements of transition metal catalysis and organocatalysis over the years. As one major pillar of asymmetric catalysis, biocatalysis has emerged as a powerful tool for the sustainable production of active pharmaceutical ingredients (APIs), largely due to the development of genome mining and directed evolution.3 Biocatalysis offers some advantages over classical chemical methods, including higher efficiency and selectivity, environmental friendliness, and better safety profiles. In addition, metabolic engineering allows for streamlining multiple enzymatic reactions inside chassis cells, thus providing rapid access to structurally complex molecules.4 Despite these advantages, enzymatic reactions have not been widely used in natural product synthesis largely for two reasons. First, guidelines for merging enzyme catalysis with traditional chemical methods in synthesis design, namely, biocatalytic retrosynthesis,5 are underdeveloped. Second, it is highly desirable to enrich the toolbox of biocatalysts for a broad range of transformations and substrates.6 Previously, we have reported the efficient synthesis of bioactive natural products by combining enzymatic reactions with chemical methods.7 Herein, we describe a unified synthetic approach to the drimane meroterpenoids by combining metabolic engineering, enzymatic hydroxylation, and transition metal catalysis.Meroterpenoids are a diverse family of hybrid natural products that have been isolated from plants, fungi, bacteria, and animals.8 Meroterpenoids possess a broad range of biological activities and supply several medicinal drugs or promising lead compounds. For example, hongoquercins A (1) and B (2) (Fig. 1A) were isolated from an unidentified terrestrial fungus by Roll and co-workers and exhibit antibacterial activities toward vancomycin-resistant Enterococcus faecium and methicillin-resistant Staphylococcus aureus.9 (+)-ent-Chromazonarol (3) was isolated from marine sponge Disidea pallescens and shows antitumor activities.10 These compounds feature a drimane scaffold that is connected to an aromatic ring by C–C and C–O bonds, and belong to the drimane meroterpenoid family. Due to their remarkable biological activities and structural diversity, intensive efforts have been made to achieve the chemical synthesis of drimane meroterpenoids. Two general strategies, including the biomimetic cyclization approach and the chiral pool approach, have been developed to this end.11,12 For instance, Yamamoto and co-workers have achieved the enantioselective synthesis of (−)-chromazonarol (7) via a biomimetic cyclization induced by a Lewis acid-assisted chiral Brønsted acid (chiral LBA) (Fig. 1B).11a This approach offers the cyclization product with good enantioselectivity (88% ee), albeit with poor diastereoselectivity (69![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :31 d.r.). Later, Barrett and co-workers developed a highly efficient synthesis of (+)-hongoquercin B (2) via a substrate-controlled cationic epoxy-ene tricyclization (Fig. 1C).11b,c For the chiral pool approach, the groups of Baran, Lei, Sau, Wu, Li, and Anderson respectively used (+)-sclareolide (8) as the starting material in the synthesis of (+)-hongoquercin A (1), (+)-ent-chromazonarol (3), 8-epi-puupehenol (4), and (−)-pelorol (5) (Fig. 1D).12 Notably, Renata and co-workers reported a hybrid synthetic strategy by combining the P450-catalyzed selective hydroxylation of (+)-sclareolide with radical-based transformations, which provides facile access to five C-3 oxidized drimane metroterpenoids.13 Despite these advances, a unified and efficient approach for the divergent synthesis of drimane meroterpenoids is highly desirable.
:31 d.r.). Later, Barrett and co-workers developed a highly efficient synthesis of (+)-hongoquercin B (2) via a substrate-controlled cationic epoxy-ene tricyclization (Fig. 1C).11b,c For the chiral pool approach, the groups of Baran, Lei, Sau, Wu, Li, and Anderson respectively used (+)-sclareolide (8) as the starting material in the synthesis of (+)-hongoquercin A (1), (+)-ent-chromazonarol (3), 8-epi-puupehenol (4), and (−)-pelorol (5) (Fig. 1D).12 Notably, Renata and co-workers reported a hybrid synthetic strategy by combining the P450-catalyzed selective hydroxylation of (+)-sclareolide with radical-based transformations, which provides facile access to five C-3 oxidized drimane metroterpenoids.13 Despite these advances, a unified and efficient approach for the divergent synthesis of drimane meroterpenoids is highly desirable.
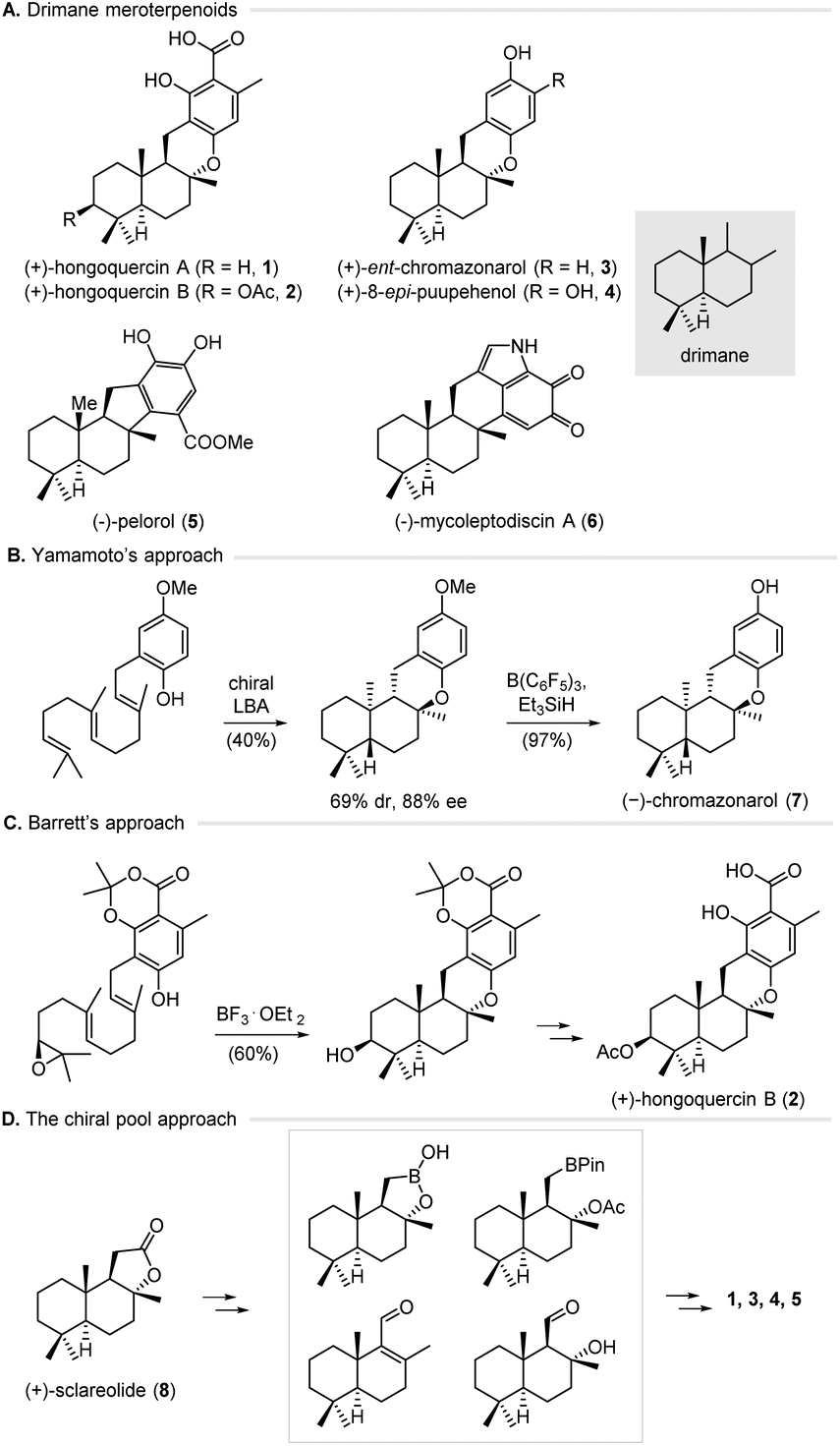 | ||
| Fig. 1 Structure and chemical synthesis of drimane meroterpenoids. (A) Structure of hongoquercins A (1) and B (2), (+)-ent-chromazonarol (3), 8-epi-puupehenol (4), (−)-pelorol (5), and (−)-mycoleptodiscin A (6). (B) Synthesis of (−)-chromazonarol (7) was achieved by Yamamoto and co-workers via a biomimetic polyene cyclization catalyzed by a Lewis acid-assisted chiral Brønsted acid. (C) Synthesis of (+)-hongoquercin B (2) was achieved by Barrett and co-workers via a substrate-controlled epoxy-ene cyclization. (D) The chiral pool approach for the synthesis of drimane meroterpenoids by Baran, Lei, Sau, Wu, Li, and Anderson. | ||
Results and discussion
Synthetic design
Biosynthetically, meroterpenoids are produced via a hybrid machinery that starts with the construction of a non-terpenoid moiety, followed by prenylation, epoxidation, and terpene cyclization.14 In contrast to the cyclization in the late stage during biosynthesis, we envisioned that the drimane moiety can be prepared in the early stage via heterologous biosynthesis, followed by connecting to the aromatics by transition metal-catalyzed C–C bond formation. Therefore, the target molecules, such as 1–4, are retrosynthetically disconnected from the drimane fragments 9 and 10 with their cognate aromatics (Fig. 2). Compounds 9 and 10 can be traced back to drimenol (11) and 3-(OH)-drimenol (12), respectively. To introduce the C-3 hydroxyl group, we planned to engineer a cytochrome P450 for the selective oxidation of drimenol. Finally, we planned to prepare drimenol by engineering a biosynthetic pathway in Escherichia coli. This unified approach harnesses enzymatic terpene cyclization and C–H hydroxylation to construct the drimane fragments on the gram scale, which otherwise has to be achieved via multi-step chemical synthesis. Moreover, it offers a concise and divergent approach to the congeners in the drimane meroterpenoid family for evaluating their biological activities.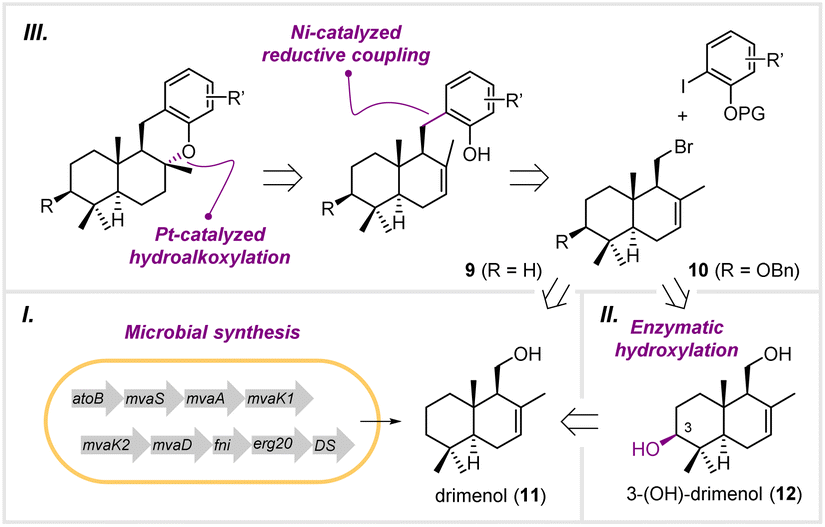 | ||
| Fig. 2 The synthetic strategy for drimane meroterpenoids. | ||
Heterologous synthesis of drimenol
To develop a strain for microbial production of drimenol, we first examined the drimenol synthases from four different species, including PhDS, VoDS, AsDMS, and DrtB.15 PhDS from Persicaria hydropiper and VoDS from Valeriana officinalis belong to plant sesquiterpene synthases; AsDMS and DrtB were identified from Aquimarina spongiae and Aspergillus calidoustus, respectively. By using our reported method, the in vivo activities of these synthases were evaluated in E. coli (Fig. S1†). As expected, both PhDS and VoDS provided drimenol after 72 hours of fermentation at a titer of 276 mg L−1 and 371 mg L−1, respectively (Fig. 3A). To our delight, DrtB afforded the highest titer (1.5 g L−1), which was 5.6-fold relative to PhDS. The specific yield was even 8.6-fold that of PhDS. The products were purified to verify their absolute configuration. We also examined the C-terminal truncated form of DrtB (53 amino acid residues), which was reported to be the only effectively expressible form rather than the full-length protein in E. coli. However, it exhibited a lower titer than the full-length DrtB.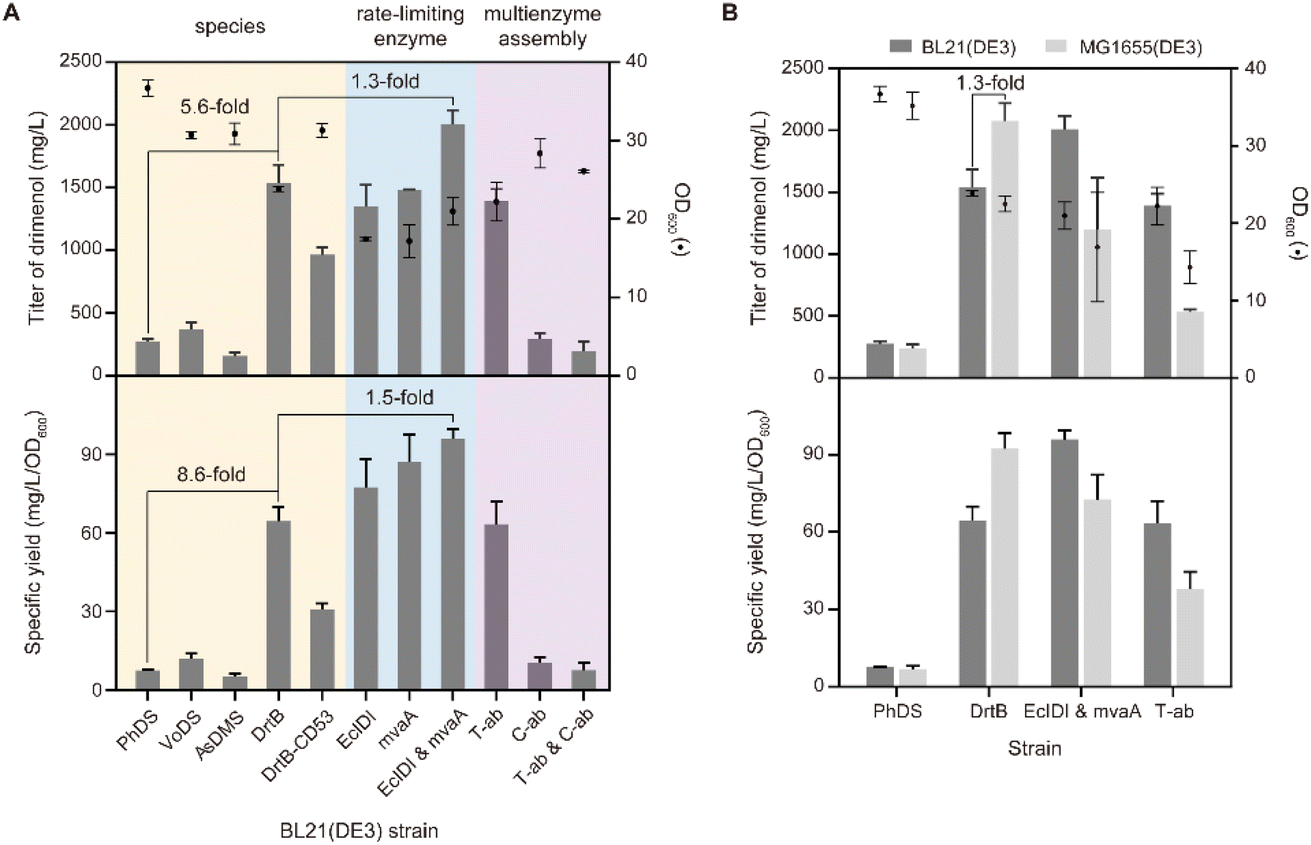 | ||
| Fig. 3 Heterologous biosynthesis of drimenol in E. coli. (A) The BL21(DE3) strain harboring MVA pathway genes, FPPS gene (ERG20), and drimenol synthase gene from 4 species (yellow background); additional copy of EcIDI and mvaA (blue background) and multienzyme assembly containing the AtoB–mvaS–mvaA assembly (T-ab) and ERG20–EcIDI–DrtB assembly (C-ab) (purple background) based on a DrtB overexpressing strain. (B) Comparison of drimenol production between BL21(DE3) and MG1655(DE3) strain which harbored comparative good construct in terms of species of drimenol synthase (DrtB), rate-limiting enzyme of metabolic pathway (EcIDI & mvaA) and multienzyme assembly (T-ab), respectively, and PhDS as control. The data represent the averages ± standard deviations of three independent colonies. | ||
To improve the production of drimenol, metabolic engineering was employed to balance host cell growth and carbon flux into terpene production. It has been suggested that isopentenyl pyrophosphate isomerase (IDI) and HMG-CoA reductase (HMGR) in the mevalonate (MVA) pathway are the key rate-limiting enzymes in that isopentenyl pyrophosphate (IPP) and HMG-CoA cause cell growth inhibition.16 Therefore, we overexpressed a second copy of EcIDI and mvaA gene individually and simultaneously in the DrtB strain. The highest titer and specific yield were achieved by combination of these two rate-limiting enzymes with 30% and 50% increases, respectively.
We next attempted to further improve the titer of drimenol by the multienzyme assembly strategy.17 Recently, Xia and co-workers have utilized orthogonal protein reactions (SnoopTag/SnoopCatcher and SpyTag/SpyCatcher pair) to covalently assemble three key enzymes in the mevalonate/terpene biosynthetic pathway.18 The Jiang group have also developed a modified Tag/Catcher system (NGTag/NGCatcher pair) by integrating covalent bonds and noncovalent interactions for highly efficient protein self-assembly.19 Both methods have been successfully used to improve terpenoid production in E. coli. Thus, we designed (1) a top MVA pathway assembly (AtoB–mvaS–mvaA three enzymes assembly, namely T-ab) by the SnoopTag/SnoopCatcher and SpyTag/SpyCatcher pair; and (2) cross-linking of the MVA-drimenol pathway (ERG20–EcIDI–DrtB three enzyme assembly, namely C-ab) by the NGTag/NGCatcher pair and peptide interaction via a caveolar membrane scaffold (Fig. S2†). Unfortunately, neither assembly strategy afforded a higher titer of drimenol. In contrast, the titer decreased dramatically by means of the second approach. Last, we compared the impact of the E. coli strain on the drimenol titer.
As shown in Fig. 3B, the MG1655(DE3) strain harboring DrtB afforded the highest titer (2.1 g L−1), which was 1.3-fold relative to BL21(DE3). In contrast, the titer and specific yield of drimenol from MG1655(DE3) harboring DrtB with another copy of EcIDI and mvaA are lower than those of BL21(DE3). Therefore, we used the MG1655(DE3) strain harboring DrtB for drimenol production.
Engineering of P450BM3 for selective oxidation of drimenol
With a few grams of drimenol in hand, we carried out biocatalytic oxidation of drimenol by engineering P450BM3 (CYP102A1) from Bacillus megaterium. P450BM3 is highly expressible in E. coli and has one of the highest CYP turnover rates among the reported cytochrome P450s.20 Several groups hence have engineered P450BM3 to catalyze the regio- and stereoselective C–H hydroxylation of cyclic substrates by means of directed evolution.21 However, to the best of our knowledge, selective oxidation of drimenol by this enzyme has not been reported thus far. To this end, we first investigated this reaction using the purified P450BM3 (F87A) (Fig. 4A). To our delight, the desired C3-hydroxylation product 12 was obtained as the main product (Fig. 4B). Its structure was confirmed by 2D NMR and X-ray crystallography (CCDC 2307775, Table S3†). Interestingly, a side product 13 was detected and structurally characterized. We reasoned that 12 was produced through selective epoxidation of compound 12, which was proved by an in vitro enzymatic assay of P450BM3 (F87A) using compound 12 as the substrate (Fig. S3†).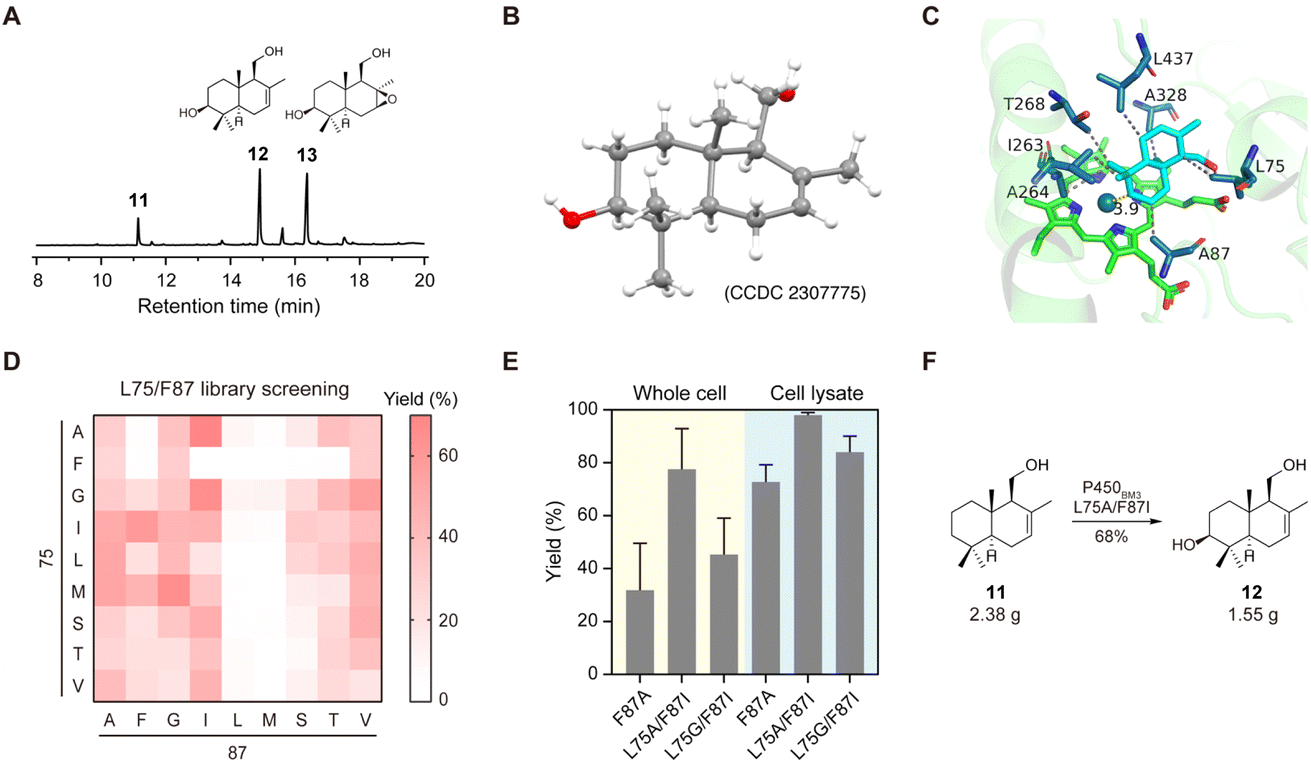 | ||
| Fig. 4 C3-hydroxylation of drimenol was catalyzed by P450BM3 variants. (A) Purified P450BM3 (F87A) catalyzed C–H hydroxylation and C–C epoxidation of 1 mM and 2 mM drimenol (yellow background) to produce compounds 12 (blue background) and 13 (purple background). (B) X-ray crystallography of 3-(OH)-drimenol (12). (C) Docking model of drimenol (cyan) in the active site of P450BM3 (F87A) (green). Several residues (dark blue) show hydrophobic interactions (gray line) with drimenol. (D) Heat map of the yield of compound 12 by screening the P450BM3 L75/F87 mutant library. The yield was determined by GC-MS analysis. (E) Whole cell catalysis (yellow background) and cell lysate catalysis (blue background) by P450BM3 (F87A, L75A/F87I, L75G/F87I). (F) The gram-scale oxidation of drimenol with P450BM3 (L75A/F87I). | ||
To avoid overoxidation and improve the yield of compound 12, we designed and constructed a focused mutagenesis library of P450BM3. Based on the docking model of drimenol in the active site of P450BM3 (F87A), we selected the positions of F87 and L75 for site-directed mutagenesis (Fig. 4C). NNK codon degeneracy that encodes all 20 amino acids has been widely used for saturation mutagenesis. The screening of ∼3000 transformants is required to achieve 95% library coverage of a two-residue mutagenesis library. Reetz and co-workers reported the use of NDC codon degeneracy encoding only 12 amino acids to reduce the transformant number to 430.21b Based on the structure of drimenol, we envisaged that mutation of the key residues in the active site to hydrophobic amino acids or those capable of forming a hydrogen bond would improve enzyme–substrate interactions. Therefore, F87 and L75 were mutated to nine amino acids (Ala, Phe, Gly, Ile, Leu, Met, Ser, Thr, Val) to generate a focused library containing 81 mutants (Fig. 4D). After screening, 6 mutants showed higher activity than P450BM3 (F87A). Two mutants, P450BM3 (L75A/F87I) and P450BM3 (L75G/F87I), were further validated with whole-cell catalysis and cell lysate. As shown in Fig. 4E, both variants showed higher conversions of drimenol and higher yields of compound 12 than P450BM3 (F87A). Previously, Renata and co-workers reported that an engineered P450BM3 variant MERO1 could efficiently transform sclareolide into 3-hydroxysclareolide.13 MERO1 contains 10 mutations at positions that do not interact directly with the substrate. Considering these mutations might have an impact on the catalytic activity, we introduced them into P450BM3 (L75A/F87I). Unfortunately, no increase in the yield of compound 12 was observed (Fig. S4A†). We next optimized the reaction conditions by changing the percentage of the co-solvent. We discovered that in the presence of 5% DMF, a GC yield of 80% was afforded at an analytical scale (Fig. S4B and C†). The gram-scale oxidation of drimenol with P450BM3 (L75A/F87I) was performed under this condition, and afforded compound 12 in an isolated yield of 68% (Fig. 4F).
Synthesis of meroterpenoid natural products
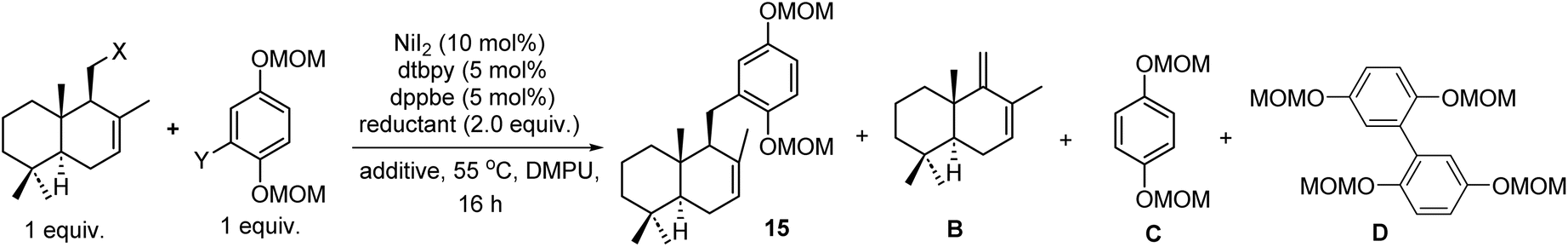
We firstly targeted natural products (+)-ent-chromazonarol (3), 8-epi-puupehenol (4), (−)-pelorol (5), and (−)-mycoleptodiscin (6) (Scheme 1). As shown in Table 1, Ni-catalyzed reductive cross-coupling between drimane halides and aryl halides was evaluated.22 According to Weix's seminal contribution of Ni-catalyzed reductive coupling of alkyl halides with aryl halides, we initially tested the cross-coupling of two iodides under similar conditions, with pyridine as an effective additive to inhibit β-H elimination (Table 1, entry 1). However, the desired product 15 was only obtained in 6% yield. The elimination product B was found to be the major side-product, accompanied with the reduced aryl compound C and the homocoupling product D. We then used drimanyl bromide which is less prone to elimination in the reductive coupling (Table 1, entry 2). To our delight, the yield of the elimination product B was significantly decreased, while the yield of the desired product 15 was increased to 40%. As a comparison, the cross-coupling of drimanyl iodide with the aryl bromide afforded compound 15 only in 3% yield with a 75% yield of compound B (Table 1, entry 3). Interestingly, when both the drimanyl bromide and the aryl bromide were used, compound 15 was obtained in 49% yield (Table 1, entry 4). To further increase the yield of 15, CoII(Pc) was added as a cocatalyst to promote alkyl radical formation for the subsequent reductive cross-coupling.22d–g Pleasingly, the yield of 15 was increased to 57% (Table 1, entry 5). The aryl iodide was also tested under the Ni/Co cocatalytic system, and the yield of 15 was further increased to 62%. The use of other reducing agents in place of Mn(0), such as Zn(0) or 1,1,2,2,-tetrakis(dimethylamino)ethylene (TDAE), still produces an appreciable amount of the eliminated product B, respectively (Table 1, entries 7 and 8). Thus, under the optimized conditions in Table 1, entry 5, compounds 15, 18, 22, and 25 (Scheme 1) were obtained successfully in 57%, 48%, 57% and 45% yields, respectively. Deprotection of the MOM group of 15 and the acetyl group of 18 afforded intermediates 16 and 19, respectively (Scheme 1A). Under the catalysis of the platinum catalyst and silver triflate,2316 was transformed into (+)-ent-chromazonarol (3) in 90% yield as a single diastereomer. 19 was subjected to the same conditions and compound 20 was provided in 90% yield. After deprotection of the benzyl group of 20, (+)-8-epi-puupehenol (4) was obtained in 95% yield. Hence, (+)-ent-chromazonarol (3) and (+)-8-epi-puupehenol (4) were efficiently synthesized over 4 steps (36% overall yield) and 5 steps (31% overall yield), respectively. For the synthesis of (−)-pelorol (5) and (−)-mycoleptodiscin A (6), the corresponding reductive coupling products 22 and 25 respectively underwent TMSOTf-mediated hydroarylation reactions to provide the core structures 23 and 26, which can be converted to the natural products (−)-pelorol (5) and (−)-mycoleptodiscin A (6) according to the literature.12f,24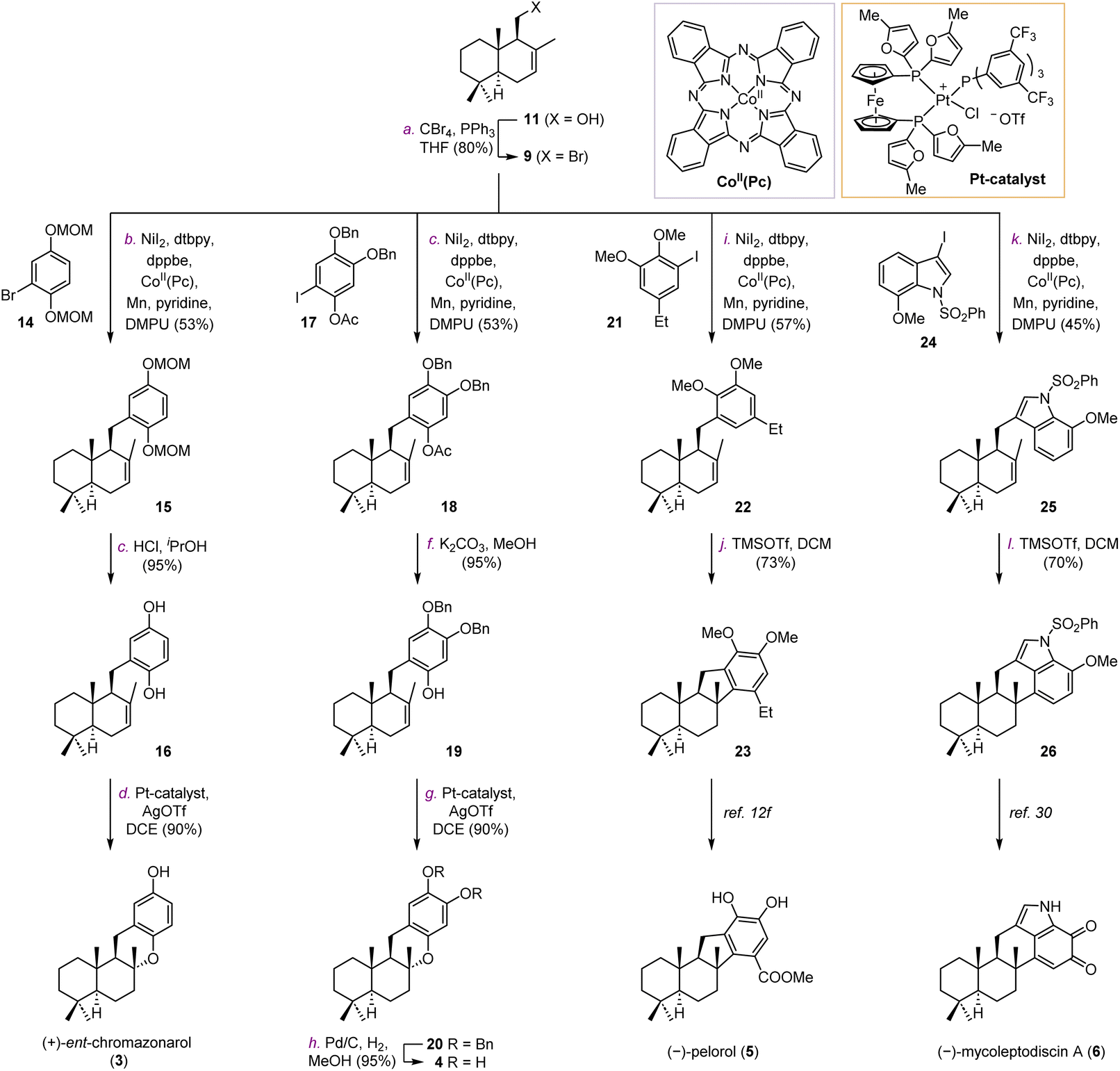 | ||
| Scheme 1 Synthesis of (+)-ent-chromazonarol (3), (+)-8-epi-puupehenol (4), (−)-pelorol (5), and (−)-mycoleptodiscin A (6). Reagents and conditions: (a) CBr4, PPh3, THF, 0 °C, 80%; (b) NiI2 (10 mol%), dtbpy (5 mol%), dppbe (5 mol%), CoII(Pc) (5 mol%), Mn, pyridine, DMPU, 55 °C, 53%; (c) HCl, iPrOH, 55 °C, 95%; (d) Pt-catalyst (3 mol%), AgOTf (6 mol%), DCE, rt, 90%; (e) NiI2 (10 mol%), dtbpy (5 mol%), CoII(Pc) (5 mol%), Mn, pyridine, DMPU, 48%; (f) K2CO3, MeOH, rt, 95%; (g) Pt-catalyst (3 mol%), AgOTf (6 mol%), DCE, rt, 90%; (h) Pd/C, H2, MeOH, rt, 95%. (i) NiI2 (10 mol%), dtbpy (5 mol%), dppbe (5 mol%), CoII(Pc) (5 mol%), Mn, pyridine, DMPU, 55 °C, 57%; (j) TMSOTf, DCM, 0 °C, 73%; (k) NiI2 (10 mol%), dtbpy (5 mol%), dppbe (5 mol%), CoII(Pc) (5 mol%), Mn, pyridine, DMPU, 55 °C, 45%; (l) TMSOTf, DCM, 0 °C, 70%. MOM = methoxymethyl, DCE = 1,2-dichloroethane, TMSOTf = trimethylsilyl triflate. | ||
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entries | X | Y | Reductant | Additive | Yield of 15b | Yield of Bb | Yield of Cc | Yield of Dc |
| a dtbpy = 4,4-di-tert-butyl bipyridine, dppbe = 1,2-bis(diphenylphosphino)benzene, DMPU = 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone, TDAE = tetrakis(dimethylamino)ethylene. The yields of isolated products are given. b Yields of 15 and B were calculated based on the drimane moiety. c Yields of C and D were calculated based on the aryl halide moiety. | ||||||||
| 1 | I | I | Mn | Pyridine (12 mol%) | 6% | 71% | 32% | 12% |
| 2 | Br | I | Mn | Pyridine (12 mol%) | 40% | 38% | 16% | 18% |
| 3 | I | Br | Mn | Pyridine (12 mol%) | 3% | 75% | 30% | 10% |
| 4 | Br | Br | Mn | Pyridine (12 mol%) | 49% | 35% | 5% | 13% |
| 5 | Br | Br | Mn | Pyridine (12 mol%) + CoIIPc (5 mol%) | 57% | 25% | 5% | 8% |
| 6 | Br | I | Mn | Pyridine (12 mol%) + CoIIPc (5 mol%) | 62% | 20% | 8% | 7% |
| 7 | Br | I | Zn | Pyridine (12 mol%) + CoIIPc (5 mol%) | 26% | 46% | 15% | 12% |
| 8 | Br | I | TDAE | Pyridine (12 mol%) + CoIIPc (5 mol%) | 35% | 40% | 12% | — |
We next investigated the synthesis of hongoquercins A (1) and B (2) (Scheme 2). We prepared compound 10 from 3-(OH)-drimenol (12) via four steps (Scheme 2A). The primary hydroxyl group was selectively protected with a TBS group in 90% yield. After benzylation of the C-3 hydroxyl, TBS was deprotected to give intermediate 28 in 85% yield over two steps. In the presence of CBr4 and PPh3, compound 28 was converted into bromide 10 in 62% yield. With compounds 9 and 10 in hand, we first tested the cross-coupling reaction with iodide 29. Unfortunately, no desired cross-coupling product was obtained under the same conditions, possibly due to the steric hindrance of the aromatic fragment. Therefore, we decided to introduce the hydroxyl group in the later stage by the palladium-catalyzed C–H oxidation developed by Yu and co-workers.25 Towards this end, compounds 9 and 10 were coupled with iodide 30 to afford intermediates 31 and 32 in 48% and 48% yields, respectively (Scheme 2B). After deprotection of the MOM group under acidic conditions, both intermediates were subjected to platinum-catalyzed hydroalkoxylation.23 Compounds 33 and 34 were obtained in 80% and 85% yield, respectively. The methyl esters of 33 and 34 were hydrolyzed by lithium hydroxide to provide 35 and 36. The resulting products underwent palladium-catalyzed C–H oxidation to provide (+)-hongoquercin A (1) and compound 37 successfully. To complete the synthesis of (+)-hongoquercin B (2), compound 37 was debenzylated with Pd/C and hydrogen. After protection and selective deprotection, (+)-hongoquercin B (2) was afforded.
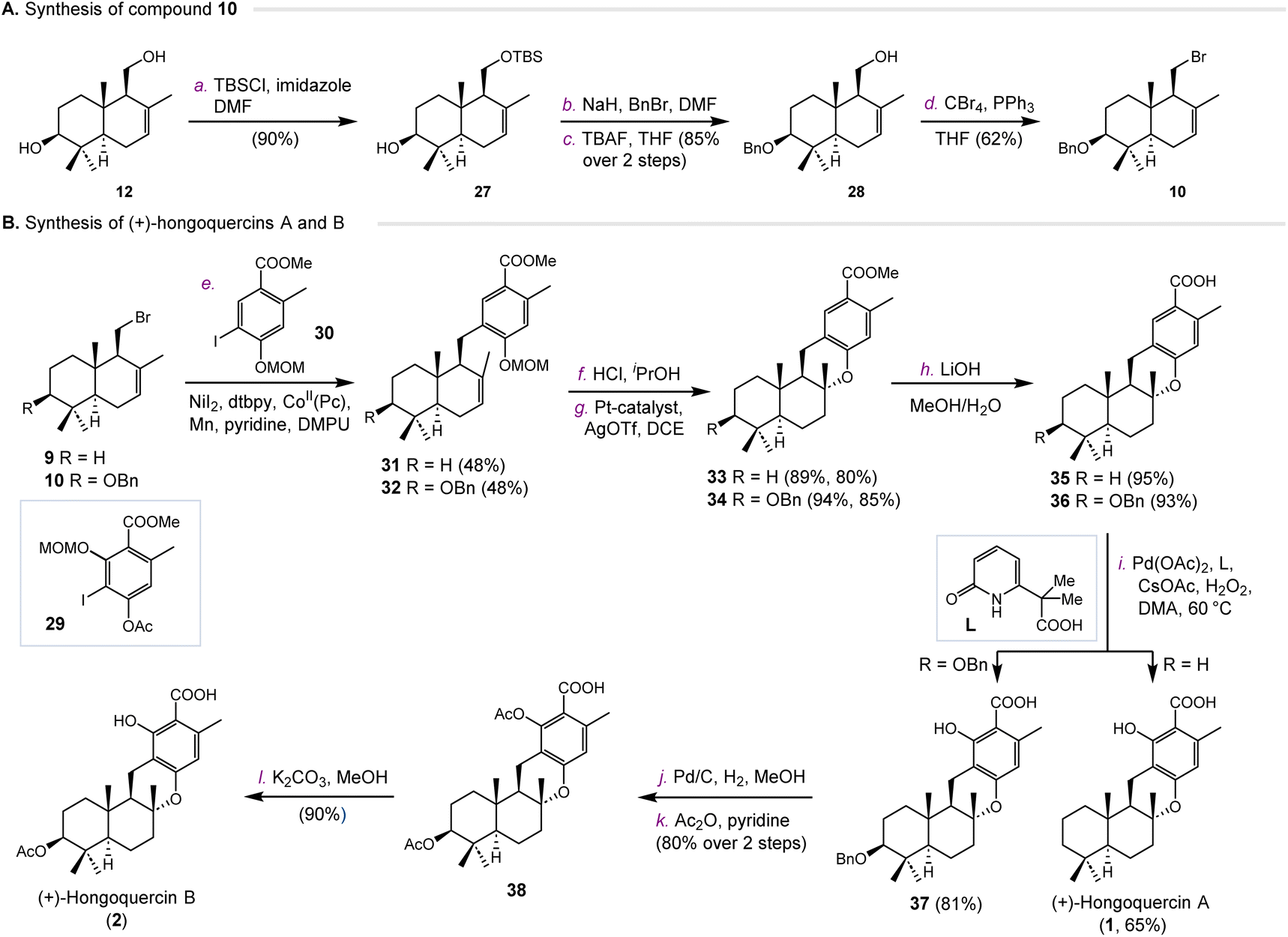 | ||
| Scheme 2 Synthesis of hongoquercins A (1) and B (2). Reagents and conditions: (a) TBSCl, imidazole, DMF, rt, 90%; (b) NaH, BnBr, DMF, rt; (c) TBAF, THF, 60 °C, 85% over 2 steps; (d) CBr4, PPh3, THF, 0 °C, 62%; (e) NiI2 (10 mol%), dtbpy (5 mol%), dppbe (5 mol%), CoII(Pc) (5 mol%), Mn, pyridine, DMPU, 55 °C, 48% for 31, 48% for 32; (f) HCl, iPrOH, 65 °C, 89%; (g) Pt-catalyst (3 mol%), AgOTf (6 mol%), DCE, rt, 80%; (h) LiOH, MeOH/H2O, reflux, 95%; (i) Pd(OAc)2, L, CsOAc, H2O2 (35% aq.), DMA, 60 °C, 65%; (j) Pd/C, H2, MeOH, rt; (k) Ac2O, pyridine, rt, 80% over 2 steps; (l) K2CO3, MeOH, rt, 90%. TBS = tert-butyldimethylsilyl, TBAF = tetrabutylammonium fluoride, DMA = N,N-dimethylacetamide. | ||
Conclusion
Remarkable progress has been achieved in the fields of chemo- and biocatalysis over the past few decades. However, harnessing the benefits of both technologies in natural product synthesis is underexplored. To this end, biocatalytic retrosynthesis has been proposed to aid synthetic chemists in designing synthetic routes with enzyme-catalyzed reactions.5 Based on this concept, we demonstrated that metabolic engineering, enzymatic hydroxylation, and transition metal catalysis can be orchestrated in natural product synthesis. Our strategy harnesses the biosynthetic machinery of terpenoids for rapid construction of the drimane scaffold. By using an engineered P450BM3 variant, the hydroxyl group was site- and stereoselectively introduced at the C-3 position of drimenol. The drimane and aromatic fragments were assembled by transition metal-catalyzed C–C and C–O bond formation reactions. Six drimane meroterpenoids, (+)-hongoquercins A (1) and B (2), (+)-ent-chromazonarol (3), 8-epi-puupehenol (4), (−)-pelorol (5) and (−)-mycoleptodiscin A (6), were synthesized in 4–12 chemical steps. We envisioned that this hybrid approach can also be applied to the synthesis of other bioactive terpenoid natural products and their analogues.Data availability
All experimental procedures and associated data are provided in the ESI.† Crystallographic data for compound 12 has been deposited at the CCDC under 2307775. For ESI and crystallographic data in CIF see DOI: https://doi.org/10.1039/d4sc06060a.Author contributions
C. X. and Z. X. supervised the project and wrote the manuscript; Y. Y. performed the chemical synthesis experiments; X. Z., W. X. and T. K. performed the biosynthesis experiments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22171128), Key-Area Research and Development Program of Guangdong Province (2020B0303070002), Guangdong Basic and Applied Basic Research Foundation (2023B1515020019), and Shenzhen Science and Technology Program (20231120100305001).References
- (a) E. J. Corey and X.-M. Cheng, The Logic of Chemical Synthesis, Wiley, 1995 Search PubMed; (b) S. Warren and P. Wyatt, Organic Synthesis: The Disconnection Approach, Wiley, 2nd edn, 2008 Search PubMed.
- (a) Z. G. Brill, M. L. Condakes, C. P. Ting and T. J. Maimone, Chem. Rev., 2017, 117, 11753–11795 CrossRef CAS PubMed; (b) D. Urabe, T. Asaba and M. Inoue, Chem. Rev., 2015, 115, 9207–9231 CrossRef CAS PubMed; (c) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134–7186 CrossRef CAS PubMed; (d) R. M. Wilson and S. J. Danishefsky, J. Org. Chem., 2007, 72, 4293–4305 CrossRef CAS PubMed.
- (a) U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed; (b) P. N. Devine, R. M. Howard, R. Kumar, M. P. Thompson, M. D. Truppo and N. J. Turner, Nat. Rev. Chem, 2018, 2, 409–421 CrossRef; (c) S. Simić, E. Zukić, L. Schmermund, K. Faber, C. K. Winkler and W. Kroutil, Chem. Rev., 2022, 122, 1052–1126 CrossRef PubMed; (d) S. P. France, R. D. Lewis and C. A. Martinez, JACS Au, 2023, 3, 715–735 CrossRef CAS PubMed.
- (a) V. Courdavault, S. E. O'Connor, M. K. Jensen and N. Papon, Nat. Prod. Rep., 2021, 38, 2145–2153 RSC; (b) J. J. Hug, D. Krug and R. Müller, Nat. Rev. Chem, 2020, 4, 172–193 CrossRef CAS PubMed.
- (a) N. J. Turner and E. O'Reilly, Nat. Chem. Biol., 2013, 9, 285–288 CrossRef CAS PubMed; (b) M. Hönig, P. Sondermann, N. J. Turner and E. M. Carreira, Angew. Chem., Int. Ed., 2017, 56, 8942–8973 CrossRef PubMed; (c) R. O. M. A. de Souza, L. S. M. Miranda and U. T. Bornscheuer, Chem.–Eur. J., 2017, 23, 12040–12063 CrossRef CAS PubMed.
- C.-I. Lin, R. M. McCarty and H. Liu, Angew. Chem., Int. Ed., 2017, 56, 3446–3489 CrossRef CAS PubMed.
- (a) K.-Y. Chen, H.-Q. Wang, Y. Yuan, S.-B. Mou and Z. Xiang, Angew. Chem., Int. Ed., 2023, 62, e202307602 CrossRef CAS PubMed; (b) W. Xiao, S.-J. Wang, M.-Z. Yu, X.-J. Zhang and Z. Xiang, Org. Biomol. Chem., 2023, 21, 5527–5531 RSC; (c) S.-B. Mou, W. Xiao, H.-Q. Wang, K.-Y. Chen and Z. Xiang, Org. Lett., 2021, 23, 400–404 CrossRef CAS PubMed; (d) S.-B. Mou, W. Xiao, H.-Q. Wang, S.-J. Wang and Z. Xiang, Org. Lett., 2020, 22, 1976–1979 CrossRef CAS PubMed.
- (a) M. Jiang, Z. Wu, L. Liu and S. Chen, Org. Biomol. Chem., 2021, 19, 1644–1704 RSC; (b) M. Zhao, Y. Tang, J. Xie, Z. Zhao and H. Cui, Eur. J. Med. Chem., 2021, 209, 112860 CrossRef CAS PubMed; (c) R. Geris and T. J. Simpson, Nat. Prod. Rep., 2009, 26, 1063–1094 RSC.
- D. M. Roll, J. K. Manning and G. T. Carter, J. Antibiot., 1998, 51, 635–639 CrossRef CAS PubMed.
- (a) G. Cimino, S. De Stefano and L. Minale, Experientia, 1975, 31, 1117–1118 CrossRef CAS; (b) A. F. Barrero, E. J. Alvarez-Manzaneda, M. M. Herrador, R. Chahboun and P. Galera, Bioorg. Med. Chem. Lett., 1999, 9, 2325–2328 CrossRef CAS PubMed.
- (a) H. Ishibashi, K. Ishihara and H. Yamamoto, J. Am. Chem. Soc., 2004, 126, 11122–11123 CrossRef CAS PubMed; (b) T. N. Barrett and A. G. M. Barrett, J. Am. Chem. Soc., 2014, 136, 17013–17015 CrossRef CAS PubMed; (c) T.-K. Ma, D. C. Elliott, S. Reid, A. J. P. White, P. J. Parsons and A. G. M. Barrett, J. Org. Chem., 2018, 83, 13276–13286 CrossRef CAS PubMed.
- (a) D. D. Dixon, J. W. Lockner, Q. Zhou and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 8432–8435 CrossRef CAS PubMed; (b) J. Huang and X. Lei, Sci. China: Chem., 2013, 56, 349–353 CrossRef CAS; (c) D. H. Dethe, G. M. Murhade, B. D. Dherange and S. K. Sau, Eur. J. Org Chem., 2017, 1143–1150 CrossRef CAS; (d) H.-S. Wang, H.-J. Li, Z.-G. Zhang and Y.-C. Wu, Eur. J. Org Chem., 2018, 915–925 CrossRef CAS; (e) X. Wang, S. Zhang, P. Cui and S. Li, Org. Lett., 2020, 22, 8702–8707 CrossRef CAS PubMed; (f) L. Yang, D. E. Williams, A. Mui, C. Ong, G. Krystal, R. van Soest and R. J. Andersen, Org. Lett., 2005, 7, 1073–1076 CrossRef CAS PubMed.
- J. Li, F. Li, E. King-Smith and H. Renata, Nat. Chem., 2020, 12, 173–179 CrossRef CAS PubMed.
- (a) L. Barra and I. Abe, Nat. Prod. Rep., 2021, 38, 566–585 RSC; (b) Y. Matsuda, T. Awakawa, T. Mori and I. Abe, Curr. Opin. Chem. Biol., 2016, 31, 1–7 CrossRef CAS PubMed.
- (a) M. G. L. Henquet, N. Prota, J. J. J. van der Hooft, M. Varbanova-Herde, R. J. M. Hulzink, M. de Vos, M. Prins, M. T. J. de Both, M. C. R. Franssen, H. Bouwmeester and M. Jongsma, Plant J., 2017, 90, 1052–1063 CrossRef CAS PubMed; (b) M. Kwon, S. A. Cochrane, J. C. Vederas and D.-K. Ro, FEBS Lett., 2014, 588, 4597–4603 CrossRef CAS PubMed; (c) N. N. Q. Vo, Y. Nomura, K. Kinugasa, H. Takagi and S. Takahashi, ACS Chem. Biol., 2022, 17, 1226–1238 CrossRef CAS PubMed; (d) Y. Huang, S. Hoefgen and V. Valiante, Angew. Chem., Int. Ed., 2021, 60, 23763–23770 CrossRef CAS PubMed.
- (a) V. J. J. Martin, D. J. Pitera, S. T. Withers, J. D. Newman and J. D. Keasling, Nat. Biotechnol., 2003, 21, 796–802 CrossRef CAS PubMed; (b) D. J. Pitera, C. J. Paddon, J. D. Newman and J. D. Keasling, Metab. Eng., 2007, 9, 193–207 CrossRef CAS PubMed.
- M. Liu, Y. Wang, H. Jiang, Y. Han and J. Xia, ChemBioChem, 2023, 24, e202200518 CrossRef CAS PubMed.
- (a) J. Qu, S. Cao, Q. Wei, H. Zhang, R. Wang, W. Kang, T. Ma, L. Zhang, T. Liu, S. Wing-Ngor Au, F. Sun and J. Xia, ACS Nano, 2019, 13, 9895–9906 CrossRef CAS PubMed; (b) Q. Wei, Y. Wang, Z. Liu, M. Liu, S. Cao, H. Jiang and J. Xia, ACS Catal., 2022, 12, 8372–8379 CrossRef CAS.
- Y. Chen, D. Ming, L. Zhu, H. Huang and L. Jiang, Biomacromolecules, 2022, 23, 3936–3947 CrossRef CAS PubMed.
- C. J. C. Whitehouse, S. G. Bell and L.-L. Wong, Chem. Soc. Rev., 2012, 41, 1218–1260 RSC.
- (a) R. Fasan, ACS Catal., 2012, 2, 647–666 CrossRef CAS; (b) S. Kille, F. E. Zilly, J. P. Acevedo and M. T. Reetz, Nat. Chem., 2011, 3, 738–743 CrossRef CAS PubMed; (c) K. Zhang, S. El Damaty and R. Fasan, J. Am. Chem. Soc., 2011, 133, 3242–3245 CrossRef CAS PubMed; (d) K. Zhang, B. M. Shafer, M. D. I. Demars, H. A. Stern and R. Fasan, J. Am. Chem. Soc., 2012, 134, 18695–18704 CrossRef CAS PubMed; (e) P. Le-Huu, T. Heidt, B. Claasen, S. Laschat and V. B. Urlacher, ACS Catal., 2015, 5, 1772–1780 CrossRef CAS; (f) S. A. Loskot, D. K. Romney, F. H. Arnold and B. M. Stoltz, J. Am. Chem. Soc., 2017, 139, 10196–10199 CrossRef CAS PubMed; (g) P. Le-Huu, D. Rekow, C. Krüger, A. Bokel, T. Heidt, S. Schaubach, B. Claasen, S. Hölzel, W. Frey, S. Laschat and V. B. Urlacher, Chem.–Eur. J., 2018, 24, 12010–12021 CrossRef CAS PubMed; (h) F. Li and H. Renata, J. Am. Chem. Soc., 2021, 143, 18280–18286 CrossRef CAS PubMed; (i) F. Li, H. Deng and H. Renata, J. Am. Chem. Soc., 2022, 144, 7616–7621 CrossRef CAS PubMed; (j) J. Li, F. Chen and H. Renata, J. Am. Chem. Soc., 2022, 144, 19238–19242 CrossRef CAS PubMed.
- (a) D. A. Everson, R. Shrestha and D. J. Weix, J. Am. Chem. Soc., 2010, 132, 920–921 CrossRef CAS PubMed; (b) D. A. Everson and D. J. Weix, J. Org. Chem., 2014, 79, 4793–4798 CrossRef CAS PubMed; (c) X. Wang, S. Wang, W. Xue and H. Gong, J. Am. Chem. Soc., 2015, 137, 11562–11565 CrossRef CAS PubMed; (d) L. K. G. Ackerman, L. L. Anka-Lufford, M. Naodovic and D. J. Weix, Chem. Sci., 2015, 6, 1115–1119 RSC; (e) J. L. Hofstra, A. H. Cherney, C. M. Ordner and S. E. Reisman, J. Am. Chem. Soc., 2018, 140, 139–142 CrossRef CAS PubMed; (f) K. Komeyama, T. Michiyuki and I. Osaka, ACS Catal., 2019, 9, 9285–9291 CrossRef CAS; (g) D. J. Charboneau, E. L. Barth, N. Hazari, M. R. Uehling and S. L. Zultanski, ACS Catal., 2020, 10, 12642–12656 CrossRef CAS PubMed; (h) W. Zhu, Q. Yin, Z. Lou and M. Yang, Nat. Commun., 2022, 13, 6633 CrossRef CAS PubMed; (i) X. Ying, Y. Li, L. Li and C. Li, Angew. Chem., Int. Ed., 2023, 62, e202304177 CrossRef CAS PubMed.
- Y. Zhou, X. Xu, H. Sun, G. Tao, X.-Y. Chang, X. Xing, B. Chen and C. Xu, Nat. Commun., 2021, 12, 1953 CrossRef CAS PubMed.
- D. H. Dethe, S. K. Sau and S. Mahapatra, Org. Lett., 2016, 18, 6392–6395 CrossRef CAS PubMed.
- Z. Li, H. S. Park, J. X. Qiao, K.-S. Yeung and J.-Q. Yu, J. Am. Chem. Soc., 2022, 144, 18109–18116 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2307775. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06060a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |