
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optical resolution via chiral auxiliaries of curved subphthalocyanine aromatics†
Giulia
Lavarda‡
 a,
Lara
Tejerina‡
a,
Tomás
Torres
*abc and
M. Victoria
Martínez-Díaz
*ab
a,
Lara
Tejerina‡
a,
Tomás
Torres
*abc and
M. Victoria
Martínez-Díaz
*ab
aDepartment of Organic Chemistry, Universidad Autónoma de Madrid, Madrid, 28049, Spain. E-mail: victoria.martinez@uam.es; tomas.torres@uam.es
bInstitute for Advanced Research in Chemical Sciences, Universidad Autónoma de Madrid, Madrid, 28049, Spain
cIMDEA-Nanociencia, c/Faraday, 9, Cantoblanco, Madrid, 28049, Spain
First published on 28th October 2024
Abstract
Chiral conjugated materials with curved topologies hold significant promise for advanced optoelectronic applications. Among these, bowl-shaped subphthalocyanine (SubPc) aromatics are particularly noteworthy due to their superb optoelectronic properties and synthetic versatility. Despite their potential, the development and application of inherently chiral SubPcs as functional materials have been hampered by the scalability and feasibility limitations of current high-performance liquid chromatography methods. In this work, we employ axial derivatization with BINOL-based chiral auxiliaries to achieve the optical resolution of C3-symmetric SubPcs. This approach allows us to obtain optically active meta and ortho-substituted SubPc derivatives in high yields and enantiomeric excess through straightforward organic chemistry protocols. In addition, we serendipitously observe unprecedented bowl-to-bowl inversion of the SubPc macrocycle upon removal of the derivatizing ligand under specific experimental conditions. These findings represent a significant milestone in the study of chirality in curved aromatics.
Introduction
Inspired by the homochiral nature of life, the study of chirality is a major focus in chemical research.1 While its critical role in biomedical applications is well established, this symmetry property has only recently emerged as a design principle for advanced functional materials in organic optoelectronics.2 In this context, the implementation of chiral elements endows molecular systems with unique properties such as chiroptical responses3–5 and chiral-induced spin selectivity.6,7 In addition, chirality-dependent organization allows for unprecedented control over conductivity and exciton diffusion by simply tuning the optical purity of optoelectronic materials.2,4,8–12Conjugated molecules – systems of choice for technological applications due to their mechanical, optical and electronic properties – pose a challenge in achieving inherently chiral structures.2,4,13,14 Introducing curvature into π-systems is an elegant strategy to expand the chemical space for chiral photo- and electroactive materials.15–19 Among the few known examples of bowl-shaped aromatics, subphthalocyanines (SubPcs) stand out for their superb optoelectronic properties and synthetic versatility.20,21 Their 14π-electron aromatic core and non-planar geometry – conferred by the tetrahedral coordination of the central boron atom – result in robust light-harvesting features, intense fluorescence emission, and excellent charge transport properties, the latter resulting from a permanent dipole moment and the ability to organize into ordered columnar assemblies.12,21–26 By virtue of these unique features, SubPcs have emerged as high-performing molecular materials in many fields of application, ranging from photovoltaics to emission technologies.21,24 Moreover, the cone-shaped geometry of these contracted porphyrinoids renders them intrinsically non-centrosymmetric. Thus, inherently chiral SubPcs are obtained from cyclotrimerization of non-C2v phthalonitrile precursors (e.g., meta- or ortho-monosubstituted dicyanobenzenes) as racemic mixtures of enantiomers (namely, P and M for the C3 regioisomer, Fig. 1).27
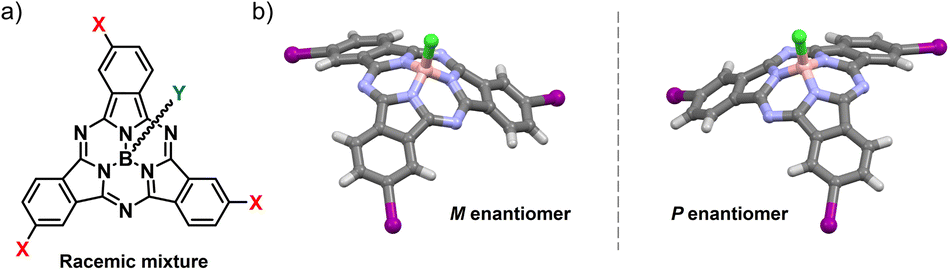 | ||
| Fig. 1 (a) Molecular structure of a racemic meta-substituted, C3-symmetric SubPc. (b) M and P enantiomers of meta-substituted, C3-symmetric I3SubPc-Cl. | ||
Due to their unique structural, optical and electronic properties, optically pure SubPcs are highly attractive value-added materials for advanced applications.12,28 Since the first report on the analytical resolution of racemic SubPcs by chiral high-performance liquid chromatography (HPLC),29 most efforts to obtain enantiopure SubPcs have focused on optimizing the scalability of the HPLC methodology.12,21,30,31 However, the intrinsic limitations of the HPLC technique (low throughput, cost, time consumption due to the need for multiple runs and method development), together with specific solubility issues of the SubPc framework, hamper scalability and feasibility, thus hindering the exploitation of curved SubPc aromatics as chiral materials. On the other hand, the exploration of alternative procedures (namely, optical resolution with chiral auxiliaries or de novo asymmetric synthesis32,33) for the preparation of enantiopure SubPcs by common organic laboratory techniques remains a pending task. Indeed, the former approach has received little attention in the literature in the frame of chiral buckybowls and contracted porphyrinoid aromatics.
Here, we report the efficient optical resolution of inherently chiral C3-symmetric SubPcs via axial derivatization with BINOL-based chiral auxiliaries. Moreover, unprecedented evidence for bowl-to-bowl inversion of the SubPc framework is serendipitously observed upon removal of the derivatizing ligand under specific conditions. Besides their interest as functional materials, enantiopure SubPcs thus also emerge as a precious tool for the in-depth study of SubPc reactivity, which mechanistic aspects have only rarely been addressed.21,34–36
Results and discussion
Chiral SubPc platforms and derivatization strategy
C 3-symmetric, meta- and ortho-substituted I3SubPc-Cl m-1 and o-1 were selected as model chiral SubPc derivatives. The halogen atom at the axial position of the macrocycle is known to play a key role in imparting directional columnar packing of the SubPc framework, which is crucial for charge- and exciton transport applications.12,22,23 In addition, I3SubPc-Cl precursors serve as ideal platforms for the preparation of targeted SubPc-based systems through post-functionalization of the axial and peripheral positions via straightforward methodologies, making their optical resolution of strategic importance.21 On the other hand, axially chiral BINOL derivatives were exploited as resolving agents.37,38 Due to the high configurational stability of the enantiopure atropoisomers, binaphthyls are compounds of choice in asymmetric synthesis and catalysis.39,40 As a derivatization strategy, axial ligand exchange was preferred over peripheral functionalization, as both the replacement of the apical halogen and the subsequent removal of the resolving agent can be achieved via simple chemical protocols.21,41–43SubPcs m-1 and o-1 were prepared as racemic mixtures by cyclotrimerization of 4-iodophthalonitrile and 3-iodophthalonitrile, respectively, in the presence of BCl3 (see ESI, Section 4† for the analysis of inherent chirality of C3-symmetric SubPcs).12,44
Optical resolution of meta-substituted SubPc
The optical resolution of m-1 was first attempted by replacing the chlorine ligand with (R)-BINOL as a chiral auxiliary after prior activation of the SubPc axial position with AlCl3 (Scheme S1†).42 This approach allows for milder reaction conditions compared to one-step axial substitution reactions, thus preserving the optical purity of the resolving agent.45 The formation of I3SubPc-BINOL m-2 (46% yield) as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers was evidenced by the presence of two distinct signal sets in the corresponding 1H NMR spectrum and their integral ratio (Fig. S1†).46 However, separation of the diastereomers by column chromatography on silica gel proved infeasible (further details in ESI, Section 2†).
:1 mixture of diastereomers was evidenced by the presence of two distinct signal sets in the corresponding 1H NMR spectrum and their integral ratio (Fig. S1†).46 However, separation of the diastereomers by column chromatography on silica gel proved infeasible (further details in ESI, Section 2†).
On the other hand, derivatization of m-1 with (R)-(+)-3,3′-dibromo-1,1′-bi-2-naphthol as chiral auxiliary afforded m-3 (62% yield, Fig. 2a and Scheme S2†), which showed two distinct spots on thin layer chromatography (Rf = 0.36 and 0.31 in dichloromethane/heptane 3:1).47 As observed for m-2, the 1H NMR spectrum of m-3 confirmed the formation of a 1:1 mixture of diastereomers (Fig. S7†). Accordingly, the HPLC chromatogram of the diastereomeric mixture revealed two distinct peaks with similar relative areas (Fig. S41†). The two diastereomers can be distinguished within the asymmetric unit in the single crystal structure of m-3, as evidenced by X-ray diffraction analysis (Fig. 2b and Tables S3 and S4†).48 Separation of the two diastereomers was achieved by column chromatography on silica gel yielding m-3a (first eluted diastereomer) and m-3b (second eluted diastereomer) in 28% and 27% yield, respectively (Fig. S6†).49 While m-3a was obtained as 97.2% pure, the second collected fraction (enriched in m-3b) appeared to contain 10.6% m-3a, as determined by HPLC (Fig. S42 and S43†). Further purification of the second eluted fraction through a second chromatographic column yielded m-3b as a pure fraction (Fig. S44†). The NMR spectra of the isolated m-3a and m-3b diastereomers are shown in Fig. 3 and S8–S18.†
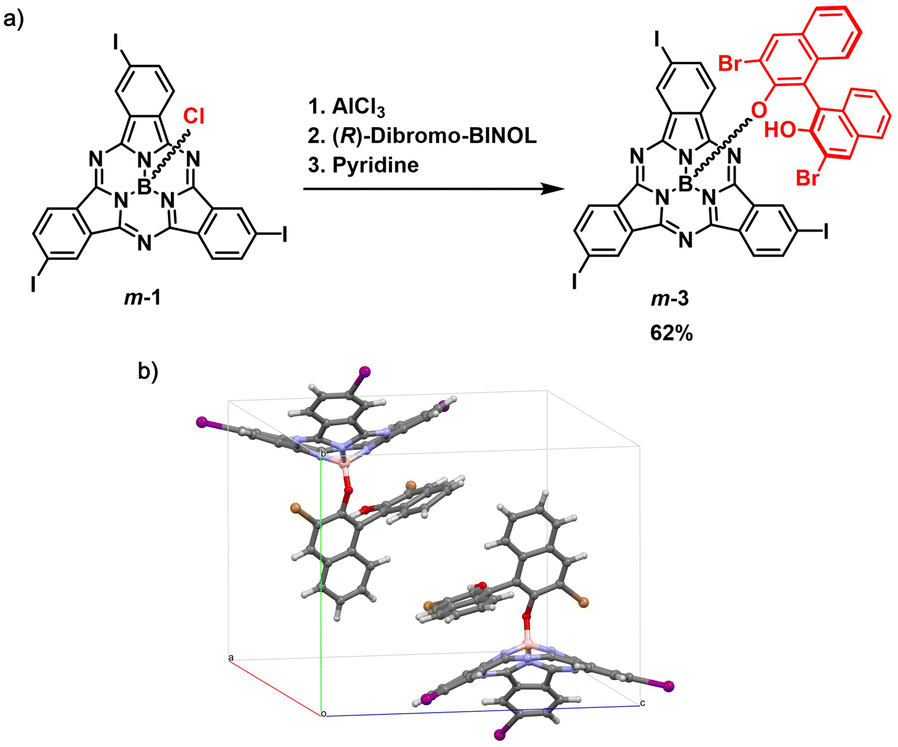 | ||
| Fig. 2 (a) Synthetic scheme for the preparation of m-3 as a mixture of diastereomers from racemic m-1via activation of the axial position with AlCl3. (b) Asymmetric unit of the crystal structure of m-3 showing the presence of two diastereomeric species. Toluene molecule of crystallization has been omitted for clarity. | ||
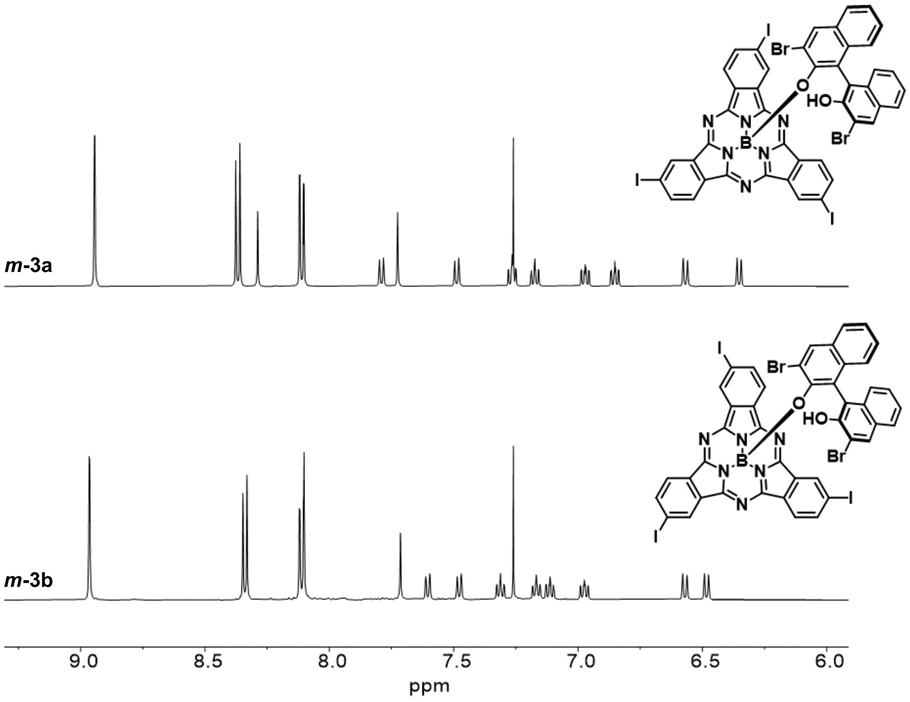 | ||
| Fig. 3 1H NMR spectra (500 MHz, chloroform-d1) of m-3a and m-3b diastereomers. | ||
At this point, the resolving agent was removed by treating the separated m-3a and m-3b species with an excess of BCl3 (10 equiv) at 50 °C in toluene (Fig. 4a and Schemes S5 and S6†). Under these conditions, m-1P and m-1M were obtained from m-3a and m-3b, respectively, in excellent yield (namely 96%) and high enantiomeric excess (e.e. 95.2% and 82.0%, Fig. 4b, S45 and S46†). The slightly lower purity of m-1M is consistent with that of its diastereomeric precursor m-3b, obtained as the second eluted fraction from the first column chromatography (vide supra). Comparison of the retention time of the I3SubPc-Cl enantiomers obtained by removal of the chiral auxiliary from m-3a and m-3b (Fig. 4b) with those of the m-1 enantiomers resolved by chiral HPLC,12 allows assignment of the absolute configuration of the separated species. The enantiomeric relationship between the m-1P and m-1M fractions obtained by axial ligand exchange from m-3a and m-3b was further confirmed by CD spectroscopy (Fig. 4c). From a qualitative comparison of the CD profiles of the isolated diastereomers (m-3a and m-3b) and the resulting enantiomers (m-1P and m-1M), and taking into account the similar purity of the diastereomeric precursors and enantiomeric products, we infer that the absolute configuration is retained upon removal of the chiral auxiliary under the conditions investigated.50 Thus, the exchange of the axial binaphthoxy ligand is likely to occur by interaction of the boron halide with the central boron atom of the SubPc framework from the convex face of the cone-shaped macrocycle. This is consistent with the bimolecular σ-bond metathesis mechanism postulated for axial ligand exchange between SubPc-Cl derivatives and phenols.35
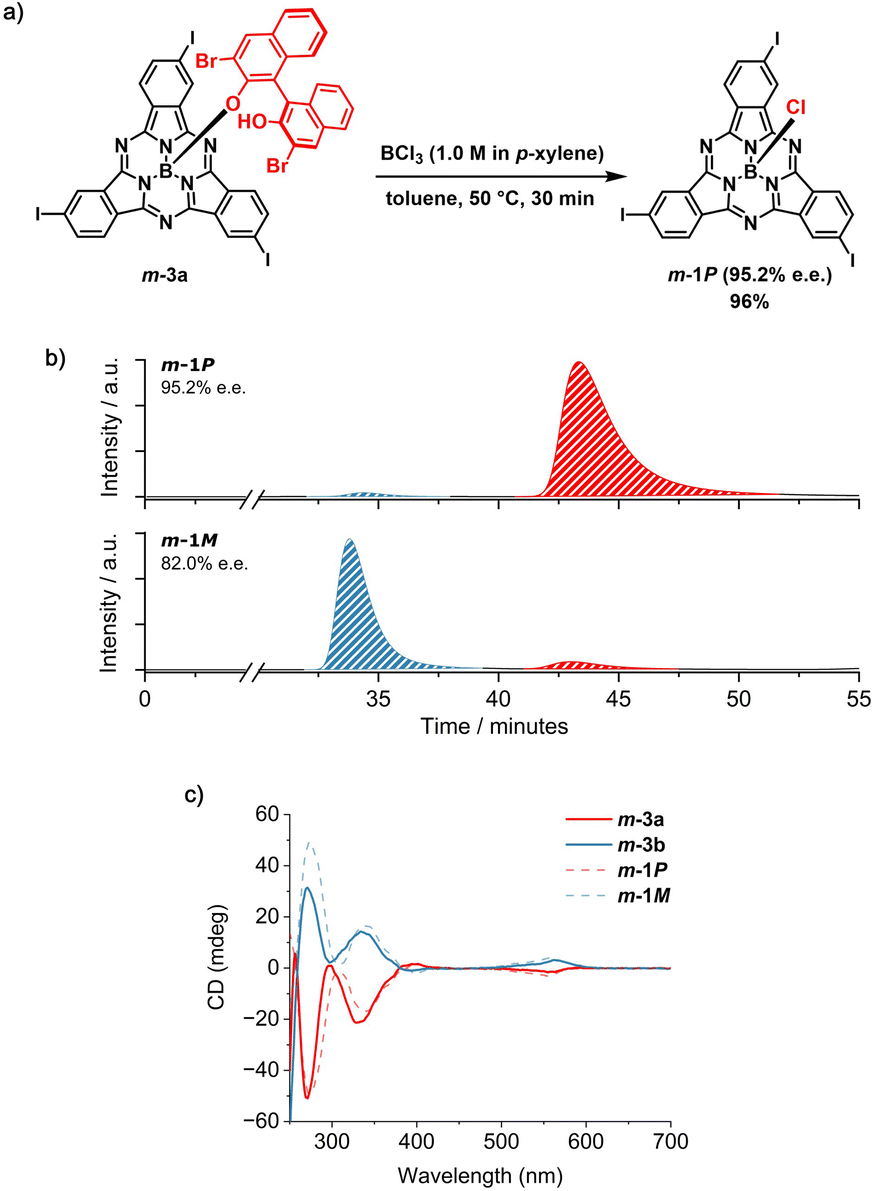 | ||
| Fig. 4 (a) Synthetic scheme for the removal of the BINOL-based chiral auxiliary from m-3a in the presence of BCl3 to afford m-1P (95.2% e.e.). (b) HPLC chromatograms of the products obtained from the removal of the chiral auxiliary from m-3a (top) and m-3b (bottom). The blue trace indicates the peak corresponding to the M enantiomer, whereas the red trace indicates the peak corresponding to the P enantiomer. Eluting solvent: toluene/n-hexane 50:50 (v/v); flow rate = 1.2 mL min−1; temperature = 10 °C; detection wavelength = 570 nm). (c) CD spectra of m-3a (red spectrum, full line), m-3b (blue spectrum, full line), m-1P (red spectrum, dotted line) and m-1M (blue spectrum, dotted line) in CHCl3 (2.0 × 10−5 M). | ||
Optical resolution of ortho-substituted SubPc
Different results were obtained for the ortho-substituted SubPc analog o-1. In this case, axial derivatization of the racemic I3SubPc-Cl precursor with (R)-BINOL via AlCl3 activation led to the formation of o-2 (52% overall yield) as a mixture of diastereomers (o-2a and o-2b) which were readily separated by column chromatography on silica gel (Rfo-2a = 0.38 and Rfo-2b = 0.30 in toluene) in 24% yield each (Scheme S3 and Fig. S23–S32†).51 At this point, removal of the chiral auxiliary from the isolated diastereomers was expected to yield the enantiomerically pure P and M I3SubPc-Cl derivatives, as observed for meta-substituted m-3. However, treatment of o-2b with 20 equiv. BCl3 at 70 °C for 1 hour afforded o-1b with an e.e. of 68.2% (Scheme S7 and Fig. S47†). This low e.e., compared to the purity of the diastereomeric precursor,52 indicates that a partial bowl-to-bowl inversion of the macrocycle occurs concomitantly with the axial nucleophilic substitution under the conditions investigated.53,54 These findings provide the first experimental evidence for bowl-to-bowl inversion of SubPc derivatives in solution.55–57At this point, the configurational stability of the ortho-substituted derivatives was tested in the presence of BF3·Et2O as a Lewis acid. Interestingly, by removing the axial BINOL ligand with an excess of BF3·Et2O (25 equiv) in refluxing toluene, o-3a and o-3b were obtained in 90% yield and e.e. of 98.0% and 93.4%, respectively (Schemes S8, S9, Fig. 5a, b, S33–S38, S51 and S52†).58,59 As observed for m-1P and m-1M, the CD spectra of o-3a and o-3b are mirror images, with the signs of the bands remaining unchanged when diastereomeric (i.e., o-2a and o-2b) and resulting enantiomeric fractions (o-3a and o-3b) are compared (Fig. 5c, S54 and S55†). This suggests that the axial nucleophilic substitution for the ortho-substituted o-2 in the presence of BF3·Et2O under the investigated conditions proceeds with retention of the configuration.
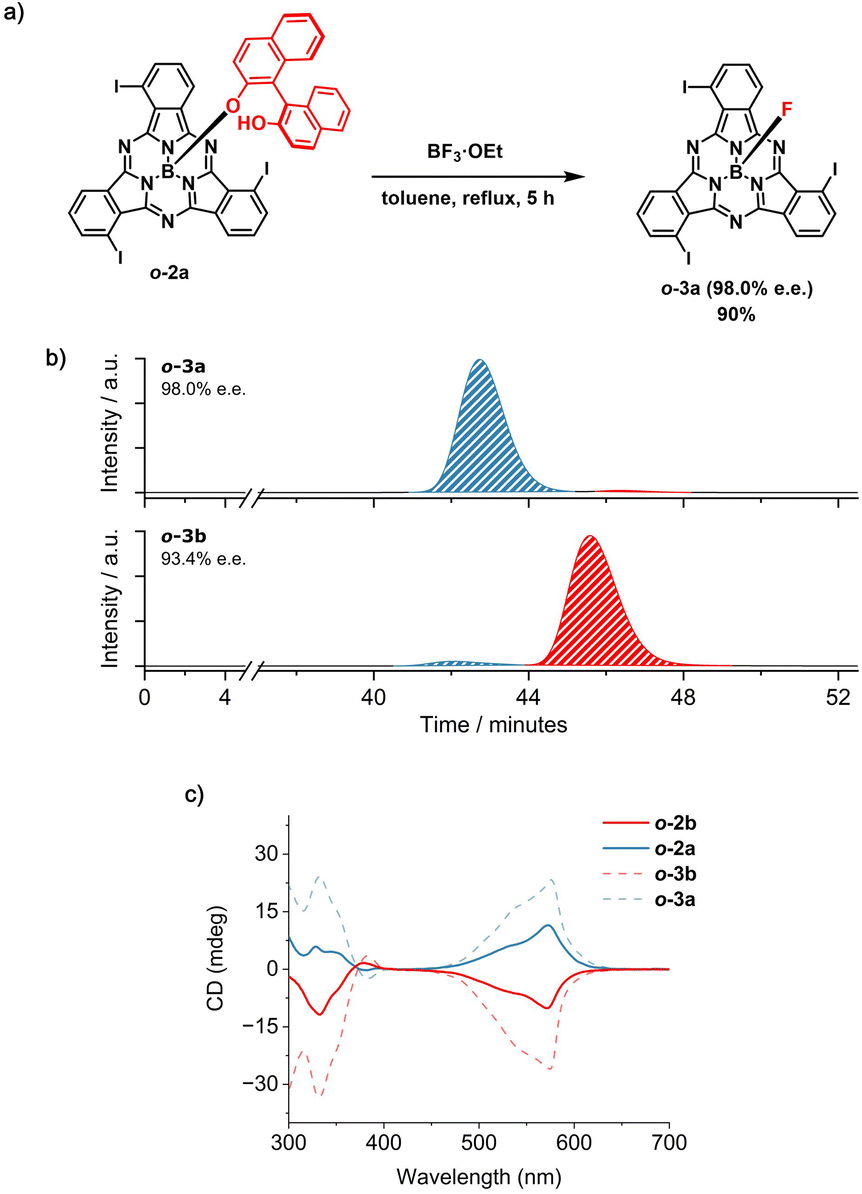 | ||
| Fig. 5 (a) Synthetic scheme for the removal of the BINOL chiral auxiliary from o-2a in the presence of BF3·Et2O to afford o-3a (98.0% e.e.). The absolute configuration of the SubPc macrocycle in the starting material and in the final product is not known.59 For visualization purposes, the M configuration has been depicted. (b) HPLC chromatograms of the products obtained from the removal of the chiral auxiliary from o-2a (top) and o-2b (bottom) in the presence of BF3·Et2O. Eluting solvent: toluene/n-hexane 80:20 (v/v); flow rate = 0.6 mL min−1; temperature = 20 °C; detection wavelength = 580 nm. (c) CD spectra of o-2a (blue spectrum, full line), o-2b (red spectrum, full line), o-3a (blue spectrum, dotted line) and o-3b (red spectrum, dotted line) in CHCl3 (5.0 × 10−5 M). | ||
Conclusions
In conclusion, we report herein the unprecedent optical resolution of inherently chiral bowl-shaped SubPc aromatics by derivatization with chiral auxiliaries, along with the first experimental evidence of bowl-to-bowl inversion of the SubPc macrocycle in solution. The chiral resolution of triiodo-substituted, C3-symmetric SubPc derivatives was successfully achieved by functionalization with enantiopure BINOL-based ligands upon activation of the apical position of the macrocycle, allowing for a facile separation of the resulting diastereomers by column chromatography on silica gel. Subsequent removal of the chiral auxiliary from the isolated diastereomeric fractions in the presence of boron halides afford optically active I3SubPc-X (X = Cl or F) in high yield (96%) and purity (97.7% and 99.0% for m-1M and o-3a, respectively). This approach, which has never been exploited for the optical resolution of contracted porphyrinoids, constitutes a feasible and efficient method for the preparation of enantiopure SubPcs using simple organic chemistry protocols. Remarkably, experimental evidence of bowl inversion of the SubPc framework was serendipitously observed in the axial ligand exchange on the ortho-substituted derivative in the presence of BCl3 as a Lewis acid. These path-breaking results open the door for in-depth mechanistic studies of SubPc reactivity and pave the way for the exploitation of enantiopure SubPcs as functional materials.Data availability
All experimental procedures, and compound characterization data are provided in the ESI.† Additional datasets and materials related to this study are available from the corresponding authors upon reasonable request.Author contributions
M. V. M.-D. and T. T. designed and supervised the research. G. L. carried out the experiments related to the meta-substituted SubPcs. L. T. performed the experimental work related to the ortho-substituted SubPcs. G. L. wrote the manuscript. M. V. M.-D. and T. T. corrected it. Overall, G. L. and L. T. contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Spanish MCIN/AEI/10.13039/501100011033 and European Union NextGenerationEU/PRTR (PID2020-116490GB-I00, TED2021-131255B-C43), MCIU/AEI/10.13039/501100011033/FEDER, UE (PID2023-151167NB-I00), the Comunidad de Madrid and the Spanish State through the Recovery, Transformation and Resilience Plan [“Materiales Disruptivos Bidimensionales (2D)” (MAD2D-CM) (UAM1)-MRR Materiales Avanzados], and the European Union through the Next Generation EU funds. IMDEA Nanociencia acknowledges support from the “Severo Ochoa” Programme for Centres of Excellence in R&D (MINECO, Grant SEV2016-0686). T. T. also acknowledges the Alexander von Humboldt Foundation (Germany) for the A. v. Humboldt – J. C. Mutis Research Award 2023 (ref. 3.3 – 1231125 – ESP-GSA). The authors acknowledge the DRX Monocrystal Laboratory of SIdI (UAM) for the crystallographic analysis.Notes and references
- J. E. Hein and D. G. Blackmond, Acc. Chem. Res., 2012, 45, 2045–2054 CrossRef PubMed.
- J. R. Brandt, F. Salerno and M. J. Fuchter, Nat. Rev. Chem., 2017, 1, 1–12 CrossRef.
- J. Han, S. Guo, H. Lu, S. Liu, Q. Zhao and W. Huang, Adv. Optical Mater., 2018, 6, 1800538 CrossRef.
- G. Albano, G. Pescitelli and L. Di Bari, Chem. Rev., 2020, 120, 10145–10243 CrossRef PubMed.
- D.-W. Zhang, M. Li and C.-F. Chen, Chem. Soc. Rev., 2020, 49, 1331–1343 RSC.
- R. Naaman, Y. Paltiel and D. H. Waldeck, Nat. Rev. Chem., 2019, 3, 250–260 CrossRef CAS.
- Y.-H. Kim, Y. Zhai, H. Lu, X. Pan, C. Xiao, E. A. Gaulding, S. P. Harvey, J. J. Berry, Z. Valy Vardeny, J. M. Luther and M. C. Beard, Science, 2021, 371, 1129–1133 CrossRef CAS PubMed.
- T. Hatakeyama, S. Hashimoto, T. Oba and M. Nakamura, J. Am. Chem. Soc., 2012, 134, 19600–19603 CrossRef CAS PubMed.
- F. Pop, P. Auban-Senzier, A. Frackowiak, K. Ptaszynski, I. Olejniczak, J. D. Wallis, E. Canadell and N. Avarvari, J. Am. Chem. Soc., 2013, 135, 17176–17186 CrossRef CAS.
- A. T. Haedler, S. C. J. Meskers, R. H. Zha, M. Kivala, H.-W. Schmidt and E. W. Meijer, J. Am. Chem. Soc., 2016, 138, 10539–10545 CrossRef CAS.
- G. Long, R. Sabatini, A. Rasmita, M. I. Saidaminov, X. Liu, E. H. Sargent, G. Lakhwani and W. Gao, Nat. Rev. Mater., 2020, 5, 423–439 CrossRef.
- J. Labella, G. Lavarda, L. Hernández-López, F. Aguilar-Galindo, S. Díaz-Tendero, J. Lobo-Checa and T. Torres, J. Am. Chem. Soc., 2022, 144, 16579–16587 CrossRef CAS.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed.
- J. Li and K. Pu, Chem. Soc. Rev., 2019, 48, 38–71 RSC.
- Y.-T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843–4867 CrossRef CAS PubMed.
- A. Szumna, Chem. Soc. Rev., 2010, 39, 4274–4285 RSC.
- Y. Shen and C.-F. Chen, Chem. Rev., 2012, 112(3), 1463–1535 CrossRef CAS.
- M. Ball, Y. Zhong, Y. Wu, C. Schenck, F. Ng, M. Steigerwald, S. Xiao and C. Nuckolls, Acc. Chem. Res., 2015, 48, 267–276 CrossRef PubMed.
- M. Rickhaus, M. Mayor and M. Juríček, Chem. Soc. Rev., 2017, 46, 1643–1660 RSC.
- C. G. Claessens, D. González-Rodríguez, M. S. Rodríguez-Morgade, A. Medina and T. Torres, Chem. Rev., 2014, 114, 2192–2277 CrossRef PubMed.
- G. Lavarda, J. Labella, M. V. Martínez-Díaz, M. Salomé Rodríguez-Morgade, A. Osuka and T. Torres, Chem. Soc. Rev., 2022, 51, 9482–9619 RSC.
- J. Guilleme, M. J. Mayoral, J. Calbo, J. Aragó, P. M. Viruela, E. Ortí, D. González-Rodríguez and T. Torres, Angew. Chem., Int. Ed., 2015, 54, 2543–2547 CrossRef.
- J. Guilleme, E. Cavero, T. Sierra, J. Ortega, C. L. Folcia, J. Etxebarria, T. Torres and D. González-Rodríguez, Adv. Mater., 2015, 27, 4280–4284 CrossRef PubMed.
- G. Lavarda, J. Zirzlmeier, M. Gruber, P. R. Rami, R. R. Tykwinski, T. Torres and D. M. Guldi, Angew. Chem., Int. Ed., 2018, 57, 16291–16295 CrossRef PubMed.
- C. Zhang, Y. Guo, D. He, J. Komiya, G. Watanabe, T. Ogaki, C. Wang, A. Nihonyanagi, H. Inuzuka, H. Gong, Y. Yi, K. Takimiya, D. Hashizume and D. Miyajima, Angew. Chem., Int. Ed., 2021, 60, 3261–3267 CrossRef.
- L. Labella and T. Torres, Trends Chem., 2023, 5, 353–366 CrossRef.
- The absolute configuration of inherently chiral SubPcs can be assigned using the P/M stereodescriptors following the rules commonly employed to designate the chirality of buckybowl compounds (further details in ESI,† section 4), see also: K. A. Winterfeld, G. Lavarda, J. Guilleme, D. M. Guldi, T. Torres and G. Bottari, Chem. Sci., 2019, 10, 10997–11005 RSC.
- J. Labella, D. Kumar Bhowmick, A. Kumar, R. Naaman and T. Torres, Chem. Sci., 2023, 14, 4273–4277 RSC.
- C. G. Claessens and T. Torres, Tetrahedron Lett., 2000, 41, 6361–6365 CrossRef.
- N. Kobayashi and T. Nonomura, Tetrahedron Lett., 2002, 43, 4253–4255 CrossRef.
- The best literature HPLC conditions allow to achieve a separation rate of less than 10 mg/24 h (ref. 12)..
- S. Higashibayashi and H. Sakurai, J. Am. Chem. Soc., 2008, 130, 8592–8593 CrossRef PubMed.
- Q. Tan, S. Higashibayashi, S. Karanjit and H. Sakurai, Nat. Commun., 2012, 3, 891 CrossRef.
- J. Guilleme, L. Martínez-Fernández, I. Corral, M. Yanez, D. González-Rodríguez and T. Torres, Org. Lett., 2015, 17, 4722–4725 CrossRef PubMed.
- J. Guilleme, L. Martínez-Fernández, D. González-Rodríguez, I. Corral, M. Yanez and T. Torres, J. Am. Chem. Soc., 2014, 136, 14289–14298 CrossRef.
- L. Tejerina, J. Labella, L. Martínez-Fernández, I. Corral, M. V. Martínez-Díaz and T. Torres, Chem.–Eur. J., 2021, 27, 12058–12062 CrossRef.
- Optical resolution via auxiliaries with point chirality (namely L-menthol and Boc-L-tyrosine methyl ester) was also attempted, but the separation of the resulting diastereomers by column chromatography turned out to not be feasible..
- (a) Enantiopure BINOL has previously been used as axial bridging unit or peripheral substituent for the preparation of optically active SubPc and SubPc dimers, respectively, exhibiting induced CD in the SubPc absorption region. See T. Fukuda, M. M. Olmstead, W. S. Durfee and N. Kobayashi, Chem. Commun., 2003, 1256–1257 RSC; (b) L. Zhao, K. Wang, T. Furuyama, J. Jiang and N. Kobayashi, Chem. Commun., 2014, 50, 7663–7665 RSC.
- J. M. Brunel, Chem. Rev., 2005, 105, 857–898 CrossRef.
- S. Kirsch, Angew. Chem., Int. Ed., 2009, 48, 2450–2451 CrossRef.
- J. Guilleme, D. González-Rodríguez and T. Torres, Angew. Chem., Int. Ed., 2011, 50, 3506–3509 CrossRef PubMed.
- G. E. Morse and T. P. Bender, Inorg. Chem., 2012, 51, 6460–6467 CrossRef PubMed.
- H. Gotfredsen, M. Jevric, S. L. Broman, A. U. Petersen and M. B. Nielsen, J. Org. Chem., 2016, 81, 1–5 CrossRef.
- C. G. Claessens, D. González-Rodríguez, B. del Rey, T. Torres, G. Mark, H. P. Schuchmann, C. von Sonntag, J. G. MacDonald and R. S. Nohr, Eur. J. Org Chem., 2003, 2547–2551 CrossRef.
- When the axial ligand exchange reaction is carried out by heating the mixture of the I3SubPc-Cl and the phenol in a high boiling solvent (namely chlorobenzene) in the presence of a base, racemization of the chiral derivatizing agent is observed (further details in ESI,† Sections 2 and 5.1)..
- A similar yield (namely 44%) was obtained by axial substitution reaction via generation of a SubPc-OTf intermediate through reaction of m-1 with AgOTf, following the procedure reported in ref. 41..
- (R)-3,3′-Di-9-phenanthrenyl-1,1′-bi-2-naphthol and (S)-vanol were also tested as chiral auxiliaries, but the separation of the resulting diastereomers by column chromatography proved to be not feasible..
- Deposition numbers CCDC 2376453 and 2376454 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service..
- The yield of m-3b is calculated after the second column chromatography..
- Unfortunately, single crystals of the separated diastereomers m-3a and m-3b with sizes suitable for X-ray diffraction analysis could not be obtained..
- The α-substitution pattern of the SubPc macrocycle is known to lead to improved chromatographic separations of C3 and C1 structural isomers with respect to the β-substitution pattern: C. G. Claessens and T. Torres, Eur. J. Org Chem., 2000, 1603–1607 CrossRef.
- The purity of the separated o-2a and o-2b diastereomers was confirmed by NMR (Fig S23-S29†)..
- To further prove that racemization of the inherently chiral SubPc scaffold is operative in the presence of BCl3, the I3SubPc-Cl o-1b fraction obtained by removal of the derivatizing agent from o-2b (with 68.2% e.e.) was further treated with BCl3 (20 equiv.) at 70°C. A substantial decrease in e.e. to 50.0% and 36.1% was observed after 1 and 2 h, respectively (Fig. S48 and S49†)..
- A significant impact of peripheral substituents on inversion barriers has been previously observed for corannulene and sumanene buckybowls, see for example ref. 32 and T. Jon Seiders, K. K. Baldridge, G. H. Grube and J. S. Siegel, J. Am. Chem. Soc., 2001, 123, 517–525 CrossRef.
- K. Yoshida and A. Osuka, Chem.–Eur. J., 2015, 21, 11727–11734 CrossRef PubMed.
- T. Kato, F. S. Tham, P. D. W. Boyd and C. A. Reed, Heteroatom Chem., 2006, 17, 209 CrossRef CAS.
- Surface-catalyzed bowl-to-bowl inversion of SubPc-Cl on Au(111) has been recently reported by us (ref. 12). Based on quantum chemistry simulations, a reaction pathway involving Cl-cleavage in SubPc units adopting a Cl-down configuration had been proposed..
- Recrystallization from dichloromethane/methanol allows to enhance the e.e. of o-3a and o-3b from 92.6% and 84.0% to 98.0% and 93.4%, respectively..
- Differently from what reported for the meta-substituted derivatives, the absolute configuration of the ortho-substituted I3SubPc-Cl enantiomers obtained by removal of the chiral auxiliary from o-2a and o-2b cannot be assigned as the corresponding crystal structures could not be resolved..
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental methods, synthetic procedures and characterization data of new compounds, additional data from X-ray diffraction analysis, HPLC chromatograms, circular dichroism and UV-vis absorption spectra. See DOI: https://doi.org/10.1039/d4sc06241h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |