
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of the Pt/ionomer/water interface on the oxygen reduction reaction: insights into the micro-three-phase interface†
Shangkun
Jiang‡
 abc,
Qiong
Xiang‡
ab,
Zhuoyang
Xie
ab,
Na
Yang
d,
Jiawei
Liu
e,
Li
Li
*ab and
Zidong
Wei
*ab
abc,
Qiong
Xiang‡
ab,
Zhuoyang
Xie
ab,
Na
Yang
d,
Jiawei
Liu
e,
Li
Li
*ab and
Zidong
Wei
*ab
aState Key Laboratory of Advanced Chemical Power Sources (Chongqing University), China. E-mail: liliracial@cqu.edu.cn; zdwei@cqu.edu.cn
bSchool of Chemistry and Chemical Engineering, Chongqing University, Chongqing, 400044, China
cCollege of Chemistry and Chemical Engineering, Chongqing University of Science &Technology, Chongqing, 401331, China
dSchool of Materials and Energy, University of Electronic Science and Technology of China, Chengdu, 611731, China
eInstitute of Sustainability for Chemicals, Energy and Environment (ISCE2), Agency for Science, Technology and Research (A*STAR), 627833, Singapore
First published on 5th November 2024
Abstract
Understanding the Pt/ionomer/water interface structure and its impact on the oxygen reduction reaction (ORR) activity is essential for enhancing catalyst utilization and performance of fuel cells. This study aimed to investigate the influence of sulfonic acid groups on the Pt/ionomer/water interface and the ORR mechanism. By using a combination of DFT, AIMD, and microkinetic simulations, the results showed that when the sulfonic acid group is located at the edge of the Helmholtz plane, it creates an optimal three-phase interface, providing more available active sites, a stronger interfacial electric field, and a more continuous H-bond network. This configuration results in the *OOH dissociation becoming the rate-determining step, demonstrating significantly higher intrinsic ORR activity with a much lower theoretical overpotential of 0.11 V. Conversely, when the sulfonic acid group is in contact with the Pt surface, it causes the Pt surface's d-band center to shift down, weakens the interfacial electric field, and disrupts the H-bond network, resulting in a blocking effect on the ORR with an overpotential of 0.23 V. These insights shed light on the role of solid–solid–liquid interfaces in the ORR performance and provide valuable information for the rational design of catalyst interfaces.
1. Introduction
The commercialization of proton exchange membrane fuel cells (PEMFCs) heavily relies on the efficiency of the oxygen reduction reaction (ORR) at the cathode. This critical process is predominantly catalyzed by precious metals, particularly platinum (Pt)-based catalysts.1 Although significant advancements have been made in developing novel catalysts to enhance catalytic activity through alloying2–4 and nanotechnology,2,5–7 the challenge remains to reduce Pt usage in PEMFCs without affecting their power density and stability.8–11 One major hurdle is the gap between the evaluated catalytic activity of catalysts in the three-electrode test and their operating performance in the membrane electrode assembly (MEA).12,13 Catalysts that perform well under lab conditions often have much lower catalytic performance under working conditions. This is because the ORR occurs in the MEA at three-phase interfaces involving oxygen, water, catalyst, and Nafion ionomers. This differs from the catalyst/water interfaces in the three-electrode test.The sulfonic acid groups inherent in Nafion ionomers are vital in enhancing proton transfer at the three-phase interfaces within the MEA. However, during the ORR, Nafion's adsorption and subsequent coverage can impede the crucial transport and diffusion of oxygen across the catalyst surface. This obstruction ultimately diminishes the catalytic efficiency, leading to a decrease in both the mass activity and peak power density of the MEA.14–16 Wang et al.17 quantified both local and bulk resistances of O2 by employing limiting current measurements and linear extrapolation, respectively. Their findings indicate that local resistance of O2 dominates the overall resistance of O2 within the MEA. Furthermore, the sulfonic acid groups in Nafion may occupy the active sites present on the Pt catalyst surface, leading to catalyst poisoning and a decrease in catalytic activity.18 Kodama et al.19 demonstrated that the dehydration of the ionomers enhances the adsorption of sulfonic acid groups on the Pt(111) surface, severely impairing the ORR performance under operating conditions, especially in comparison to well-hydrated conditions. Additionally, strong adsorption of these groups impedes the mobility of Nafion's side chains, further increasing the mass transfer resistance for water.20,21
To mitigate the catalyst poisoning caused by sulfonic acid groups and enhance the oxygen transport rate, strategies have been devised, including solvent regulation,22 ionomer modification,15,23 and innovative support materials.24,25 Chen et al.22 effectively reduced the adsorption of sulfonic acid groups on Pt surfaces using cyclohexanol, creating a non-contact interface between Pt and Nafion. This method significantly decreased sulfonic acid group coverage on Pt, improving reaction kinetics and reducing mass transfer resistance. Zhang et al.26 employed steric hindrance through a porous covalent organic framework to lessen the ionomers' encapsulation of the Pt/C catalyst and minimize direct interactions between sulfonic acid groups and Pt, thus enhancing mass activity and peak power density by a factor of 1.6. Cheng et al.27 demonstrated that the introduction of nanopores onto/within the Nafion film significantly reduced the local oxygen transport resistance by 78%, from 0.37 s cm−1 to 0.08 s cm−1, ultimately enhancing the performance of fuel cells. Additionally, Yarlagadda et al.24 used ordered mesoporous carbon as a carrier to prevent Nafion coverage on Pt surfaces, effectively reducing mass transfer resistance for protons and oxygen. However, the specific mechanisms of how sulfonic acid groups interact with catalysts and their role in inducing catalyst poisoning, as well as their impact on the structure and performance of the three-phase interface, remain largely unexplored at present. Additionally, the presence of ionomers at the three-phase interfaces significantly impacts the electrical double layer28–33 and alters the electronic structure of the catalyst surface.34,35 This complicates the ionomer–catalyst interfaces, making it difficult to comprehend their interactions and catalytic processes. Therefore, more research is needed to better understand the ionomer–catalyst interactions, reveal its underlying block mechanism for the ORR, and then develop effective strategies for regulating ionomer distribution in the MEA.
This work combines density functional theory (DFT) and ab initio molecular dynamics (AIMD) simulations to investigate how the sulfonic acid groups of ionomer side chains influence the three-phase interfaces of Pt/ionomers/water. The focus is on their impact on the electric double-layer structure and ORR activities. The configurations of ionomer side chains at different distances from the Pt(111) surface are screened to determine their interaction. Then, the electronic structure of the Pt(111) surface, along with the distribution of water molecules and hydrogen bonds (H-bonds) within the Helmholtz plane(HP), is analyzed. In the end, the free energy of the elementary steps and the linear scanning curve (LSV) of the ORR are simulated to evaluate the activity of the Pt/ionomer/water interface. The study identifies an ideal Pt/ionomer/water interface structure with high intrinsic ORR activity, provides valuable insights into the underlying blocking effect of Pt–ionomer interaction on the ORR, and underscores the crucial role of the solid–solid–liquid interfaces in ORR performance.
2. Computational models and parameters
The vacuum model of Pt(111)–Nafion was constructed with a four-layer (4 × 4) periodic Pt(111) slab and Nafion ionomer representative fragment to replicate the catalyst surface, augmented by a 20 Å vacuum gap vertically. Among them, the Nafion side chain with hydrophilic sulfonic acid was selected, retaining the two carbon atoms on the main chain and their bonded fluorine atoms to ensure accuracy and structural integrity. Implicit solvation effects are simulated using the VASPsol method.36 A continuum of Pt–Nafion ionomer interaction structures is established by modulating the proximity between Nafion and the Pt surface, beginning from Nafion's equilibrium adsorption distance on Pt(111) and incrementally extending in the Z-direction. Throughout the structural optimization phase, the bottom two layers of Pt atoms, the top two carbon atoms in Nafion, and their bonded fluorine atoms remain fixed, permitting relaxation of the remaining atoms. In the explicit solvation model, water with a density of 1 g cm−3 fills the structure's vacuum space based on these thermodynamically derived structures.37,38 Protons are added to neutralize the negative charge of the sulfonic acid group in the Nafion ionomer to maintain the electric neutrality of the system.DFT was implemented in the Vienna Ab Initio Simulation Package (VASP) code.39 The projector-augmented wave (PAW) pseudopotential method was used to describe the electron–ion interactions,40 and the generalized gradient approximation (GGA) of the revised Perdew–Burke–Ernzerhof (RPBE) functional determined the electron exchange–correlation energy.41 The cutoff energy of the basis set of plane waves was 450 eV, and Monkhorst–Pack k-point meshes of (3 × 3 × 1) were selected for k-space integration. The calculation cannot be stopped until the system energy and force converge within 10−5 eV and 0.01 eV Å−1, respectively. AIMD is based on a canonical (NVT) ensemble, maintaining a constant temperature of 400 K by the Nosé–Hoover thermostat method, where the timestep was set to 1 fs. Initial dynamic pre-optimization over 2 ps ensures kinetic equilibrium, followed by a 10 ps steady-state simulation, from which 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 frames are statistically analyzed.
000 frames are statistically analyzed.
3. Results and discussion
3.1 The structure of the Pt/ionomer/water interface
To investigate the interaction between Pt(111) and the side chains of Nafion ionomers, a series of Pt/ionomer structures were created and optimized (Fig. S1†). In Fig. 1a, the energy of the system and the structure of the ionomer side chains change significantly as the distance between Pt(111) and the Nafion ionomer main chain increases. The distance between Pt(111) and the sulfonic acid group (DPt–S) initially remains constant, but then abruptly changes and continues to increase thereafter. Additionally, the length of the ionomer side chains exhibits a pattern of initial increase, followed by a slight decrease. The observed sudden change in the energy and structure is attributed to the transition of the sulfonic acid group on Pt(111) from adsorption to non-adsorption. The increase in the distance of the main chain led to the stretching of the ionomer side chains when the O atoms of the sulfonic acid group bonded with Pt. Conversely, the ionomer side chain contracted to its free length when bonding did not occur. Then, the Pt/ionomer system can be classified into contact and non-contact structures based on the interaction of the sulfonic acid group with the Pt(111).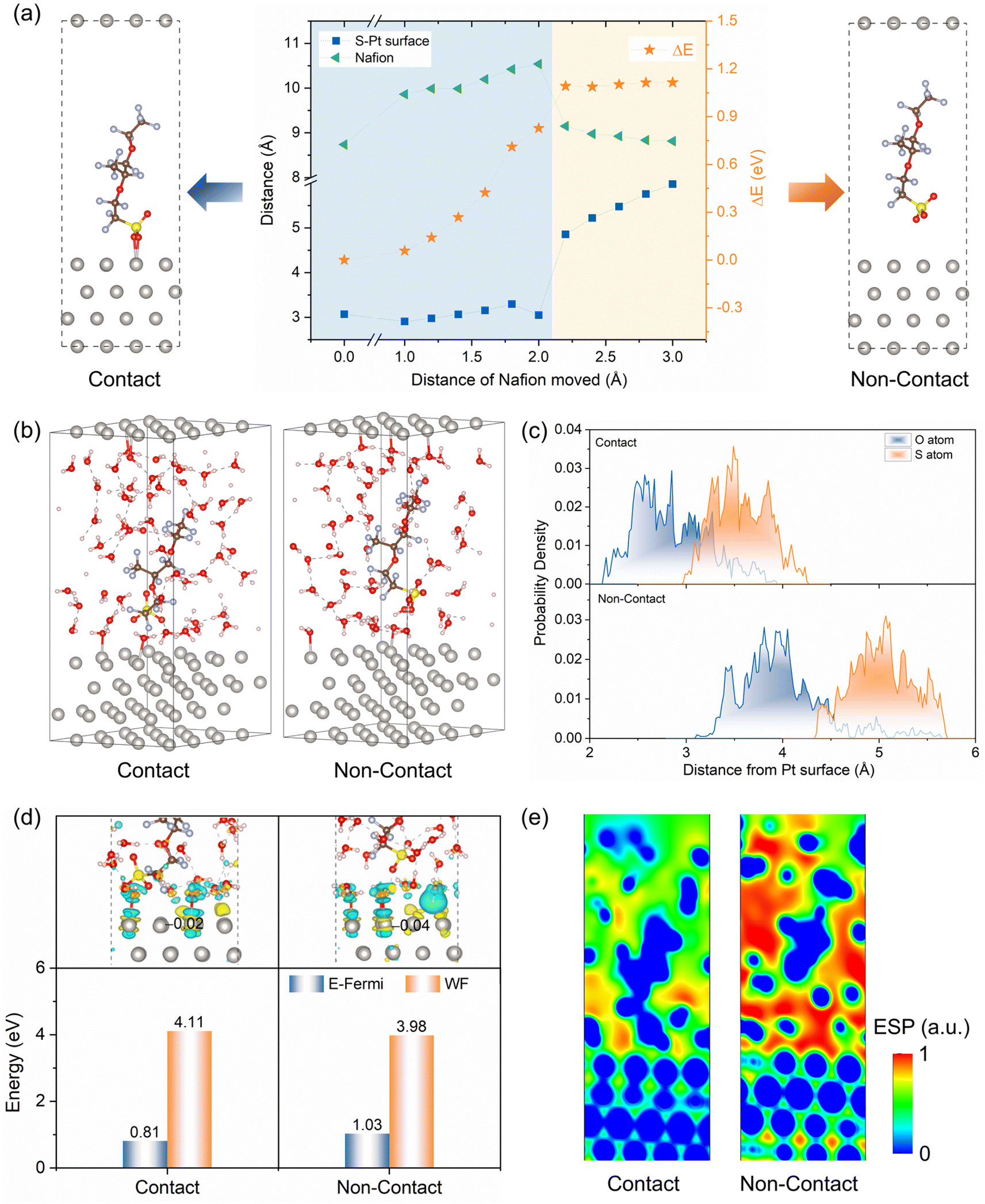 | ||
| Fig. 1 (a) Energy of the Pt–Nafion structure, the distance between the S–Pt surface, and the length of the Nafion ionomer as a function of the distance from the Nafion ionomer to the Pt surface; (b) Pt/ionomer/water systems in both contact and non-contact structures; (c) statistical distance between the sulfur (S) and oxygen (O) atoms of the sulfonic acid groups and the Pt surface; (d) difference in charge density (iso-surface levels are 0.0003 e Å−3; cyan-green indicates electron deficiency, while yellow indicates electron accumulation), average Bader charge on the Pt surface, Fermi energy (E-Fermi), and work function (WF); (e) distribution of electrostatic potential in the contact and non-contact structures. | ||
Subsequently, solvent water molecules are involved in the system to simulate the Pt/ionomer/water interfaces. After conducting AIMD steady-state simulation (Fig. S2†), shown in Fig. 1b, the water molecules tend to interact with Pt atoms and the sulfonic acid group, leading to a slight increase in the distance between Pt and the sulfonic acid group. In Fig. 1c, the statistical distance between Pt and the sulfonic acid group shows that in the contact structure, the DPt–S fluctuates between 3.0 Å and 4.2 Å and is mainly located at 3.6 Å, and the DPt–O varies between 1.8 Å and 3.8 Å, with peak location of 2.8 Å. They are much shorter than those in the non-contact structure; that is, the DPt–S and DPt–O are mainly located at 5.0 Å and 4.2 Å, respectively. This indicates that the sulfonic acid group in the contact structure still interacts more strongly with Pt than it does in the non-contact structure.
The charge transfer, E-Fermi, and WF further confirm the interaction mentioned above. In Fig. 1d, the interaction between Pt(111) and the sulfonic acid group in the contact structure leads to a transfer of charge from Pt(111) to the sulfonic acid group, resulting in more electron deficiency of surface Pt atoms, exhibiting less negative Bader charge (−0.02 e per atom) and a downshifted d-band center (Fig. S3(b)†). In the non-contact structure, only the charge transfer between Pt and water causes the surface Pt atoms to show a more negative Bader charge (−0.04 e per atom) with less electron deficiency and a higher d-band center. Correspondingly, the contact structure displays a lower E-Fermi and a larger WF due to the fewer redundant electrons on the surface Pt atoms compared to the non-contact structure. At the same time, as shown in Fig. 1e, the interaction of the Pt–sulfonic acid group reduces the difference in electrostatic potential between Pt and electrolyte, while the non-contact structure shows an increased electric field strength. In short, the electronic structures observed under vacuum and solvent conditions show a similar trend. Solvent models differ slightly due to explicit water, weakening Pt–sulfonic acid group interactions, and reducing charge transfer.
3.2 The electric double layer structure of the Pt/ionomer/water interface
The radial distribution function (RDF) shown in Fig. 2a shows the distance of water from the Pt surface within the Pt/ionomer/water interface. Two distinct peaks in the RDF correspond to the adsorbed water layer in the inner Helmholtz plane (IHP) and the interfacial water layer in the outer Helmholtz plane (OHP). By comparing contact and non-contact structures, we observe that the non-contact structure contains a higher amount of water within the IHP due to the higher Pt–O peak. The adsorbed water layer is slightly closer to the Pt surface in the non-contact structure, as indicated by a slightly smaller peak position. Fig. 2b displays the water number density, revealing the width of the IHP (2.0 Å to 2.6 Å) and OHP (2.6 Å to 4.6 Å) and confirming that the HP of the non-contact structure possesses more water.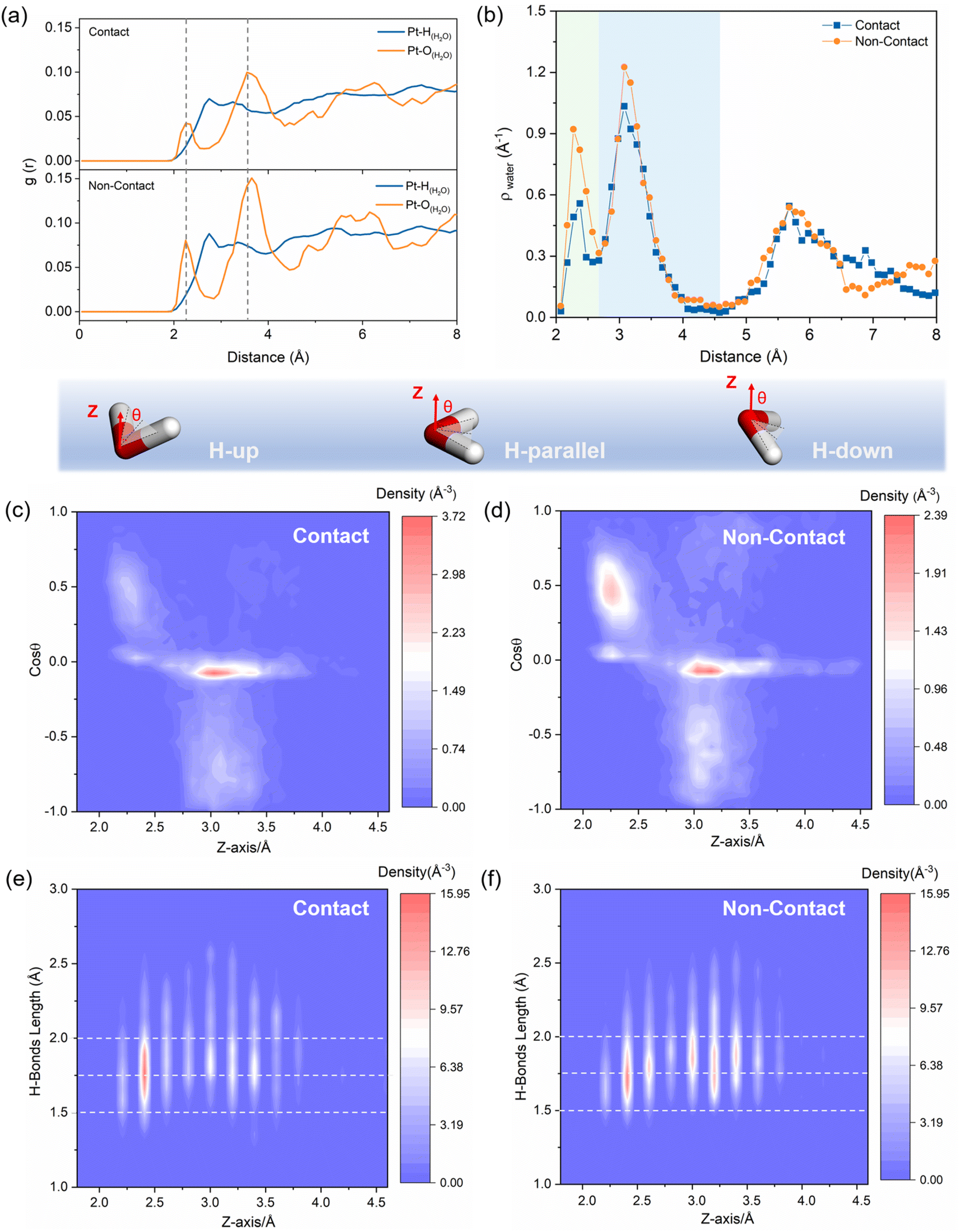 | ||
| Fig. 2 (a) RDF of Pt–H(H2O) and Pt–O(H2O); (b) distribution of water number density and its proportion across different ranges; the two-dimensional density profile of the cosθ of H2O molecules in the contact (c) and non-contact (d) structures; two-dimensional density distribution profiles of H-bond lengths in the contact (e) and non-contact (f) structures. | ||
These variations in the Pt/ionomer/water interface are primarily influenced by the positioning of the sulfonic acid groups. In Fig. S4,† the RDF peaks of the sulfonic acid group in the contact structure suggest that the group is positioned within the IHP, leading to a reduction in water density and weakened water adsorption. This can be attributed to the large charge transfer from the Pt to the sulfonic acid group in the contact structure, which downshifts the d-band center of Pt(111), weakens species adsorption, and reduces the density of the active sites (Fig. S5†). Conversely, in the non-contact structure, the sulfonic acid groups are predominantly situated on the edge of the OHP, exerting less influence on the water structure in the HP.
Furthermore, the orientation distributions of water in the HP are depicted in Fig. 2c and d. In the IHP, the adsorbed water molecules predominantly align with H-up (θ = 0° to 60°) and H-parallel (θ = 60° to 120°) orientations due to the interaction between Pt and water. On the other hand, the interfacial water molecules are predominantly characterized by H-parallel and H-down (θ = 120° to 180°) orientations. The concentration and orientation of water molecules differ between the contact and non-contact structures. In Fig. S6,† the contact structure contains fewer water molecules and a relatively concentrated H-parallel orientation. The non-contact structure has a larger number of water molecules with a more dispersed orientation, mainly concentrated in the H-up to the H-parallel orientations.
Additionally, 2D density distribution maps of the H-bond in Fig. 2e and f show that the H-bond of the contact structure is mainly concentrated in the IHP, while the non-contact structure has a higher number of H-bonds in both the IHP and the OHP, resulting in a wider distribution and continuous H-bond network. Specifically, the number of H-bonds in the HP of non-contact structures is approximately double that of contact structures, as shown in Fig. S7.† The length of the H-bond in the IHP of the non-contact structure is slightly more concentrated and shorter than that of the contact structure. This discrepancy is also attributed to the interaction of the sulfonic acid group in the contact structure with Pt and adsorbed water, which weakens the H-bond between them and disrupts the H-bond network.
3.3 The ORR activity of the Pt/ionomer/water interface
To investigate the influence of sulfonic acid groups on the ORR mechanism, we simulate the free energy changes of the elementary steps of the ORR, including oxygen diffusion, adsorption, activation, protonation, and desorption as shown in Fig. 3a. The energy change of elementary steps is closely related to the electronic structure and the electric double layer of the Pt/ionomer/water interface structure. | ||
| Fig. 3 In the contact and non-contact structures, the diagram of the ORR elementary step pathway (a) and the evolution of free energy and bond lengths for each elementary step: (b) O2 diffusion; (c) *OO + H+ + e− → *OOH; (d) *OOH + H+ + e− → *O + H2O; (e) *O + H+ + e− → *OH; (f) *OH + H+ + e− → H2O. | ||
Fig. 3b illustrates the change in free energy for the diffusion of O2 from the outside of the HP, at a distance of 5.0 Å, to the inside of the HP, at a distance of 2.5 Å. The corresponding snapshots of solvated structures are shown in Fig. S8 and S9.† The free energy first increases and then decreases as O2 moves closer to the Pt surface, indicating that overcoming a barrier is necessary for oxygen diffusion into the HP. The O–O bond length oscillates around the equilibrium bond length of 1.23 Å without activation. The energy barriers for O2 diffusion in the contact and non-contact structures are 0.18 eV and 0.15 eV, respectively. This suggests that there is a relatively small difference between these two structures in the diffusion resistance by the HP. However, the lower final state energy in the non-contact structure indicates a stronger interaction of its Pt surface with O2. In other words, the sulfonic acid group in the contact structure blocks the Pt surface from attracting and stabilizing reactants.
Due to the interaction between the Pt surface and O2, as shown in Fig. 3c, the adsorption of O2 is a spontaneous process with a slight decrease in the free energy. Subsequently, the protonation of *O2 to form *OOH requires overcoming the energy barrier accompanied by the elongation of the O–O bond from 1.23 to 1.40 Å. The formation of *OOH in the non-contact structure exhibits much lower energy barriers and a more stable final state compared to that in the contact structure. The snapshots of O2 protonation structures are shown in Fig. S10 and S11.† It can be attributed to the strong interaction between Pt and species and the well-connected H-bond network in the non-contact structure, which benefits proton transfer and stabilization of intermediates.
For the same reason, in the non-contact structure, the following ORR steps, including the dissociation of *OOH, the protonation of *O, and the desorption of *OH, have significantly lower energy barriers compared to the contact structure. For example, in Fig. 3d, the dissociation energy barrier of *OOH in the non-contact structure is 0.29 eV because the well-connected H-bond network benefits the proton transfer and the strengthened interfacial electric field improves the activation of the *O–OH bond, whereas, in the contact structure, it is as high as 0.60 eV. The snapshots of the dissociation of *OOH structures are shown in Fig. S12 and S13.† Additionally, in Fig. 3e and f, the protonation of *O and *OH and the following desorption in the non-contact structure (Fig. S15 and S17†) occur almost spontaneously with energy barriers of less than 0.05 eV, while in the contact structure (Fig. S14 and S16†), these steps require 0.31 and 0.15 eV, respectively.
Fig. 4a summarizes all energy barriers of the ORR elementary step in both contact and non-contact structures. Contrary to what thermodynamic calculations in vacuum suggest about the ORR's potential-determining step (PDS) being the desorption of OH,42 the rate-determining step (RDS) at the Pt/ionomer/water interface is the protonation and dissociation step of *OOH. This means that the presence of a solution can decrease the adsorption strength of species on the Pt surface and promote the protonation and desorption of OH during the ORR. In the non-contact structure, the energy barriers for each step are consistently lower than those in the contact structure. However, the steps involving adsorption and proton transfer in the contact structure have much higher reaction energy barriers. Utilizing the RDS energy barriers, exchange current densities are calculated and presented in Fig. 4b. The exchange current density for the non-contact structure is 2.87 × 10−5 A cm−2, approximately 100 times higher than that in the contact structure (3.21 × 10−7 A cm−2), underscoring the superior ORR activity of the non-contact structure. Considering the cathodic potential, species coverage, and active site density, the LSV curves were fitted by microkinetic simulations shown in Fig. 4c. The half-wave potential and onset potential for the contact structure are 0.93 V and 1.00 V (vs. RHE), respectively. The half-wave potential and onset potential for the non-contact structure are increased to 1.05 V and 1.12 V (vs. RHE), respectively. The much lower theoretical overpotential demonstrates that the non-contact structure is the ideal three-phase interface exhibiting much higher intrinsic ORR activity.
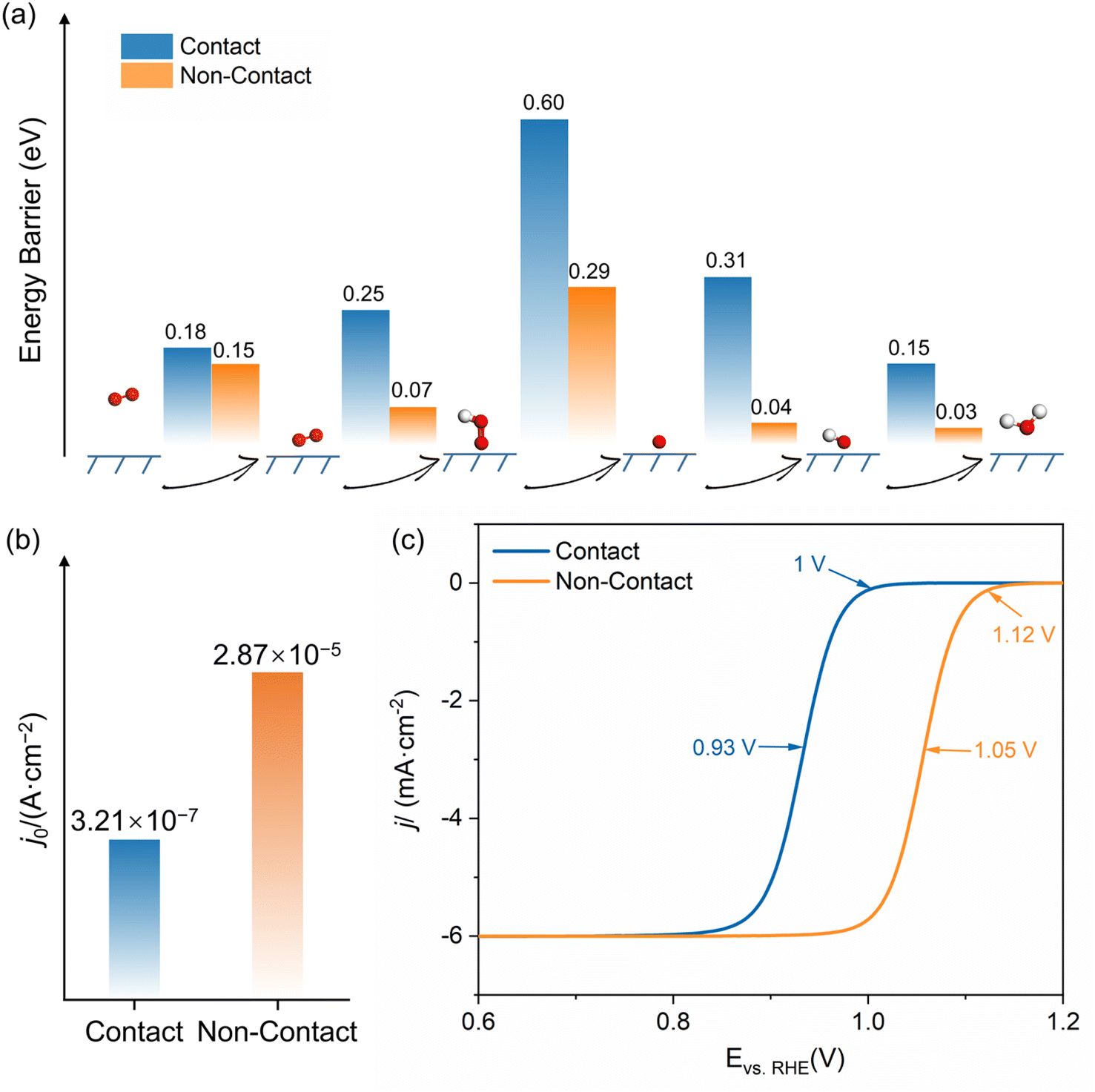 | ||
| Fig. 4 (a) Energy barriers for each step of the ORR; (b) exchange current density; (c) the simulated LSV curve for both contact and non-contact structures. | ||
Therefore, the ideal Pt/ionomer/water interface involves having the ionomer located at the edge of the OHP. This arrangement helps to reduce the poisoning of sulfonic acid groups on active sites while also strengthening the interfacial electric field and forming a more continuous H-bond network. These factors are conducive to oxygen adsorption, activation, and protonation. Specific strategies, including selecting suitable dispersants, adjusting the pore structure of carbon supports, and modifying the structure of sulfonic acid groups, among others, can regulate the distance between the sulfonic acid groups and the catalyst surface. If the ionomer is located within the IHP, it has a blocking effect on the ORR by decreasing the surface redundant electrons, lowering the d-band center, weakening the interfacial electric field, and disrupting the H-bond network of the HP. These changes hinder species adsorption and O–O bond activation, impede proton transfer, and ultimately reduce the intrinsic activity of the ORR.
4. Conclusion
In this research, we systematically investigated the impact of the positioning of sulfonic acid groups on the electronic structure, electric double-layer structures, H-bond networks, and the activity mechanism of the ORR at Pt/ionomer/water interfaces. The combination of DFT and AIMD simulations revealed that sulfonic acid groups within the IHP in contact with the Pt(111) surface hinder oxygen adsorption, activation, and protonation, resulting in a high energy barrier of 0.6 eV for the RDS of *OOH activation, an onset potential of 1.0 V, and an exchange current density of 2.87 × 10−5 A cm−2. This blocking effect of the sulfonic acid groups is attributed to electron depletion on the Pt surface, a reduced d-band center, a weak interfacial electric field, decreased water density in the HP, and fewer H-bonds in HP. Conversely, in the non-contact structure, sulfonic acid groups positioned at the edge of the OHP create an ideal Pt/ionomer/water interface with more active sites, a strong interfacial electric field, and a continuous H-bond network in the HP, resulting in significantly higher intrinsic activity for the ORR. The energy barrier for the RDS decreases to 0.29 eV, the onset potential increases to 1.12 V, and the exchange current density increases to 3.21 × 10−7 A cm−2. Overall, this study provides valuable insights into the influence of solid–solid–liquid interfaces on ORR performance, which can provide information for the design and precise manipulation of micro-three-phase interfaces.Data availability
Data will be made available on request.Author contributions
S. J. conducted the simulation, while S. J. and Q. X. were responsible for data collection, analysis, and the writing of the original draft. Z. X. assisted with data analysis, and N. Y. and J. L. contributed to the discussion of the data. L. L. handled the conceptualization, funding, and revision of the paper. Z. W. provided the necessary resources. All authors discussed the results and contributed to the final manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that might have influenced the work reported in this paper.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2021YFA1501001) and the National Natural Science Foundation of China (grant no. 22179013 and 21822803).References
- J. Liang, X. Liu and Q. Li, Principles, Strategies, and Approaches for Designing Highly Durable Platinum-based Catalysts for Proton Exchange Membrane Fuel Cells, Acta Phys.-Chim. Sin., 2020, 2010072 Search PubMed.
- X. L. Tian, X. Zhao, Y. Q. Su, L. J. Wang, H. M. Wang, D. Dang, B. Chi, H. F. Liu, E. J. M. Hensen, X. W. Lou and B. Y. Xia, Engineering bunched Pt-Ni alloy nanocages for efficient oxygen reduction in practical fuel cells, Science, 2019, 366, 850–856 CrossRef CAS PubMed.
- S. Zaman, Y. Q. Su, C. L. Dong, R. J. Qi, L. Huang, Y. Y. Qin, Y. C. Huang, F. M. Li, B. You, W. Guo, Q. Li, S. J. Ding and B. Y. Xia, Scalable molten salt synthesis of platinum alloys planted in metal-nitrogen-graphene for efficient oxygen reduction, Angew. Chem., Int. Ed., 2022, 61, e202115835 CrossRef CAS.
- W. Z. Tu, W. J. Luo, C. L. Chen, K. Chen, E. B. Zhu, Z. P. Zhao, Z. L. Wang, T. Hu, H. C. Zai, X. X. Ke, M. L. Sui, P. W. Chen, Q. S. Zhang, Q. Chen, Y. J. Li and Y. Huang, Tungsten as “adhesive” in Pt2CuW0.25 ternary alloy for highly durable oxygen reduction electrocatalysis, Adv. Funct. Mater., 2020, 30, 1908230 CrossRef CAS.
- C. Chen, Y. J. Kang, Z. Y. Huo, Z. W. Zhu, W. Y. Huang, H. L. L. Xin, J. D. Snyder, D. G. Li, J. A. Herron, M. Mavrikakis, M. F. Chi, K. L. More, Y. D. Li, N. M. Markovic, G. A. Somorjai, P. D. Yang and V. R. Stamenkovic, Highly crystalline multimetallic nanoframes with three-dimensional electrocatalytic surfaces, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- M. F. Li, Z. P. Zhao, T. Cheng, A. Fortunelli, C. Y. Chen, R. Yu, Q. H. Zhang, L. Gu, B. V. Merinov, Z. Y. Lin, E. B. Zhu, T. Yu, Q. Y. Jia, J. H. Guo, L. Zhang, W. A. Goddard, Y. Huang and X. F. Duan, Ultrafine jagged platinum nanowires enable ultrahigh mass activity for the oxygen reduction reaction, Science, 2016, 354, 1414–1419 CrossRef CAS PubMed.
- N. Jung, Y. Sohn, J. H. Park, K. S. Nahm, P. Kim and S. J. Yoo, High-performance PtCux@Pt core-shell nanoparticles decorated with nanoporous Pt surfaces for oxygen reduction reaction, Appl. Catal., B, 2016, 196, 199–206 CrossRef CAS.
- L. Chong, J. G. Wen, J. Kubal, F. G. Sen, J. X. Zou, J. Greeley, M. Chan, H. Barkholtz, W. J. Ding and D. J. Liu, Ultralow-loading platinum-cobalt fuel cell catalysts derived from imidazolate frameworks, Science, 2018, 362, 1276–1281 CrossRef PubMed.
- X. X. Wang, M. T. Swihart and G. Wu, Achievements, challenges and perspectives on cathode catalysts in proton exchange membrane fuel cells for transportation, Nat. Catal., 2019, 2, 578–589 CrossRef.
- C. Y. Ahn, J. E. Park, S. Kim, O. H. Kim, W. Hwang, M. Her, S. Y. Kang, S. Park, O. J. Kwon, H. S. Park, Y. H. Cho and Y. E. Sung, Differences in the electrochemical performance of Pt-based catalysts used for polymer electrolyte membrane fuel cells in liquid half- and full-cells, Chem. Rev., 2021, 121, 15075–15140 CrossRef PubMed.
- H. Zhu, M. C. Luo, Y. Z. Cai and Z. N. Sun, Core-Shell Structured Electrocatalysts for the Cathodic Oxygen Reduction Reaction in Proton Exchange Membrane Fuel Cells, Acta Phys.-Chim. Sin., 2016, 32, 2462–2474 Search PubMed.
- A. Kongkanand and M. F. Mathias, The priority and challenge of high-power performance of low-platinum proton-exchange membrane fuel cells, J. Phys. Chem. Lett., 2016, 7, 1127–1137 CrossRef PubMed.
- M. Ma, L. Shen, Z. Zhao, P. Guo, J. Liu, B. Xu, Z. Zhang, Y. Zhang, L. Zhao and Z. Wang, Activation methods and underlying performance boosting mechanisms within fuel cell catalyst layer, eScience, 2024, 100254 CrossRef.
- J. Tymoczko, F. Calle Vallejo, V. Colic, M. T. M. Koper, W. Schuhmann and A. S. Bandarenka, Oxygen reduction at a Cu-modified Pt(111) model electrocatalyst in contact with Nafion polymer, ACS Catal., 2014, 4, 3772–3778 CrossRef.
- K. Kodama, K. Motobayashi, A. Shinohara, N. Hasegawa, K. Kudo, R. Jinnouchi, M. Osawa and Y. Morimoto, Effect of the side-chain structure of perfluoro-sulfonic acid ionomers on the oxygen reduction reaction on the surface of Pt, ACS Catal., 2018, 8, 694–700 CrossRef.
- S. Ott, A. Orfanidi, H. Schmies, B. Anke, H. N. Nong, J. Hubner, U. Gernert, M. Gliech, M. Lerch and P. Strasser, Ionomer distribution control in porous carbon-supported catalyst layers for high-power and low Pt-loaded proton exchange membrane fuel cells, Nat. Mater., 2020, 19, 77–85 CrossRef CAS PubMed.
- C. Wang, X. J. Cheng, J. B. Lu, S. Y. Shen, X. H. Yan, J. W. Yin, G. H. Wei and J. L. Zhang, The experimental measurement of local and bulk oxygen transport resistances in the catalyst layer of proton exchange membrane fuel cells, J. Phys. Chem. Lett., 2017, 8, 5848–5852 CrossRef PubMed.
- F. Zhou, H. Zhang, S. Guan, G. Li, L. Xia and M. Pan, Experimental probing of the effect of PFSA ionomer poisoning at different Pt loadings in a PEMFC, J. Catal., 2022, 414, 330–335 CrossRef.
- K. Kodama, R. Jinnouchi, T. Suzuki, H. Murata, T. Hatanaka and Y. Morimoto, Increase in adsorptivity of sulfonate anions on Pt(111) surface with drying of ionomer, Electrochem. Commun., 2013, 36, 26–28 CrossRef.
- K. A. Page, A. Kusoglu, C. M. Stafford, S. Kim, R. J. Kline and A. Z. Weber, Confinement-driven increase in ionomer thin-film modulus, Nano Lett., 2014, 14, 2299–2304 CrossRef PubMed.
- Y. J. Xu, L. L. Zhang, W. Chen, H. W. Cui, J. Cai, Y. X. Chen, J. M. Feliu and E. Herrero, Boosting Oxygen Reduction at Pt(111)|Proton Exchange Ionomer Interfaces through Tuning the Microenvironment Water Activity, ACS Appl. Mater. Interfaces, 2024, 16, 4540–4549 CrossRef.
- F. D. Chen, S. G. Chen, A. X. Wang, M. Wang, L. Guo and Z. D. Wei, Blocking the sulfonate group in Nafion to unlock platinum's activity in membrane electrode assemblies, Nat. Catal., 2023, 6, 392–401 CrossRef.
- R. Jinnouchi, K. Kudo, K. Kodama, N. Kitano, T. Suzuki, S. Minami, K. Shinozaki, N. Hasegawa and A. Shinohara, The role of oxygen-permeable ionomer for polymer electrolyte fuel cells, Nat. Commun., 2021, 12, 230821 Search PubMed.
- V. Yarlagadda, N. Ramaswamy, R. S. Kukreja and S. Kumaraguru, Ordered mesoporous carbon supported fuel cell cathode catalyst for improved oxygen transport, J. Power Sources, 2022, 532, 231349 CrossRef.
- Y. C. Park, H. Tokiwa, K. Kakinuma, M. Watanabe and M. Uchida, Effects of carbon supports on Pt distribution, ionomer coverage and cathode performance for polymer electrolyte fuel cells, J. Power Sources, 2016, 315, 179–191 CrossRef.
- Q. Zhang, S. Dong, P. Shao, Y. Zhu, Z. Mu, D. Sheng, T. Zhang, X. Jiang, R. Shao, Z. Ren, J. Xie, X. Feng and B. Wang, Covalent organic framework-based porous ionomers for high-performance fuel cells, Science, 2022, 378, 181–186 CrossRef.
- X. Cheng, J. You, S. Shen, G. Wei, X. Yan, C. Wang and J. Zhang, An ingenious design of nanoporous Nafion film for enhancing the local oxygen transport in cathode catalyst layers of PEMFCs, Chem. Eng. J., 2022, 439 Search PubMed.
- P. Li, Y. L. Jiang, Y. C. Hu, Y. N. Men, Y. W. Liu, W. B. Cai and S. L. Chen, Hydrogen bond network connectivity in the electric double layer dominates the kinetic pH effect in hydrogen electrocatalysis on Pt, Nat. Catal., 2022, 5, 900–911 CrossRef.
- O. M. Magnussen and A. Gross, Toward an atomic-scale understanding of electrochemical interface structure and dynamics, J. Am. Chem. Soc., 2019, 141, 4777–4790 CrossRef PubMed.
- A. Bouzid and A. Pasquarello, Atomic-scale simulation of electrochemical processes at electrode/water interfaces under referenced bias potential, J. Phys. Chem. Lett., 2018, 9, 1880–1884 CrossRef PubMed.
- S. J. Shin, D. H. Kim, G. Bae, S. Ringe, H. Choi, H. K. Lim, C. H. Choi and H. Kim, On the importance of the electric double layer structure in aqueous electrocatalysis, Nat. Commun., 2022, 13, 174 CrossRef PubMed.
- Y. H. Wang, S. S. Zheng, W. M. Yang, R. Y. Zhou, Q. F. He, P. Radjenovic, J. C. Dong, S. N. Li, J. X. Zheng, Z. L. Yang, G. Attard, F. Pan, Z. Q. Tian and J. F. Li, In situ Raman spectroscopy reveals the structure and dissociation of interfacial water, Nature, 2021, 600, 81–85 CrossRef PubMed.
- K. Ojha, K. Doblhoff-Dier and M. T. M. Koper, Double-layer structure of the Pt(111)-aqueous electrolyte interface, Proc. Natl. Acad. Sci., 2022, 119, e2116016119 CrossRef PubMed.
- N. Ramaswamy, S. Ghoshal, M. K. Bates, Q. Y. Jia, J. K. Li and S. Mukerjee, Hydrogen oxidation reaction in alkaline media: Relationship between electrocatalysis and electrochemical double-layer structure, Nano Energy, 2017, 41, 765–771 CrossRef.
- I. Ledezma-Yanez, W. D. Z. Wallace, P. Sebastian-Pascual, V. Climent, J. M. Feliu and M. T. M. Koper, Interfacial water reorganization as a pH-dependent descriptor of the hydrogen evolution rate on platinum electrodes, Nat. Energy, 2017, 2, 17031 CrossRef.
- L. S. Li, J. M. P. Martirez and E. A. Carter, Prediction of highly selective electrocatalytic nitrogen reduction at low overpotential on a Mo-doped g-GaN monolayer, ACS Catal., 2020, 10, 12841–12857 CrossRef.
- B. Tang, Y. Fang, S. Zhu, Q. Bai, X. Li, L. Wei, Z. Li and C. Zhu, Tuning hydrogen bond network connectivity in the electric double layer with cations, Chem. Sci., 2024, 15, 7111–7120 RSC.
- M. Li, L. Li, X. Huang, X. Qi, M. Deng, S. Jiang and Z. Wei, Platinum-Water Interaction Induced Interfacial Water Orientation That Governs the pH-Dependent Hydrogen Oxidation Reaction, J. Phys. Chem. Lett., 2022, 13, 10550–10557 CrossRef.
- G. Kresse and J. Furthmuller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef PubMed.
- P. E. Blochl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- B. Hammer, L. B. Hansen and J. K. Norskov, Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- J. K. Norskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, Origin of the overpotential for oxygen reduction at a fuel-cell cathode, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06600f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |