
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioinspired and biomimetic nucleobase-containing polymers: the effect of selective multiple hydrogen bonds
Nan
Yao
a,
Jiang
Wu
b,
Guangming
Liu
 *b and
Zan
Hua
*a
*b and
Zan
Hua
*a
aThe Key Laboratory of Functional Molecular Solids, Ministry of Education, Department of Materials Chemistry, School of Chemistry and Materials Science, Anhui Normal University, Wuhu 241002, P. R. China. E-mail: zanhua23@ahnu.edu.cn
bHefei National Research Center for Physical Sciences at the Microscale, Department of Chemical Physics, Key Laboratory of Surface and Interface Chemistry and Energy Catalysis of Anhui Higher Education Institutes, University of Science and Technology of China, Hefei 230026, P. R. China. E-mail: gml@ustc.edu.cn
First published on 5th November 2024
Abstract
Bioinspired and biomimetic nucleobase-containing polymers are a series of polydisperse nucleic acid analogs, mainly obtaining through highly efficient and scalable step-growth or chain polymerizations. The combination of pendant nucleobase groups and various backbones endows the polymers/materials with selective multiple H-bonds under distinct conditions, demonstrating the broad applicability of this new family of polymeric materials. In this perspective, we critically summarize recent advances of bioinspired and biomimetic nucleobase-containing polymers and materials in both solution and the bulk. Then, we discuss the effect of multiple H-bonds between complementary nucleobases on the structures and properties of the nucleobase-containing polymers and materials. Selective multiple H-bonds between complementary nucleobases are feasible to modulate the polymer sequence and self-assembly behaviour, achieve templated polymerization, tune nanostructure morphologies and functions, and selectively bind with nucleic acids in various solutions. Meanwhile, bioinspired and biomimetic nucleobase-containing polymers are capable of forming robust polymeric materials such as hydrogels, bioplastics, elastomers, adhesives, and coatings by optimizing the inter- and intramolecular multiple H-bonding interactions. Further, the conclusions and outlook for future development and challenges of bioinspired and biomimetic nucleobase-containing polymers are also presented. This perspective presents useful guidelines for fabricating novel bioinspired and biomimetic polymers and materials through rational design of multiple H-bonds nucleobase interactions.
 Nan Yao | Nan Yao received his MS degree from Anhui Agricultural University in 2024. Now, he is pursuing his PhD at the Anhui Normal University under the supervision of Prof. Zan Hua. His main research interests are the construction and properties of high performance bioinspired nucleobase-containing materials. |
 Jiang Wu | Jiang Wu graduated from Anhui Agricultural University with bachelor and master degrees in 2019 and 2022, respectively. He is currently pursuing his doctor degree in the Department of Chemical Physics at University of Science and Technology of China. His main research interests are the syntheses and applications of functional polymers. |
 Guangming Liu | Prof. Guangming Liu received his BS degree from Anhui Normal University in 2002 and his PhD degree from University of Science and Technology of China in 2007. He worked as a research assistant at The Hong Kong University of Science and Technology from 2005 to 2006 and worked as a postdoctoral fellow at The Australian National University from 2008 to 2010. He is currently a professor at University of Science and Technology of China. His present research interests focus on the high-performance polymeric materials. |
 Zan Hua | Prof. Zan Hua obtained his MChem degree from the University of Science and Technology of China in 2014. He received his PhD degree from the University of Warwick working with Prof. Rachel K. O'Reilly in 2018. Then he worked in the Department of Material Science and Engineering at Anhui Agricultural University until the end of 2022. In 2023, he started as a full professor in the Department of Materials Chemistry at Anhui Normal University. His main research interests are syntheses, properties, and applications of new bioinspired and biomimetic nucleobase-containing polymers and materials. |
1. Introduction
Multiple hydrogen bonds are some of the most widely used noncovalent interactions for fabricating supramolecular polymers and materials.1,2 In contrast to π–π stacking and van der Waals forces, hydrogen bonds are directional and strong, achieving controlled interaction and precise molecular recognition. Although the bond energy of a hydrogen bond is only 5–10% of that of covalent or ionic bonds, multiple hydrogen bonds of macromolecules endow the system with robust and dynamically reversible properties. Nature has elegantly selected hydrogen bonds to implement the significant functionality of biomolecules including nucleic acids and proteins. The evolutionary selection of multivalent low affinity hydrogen bonds over specific high affinity interactions has several advantages. Multivalency makes it possible to improve the binding affinity over a dynamic range rather than a simple on/off switch. In addition, it is surmised that there is an evolutionary efficiency to employing existing low affinity interaction rather than fabricating new ones. Depending on the high fidelity of hydrogen bonds between complementary nucleobases (adenine (A) with thymine (T) or uracil (U), and guanine (G) with cytosine (C)), nucleic acids such as DNAs and RNAs have precisely achieved the biological functions of replication, transcription, and translation processes of genetic information. Further, selective multiple hydrogen bonds between nucleic acids have enabled the development of gene editing,3,4 RNA interference (RNAi),5,6 and DNA nanotechnology.7–9Nucleic acids as a kind of sequence-defined biopolymers are superior to common polymers, programming the formation of complex structures and realizing the advanced function through hydrogen bonds. Currently, the syntheses of nucleic acids mainly rely on solid-phase synthesis or polymerase-mediated synthesis, giving rise to high cost and limited scalability. Meanwhile, nucleic acids have the labile phosphodiester backbone of negative charges, restricting their wide applicability (Fig. 1a). In order to achieve enhanced stability, bioavailability, and distinct protein binding properties, various modifications and structural variations of nucleic acids have been explored.10–13 Hundreds of chemical modifications for nucleic acids have been reported and the development of polymers with nucleobase functionalities has also been intensively studied,14–18 both of which make it necessary to clarify how new synthetic nucleobase-containing macromolecules should be classified. According to the synthetic approaches, there are basically two kinds of important nucleobase-containing macromolecules, i.e. ‘xeno-nucleic acids’ and nucleobase-containing polymers (Fig. 1b and c). ‘Xeno-nucleic acids’ abbreviated as XNAs are synthesized through step-by-step, highly-efficient organic reactions to yield monodisperse and sequence-defined nucleic acid analogs, which usually have chemical backbone motifs differing from deoxyribose and ribose.19,20 Representative examples include peptide nucleic acid (PNA),10,21 threose nucleic acid (TNA),11,22 and glycol nucleic acid (GNA)12 (Fig. 1b). Synthetic biologists might have more elaborate naming and classification of XNAs,20 which is out of the scope for the current perspective. In contrast, nucleobase-containing polymers are mainly prepared through high-efficient and scalable step-growth or chain polymerizations to generate polydisperse nucleic acid analogs (Fig. 1c).23–32 Alternatively, post-polymerization modifications of polymers with nucleobases are also feasible to yield polydisperse nucleobase-containing polymers.33–36 They have various neutral backbones of carbon–carbon bond, phosphoester, thioether, and so on, expanding their applications in chemistry and materials science.
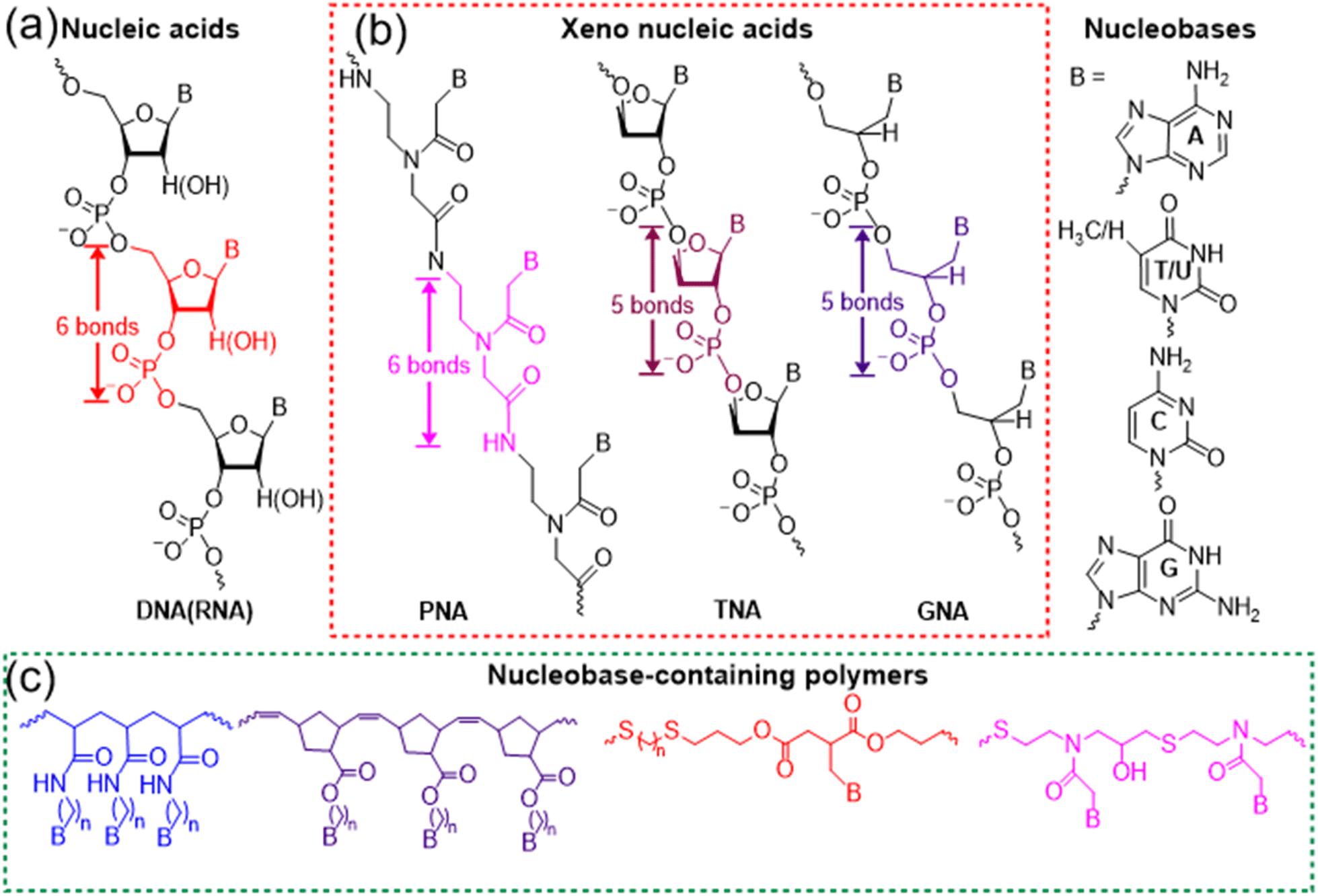 | ||
| Fig. 1 Molecular structures and classification of various nucleobase-containing macromolecules. (a) Nucleic acids; (b) representative xeno-nucleic acids; (c) representative nucleobase-containing polymers. | ||
Importantly, bioinspired and biomimetic nucleobase-containing polymers retain the same pendant nucleobases as nucleic acids, which are capable of forming selective multiple hydrogen bonds and bridging the gap between materials/polymer chemistry and biology. Therefore, exploring and developing novel nucleobase-containing polymers lay a solid foundation for their widespread applications in many areas including nanomedicines,37–41 nanotechnology,42,43 and high performance polymeric materials.44–46 For example, hydrogen bonding interactions between nucleobase-containing polymers and nucleoside drugs or therapeutic nucleic acids provide a novel and dynamic bonding for drug/gene loading and delivery.47 Simultaneously, the selective hydrogen bonds between complementary nucleobases can be used to mediate the polymerization process for achieving the control over chain length and sequence of polymers.27 The multiple hydrogen bonds between complementary nucleobase-containing polymers enable us to modulate the structures and properties of relevant materials.
Several recent reviews have mainly summarized the synthesis, self-assembly and potential applications of nucleobase-containing polymers.15,16,18 Notably, the bulk properties and the structure–property relationship of nucleobase-containing polymers were not involved. Besides, the systematic discussion and analyses between the selective multiple hydrogen bonds and the structures and properties of bioinspired nucleobase-containing polymers are lacking, which impedes the rapid development and rational design of new bioinspired nucleobase-containing polymers and materials. In this perspective, we summarize the recent advances in the renaissance research area of bioinspired and biomimetic nucleobase-containing polymers. We focus on the effect of multiple H-bonds on the structures and properties of the nucleobase-containing polymers and materials in both solution and the bulk. Further, the conclusions and outlook for future development and challenges of bioinspired and biomimetic nucleobase-containing polymers are also presented. An updated perspective relating to synthetic nucleobase-containing polymers provides new insights in designing remarkable bioinspired and biomimetic polymers and materials by optimizing supramolecular selective hydrogen bonding interactions.
2. Bioinspired and biomimetic nucleobase-containing polymers in solution
The initial motivation of preparing nucleobase-containing polymers is to mimic nucleic acids in aqueous solutions. The scientists have made many valuable attempts on the synthesis of nucleobase-containing polymers up to 1990s.48,49 The major synthetic approaches include conventional free radical polymerization (FRP), ring opening polymerization (ROP), and post-polymerization modifications of polyethyleneimine and poly(α-amino acids). The hydrogen bonding interaction of nucleobase-containing polymers in solutions was investigated at different conditions such as solvents, temperature, and so on. However, the ill-defined nucleobase-containing polymers compromise the selective hydrogen bonding interaction, as FRP gives poor control over the polymerization and both ROP and post-polymerization modifications only give limited incorporation of nucleobases. With the rapid development of controlled polymerization techniques in the past three decades, bioinspired and biomimetic nucleobase-containing polymers have garnered significant attentions.16,50,51 The combination of efficient polymerizations and the selective hydrogen bonding interaction enables the synthesis of new nucleobase-containing polymers with novel properties. In this section, we will discuss the effect of selective multiple H-bonds between complementary nucleobases on the structures and properties of the relevant polymers and materials in solution.2.1 Effect of multiple H-bonds between complementary nucleobase-containing monomers
Nucleic acids are essential components of all life processes relying on the precise sequences of only five nucleotides. Synthetic polymer chemists are inspired by the unique structures and properties of nucleic acids to produce nucleobase-containing polymers. Notably, the basic building block of nucleic acids, i.e. nucleotides with the phosphate group have good solubility in water but very limited solubility in common organic solvents with hydrogen-bond supporting ability. However, double or triple hydrogen bonds between individual nucleotides are not sustained in aqueous solutions, which prevents the control over the structures of nucleotides using hydrogen bonds. In contrast, the neutral nucleobase-containing monomers for producing nucleobase-containing polymers are inclined to be dissolved in various solvents, providing the possibility of controlling hydrogen bonds of polymerizable motifs. Consequently, the properties of the relevant nucleobase-containing polymers can be elegantly modulated through this strategy.Early work to study the effect of multiple H-bonds between complementary nucleobase-containing monomers on copolymers was reported by O'Reilly et al.52 They explored the RAFT polymerization of complementary nucleobase-containing methacrylate monomers in solvents with different H-bonding tolerance (Fig. 2a). In chloroform (CHCl3), an alternated copolymer was yielded after copolymerization due to the strong intermolecular hydrogen bonds between adenine- and thymine-containing monomers. In stark contrast, a statistical copolymer was formed due to the absence of nucleobase interactions in dimethylformamide (DMF). Therefore, the hydrogen bonding interaction between nucleobases can be used to manipulate the monomer sequence of copolymers. Interestingly, the statistical or alternated sequences of nucleobases dramatically influenced the self-assembly behavior of copolymers. The diblock copolymer with the solvophobic block of alternated nucleobase sequence tended to form spherical nanostructures, while one with the statistical nucleobase sequence tended to generate worm-like micelles.
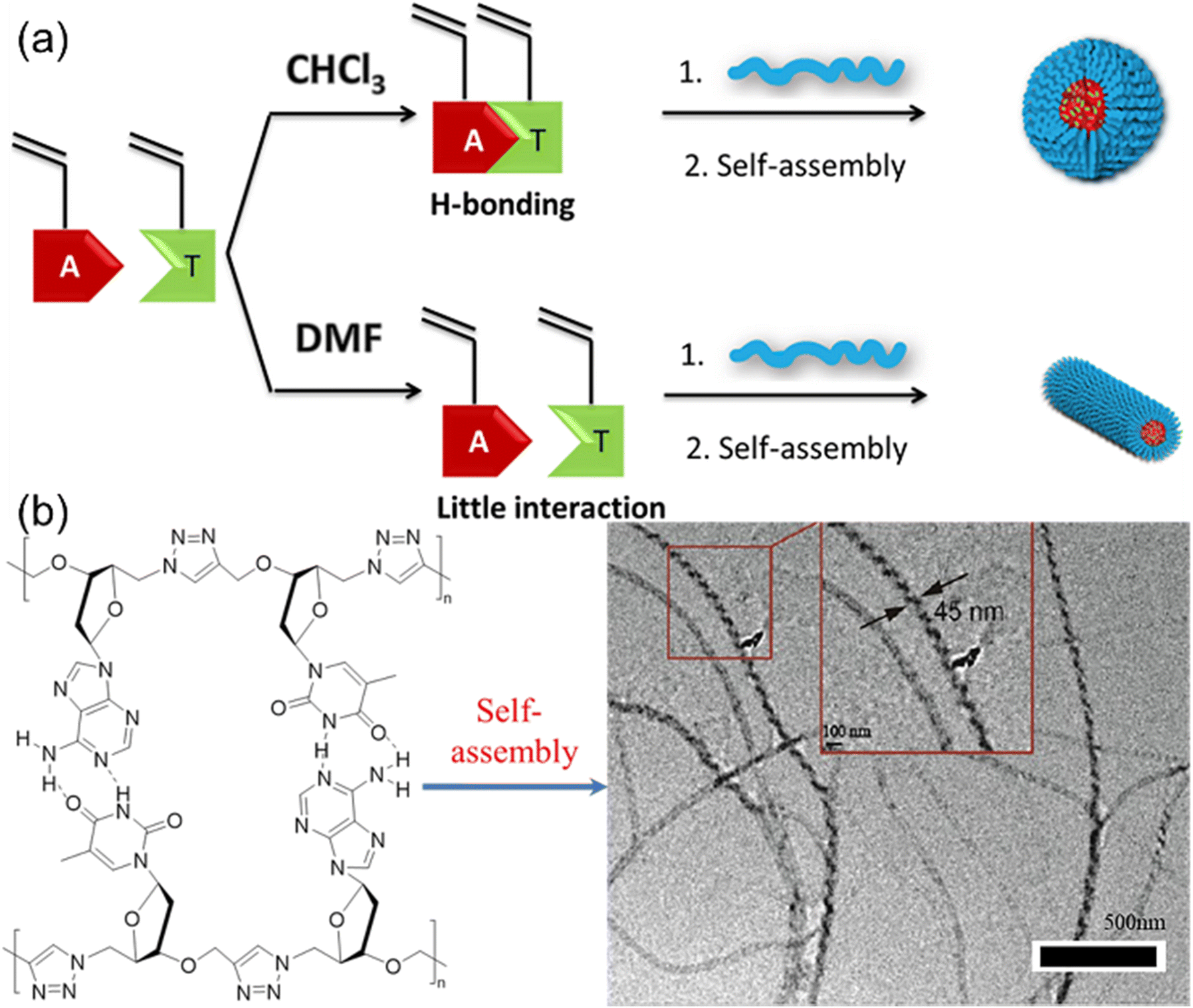 | ||
| Fig. 2 Control of polymerization and solution self-assembly using hydrogen bonds between complementary nucleobase-containing monomers. (a) Schematic representation of the synthesis of block polymers from nucleobase-containing monomers in the presence or absence of hydrogen bonds.52 Adapted from ref. 52 with permission from American Chemical Society, copyright 2013. (b) Synthesis and self-assembly of alternating heterodinucleoside polytriazoles.53 Adapted from ref. 53 with permission from American Chemical Society, copyright 2021. | ||
Further, multiple H-bonds between complementary nucleobase-containing monomers were employed to fabricate supramolecular double-stranded structures and nanostructures. Yang and the coworkers53,54 prepared two alternating heterodinucleoside polytriazoles from the hydrogen-bonded azide/alkyne functionalized heterodinucleoside monomers through 1,4-regioregular CuAAC polymerization method (Fig. 2b). Meanwhile, the self-assembly behavior of heterodinucleoside polytriazole and its monomers indicated that the 1,4-regioregular polytriazole backbone could not only constrain and regulate the complementary hydrogen bonds of nucleobases, but also transfer the molecular chirality of monomers to supramolecular nanostructures, thereby inducing the formation of spiral nanofibers. This approach can be further extended to the iterative polymerization of nucleoside monomers with more complex nucleobase sequences, providing a new way for the development of new artificial nucleobase-containing polymers.
2.2 Effect of multiple H-bonds between nucleobase-containing polymers and small molecules
Although individual complementary nucleotides are not able to form stable H-bonds in aqueous solutions, the hydrophobic domain of macromolecular nucleic acids facilitates the formation of stronger H-bonds between complementary nucleobases of nucleotides and nucleic acids. This eminent nature builds the molecular foundation for the precise replication and transcription of nucleic acids with nucleotides as feedstocks through the polymerase-mediated reactions. Meanwhile, the segregated and concentrated monomers in the microenvironment are favourable for accelerating the polymerization reactions. These interesting and ingenious processes in the biological systems are eagerly pursued by polymer chemists. Indeed, nucleobase-containing small molecules are capable of forming strong hydrogen bonds with complementary nucleobase-containing polymers. The unique properties can be employed to achieve templated polymerization and load nucleobase-type drugs.Templated polymerization/synthesis have garnered great attention in polymer chemistry, especially with the synthesis of nucleobase-containing polymers.24,25,27,33,55–65 However, the homogeneous nature of most templating approaches only achieves limited control over the formation of daughter nucleobase-containing polymers in a controlled fashion. An important progress has been made by McHale and coworkers66 by combining segregation and templating strategies to afford well-defined high molecular weight polymers (Fig. 3a). Low molecular weight block copolymer polystyrene-b-poly(vinylbenzyl thymine) (PSt-b-PVBT) was firstly synthesized and self-assembled into core–shell micelles with thymine units in the core. The utilization of well-defined thymine-containing micelles represented an obvious advantage through simultaneously templating and segregating complementary adenine-containing monomers as the polymerization propagated. Conventional radical polymerization in such an environment substantially eliminated or reduced the undesired bimolecular termination, generating the daughter polymer with high molecular weight (up to ∼400 kg mol−1) and low polydispersity (Đ ≤ 1.08). It was clearly demonstrated that the combined templating and segregation approaches through nucleobase interactions imparted unprecedented control of polymerization to yield well-defined polymers.
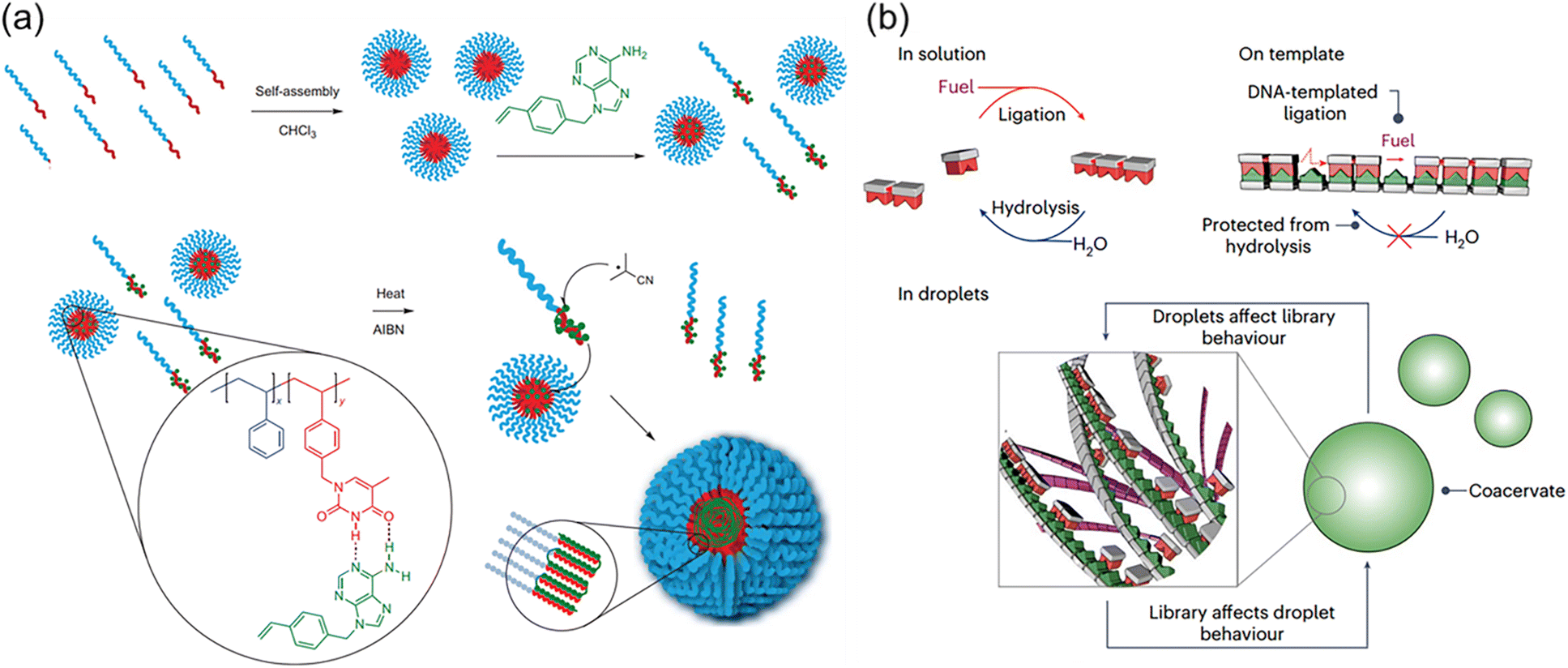 | ||
| Fig. 3 Complementary multiple H-bonds-mediated templated polymerization. (a) PSt-b-PVBT in CHCl3 as a template with the addition of complementary adenine monomers (VBA) enables the preparation of a high MW, low PDI daughter polymer PVBA.66 Adapted from ref. 66 with permission from Springer Nature, copyright 2012. (b) Template-based replication for the selection in the chemical library and its influence on the physical properties of the macromolecular coacervate.67 Adapted from ref. 67 with permission from Springer Nature, copyright 2024. | ||
Subsequently, Marsh et al. have reported template-directed radical polymerization on solid supports based on complementary nucleobase interaction in nonpolar solvents.68 The templates attached on functional Wang resins were synthesized through copper-mediated “living” radical polymerization from uridine, adenosine, cytidine, and guanosine-containing methacrylates. The covalent linkage of the parent polymer to the solid support provided a great advantage for the subsequent separation of the templated daughter polymer. A degree of control over templated polymerization of the complementary nucleobase-containing monomers was observed in the presence of the template but not observed in the absence. Uridine-functionalized templates with controlled length and dispersity were found to provide good fidelity of replication by incorporating mainly adenosine monomer in the final polymer, from a mixture of monomers. This particular templated polymerization indicated that solid-supported polymerizations provided good control over the polymerization process through multiple H-bonds of complementary nucleobases.
Recently, Kriebisch et al. presented a chemically fueled dynamic combinatorial library to mimic RNA oligomerization and deoligomerization and offered new insights on selection and purification mechanisms under kinetic control (Fig. 3b).67 The molecular basis was the combination of selective multiple H-bonds between nucleobase-containing monomers and DNA/RNA templates with the diacid condensation reactions to produce the oligomer. It was found that hybridization through multiple H-bonds accelerated oligomerization by selectively concentrating longer oligomers. Simultaneously, it decelerated deoligomerization through a protection mechanism. These feedback mechanisms were able to select oligomer molecules of specific lengths and sequences, which were used to purify oligomers. In addition, template-assisted formation of nucleobase-containing oligomers within coacervate-based protocells was capable of altering its compartment's physical properties, such as their ability to fuse. Such reciprocal coupling between oligomer production and physical properties is an important step towards synthetic life, providing valuable understanding of the fundamental mechanisms of minimalistic life.
2.3 Effect of multiple H-bonds between complementary nucleobase-containing polymers
Although the bond energy of a single hydrogen bond is about 5–20 kJ mol−1, multiple H-bonds of complementary nucleic acids over 10 kDa are several times stronger than common covalent bonds simply considering the primary attractive hydrogen bonds. When taking into consideration the secondary electrostatic interactions between proton donors and acceptors, the multiple H-bonds are even more robust. More importantly, the multiple H-bonds between nucleic acids are dynamically reversible, enabling the repeated execution of advanced function. For example, the CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9) system has been developed into a revolutionary technology for editing and modulating genomes.69,70 It relies on the precise binding of single-guide RNA to targeted DNA through multiple H-bonds. Likewise, the DNA origami to form well-defined nanostructures also depend on the accurate multiple H-bonds between short stapler DNA chains and the long DNA scaffold.71,72 Inspired by the multiple H-bonds of macromolecular nucleic acids, the similar strategy is feasible to achieve precise control over morphologies and properties of nanostructures from nucleobase-containing polymers.73–77Block copolymers with complementary nucleobases are capable of forming selective multiple H-bonds, which is the effective way to modulate the morphologies and properties of nanostructures.78 We have exploited supramolecular multiple H-bonds between adenine and thymine-containing polymers to generate anisotropic polymer nanoparticles with well-defined aspect ratios from isotropic seeds (Fig. 4a). The polymer PT was employed to form spherical nanoparticles NT with a thymine-containing core via self-assembly, and the introduction of the adenine-containing polymer PA induced the morphological change at different A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :T molar ratios. The results indicated that the length and the diameter of the worms were governed by the molar ratios of adenine/thymine. Building on the selective multiple H-bonds between PA and NT, the global transformation process made it straightforward to introduce additional functional groups into various nanostructures, as demonstrated by the stepwise incorporation of fluorescent tags. Nucleobase interactions-driven morphological transformation presents a valuable tool for controlling nanoscale self-assembly, and for the production of functional nanostructures with diverse potential applications.
:T molar ratios. The results indicated that the length and the diameter of the worms were governed by the molar ratios of adenine/thymine. Building on the selective multiple H-bonds between PA and NT, the global transformation process made it straightforward to introduce additional functional groups into various nanostructures, as demonstrated by the stepwise incorporation of fluorescent tags. Nucleobase interactions-driven morphological transformation presents a valuable tool for controlling nanoscale self-assembly, and for the production of functional nanostructures with diverse potential applications.
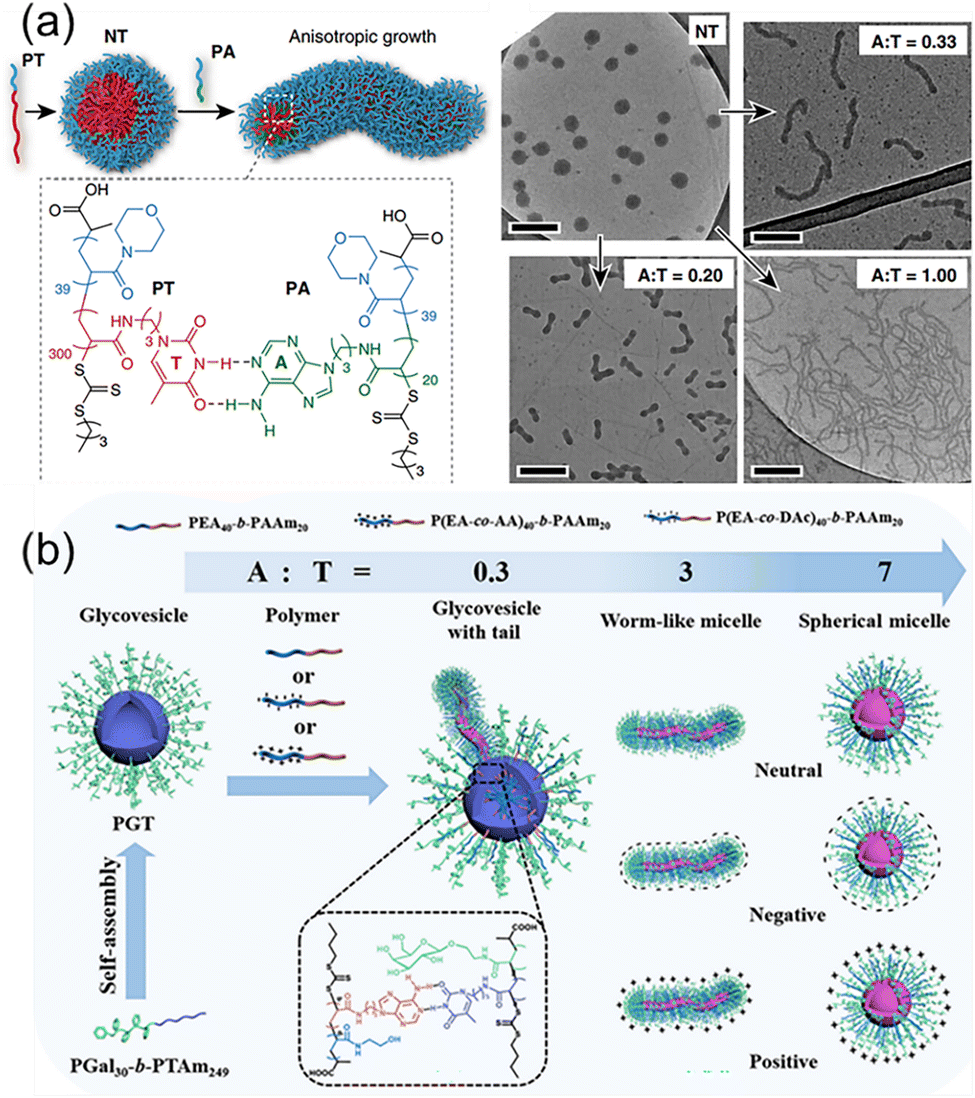 | ||
| Fig. 4 Modulation of polymer self-assembly morphology and surface properties through hydrogen bonding interactions between complementary nucleobase-containing polymers. (a) Transformation of spherical nanoparticles into different anisotropic morphologies.78 Adapted from ref. 78 with permission from Springer Nature, copyright 2019. (b) Fabrication of sugar vesicles and the utilization of complementary hydrogen bonding for morphological transformation and surface modification.79 Adapted from ref. 79 with permission from American Chemical Society, copyright 2024. | ||
In addition to the morphologies, the nucleobase-containing polymer interactions were also used to control the surface properties of self-assembled structures. Recently, cascade morphological transformation and surface engineering of nucleobase-containing glycovesicles were achieved by Yang and coworkers (Fig. 4b).79 Firstly, thymine-containing glycoconjugates (PGal30-b-PTAm249) was synthesized and self-assembled into well-defined glycovesicles. Several neutrally, positively, and negatively charged amphiphilic adenine-containing block copolymers were synthesized, which could be used to modulate the surface properties of nanostructures. For example, neutral amphiphilic adenine-containing block copolymers were successively added to thymine-containing sugar vesicles. It can be observed that as the ratio of A:T increased, the sugar vesicles underwent a morphological transition from ruptured vesicles with tails, to worm-like micelles, and finally to spherical micelles. Simultaneously, the surface charged properties of polymeric glyconanoobjects were modulated by doping with charged adenine-containing polymers. Thus, selective multiple H-bonds between complementary nucleobase-containing polymers are feasible for fabricating glyconanoparticles with different morphologies and surface properties as new tailor-made biomaterials.
Further, O'Reilly and coworkers have exploited the nucleobase interactions to achieve the self-assembly of amphiphilic core–shell bottlebrush polymers (BBPs) in an end-to-end fashion.80 Ring-opening metathesis polymerization (ROMP) and reversible addition-fragmentation chain transfer (RAFT) polymerization were employed to synthesize core–shell BBPs containing complementary nucleobase functionalities using a “grafting from” approach, and hierarchical self-assembly was performed in aqueous media. Interestingly, BBPs containing complementary nucleobases were observed to self-assemble into cylindrical supramolecules only when the sample was heated above a certain threshold, corresponding to the lower critical solution temperature (LCST) of the corona-forming poly(4-acryloymorpholine) block (Fig. 5a). Furthermore, this self-assembly behavior was only observed when equal amounts of BBPs containing complementary thymine and adenine functions were mixed and heated at elevated temperatures. Therefore, the nucleobase functionality within the core compartment facilitated interfacial interactions between BBPs in the assemblies dominated by multiple hydrogen bonding interactions between complementary nucleobases.
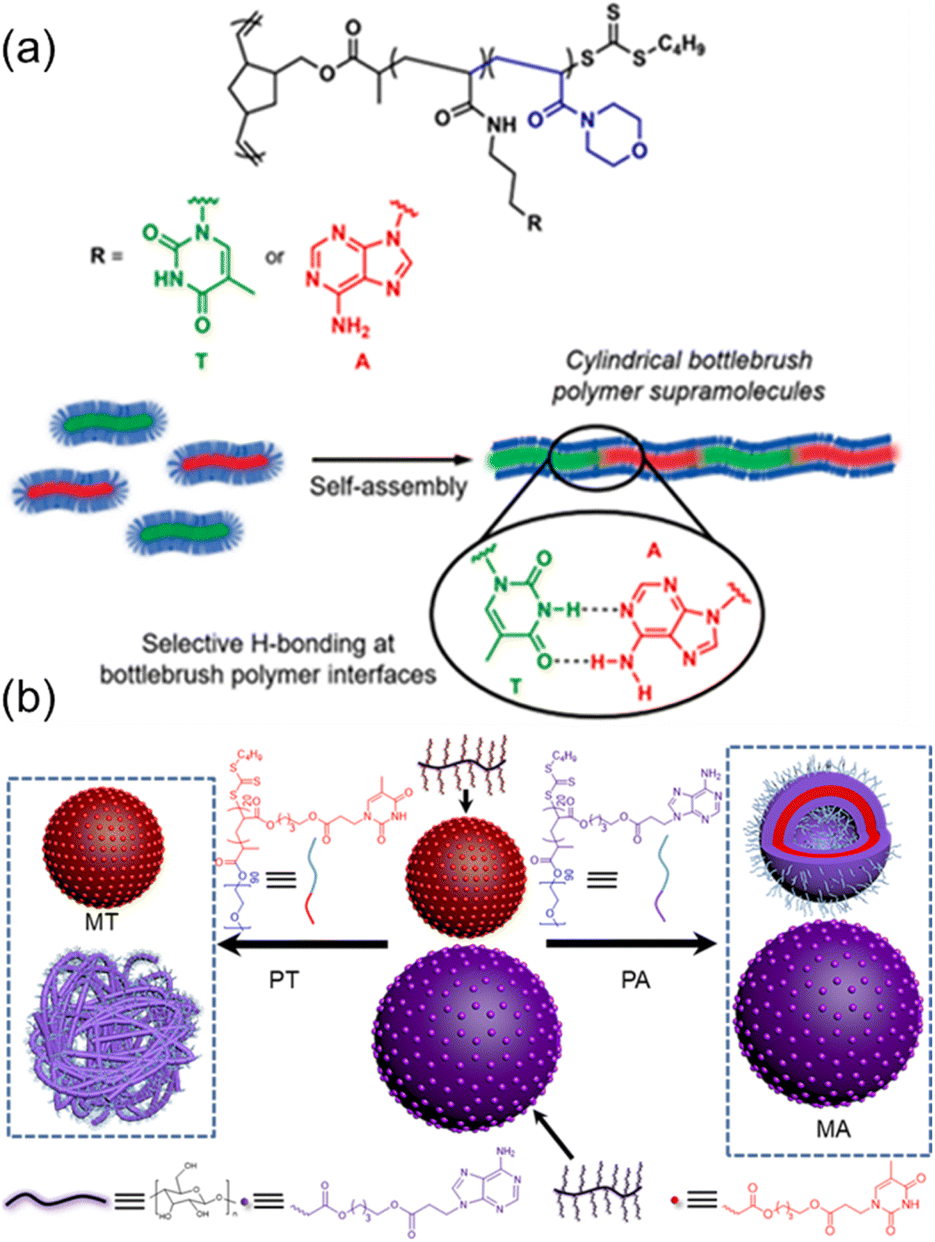 | ||
| Fig. 5 Tunable bottlebrush polymer self-assembly morphology and surface properties through hydrogen bonding interactions between complementary nucleobase-containing polymers. (a) Complementary nucleobase interactions achieving hierarchical self-assembly of core–shell bottlebrush polymers into cylindrical supramolecules.80 Adapted from ref. 80 with permission from American Chemical Society, copyright 2020. (b) Selective morphological transformation of mixed nanostructures from cellulose-grafted bottlebrush polymers.81 Adapted from ref. 81 with permission from American Chemical Society, copyright 2022. | ||
Recently, we have achieved the programmable morphological transformations of mixed nanostructures from cellulose-grafted bottlebrush polymers based on multiple H-bonds between linear and bottlebrush polymers. Both adenine and thymine-containing cellulose-grafted-polymers (Cell-g-PAAc20 and Cell-g-PTAc20) were obtained by using surface-initiated RAFT polymerization (Fig. 5b).81 The self-assembly of Cell-g-PAAc20 and Cell-g-PTAc20 generated the spherical micelles MA and MT, which were not able to interact through H-bonds due to the enthalpy-unfavorable chain extension and steric repulsion of cellulose-grafted bottlebrush polymers. Interestingly, the introduction of linear diblock copolymers, polyethylene glycol-b-poly(4-((3 (adenin-9-yl)propanoyl)oxy)butyl acrylate) (PEG90-b-PAAc20, PA) and polyethylene glycol-b-poly(4-((3-(thymin-l-yl) propanoyl)oxy)butyl acrylate) (PEG90-b-PTAc20, PT), triggered the morphological transformations of MT or MA mediated by multiple H-bonds of complementary nucleobases, respectively. The distinct morphological transformations for MA and MT were caused by the variation of intermolecular interactions between cellulose-grafted bottlebrush polymers and linear diblock copolymers. Depending on the unique properties of nanostructures from cellulose-grafted bottlebrush polymers, the bioinspired selective responsiveness of mixed nanostructures was realized by employing complementary hydrogen bonds of nucleobases.
2.4 Effect of multiple H-bonds between nucleobase-containing polymers and nucleic acids
Nucleic acids have great potential for gene silencing and elimination of drug resistance in diseased cells such as cancer, but constructing effective delivery carriers is necessary and challenging.82,83 The delivery of nucleic acids to targeted cells is difficult as the process substantially needs to overcome several obstacles, such as in vivo nuclease susceptibility, the rapid clearance by phagocytic cells, and limited cellular uptake of negatively charged nucleic acids.84 Cationic polymers and lipids or DNA nanostructures have been widely explored to load nucleic acids due to high attachment on an anionic cell surface and favourable gene transportation into the cells.85–87 However, several issues should be addressed including immunogenicity, potential cell damage, failure in nucleic acid release, cost, and so on. Thus, developing new nucleobase-containing polymers are highly desired, which is to allow the specific hydrogen bonding interaction with nucleic acids and efficient delivery into the cell.Nucleic acids have a six-atom backbone spacing between two adjacent nucleobases, prompting the elegant structural design of nucleobase-containing polymers. The pioneering works were carried out by the Bowman group,29,40 developing a variety of clickable nucleic acids (CNAs). Actually, CNAs are polydisperse nucleobase-containing polymers obtained from step-growth thiol–ene polymerization. Cha et al. have demonstrated that significant amounts of single stranded DNA were encapsulated within triblock copolymer nanoparticles through the multiple H-bonds with CNAs.41 Firstly, poly(ethylene glycol)-block-clickable nucleic acid-block-poly(lactic-co-glycolic acid) (PEG-CNA-PLGA) triblock copolymer (Fig. 6a) was synthesized and formulated into nanoparticles using an O/W emulsion method. The results showed that particles containing CNAs with complementary nucleobases presented a high degree of encapsulation with DNAs. In stark contrast, the nanoparticles of PEG-PLGA alone showed minimal DNA encapsulation, and DNA strands with non-complementary nucleobases were not encapsulated into the PEG-CNA-PLGA nanoparticles. In addition, longer and more complex DNA sequences with A10 DNA as the tail was also encapsulated by sequence complementary CNAs, indicating the potential utility of CNAs as efficient nucleic acid drug carriers.
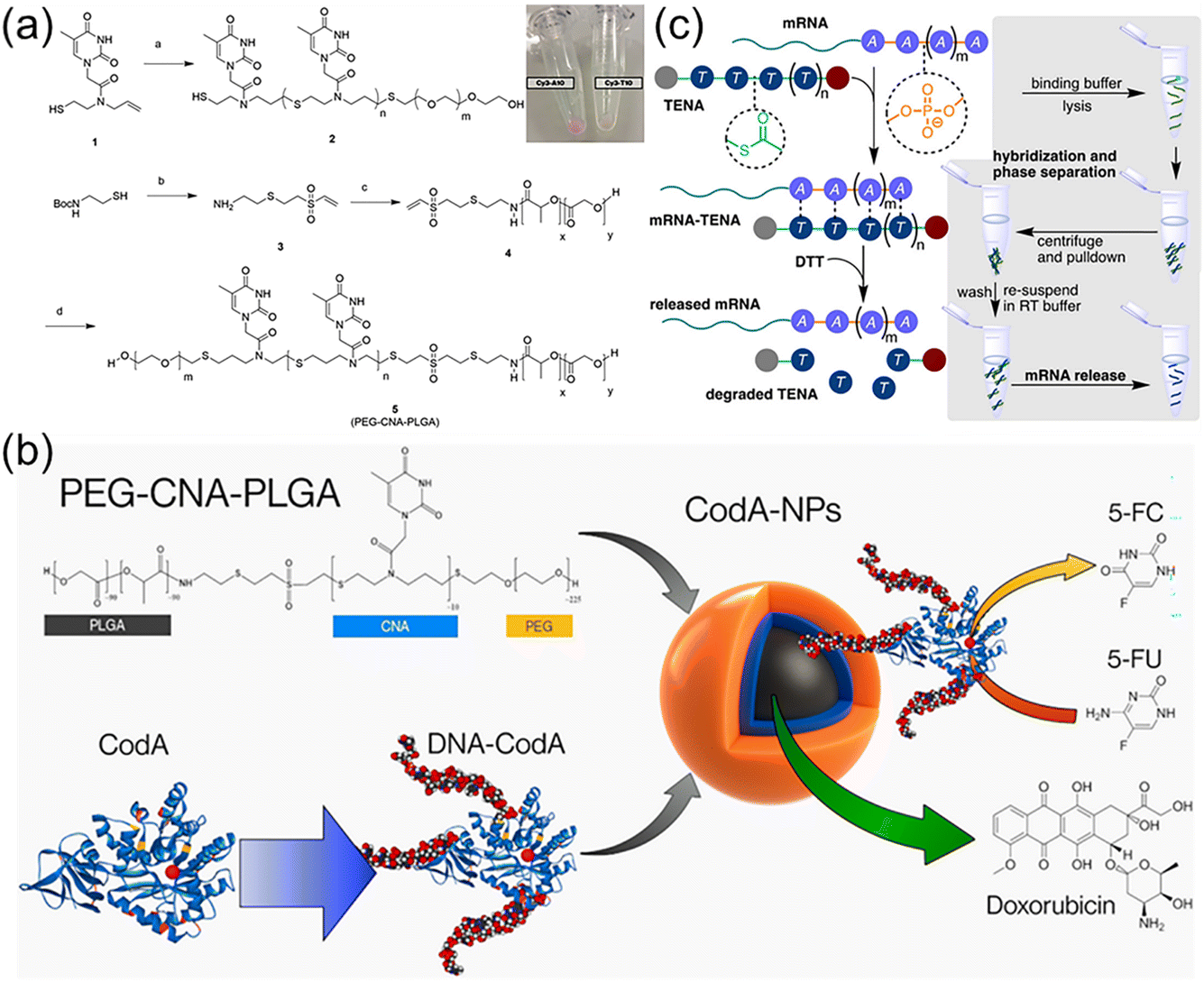 | ||
| Fig. 6 Nucleobase-containing polymers bind to nucleic acids through hydrogen bonding interactions. (a) Scheme of the synthesis of the triblock nucleobase-containing polymer PEG-CNA(T10)-PLGA.88 Adapted from ref. 88 with permission from Wiley-VCH, copyright 2017. (b) Schematic illustration of PEG-CNA-PLGA nanoparticles loaded with both DNA-CodA and DOX.89 Adapted from ref. 89 with permission from American Chemical Society, copyright 2019. (c) Schematic representation of TENA binding complementary mRNA through hydrogen bonding interactions and thermally degrading to enrich mRNA under ambient conditions.90 Adapted from ref. 90 with permission from Wiley-VCH, copyright 2021. | ||
Furthermore, the same group have achieved the loading of the prodrug activating enzymes (cytosine deaminase, CodA) and chemotherapy drugs (doxorubicin, DOX) in a single polymer nanoparticle concomitantly (Fig. 6b).89 This is of great importance as the simultaneous delivery of multiple therapeutics is highly promising for cancer targeting and treatment. CodA was appended with complementary DNA sequences and fluorescent dyes to achieve its encapsulation into PEG-CNA-PLGA nanoparticles, which was compatible with the loading of DOX in the hydrophobic core. Importantly, using a modified fluorescent assay it was found that the DNA-modified CodA retained its enzyme activity for converting prodrug 5-fluorocytosine (5-FC) to active 5-fluorouracil (5-FU). Subsequently, cytotoxicity assay of MDA-MB-468 breast cancer cells after encapsulation of DNA-CodA in PEG-CNA-PLGA nanoparticles revealed that the toxicity of the particles alone was low in the presence of 5-FC. It was worth noting that the cell-killing effect was significantly increased with the addition of DNA-CodA. In addition, the toxic effect was enhanced when the hydrophobic drug DOX was also encapsulated, suggesting its potential in codelivery of multiple chemotherapeutic drugs.
Recently, the Bowman group have utilized the selective multiple H-bonds between nucleobase-containing polymers and RNA for extracting targeted mRNA from the total RNA mixture (Fig. 6c).90 Oligo(thymine) thioester linked nucleic acids (oT TENAs) was designed and synthesized, presenting a six-atom spacing between nucleobases for selectively base-pairing with polyadenine RNA. The attained TENAs had a significant merit, i.e. self-immolative degradation under ambient temperature. The neutral oT TENA with an added hydrophobicity of the octane thiol end group was capable of pulling down ∼93% of EGFP mRNA by selectively binding to the poly(A) tail. Upon exposure to the nucleophilic buffer components such as Tris and DTT under ambient temperature, the dynamic thioester backbone was observed to degrade via a self-immolative mechanism recovering ∼65% of EGFP mRNA. The optimized protocol by using TENA was able to recover mRNA from total RNA, which was not statistically different in comparison to a commercial kit. The following gene expression as measured by RT-qPCR was comparable for both enrichment methods. The results suggested that the mild conditions required for enrichment of mRNA using oT TENA were compatible with RT-qPCR and other downstream molecular biology applications.
3. Bioinspired nucleobase-containing polymers in the bulk
Nucleic acids especially DNAs have been widely utilized as biodegradable macromolecules for the fabrication of a variety of bulk polymeric materials such as hydrogels and bioplastics.91–94 However, nucleic acids have rigid and charged backbones with large pendant groups, leading to brittle and weak mechanical properties of relevant materials. Meanwhile, bioplastics obtained from nucleic acids have poor stability under ambient conditions due to their high hydrophilicity and moisture sensitiveness. By contrast, the properties of bioinspired nucleobase-containing polymers can be modulated through the variation of the backbone structure, enabling the fabrication of various polymeric materials such as hydrogels,95–97 bioplastics,98–100 elastomers,101,102 adhesives,103–108 coatings,109 and so on.110–112 The introduction of flexible backbone endows the supramolecular nucleobase interactions with high dynamic and tunable characters. Therefore, rational design of selective multiple H-bonds of nucleobases provides an efficient route to constructing outstanding bioinspired nucleobase-containing polymeric materials.3.1 Effect of multiple H-bonds between nucleobase-containing monomers
Nature provides many excellent examples of polymeric materials, such as spider silks with high toughness, mussel mucins with strong underwater adhesion, to name just a few. The superior properties of these materials are determined by the precise structures obtained from natural evolution mainly including primary, secondary, and tertiary structures. Undoubtedly, the elegant design of the monomer sequence is of great importance, determining the primary macromolecular chain sequence and their higher structures. There are plenty of toolkits in biological systems to achieve precise control over the monomer sequence via the highly efficient enzyme-catalyzed reactions. Synthetic chemists inspired by nature have also developed various strategies to synthesize sequence-controlled polymers and their self-assemblies.113–116 However, the tedious purification processes and the accessibility of only short chain lengths often hinder the application of obtained polymers as robust and tough materials. In stark contrast, the selective multiple H-bonds of nucleobase-containing monomers provide an efficient strategy to achieve the precise control over polymer structures at the molecular level. Depending on fast polymerization and efficient nucleobase interactions, strong and tough polymeric materials such as elastomers and adhesives can be obtained.Our group have demonstrated that selective multiple H-bonds between nucleobase-containing monomers bestowed the attained supramolecular polymer network with prominent mechanical properties, including high toughness, outstanding energy dissipation, excellent damping capacity, and good crack resistance (Fig. 7a).101 Bio-based methyl 10-undecenoate was derived to yield adenine and thymine-functionalized monomers (UDA-A and UDA-T) with long alkyl chains, endowing good monomer solubility and chain flexibility for polymer networks. NMR analyses indicated that efficient binding between UDA-A and UDA-T in chloroform gave an association constant of 36.0 M−1, which was drastically disrupted in DMF. As a result, the covalent “locking” of the precise multiple H-bonds between complementary nucleobases dramatically improved the cross-linking density and activation energy of the formed polymer network. The polymer network prepared in chloroform displayed more eminent mechanical properties, in comparison to the one made in DMF with the same composition but different hydrogen bonding interaction. Thus, precise control over the arrangement of nucleobase-containing monomers in polymer networks is a feasible method to engineering robust polymer materials.
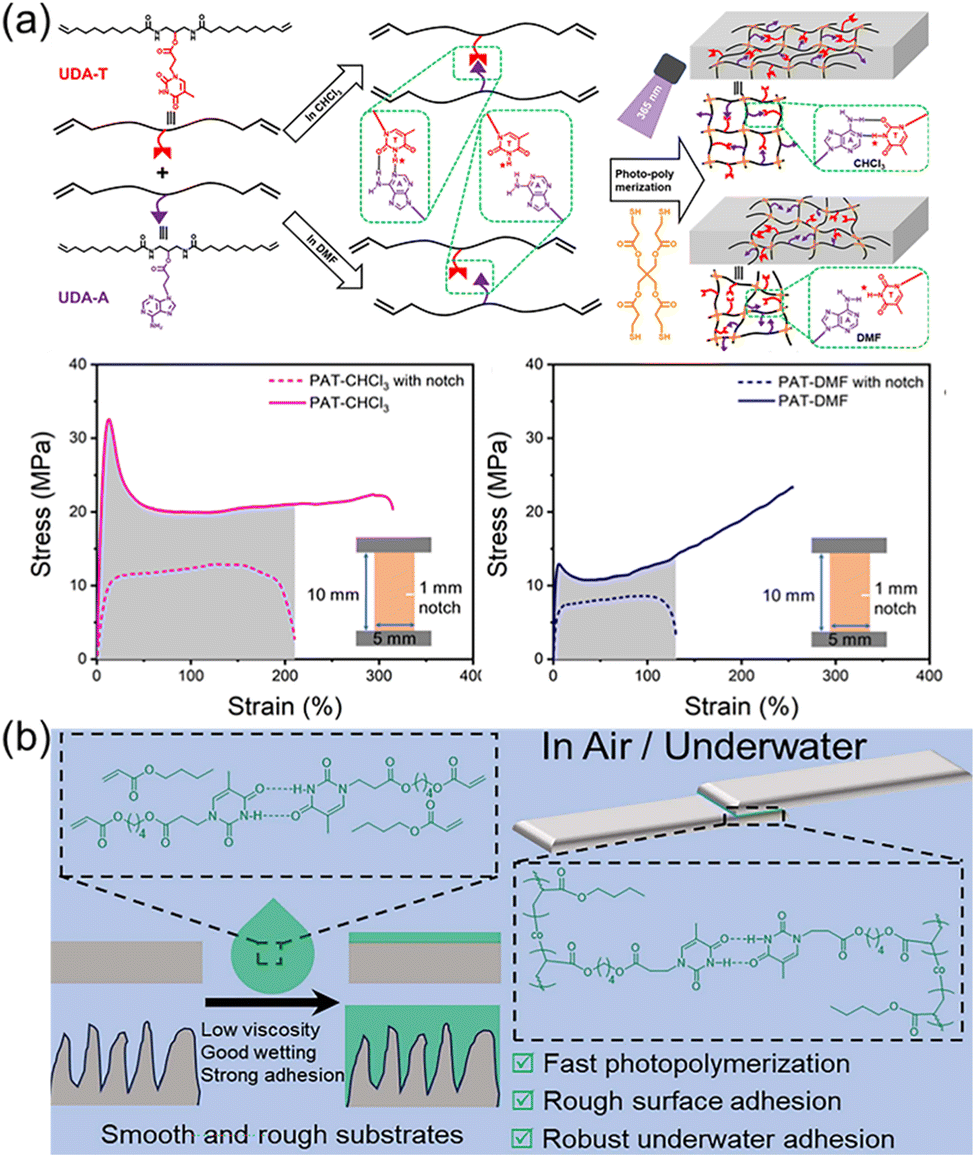 | ||
| Fig. 7 (a) Schematic representation of the nucleobase-containing networks made in the presence or absence of selective hydrogen bonds.101 Adapted from ref. 101 with permission from American Chemical Society, copyright 2024. (b) Schematic illustration showing efficient adhesion on smooth and rough surfaces through utilizing the hydrogen bonds of nucleobases.107 Adapted from ref. 107 with permission from Royal Society of Chemistry, copyright 2024. | ||
Similarly, we have recently developed strong photopolymerized supramolecular adhesives based on the preorganized thymine-containing monomers.107 The nucleobase thymine usually tends to form hydrogen bonds with the complementary nucleobase adenine, but non-complementary hydrogen bonds between thymine and thymine have also been observed in biological systems, such as the wobble region in tRNA recognition. Strong adhesion for both smooth and rough surfaces in air and underwater was achieved by taking full advantage of interfacial wetting properties and supramolecular interactions of nucleobases (Fig. 7b). Strong supramolecular adhesives were fabricated via in situ fast photopolymerization of precursors with well-defined multiple H-bonds between nucleobases. The low viscosity and high monomer concentration of supramolecular precursors not only facilitated the wetting of the substrate, but also were beneficial for the formation of adhesive polymers with high molecular weights. The combination of selective multiple H-bonds and good interfacial wetting of nucleobase monomers is efficient for the formation of strong adhesion under different conditions, opening up a unique avenue for designing robust adhesives.
Inspired by the multiple complementary hydrogen bonds interactions in RNA, Gao et al. improved the adhesion performance of hydrogels by introducing complementary nucleobase interactions.117 They formed polyacrylamide hydrogels with complementary nucleobases by copolymerizing adenine- and uracil-containing acrylate monomers with acrylamide. During the polymerization process, hydrogen bonds were formed between complementary nucleobases. The results showed that as the A and U components increased, the hydrogen bond interactions enhanced the internal cohesion strength of the hydrogels. However, with the excess of A and U, the bonding force tended to decrease, resulting in a decrease in apparent bonding strengths. This was due to dense hydrogen bonds in the hydrogel for strong cohesion, leading to weak interface adhesion and low bonding strength. In addition, the complementary nucleobase-containing hydrogels had remarkable adhesion properties to a variety of materials, including polytetrafluoroethylene (PTFE), plastic, rubber, glass, metal, ceramics, and wood, and can easily and directly adhere to wet biological tissues without any additional processes.
3.2 Effect of multiple H-bonds between complementary nucleobase-containing polymers
Multiple H-bonds between macromolecular chains can simultaneously toughen and enhance polymeric materials through the dynamic dissociation and reformation of H-bonds. However, the synthesis of materials with increasing number of H-bond acceptors and donors is exceedingly difficult.2 Meanwhile, the poor solubility and rigidity of H-bond domain also restrict the fabrication of strong polymer materials limited to mainly double, triple, and quadruple H-bonds. It should be noted that strong multiple H-bonds often drastically compromise and impede the chain mobility. In biological systems, proteins and nucleic acids employ either single or double and triple H-bonds, which endow high chain dynamics for enhanced properties and precise functions. Inspired by this, multiple H-bonds between complementary nucleobase-containing polymers is highly promising to building robust polymeric materials through the rational design of intermolecular and intramolecular selective multiple H-bonds.Many pioneering works about nucleobase-containing polymers in the bulk were carried out by the Long group, utilizing as supramolecular blends and adhesives.28,63,103,110 The copolymerization of n-butyl acrylates (nBA) and thymine- or adenine-containing acrylates monomers was widely explored and the effect of supramolecular multiple H-bonds on the microphase-separated morphologies was thoroughly investigated. In order to incorporate more supramolecular nucleobase interactions, we have incorporated flexible plant oil-based monomers to produce strong adhesives (Fig. 8a).106 The high hydrophobicity of plant oil-based monomers endowed the adhesives with instant strong and responsive underwater adhesion. These bioinspired hydrophobic supramolecular adhesives efficiently removed the interfacial water layer, leading to an immediately strong underwater adhesion. Multiple H-bonds interactions between complementary nucleobase-containing polymers imparted strong cohesive and adhesive capacity to the adhesives within 10 s, avoiding a long curing time. The hydrophobic complementary nucleobase-containing adhesives achieved an efficient adhesion on seven distinct substrates and under various underwater environments including seawater, different pHs, and in the presence of organic solvents. We have also demonstrated that the instant strong and responsive supramolecular adhesives based on nucleobase interactions are promising for urgent underwater adhesion and repair.
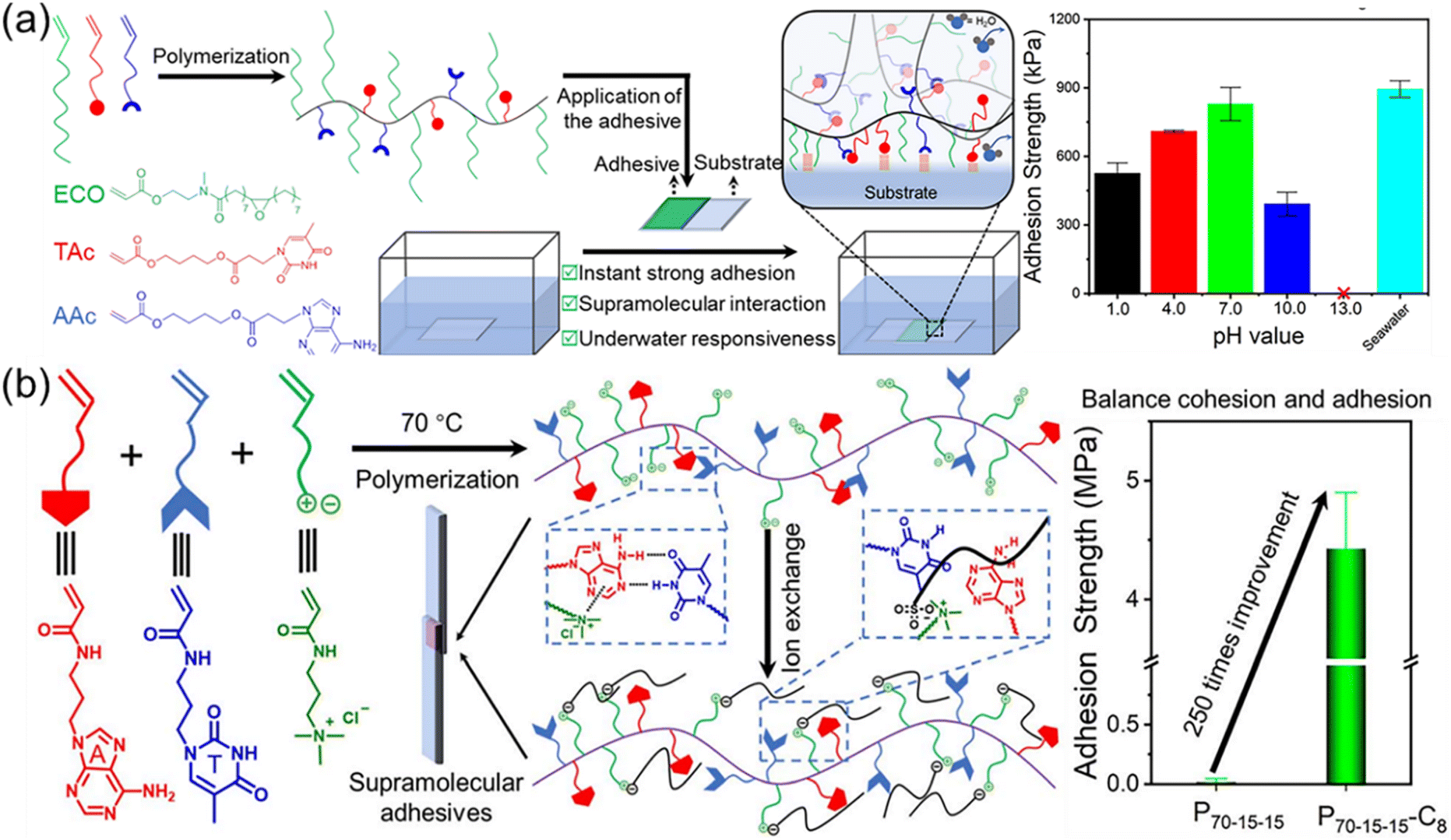 | ||
| Fig. 8 Improvement of adhesion properties through selective multiple hydrogen bonds between complementary nucleobase-containing polymers. (a) Scheme of the preparation of underwater nucleobase-containing supramolecular adhesives with instant strong and responsive bonding.106 Adapted from ref. 106 with permission from American Chemical Society, copyright 2019. (b) Schematic illustration of the fabrication of nucleobase-containing adhesives with enhanced adhesive properties via counter-ion exchange of positively charged polyelectrolytes.104 Adapted from ref. 104 with permission from Royal Society of Chemistry, copyright 2023. | ||
Further, multiple H-bonds between complementary nucleobase-containing polymers were combined with cationic polyelectrolytes to fabricate robust and tunable adhesives (Fig. 8b).104 Inspired by the cooperativity of catechols and positively charged amino acids in the adhesive proteins, we have prepared nucleobase-containing polyelectrolyte adhesives. The counterion exchange of the polyelectrolyte provided an additional handle for optimizing the adhesive and cohesive properties. Hydrogen bonds between complementary nucleobases can also be used to improve the performance of hydrogels from peptides.118 Additionally, Yu et al. synthesized complementary nucleobases (A, T, and U)-containing polymers to tackify hydrogels with degradable polyphosphate mainchains.35 The adhesion tests showed that the adhesion strength of the nucleobase-containing polymer bonded hydrogel was 2–5 times higher than that of the original polyphosphate hydrogel. As the main chain of the polymers was polyphosphate, the hydrogels had good biodegradability. Due to the multiple hydrogen bonds and hydrophobic interactions of purine rings and pyrimidine functional groups, these hydrogels were able to attach to a variety of substrate surfaces and easily peel off without leaving residues when adhesion behavior was no longer required. In addition, polyphosphate hydrogels from polymers with complementary nucleobases were non-toxic and non-irritating to human skin. Cell viability tests showed that these hydrogels were beneficial to cell proliferation, and had great application potentials in wound dressing and tissue sealing.
In addition to polymer adhesives, strong and tough polymer bioplastics can also be attained by employing multiple H-bonds between complementary nucleobase-containing polymers. We have developed a family of nucleobase-containing polymers with homogeneous and efficient energy dissipation networks achieving polymeric materials of simultaneously enhanced stiffness and toughness (Fig. 9).100 A series of nucleobase-containing polymers PA/PT/P(T-co-A) were obtained via an efficient thiol–ene addition polymerization. The statistical copolymers possessing both inter- and intramolecular complementary multiple H-bonds, manifested higher Young's modulus and toughness in contrast to individual thymine-containing polymer PT and the supramolecular mixtures of PA and PT. The excellent properties of the polymeric materials were attributed to the dynamic breakage and reformation of H-bond networks. Rheological analyses suggested that the homogeneous network formed by the copolymer containing complementary nucleobases was more stable and had a higher activated energy than that of the thymine-containing polymer. Therefore, multiple H-bonds between complementary nucleobases imparted outstanding energy dissipation and damping capacity to the polymer materials. Rational design of multiple H-bonds nucleobase-containing polymers provides a bioinspired route to fabricating robust and tough polymeric materials.
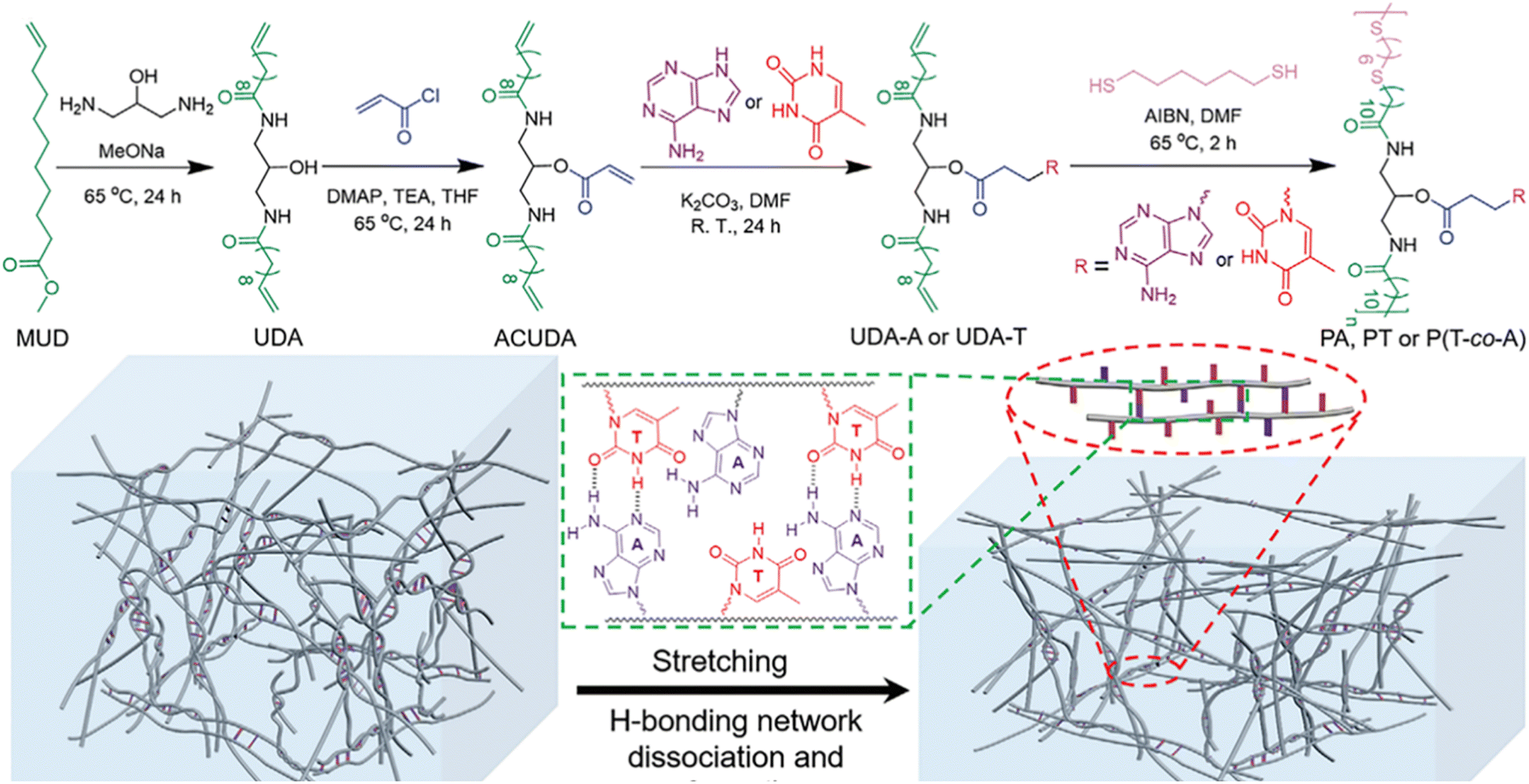 | ||
| Fig. 9 Synthesis of nucleobase-containing polymers and the dissociation and reorganization of hydrogen-bonding networks of nucleobase-containing flexible long-chain copolymers under elongation.100 Adapted from ref. 100 with permission from Wiley-VCH, copyright 2022. | ||
3.3 Effect of multiple H-bonds between nucleobase-containing polymers and nanoparticles
Nature in the long-term evolution has mastered the construction of outstanding nanocomposites such as nacres and bones consisting of nanoparticles and biopolymers. The unique properties such as high strength and toughness offer good inspiration for optimizing materials design. Mimicking exquisite microstructures of natural materials is an efficient approach to fabricating robust synthetic nanocomposites. Additionally, the variety of synthetic nanoparticles also impart additional advantages and properties to the formed nanocomposites. The formation of the strong interfacial interaction in nanocomposites is critical for effective energy dissipation and stress transfer. The supramolecular multiple H-bonds between nucleobases are capable of undergoing fast breakage and reformation upon deformation. Therefore, elaborate interfacial interactions based on nucleobases are supposed to enable the formation of outstanding polymer nanocomposites.Liu et al. have reported an interesting work about inverted organic solar cells (OSCs) by using nanocomposites as cathode interlayer materials (Fig. 10a),119 which were composed of zinc oxide (ZnO) nanoparticles and a nucleobase adenine-containing polymer (PA). Although ZnO nanoparticles were widely used as cathode interlayer materials in inverted OSCs, low temperature processed ZnO nanoparticle films always had large amount of defect states, decreasing the device efficiency and stability. The nucleobase adenine is well known to be adsorbed on metal oxide surfaces but not suitable for the manufacture of solar cells due to its leakage. PA obtained from RAFT polymerization of adenine-containing monomer served as an efficient modifier to passivate the defects, enhance the conductivity, and decrease the work function of the ZnO nanoparticle films. Devices containing ZnO nanoparticle interlayers modified with an optimal PA weight ratio (∼25%) yielded a maximum efficiency of 16.2%, which is among the highest efficiencies reported for inverted OSCs with the non-fullerene active layer. Due to multiple H-bonds between ZnO and PA, the resultant organic–inorganic hybrid composite presented strong power on interfacial modification and energy level alignment at the device interface.
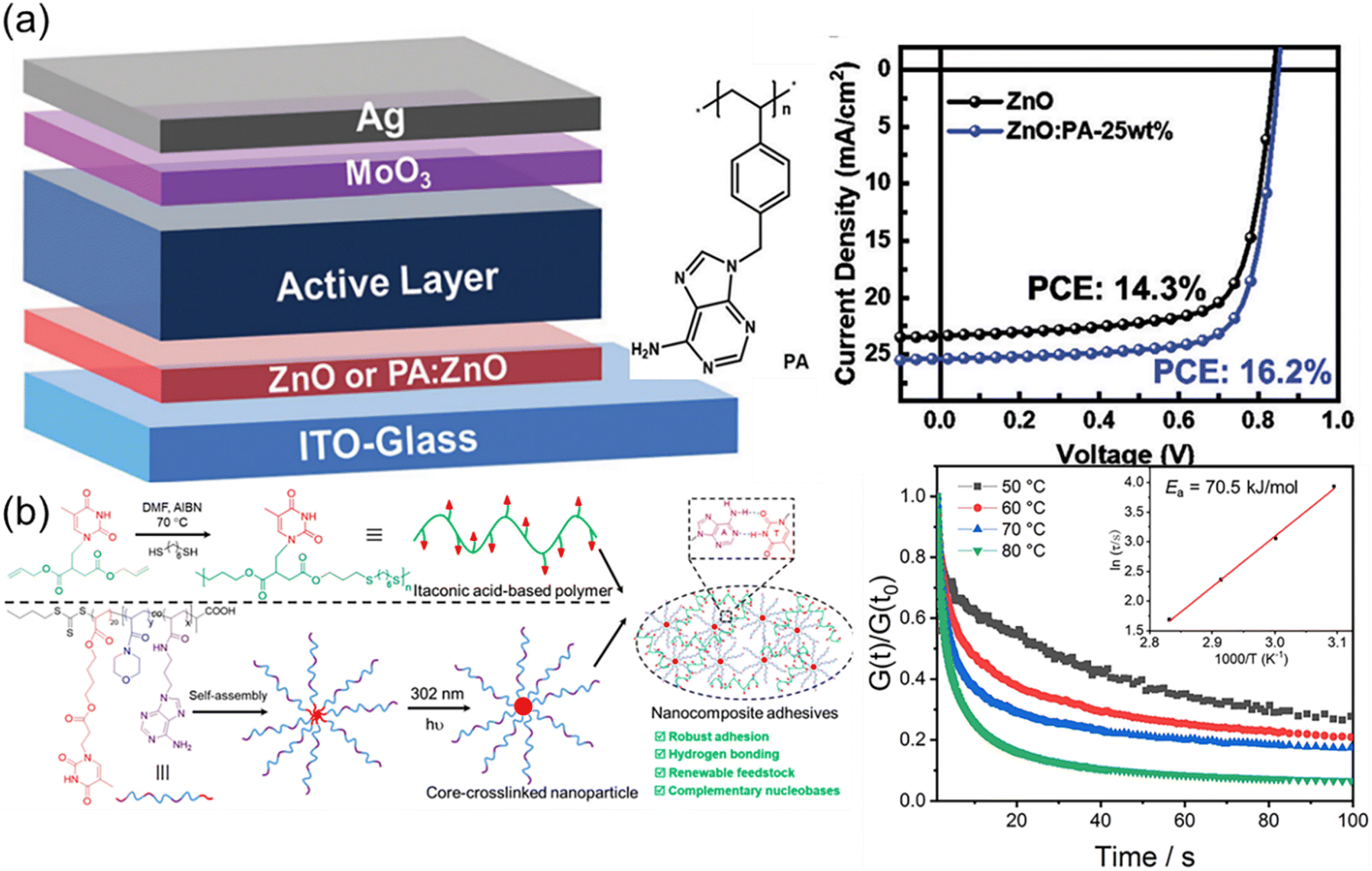 | ||
| Fig. 10 Nanocomposites based on nucleobase-containing polymers. (a) Nanocomposites consisting of adenine-containing polymer PA and ZnO nanoparticles for inverted organic solar cells.119 Adapted from ref. 119 with permission from Royal Society of Chemistry, copyright 2021. (b) Preparation of nucleobase-containing itaconic acid-based polymers and the fabrication of nanocomposite adhesives.120 Adapted from ref. 120 with permission from American Chemical Society, copyright 2023. | ||
Our group have developed robust itaconic acid-based polymer adhesive nanocomposites by utilizing bioinspired multiple H-bonds at the polymer–nanofiller interface (Fig. 10b).120 Itaconic acid, as one of the top 12 biomass-derived molecules, provides a renewable feedstock for the construction of sustainable polymeric materials. Nucleobase-containing itaconic acid-based polymers prepared via thiol–ene addition polymerization only presented moderate adhesive strengths, which could be used as the polymer matrix. Adhesive properties were enhanced by introducing core-cross-linked nanoparticles with variable complementary nucleobases in the corona, giving rise to the adhesive strength as high as 10.6 MPa. The complementary nucleobases at the polymer–nanofiller interface endowed the adhesive nanocomposites with plenty of supramolecular multiple H-bonds to relax the stress. The robust adhesive properties of the nanocomposites were mainly attributed to the high activation energy of the supramolecular network. Therefore, the significance of the rationally designed interfacial interaction was underscored for enhancing the properties of adhesive nanocomposites.
4. Conclusions and outlook
In summary, we have presented the effect of selective multiple H-bonds between complementary nucleobases on the structures and properties of bioinspired and biomimetic nucleobase-containing polymers and materials in both solution and the bulk. Controlling H-bonds between complementary nucleobases in various solutions enables us to modulate the polymer sequence and self-assembly behaviour, achieve templated polymerization, tune nanostructure morphologies and functions, and selectively bind with nucleic acids. Meanwhile, strong and robust polymeric materials including hydrogels, bioplastics, elastomers, adhesives, and coatings can be obtained by optimizing the inter- and intramolecular multiple H-bonds of bioinspired and biomimetic nucleobase-containing polymers. Compared with nucleic acids and ‘XNAs’, various nucleobase-containing polymers are much more straightforward and facile to prepare, modify and scale-up through step-growth or chain polymerizations. Bioinspired and biomimetic nucleobase-containing polymers are highly promising to surrogate nucleic acids and ‘XNAs’ for many applications in nanotechnology, biological science, materials chemistry, to name just a few. However, there are still certain challenges and opportunities in order to achieve more significant impact and more widespread applications of bioinspired and biomimetic nucleobase-containing polymers.Compositional and structural diversity of nucleobase-containing polymers should be further expanded. Currently, the majority of bioinspired and biomimetic nucleobase-containing polymers is based on A:T/U nucleobase interactions. In contrast, the stronger binding of C:G pairs with triple H-bonds is difficult to be incorporated. The strong self-dimerization and poor solubility of guanine in common solvents often give rise to poor control over polymerization. These issues can be addressed either through copolymerization with other soluble monomers or through the efficient polymerization of protected nucleobase monomers followed with deprotection. The incorporation of both A:T/U and C:G nucleobase pairs dramatically increases the molecular design and tunable properties of nucleobase-containing polymers. Furthermore, in addition to linear and bottlebrush polymers, nucleobase-containing polymers with distinct topological architectures can be prepared including multi-arm star polymers, hyperbranched polymers, and so on. Another great challenge is the preparation of sequence-defined nucleobase-containing polymers. Although sequence-defined nucleobase-containing oligomers can be synthesized via RAFT polymerization through single monomer addition, it is still limited to short nucleobase DP of less than 10 depending on tedious chromatography purifications. The high-efficient polymerization with well-defined DNAs as templates might provide a promising route to generate sequence-defined nucleobase-containing polymers just mimicking DNA transcription to RNA.
Further, nucleobase-containing polymers can be explored as effective delivery carriers of nucleoside-type drugs in aqueous solution. Although the loading and delivery of nucleic acids with nucleobase-containing polymers are achieved with high efficiency and low toxicity, the selective multiple H-bonds between nucleobase-containing polymers and nucleoside-type drugs have not been explored in-depth. Notably, nucleoside-type drugs are an important class of drugs widely used for cancer or antiviral therapy such as clofarabine, gemcitabine, doxifluridine, and so on. In contrast to the drug loading based on hydrophobic interaction, multiple H-bonds between nucleoside-type drugs and nucleobase-containing polymer carriers might improve the loading capacity and achieve the controlled release at the targeted site. Meanwhile, the diversity of polymerization techniques also makes it possible to produce hydrophilic nucleobase-containing polymers for loading nucleoside-type drugs.
Nucleobase-containing polymers serve as robust and tough materials including hydrogels, bioplastics, elastomers, adhesives, and coatings, for which the degradation and/or recyclability are also highly pursued to overcome the tremendous environmental challenges. Currently, most of nucleobase-containing polymers have stable backbones, which show strong resistance to degradation. Distinct strategies can be employed to resolve the issues for the development of sustainable nucleobase-containing polymers or materials. The simple way should be the copolymerization of nucleobase-containing vinyl monomers with degradable monomers including cyclic ketene acetals and dibenzo-[c,e]-oxepane-5-thione. The inserted ester or thioester groups in the backbone facilitate the breakage of the formed polymers. In addition, new nucleobase-containing monomers from lipoic acids or lactones are also good candidates to produce degradable nucleobase-containing polymers and materials using ring opening polymerization. Meanwhile, the recyclability of the obtained polymer into the cyclic feedstocks is expected to be achieved in a closed-loop manner. In addition to the low environmental burden, multiple H-bonds of degradable nucleobase-containing polymers endows the relevant materials with outstanding mechanical properties.
Looking forward, selective multiple H-bonds of nucleobase-containing polymers can also be employed to reversibly alter surface properties. Grafting of polymers is widely utilized to modify surface properties such as wettability, adhesion, responsiveness, compatibility, and so on. The properties of the obtained surface are determined by the chemical composition of attached polymers, which is difficult to change, limiting the design of smart surfaces and materials. To address the issue, rewritable polymer-attached surfaces with cleavable/dynamic covalent bonds or common supramolecular host–guest interactions were developed. However, the large steric hindrance of small reactive groups usually gives rise to the low grafting density and efficiency. Multiple H-bonds of nucleobase-containing polymers is capable of achieving rewritable surface properties by attaching complementary nucleobase-containing polymers on the surface. The large H-bonding interaction domain facilitates the efficient interaction for bonding. Meanwhile, multiple H-bonds between nucleobase-containing polymers is expected to generate the strong bonding over common covalent bonds. Rewritable surface properties will be achieved by using solvents with different hydrogen-bond supporting ability. Collectively, multiple H-bonds of bioinspired and biomimetic complementary nucleobases provide various efficient approaches to modulating the properties of polymers and materials. We expect that by rationally designing multiple H-bonds nucleobase interactions this kind of new polymers and materials are able to find more widespread applications in chemistry, materials science, biomedicines, and so on.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
N. Yao, J. Wu and Z. Hua performed the literature search, analyzed the published results, and wrote the manuscript. G. Liu and Z. Hua provided key advice and supervised the preparation of the text.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22373003, 22273098, 22103002, 52033001). Z. H. also acknowledged the financial support of Anhui Provincial Natural Science Foundation (No. 2408085Y004).Notes and references
- L. L. Yang, X. X. Tan, Z. Q. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef PubMed.
- Y. X. Liu, L. L. Wang, L. Zhao, Y. G. Zhang, Z. T. Li and F. H. Huang, Chem. Soc. Rev., 2024, 53, 1592–1623 RSC.
- M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna and E. Charpentier, Science, 2012, 337, 816–821 CrossRef PubMed.
- H. Nishimasu, F. A. Ran, P. D. Hsu, S. Konermann, S. I. Shehata, N. Dohmae, R. Ishitani, F. Zhang and O. Nureki, Cell, 2014, 156, 935–949 CrossRef PubMed.
- A. Fire, S. Q. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver and C. C. Mello, Nature, 1998, 391, 806–811 CrossRef.
- V. Jadhav, A. Vaishnaw, K. Fitzgerald and M. A. Maier, Nat. Biotechnol., 2024, 42, 394–405 CrossRef.
- N. C. Seeman, J. Theor. Biol., 1982, 99, 237–247 CrossRef.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS.
- M. Madsen and K. V. Gothelf, Chem. Rev., 2019, 119, 6384–6458 CrossRef CAS.
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497–1500 CrossRef CAS PubMed.
- K. U. Schöning, P. Scholz, S. Guntha, X. Wu, R. Krishnamurthy and A. Eschenmoser, Science, 2000, 290, 1347–1351 CrossRef.
- L. L. Zhang, A. Peritz and E. Meggers, J. Am. Chem. Soc., 2005, 127, 4174–4175 CrossRef CAS PubMed.
- I. K. Astakhova, L. H. Hansen, B. Vester and J. Wengel, Org. Biomol. Chem., 2013, 11, 4240–4249 RSC.
- R. McHale and R. K. O'Reilly, Macromolecules, 2012, 45, 7665–7675 CrossRef.
- L. V. Arsenie, V. Ladmiral, P. Lacroix-Desmazes and S. Catrouillet, Polym. Chem., 2022, 13, 5798–5810 RSC.
- A. Sikder, C. Esen and R. K. O'Reilly, Acc. Chem. Res., 2022, 55, 1609–1619 CrossRef PubMed.
- J. J. Li, Z. K. Wang, Z. Hua and C. B. Tang, J. Mater. Chem. B, 2020, 8, 1576–1588 RSC.
- F. B. Ilhami, Y. S. Birhan and C. C. Cheng, ACS Biomater. Sci. Eng., 2023, 10, 234–254 CrossRef.
- P. Herdewijn and P. Marlière, Chem. Biodivers., 2009, 6, 791–808 CrossRef PubMed.
- J. C. Chaput and P. Herdewijn, Angew. Chem., Int. Ed., 2019, 58, 11570–11572 CrossRef.
- G. Singh and V. Monga, Bioorg. Chem., 2023, 141, 106860 CrossRef.
- F. Wang, L. S. Liu, C. H. Lau, T. J. H. Chang, D. Y. Tam, H. M. Leung, C. Tin and P. K. Lo, ACS Appl. Mater. Interfaces, 2019, 11, 38510–38518 CrossRef.
- H. S. Bazzi and H. F. Sleiman, Macromolecules, 2002, 35, 9617–9620 CrossRef.
- J. F. Lutz, A. F. Thünemann and K. Rurack, Macromolecules, 2005, 38, 8124–8126 CrossRef.
- H. J. Spijker, F. L. van Delft and J. C. M. van Hest, Macromolecules, 2007, 40, 12–18 CrossRef.
- C. R. South and M. Weck, Macromolecules, 2007, 40, 1386–1394 CrossRef.
- P. K. Lo and H. F. Sleiman, J. Am. Chem. Soc., 2009, 131, 4182–4183 CrossRef.
- S. J. Cheng, M. Q. Zhang, N. Dixit, R. B. Moore and T. E. Long, Macromolecules, 2012, 45, 805–812 CrossRef.
- W. X. Xi, S. Pattanayak, C. Wang, B. Fairbanks, T. Gong, J. Wagner, C. J. Kloxin and C. N. Bowman, Angew. Chem., Int. Ed., 2015, 54, 14462–14467 CrossRef.
- Z. Hua, A. Pitto-Barry, Y. Kang, N. Kirby, T. R. Wilks and R. K. O'Reilly, Polym. Chem., 2016, 7, 4254–4262 RSC.
- X. Han, B. D. Fairbanks, J. Sinha and C. N. Bowman, Macromol. Rapid Commun., 2020, 41, 2000327 CrossRef PubMed.
- S. Chea, K. Schade, S. Reinicke, R. Bleul and R. R. Rosencrantz, Polym. Chem., 2022, 13, 5058–5067 RSC.
- F. Ilhan, T. H. Galow, M. Gray, G. Clavier and V. M. Rotello, J. Am. Chem. Soc., 2000, 122, 5895–5896 CrossRef.
- C. W. Huang, W. Y. Ji and S. W. Kuo, Macromolecules, 2017, 50, 7091–7101 CrossRef.
- W. L. Wang, S. R. Liu, B. G. Chen, X. X. Yan, S. G. Li, X. J. Ma and X. F. Yu, Biomacromolecules, 2019, 20, 3672–3683 CrossRef.
- M. Wang, B. N. Choi, X. H. Wei, A. C. Feng and S. H. Thang, Polym. Chem., 2018, 9, 5086–5094 RSC.
- I. H. Lin, C. C. Cheng, C. W. Huang, M. C. Liang, J. K. Chen, F. H. Ko, C. W. Chu, C. F. Huang and F. C. Chang, RSC Adv., 2013, 3, 12598–12603 RSC.
- F. B. Ilhami, E. A. Bayle and C. C. Cheng, Pharmaceutics, 2021, 13, 1929 CrossRef.
- H. R. Culver, J. Sinha, T. R. Prieto, C. J. Calo, B. D. Fairbanks and C. N. Bowman, Biomacromolecules, 2020, 21, 4205–4211 CrossRef PubMed.
- Z. Z. Liu, B. Fairbanks, L. C. He, T. Liu, P. Shah, J. N. Cha, J. W. Stansbury and C. N. Bowman, Chem. Commun., 2017, 53, 10156–10159 RSC.
- A. Harguindey, D. W. Domaille, B. D. Fairbanks, J. Wagner, C. N. Bowman and J. N. Cha, Adv. Mater., 2017, 29, 1700743 CrossRef.
- I. Abeysekera, L. Maes and T. Junkers, Polym. Chem., 2024, 15, 2129–2139 RSC.
- L. V. Arsenie, M. Semsarilar, J. C. Brendel, P. Lacroix-Desmazes, V. Ladmiral and S. Catrouillet, Polym. Chem., 2022, 13, 5604–5615 RSC.
- C. C. Cheng, J. J. Huang, Z. S. Liao, S. Y. Huang, D. J. Lee and Z. Xin, Polym. Chem., 2017, 8, 1454–1459 RSC.
- X. Liu, Q. Zhang, L. J. Duan and G. H. Gao, Adv. Funct. Mater., 2019, 29, 1900450 CrossRef.
- X. Liu, Q. Zhang and G. H. Gao, Adv. Funct. Mater., 2017, 27, 1703132 CrossRef.
- X. Han, D. W. Domaille, B. D. Fairbanks, L. C. He, H. R. Culver, X. P. Zhang, J. N. Cha and C. N. Bowman, Biomacromolecules, 2018, 19, 4139–4146 CrossRef PubMed.
- W. T. Smith Jr, Prog. Polym. Sci., 1996, 21, 209–253 CrossRef.
- Y. Inaki, Prog. Polym. Sci., 1992, 17, 515–570 CrossRef.
- D. Fong, Z. Hua, T. R. Wilks, R. K. O'Reilly and A. Adronov, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2611–2617 CrossRef.
- H. T. Yang and W. X. Xi, Polymers, 2017, 9, 666 CrossRef PubMed.
- Y. Kang, A. Lu, A. Ellington, M. C. Jewett and R. K. O'Reilly, ACS Macro Lett., 2013, 2, 581–586 CrossRef.
- L. Wang, S. Huang, M. Wang, Z. Y. Liu, X. M. Chen and H. Yang, Macromolecules, 2021, 54, 341–350 CrossRef.
- L. Wang, M. Wang, L. X. Guo, Y. Sun, X. Q. Zhang, B. P. Lin and H. Yang, Macromolecules, 2019, 52, 649–659 CrossRef.
- Y. Y. Tan, Prog. Polym. Sci., 1994, 19, 561–588 CrossRef.
- A. Khan, D. M. Haddleton, M. J. Hannon, D. Kukulj and A. Marsh, Macromolecules, 1999, 32, 6560–6564 CrossRef.
- A. Marsh, A. Khan, M. Garcia and D. M. Haddleton, Chem. Commun., 2000, 21, 2083–2084 RSC.
- R. J. Thibault, T. H. Galow, E. J. Turnberg, M. Gray, P. J. Hotchkiss and V. M. Rotello, J. Am. Chem. Soc., 2002, 124, 15249–15254 CrossRef PubMed.
- S. Polowinski, Prog. Polym. Sci., 2002, 27, 537–577 CrossRef.
- J. F. Lutz, A. F. Thünemann and R. Nehring, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4805–4818 CrossRef.
- H. J. Spijker, A. J. Dirks and J. C. M. van Hest, Polymer, 2005, 46, 8528–8535 CrossRef.
- H. J. Spijker, A. J. Dirks and J. C. M. Van Hest, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4242–4250 CrossRef.
- B. D. Mather, M. B. Baker, F. L. Beyer, M. A. G. Berg, M. D. Green and T. E. Long, Macromolecules, 2007, 40, 6834–6845 CrossRef.
- K. Saito, L. R. Ingalls, J. Lee and J. C. Warner, Chem. Commun., 2007, 24, 2503–2505 RSC.
- G. Kaur, S. L. Y. Chang, T. D. M. Bell, M. T. W. Hearn and K. Saito, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4121–4128 CrossRef.
- R. McHale, J. P. Patterson, P. B. Zetterlund and R. K. O'Reilly, Nat. Chem., 2012, 4, 491–497 CrossRef PubMed.
- C. M. E. Kriebisch, L. Burger, O. Zozulia, M. Stasi, A. Floroni, D. Braun, U. Gerland and J. Boekhoven, Nat. Chem., 2024, 16, 1240–1249 CrossRef.
- M. Garcia, K. Kempe, D. M. Haddleton, A. Khan and A. Marsh, Polym. Chem., 2015, 6, 1944–1951 RSC.
- F. A. Ran, P. D. Hsu, J. Wright, V. Agarwala, D. A. Scott and F. Zhang, Nat. Protoc., 2013, 8, 2281–2308 CrossRef PubMed.
- J. A. Doudna and E. Charpentier, Science, 2014, 346, 1258096 CrossRef PubMed.
- P. F. Zhan, A. Peil, Q. Jiang, D. F. Wang, S. Mousavi, Q. C. Xiong, Q. Shen, Y. X. Shang, B. Q. Ding, C. X. Lin, Y. G. Ke and N. Liu, Chem. Rev., 2023, 123, 3976–4050 CrossRef PubMed.
- J. H. Ji, D. Karna and H. B. Mao, Chem. Soc. Rev., 2021, 50, 11966–11978 RSC.
- Y. Kang, A. Pitto-Barry, M. S. Rolph, Z. Hua, I. Hands-Portman, N. Kirby and R. K. O'Reilly, Polym. Chem., 2016, 7, 2836–2846 RSC.
- Z. Hua, R. Keogh, Z. Li, T. R. Wilks, G. S. Chen and R. K. O'Reilly, Macromolecules, 2017, 50, 3662–3670 CrossRef PubMed.
- B. T. Gebeyehu, S. Y. Huang, A. W. Lee, J. K. Chen, J. Y. Lai, D. J. Lee and C. C. Cheng, Macromolecules, 2018, 51, 1189–1197 CrossRef.
- M. Wang, B. Choi, Z. H. Sun, X. H. Wei, A. C. Feng and S. H. Thang, Chem. Commun., 2019, 55, 1462–1465 RSC.
- Y. Y. Yan, C. Gao, J. J. Li, T. Zhang, G. Yang, Z. K. Wang and Z. Hua, Biomacromolecules, 2020, 21, 613–620 CrossRef.
- Z. Hua, J. R. Jones, M. Thomas, M. C. Arno, A. Souslov, T. R. Wilks and R. K. O'Reilly, Nat. Commun., 2019, 10, 5406 CrossRef.
- C. Yang, Y. Du, Q. Li, X. Gao, P. Zha, W. Zhan, K. Liu, F. Bi, Z. Hua and G. Yang, ACS Macro Lett., 2024, 13, 468–474 CrossRef.
- S. Varlas, Z. Hua, J. R. Jones, M. Thomas, J. C. Foster and R. K. O'Reilly, Macromolecules, 2020, 53, 9747–9757 CrossRef.
- Y. Yan, X. Fang, N. Yao, H. Gu, G. Yang and Z. Hua, Macromolecules, 2022, 55, 7798–7805 CrossRef.
- T. A. Tockary, W. Foo, A. Dirisala, Q. X. Chen, S. Uchida, S. Osawa, Y. Mochida, X. Y. Liu, H. Kinoh, H. Cabral, K. Osada and K. Kataoka, ACS Nano, 2019, 13, 12732–12742 CrossRef PubMed.
- S. Phunpee, S. Chirachanchai and U. R. Ruktanonchai, Langmuir, 2022, 38, 5915–5923 CrossRef PubMed.
- A. Aayush, S. Darji, K. M. Estes, E. Yeh and D. H. Thompson, Biomacromolecules, 2024, 25, 5729–5744 CrossRef PubMed.
- X. M. Cai, R. Dou, C. Guo, J. R. Tang, X. J. Li, J. Chen and J. Y. Zhang, Pharmaceutics, 2023, 15, 1502 CrossRef PubMed.
- J. E. Dahlman, C. Barnes, O. F. Khan, A. Thiriot, S. Jhunjunwala, T. E. Shaw, Y. P. Xing, H. B. Sager, G. Sahay, L. Speciner, A. Bader, R. L. Bogorad, H. Yin, T. Racie, Y. Z. Dong, S. Jiang, D. Seedorf, A. Dave, K. S. Sandhu, M. J. Webber, T. Novobrantseva, V. M. Ruda, A. K. R. Lytton-Jean, C. G. Levins, B. Kalish, D. K. Mudge, M. Perez, L. Abezgauz, P. Dutta, L. Smith, K. Charisse, M. W. Kieran, K. Fitzgerald, M. Nahrendorf, D. Danino, R. M. Tuder, U. H. von Andrian, A. Akinc, D. Panigrahy, A. Schroeder, V. Koteliansky, R. Langer and D. G. Anderson, Nat. Nanotechnol., 2014, 9, 648–655 CrossRef PubMed.
- K. E. Bujold, J. Fakhoury, T. G. W. Edwardson, K. M. M. Carneiro, J. N. Briard, A. G. Godin, L. Amrein, G. D. Hamblin, L. C. Panasci, P. W. Wiseman and H. F. Sleiman, Chem. Sci., 2014, 5, 2449–2455 RSC.
- A. Harguindey, D. W. Domaille, B. D. Fairbanks, J. Wagner, C. N. Bowman and J. N. Cha, Adv. Mater., 2017, 29, 1700743 CrossRef.
- A. Harguindey, S. Roy, A. W. Harris, B. D. Fairbanks, A. P. Goodwin, C. N. Bowman and J. N. Cha, Biomacromolecules, 2019, 20, 1683–1690 CrossRef PubMed.
- S. Mavila, H. R. Culver, A. J. Anderson, T. R. Prieto and C. N. Bowman, Angew. Chem., Int. Ed., 2022, 61, e202110741 CrossRef PubMed.
- D. Wang, J. H. Cui, M. Z. Gan, Z. H. Xue, J. Wang, P. F. Liu, Y. Hu, Y. Pardo, S. Hamada, D. Y. Yang and D. Luo, J. Am. Chem. Soc., 2020, 142, 10114–10124 CrossRef PubMed.
- J. P. Han, Y. F. Guo, H. Wang, K. Y. Zhang and D. Y. Yang, J. Am. Chem. Soc., 2021, 143, 19486–19497 CrossRef PubMed.
- Y. J. Li, R. F. Chen, B. N. Zhou, Y. C. Dong and D. S. Liu, Adv. Mater., 2024, 36, 2307129 CrossRef PubMed.
- B. N. Zhou, B. Yang, Q. Liu, L. Jin, Y. Shao, T. Y. Yuan, Y. N. Zhang, C. Wang, Z. W. Shi, X. Li, Y. F. Pan, N. Qiao, J. F. Xu, Y. R. Yang, Y. C. Dong, L. J. Xu, S. B. Gui and D. S. Liu, J. Am. Chem. Soc., 2023, 145, 8954–8964 CrossRef PubMed.
- T. Giraud, S. Bouguet-Bonnet, M. J. Stébé, L. Richaudeau, G. Pickaert, M. C. Averlant-Petit and L. Stefan, Nanoscale, 2021, 13, 10566–10578 RSC.
- H. H. Kuang, H. Y. He, Z. Y. Zhang, Y. X. Qi, Z. G. Xie, X. B. Jinga and Y. B. Huang, J. Mater. Chem. B, 2014, 2, 659–667 RSC.
- X. Ye, X. Li, Y. Shen, G. Chang, J. Yang and Z. Gu, Polymer, 2017, 108, 348–360 CrossRef CAS.
- J. J. Li, J. Q. Chen, J. Wu, H. D. Lei, Y. T. Tian, G. Yang, Z. K. Wang and Z. Hua, Polym. Chem., 2021, 12, 3533–3543 RSC.
- L. Chen, H. Peng, Y. Y. Wei, X. Y. Wang, Y. J. Jin, H. T. Liu and Y. Jiang, Macromol. Chem. Phys., 2019, 220, 1900280 CrossRef.
- Y. T. Tian, J. Wu, X. Z. Fang, L. H. Guan, N. Yao, G. Yang, Z. K. Wang, Z. Hua and G. M. Liu, Adv. Funct. Mater., 2022, 32, 2112741 CrossRef CAS.
- J. Wu, Z. Dong, X. Y. Shen, M. Y. Zhao, F. X. Zeng, Y. Tao, L. Lu, Z. Hua and G. M. Liu, Macromolecules, 2024, 57, 1761–1769 CrossRef CAS.
- J. Wu, F. X. Zeng, Z. Y. Fan, S. H. Xuan, Z. Hua and G. M. Liu, Adv. Funct. Mater., 2024, 2410518 CrossRef.
- K. Zhang, G. B. Fahs, E. Margaretta, A. G. Hudson, R. B. Moore and T. E. Long, J. Adhes., 2019, 95, 146–167 CrossRef CAS.
- Z. Dong, J. Wu, X. Shen, Z. Hua and G. Liu, Chem. Sci., 2023, 14, 3938–3948 RSC.
- J. Wu, H. Lei, J. Li, Z. Zhang, G. Zhu, G. Yang, Z. Wang and Z. Hua, Chem. Eng. J., 2021, 405, 126976 CrossRef CAS.
- J. Wu, H. Lei, X. Fang, B. Wang, G. Yang, R. K. O'Reilly, Z. Wang, Z. Hua and G. Liu, Macromolecules, 2022, 55, 2003–2013 CrossRef.
- M. Zhao, J. Wu, F. Zeng, Z. Dong, X. Shen, Z. Hua and G. Liu, Chem. Sci., 2024, 15, 6445–6453 RSC.
- N. Ishikawa, M. Furutani and K. Arimitsu, ACS Macro Lett., 2015, 4, 741–744 CrossRef PubMed.
- H. Lei, N. Yao, S. Wang, X. Fang, J. Wu, G. Yang and Z. Hua, Chem. Eng. J., 2023, 471, 144602 CrossRef.
- K. R. Zhang, M. Aiba, G. B. Fahs, A. G. Hudson, W. D. Chiang, R. B. Moore, M. Ueda and T. E. Long, Polym. Chem., 2015, 6, 2434–2444 RSC.
- K. R. Zhang, S. J. Talley, Y. P. Yu, R. B. Moore, M. Murayama and T. E. Long, Chem. Commun., 2016, 52, 7564–7567 RSC.
- K. R. Zhang, G. B. Fahs, M. Aiba, R. B. Moore and T. E. Long, Chem. Commun., 2014, 50, 9145–9148 RSC.
- J. Lawrence, S. H. Lee, A. Abdilla, M. D. Nothling, J. M. Ren, A. S. Knight, C. Fleischmann, Y. L. Li, A. S. Abrams, B. Schmidt, M. C. Hawker, L. A. Connal, A. J. McGrath, P. G. Clark, W. R. Gutekunst and C. J. Hawker, J. Am. Chem. Soc., 2016, 138, 6306–6310 CrossRef.
- Z. X. Huang, B. B. Noble, N. Corrigan, Y. Y. Chu, K. Satoh, D. S. Thomas, C. J. Hawker, G. Moad, M. Kamigaito, M. L. Coote, C. Boyer and J. T. Xu, J. Am. Chem. Soc., 2018, 140, 13392–13406 CrossRef PubMed.
- E. Maron, J. H. Swisher, J. J. Haven, T. Y. Meyer, T. Junkers and H. G. Börner, Angew. Chem., Int. Ed., 2019, 58, 10747–10751 CrossRef PubMed.
- L. Maes, D. M. Roqeuro, L. M. Pitet, P. Adriaensens and T. Junkers, Polym. Chem., 2020, 11, 2027–2033 RSC.
- X. Liu, Q. Zhang, K. Li, L. Duan and G. Gao, Chem.–Eur. J., 2018, 24, 15119–15125 CrossRef.
- T. Giraud, S. Bouguet-Bonnet, M.-J. Stébé, L. Richaudeau, G. Pickaert, M.-C. Averlant-Petit and L. Stefan, Nanoscale, 2021, 13, 10566–10578 RSC.
- Y. Wang, M. Liu, Z. Chen and Y. Liu, J. Mater. Chem. C, 2021, 9, 11851–11858 RSC.
- Z. Hua and X. Fang, Macromolecules, 2023, 56, 8047–8053 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |