
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-triggered NO release of nitrosyl complexes bearing first-row transition metals and therapeutic applications
Seungwon
Sun
 a,
Jisu
Choe
a and
Jaeheung
Cho
*ab
a,
Jisu
Choe
a and
Jaeheung
Cho
*ab
aDepartment of Chemistry, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea. E-mail: jaeheung@unist.ac.kr
bGraduate School of Carbon Neutrality, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea
First published on 7th November 2024
Abstract
In biological systems, nitric oxide (NO) is a crucial signaling molecule that regulates a wide range of physiological and pathological processes. Given the significance of NO, there has been considerable interest in delivering NO exogenously, particularly through light as a non-invasive therapeutic approach. However, due to the high reactivity and instability of NO under physiological conditions, directly delivering NO to targeted sites remains challenging. In recent decades, photo-responsive transition metal–nitrosyl complexes, especially based on first-row transition metals such as Mn, Fe, and Co, have emerged as efficient NO donors, offering higher delivery efficiency and quantum yields than heavy metal–nitrosyl complexes under light exposure. This review provides a comprehensive overview of current knowledge and recent developments in the field of photolabile first-row transition metal–nitrosyl complexes, focusing on the structural and electronic properties, photoreactivity, photodissociation mechanisms, and potential therapeutic applications. By consolidating the key features of photoactive nitrosyl complexes, the review offers deeper insights and highlights the potential of first-row transition metal–nitrosyl complexes as versatile tools for photo-triggered NO delivery.
 Seungwon Sun | Seungwon Sun completed his BS degree in Chemistry at Chungnam National University in 2020. He is currently an integrated MS–PhD candidate under the supervision of Professor Jaeheung Cho at the Ulsan National Institute of Science and Technology (UNIST). His research focuses on nitric oxide delivery and mechanistic investigations of metalloenzymes through biomimetic systems. |
 Jisu Choe | Jisu Choe received his PhD degree from Daegu Gyeongbuk Institute of Science and Technology (DGIST) in 2023. He is currently a postdoctoral researcher in the Department of Chemistry at Ulsan National Institute of Science and Technology (UNIST). His research focuses on the design and synthesis of metal–nitrosyl complexes with first-row transition metals and optimization of photolabile NO donors for applications in biological systems. |
 Jaeheung Cho | Jaeheung Cho received his PhD degree in inorganic chemistry from Kanazawa University in Japan (2005), under the supervision of Prof. Masatatsu Suzuki. He then completed a postdoctoral fellowship at the University of Delaware in US (2005–2007). Upon returning to Korea, he joined Ewha Womans University as a Full-Time Lecturer and Specially Appointed Professor (2007–2012). In 2012, he began his independent career at DGIST, where he served as an Assistant Professor and later became an Associate Professor in 2017. He joined the Department of Chemistry at UNIST as a Professor in 2020. His research focuses on synthetic bioinorganic chemistry for catalysis and drug discovery. |
1. Introduction
Nitric oxide (NO), an endogenously generated gaseous signaling molecule, plays an important role in various physiological and pathophysiological processes in biological systems. Over the past few decades, NO has attracted significant attention due to diverse biological roles. The importance of NO was highlighted in 1998 when Louis J. Ignarro, Robert F. Furchgott, and F. Murad were awarded the Nobel Prize for the discovery of NO as an endothelial-derived relaxation factor (EDRF), which regulates vasodilation in smooth muscle cells and contributes to lower blood pressure.1,2 After the discovery of the vasorelaxation effect by NO, subsequent research has expanded our understanding of the physiological functions of NO, revealing involvement in neurotransmission, immune defense, and other critical biological processes.3–5NO is tightly and precisely regulated in biological systems, where NO performs a variety of essential functions depending on its concentration. At nanomolar (nM) levels, NO modulates neurotransmission and blood pressure, while at micromolar (μM) concentrations, NO acts as an immune defense agent against invading pathogens.6,7 The multifaceted roles of NO are primarily mediated by chemical interactions with transition metal sites in metalloenzymes. In the vasodilation process, for example, NO interacts with an iron site in soluble guanylate cyclase (sGC). NO diffuses across the cell membrane and coordinates to the heme site in sGC, leading to the generation of cyclic guanosine-3′,5′-monophosphate (cGMP), which facilitates blood pressure relaxation.8,9
Beyond the roles of NO in mammals, NO regulates enzymatic processes in bacteria. A prominent example is an iron-containing nitrile hydratase (Fe-NHase), a metalloenzyme found in microorganisms such as Rhodococcus sp. N-771, N-774, and R-312. Fe-NHase catalyzes the conversion of organic nitriles into amides via hydrolysis reaction, which is photo-regulated by the reversible interaction of NO with the nonheme Fe center as shown in Scheme 1. In the absence of light, NO binds to the Fe active site to form a nitrosylated complex in a catalytically inactive form. Upon exposure to light, the Fe–NO bond is cleaved, restoring catalytic activity by generating a reactive FeIII–hydroxo (FeIII–OH) intermediate.10–12
 | ||
| Scheme 1 Photo-regulated transformation between the active and inactive forms of the Fe-NHase enzyme for catalytic reaction. NO reversibly interacts with the nonheme Fe center. | ||
The diverse biological roles of NO and photoactivation of Fe–NO bond have spurred significant interest in developing NO delivery systems for therapeutic applications.13 However, artificially controlling NO concentration in biological systems remains challenging due to the very short half-life time and high reactivity of NO under physiological conditions. To overcome the aforementioned limitation of NO delivery, various NO donors have been explored, including organic nitrates (RO-NO2), S-nitrosothiols (RSNOs), N-diazeniumdiolates (NONOates), and transition metal-based NO donors.14–21 While organic NO donors have been approved for clinical uses, spontaneous NO release in biological environments complicates the precise control of NO concentrations at target sites. In contrast, transition metal–nitrosyl (M–NO) complexes have attracted attention for the ability to offer thermodynamic stability to NO by coordinating with metal centers, allowing for selective delivery through external stimuli such as light, solvation, pH, and heat.22,23 Among the numerous transition metals, first-row transition ions are preferred for medical applications because of the relatively lower cytotoxicity compared to heavy metals such as ruthenium (Ru),24–26 platinum (Pt),27,28 palladium (Pd),29 and osmium (Os).30,31 Moreover, first-row transition M–NO complexes bearing manganese (Mn), iron (Fe), and cobalt (Co) represent a particularly promising approach, offering a non-invasiveness and easily controllable method for precise spatiotemporal control of NO delivery.
Identification and characterization of nitrosyl complexes are essential in bioinorganic chemistry to enhance our understanding of the chemical properties of M–NO complexes and photodissociation reactions. Although several review articles have examined photoactive M–NO complexes, the majority have focused primarily on ruthenium–nitrosyl (Ru–NO) complexes, with comparatively less attention given to manganese–nitrosyl (Mn–NO) and iron–nitrosyl (Fe–NO) complexes.22,26,32–35 This review aims to provide a comprehensive overview of the latest research trends on photoactive nitrosyl complexes containing first-low transition metals such as Mn, Fe, and Co. The review will cover essential geometric and electronic structures, photoreactivity, and computational studies on photodissociation mechanisms. Additionally, the factors influencing photolability will be discussed. By taking comprehensive approach to M–NO adducts, the review seeks to offer a deeper understanding of the nature and properties of light-sensitive M–NO species.
2. Synthesis, characterization, and photoreactivity of M–NO complexes
2.1 Generation of M–NO complexes
Inspired by the regulatory roles of NO in biological systems and the photolability observed in Fe-NHase enzymes, a variety of photoactive mononitrosyl complexes containing first-row transition metals such as Mn,36–40 Fe,41–54 and Co55 have been synthesized and characterized to understand physical and chemical properties, as well as photoreactivity. All supporting ligands used for the synthesis of photoactive M–NO complexes, as discussed in this review, are illustrated in Chart 1.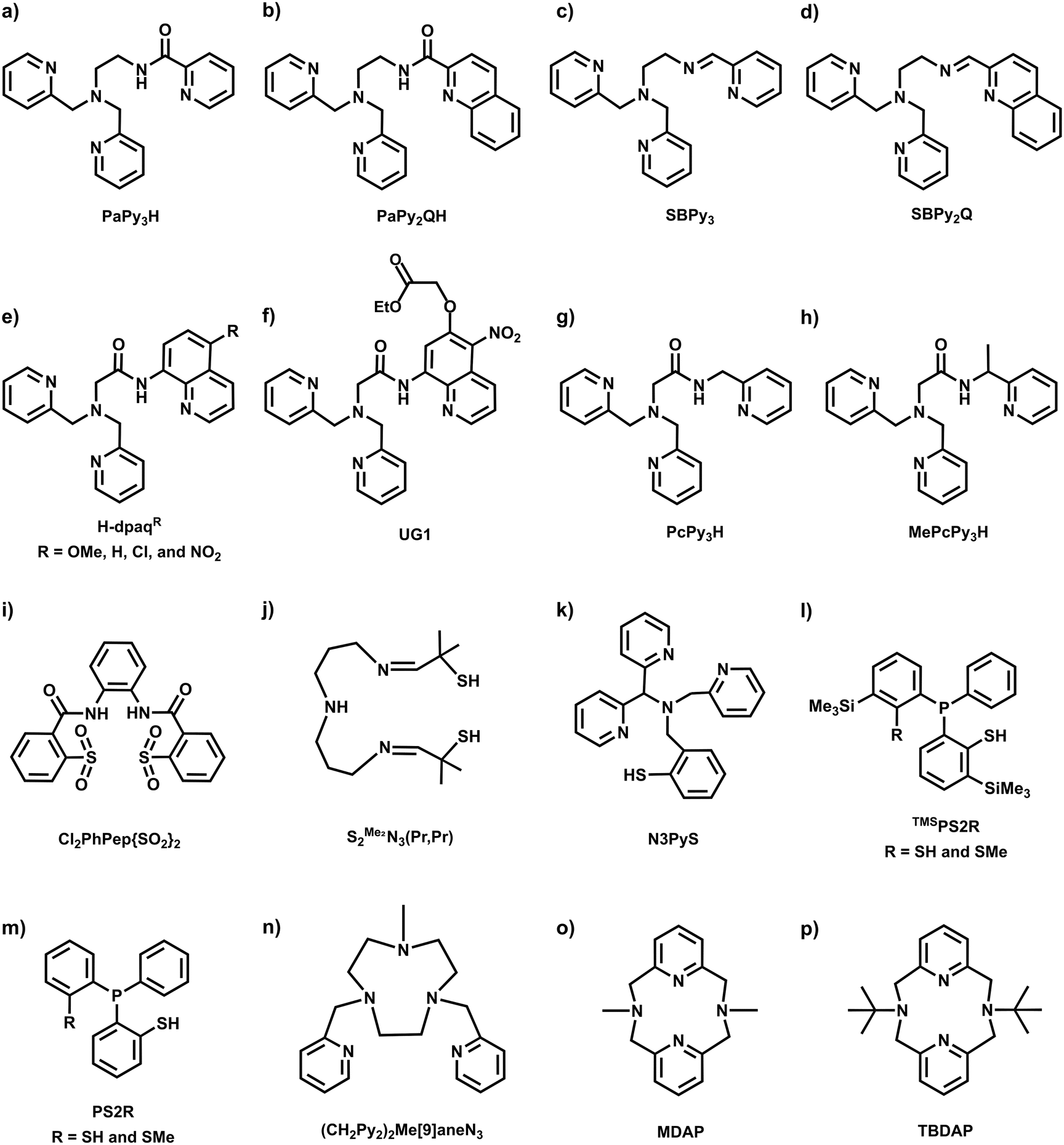 | ||
| Chart 1 Ligand structures discussed in this review. | ||
A common method for synthesizing M–NO species involves introducing purified NO gas into a solution of precursor complexes under inert conditions.36–49 Alternative strategies include employing redox reactions of existing nitrosyl complexes50 or utilizing commercially available nitrosylated metal sources.52,53 Transition metal ions in the 2+ or 3+ oxidation states are typically used to generate {MNO}n complexes, based on Enemark–Feltham notation, where n represents the total number of electrons in the metal d orbitals and NO π* orbitals (Scheme 2).56 After nitrosyl complexes are generated in solution, the resulting species are crystallized and characterized using various physicochemical methods such as ultraviolet-visible (UV-vis) spectroscopy, infrared (IR) spectroscopy, proton nuclear magnetic resonance (1H NMR) spectroscopy, electron paramagnetic resonance (EPR) spectroscopy, Mössbauer spectroscopy, and single crystal X-ray diffractometry (SC-XRD).
 | ||
| Scheme 2 Synthetic strategies for the generation of M–NO complexes. | ||
2.2 Manganese–nitrosyl complexes
Photoactive manganese–nitrosyl (Mn–NO) species have been suggested as potent NO-releasing agents. The Mn–NO complexes are synthesized from Mn(II) precursors, resulting in the formation of low-spin S = 0 {MnNO}6 species.36–40 The thermal stability of Mn–NO adducts in the absence of light has allowed researchers to scrutinize electronic and structural properties. Additionally, photoreactivity studies in various solvents have demonstrated that Mn–NO complexes can selectively release NO at targeted sites through the photodissociation of the Mn–NO bond, presenting significant potential for biomedical applications.The first photolabile complex, [Mn(PaPy3)(NO)]+ (1), bearing a deprotonated anionic pentadentate N5 carboxamide ligand (Chart 1a) was reported.36 The introduction of purified NO gas to a CH3CN solution of [MnII(PaPy3)(H2O)]+ led to the formation of complex 1, which exhibited UV-vis absorption bands at 420, 440, and 635 nm. The spin state of 1 was determined to be in the S = 0 ground state by 1H NMR spectroscopy, with displayed diamagnetic resonance peaks within the range of 0–10 ppm. Single crystal X-ray structure exhibited a mononuclear and six-coordinate distorted octahedral geometry with the NO ligand positioned trans to the anionic carboxamide nitrogen donor of the supporting ligand (Fig. 1a). The Mn–NO and N–O bond lengths were measured at 1.6601 and 1.1928 Å, respectively, with a Mn–N–O bond angle of 171.91°. The IR spectrum of 1 showed a strong NO stretching vibration band at 1745 cm−1, which is consistent with typical diamagnetic {MnNO}6 complexes (1700–1775 cm−1).
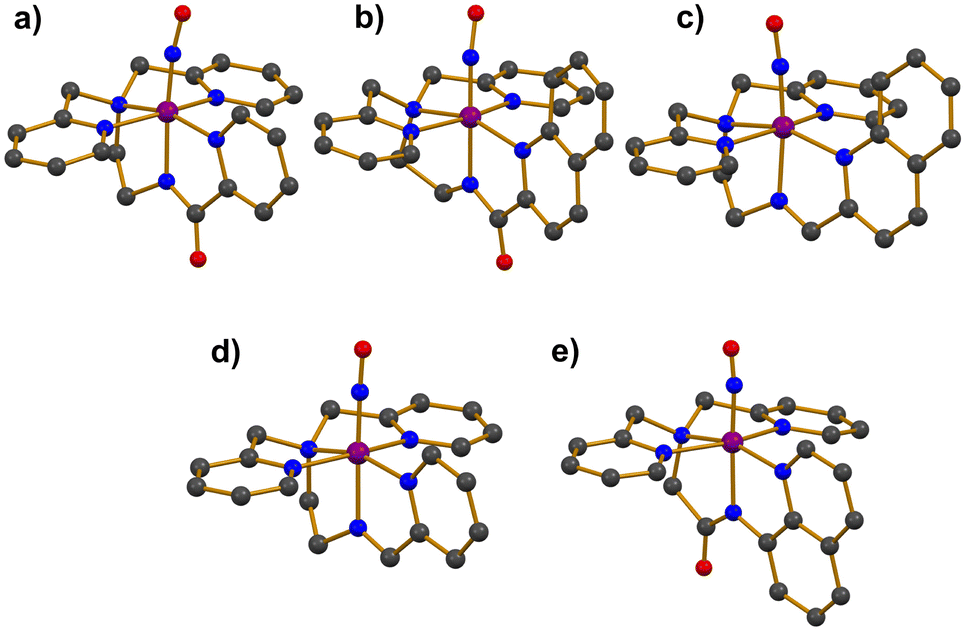 | ||
| Fig. 1 Crystal structures of Mn–NO complexes: (a) [Mn(PaPy3)(NO)]+ (1), (b) [Mn(PaPy2Q)(NO)]+ (2), (c) [Mn(SBPy3)(NO)]2+ (3), (d) [Mn(SBPy2Q)(NO)]2+ (4), and (e) [Mn(dpaqH)(NO)]+ (5H) (dark grey, C; blue, N; red, O; purple, Mn). | ||
The photolysis of 1 was investigated across a broad range of wavelengths (380–810 nm) and light intensities.36,38 The highest quantum yield was achieved with 500 nm light irradiation, resulting in the formation of a solvated Mn(III) species, which is the oxidized form of the corresponding NO-released Mn(II) complex under aerobic conditions. The reaction kinetics followed pseudo-first-order behavior in various solvents such as CH3CN, DMF, and H2O, showing different rate constants (CH3CN > DMF > H2O). Additionally, the NO dissociation rate increased proportionally with light intensity.
Another photoactive complex, [Mn(PaPy2Q)(NO)]+ (2), was synthesized by reacting excess NO gas with the Mn(II) starting complex of [MnII(PaPy2Q)(H2O)]+ in CH3CN.37 The UV-vis absorption band of 2 was red-shifted to 650 nm compared to 635 nm for 1. In addition, the extinction coefficient of 2 was nearly double that of 1 (Table 1). The red-shift and enhanced light absorption were attributed to the modification of the ligand system, where the pyridine group in 1 was replaced by a quinoline group, a more conjugated ring system (Chart 1aversus1b). Consequently, the increased π-conjugation on the supporting ligand enhanced the light responsiveness of 2 (vide infra). The S = 0 ground state of 2 was confirmed by 1H NMR spectroscopy, indicating that 2 is a diamagnetic species. The X-ray diffraction analysis of 2 revealed similar coordination geometry with 1 as shown in Fig. 1b.36 The slightly longer Mn–NO and N–O bond distances of 1.678 and 1.237 Å, respectively, were accompanied with a linear Mn–N–O angle of 171.5°. The elongation of the bond length in 2 resulted in a lower NO vibration energy, observed at 1725 cm−1.
| Mn–NO complex | UV-vis | IR [cm−1] | Bond lengths | Bond angle | Ref. |
|---|---|---|---|---|---|
| λ [nm] (ε [M−1 cm−1]) | Mn–NO/N–O [Å] | M–N–O [°] | |||
| a UV-vis absorption bands in CH3CN. b UV-vis absorption bands in MES buffer = 2-(N-morpholino)ethanesulfonic acid (pH 7.2, 5% DMSO). c UV-vis absorption bands in H2O. d IR spectrum with KBr pellet. e IR spectrum with ATR-IR. | |||||
| [Mn(PaPy3)(NO)]+ (1) | 420(3320), 440(3300), 635(220)a | 1745d | 1.6601/1.1918 | 171.91 | 36 |
| [Mn(PaPy2Q)(NO)]+ (2) | 240(29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500), 290(7700), 495(2030), 650(420)a 500), 290(7700), 495(2030), 650(420)a |
1725d | 1.678/1.237 | 171.5 | 37 |
| [Mn(SBPy3)(NO)]2+ (3) | 340(1725), 500(1990), 720(750)a | 1773d | 1.649/1.167 | 177.2 | 38 |
| [Mn(SBPy2Q)(NO)]2+ (4) | 330(12850), 550(2100), 785(1200)a |
1759d | 1.651/1.179 | 175.3 | 38 |
| [Mn(dpaqOMe)(NO)]+ (5OMe) | 398(3830), 457(4230)b | 1737e | 1.742/1.015 | 176.7 | 39 |
| [Mn(dpaqH)(NO)]+ (5H) | 357(3910), 461(3120)b | 1739e | 1.635/1.022 | 171 | 39 |
| [Mn(dpaqCl)(NO)]+ (5Cl) | 375(4560), 475(2960)b | 1743e | 1.713/1.044 | 171.3 | 39 |
| [Mn(dpaqNO2)(NO)]+ (5NO2) | 392(10300), 523(1570)b |
1744e | 1.660/1.136 | 175.8 | 39 |
| UG1NO (6) | 385(8300), 494(2700)c | 1746e | — | — | 40 |
The photo-triggered NO release from 2 was observed in both CH3CN and H2O under visible light irradiation.37,38 The UV-vis spectral change of 2 indicated that NO was dissociated from the Mn center, regenerating the Mn(II) precursor species. The photochemical kinetics obeyed pseudo-first-order behavior in all solvents, showing almost two times higher quantum yield of 1 under the same wavelength of light (Table 3).
| Fe–NO complex | Enemark–Feltham notation | UV-vis | IR [cm−1] | Spin state | Bond lengths | Bond angle | Ref. |
|---|---|---|---|---|---|---|---|
| λ [nm] (ε [M−1 cm−1]) | Fe–NO/N–O [Å] | M–N–O [°] | |||||
| a UV-vis absorption bands in CH3CN. b UV-vis absorption bands in toluene. c UV-vis absorption bands in CH2Cl2. d UV-vis absorption bands in acetone. e IR spectrum with KBr pellet. f IR spectrum with ATR-IR. g Low temperature FTIR at 15 K in CH2Cl2. h Obtained from EXAFS. i Measured at 110 K. j Measured at 293 K. | |||||||
| [FeS2Me2N3(Pr,Pr)(NO)]+ (7) | {FeNO}6 | 420(1700)a | 1822e | S = 0 | 1.676/1.161 | 172.3 | 41 |
| [Fe(PaPy3)(NO)]2+ (8) | {FeNO}6 | 365(1850), 500(1050)a | 1919e | S = 0 | 1.677/1.139 | 173.1 | 42 and 43 |
| [Fe(PcPy3)(NO)]2+ (9) | {FeNO}6 | 363(2040), 500(925)a | 1897e | S = 0 | 1.680/1.147 | 177.3 | 44 |
| [Fe(MePcPy3)(NO)]2+ (10) | {FeNO}6 | 360(2012), 505(735)a | 1918e | S = 0 | 1.678/1.147 | 177.6 | 44 |
| [Fe(PaPy2Q)(NO)]2+ (11) | {FeNO}6 | 310(83400), 330(83000), 510(10100)a |
1885e | S = 0 | 1.6817/1.1435 | 166.66 | 45 |
| [Fe(Cl2PhPep{SO2}2)(NO)(DMAP)]− (12) | {FeNO}6 | 440a | 1854e | S = 0 | — | — | 46 and 47 |
| [Fe(N3PyS)(NO)]+ (13) | {FeNO}7 | 350(5300), 440(2400), 540(970)a | 1660f | S = 1/2i | 1.7327/1.15i | 147.2i | 48 and 49 |
| 1753f | S = 3/2j | 1.747/1.14j | 161.11j | ||||
| [Fe(N3PyS)(NO)]2+ (14) | {FeNO}6 | 342(12200), 636(820)a |
1909g | S = 0 | 1.69h | — | 50 |
| [Fe((CH2Py2)2Me[9]aneN3)(NO)]2+ (15) | {FeNO}7 | 242(21000), 318(36000)a |
1660f | S = 1/2 | 1.731/1.143 | 148.3 | 51 |
| [Fe(TMSPS2)(TMSPS2H)(NO)] (16) | {FeNO}6 | 480(1810), 630(755)b | 1829e | S = 0 | 1.644/1.153 | 177.3 | 52 |
| [Fe(TMSPS2)(TMSPS2CH3)(NO)] (17) | {FeNO}6 | 480(1760), 630(705)b | 1822e | S = 0 | 1.642/1.152 | 176.1 | 52 |
| [Fe(PS2)(PS2H)(NO)] (18) | {FeNO}6 | 480(1810), 630(755)c | 1832e | S = 0 | 1.6513/1.152 | 175.56 | 53 |
| [Fe(PS2)(PS2CH3)(NO)] (19) | {FeNO}6 | 480(1760), 630(705)c | 1832e | S = 0 | 1.6449/1.150 | 175.05 | 53 |
| [Fe(TBDAP)(NO)(H2O)]2+ (20) | {FeNO}7 | 407(1100), 522(320), 745(97)d | 1781f | S = 3/2 | 1.754/1.153 | 152.9 | 54 |
| Nitrosyl complex | Solvent | λ irr [nm] | ϕ NO [mol per einst] | Ref. |
|---|---|---|---|---|
| a MES = 2-(N-morpholino)ethanesulfonic acid (pH 7.2, 5% DMSO). | ||||
| [Mn(PaPy3)(NO)]+ (1) | CH3CN | 500 | 0.326 | 36 and 38 |
| 550 | 0.309 | |||
| H2O | 500 | 0.400 | ||
| 550 | 0.385 | |||
| [Mn(PaPy2Q)(NO)]+ (2) | CH3CN | 500 | 0.623 | 37 and 38 |
| 550 | 0.579 | |||
| H2O | 500 | 0.742 | ||
| 550 | 0.694 | |||
| [Mn(SBPy3)(NO)]2+ (3) | CH3CN | 500 | 0.411 | 38 |
| 550 | 0.580 | |||
| [Mn(SBPy2Q)(NO)]2+ (4) | CH3CN | 500 | 0.393 | 38 |
| 550 | 0.434 | |||
| [Mn(dpaqOMe)(NO)]+ (5OMe) | MES buffera | 460 | 0.58 | 39 |
| 530 | 0.47 | |||
| 650 | 0.49 | |||
| [Mn(dpaqH)(NO)]+ (5H) | MES buffera | 460 | 0.61 | 39 |
| 530 | 0.51 | |||
| 650 | 0.47 | |||
| [Mn(dpaqCl)(NO)]+ (5Cl) | MES buffera | 460 | 0.66 | 39 |
| 530 | 0.66 | |||
| 650 | 0.73 | |||
| [Mn(dpaqNO2)(NO)]+ (5NO2) | MES buffera | 460 | 0.61 | 39 |
| 530 | 0.63 | |||
| 650 | 0.78 | |||
| UG1NO (6) | MES buffera | 650 | 0.74 | 40 |
| [Fe(PaPy3)(NO)]2+ (8) | CH3CN | 500 | 0.185 | 42 and 43 |
| [Fe(PaPy2Q)(NO)]2+ (11) | CH3CN | 500 | 0.258 | 45 |
| [Fe(Cl2PhPep{SO2}2)(NO)(DMAP)]− (12) | CH3CN | 450 | 0.55 | 46 and 47 |
| [Fe(TBDAP)(NO)(H2O)]2+ (20) | H2O | White light | 0.23 | 54 |
| [Co(MDAP)(NO)(CH3CN)]2+ (21) | H2O | White light | 0.78 | 55 |
Two additional complexes, [Mn(SBPy3)(NO)]2+ (3) and [Mn(SBPy2Q)(NO)]2+ (4), supported by neutral pentadentate N5 Schiff base ligands (Chart 1c and d), were synthesized by reacting NO gas with the Mn(II) precursor complexes, [MnII(SBPy3)(MeOH)]2+ and [MnII(SBPy2Q)(EtOH)]2+.38 The coordination environments around Mn center in 3 and 4 were characterized by X-ray diffraction analysis (Fig. 1c and d). In both complexes, the NO ligand was coordinated trans to the imine nitrogen donor of the supporting ligands, displaying linear Mn–N–O bond angles (Table 1). The N–O bond lengths were measured as 1.167 Å for 3 and 1.179 Å for 4, while the Mn–NO bond lengths were determined to be 1.649 and 1.651 Å, respectively. The shorter N–O bond in 3 corresponds to a higher NO stretching frequency of 1773 cm−1, compared to 1759 cm−1 for 4. The UV-vis absorption bands of 4 were red-shifted relative to those of 3 (500 and 720 nm for 3; 550 and 780 nm for 4), along with enhanced absorptivity (Table 1). The shifts in absorption bands and increased absorbance were attributed to the presence of the additional conjugated quinoline ring in the ligand system (Chart 1cversus1d), similar to the behavior observed in complexes 1 and 2.
Near-infrared (NIR) light responsiveness was observed in the photolysis of 3 and 4, which released NO under NIR light (800–950 nm) in CH3CN and aqueous solutions, differing from the photolysis of 1 and 2.36–38 The NIR responsiveness was attributed to the coordination of the imine ligand, which modulates the absorption bands to shift to longer wavelengths and increases the extinction coefficient (vide infra). Sensitivity to NIR light is noteworthy because NIR light can effectively penetrate human skin without causing damage.
The electronic effects on NO release, influenced by substituting functional groups in ligand frameworks, were systematically investigated.39 To explore the electronic effects, a series of four Mn–NO complexes, [Mn(dpaqR)(NO)]+ (5R, R = OMe, H, Cl, and NO2), containing anionic pentadentate N5 carboxamide ligands (Chart 1e) were synthesized by bubbling purified NO gas into CH3CN solutions of the precursor complexes. SC-XRD analysis revealed that all nitrosyl complexes exhibited distorted octahedral geometries. The geometric parameters and vibration energies of N–O bond are listed in Table 1. All complexes released NO under light irradiation, displaying varying dissociation rates. The impact of the substituents on the photodissociation reaction and electronic structure will be discussed (vide infra).
Later, an additional electron-donating ethyl ester group was introduced at the 6-position of the quinoline ring in the dpaqNO2 ligand of complex 5NO2 (Chart 1f) to produce the UG1NO (6).40 Upon adding purified NO gas to the Mn(II) starting complex, [MnII(UG1)]+, in a CH3CN/MeOH mixture, dark red species of 6 were precipitated. Although single crystals could not be obtained, ESI-MS analysis supported the generation of 6 with an ion peak at a mass-to-charge ratio (m/z) of 614.31, corresponding to 6. The UV-vis spectrum of 6 exhibited absorption bands at 385 and 494 nm in aqueous solution, while the IR spectrum showed a NO stretching vibration at 1746 cm−1.
Complex 6 remained stable in a buffer solution at pH 7.2 when kept in the dark. However, upon exposure to 650 nm light, 6 rapidly decomposed back to the corresponding Mn(II) starting complex due to NO loss from the Mn center with a quantum yield of 0.74 (Table 3). Complex 6 exhibited light absorption in the visible-NIR region and displayed a similar response to light irradiation as 5NO2, despite the presence of the additional electron-donating ethyl ester moiety on the quinoline ring.
2.3 Iron–nitrosyl complexes
Photolabile iron–nitrosyl (Fe–NO) complexes have garnered significant attention as promising NO transfer agents for biomedical treatments and as biomimetic models of the Fe-NHase enzymes. Accordingly, extensive research has focused on the synthetic Fe–NO complexes. As illustrated in Scheme 2, Fe–NO adducts are commonly synthesized by reacting NO gas with Fe precursors in the Fe(II) or Fe(III) oxidation states, followed by characterization using various physicochemical methods.41–54The photolabile complex, [FeS2Me2N3(Pr,Pr)(NO)]+ (7), bearing a pentadentate thiolate ligand (Chart 1j) was reported.41 Treating a solution of the precursor complex, [FeIIIS2Me2N3(Pr,Pr)]+, with excess NO gas afforded 7. Upon the addition of NO to the starting complex, a UV-vis absorption band appeared at 420 nm in CH3CN at room temperature and the redox potential decreased from −400 to −455 mV (vs. SCE), implying that the Fe(III) center was stabilized by NO coordination. X-ray crystallography revealed a distorted octahedral geometry for 7, with the NO ligand coordinated trans to the thiolate donor (Fig. 2a). The Fe–N–O bond angle was found to be linear at 172.3°, while the Fe–NO and N–O bond distances were measured as 1.676 and 1.161 Å, respectively. The NO stretching vibration band in the IR spectrum was observed at 1822 cm−1, which is close to that of the inactive NO-bound Fe-NHase enzyme (νNO = 1853 cm−1). The diamagnetic 1H NMR signal of 7 indicated antiferromagnetic coupling between the low-spin S = 1/2 Fe(III) and the S = 1/2 NO radical.
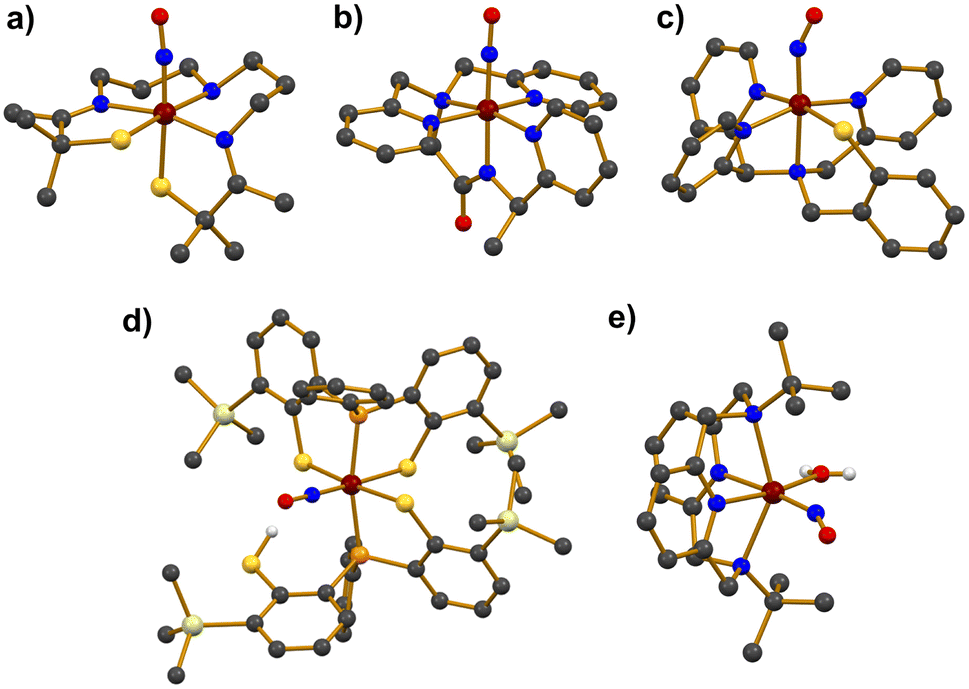 | ||
| Fig. 2 Crystal structures of Fe–NO complexes, (a) [FeS2Me2N3(Pr,Pr)(NO)]+ (7), (b) [Fe(MePcPy3)(NO)]2+ (10), (c) [Fe(N3PyS)(NO)]+ (13), (d) [Fe(TMSPS2)(TMSPS2H)(NO)] (16), and (e) [Fe(TBDAP)(NO)(H2O)]2+ (20) (dark grey, C; blue, N; red, O; yellow, S; scarlet, Fe). | ||
Photodissociation of 7 was demonstrated by illuminating a CH3CN solution of the complex under inert conditions with a Hg lamp, applied at slightly reduced pressure. Light exposure cleaved the Fe–NO bond, releasing a free NO radical and regenerating the starting Fe(III) complex. The release of NO gas was further confirmed by mass spectrometric analysis of the headspace gases.
A photoactive nitrosyl complex, [Fe(PaPy3)(NO)]2+ (8), a Fe analogue of 1, was synthesized. Addition of NO to a solution of [FeIII(PaPy3)(CH3CN)]2+ resulted in the formation of the {FeNO}6 complex.42,43 The coordination environment was found to be comparable to that of 1, featuring a distorted octahedral geometry with a linear Fe–N–O bond angle of 173.1°. The NO ligand occupied one coordination site with Fe–NO and N–O bond lengths of 1.677 and 1.139 Å, respectively. The IR spectrum displayed a NO stretching band at 1919 cm−1, which falls within the typical range for {FeNO}6 complexes (1850–1940 cm−1). The diamagnetic signal in the 1H NMR spectrum in CD3CN, along with isomer shift (δ) of −0.05 mm s−1 and quadrupole splitting (ΔEQ) of +0.85 mm s−1 in the zero-field Mössbauer spectrum at 4.2 K, indicated an FeII–NO+ electronic configuration for 8.
Photolysis of 8 under visible light (tungsten lamp, 50 W) caused a rapid color change with isosbestic points observed at 484, 392, and 334 nm in the UV-vis spectrum.43 The final UV-vis absorption band suggested the formation of an Fe(III) precursor species, indicating NO release. The photolysis kinetics in various solvents such as CH3CN, H2O, and DMF followed pseudo-first-order behavior with the rate of NO dissociation increasing proportionally to the light intensity.
Two additional complexes, [Fe(PcPy3)(NO)]2+ (9) and [Fe(MePcPy3)(NO)]2+ (10), were prepared by reacting NO gas with the Fe(III) precursor complexes, which are supported by the anionic pentadentate N5 carboxamide ligands PcPy3H (Chart 1g) and MePcPy3H (Chart 1h).44 The binding of NO to the Fe center produced diamagnetic signals of 1H NMR, indicating S = 0 ground states for both complexes. The coordination environments around the Fe center in both complexes showed similar features to that observed in 8, with NO binding to Fe in a linear Fe–N–O arrangement (177.3° for 9 and 177.6° for 10) (Fig. 2b). The Fe–NO and N–O bond lengths of 9 were measured as 1.680 and 1.147 Å, respectively. For 10, almost identical bond distances of Fe–NO and N–O were determined to be 1.678 and 1.147 Å, respectively. The NO stretching frequencies were 1897 cm−1 for 9 and 1918 cm−1 for 10, which are typical for {FeNO}6 complexes.
Under light illumination, complexes 9 and 10 demonstrated slightly faster NO release rates compared to 8 in CH3CN. The NO dissociation followed pseudo-first-order kinetics, leading to the formation of solvated Fe(III) species. The NO dissociation rates were also dependent on light intensity, showing a linear increase with higher light power.
To verify the conjugation effect seen in Mn–NO complexes 1–4, a modified complex, [Fe(PaPy2Q)(NO)]2+ (11), was synthesized by replacing the pyridine group in 8 with a quinoline ring (Chart 1b).45 Reacting NO gas with the Fe(III) precursor complex, [Fe(PaPy2Q)(EtOH)]2+, yielded a dark purple product of 11. The X-ray crystal structure showed a similarity to that of 8. The IR spectrum of 11 exhibited an NO stretching vibration energy at 1885 cm−1, close to the vibration energy of the free NO radical (1875 cm−1). The N–O bond distance in 11 (1.1435 Å) was also similar to that of the free NO radical (1.15 Å), suggesting minimal back-donation from the Fe center to the bound NO.
The photolability of NO in 11 was examined in CH3CN solution. Upon exposure to visible light (500 nm, 5 mW), the CH3CN solution of 11 decomposed back to the starting complex, [FeIII(PaPy2Q)(CH3CN)]2+, as evidenced by the disappearance of the UV-vis absorption band at 510 nm with the isosbestic points at 375 and 690 nm. To evaluate the impact of replacing the pyridine group in 8 with a quinoline group in 11, the quantum yields were compared. Complex 11 exhibited a higher quantum yield (ϕNO = 0.258) than 8 (ϕNO = 0.185), demonstrating that the increased conjugation from the quinoline group enhances light absorptivity and improves the quantum yield of the nitrosyl complex (Table 3).
An Fe–NO complex, [Fe(Cl2PhPepS)(NO)(DMAP)]−, was synthesized using a tetradentate N2S2 ligand.46,47 The complex was obtained by reacting the starting complex of [Fe(Cl2PhPepS)(DMAP)]− with NO gas in CH3CN at −40 °C. The crystal structure of [Fe(Cl2PhPepS)(NO)(DMAP)]− revealed an extremely distorted geometry with DMAP (N,N-dimethylaminopyridine) and NO ligand positioned axially. The Fe–NO and N–O bond distances were determined to be 1.612 and 1.167 Å, respectively, with a nearly linear Fe–N–O bond angle of 173.2°. The IR spectrum of [Fe(Cl2PhPepS)(NO)(DMAP)]− showed a strong νNO stretch at 1849 cm−1. While light-triggered NO release was not observed for [Fe(Cl2PhPepS)(NO)(DMAP)]−, NO dissociation occurred in coordinating solvents such as CH3CN, THF, and DMF due to ligand exchange reactions, where the NO ligand was replaced by solvent molecules. In contrast, [Fe(Cl2PhPepS)(NO)(DMAP)]− remained stable in non-coordinating solvents like CHCl3 or CH2Cl2.
To mimic the ligand backbone of the Fe-NHase enzyme, the thiolate (–S) groups in [Fe(Cl2PhPepS)(NO)(DMAP)]− were oxygenated to form sulfinates (–SO2) by treating the complex with 4 equivalents of (1S)-(+)-(10-camphorsulfonyl)oxaziridine in CHCl3 at −40 °C in the absence of light. The reaction produced a pale-orange species, [Fe(Cl2PhPep{SO2}2)(NO)(DMAP)]− (12), featuring a S-oxygenated supporting ligand (Chart 1i and Scheme 3). Complex 12 exhibited a UV-vis absorption band at 440 nm, which is very similar to the absorption band of the NO-bound Fe-NHase enzyme (400 nm). The instability of 12 prevented crystallization for X-ray diffraction analysis. However, the IR spectrum of 12 showed a νNO value of 1854 cm−1, which is close to that of the inactivated Fe-NHase (1853 cm−1), along with S-oxygenated bands at 1078, 1046, and 1007 cm−1.
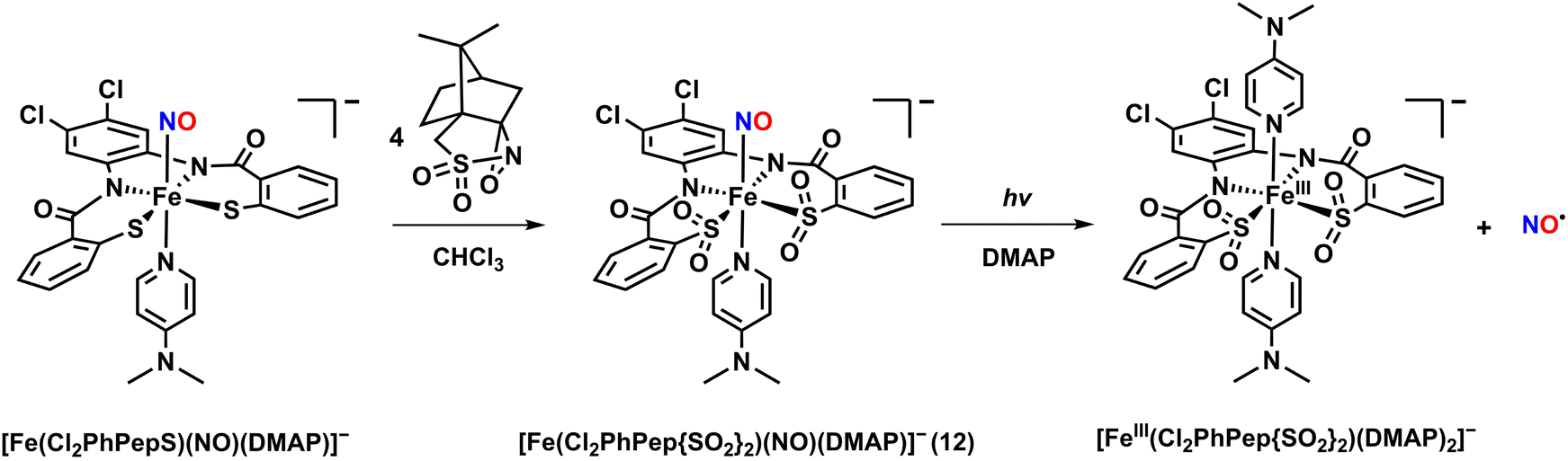 | ||
| Scheme 3 S-Oxygenation reaction to yield the photoactive {FeNO}6 complex 12 and photodissociation. | ||
Interestingly, when a CH3CN or CHCl3 solution of 12 was exposed to visible light (10 mW) at −40 °C, the UV-vis spectrum showed immediate change, appearing a new band at 650 nm, indicating the release of NO. The quantum yield (λirr = 450 nm) for NO release was determined to be 0.55 (Table 3), which is comparable to that of Fe-NHase (ϕNO = 0.48). The released free NO was detected using a NO-sensitive electrode. The X-band EPR spectrum of the NO-released species revealed g values of 2.23, 2.03, and 2.02, indicating the formation of low-spin [FeIII(Cl2PhPep{SO2}2)(DMAP)2]−.
The first temperature-dependant spin-crossover photoactive complex, [Fe(N3PyS)(NO)]+ (13), was reported, utilizing a tetradentate N3S ligand (Chart 1k).48,49 Complex 13 was synthesized by reacting [FeII(N3PyS)(CH3CN)]+ with NO gas. X-ray diffraction revealed a distorted octahedral geometry for 13 with the NO ligand positioned trans to the amine nitrogen in the axial position (Fig. 2c). Magnetic susceptibility experiments indicated that 13 exhibited a low-spin (S = 1/2) state within the temperature range of 10–150 K, while an increasing contribution of a high-spin (S = 3/2) state was observed above 150 K. The spin-crossover of 13 resulted in changes to the geometric parameters for bond lengths and NO stretching vibration energies at different temperatures. For the S = 1/2 state at 110 K, the bond distances of Fe–NO and N–O bond were 1.7327 and 1.150 Å, respectively. In contrast, at 293 K, the Fe–NO and N–O bond lengths were determined to be 1.747 and 1.14 Å, respectively (Table 2). The ATR-IR spectrum of crystalline 13 at room temperature displayed two NO stretching bands at 1753 and 1660 cm−1, indicative of a mixture of S = 3/2 and S = 1/2 species. Upon 15N18O labelling, NO vibrational frequencies shifted to lower energies of 1677 and 1587 cm−1.
The release of NO from 13 was triggered by photoillumination with visible light (λ > 400 nm, 150 W) in CH3CN. The photorelease led to the clean conversion of 13 to the NO-released species, [FeII(N3PyS)(CH3CN)]+, as evidenced by UV-vis spectral changes with an isosbestic point at 380 nm. Upon light irradiation, the characteristic absorption band of 13 at 360 nm decreased, while the bands at 418 and 493 nm, corresponding to the photoproduct, increased.
The photoactive Fe–NO complex, [Fe(N3PyS)(NO)]2+ (14), was prepared through an one-electron oxidation of the {FeNO}7 complex 13.50 The oxidation reaction was initiated by adding one equivalent of acetylferrocenium tetrafluoroborate (AcFcBF4) to a solution of 13, resulting in the appearance of new UV-vis absorption bands at 342 and 636 nm (Scheme 4). Similar results were observed with other oxidants such as tris(4-bromophenyl)ammoniumyl tetrafluoroborate and thianthrenium tetrafluoroborate. Complex 14 was unstable in CH3CN at 25 °C, decomposing within minutes. However, the stability was significantly improved when oxidation was performed at a lower temperature of −40 °C, extending the lifetime up to 8 hours. Stability was also enhanced in the non-coordinating solvent CH2Cl2, where 14 remained stable at 25 °C for 24 hours without noticeable decomposition.
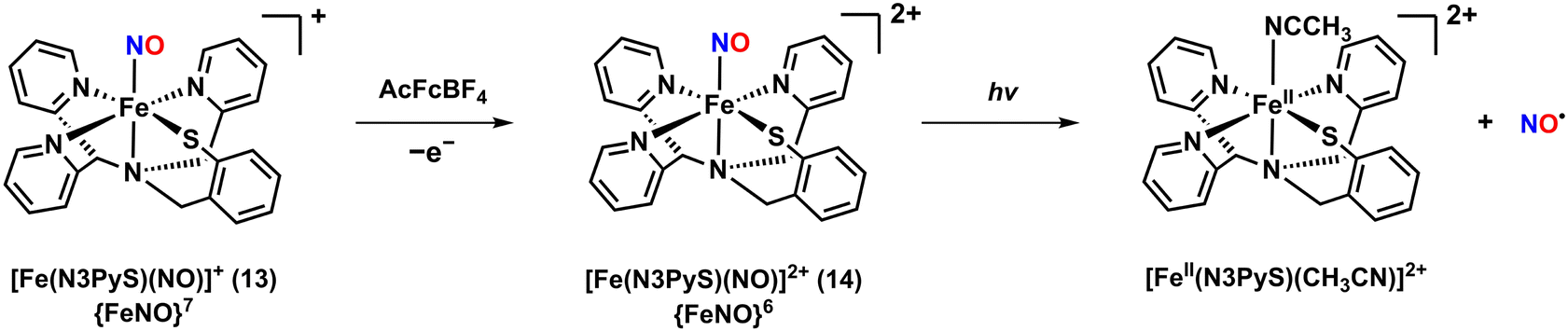 | ||
| Scheme 4 Generation of the {FeNO}6 complex 14 by one-electron oxidation from the {FeNO}7 complex 13. | ||
Although the crystal structure of 14 could not be obtained, extended X-ray absorption fine structure (EXAFS) analysis provided an Fe–NO bond length of 1.69 Å. The IR spectrum of 14 showed an NO vibrational band at 1909 cm−1, which aligns with typical values for {FeNO}6 complexes. The Mössbauer spectrum at 80 K displayed an isomer shift (δ) value of 0.03 mm s−1 and quadrupole splitting (ΔEQ) of 1.7 mm s−1, consistent with a FeII–NO+ configuration. The Mössbauer parameters are similar to those of the NO-bound Fe-NHase enzyme (δ = 0.03 mm s−1 and ΔEQ = 1.47 mm s−1).
Photorelease of NO from 14 was observed upon exposure to visible light (λ > 400 nm) in CH3CN at −40 °C, resulting in the rapid decomposition into the [FeIII(N3PyS)(CH3CN)]2+ precursor complex. However, irradiation of 14 in CH2Cl2 at both 25 and −40 °C did not lead to NO release, likely due to the rapid rebinding of NO in the non-coordinating solvent.
A photo-sensitive complex, [Fe((CH2Py2)2Me[9]aneN3)(NO)]2+ (15), was synthesized using a pentadentate N5 ligand (Chart 1n).51 The synthesis of 15 was achieved in a single step by bubbling NO gas into a solution of CH3CN/MeOH mixture containing Fe(BF4)2·5H2O and the (CH2Py2)2Me[9]aneN3 ligand, resulting in the generation of black solution of 15. Crystals suitable for X-ray diffraction analysis were obtained through slow evaporation of the resulting solution under an Ar stream. The X-ray diffraction revealed Fe–NO and N–O bond distances of 1.731 and 1.143 Å, respectively, with a bent Fe–N–O angle of 148.3°. The X-band EPR spectrum of 15 in frozen CH3CN at 30 K exhibited a rhombic signal at g value of 2.00 with hyperfine splitting due to the interaction of the Fe center with the 14N nucleus of the ligand donor atom, indicative of an S = 1/2 spin state. The IR spectrum showed a strong NO stretching peak at 1660 cm−1, typical for low-spin {FeNO}7 complexes.
The photodissociation of 15 in CH3CN resulted in the release of NO with a quantum yield of 0.52 upon exposure to a 450 nm light source. A lower quantum yield of 0.40 was observed with 365 nm light in CH3CN. The isosbestic point in the UV-vis spectrum indicated a complete conversion to the solvent-bound Fe(II) starting complex, [FeII((CH2Py2)2Me[9]aneN3)(CH3CN)]2+. The released NO was qualitatively detected by reacting NO with reduced myoglobin (Mb) that is prepared by reducing metMb with sodium dithionite. The free NO radical was transferred by an Ar stream to the metMb solution, and the spectral change in the UV-vis spectrum due to NO coordinating to the Fe(II) center of Mb was monitored.
Two photolabile complexes, [Fe(TMSPS2)(TMSPS2H)(NO)] (16) and [Fe(TMSPS2)(TMSPS2CH3)(NO)] (17), bearing a pendant thiol and thioether (Chart 1l), respectively, were reported.52 The neutral complex 16 was synthesized by treating Fe(CO)2(NO)2 in THF with two equivalents of the TMSPS2H2 ligand in the absence of light. Complex 17 was produced by methylating the pendant thiol of 16 with trimethyloxonium tetrafluoroborate (Me3OBF4), resulting in a thioether as shown in Scheme 5. X-ray crystallography revealed that both complexes feature Fe coordination environments with two phosphine donors in the axial position and three thiolate and one NO in the equatorial plane, resulting in distorted octahedral geometries. The Fe–NO and N–O bond distances were nearly identical for both complexes (Fe–NO = 1.644 Å for 16 and 1.642 Å for 17; N–O = 1.153 Å for 16 and 1.152 Å for 17). The IR spectra showed NO stretching bands at 1829 cm−1 for 16 and 1822 cm−1 for 17, which were sensitive to the 15NO isotope. The diamagnetic signals of 1H NMR indicated S = 0 ground states for 16 and 17.
 | ||
| Scheme 5 Proposed photolysis pathways of complexes 16–19. | ||
As illustrated in Scheme 5, photolysis (λ > 400 nm, 150 W, xenon lamp) of 16 in toluene at ambient temperature for 20 seconds led to the formation of a unique intermediate involving an intramolecular [SH⋯ON–Fe] interaction between NO and a pendant SH group within the ligand framework. The intramolecular interaction was evidenced by an IR band shift of the NO stretching frequency from 1833 to 1823 cm−1 and a downfield shift from 4.79 to 5.78 ppm of 1H NMR for the proton on the pendent SH group, attributable to the hydrogen bonding interaction.57–60 Prolonged irradiation led to a decrease in the intensity of the NO vibration peak at 1823 cm−1, indicating partial NO loss, along with the appearance of IR bands at 2219 and 1774 cm−1 corresponding to the formation of N2O and predicted {FeNO}7 species, respectively. The generation of N2O implies that HNO was initially formed through proton-coupled electron transfer reaction via the [SH⋯ON–Fe] interaction,61–63 which then either dimerized or reacted with partially released NO to form N2O. The release of NO was further confirmed by the formation of [Co(TPP)(NO)] adduct from [Co(TPP)] and N2O was characterized by gas chromatography (GC) analysis. The photoproduct of 16 was crystallized and characterized by SC-XRD and Mössbauer spectroscopy (δ = 0.15 mm s−1, ΔEQ = 1.99 mm s−1, and Γ = 0.29 mm s−1) to be an intermediate spin state of S = 1 Fe(IV) species. In contrast, the photolysis of 17 resulted only in NO release. The absence of a pendant SH group in 17 prevented the formation of the [SH⋯ON–Fe] intermediate, which played a crucial role in the HNO production pathway seen in 16.
Additional photolabile complexes, [Fe(PS2)(PS2H)(NO)] (18) and [Fe(PS2)(PS2CH3)(NO)] (19), were reported as analogous to complexes 16 and 17.53 Complexes 18 and 19 were prepared using the same methodology employed for 16 and 17 (Chart 1m). The both complexes exhibited distorted octahedral geometries similar to those of 16 and 17, with nearly linear bond angles of Fe–N–O units (175.56° for 18 and 175.05° for 19). The Fe–NO and N–O bond lengths in 18 were measured to be 1.6513 and 1.152 Å, respectively, while in 19, they were slightly shorter at 1.6449 and 1.150 Å. The 1H NMR spectra of both complexes indicated diamagnetic signals, suggesting low-spin {FeNO}6 complexes.
A solution IR study of NO photodissociation from 18 under xenon lamp irradiation (λ > 400 nm, 150 W) in CH2Cl2 at room temperature revealed a notable decay of the NO stretching peak at 1836 cm−1, accompanied by the appearance of a 2223 cm−1 band corresponding to N2O formation. The IR band change was similar to that observed in 16, indicating HNO formation through an intramolecular [SH⋯ON–Fe] interaction between NO and a pendant SH group. In contrast, like 17, 19 exhibited NO release under the same light conditions, evidenced by a decrease of the NO stretching band at 1823 cm−1. The UV-vis spectroscopy was also used to monitor the photolysis of 18 and 19. Upon photoillumination of 18, the UV-vis bands at 500, 570, 665, and 915 nm gradually increased, indicating that 18 is photo-sensitive to forming NO and HNO under visible light. The UV-vis absorption bands were reminiscent of photoproduct of 16 (475, 535, 670, and 865 nm), suggesting the formation of [Fe(PS2)2] as a photo-induced product. Likewise, for complex 19, the UV-vis spectra showed growth of bands at 480, 560, 705, and 885 nm comparable to the photoproduct of 17 with bands at 470, 540, 700, and 865 nm, indicating the formation of NO-released species, [Fe(PS2)(PS2CH3)].
A Fe–NO complex, [Fe(TBDAP)(NO)(H2O)]2+ (20), which contains a tetradentate N4 macrocyclic ligand (Chart 1p) was recently reported.54 Complex 20 was synthesized by reacting an excess of NO gas with a CH3CN solution of the Fe(II) precursor complex, [FeII(TBDAP)(CH3CN)2]2+. Crystallographic analysis of 20 revealed a distorted octahedral geometry with NO and H2O ligands coordinated to the Fe center at cis positions (Fig. 2e). The N–O and Fe–NO bond lengths were measured to be 1.153 and 1.754 Å, respectively, while the Fe–N–O angle was bent at 152.9°. The ATR-IR spectrum of crystalline 20 showed a NO stretching band at 1781 cm−1. To determine the electronic configuration of 20, 1H NMR Evans method and X-band EPR analysis were performed. The effective magnetic moment was found to be 4.31 μB, and the EPR spectrum in a frozen acetone at 5 K displayed g values of 4.28, 3.79, and 1.99, indicating an FeIII–NO− (S = 3/2) states with antiferromagnetic coupling between high-spin FeIII (S = 5/2) and NO− (S = 1). The CASSCF calculations on the electronic structure of 20 in the ground state also suggested that 5.46 electrons occupy the five d-orbitals of the Fe center, and 1.71 electrons occupy the two π*-orbitals of the NO ligand, representing an FeIII–NO− state.
The photodissociation reaction of an aqueous solution of 20 at 37 °C resulted in the release of NO with a quantum yield of 0.23 under the white light irradiation (λirr = 385–740 nm, 300 W, xenon lamp) (Table 3). The characteristic UV-vis absorption bands at 407, 522, and 745 nm disappeared upon illumination, resulting in the Fe(II) precursor species. The released NO was captured by [Co(TPP)], resulting in [Co(TPP)(NO)], which was confirmed by characteristic UV-vis absorption bands at 414 and 538 nm. The generation of the Fe(II) complex was further supported by electrospray ionization mass (ESI-MS) spectrometry. Additionally, the total amount of released free NO was quantified to be 0.90 using the Griess assay.
2.4 Cobalt–nitrosyl complexes
The photo-sensitive complex, [Co(MDAP)(NO)(CH3CN)]2+ (21), with a tetradentate N4 macrocyclic ligand (Chart 1o), was synthesized by exposing NO gas to a CH3CN solution of [CoII(MDAP)(CH3CN)2]2+ at −40 °C.55 Complex 21 exhibited UV-vis absorption bands at 330 and 480 nm in CH3CN. The X-ray crystal structure of 21 revealed that the coordination environment consists of four nitrogen atoms from the macrocyclic MDAP ligand, one nitrogen from CH3CN, and one nitrogen from NO, forming a distorted octahedral geometry (Fig. 3). The Co–NO and N–O bond lengths were measured to be 1.855 and 1.097 Å, respectively, and the Co–N–O unit had a significant bending angle of 125.2°. The IR spectrum of 21 showed a NO stretching vibration at 1634 cm−1, consistent with previously reported {CoNO}8 complexes. The 1H NMR signal of 21 was observed in the range of 0–10 ppm, indicating a diamagnetic low-spin {CoNO}8 ground state.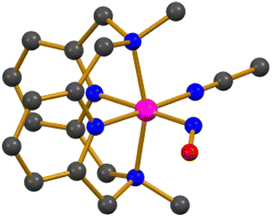 | ||
| Fig. 3 Crystal structure of the Co–NO complex, [Co(MDAP)(NO)(CH3CN)]2+ (21) (dark grey, C; blue, N; red, O; magenta, Co). | ||
In an aerobic aqueous solution, 21 demonstrated minimal NO release under dark conditions for 30 minutes. However, when the aqueous solution of 21 was exposed to white light (xenon lamp, λirr = 385–740 nm, 300 W), significant changes were observed, particularly a decrease in the absorption band at 330 nm. NO was rapidly photo-released from 21, with a half-life of 12 seconds, following first-order kinetics. The quantum yield for photo-released NO was calculated to be 0.78 using standard ferrioxalate actinometry (Table 3). In CH3CN, NO photolysis process was even faster, with a half-life of 6 seconds, also following first-order kinetics. The released NO was captured by [Co(TPP)], forming [Co(TPP)(NO)], confirmed by characteristic UV-vis absorption bands of at 414 and 538 nm. The final product after photolysis was identified as the [CoII(MDAP)(CH3CN)2]2+ precursor complex. The Griess assay further confirmed the photo-induced release of NO.
3. Discussion
3.1 Ligand effect
The design of supporting ligands plays a critical role in modulating the photoactivity of nitrosyl complexes, which is particularly important for potential biological applications, such as controlled NO release. Ligands can significantly influence the electronic structure, absorption properties, and photoreactivity. By introducing functional groups with various electronic properties, such as extended conjugation or electron-donating and electron-withdrawing substituents, the photolability and overall behavior of nitrosyl complexes can be fine-tuned. The effects of ligands on photolability have been experimentally and theoretically examined.Time-dependent density functional theory (TD-DFT) calculations further provided insight into the effects of conjugation on the red-shift of light responsiveness.64 Computational results elucidated that the level of the lowest unoccupied molecular orbital (LUMO) in 2 is significantly lower than that of 1 due to energy stabilization by the extended conjugation of the quinoline group. A comparable effect was also observed in 3 and 4. However, the highest occupied molecular orbital (HOMO) levels showed only slight changes (Fig. 4). These results indicated that quinoline substitution notably impacts the LUMO levels of 2 and 4, leading to narrower energy gaps between HOMO and LUMO, which are associated with electronic transitions. Consequently, smaller energy differences between HOMO and LUMO levels induce electronic excitation at lower energy, resulting in the red-shifted UV-vis absorption bands observed in 2 and 4.
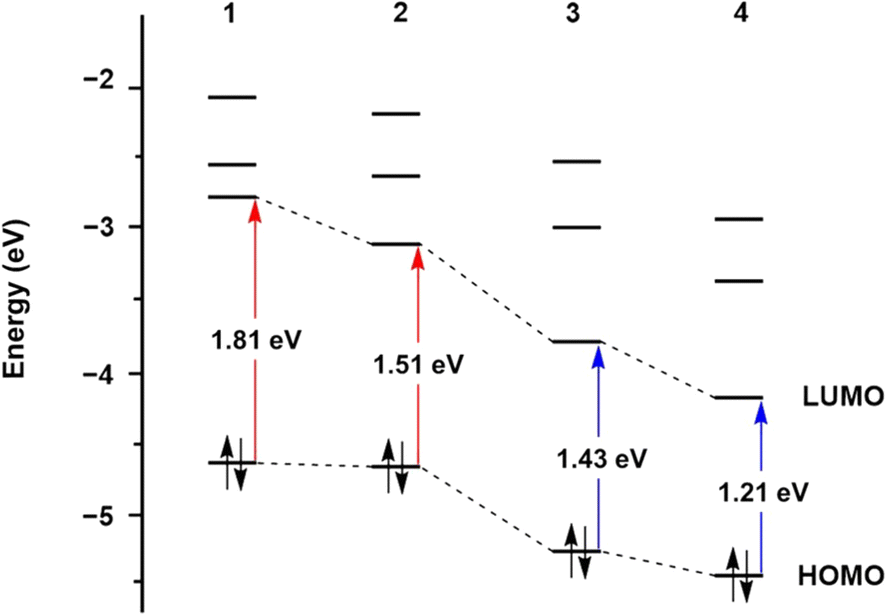 | ||
| Fig. 4 Time-dependent density functional theory (TD-DFT) results showing the molecular orbital energy levels for complexes 1–4. | ||
As shown in Chart 1e, the electrochemical properties of the H-dpaqR ligand (R = OCH3, H, Cl, and NO2) were systematically altered.39 The cyclic voltammograms of all the complexes exhibited quasi-reversible behavior associated with the {MnNO}5/6 redox couple. The redox potentials (E1/2vs. Fc+/Fc) showed a linear trend in the order of 5NO2 (0.63 V) > 5Cl (0.56 V) > 5H (0.52 V) > 5OMe (0.49 V). The increase in redox potentials with stronger electron-withdrawing substituents indicates that π-backdonation from the Mn center to the NO ligand decreases as the electron density at the Mn center diminishes. In IR spectroscopy, the NO stretching frequencies shifted to higher energies with electron-withdrawing groups. The shift signifies a reduction in π-backdonation from Mn to NO, weakening the bond between Mn and the NO ligand.
The electronic effects also influenced the UV-vis absorption spectra of the complexes. As the electron-withdrawing character of the substituents increased, the absorption bands of the nitrosyl complexes exhibited a red-shift (Table 1). Notably, the 5NO2 complex displayed a distinctive absorption profile with an extended tail into the NIR region up to 700 nm due to the stabilization of the quinoline π* orbitals by the nitro group. The observed shifts in absorption bands induced by the electronic nature of substituent groups significantly affected the photolysis rates of the nitrosyl complexes under different light conditions.
As shown in Table 4, the rate constants for photolysis decreased with electron-withdrawing substituents under 460 nm light, indicating a slower NO release. However, under longer wavelength light exposure at 530 and 650 nm, the opposite trend was observed. Remarkably, the photolysis rate of 5NO2 was significantly enhanced under 650 nm light compared to the other derivatives. The enhancement is attributed to the extended absorption band into the NIR region. Consequently, the electronic effects of the substituent groups on supporting ligands can modulate the UV-vis absorption bands, and the changes in UV-vis features alter light absorptivity at specific wavelengths, which in turn affects the rate of NO release.
| Complex | Extinction coefficients [mM−1 cm−1] | Rate constant [μM s−1] | ||||
|---|---|---|---|---|---|---|
| 460 nm | 530 nm | 650 nm | 460 nm | 530 nm | 650 nm | |
| 5 OMe | 4.20 | 0.370 | 0.111 | 1.17 | 0.254 | 0.0835 |
| 5 H | 3.11 | 0.363 | 0.123 | 1.79 | 0.300 | 0.0999 |
| 5 Cl | 2.69 | 0.707 | 0.113 | 1.26 | 0.494 | 0.0988 |
| 5 NO2 | 1.77 | 1.56 | 0.493 | 0.84 | 0.809 | 0.252 |
3.2 Computational studies on photodissociation mechanisms
To further elucidate the mechanism of photodissociation in iron and cobalt nitrosyl complexes, complete active space self-consistent field (CASSCF) calculations were performed on 20 and 21.54 For 20, the CASSCF results suggested a resonance between FeI–NO+, FeII–NO˙, and FeIII–NO− configurations in the ground state, with FeIII–NO− being the dominant form. The low-lying excited states Q1 and Q2 were mainly composed of the FeII–NO˙ state, which can be accessed via metal-to-ligand charge transfer (MLCT) from the FeI–NO+ configuration at 740 nm. The transitions involve electron transfer from Fe-dominated Fe–NO bonding orbitals to NO-π*-dominated Fe–NO antibonding orbitals, leading to Fe–NO bond cleavage and the release of NO.
Similarly, in 21, the CASSCF results indicated that the absorption band at 480 nm is associated with an MLCT transition from CoI–NO+ to CoII–NO˙ configuration, resulting in NO photodissociation. Notably, 21 is more likely to reach the MLCT than 20 because the contribution of MI–NO+ state to MLCT showed higher probability (41.86% for 21 and 32.57% for 20) (Fig. 5). These probabilities indicate the likelihood of undergoing MLCT. Due to the higher percentage for complex 21, it has a greater quantum yield (0.78 for 21versus 0.23 for 20) and a faster photolysis rate with a half-life of 24 seconds for 21 compared to 255 seconds for 20.
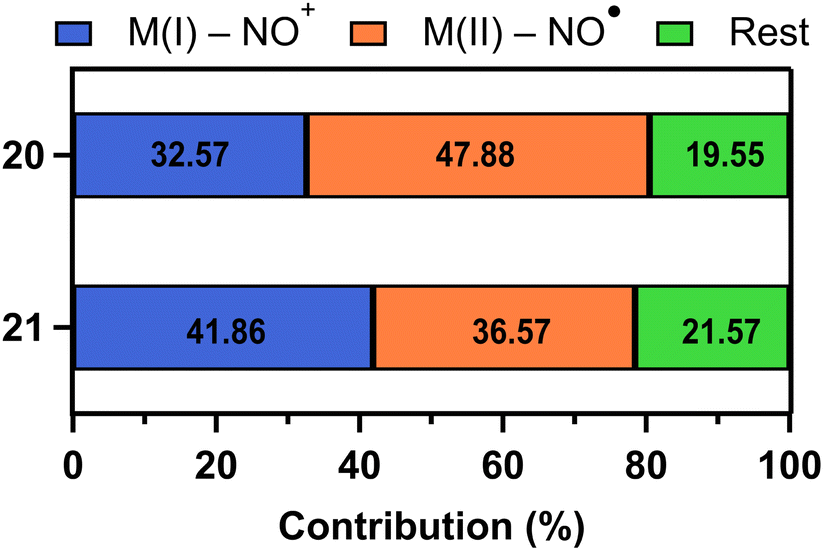 | ||
| Fig. 5 Bar graphs highlight the MI–NO+ configuration percentage of complexes 20 and 21 involved in reaching the MLCT state. | ||
3.3 Structural and spectroscopic correlations in the Fe–N–O unit
The relationships between structural and spectroscopic properties offer valuable insights into the electronic structures of transition metal complexes. In metal–carbonyl (M–CO) complexes, there are well-established correlations between C–O bond length and stretching frequency. Additionally, the inverse relationship between M–CO bond and C–O bond distance, caused by π-backdonation, is widely accepted.68–70Similarly, in nitrosyl complexes, the bonding between the metal and NO involves a combination of σ-donation from NO to the metal and π-backdonation from the metal to the NO-π* orbital. Complexes with linear bond angles (170–180°) are considered more capable of donating electron density from the metal to the NO-π* orbital. In contrast, complexes with bent angles (120–170°) are more restricted to provide electron density through π-backdonation.71 Despite numerous studies on nitrosyl complexes based on the general understanding, the structural and spectroscopic relationships in M–NO complexes often display irregularities that challenge conventional expectations.72
In Mn–NO species, to validate the correlation between NO stretching vibration energies and N–O bond lengths, {MnNO}6 complexes supported by anionic pentadentate N5 carboxamide ligands that can provide comparable coordination environments were plotted. Only complexes with the same spin state (S = 0) and a linear bond angle (170–180°) were considered.36–39 However, as shown in Fig. 6a, no linear correlation was observed between the vibrational energy and N–O bond strength. Additionally, the plot of the relationship between N–O bond length and Mn–NO distance also indicated no clear correlation (Fig. 6b). In contrast, for S = 0 {FeNO}6 complexes, a correlation between NO stretching vibration energies and N–O bond lengths follows the expected trend where the IR frequency increases as the N–O bond shortens, although the coordination environments are not considered.41–45,52,53 Moreover, the N–O bond length decreases when the Fe–NO bond weakens, demonstrating the influence of π-backdonation in the Fe–N–O unit.
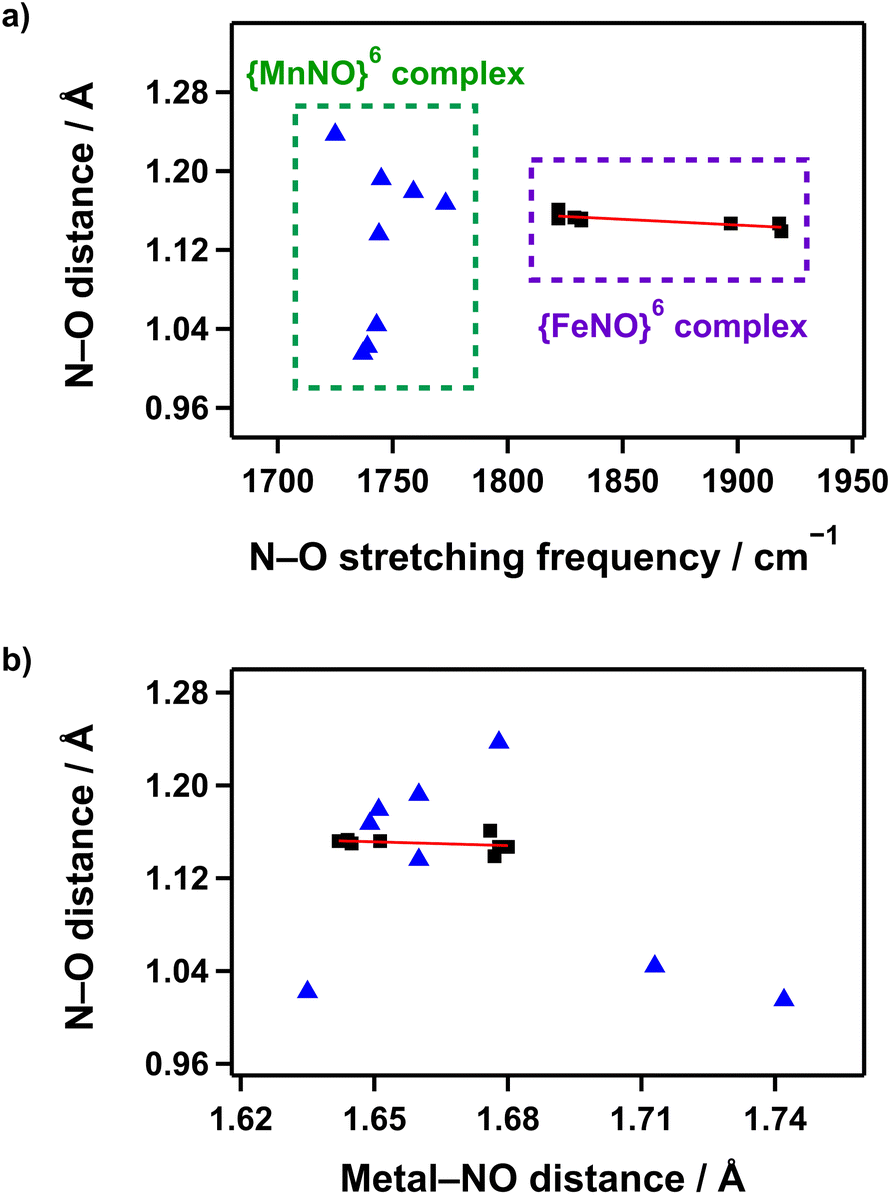 | ||
| Fig. 6 (a) Plot of the N–O bond distance (Å) against the N–O bond stretching frequency (cm−1) for {MnNO}6 and {FeNO}6 complexes. (b) Plot of the N–O bond distance (Å) against the M–NO bond length (Å). Triangle (blue) and rectangle (black) represent {MnNO}6 and {FeNO}6 complexes, respectively. The solid red line shows a least-squares linear fit of the data. | ||
Overall, the structural and spectroscopic relationships in the M–N–O unit remain challenging to fully understand. The irregularities observed in Mn–NO and Fe–NO complexes suggest that the bonding interactions between metal and NO are highly sensitive to metal identity, coordination geometry, and environmental factors. Further systematic and comprehensive investigations are necessary to clarify the relationships and develop a more complete understanding of the underlying principles governing metal–nitrosyl chemistry.
4. Application
4.1 Vasodilation and reperfusion
Endogenously produced NO plays an essential role in vascular regulation. NO diffuses into smooth muscle cells and stimulates the conversion of guanosine-5′-triphosphate (GTP) into cyclic guanosine-3′,5′-monophosphate (cGMP) through the activation of the soluble guanylyl cyclase (sGC) enzyme. Elevated cGMP levels trigger the activation of protein kinase G (PKG), leading to the subsequent activation of extracellular signal-regulated kinases (ERKs). The cascade ultimately promotes vasodilation through the relaxation of smooth muscle cells.8,9Mascharak and co-workers demonstrated the potential of a photo-responsive Mn–NO complex 1 to induce vasodilation. Under light irradiation, 1 dramatically increases sGC activation in physiological environment. In rat aorta smooth muscle cells, 1 triggered vasorelaxation when exposed to visible light.73 The vasodilatory effect was reduced by the sGC inhibitor, 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), supporting that 1 can effectively activate sGC in the presence of light.
Cho and co-workers showed that ERK pathways in live cells can be selectively activated through light-triggered NO delivery using Co–NO complex 21, revealing distinct kinetic differences between endogenous and exogenous NO delivery.55 A kinase translocation reporter (KTR) cell line was developed to visualize the ERK signaling pathway in real-time, enabling quantitative comparison of endogenous and exogenous NO delivery on ERK activation. Additionally, the study confirmed that NO-induced ERK activation did not result from non-specific perturbation of membrane receptors, including the epidermal growth factor receptor (EGFR), validating that NO regulates ERK signaling with distinct dynamics based on spatial regions at the single-cell levels.
Furthermore, the therapeutic potential of the Fe–NO (20) and Co–NO (21) complexes for retinal vascular occlusion (RVO), a disease that impairs blood flow in the retina, was investigated.5420 and 21 were intravitreally injected into the eyes of mice with transient light exposure, resulting in significant dilation of retinal blood vessels. Notably, in the artificially induced mouse RVO model, only complex 20 rapidly opened occluded sites and successfully facilitated reperfusion. The research speculated that the superior therapeutic effect of complex 20 can be attributed to the relatively slow photoresponse, resulting in a sustained NO release that effectively induces vasodilation and reperfusion. Additionally, NO-releasing donors such as diethylenetriamine diazeniumdiolate (DETA-NONOate) and sodium nitroprusside (SNP) were employed under identical conditions as positive controls. For DETA-NONOate and SNP, however, adverse effects like exudative retinal detachment occurred 10 minutes after injection. These findings suggest that the treatment strategy using M–NO with light exposure can be extended to effectively treat other vascular diseases.
4.2 Immune response and antibiotic effect
NO is a powerful bioactive molecule that plays a multifaceted role in immune system, by interacting with transition metal ions or reactive oxygen species (ROS), including superoxide, peroxide, and dioxygen, to form reactive nitrogen species (RNS) such as peroxynitrite (ONOO−) and nitrogen dioxide (NO2). The RNS exerts nitrosative stress on pathogen, targeting DNA, proteins, and lipids, which helps eradicate invading bacteria and enhances the body's defense mechanism.74To harness NO's antimicrobial properties, light-responsive NO delivery systems targeting drug-resistant bacteria was employed.73,75,76 One such system incorporated 1 into a sol–gel matrix that allowed precise NO release under visible light, which effectively reduced bacterial loads of Pseudomonas aeruginosa (P. aeruginosa, Gram-negative), Escherichia coli (E. Coli, Gram-negative), Staphylococcus aureus (S. aureus, Gram-positive), and methicillin-resistant Staphylococcus aureus (MRSA, antibiotic-resistant). This approach demonstrated the potential for localized treatment of infections, offering a significant advantage over conventional antibiotic treatment.
The polyurethane-based composite (PUX-NO) films with embedded NO-releasing silica particles containing 1 was developed.77 The films exhibited high stability and capacity of NO release over extended periods, which is suitable for applications as wound dressings. The PUX-NO films showed effective reduction of bacterial loads of both Gram-positive and Gram-negative bacteria, including MRSA and S. aureus, under controlled light exposure, indicating the potential for treatment of skin and soft-tissue infections (SSTI).
A porous material MCM-41 loading complex 1 was developed to eradicate the drug-resistant bacterium Acinetobacter baumannii (A. baumannii), which exhibits a high transmission rate of infection in hospital settings.78 The loading efficiency was confirmed by using powder X-ray diffraction (PXRD) and N2 adsorption/desorption isometry. In the absence of visible light, the material demonstrated significant stability with minimal leaching of 1 in physiological saline. Upon exposure to low-power (10–100 mW) visible light, rapid NO release was observed while the photoproducts were retained within the host structure, decreasing the risk of toxicity from the material. This strategy reveals the potential of photo-triggered NO delivery systems for effective treatment of multidrug-resistant bacterial infections in challenging clinical environments.
5. Conclusions and outlook
In this review, we have summarized the recent progress in the development of photo-triggered NO release from first-low transition metal–nitrosyl complexes, focusing on the geometric and electronic structures that determine photoreactivity and NO release efficiency. Key advancements have emerged from the modulation of ligand frameworks through the introduction of π-conjugation systems and electronic substituents, allowing for more precise control over light responsiveness. The modifications have enhanced the potential of nitrosyl complexes for biomedical applications. Spectroscopic and computational studies have provided crucial insights into the mechanisms of photodissociation, revealing that the electronic transitions from bonding to antibonding orbitals play a pivotal role in facilitating NO release.Despite advancements, several challenges remain. One of the most significant barriers to clinical application is optimizing key photophysical properties such as quantum yield and activation wavelengths. To overcome obstacles, future research must focus on developing ligand frameworks that improve light absorption in the NIR region, enhance stability under physiological conditions, and minimize toxicity. The integration of experimental results with computational modeling is also essential to predict and refine photoreactivity, enabling the development of more effective NO donors. Such innovations could significantly broaden the therapeutic potential of nitrosyl complexes, enabling their use in a wider range of medical applications such as cardiovascular disease treatment and targeted eradication of bacterial infections. Photo-responsive NO donors may become a key tool in advanced therapeutic strategies, offering precise control over NO delivery with minimal side effects.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
S. S. and J. Ce. wrote the manuscript. J. C. revised and supervised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was supported by National Research Foundation funded by the Ministry of Science, ICT and Future Planning (RS-2024-00333606) and the Ministry of Health and Welfare (RS-2023-00217242) of Korea.References
- R. M. J. Palmer, A. G. Ferrige and S. Moncada, Nature, 1987, 327, 524–526 CrossRef CAS PubMed.
- C. Farah, L. Y. M. Michel and J.-L. Balligand, Nat. Rev. Cardiol., 2018, 15, 292–316 CrossRef CAS.
- E. Culotta and D. E. Koshland, Science, 1992, 258, 1862–1865 CrossRef CAS.
- S. H. Snyder, Science, 1992, 257, 494–496 CrossRef CAS.
- C. Bogdan, Nat. Immunol., 2001, 2, 907–916 CrossRef CAS PubMed.
- S. M. Andrabi, N. S. Sharma, A. Karan, S. M. S. Shahriar, B. Cordon, B. Ma and J. Xie, Adv. Sci., 2023, 10, 2303259 CrossRef CAS PubMed.
- M. A. Cinelli, H. T. Do, G. P. Miley and R. B. Silverman, Med. Res. Rev., 2020, 40, 158–189 CrossRef CAS PubMed.
- Y. Kang, R. Liu, J.-X. Wu and L. Chen, Nature, 2019, 574, 206–210 CrossRef CAS PubMed.
- R. Liu, Y. Kang and L. Chen, Nat. Commun., 2021, 12, 5492 CrossRef CAS PubMed.
- I. Endo, M. Nojiri, M. Tsujimura, M. Nakasako, S. Nagashima, M. Yohda and M. Odaka, J. Inorg. Biochem., 2001, 83, 247–253 CrossRef CAS PubMed.
- I. Endo, M. Odaka and M. Yohda, Trends Biotechnol., 1999, 17, 244–248 CrossRef CAS PubMed.
- M. Odaka, K. Fujii, M. Hoshino, T. Noguchi, M. Tsujimura, S. Nagashima, M. Yohda, T. Nagamune, Y. Inoue and I. Endo, J. Am. Chem. Soc., 1997, 119, 3785–3791 CrossRef CAS.
- N. Lehnert, E. Kim, H. T. Dong, J. B. Harland, A. P. Hunt, E. C. Manickas, K. M. Oakley, J. Pham, G. C. Reed and V. S. Alfaro, Chem. Rev., 2021, 121, 14682–14905 CrossRef CAS PubMed.
- P. G. Wang, M. Xian, X. Tang, X. Wu, Z. Wen, T. Cai and A. J. Janczuk, Chem. Rev., 2002, 102, 1091–1134 CrossRef CAS PubMed.
- R. Scatena, P. Bottoni, G. Martorana and B. Giardina, Expert Opin. Invest. Drugs, 2005, 14, 835–846 CrossRef CAS PubMed.
- D. A. Riccio and M. H. Schoenfisch, Chem. Soc. Rev., 2012, 41, 3731–3741 RSC.
- J. A. Hrabie and L. K. Keefer, Chem. Rev., 2002, 102, 1135–1154 CrossRef CAS PubMed.
- V. N. Varu, N. D. Tsihlis and M. R. Kibbe, Vascular and Endovascular Surgery, 2009, 43, 121–131 CrossRef PubMed.
- J. Saraiva, S. S. Marotta-Oliveira, S. A. Cicillini, J. D. O. Eloy and J. M. Marchetti, J. Drug Delivery, 2011, 2011, 936438 Search PubMed.
- S. Sortino, Chem. Soc. Rev., 2010, 39, 2903–2913 RSC.
- P. G. Wang, M. Xian, X. Tang, X. Wu, Z. Wen, T. Cai and A. J. Janczuk, Chem. Rev., 2002, 102, 1091–1134 CrossRef CAS PubMed.
- R. Weinstain, T. Slanina, D. Kand and P. Klán, Chem. Rev., 2020, 120, 13135–13272 CrossRef CAS PubMed.
- H. M. Elbeheiry and M. Schulz, Coord. Chem. Rev., 2024, 515, 215921 CrossRef CAS.
- N. L. Fry and P. K. Mascharak, Acc. Chem. Res., 2011, 44, 289–298 CrossRef CAS PubMed.
- I. Stepanenko, M. Zalibera, D. Schaniel, J. Telser and V. B. Arion, Dalton Trans., 2022, 51, 5367–5393 RSC.
- H.-J. Xiang, M. Guo and J.-G. Liu, Eur. J. Inorg. Chem., 2017, 2017, 1586–1595 CrossRef CAS.
- J. J. Becker, P. S. White and M. R. Gagné, Inorg. Chem., 1999, 38, 798–801 CrossRef CAS PubMed.
- I. Ara, J. Forniés, M. A. García-Monforte, B. Menjón, R. M. Sanz-Carrillo, M. Tomás, A. C. Tsipis and C. A. Tsipis, Chem.–Eur. J., 2003, 9, 4094–4105 CrossRef CAS PubMed.
- M. J. G. Sinclair, N. Roig, M. R. Gyton, N. Tsoureas, F. G. N. Cloke, M. Alonso and A. B. Chaplin, Inorg. Chem., 2024, 63, 1709–1713 CrossRef CAS.
- J. Xiang, Q. Wang, S.-M. Yiu, W.-L. Man, H.-K. Kwong and T.-C. Lau, Inorg. Chem., 2016, 55, 5056–5061 CrossRef CAS PubMed.
- J. Xiang, Q. Wang, S.-M. Yiu and T.-C. Lau, Inorg. Chem., 2017, 56, 2022–2028 CrossRef CAS.
- R. Bhowmik and M. Roy, Eur. J. Med. Chem., 2024, 268, 116217 CrossRef CAS PubMed.
- J. M. Mir, B. A. Malik and R. C. Maurya, Rev. Inorg. Chem., 2019, 39, 91–112 CAS.
- M. J. Rose and P. K. Mascharak, Curr. Opin. Chem. Biol., 2008, 12, 238–244 CrossRef CAS PubMed.
- B. Heilman and P. K. Mascharak, Philos. Trans. R. Soc., A, 2013, 371, 20120368 CrossRef.
- K. Ghosh, A. A. Eroy-Reveles, B. Avila, T. R. Holman, M. M. Olmstead and P. K. Mascharak, Inorg. Chem., 2004, 43, 2988–2997 CrossRef CAS PubMed.
- A. A. Eroy-Reveles, Y. Leung, C. M. Beavers, M. M. Olmstead and P. K. Mascharak, J. Am. Chem. Soc., 2008, 130, 4447–4458 CrossRef CAS PubMed.
- C. G. Hoffman-Luca, A. A. Eroy-Reveles, J. Alvarenga and P. K. Mascharak, Inorg. Chem., 2009, 48, 9104–9111 CrossRef CAS PubMed.
- Y. Hitomi, Y. Iwamoto and M. Kodera, Dalton Trans., 2014, 43, 2161–2167 RSC.
- Y. Iwamoto, M. Kodera and Y. Hitomi, Chem. Commun., 2015, 51, 9539–9542 RSC.
- D. Schweitzer, J. J. Ellison, S. C. Shoner, S. Lovell and J. A. Kovacs, J. Am. Chem. Soc., 1998, 120, 10996–10997 CrossRef CAS.
- A. K. Patra, R. Afshar, M. M. Olmstead and P. K. Mascharak, Angew. Chem., Int. Ed., 2002, 41, 2512–2515 CrossRef CAS PubMed.
- A. K. Patra, J. M. Rowland, D. S. Marlin, E. Bill, M. M. Olmstead and P. K. Mascharak, Inorg. Chem., 2003, 42, 6812–6823 CrossRef CAS PubMed.
- R. K. Afshar, A. K. Patra, M. M. Olmstead and P. K. Mascharak, Inorg. Chem., 2004, 43, 5736–5743 CrossRef CAS PubMed.
- A. A. Eroy-Reveles, C. G. Hoffman-Luca and P. K. Mascharak, Dalton Trans., 2007, 5268–5274 RSC.
- M. J. Rose, N. M. Betterley and P. K. Mascharak, J. Am. Chem. Soc., 2009, 131, 8340–8341 CrossRef CAS PubMed.
- M. J. Rose, N. M. Betterley, A. G. Oliver and P. K. Mascharak, Inorg. Chem., 2010, 49, 1854–1864 CrossRef CAS.
- A. C. McQuilken, Y. Ha, K. D. Sutherlin, M. A. Siegler, K. O. Hodgson, B. Hedman, E. I. Solomon, G. N. L. Jameson and D. P. Goldberg, J. Am. Chem. Soc., 2013, 135, 14024–14027 CrossRef CAS PubMed.
- A. C. McQuilken, H. Matsumura, M. Dürr, A. M. Confer, J. P. Sheckelton, M. A. Siegler, T. M. McQueen, I. Ivanović-Burmazović, P. Moënne-Loccoz and D. P. Goldberg, J. Am. Chem. Soc., 2016, 138, 3107–3117 CrossRef CAS PubMed.
- A. Dey, A. M. Confer, A. C. Vilbert, P. Moënne-Loccoz, K. M. Lancaster and D. P. Goldberg, Angew. Chem., Int. Ed., 2018, 57, 13465–13469 CrossRef CAS PubMed.
- N. Levin, J. Perdoménico, E. Bill, T. Weyhermüller and L. D. Slep, Dalton Trans., 2017, 46, 16058–16064 RSC.
- C.-K. Chiang, K.-T. Chu, C.-C. Lin, S.-R. Xie, Y.-C. Liu, S. Demeshko, G.-H. Lee, F. Meyer, M.-L. Tsai, M.-H. Chiang and C.-M. Lee, J. Am. Chem. Soc., 2020, 142, 8649–8661 CrossRef CAS PubMed.
- H.-C. Chen, G.-H. Lee, S.-Y. Chien and C.-M. Lee, J. Chin. Chem. Soc., 2023, 70, 1125–1135 CrossRef CAS.
- J. Choe, S. J. Kim, J.-H. Kim, M.-H. Baik, J. Lee and J. Cho, Chem, 2023, 9, 1309–1317 CAS.
- S. Shin, J. Choe, Y. Park, D. Jeong, H. Song, Y. You, D. Seo and J. Cho, Angew. Chem., Int. Ed., 2019, 58, 10126–10131 CrossRef CAS PubMed.
- J. H. Enemark and R. D. Feltham, Coord. Chem. Rev., 1974, 13, 339–406 CrossRef CAS.
- J. S. Southern, G. L. Hillhouse and A. L. Rheingold, J. Am. Chem. Soc., 1997, 119, 12406–12407 CrossRef CAS.
- R. Lin and P. J. Farmer, J. Am. Chem. Soc., 2000, 122, 2393–2394 CrossRef CAS.
- Y. Ling, C. Mills, R. Weber, L. Yang and Y. Zhang, J. Am. Chem. Soc., 2010, 132, 1583–1591 CrossRef CAS PubMed.
- E. G. Abucayon, R. L. Khade, D. R. Powell, Y. Zhang and G. B. Richter-Addo, J. Am. Chem. Soc., 2016, 138, 104–107 CrossRef CAS PubMed.
- S. A. Suarez, N. I. Neuman, M. Muñoz, L. a. Álvarez, D. E. Bikiel, C. D. Brondino, I. Ivanović-Burmazović, J. L. Miljkovic, M. R. Filipovic, M. A. Martí and F. Doctorovich, J. Am. Chem. Soc., 2015, 137, 4720–4727 CrossRef CAS PubMed.
- S. A. Suarez, M. Muñoz, L. Alvarez, M. F. Venâncio, W. R. Rocha, D. E. Bikiel, M. A. Marti and F. Doctorovich, J. Am. Chem. Soc., 2017, 139, 14483–14487 CrossRef CAS PubMed.
- I. Ivanovic-Burmazovic and M. R. Filipovic, Inorg. Chem., 2019, 58, 4039–4051 CrossRef CAS PubMed.
- W. Zheng, S. Wu, S. Zhao, Y. Geng, J. Jin, Z. Su and Q. Fu, Inorg. Chem., 2012, 51, 3972–3980 CrossRef CAS PubMed.
- N. L. Fry and P. K. Mascharak, Dalton Trans., 2012, 41, 4726–4735 RSC.
- N. L. Fry, X. P. Zhao and P. K. Mascharak, Inorg. Chim. Acta, 2011, 367, 194–198 CrossRef CAS.
- A. C. Merkle, N. L. Fry, P. K. Mascharak and N. Lehnert, Inorg. Chem., 2011, 50, 12192–12203 CrossRef CAS PubMed.
- G. Frenking, I. Fernández, N. Holzmann, S. Pan, I. Krossing and M. Zhou, JACS Au, 2021, 1, 623–645 CrossRef CAS PubMed.
- A. J. Lupinetti, S. Fau, G. Frenking and S. H. Strauss, J. Phys. Chem. A, 1997, 101, 9551–9559 CrossRef CAS.
- A. S. Goldman and K. Krogh-Jespersen, J. Am. Chem. Soc., 1996, 118, 12159–12166 CrossRef CAS.
- D. M. P. Mingos, in Nitrosyl Complexes in Inorganic Chemistry, Biochemistry and Medicine I, ed. D. M. P. Mingos, Springer, Berlin, Heidelberg, 2014, pp. 1–44, DOI:10.1007/430_2013_116.
- H. Lewandowska, in Nitrosyl Complexes in Inorganic Chemistry, Biochemistry and Medicine I, ed. D. M. P. Mingos, Springer, Berlin, Heidelberg, 2014, pp. 117–124, DOI:10.1007/430_2013_109.
- M. Madhani, A. K. Patra, T. W. Miller, A. A. Eroy-Reveles, A. J. Hobbs, J. M. Fukuto and P. K. Mascharak, J. Med. Chem., 2006, 49, 7325–7330 CrossRef CAS PubMed.
- P. K. Mascharak, J. Inorg. Biochem., 2022, 231, 111804 CrossRef CAS PubMed.
- A. A. Eroy-Reveles, Y. Leung and P. K. Mascharak, J. Am. Chem. Soc., 2006, 128, 7166–7167 CrossRef CAS PubMed.
- G. M. Halpenny, K. R. Gandhi and P. K. Mascharak, ACS Med. Chem. Lett., 2010, 1, 180–183 CrossRef CAS PubMed.
- B. J. Heilman, G. M. Halpenny and P. K. Mascharak, J. Biomed. Mater. Res., Part B, 2011, 99, 328–337 CrossRef PubMed.
- B. J. Heilman, J. St. John, S. R. J. Oliver and P. K. Mascharak, J. Am. Chem. Soc., 2012, 134, 11573–11582 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |