
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Graphene and graphene–cellulose nanocrystal composite films for sustainable anodes in biophotovoltaic devices†
Sara
Lund‡
 abc,
Laura T.
Wey‡
c,
Jouko
Peltonen
a,
Johan
Bobacka
ab,
Rose-Marie
Latonen
*ab and
Yagut
Allahverdiyeva
*c
abc,
Laura T.
Wey‡
c,
Jouko
Peltonen
a,
Johan
Bobacka
ab,
Rose-Marie
Latonen
*ab and
Yagut
Allahverdiyeva
*c
aLaboratory of Molecular Science and Engineering, Faculty of Science and Engineering, Åbo Akademi University, Henriksgatan 2, 20500 Turku (Åbo), Finland. E-mail: rose-marie.latonen@abo.fi
bJohan Gadolin Process Chemistry Centre (PCC), Åbo Akademi University, Henriksgatan 2, 20500 Turku (Åbo), Finland
cPhotomicrobes Research Group, Molecular Plant Biology Unit, Department of Life Technologies, University of Turku, 20014 Turku, Finland. E-mail: allahve@utu.fi
First published on 1st December 2023
Abstract
The urgent need for renewable energy technologies has fuelled the exploration of biophotovoltaic devices (BPVs) that harness photosynthetic microorganisms, such as cyanobacteria, for solar-to-electricity conversion. To address the need for sustainable and scalable BPV power generation, the development of suitable electrode materials is crucial. In this study, we investigated electrically conducting few-layer graphene films and composites of graphene and cellulose nanocrystals (CNC) as potential BPVs anodes. Graphene and graphene–CNC electrodes were fabricated using a green liquid-phase shear exfoliation method in aqueous environments, employing sodium cholate (SC) surfactant solution or a CNC suspension, respectively, followed by spray-coating onto non-conductive glass substrates. Both CNC and SC are non-toxic, naturally derived, and renewable. Surface characterisation revealed hydrophilic films with nanoscale roughness, ideal for interfacing cyanobacterial cells. Cyclic voltammetry experiments demonstrated the electroactivity and stability of the electrodes in aqueous electrolyte solutions compatible with cyanobacteria. The photoelectrochemical performance of cyanobacterial cells on these electrodes was evaluated using a three-electrode electrochemical set-up. The graphene and graphene–CNC electrodes harvested photocharge densities over a 5 min period of 86.0 ± 32.0 μC cm−2 and 52.8 ± 23.2 μC cm−2, respectively; and with ferricyanide 339 ± 139 μC cm−2 and 134 ± 79 μC cm−2, respectively (photocurrent densities with ferricyanide of 2.17 ± 0.74 μA cm−2 and 1.11 ± 0.60 μA cm−2, respectively). Due to their abundant source materials and efficient fabrication method, few-layer graphene and graphene–CNC composites present a sustainable solution as anodes for renewable electricity generation in BPVs. This research provides a foundation for the advancement of cost-effective and environmentally friendly BPV technologies, thereby contributing to the reduction of fossil fuel dependence in energy generation.
1 Introduction
Addressing the global energy crisis and mitigating climate change requires the widespread adoption of advanced clean energy technologies. Biophotovoltaic devices (BPVs) present a promising component of this solution, whereby oxygenic photosynthetic microorganisms (cyanobacteria and algae) are deployed as living photocatalysts to generate renewable electricity from sunlight and water.1–3 The electricity comes from their innate ability to perform exoelectrogenesis, a phenomenon where they export electrons liberated from photosynthesis to the environment under illumination.2 In a BPV, the photosynthetic microorganisms are interfaced with an anode electrode that harvests the resulting current under illumination (i.e., the photocurrent, or photocharge over a period of time), and is connected to a cathode electrode in an external circuit. Analysis of exoelectrogenesis and different electrodes for future BPV applications are conducted in three-electrode electrochemical set-ups,2 in which the photosynthetic microorganisms are interfaced with the working electrode.Efforts to enhance photocurrent outputs in BPVs have predominantly focused on the development of an efficient anode.2 Indium tin oxide (ITO) electrodes with a micropillar structure have demonstrated the highest photocurrent outputs to date.4 ITO-based electrodes have been a popular choice as anodes in BPVs5–7 due to their nanoscale surface roughness, electrical conductivity and optical transparency that facilitates light penetration through the substrate to reach the photosynthetic microorganisms. Furthermore, the availability of ITO in nanoparticle form has enabled fabrication of ITO electrodes with complex architectures, a key element in achieving high photocurrent outputs, for example by aerosol jet printing.2,4,6 However, the scarcity and high cost of indium as a rare earth metal and the intensive processing requirements such as high vacuum and elevated processing temperatures8 necessitate the development of sustainable and cost-effective electrode alternatives with easy fabrication for large-scale BPV power generation.9
Carbon nanomaterials, particularly graphene, are emerging as a potential replacement for rare and expensive metals, including indium, gallium, platinum, and silver, in electronic applications.10 These carbon nanomaterials can be solution-processed as dispersions at room temperature by exfoliating graphite in a liquid-phase, and used, for example, in inkjet printing,11 but are considered as a more sustainable alternative to scarce metals due to the abundance of carbon sources.10,12 While in BPVs carbon-based anodes like carbon fibre, carbon cloth and graphite have been extensively utilised due to their inert electrochemistry, electrical conductivity and cost-effectiveness,9,13 graphene as an anode material remains relatively unexplored. Graphene exhibits desirable properties of high specific surface area, excellent electrical conductivity and good mechanical strength.14 Reduced graphene oxide (RGO) has been the form of graphene used as anode in BPVs to date (Tables S1 and S2†).15–21 RGO is obtained through reduction of graphene oxide (GO) using chemical agents such as hydrazine15,22,23 and sodium borohydride.23,24 The precursor, GO, is prepared by oxidising graphite using strong oxidising agents in an acidic environment via Hummers' method22,25 or its modifications.15,16 After oxidation, the GO sheets are typically separated via sonication.22–24 However, the aggressive oxidation process brings defects to the carbon lattice which are not restored after reduction22,26 even if the conductivity is improved by the reduced amount of oxygen functionalities.23,24 The strong oxidising and reducing reagents pose safety and environmental risks27 and these complicated fabrication procedures are also energy-intensive and time consuming.
We recently reported a fast and efficient method of fabricating unoxidised graphene that has yet to be employed as an anode in BPVs. Few-layer graphene in liquid-phase was fabricated by shear exfoliation of natural flake graphite in a sodium cholate (SC) surfactant solution,28 and this method was further extended to produce composites of graphene and cellulose nanocrystals (CNC) by shear exfoliating graphite directly in a CNC suspension.29 The shear exfoliation method offers advantages of simplicity, cost effectiveness, high yield and scalability.30 Furthermore, the fabrication of graphene dispersions in an SC surfactant solution or CNC suspension eliminates the need for hazardous chemicals and produces defect-free few-layer graphene. Both SC and CNC are non-toxic materials readily available from renewable sources: CNC is derived from cellulose, the most abundant polymer on Earth, and SC is the bile salt of cholic acid from liver. SC as an ionic surfactant and an amphiphile stabilises the hydrophobic graphene in an aqueous environment. CNC has hydrophilic –OH groups and hydrophobic –CH moieties through which it can act as a dispersant.31,32 Due to its nearly perfect crystalline structure resulting from the removal of the amorphous regions in cellulose segments,33 CNC possesses a high mechanical strength. As a result, it is often used in composite materials to enhance their mechanical properties.33,34 In a previous study, we also confirmed that both SC- and CNC-stabilised graphene dispersions prepared by shear exfoliation are biocompatible with the model cyanobacterium Synechocystis sp. PCC 6803 (hereafter Synechocystis) performing efficient photosynthesis.29
In this study, we explored the utilisation of few-layer graphene and graphene–CNC films, which were fabricated using environmentally friendly water-based shear exfoliation method, for their potential as anodes in BPVs. These films, composed of highly concentrated dispersions of SC- and CNC-stabilised few-layer graphene (hereafter graphene and graphene–CNC), offer an easy route to electrode preparation by spray-coating, and they are produced without using any costly or hazardous chemicals. We characterised the electrodes for their suitability for interfacing with cyanobacterial cells, which included an examination of the surface properties such as roughness and hydrophilicity. Electrochemical characterisation was conducted using cyclic voltammetry to investigate the electron transfer properties of the electrodes in electrolyte favourable for the cyanobacteria. We quantified the photoelectrochemical performance of biofilms of Synechocystis cells loaded onto the electrodes in an analytical three-electrode set-up. To characterise the scope of performance, we explored different electrodes (including variations in composition involving CNC and film thickness) and biocatalyst loadings (such as biofilm formation duration and cell loading).
2 Results and discussion
2.1 Characterisation of the graphene and graphene–CNC films
 | ||
| Fig. 1 Surface imaging and characterisation of graphene and graphene–CNC films. Top-view topographical atomic force micrograms of (a) graphene and (b) graphene–CNC films on glass. Scale bar 1 μm indicates x–y plane; whole image is 5.0 μm × 5.0 μm; colour scale bar on right indicates z-height. (c) Scanning electron microgram of a Synechocystis cyanobacterium cell on a graphene film. (d) Root-mean-square (RMS) – roughness (Sq) of graphene and graphene–CNC films, as well as ITO-coated glass. (e) Density of summits (Sds) and (f) surface area ratios (Sdr) of graphene and graphene–CNC films measured from 5.0 μm × 5.0 μm AFM images. Data presented as mean ± standard deviation (n = 3 films, 3 images/film), statistical significance by t-test P ≤ 0.001 denoted by ***, P ≤ 0.01 denoted by **, P > 0.05 denoted by n.s. (not significant). | ||
The root-mean-square roughness parameter (Sq) represents the standard deviation of surface heights from the mean level. For context, the Sq value of a smooth silicon wafer is 1.35 nm (10 μm × 10 μm image).36 The Sq values for the graphene and graphene–CNC films were 44.1 ± 13.4 nm and 40.8 ± 3.2 nm, respectively, (n = 3, P = 0.700) indicating that both films possess similar levels of nanoscale roughness (Fig. 1d). These Sq values are an order of magnitude larger than for ITO-coated glass (3.6 ± 0.5 nm), indicating that both graphene-based films are rougher (n = 3, graphene: P = 0.006, graphene–CNC: P = 0.0001). The ten-point height parameter (S10z) was used to further evaluate the nanoscale roughness. The S10z parameter represents the average height of the five highest local maxima plus the average height of the five lowest local minima. The S10z values for graphene, graphene–CNC, and ITO were 238 ± 76, 334 ± 21, and 45.6 ± 6.2 nm, respectively, confirming that the graphene-based films possess more nanoscale roughness than ITO. Analysis of roughness parameters such as skewness (Ssk) and kurtosis (Sku) provide information about the distribution of height values. The Ssk values for both films were close to zero, indicating a symmetrical height distribution around the average height and the absence of peak- or valley-dominated roughness. The Sku values for both films were close to 3, indicating a Gaussian distribution of the height values.
The density of summits parameter (Sds), which describes the density of local maxima, provides insight into the fine texture of the film surface. The Sds value was ca. 6-fold higher for the graphene–CNC film (163.5 ± 21.3 μm−2) than for the graphene film (29.0 ± 6.7 μm−2) (Fig. 1e) indicating that the graphene–CNC film exhibits a finer texture (n = 3, P = 0.0005). The surface area ratio parameter (Sdr) describes the relative (percentual) increase in surface area caused by roughness compared to the area of an ideally smooth surface (i.e., the projected area). Roughness increased the surface areas of graphene and graphene–CNC films by 1.6 ± 1.3% and 16.6 ± 0.7%, respectively (Fig. 1f). Although the root-mean-square roughness was similar for both film types, the Sdr value was ca. 10-fold higher for the graphene–CNC film than for the graphene film (n = 3, P = 0.0001) (Fig. 1f). This could be attributed to graphene–CNC films having a finer texture than the graphene films.
Micron-scale surface morphology has been recognised as a favourable property for adsorption of cells of corresponding size.2,19 In line with this, we analysed the cavities (>0.5 μm diameter) observed in the topographical AFM images (Fig. 1a, b and S1†). Graphene–CNC films had more and smaller cavities than the graphene films. The graphene–CNC films had cavities with a diameter of 900 ± 300 and a depth of 100 ± 30 nm. Considering a Synechocystis cell with a diameter of ca. 1.5 μm and a dome cap width of 750 nm at 100 nm from the cell base (Fig. 1c), then some cells would be secured in some cavities in the graphene–CNC films. The graphene films had cavities with a diameter 1400 ± 700 nm and a depth of 70 ± 50 nm. Therefore, the Synechocystis cells would have less contact with the graphene film surface in comparison to the graphene–CNC films. These findings indicate that the graphene–CNC films are better suited for the adsorption of cyanobacterial cells than the graphene films. This conclusion is further supported by the larger value of the Sdr parameter observed for the graphene–CNC composite film.
The Young's contact angle (ΘY), which deconvolutes the effects of surface roughness, is the best parameter for comparing hydrophilicity (Table 1). A contact angle below 90° indicates hydrophilicity.38 The ΘY values of the graphene and graphene–CNC films were 13.9 ± 2.7° and 64.0 ± 1.7° (n = 3, P = 0.0001), respectively, showing that graphene films are more hydrophilic than graphene–CNC films. The ΘY value of ITO was found to be 96.7 ± 1.3°, consistent with a previous study that reported 92 ± 2°,39 suggesting that the ITO glass is slightly hydrophobic.
| Film material | Apparent contact angle, Θapp (°) | Roughness factor, r | Young's contact angle, ΘY (°) |
|---|---|---|---|
| Graphene | 10.0 ± 0.8 | 1.026 | 13.9 ± 2.7 |
| Graphene–CNC | 59.1 ± 2.3 | 1.169 | 64.0 ± 1.7 |
The hydrophilicity of the graphene films can be attributed to the SC surfactant, which has three hydroxyl groups on the steroid ring and one ionic head of a carboxyl group. The graphene dispersions are dialysed after exfoliation, but this process does not remove all SC.28 If this were the case, the hydrophobic graphene sheets would restack building large aggregates in the aqueous solution. The hydrophilicity of graphene–CNC arises from CNC, which has three hydroxyl groups on each glucopyranose unit.33 SC is also hygroscopic, which could account for the higher hydrophilicity of graphene films than graphene–CNC films, despite both containing a similar graphene content. The graphene–CNC films contain ca. 26% (m m−1) CNC, while the graphene films consist of ca. 27% (m m−1) SC as estimated by thermogravimetric analysis.29
First, we examined the electrochemical performance of the graphene films in two scenarios: when used as an electrode on non-conductive glass and when employed as a mediator on a flat ITO electrode. CV measurements were performed in the presence of 1.0 mM Fe(CN)63−/4− redox couple, which is a commonly used artificial electron mediator in BPVs,40,41 in 1.0 M KNO3 electrolyte solution (Fig. 2a). No discernible difference in the electrochemical performance was observed between the two configurations, indicating that the contribution of the ITO to the observed current under these conditions was minimal. This result shows that it is not necessary to use ITO as a substrate for the graphene-based films in this 2D flat structure. For subsequent experiments, we used graphene and graphene–CNC films on non-conductive glass.
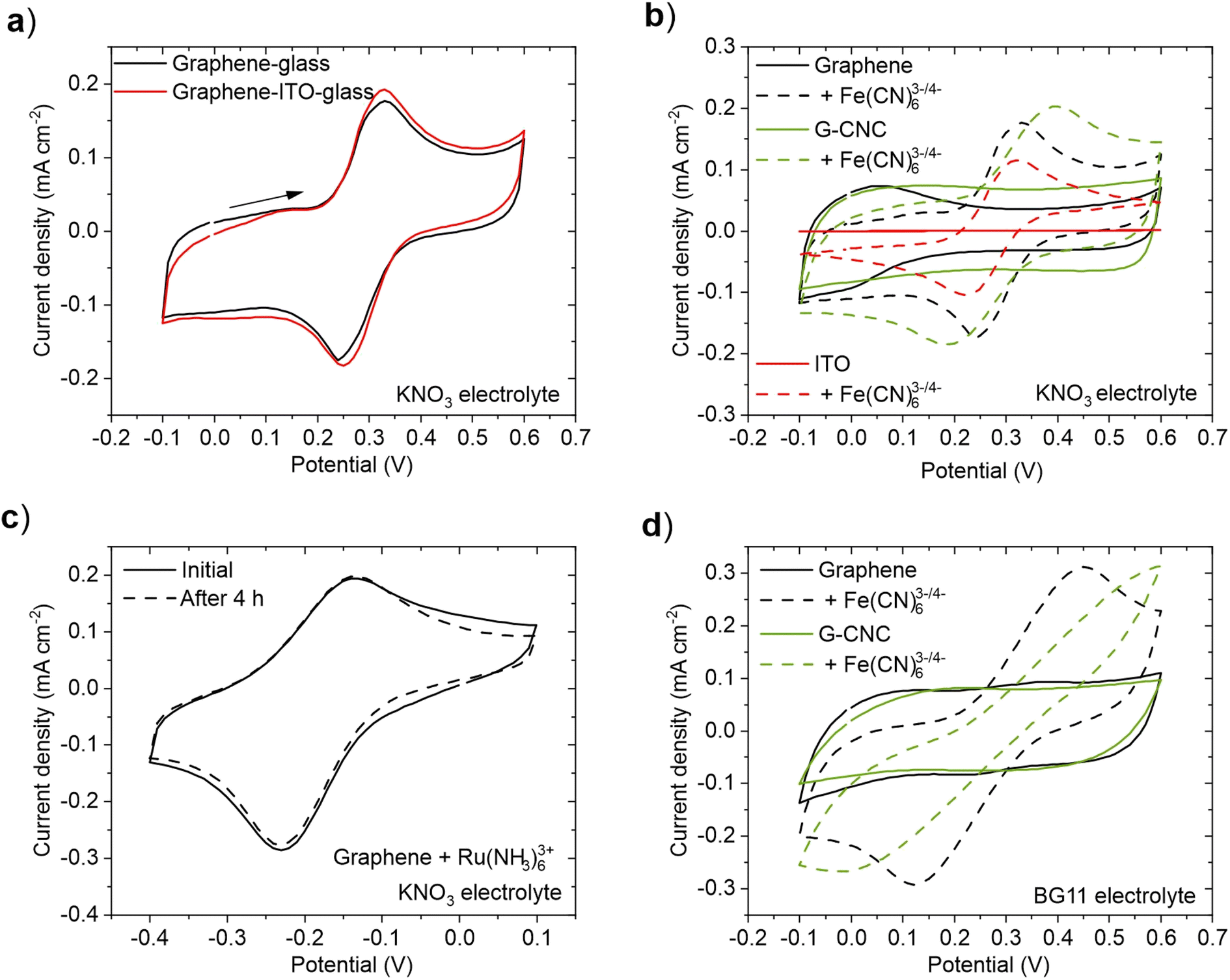 | ||
| Fig. 2 Electrochemical performance of graphene and graphene–CNC films. (a) Cyclic voltammograms of the graphene films on non-conductive glass (black line) and ITO-coated glass (red line) in 0.5 mM K3[Fe(CN)]6 and 0.5 mM K4[Fe(CN)]6 with 1.0 M KNO3 as the background electrolyte. (b) Cyclic voltammograms of graphene on non-conductive glass (black line), graphene–CNC on non-conductive glass (green line), and ITO-coated glass (red line) in 1.0 M KNO3 electrolyte. Dashed lines indicate addition of redox couple 0.5 mM K3[Fe(CN)]6 and 0.5 mM K4[Fe(CN)]6. (c) Cyclic voltammograms of a graphene electrode before and after 4 h of continuous cycling in 1.0 mM [Ru(NH3)6]Cl3 with 1.0 M KNO3 electrolyte. (d) Cyclic voltammograms of graphene (black line) and graphene–CNC (green line) films on non-conductive glass in BG11 electrolyte. Dashed lines indicate addition of redox pair 0.5 mM K3[Fe(CN)]6 and 0.5 mM K4[Fe(CN)]6. Scan rate for all was 50 mV s−1. In panels (a), (b) and (d), the last cycle of five cycles is shown; panel (c) the last cycle after 4 h of continuous cycling is shown. The scanning direction which applies for all CVs is indicated with an arrow in panel (a). The starting potential was 0.0 V. | ||
To investigate the electrochemical performance of graphene and graphene–CNC electrodes, we conducted cyclic voltammetry in 1.0 M KNO3 electrolyte (aqueous, but simpler than BG11 medium used for cyanobacteria culturing). For comparison, we also analysed ITO-coated glass electrodes. In the absence of a redox couple, both graphene-based electrodes demonstrated high capacitive currents due to the double layer charging (Fig. 2b – solid lines) whereas this was not observed with the ITO electrode. This result indicates that the graphene-based electrodes have a higher active surface area than the ITO electrode. This is in accordance with the surface roughness analysis which indicated that the graphene-based films had a rougher surface than ITO (Fig. 1d). Furthermore, the graphene–CNC electrodes display an even higher capacitive current than the graphene electrodes. This is also in line with the surface roughness analysis that graphene–CNC electrodes had a higher surface area ratio (Sdr), which was attributed to the graphene–CNC having more fine texture (Sds) than the graphene films (Fig. 1e). When designing BPVs, low capacitance electrodes are advantageous for achieving high power densities, which is related to fast charge transfer kinetics. High capacitance electrodes are suitable for extending BPVs to have energy storage capabilities as they can store a larger amount of charge,42,43 and better stability as they can reduce voltage fluctuations.44
In the presence of a negatively charged redox couple, 1.0 mM Fe(CN)63−/4− in 1.0 M KNO3, well-defined reduction and oxidation peaks were observed with all electrodes (Fig. 2b – dashed lines). The faradaic peak currents are similar for all electrodes (Table S4†), indicating similar electron transfer capacities. The peak separation, however, is smaller for graphene (0.090 V) and ITO (0.091 V) electrodes than graphene–CNC (0.21 V), indicating that the redox reaction is more reversible on the graphene than the graphene–CNC electrode (Table S4†). The fact that the graphene and ITO electrodes have similar peak separations means that the electrochemical response on the graphene electrode is not limited by the lower conductivity of graphene (ca. 8900 S m−1) compared to ITO (ca. 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–500000 S m−1, calculated with the conductive coating thickness of 150–200 nm). The electrical conductivities of graphene and graphene–CNC (ca. 1500 S m−1) are in the same range and yet the electrochemical reaction on graphene–CNC is less reversible.
000–500000 S m−1, calculated with the conductive coating thickness of 150–200 nm). The electrical conductivities of graphene and graphene–CNC (ca. 1500 S m−1) are in the same range and yet the electrochemical reaction on graphene–CNC is less reversible.
To investigate if the difference in the peak separation between graphene and graphene–CNC is caused by the possible differences in the surface charges of these electrodes, we performed CV in the presence of positively charged redox species 1.0 mM Ru(NH3)63+. Again, well-defined reduction and oxidation peaks were observed with both electrodes (Fig. S2†), and the peak separation was smaller with graphene (0.10 V) than graphene–CNC (0.25 V) (Table S4†), indicating that the surface charge was not causing the difference in peak separation. It could be speculated that even though the bulk conductivities of the graphene-based electrodes are in the same range, the graphene–CNC electrode might have a higher resistance on the outermost surface depending on the orientation and location of the electrically insulating CNC nanorods. However, when analysing the relationship between the peak currents and the square root of the scan rate (from 5 to 100 mV s−1), both electrodes show a linear dependence (Fig. S3†), indicating a diffusion controlled redox reaction with fast electron transfer kinetics. The correlation is slightly higher for the graphene electrode which is also consistent with the smaller peak separation of the graphene electrode. Nevertheless, the linear dependence implies that the electron transfer with the redox couple in solution occurs predominantly at the electrode/electrolyte interface for both electrode types. In BPV design, electrodes with efficient electron transfer capacity with high electron transfer reversibility are desirable for achieving high power densities from loaded photosynthetic microorganisms. In this sense, both electrodes show potential, although the electrochemical response on the graphene electrodes is more reversible.
To investigate the electrochemical stability of the films in aqueous electrolyte, the electrochemical performance of the films was measured continuously for 4 h of exposure to the 1.0 M KNO3 electrolyte with 1.0 mM Ru(NH3)63+. For graphene films, we observed no degradation of electroactivity as the cyclic voltammogram remained unchanged over the entire measurement (Fig. 2c). This indicates that despite the high hydrophilicity of the graphene films, as determined by contact angle measurements (Table 1), they do not disintegrate when exposed to an aqueous electrolyte solution. The electrochemical stability of the graphene–CNC electrode was previously examined under comparable conditions.29 Although both films featured electrochemical stability under 4 h of cycling, it was noticed that the graphene films peeled off from the area under the O-ring after the electrochemical measurements, whereas such issues were not observed with the graphene–CNC films (Fig. S4†). This observation aligns with the greater mechanical stability and durability of graphene–CNC electrodes, attributed to the crystalline structure of CNC. When designing BPVs, it becomes crucial to consider the long-term stability of the electrodes for achieving extended usage. Longest-performing BPVs have recently operated for over 6 months, whereas earlier benchmarks only reached tens of days.45–47 The inclusion of CNC into the graphene films stand out as a suitable approach to increase their durability.
Finally, we investigated the electrochemical performance of the electrodes using the growth medium of the cyanobacterium – BG11 medium (pH 8.2) – as the electrolyte.48 BG11 medium is typically used as the electrolyte in BPV experiments with cyanobacteria.2 However, certain components of BG11 have been identified as potential sources of interferences in background current during electrochemical experiments.45 In the presence of 1.0 mM Fe(CN)63−/4−, both films showed a clear redox response (Fig. 2d), indicating that both films have electron transfer capacity in BG11. However, the peak separation is higher with both electrodes in BG11 than in 1.0 M KNO3 (Table S4†). This distinction can be attributed to the lower conductivity of BG11 (ρ = 3.3 mS cm−1) than 1.0 M KNO3 (ρ = 83.4 mS cm−1), which impacts the electron transfer process and results in a more pronounced iR-drop. However, the high salt concentration of 1.0 M KNO3 or BG11 medium with additional salts45 would not be suitable for freshwater cyanobacteria species including Synechocystis, as it would decrease the cell viability and photosynthetic performance (source of electrons in exoelectrogenesis).49 Therefore, we used BG11 as the electrolyte solution in subsequent photoelectrochemical experiments.
2.2 Photoelectrochemical performance of cyanobacterial cells on graphene and graphene–CNC films
 | ||
| Fig. 3 Photoelectrochemical performance of Synechocystis cyanobacterial cells on graphene and graphene–CNC electrodes. Representative photocurrent profiles of cyanobacteria on (a) graphene and (b) graphene–CNC (G–CNC, thickness 1590 ± 460 nm) electrodes. (c) Photocurrent profile with the current density in the dark subtracted showing calculated charges (shaded areas under the trace). ‘Photocharge’ is the difference between the charges in the light (yellow) and dark (dark grey) periods, each 5 min. All photocharges in this study are calculated this way. (d) Photocharge density from cyanobacteria on graphene–CNC electrodes before and after treatment with 1 mM DCMU, and a cell-free control experiment. Cell loading was 60 nmolChl for panels (a), (b) and 45 nmolChl for panel (c). Biofilms were formed for 17 h. Electrolyte was BG11 (pH 8.2). Applied potential was 0.1 V vs. Ag/AgCl. Light/dark cycles were 5 min/5 min with light intensity 100 μmolphotons m−2 s−1 and wavelength 660 nm. Data in panel (d) presented as mean ± standard error of the mean (n = 3), statistical significance by t-test P ≤ 0.01 denoted by **, P ≤ 0.05 denoted by *. | ||
Stepped chronoamperometry under light/dark cycles was performed using the graphene electrode, yielding positive/anodic photocharges at applied potentials equal to and more positive than +0.1 V vs. Ag/AgCl (3 M KCl) reference electrode (equivalent to +0.3 V vs. Standard Hydrogen Electrode (SHE)) (Fig. S5†). At higher potentials, the photocharge was not significantly higher (Fig. S5†). Similarly, an applied potential of +0.1 vs. Ag/AgCl the maximum photocurrent was reached for Synechocystis cells in a previous study using ITO electrodes.5 Therefore, subsequent chronoamperometry experiments (without addition of an artificial electron mediator) were conducted at an applied potential of +0.1 vs. Ag/AgCl.
To investigate whether the electrode material itself contributed to the light-induced current output, we performed cell-free control experiments on both electrode types. In the absence of cyanobacterial cells, both electrode types yielded low photocharges of 0.53 μC cm−2 (n = 2) and 0.45 ± 0.17 μC cm−2 (n = 3) for graphene and graphene–CNC electrodes, respectively. When the graphene–CNC electrodes were interfaced with cyanobacterial cells, they yielded a 7-fold higher photocharge density of 3.30 ± 0.47 μC cm−2 (n = 3, P = 0.0046) (Fig. 3d). Therefore, the photocharge output was predominantly from the cyanobacteria.
To attribute the current increase under illumination to the photosynthesis performed by cyanobacteria, we tested the effect of adding the herbicide 3-(3,4-dichlorophenyl)-1,1-dimethylurea (DCMU). DCMU inhibits photosynthesis by binding to the QB pocket of photosystem II (PSII), thus blocking the flow of electrons from water photo-oxidation into the photosynthetic electron transport chain.51 We found that the introduction of DCMU reduces the photocharge by 75% (n = 3, P = 0.038) on the graphene–CNC electrode as the photocharge density was 0.82 ± 0.30 μC cm−2 after DCMU treatment (Fig. 3d). This decrease is consistent with previous studies,40,41,52 confirming that water-splitting photosynthesis is indeed the primary source of the photocharge. It is important to note that the remaining cell-based photocharge may originate from respiratory and other metabolic processes.40
000–500000 S m−1) than the graphene (8900 ± 1300 S m−1) and graphene–CNC (1500 ± 400 S m−1) electrodes. We speculate that the higher photocharges from the graphene and graphene–CNC electrodes is due to their more favourable surface properties for interfacing with cyanobacterial cells than ITO: more hydrophilicity for biofilm wetting and nanoscale roughness facilitating cell adhesion (Fig. 1d and Table 1).
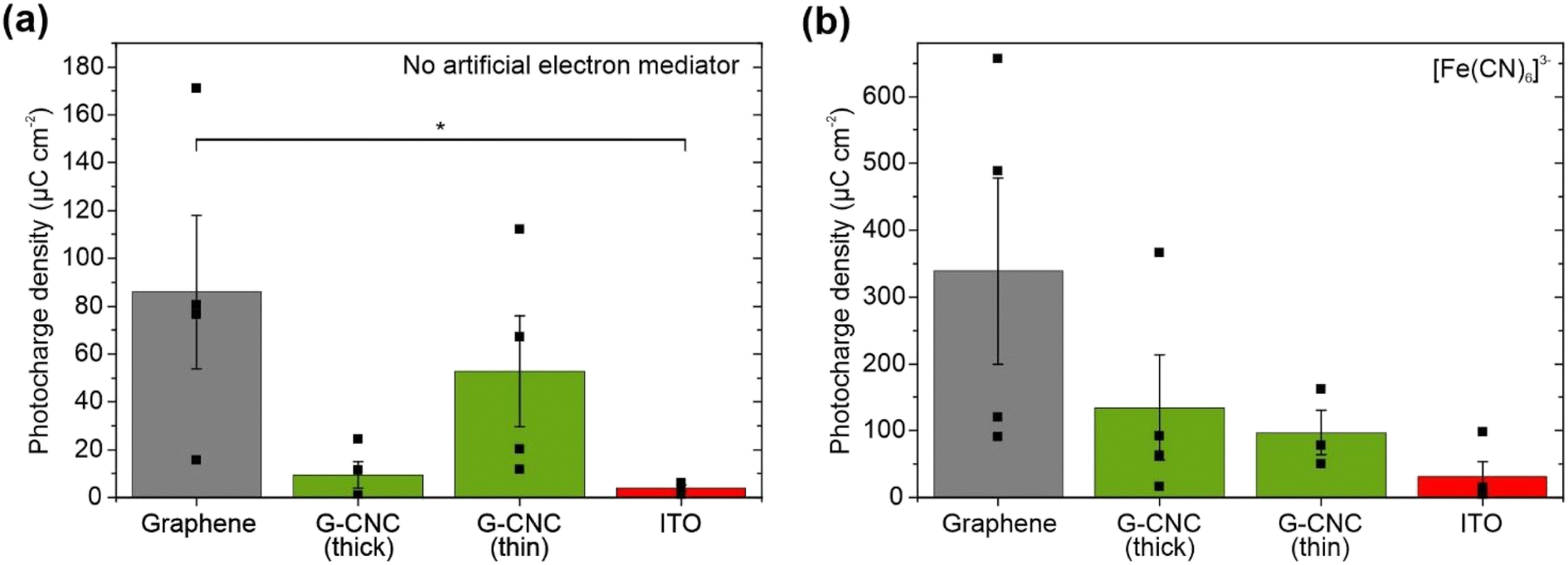 | ||
| Fig. 4 Photocharge densities from cyanobacteria on different electrode materials. Photocharge densities in graphene, thick and thin graphene–CNC (G–CNC) electrodes, and ITO-coated glass (a) in the absence of an artificial electron mediator at 0.1 V vs. Ag/AgCl, and (b) in the presence of 1.0 mM potassium ferricyanide ([Fe(CN)6]3−) at 0.5 V vs. Ag/AgCl. Cell loading was 60 nmolChl and biofilms were formed for 17 h. Electrolyte was BG11 (pH 8.2). Light/dark cycles were 5 min/5 min with light intensity 100 μmolphotons m−2 s−1 and wavelength 660 nm. Photocharge was calculated as area under the current–time trace in light minus the area in dark, each for 5 min. Data presented as mean ± standard error of the mean (n = 4, thin graphene electrodes with ferricyanide n = 3), statistical significance by t-test P ≤ 0.05 denoted by *. | ||
Most graphene electrodes (three of n = 4) harvested at least 4-fold higher photocharge from the cyanobacteria than the thick graphene–CNC electrodes (n = 4) and some thin graphene–CNC electrodes (two of n = 4). This was consistent with the more efficient electron transfer of the graphene electrodes than the graphene–CNC electrodes, as indicated by the CV experiments where the graphene electrodes had a smaller peak separation (Fig. 2 and Table S4†). However, the difference in the average photoelectrochemical performance of graphene and graphene–CNC electrodes with cyanobacterial cells was not statistically significant (graphene versus thick graphene–CNC: P = 0.057, graphene versus thin graphene–CNC: P = 0.43) due to the large variability between replicates.
In the CV experiments, it was noticed that some thicker graphene–CNC films showed a higher capacitive current than the thinner ones (Fig. S6†). It was speculated that this could translate as the thick films having a higher active surface area than the thinner ones which would provide more contact area for the adsorption of cyanobacterial cells. In the cell experiments, some thin graphene–CNC films (two of n = 4) harvested at least 4-fold higher photocharge from the cyanobacterial cells than the thick graphene–CNC electrodes (n = 4). Again, the difference in average photoelectrochemical performance of thick and thin graphene–CNC electrodes with cyanobacterial cells was not statistically significant (P = 0.120) also due to large variability between replicates. Therefore, the higher capacitive current of the thicker graphene–CNC films (Fig. S6†) did not lead to an increase in the photocharge harvested from the cyanobacterial cells. This is consistent with the cyanobacteria cells exporting an endogenous electron mediator that is oxidised at the surface of the electrode.
The reason for the large variability between the replicates, that makes comparisons between electrodes challenging, is unknown. We do not suspect differences in biological traits between cyanobacterial cultures, as our previous study showed clustered photosynthetic performance of these cells when interfaced with these electrodes.29 Similarly, we do not suspect differences in the fabrication of distinct films because, in the CV experiments (Table S4†), the faradaic responses were consistent across all film variations for both types, indicating a consistent electron transfer capacity, a crucial trait in electrode materials. Therefore, we hypothesise that the variability might stem from uncontrolled differences in environmental conditions, such as humidity, during cells incubating on electrodes (Fig. S7†). Indeed, the most significant differences were observed when we explored different cell loadings on our flat electrodes (see Section 2.2.3).
To evaluate the maximum photocharge harvesting capabilities of the electrodes, we added the artificial electron mediator potassium ferricyanide, which efficiently harvests electrons from the periplasmic space in the outermost compartment of the cyanobacterial cell.40 Chronoamperometry was performed at an applied potential of +0.5 V vs. Ag/AgCl (+0.7 V vs. SHE equivalent) to maximise mediation by ferricyanide (Em = 0.420 V vs. SHE).54 With graphene electrodes and ferricyanide, we recorded the highest photocharge in this study of 339 ± 139 μC cm−2 (Fig. 4b), which is a 4-fold boost of the non-mediated output (Fig. 4a). This corresponds to a photocurrent of 2.17 ± 0.74 μA cm−2 (Table S1†), with one electrode yielding the highest recorded photocurrent in this study of 3.53 μA cm−2. Graphene–CNC electrodes and ferricyanide harvested intermediate photocharges of 134 ± 79 μC cm−2 and 97.2 ± 33.5 μC cm−2 for thick and thin electrodes, respectively (Fig. 4b), which are ca. 14- and 2-fold boosts of the non-mediated output (Fig. 4a). These values correspond to photocurrent outputs of 1.11 ± 0.60 and 0.60 ± 0.12 μA cm−2 for thick and thin graphene–CNC electrodes, respectively (Table S1†). With ferricyanide, the ITO electrodes harvested the smallest photocharge of 31.4 ± 22.2 μC cm−2.
Comparing the performance of electrodes from different BPV studies is challenging due to different experimental parameters, including electrochemical set-ups and biocatalysts.2,53 Nevertheless, we compared the photo-outputs generated by our graphene electrodes loaded with Synechocystis cells with other studies involving graphene-based electrodes (such as graphene oxide electrodes or graphene oxide coated over ITO) combined with a variety of photosynthetic materials (Tables S1 and S2†). In the other studies that also used analytical three-electrode set-ups for electrode characterisation (Table S1†), isolated PSII15,16 and isolated photosynthetic thylakoid membranes18,55,56 were employed. These photosynthetic components are capable of direct electron transfer to electrodes, resulting in higher photocurrents than whole cells that perform indirect electron transfer.5,50,57 Nevertheless, the photocurrents we report are in the same order of magnitude as isolated PSII without an artificial mediator on flat RGO electrodes.15 Furthermore, the photocurrents we report are only an order of magnitude lower than isolated PSII without an artificial mediator on 3D inverse-opal structured GO electrodes.16 Graphene-based electrode studies that used whole photosynthetic microorganisms (green alga Chlorella vulgaris17,19,20 or a different cyanobacterium Synechococcus elongatus21) used two-electrode BPV set-ups (Table S2†).
The benchmark electrodes that yield the highest photocurrent have been fabricated from ITO and with a 3D, 4th generation micropillar architecture (up to 245 μA cm−2 for exogenously mediated electron transfer with DCBQ, and up to 1.93 μA cm−2 naturally from Synechocystis with no exogenous mediators added),4 and prior to that a 3D, 3rd generation inverse-opal architecture (14.7 μA m−2 for exogenously mediated electron transfer with DCBQ5 and ca. 0.95 μA m−2 naturally from Synechocystis with no exogenous mediators added).6 Our highest photocurrent from 2D flat architecture graphene electrodes with ferricyanide of 3.5 μA cm−2, are of the same order of magnitude as achieved by the inverse-opal-ITO electrodes without a mediator.
To investigate the effect of biofilm formation duration, we loaded cyanobacterial cells (60 nmolChl) onto the electrodes and incubated them for either 2 h or 17 h before adding electrolyte and performing photoelectrochemical experiments. Longer biofilm formation times led to higher photocharges with both electrode types (Fig. 5a). The graphene electrodes harvested ca. 75-fold higher photocharge when the biofilm formation was extended from 2 h (1.17 ± 0.66 μC cm−2) to 17 h (86.0 ± 32.0 μC cm−2) (n = 4, P = 0.038). The graphene–CNC electrodes harvested higher photocharge from 17 h biofilm formation (9.53 ± 5.60 μC cm−2) compared to 2 h biofilm formation (0.519 ± 0.171 μC cm−2), although the difference was not significant (n = 4, P = 0.16).
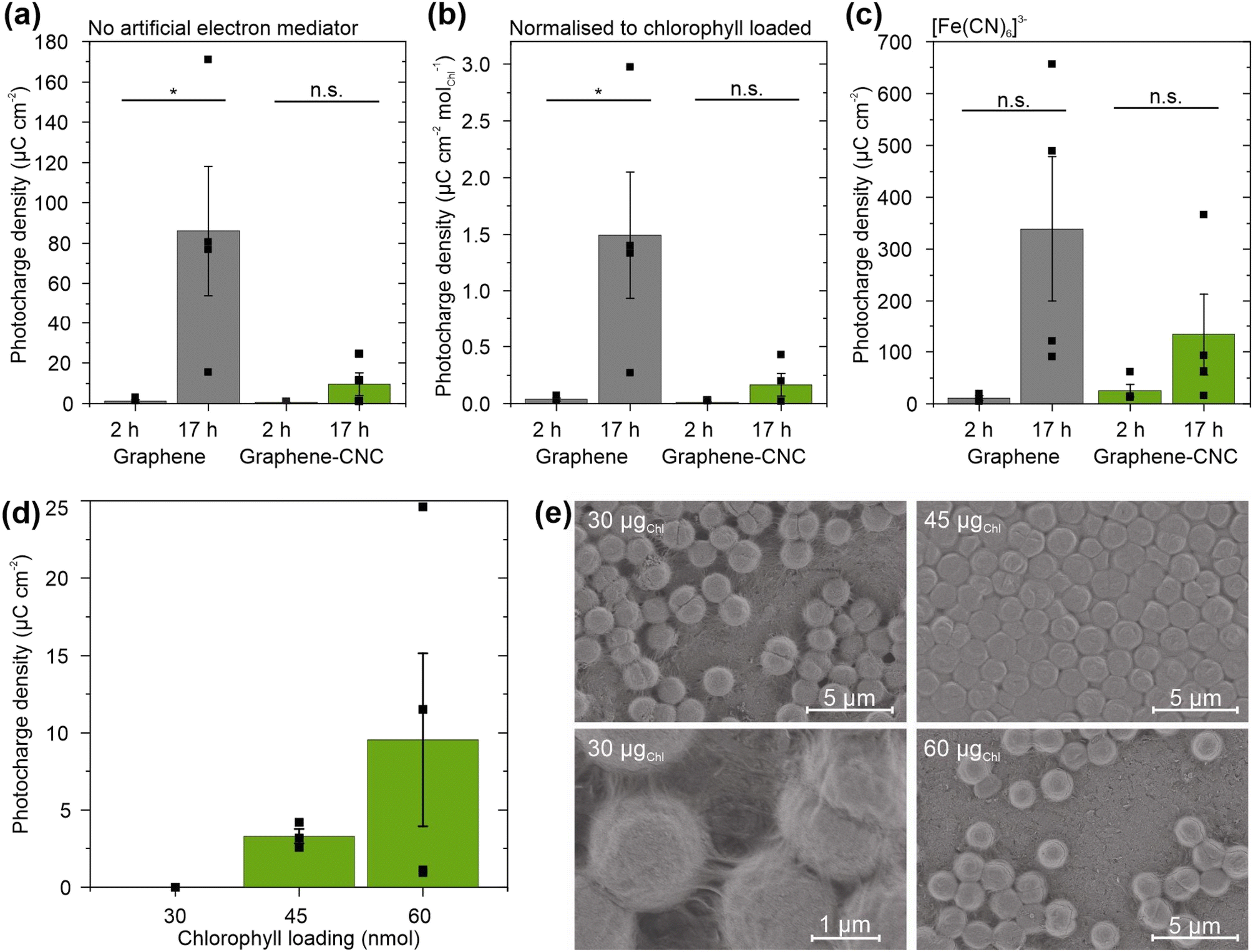 | ||
| Fig. 5 Photocharges harvested by graphene (grey) and thick graphene–CNC (green) electrodes from Synechocystis cyanobacteria variably loaded. (a) Photocharge densities from biofilms formed for 2 or 17 h before addition of electrolyte. No addition of an artificial electron mediator and applied potential 0.1 V vs. Ag/AgCl. (b) Photocharge densities in panel (a) normalised to Chl amount in cells that remained tightly adhered onto the electrode after photoelectrochemical experiments. (c) Photocharge densities with addition of 1.0 mM potassium ferricyanide ([Fe(CN)6]3−) at 0.5 V vs. Ag/AgCl. (d) Photocharge densities from different numbers of cyanobacterial cells (measured by total Chl content) loaded on thick graphene–CNC electrodes. For panels (a)–(c) cell loading was 60 nmolChl; for panel (d) biofilms were formed for 17 h. Electrolyte was BG11 (pH 8.2). Light/dark cycles were 5 min/5 min with light intensity 100 μmolphotons m−2 s−1 and wavelength 660 nm. Photocharge was calculated as area under the current–time trace in light minus the area in dark, each for 5 min. Data presented as mean ± standard error of the mean (panels (a)–(c) n = 4, except n = 3 for graphene 2 h; panel (d) n = 1 for 30 nmolChl, n = 3 for 45 nmolChl, n = 4 for 60nmolChl), statistical significance by t-test P ≤ 0.05 denoted by *, P > 0.05 denoted by n.s. (not significant). (e) Scanning electron micrograms of biofilms from different numbers of cyanobacterial cells (measured by total Chl) loaded on graphene electrodes. | ||
To understand the performance difference between biofilms formed for different durations, we considered the quantity of cyanobacterial cells interfacing tightly with the electrode. We estimated this by measuring the non-adherent cells floating in the electrolyte using UV-vis spectroscopy of chlorophyll a (Chl) photosynthetic pigment. Despite loading the same number of cells on the electrodes (60 nmolChl), ca. 10-fold fewer cells detached from the electrodes when the biofilm was allowed to form for 17 h compared to 2 h for both electrode types (Fig. S8†). For the graphene electrodes, the detached Chl amounts were 2.48 ± 1.34 nmolChl (n = 3) and 19.1 ± 1.0 nmol (n = 4, P = 0.0002) after 17 h and 2 h loadings, respectively. For the graphene–CNC electrodes, 2.63 ± 0.55 nmol (n = 6) and 20.0 ± 0.55 nmol (n = 2, P = 0.0001) detached after 17 and 2 h loadings, respectively. The detached Chl amounts were independent of the electrode type (P = 0.58 and 0.90 for the 2 h and 17 h films, respectively).
Nevertheless, when we normalised the photocharges to the amount of Chl adherent on the electrodes, the 17 h biofilms still yielded significantly higher photocharges than the 2 h biofilms on the graphene electrodes (2 h: 0.0366 ± 0.0136 μC cm−2 nmolChl−1, 17 h: 1.49 ± 0.56 μC cm−2 nmolChl−1, n = 4, P = 0.040) (Fig. 5b). Therefore, the number of cyanobacterial cells interfacing tightly with the electrode was not the only reason that the 17 h biofilms perform better than the 2 h biofilms. For the graphene–CNC electrode, the Chl-normalised photocharge densities were still similar for 2 h biofilms 0.0130 ± 0.00429 μC cm−2 nmolChl−1 and 17 h biofilms 0.167 ± 0.0986 μC cm−2 nmolChl−1 (P = 0.16, n = 4).
To investigate the feasibility of harvesting electrons from cyanobacterial cells in the electrolyte, we added the artificial electron mediator potassium ferricyanide.40 Ferricyanide boosted the photocharges of all samples (Fig. 5c). With 2 h biofilm loading, ferricyanide increased the photocharge ca. 10-fold for graphene electrodes to 10.3 ± 4.8 μC cm−2 and 50-fold for graphene–CNC electrodes to 25.2 ± 12.2 μC cm−2. Nevertheless, the average photocharges for 17 h loaded biofilms were still ca. 30-fold and 5-fold higher than 2 h loaded biofilms on graphene and graphene–CNC electrodes, respectively. The average photocharges with ferricyanide after 2 and 17 h loading times were 10.3 ± 4.8 μC cm−2 and 339 ± 139 μC cm−2 for the graphene electrode and 25.2 ± 12.2 μC cm−2 and 134 ± 78.8 μC cm−2 for the graphene–CNC electrode.
To investigate the impact of cyanobacterial cell quantity on electrode performance, we loaded different amounts of Chl (a measure of cell quantity) onto the electrodes and incubated them for 17 h before adding electrolyte and performing photoelectrochemical experiments (Fig. 5d). Loading up to 30 nmolChl yielded negligible photocharge, whereas loading 45 and 60 nmolChl yielded photocharges of 3.30 ± 0.47 (n = 3) and 9.53 ± 5.60 μC cm−2 on thick graphene–CNC, respectively. These photocharges were not significantly different (n = 4, P = 0.39), indicating that the cell loading on the graphene-based electrodes was already optimized for harvesting photocharge.
To examine the arrangement of differently loaded cells on the graphene films, the cell-loaded electrodes were visualised by scanning electrochemical microscopy (SEM). The 45 nmolChl resulted in an even biofilm across the surface of the graphene film, with cells tightly packed together, whereas the 30 and 60 nmolChl loadings led to sparser coverage of the graphene films with cells (Fig. 5e). The 30 nmolChl loading also showed the extracellular matrix of exopolysaccharides and some type IV pili (Fig. 5e), that do not contribute to exoelectrogenesis.50 These images suggest that in the case of 60 nmolChl loading, the cells adhered more strongly to each other than to the electrode. As a result, the biofilm lifted away from the electrode during the sample preparation for SEM (or electrochemistry). Most of the photoelectrochemistry experiments in this study (Section 2.2.2) were performed with the 60 nmolChl loading, which, along with noted variability in environmental conditions, may have contributed to the increased variability between replicates.
The results obtained indicate that strong biofilm formation plays a crucial role in establishing efficient electronic connectivity between cyanobacterial cells and the graphene-based electrodes, surpassing the influence of any other parameter tested in this study. Previous advancements in electrode architecture have yielded significant improvements in photo-outputs from photosynthetic microorganisms.2,4 Therefore, we propose that the bottleneck of converting more cell loading on our electrodes into harvesed photocharges lies in the 2D architecture of the few-layer graphene film electrodes, which are flat with only nano-roughness. In future investigations, our focus will be on improving the architecture of our graphene-based electrodes to increase the electronic connectivity between the cyanobacteria and the electrode.
One approach to augment the electroactive surface area of graphene electrodes could be to spray coat the graphene dispersions onto rough substrates, such as electrochemically roughened metal/etching glass. The development of 3D graphene electrodes is feasible with our graphene material, as demonstrated by the creation of inverse opal (IO) graphene oxide electrodes.16 However, it is crucial to address the challenge of light management, which emerges as the next limiting factor following the achievement of a 3D architecture.4 Light management presents a hurdle for carbon-based electrodes due to their opacity, potentially resulting in limited light penetration to the photosynthetic biofilm. However, single-layer graphene films have been considered to hold a great promise as a suitable material for various photonic devices requiring transparency,8,58,59 for example a monosheet of graphene has 97.7% transmission.60 Furthermore, liquid-phase exfoliated few-layer graphene films can maintain >85% transmission while also featuring a relatively high electrical conductivity after annealing the films.11,59,61 Composites of graphene-based materials and CNC can also be made transparent as nanocellulose itself is transparent.34 This indicates the feasibility of graphene and graphene–CNC films as transparent, conductive, 3D electrodes for BPVs.
3 Conclusion
In this work, we showcased the potential of few-layer graphene and graphene–CNC films, fabricated by shear exfoliation of natural flake graphite, as efficient electrodes for harvesting current from cyanobacteria. The fabrication process employed was not only environmentally friendly, but also fast and simple, avoiding the use of toxic or hazardous chemicals. Surface characterization of the films revealed favourable qualities such as hydrophilicity and surface roughness. Notably, the graphene electrodes exhibited higher conductivities, and the graphene–CNC composite films exhibited higher nano-roughness and mechanical durability. Electrochemical assessments showed substantial electron transfer capabilities for both film types. Biophotoelectrochemical investigations validated the capacity of both electrode types to harvest current from cyanobacterial cells under illumination. The graphene electrodes yielded the highest photocharges, especially when biofilms were formed over a long period of time and with an artificial electron mediator. The shear exfoliated few-layer graphene and graphene–CNC films hold immense potential as electrode materials in BPVs, offering sustainability and the potential for architectural advancements.4 Materials and methods
4.1 Materials
Natural flake graphite (d < 125 μm) from Haapamäki, Finland was used in this work. The graphite ore was enriched in an in-house process to a concentrate containing 99.3 ± 0.5% (m m−1) carbon.28 The CNC suspension used for the graphene–CNC composite fabrication was prepared from cellulose microcrystals (CMC) by sulfuric acid (64% (v/v)) hydrolysis as previously reported.29 Sodium cholate (SC) powder used for the preparation of the surfactant solutions was received from Acros Organics and it was dried overnight in oven at 100 °C prior to use. Hexaammineruthenium(III) chloride ([Ru(NH3)6]Cl3), 3-(3,4-dichlorophenyl)-1,1-dimethylurea (DCMU) and 2,6-dichloro-1,4-benzoquinone (DCBQ) were used as purchased from Sigma-Aldrich, likewise potassium hexacyanoferrate(II) (K4[Fe(CN)6]), potassium hexacyanoferrate(III) (K3[Fe(CN)6]), and potassium nitrate from Fluka. Blue-green algae number 11 (BG11)48 cell growth medium buffered at pH 8.2 was used for cell culture, cell resuspension and as electrolyte in all biophotoelectrochemical experiments. Deionised MilliQ water (resistivity 18 MΩ cm) was used throughout this work.4.2 Preparation of graphene dispersions
Few-layer graphene and graphene–CNC dispersions were prepared by high-shear exfoliation using a POLYTRON® immersion disperser (Kinematica AG, Switzerland) with a rotor-stator combination. A PT 10-35 GT drive unit was coupled with a PT-DA 20 dispersing aggregate with a rotor diameter of 15.0 mm and a rotor-stator gap of 0.30 mm. In a typical experiment, 30.0 ml of either 5 mM sodium cholate solution or 2.0 mg ml−1 CNC suspension were first combined with 3.00 g of graphite. The starting graphite-to-CNC concentration ratio was 50:1. For both dispersion types, the initial graphite concentration (C(i)) was 100 mg ml−1 (±0.05 mg ml−1), mixing time (t) 3 h and rotor speed (N) 22000 rpm. A temperature regulation system with a Heto DT1 water bath circulator and Hetofrig (Heto, Birkerod, Denmark) water bath cooler was applied to maintain a constant temperature of 10.0 °C (±0.4 °C) and 18.0 °C (±0.4 °C) for the SC- and CNC-stabilised dispersions, respectively. After exfoliation, the resultant dispersions were left standing overnight and on the next day, the dispersions were centrifuged for 1 h to separate and remove any unexfoliated graphite. A Hermle Z 36HK tabletop centrifuge with a rotor no. 221.22 and speed of 1700 rpm corresponding to a relative centrifugal force (RCF) of 270g was used for this purpose.
UV-vis spectra were recorded with a Shimadzu UV-2501PC spectrophotometer to determine the concentration of the resultant dispersions (absorbance spectra in Fig. S10†). The concentration (C) was calculated according to Beer–Lambert's law, A (absorbance) = εbC (b = path length) using the absorbance value at 660 nm. The value of the extinction coefficient, ε, was previously determined to be 6600 ml mg−1 m−1.28
To decrease the amount of sodium cholate in the dispersions, the SC-stabilised dispersions were dialyzed for 2.5 h under magnetic stirring using Spectra/Por® 3 dialysis membranes with a molecular weight cut-off of 3500 Da. The volume ratio of the sample and the dialysate (water) was 1:250. To estimate the graphene and SC ratio in the samples, thermogravimetric analysis (TGA) was performed with SDT Q600 apparatus from TA Instruments (New Castle, DE, USA). The analysis was conducted using nitrogen gas with a flow rate of 60 ml min−1 in an oxygen-free environment up to 900 °C with a heating rate of 10 K min−1. The dispersions were oven-dried at 60 °C overnight prior to the analysis. A pure SC sample was also analysed as a reference.
4.3 Film preparation
A standard airbrush pen was used to manually spray-coat the dispersions on glass substrates. Spray coating was performed on a heating plate which maintained a temperature of 250 °C and 100 °C for graphene and graphene–CNC films, respectively to speed up the evaporation of water from the dispersions. For comparison, some films were also prepared on ITO glass (Part No. CB-40IN-0107, Delta Technologies, Ltd, Stillwater, MN, USA) with RS = 4–10 Ω and σ = 400000–500000 S m−1 for the CV experiments. The prepared films were ca. 1–2 μm thick as measured with atomic force microscopy (AFM). The film thickness was calculated based on three 100 μm × 100 μm AFM images (256 × 256 pixels). ITO glass was cleaned by sonicating for 10 min in chloroform and for 10 min in acetone prior to measurements.
4.4 Film characterisation
4.5 Cell culture and growth conditions
We used the wild-type strain Synechocystis sp. PCC 6803 (Synechocystis) from the Howe Lab.66 Cultures were always grown in 30 ml batches of BG11 medium, pH 8.2 (ref. 48) at 30 °C under continuous white light of 50 μmolphotons m−2 s−1 with shaking 120 rpm. At the logarithmic growth phase, cells were harvested and resuspended in fresh BG11 at OD750 of 0.1 for sub-culture. Culture chlorophyll a concentration (nmolChl ml−1) was calculated from absorbances at 680 and 750 nm: (A680–A750) 10.814.67 All measurements were taken using a UV-1800 Spectrophotometer (Shimadzu). Cultures of early stationary phase cells at OD750 of ca. 1 were concentrated by centrifugation at 6000g for 20 min, the supernatant removed, and the pellet resuspended in fresh BG11 medium (pH 8.2) to a concentration of 600 nmol Chl a ml−1.4.6 Biophotoelectrochemical measurements
The photoelectrochemical measurements were performed in a three-electrode electrochemical cell described above (Section 4.4.2) using the graphene and graphene–CNC films as electrodes coated with the biofilm of Synechocystis. In a typical experiment, 100 μl of 600 nmolChl a ml−1 was drop-cast onto the spray-coated films and left on the lab bench at room temperature and ca. 40–50% R.H. for either 2 or 17 h to allow cell adhesion. If the humidity was outside the range of 40–50% R.H., a desiccator was used to control the humidity under biofilm incubation. The cells were kept in the dark to minimise the effects of any differences in light-treatment between replicates. Unless otherwise stated, chronoamperometry experiments without a mediator were performed at an applied potential of 0.1 V vs. Ag/AgCl (3 M KCl). Chronoamperometry experiments with 1.0 mM potassium ferricyanide artificial electron mediator were performed at an applied potential of 0.5 V vs. Ag/AgCl, respectively. All experiments were performed in 15 ml BG11 (pH 8.2)48 electrolyte solution. During chronoamperometry, the cells were illuminated with a Modulight 2 programmable light source (Ivium Technologies, The Netherlands) using a wavelength of 660 nm and light intensity 100 μmolphotons m−2 s−1. The cyanobacterial cells on the electrode were illuminated from above. Photocurrent profiles were measured under six 5 min light/5 min dark cycles and the final three cycles were used for analysis. To quantify the light-induced output, we measured the photocurrent (current in light minus the current in dark) and the photocharge (area under the current–time trace in light minus the area in dark, each for a 5 min period) (Fig. 3c). The calculated outputs were normalised to the nominal exposure area of the electrode (1.0 cm2) that was covered with cells and exposed to electrolyte and light to obtain densities.4.7 Cell quantification
The Chl a content of the bio-loaded electrode was determined by analysing the Chl a concentration in the electrolyte solution. After chronoamperometry, the electrolyte solution was collected from the electrochemical cell and stored frozen in the dark. The sample was analysed after maximum 3 days from collection by diluting it in 90% (v/v) methanol followed by 1 h sonication and 3 min centrifugation at 12000g. After centrifugation, the supernatant was analysed by UV-vis spectroscopy to determine the Chl a concentration. The extinction coefficient of Chl a at 665.0 nm in 90% (v/v) methanol containing also DCBQ, ferricyanide and BG11 was determined to be 222.7 [molChl a]−1 dm3 cm−1 (Fig. S9†).
4.8 Scanning electron microscopy
Cells were loaded onto graphene electrodes and left for a set amount of time under darkness as per the protocol for photoelectrochemistry. The electrochemical chamber was filled with BG11 electrolyte, which was then gently decanted to rinse the loaded electrodes of non-adherent cells. The loaded electrodes were dried in room conditions, completely dried in a plasma cleaner and stored in a desiccator until imaging. The loaded electrodes were mounted on aluminium stubs with copper tape and coated with 10 nm platinum using a Quorum Q150V ES+ sputter coater. The sample was stored in a desiccator until imaged using a Apreo Scanning Electron Microscope (Thermo Fisher) with a 2 kV beam acceleration.4.9 Statistical analysis
Results are presented as the mean of at least three replicates, and errors are the standard error of the mean. Where experiments involve living cells, replicates are biological not just technical replicates. Different treatments were compared by Student's t-test and P values are analysed as not statistically significant (n.s.) > 0.05, or statistically significant: * < 0.05, ** < 0.01, *** < 0.001.Author contributions
S. L. and L. T. W. contributed equally to the work. S. L.: conceptualization, methodology, investigation, formal analysis, visualization, writing – original draft, writing – review & editing, funding acquisition. L. T. W.: conceptualization, methodology, investigation, formal analysis, visualization, writing – original draft, writing – review & editing, supervision, funding acquisition. J. P.: methodology, resources, writing – review & editing, supervision. J. B.: methodology, resources, writing – review & editing, supervision. R.-M. L.: conceptualization, methodology, resources, writing – review & editing, supervision. Y. A.: conceptualization, resources, writing – review & editing, supervision, funding acquisition.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This project was supported by the Novo Nordisk Foundation (PhotoCat, project no. NNF20OC0064371 to Y. A.), by the Academy of Finland (AlgaLEAF, project no. 322754 to Y. A.), by the NordForsk Nordic Center of Excellence ‘NordAqua’ (no. 82845 to Y. A.). We acknowledge funding from the Waldemar von Frenckell's foundation and Magnus Ehrnrooth Foundation (S. L.), Maj and Tor Nessling Foundation (no. 202300115 to L. T. W.). Biophysical experiments were performed within the PHOTOSYN infrastructure at the University of Turku. The ‘SmartBio’Biocity Turku Research program is acknowledged for stimulating multidisciplinary collaboration. We would like to acknowledge Ermei Mäkilä for scanning electron microscopy sample preparation and training. MSc Qingbo Wang and Dr Xiaoju Wang from The Laboratory of Natural Materials Technology at Åbo Akademi University are acknowledged for providing the CNC suspensions for this work. Authors would like to thank Dr Dmitry Shevala (ShevelaDesign AB, Sweden) for the preparation of the graphical abstract.References
- C. J. Howe and P. Bombelli, PLoS Biol., 2023, 21, e3001970 CrossRef CAS PubMed.
- L. T. Wey, P. Bombelli, X. Chen, J. M. Lawrence, C. M. Rabideau, S. J. L. Rowden, J. Z. Zhang and C. J. Howe, ChemElectroChem, 2019, 6, 5375–5386 CrossRef CAS PubMed.
- Y. Shlosberg, G. Schuster and N. Adir, Bioelectricity, 2023, 5, 132–138 CrossRef.
- X. Chen, J. M. Lawrence, L. T. Wey, L. Schertel, Q. Jing, S. Vignolini, C. J. Howe, S. Kar-Narayan and J. Z. Zhang, Nat. Mater., 2022, 21, 811–818 CrossRef CAS PubMed.
- J. Z. Zhang, P. Bombelli, K. P. Sokol, A. Fantuzzi, A. W. Rutherford, C. J. Howe and E. Reisner, J. Am. Chem. Soc., 2018, 140, 6–9 CrossRef CAS PubMed.
- T. Wenzel, D. Härtter, P. Bombelli, C. J. Howe and U. Steiner, Nat. Commun., 2018, 9, 1299 CrossRef PubMed.
- F.-L. Ng, S.-M. Phang, V. Periasamy, K. Yunus and A. C. Fisher, PLoS One, 2014, 9, e97643 CrossRef PubMed.
- U. Shankar, D. Oberoi and A. Bandyopadhyay, Polym. Adv. Technol., 2022, 33, 3078–3111 CrossRef CAS.
- M. Anam, H. I. Gomes, G. Rivers, R. L. Gomes and R. Wildman, Sustainable Energy Fuels, 2021, 5, 4209–4232 RSC.
- R. Arvidsson and B. A. Sandén, J. Cleaner Prod., 2017, 156, 253–261 CrossRef CAS.
- S. Majee, M. Song, S.-L. Zhang and Z.-B. Zhang, Carbon, 2016, 102, 51–57 CrossRef CAS.
- K. H. Wedepohl, Geochim. Cosmochim. Acta, 1995, 59, 1217–1232 CrossRef CAS.
- G. Saper, D. Kallmann, F. Conzuelo, F. Zhao, T. N. Tóth, V. Liveanu, S. Meir, J. Szymanski, A. Aharoni, W. Schuhmann, A. Rothschild, G. Schuster and N. Adir, Nat. Commun., 2018, 9, 2168 CrossRef PubMed.
- K. S. Novoselov, V. I. Fal'ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- P. Cai, X. Feng, J. Fei, G. Li, J. Li, J. Huang and J. Li, Nanoscale, 2015, 7, 10908–10911 RSC.
- X. Fang, K. P. Sokol, N. Heidary, T. A. Kandiel, J. Z. Zhang and E. Reisner, Nano Lett., 2019, 19, 1844–1850 CrossRef CAS PubMed.
- N. Senthilkumar, S. Sheet, Y. Sathishkumar, Y. S. Lee, S.-M. Phang, V. Periasamy and G. G. Kumar, Appl. Phys. A, 2018, 124, 769 CrossRef CAS.
- G. Pankratova, D. Pankratov, C. Di Bari, A. Goñi-Urtiaga, M. D. Toscano, Q. Chi, M. Pita, L. Gorton and A. L. De Lacey, ACS Appl. Energy Mater., 2018, 1, 319–323 CrossRef CAS.
- F.-L. Ng, M. M. Jaafar, S.-M. Phang, Z. Chan, N. A. Salleh, S. Z. Azmi, K. Yunus, A. C. Fisher and V. Periasamy, Sci. Rep., 2014, 4, 7562 CrossRef CAS PubMed.
- S. A. Ibrahim, M. M. Jaafar, F.-L. Ng, S.-M. Phang, G. G. Kumar, W. H. A. Majid and V. Periasamy, Appl. Phys. A, 2017, 124, 59 CrossRef.
- F.-L. Ng, S.-M. Phang, V. Periasamy, J. Beardall, K. Yunus and A. C. Fisher, J. Appl. Phycol., 2018, 30, 2981–2988 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679–1682 CrossRef CAS PubMed.
- H.-J. Shin, K. K. Kim, A. Benayad, S.-M. Yoon, H. K. Park, I.-S. Jung, M. H. Jin, H.-K. Jeong, J. M. Kim, J.-Y. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987–1992 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol., 2008, 3, 270–274 CrossRef CAS PubMed.
- Y. Wei and Z. Sun, Curr. Opin. Colloid Interface Sci., 2015, 20, 311–321 CrossRef CAS.
- S. Lund, J. Kauppila, S. Sirkiä, J. Palosaari, O. Eklund, R.-M. Latonen, J.-H. Smått, J. Peltonen and T. Lindfors, Carbon, 2021, 174, 123–131 CrossRef CAS.
- S. Lund, E. Björnvik, Q. Wang, X. Wang, S. Vajravel, L. T. Wey, Y. Allahverdiyeva, J. Kauppila, J.-H. Smått, J. Peltonen, R.-M. Latonen and T. Lindfors, Carbon Trends, 2022, 9, 100210 CrossRef CAS.
- K. R. Paton, E. Varrla, C. Backes, R. J. Smith, U. Khan, A. O'Neill, C. Boland, M. Lotya, O. M. Istrate, P. King, T. Higgins, S. Barwich, P. May, P. Puczkarski, I. Ahmed, M. Moebius, H. Pettersson, E. Long, J. Coelho, S. E. O'Brien, E. K. McGuire, B. M. Sanchez, G. S. Duesberg, N. McEvoy, T. J. Pennycook, C. Downing, A. Crossley, V. Nicolosi and J. N. Coleman, Nat. Mater., 2014, 13, 624–630 CrossRef CAS PubMed.
- R. Koshani, M. Tavakolian and T. G. M. van de Ven, J. Mater. Chem. B, 2020, 8, 10502–10526 RSC.
- Y. Chu, Y. Sun, W. Wu and H. Xiao, Carbohydr. Polym., 2020, 250, 116892 CrossRef CAS PubMed.
- J. George and S. N. Sabapathi, Nanotechnol., Sci. Appl., 2015, 8, 45–54 CrossRef CAS PubMed.
- R. Xiong, K. Hu, A. M. Grant, R. Ma, W. Xu, C. Lu, X. Zhang and V. V. Tsukruk, Adv. Mater., 2016, 28, 1501–1509 CrossRef CAS PubMed.
- R. Sekar, V. P. Venugopalan, K. K. Satpathy, K. V. K. Nair and V. N. R. Rao, Hydrobiologia, 2004, 512, 109–116 CrossRef.
- L. Zhang, B. Hu, H. Song, L. Yang and L. Ba, Sci. Rep., 2017, 7, 12939 CrossRef PubMed.
- A. Choudhury, L. Barbora, D. Arya, B. Lal, S. Subudhi, S. V. Mohan, S. Z. Ahammad and A. Verma, Eng. Life Sci., 2017, 17, 186–192 CrossRef CAS PubMed.
- J. N. Israelachvili, in Intermolecular and Surface Forces, ed. J. N. Israelachvili, Academic Press, San Diego, 3rd edn, 2011, pp. 191–204 Search PubMed.
- H.-Y. Tsai, C.-N. Hsu, C.-R. Li, Y.-H. Lin, W.-T. Hsiao, K.-C. Huang and J. A. Yeh, Micromachines, 2021, 12, 44 CrossRef PubMed.
- P. Bombelli, R. W. Bradley, A. M. Scott, A. J. Philips, A. J. McCormick, S. M. Cruz, A. Anderson, K. Yunus, D. S. Bendall, P. J. Cameron, J. M. Davies, A. G. Smith, C. J. Howe and A. C. Fisher, Energy Environ. Sci., 2011, 4, 4690–4698 RSC.
- A. Cereda, A. Hitchcock, M. D. Symes, L. Cronin, T. S. Bibby and A. K. Jones, PLoS One, 2014, 9, e91484 CrossRef PubMed.
- A. Deeke, T. H. J. A. Sleutels, H. V. M. Hamelers and C. J. N. Buisman, Environ. Sci. Technol., 2012, 46, 3554–3560 CrossRef CAS PubMed.
- K. L. Saar, P. Bombelli, D. J. Lea-Smith, T. Call, E.-M. Aro, T. Müller, C. J. Howe and T. P. J. Knowles, Nat. Energy, 2018, 3, 75–81 CrossRef CAS.
- L. Koók, N. Nemestóthy, K. Bélafi-Bakó and P. Bakonyi, Bioresour. Technol., 2020, 309, 123313 CrossRef PubMed.
- B. Lai, H. Schneider, J. Tschörtner, A. Schmid and J. O. Krömer, Biotechnol. Bioeng., 2021, 118, 2637–2648 CrossRef CAS PubMed.
- P. Bombelli, A. Savanth, A. Scarampi, S. J. L. Rowden, D. H. Green, A. Erbe, E. Årstøl, I. Jevremovic, M. F. Hohmann-Marriott, S. P. Trasatti, E. Ozer and C. J. Howe, Energy Environ. Sci., 2022, 15, 2529–2536 RSC.
- H. Zhu, H. Meng, W. Zhang, H. Gao, J. Zhou, Y. Zhang and Y. Li, Nat. Commun., 2019, 10, 4282 CrossRef PubMed.
- R. Y. Stanier, R. Kunisawa, M. Mandel and G. Cohen-Bazire, Bacteriol. Rev., 1971, 35, 171–205 CrossRef CAS PubMed.
- M. Hagemann, FEMS Microbiol. Rev., 2011, 35, 87–123 CrossRef CAS PubMed.
- L. T. Wey, J. M. Lawrence, X. Chen, R. Clark, D. J. Lea-Smith, J. Z. Zhang and C. J. Howe, Electrochim. Acta, 2021, 395, 139214 CrossRef CAS.
- A. Trebst, Z. Naturforsch., C: J. Biosci., 1987, 42, 742–750 CrossRef CAS.
- J. M. Pisciotta, Y. Zou and I. V. Baskakov, Appl. Microbiol. Biotechnol., 2011, 91, 377–385 CrossRef CAS PubMed.
- J. Tschörtner, B. Lai and J. O. Krömer, Front. Microbiol., 2019, 10, 866 CrossRef PubMed.
- J. E. O'Reilly, Biochim. Biophys. Acta, Bioenerg., 1973, 292, 509–515 CrossRef PubMed.
- P. Cai, G. Li, Y. Yang, X. Su and Z. Zhang, Colloids Surf., A, 2018, 555, 37–42 CrossRef CAS.
- H. Shin, T. Kim, I. Seo, S. Kim, Y. J. Kim, H. Hong, Y. Park, H. M. Jeong, K. Kim and W. Ryu, Appl. Surf. Sci., 2019, 481, 1–9 CrossRef CAS.
- T. K. Baikie, L. T. Wey, J. M. Lawrence, H. Medipally, E. Reisner, M. M. Nowaczyk, R. H. Friend, C. J. Howe, C. Schnedermann, A. Rao and J. Z. Zhang, Nature, 2023, 615, 836–840 CrossRef CAS PubMed.
- S. Pang, Y. Hernandez, X. Feng and K. Müllen, Adv. Mater., 2011, 23, 2779–2795 CrossRef CAS PubMed.
- P. Blake, P. D. Brimicombe, R. R. Nair, T. J. Booth, D. Jiang, F. Schedin, L. A. Ponomarenko, S. V. Morozov, H. F. Gleeson, E. W. Hill, A. K. Geim and K. S. Novoselov, Nano Lett., 2008, 8, 1704–1708 CrossRef PubMed.
- R. R. Nair, P. Blake, A. N. Grigorenko, K. S. Novoselov, T. J. Booth, T. Stauber, N. M. R. Peres and A. K. Geim, Science, 2008, 320, 1308 CrossRef CAS PubMed.
- K. Parvez, Z.-S. Wu, R. Li, X. Liu, R. Graf, X. Feng and K. Müllen, J. Am. Chem. Soc., 2014, 136, 6083–6091 CrossRef CAS PubMed.
- A. Marmur, Colloids Surf., A, 1996, 116, 55–61 CrossRef CAS.
- R. N. Wenzel, Ind. Eng. Chem., 1936, 28, 988–994 CrossRef CAS.
- J. Peltonen, M. Järn, S. Areva, M. Linden and J. B. Rosenholm, Langmuir, 2004, 20, 9428–9431 CrossRef CAS PubMed.
- F. M. Smits, Bell Syst. Tech. J., 1958, 37, 711–718 CrossRef.
- D. Solymosi, L. Nikkanen, D. Muth-Pawlak, D. Fitzpatrick, R. Vasudevan, C. J. Howe, D. J. Lea-Smith and Y. Allahverdiyeva, Plant Physiol., 2020, 183, 700–716 CrossRef CAS PubMed.
- D. J. Lea-Smith, N. Ross, M. Zori, D. S. Bendall, J. S. Dennis, S. A. Scott, A. G. Smith and C. J. Howe, Plant Physiol., 2013, 162, 484–495 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3se01185b |
| ‡ These authors contributed equally: Sara Lund, Laura T. Wey. |
| This journal is © The Royal Society of Chemistry 2024 |