
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of urea from CO2 and N2 fixation under mild conditions using polarized hydroxyapatite as a catalyst†
Jordi
Sans
 a,
Marc
Arnau
a,
Ricard
Bosque
a,
Pau
Turon
*b and
Carlos
Alemán
*ac
a,
Marc
Arnau
a,
Ricard
Bosque
a,
Pau
Turon
*b and
Carlos
Alemán
*ac
aDepartament d'Enginyeria Química, Barcelona Research Center in Multiscale Science and Engineering, Universitat Politècnica de Catalunya, EEBE, C/Eduard Maristany 10-14, 08019, Spain. E-mail: carlos.aleman@upc.edu
bB. Braun Surgical, S.A.U., Carretera de Terrassa 121, Rubí, 08191, Barcelona, Spain. E-mail: pau.turon@bbraun.com
cInstitute for Bioengineering of Catalonia (IBEC), The Barcelona Institute of Science and Technology, Baldiri Reixac 10-12, 08028 Barcelona, Spain
First published on 13th February 2024
Abstract
Polarized hydroxyapatite (p-HAp) has been used as a catalyst for the synthesis of urea coupling N2, CO2 and water under mild reaction conditions when compared to classical nitrogen fixation reactions, such as the Haber–Bosch process. The reaction of 3 bar of N2 and 3 bar of CO2 under UV illumination at 120 °C (for 48 h) results in a urea yield of 1.5 ± 0.1 mmol per gram of catalyst (gc) with a selectivity close to 80%, whereas the reaction is not successful without UV irradiation. However, the addition of small amounts of NO (314 ppm) produces 15.2 ± 0.6 and 4.6 ± 0.4 mmol gc−1 with and without UV illumination, respectively, with the selectivity in both cases being close to 100%. As nitrogen fixation without UV irradiation using p-HAp as a catalyst is a challenge, studies with NO have been conducted varying the reaction conditions (time, pressure and temperature). The results suggest a mechanism based on the production of NH4+ through the oxidation of N2.
Introduction
About a half the worldwide produced ammonia is currently converted into urea (CO(NH2)2), one of the most important nitrogen fertilizers.1–3 In 2020, the global demand for urea reached almost 188 million Tm, and it is expected to surpass 211 million Tm by 2026.4 Urea is gaining importance as a green source for H2 production.5–7 Although water splitting is the most used electrolysis method for H2 production,8 the high electrical energy consumption required for reaching the electrochemical voltage (1.23 V) is a major limitation for such a process. However, the voltage necessary for urea splitting is much lower (0.37 V), requiring much lower energy consumption.9,10 On an industrial scale, urea is synthesized by combining NH3 (i.e. consumes 80% of the NH3 produced globally) and CO2 at high pressure to form ammonium carbamate (NH2COONH4). By raising the temperature, the latter compound decomposes forming urea and water. Accordingly, the industrial synthesis of urea requires a significant energy supply (350–550 °C and 150–350 bar) and emits a massive amount of CO2.11,12In a recent study, Wang and co-workers13 proposed the synthesis of urea by coupling N2 and CO2 in H2O under ambient conditions. The process was conducted using an electrocatalyst consisting of PdCu alloy nanoparticles on TiO2 nanosheets. Since then, the electrocatalytic activation of N2 and CO2 with the desirable C–N coupling reaction to urea has received widespread attention and some complex electro- and photocatalysts have been engineered for this process (e.g. InOOH nanocrystals,14 Mott–Schottky Bi–BiVO4 heterostructures,15 and coal-based carbon nanotubes with an Fe-core-supported Ti3+–TiO2 composite catalyst).16 Nevertheless, the urea electrosynthesis technology is typically associated with low selectivity and reactivity. Also, gold nanosheets,17 TiO2-immobilized reversible single-atom copper,18 palladium-decorated CeO2 (ref. 19) and 2D-CdS@3D-BiOBr composites20 have been recently used for the photoreduction of N2 and CO2 into urea by applying a potential and visible-NIR illumination.
In this work, we present a green ceramic catalyst able to produce urea using mild reaction conditions without requiring an external potential. Indeed, the conditions used are particularly noticeable when compared with those employed in classical N2 fixation processes, such as the Haber–Bosch process (i.e. such a process uses pressures ranging from 200 to 400 atm and at temperatures ranging from 400 to 650 °C). The catalyst, which is heavy metal free, consists of nanoporous permanently polarized hydroxyapatite (p-HAp).21 It should be noted that p-HAp was recently used to convert CO2 and water into ethanol by promoting the reduction of CO2 and the formation of C–C bonds, as well as to convert N2 and water into NH4+.21 However, no study combining N2, CO2 and water to produce urea has been reported yet.
Two electrothermal approaches (A and B) have been followed to obtain urea using p-HAp as a catalyst and N2, CO2 and water as reactants. In approach A, which is closer to that in recently reported studies,13–20 UV illumination was applied achieving high selectivity (∼80%) and reactivity. In approach B, a very small amount of nitric oxide (NO) was added to the feed mixture of gases with and without UV illumination. This work has mainly focused on approach B, due to the novelty of NO as an activating agent in catalytic reactions and the unexpectedly high selectivity (almost 100%) towards urea.
Methods
The methods used to prepare p-HAp were reported in previous work21 and, therefore, have been briefly described in the ESI.† Also, the procedures and equipment used to characterize the catalyst are provided in the ESI.†Synthesis of urea
The reactor consisted of an inert reaction chamber coated with a perfluorinated polymer (120 mL), in which both the catalyst and water (20 mL) were incorporated. The reactor was equipped with an inlet valve for the entrance of N2 and CO2 and an outlet valve to recover the gaseous reaction products (Fig. S1†). A UV lamp (GPH265T5L/4, 253.7 nm) was also placed in the middle of the reactor to irradiate the catalyst directly, the lamp being protected by a UV transparent quartz tube. All surfaces were coated with a thin film of a perfluorinated polymer in order to avoid any contact between the reaction medium and the reactor surfaces, thus discarding other catalyst effects.Reactions were performed at temperatures ranging from 60 to 120 °C for a reaction time between 6 and 48 h. Both the p-HAp catalyst and 20 mL of de-ionized liquid water were initially incorporated into the reaction chamber (reactions were performed separately for each catalyst). The average mass of each catalyst was m = 31 ± 4 mg. A vacuum pump was used in order to eliminate the initial air content of the reaction chamber. Each selected gas was introduced to increase the reaction chamber pressure (measured at room temperature) to the target pressure. The chamber pressure was increased to a pressure between 1 and 6 bar by introducing sequentially each feed reaction gas (half and half). Blank reactions were conducted without the catalyst and with UV light radiation, while control reactions were performed using p-HAp as the catalyst but without UV. On the other hand, it is worth mentioning that non-polarized HAp has not been employed as a blank in this work since previous studies demonstrated that the polarization treatment is essential to exhibit catalytic activity (i.e. non-polarized HAp is not catalytically active).22,23
Analyses of the reaction products
The reaction products were analyzed by 1H NMR spectroscopy. All 1H NMR spectra were acquired with a Bruker Avance-II+ spectrometer operating at 600 MHz. The chemical shift was calibrated using a tetramethylsilane (TMS) internal standard. 512 scans were recorded in all cases. In order to remove the reaction products from the catalyst, 10 mg of the reacted catalyst were dissolved in 15 mL of water with pH adjusted to 2.1 ± 0.2 using 7.6 mM H2SO4, to promote the conversion of ammonia to NH4+, and 4 cycles were applied that involved sonication (5 min) and stirring (1 min) steps. Then, for the 1H NMR sample preparation, 500 μL of the reacted catalyst solution were mixed with 100 μL of DMSO-d6 instead of solvents with labile deuterons (i.e. D2O) to avoid the formation of ammonium deuterated analogues, not desired for quantitative analysis. The same treatment was applied to the water supernatant.Initially, 1H NMR spectra were acquired using the watergate suppression method, which was reported to be very useful to identify products coming from N2 fixation, for example NH4+. Unfortunately, the urea signal almost overlapped the water signal and, therefore, the application of strong water signal suppression (zgcppr pulse sequence) was necessary to quantify urea. However, zgcppr was too aggressive for the rest of the products, which were lost. Therefore, spectra were recorded in duplicate using both the watergate and the zgcppr methods. Additionally, reaction gas products for specific reactions were captured using Tedlar® sampling bags (Screw Cap Valve, 1 Liter, 30272-U) and later analyzed by means of gas chromatography (GC) using a micro-chromatograph AGILENT 3000 (μPGC3000) to corroborate the mechanism behind the urea synthesis.
In order to compare the different products obtained from the studied reaction, the areas associated with the proton contribution were normalized and calibrated through external references. In all cases the yield of reaction products was normalized per gram of catalyst (gc). The selectivity was calculated as the ratio between the yield of urea and the sum of the yields of all products detected in the liquid phase.
Reaction with engine exhaust gases
Engine exhaust gases were collected from the exhaust pipe of a gasoline passenger car manufactured in 2007 (Mazda 3) by means of a MSHA portable pump and stored in a Tedlar® gas sampling bag (LB-2 Septa). According to the European Regulation EURO4, the CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NOx ratio in the emissions of such a vehicle is 1962.5. The extraction of gases from the exhaust pipe was carried out by revolutionizing the engine of the car at approximately 3500 rpm. After incorporating the catalyst and 20 mL of de-ionized liquid water in the reactor described above, the collected engine exhaust gas was introduced into the reaction chamber (1 bar). The reaction was conducted at 120 °C for 24 h. The reaction products were analyzed by 1H NMR spectroscopy, following the procedures described for the urea reactions.
:NOx ratio in the emissions of such a vehicle is 1962.5. The extraction of gases from the exhaust pipe was carried out by revolutionizing the engine of the car at approximately 3500 rpm. After incorporating the catalyst and 20 mL of de-ionized liquid water in the reactor described above, the collected engine exhaust gas was introduced into the reaction chamber (1 bar). The reaction was conducted at 120 °C for 24 h. The reaction products were analyzed by 1H NMR spectroscopy, following the procedures described for the urea reactions.
Results and discussion
p-HAp was prepared using a four-step process:21 (1) hydroxyapatite (HAp) powder, which was synthesized by regulating the shape and size of the crystals using organic solvent and the controlling of the pH,24 was homogeneously mixed with 60% wt. commercial Pluronic® F-127 hydrogel at low temperature; (2) after aging for 24 h, the resulting white paste, which is a printable ink, was shaped as desired; (3) the shaped paste was calcined in air (1000 °C for 2 h) to remove the hydrogel (i.e. the composition of the calcined samples was checked and, as expected, no carbon-based impurities were observed after calcination at such high temperature); and (4) an external DC electric field of 3 kV cm−1 was imposed at 1000 °C for 1 h to the calcined sample (thermal polarization step). Details are in the ESI.† The overall catalytic activation process results in the enhancement of the activity of the acidic (Ca2+) and basic (PO43− and OH−) binding sites present in the HAp structure.21,25Representative scanning electron microscopy (SEM) images of p-HAp (Fig. 1a) showed a nanoporous structure made of coalesced spherical nanoparticles (average diameter: 152 ± 40 nm), defining a 3D network of nanometric pores (average size: 168 ± 48 nm). High-resolution transmission electron microscopy (HRTEM) images confirmed the presence of highly crystalline HAp grains with well-defined lattice fringes (Fig. 1b). Thus, the Fourier transform image showed well-resolved fringes at 2.8, 3.4 and 6.8 Å, which correspond to the (211), (002) and (001) crystallographic planes of HAp, evidencing a single crystalline domain with high crystallinity. It should be noted that the existence of a (001) plane, which did not appear in the Fourier image transform of non-polarized HAp due to extinction conditions, is only identified when polarization conditions cause lattice re-arrangements and produce catalytically active p-HAp, as was shown in previous work.26,27
 | ||
| Fig. 1 (a) Low to high SEM micrographs and (b) HRTEM images of p-HAp. Fourier transform of the image is also displayed in (b). | ||
The Raman spectrum of HAp (Fig. S2†) was dominated by PO43− characteristic vibrations (ν1–ν4) and the OH− characteristic stretching vibration at νOH = 3574 cm−1.26–28 After catalytic activation by applying the calcination and the electro-thermal polarization steps, the crystallinity and the generated OH− vacancies increased. Furthermore, the Raman spectrum of p-HAp (Fig. S2†) reflected the apparition of three peaks characteristic of brushite (CaHPO4·2H2O).26,27 Previous studies showed that the incorporation of the brushite phase at the surface of p-HAp creates synergistic effects that increased the efficiency and selectivity of HAp-based catalysts.26,27 Accordingly, the distribution of brushite on the surface of p-HAp was examined by acquiring several Raman spectral grids of 2 × 3 (a representative map is depicted in Fig. S3†). The results showed the consistent presence of HAp, while brushite was distributed heterogeneously which is in complete agreement with the results reported in the literature. Electrochemical impedance spectroscopy (EIS) measurements (Fig. S4†), which were conducted on wetted p-HAp and HAp samples using an adapted electrochemical cell,29 evidenced the success of the thermal polarization. The resistance obtained by adjusting experimental parameters to an equivalent electrical circuit30 (Fig. S5†) was lower for p-HAp than for HAp (Rb = 1.6 and 9.7 MΩ cm−2). Details on the Raman and EIS characterization results are provided in the ESI.†
The catalytic conversion of CO2 and N2 gases and liquid water into urea using nanoporous p-HAp was performed using the reactor described in the Methods section. After eliminating the air content with a vacuum pump, the reaction chamber was filled with CO2 and N2 (3 bar each) and a volume of 20 mL of de-ionized water, which was placed in contact with the non-irradiated side of the p-HAp catalyst (Scheme 1). In this case (approach A), the reaction was conducted for 48 h at 120 °C using UV radiation, which was required to catalyze the rupture of the nitrogen–nitrogen triple bond at such low temperature.31 It should be emphasized that no external electrical field was applied across p-HAp, thus minimizing the energy requirements. Therefore, the catalytic activity of p-HAp comes from the lattice rearrangement caused by the thermal polarization process, which results in a permanent polarization step, consisting of a highly crystalline HAp with OH− vacancies (available electric charge) and with the remaining OH− aligned pointing in a specific direction.26,27 These changes resulted in a significant increment of the crystallinity, enhanced proton conductivity and customized surface charge accumulation with electrical charge delocalization along the microscopic HAp grains.30
 | ||
| Scheme 1 Reactor used for the processes described in this work. | ||
After 48 h, the reaction products on the catalyst surface, and those transferred from the solid catalyst to the liquid water (henceforth “supernatant”), were identified by 1H NMR spectroscopy using DMSO-d6 for recognition and quantification of NH4+ and urea.31,32 Initially, the urea signal almost overlapped the water signal and the application of strong water signal suppression (zgcppr pulse sequence) was necessary to quantify urea. Fig. 2a and b show representative 1H NMR spectra acquired for the supernatant, while Fig. S6† displays the spectra recorded for the dissolved catalyst. The yields of the different reaction products obtained for approach A, which are expressed as mmol or μmol of product per gram of catalyst (mmol gc−1 or μmol gc−1), are listed in Table 1 for both the dissolved catalyst and the supernatant.
 | ||
| Fig. 2 Representative 1H NMR spectra used to identify the reaction products in the supernatant: (a) spectrum with a suppressed water signal (zgcppr pulse sequence); and (b) spectrum with an attenuated water signal (watergate). The reaction was conducted at 120 °C for 48 h using a mixture of CO2 and N2 (3 bar each), 20 mL of de-ionized water, and p-HAp as the catalyst. | ||
| Reactants | N2 + CO2 (approach A) | N2/NO + CO2 (approach B with UV) | N2/NO + CO2 (approach B without UV) | |||
|---|---|---|---|---|---|---|
| Conditions | 6 bar (3 bar each gas), 120 °C, UV radiation, p-HAp and 48 h | 6 bar (3 bar each gas; 314 ppm NO in N2), 120 °C, UV radiation, p-HAp and 48 h | 6 bar (3 bar each gas; 314 ppm NO in N2), 120 °C, without UV radiation, p-HAp and 48 h | |||
| Products | Catalyst | Supernatant | Catalyst | Supernatant | Catalyst | Supernatant |
| a Total (catalyst + supernatant) selectivity towards urea. | ||||||
| Urea (mmol g−1) | 0.50 ± 0.02 | 0.98 ± 0.07 | 0.82 ± 0.06 | 14.40 ± 0.49 | 0.19 ± 0.02 | 4.42 ± 0.37 |
| NH4+ (μmol g−1) | — | 95 ± 5 | — | 46 ± 3 | — | <0.1 |
| EtOH (μmol g−1) | 6 ± 1 | 45 ± 3 | 0.6 ± 0.1 | 12 ± 1 | <0.1 | <0.1 |
| Acetic acid (μmol g−1) | 1.1 ± 0.1 | 138 ± 7 | 0.7 ± 0.1 | — | — | <0.1 |
| Acetone (μmol g−1) | 2.3 ± 0.2 | 50 ± 5 | 0.3 ± 0.1 | 70 ± 6 | <0.1 | — |
| Formic acid (μmol g−1) | — | 36 ± 3 | — | 42 ± 2 | <0.1 | <0.1 |
| Selectivitya | 79.9% | 98.9% | 99.9% | |||
In addition to the characteristic signal of H2O at 4.6 ppm and the DMSO-d6 fingerprint at 3.5 ppm, the 1H NMR spectrum collected with the solvent-suppression pulse sequence allowed the clear detection of the presence of urea as a sharp singlet (Fig. 2a). It is worth noting that the peak of urea shifts from 5.42 ppm (neutral pH) to 6.68 ppm in the acid solution used to dissolve the catalyst (Fig. S7†). The study of the shift of the urea peak with the solution acidity is provided in Fig. S8.† This peak was much less defined in the spectrum acquired by applying the widely used watergate technique (Fig. 2b), which instead allowed identification of NH4+, ethanol (EtOH), acetic acid, acetone and formic acid as reaction products. Thus, Fig. 2b reveals the formation of NH4+ at around 7 ppm as a relatively sharp 1:1:1 triplet due to spin coupling to 14N.
The production of NH4+ from pure N2, which was demonstrated by 15N2 isotope-labeling experiments, was recently reported using a partially porous catalyst and identical reaction conditions.31 The NH4+ yield achieved in that reaction was one order of magnitude lower (155 ± 26 μmol gc−1) than the urea yield reported in Table 1 (1.5 ± 0.9 mmol gc−1). This significant increment was due to the improvement in the preparation of the p-HAp catalyst, which was shaped under cold conditions to minimize friction and adhesion forces and, therefore, to maximize the porosity at the surface.
Ethanol (EtOH), acetic acid, acetone and formic acid were also identified (Fig. 2b). EtOH, acetic acid, acetone and formic acid were previously obtained by fixing pure CO2 and under similar reaction conditions, although in that case the yields were lower than 10 μmol gc−1 because p-HAp was prepared as a compact disc rather than an ultraporous 3D shaped cuboid.22 Thus, the yield of C2 products increased considerably (51 ± 3 and 139 ± 7 μmol gc−1 for EtOH and acetic acid, respectively) when the Pluronic® hydrogel was used to induce porosity. The reaction products displayed in Table 1 reflect that the yield of urea coming from the reaction of N2 and CO2 is significantly higher than the sum of the yields of the different products obtained from each of the gases individually, suggesting the existence of a synergistic effect.
The yield of the different reaction products was null in the absence of the catalyst (blank reaction) and very low (<2 μmol g−1) when p-HAp was not irradiated with UV light (control reaction). The latter observation indicates that obtaining ammonia is crucial for the formation of urea, allowing mechanisms to be discarded based on precursors formed through the direct coupling of N2 to CO2, as reported for other electrocatalysts.13–17,33–35 Based on the experimental results and previously reported data for p-HAp,22,31,36,37 a reaction mechanism is proposed for the conditions used in approach A (Scheme 2).
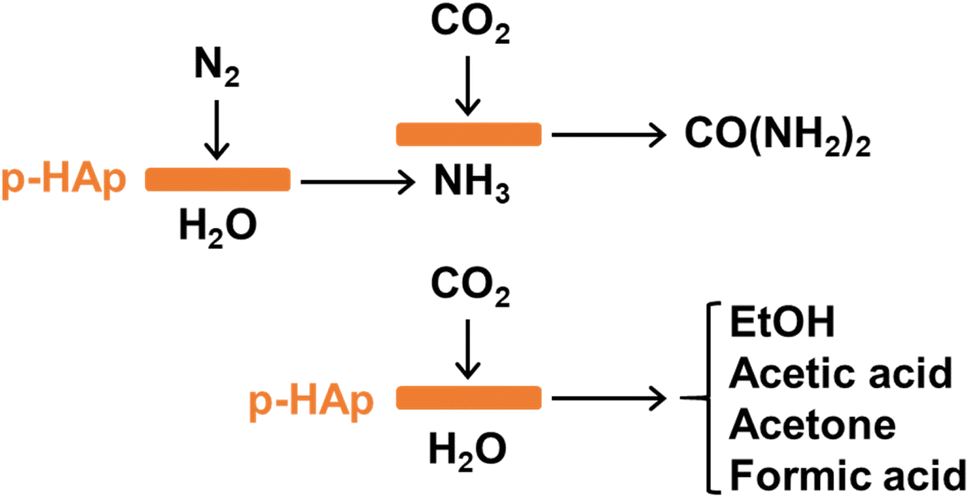 | ||
| Scheme 2 Mechanism proposed for the formation of urea (top) and other products (bottom) under the reaction conditions defined in approach A: N2 (3 bar) + CO2 (3 bar) + H2O (20 mL) at 120 °C with UV illumination. | ||
In the first step, N2 and liquid water are converted into ammonia, with UV illumination being essential to overcome the activation energy barrier for p-HAp catalyzed nitrogen-fixation.31 The resulting ammonia reacts with CO2 to yield urea, probably following the classical two equilibrium reactions:38 the formation of ammonium carbamate (eqn (1)) and the subsequent decomposition of ammonium carbamate into urea and water (eqn (2)).
| 2NH3 + CO2 ⇆ NH2COONH4 | (1) |
| NH2COONH4 ⇆ CO(NH2)2 + H2O | (2) |
The reaction of N2 and H2O through an independent process results in an excess of ammonia to shift the equilibrium towards urea formation, achieving high selectivity towards urea (around 80%). The reaction catalyzed by p-HAp operates at much lower temperature and, especially, pressure than those typically employed for this industrial process.31,38 Besides, secondary reactions give rise to the formation of the other carbonaceous products through the reduction of CO2 catalyzed by p-HAp.
To further increase both the yield and selectivity towards urea, the addition of a small amount of another gas, NO, to the feed mixture was considered. Although the preferential electroreduction of NO over CO2 in a mixture containing such two gases could decrease CO2 reduction performance of the electrocatalyst, no significant detrimental effect was reported when the amount of NO was very small.39 The attainment of ammonia, as a primary product from the electroreduction of NO, using a source different from N2 fixation, which is limited by the triple bond,37 could have three advantages: (1) promoting the formation of urea at the initial stages of the reaction catalyzed by p-HAp and favoring the equilibrium towards urea; (2) minimizing the transformation of CO2 in reaction products different from urea; and (3) reducing the activation energy required to break the N–N triple bond and facilitating the transformation of N2 into NH3. Accordingly, the reaction was repeated using experimental conditions identical to those described above (120 °C, UV radiation, 20 mL of water and p-HAp as the catalyst) but using the following feed gas composition: 3 bar of CO2 and 3 bar of a N2 and NO mixture, in which the content of NO was as low as 314 ppm (hereafter named N2/NO mixture). The reaction products obtained after 48 h, which were quantified by 1H NMR spectroscopy (Fig. S9 and S10†), are listed in Table 1.
The total yield of urea (i.e. catalyst + supernatant) significantly increased from 1.48 ± 0.09 mmol gc−1 to 15.2 ± 0.55 mmol gc−1 upon the addition of a small amount of NO to the feed gas mixture. In contrast, the total yield of C2 products (EtOH and acetic acid) decreased by one order of magnitude (from ∼190 to ∼14 μmol gc−1), while that of C1 (formic acid), C3 (acetone) and NH4+ remained approximately the same. Overall, the addition of a small amount of NO to the feed gas mixture increases the selectivity towards urea (from 79.9% to 98.9%), which confirms some of our previous hypothesized advantages. Indeed, comparison with the yields obtained for approach A suggests that the mechanism in the synthesis of urea is different when a small amount of NO is present in the feed gas mixture.
In order to corroborate that the presence of NO contributes directly or indirectly to lowering the activation energy required for the cleavage of the N–N triple bond, the reaction was repeated without UV illumination keeping the rest of the experimental conditions identical. The yields of the different reaction products after 48 h are included in Table 1, while Fig. S11 and S12† show representative 1H NMR spectra. Although the total yield of urea was much lower compared with that under UV radiation (4.61 vs. 15.22 mmol gc−1), the reaction was not only successful but also showed a total yield of urea higher than the reaction with UV radiation but without NO in the feed gas (4.61 vs. 1.48 mmol gc−1). Another important feature was the very high selectivity towards urea since, as is reflected in Table 1, the rest of the reaction products were only intuited. Thus, the concentration of NH4+, EtOH, acetic acid, acetone and formic acid was so low (<0.1 μmol gc−1) that a more precise quantification was not possible.
Other photocatalysts have recently been used to produce urea from N2, CO2 and H2O.16,40 For example, the yield of urea reported for pure TiO2, TiO2-immobilized reversible single-atom copper (denoted as Cu SA–TiO2) and coal-based carbon nanotubes with Fe-core-supported Ti3+–TiO2 composite catalysts was 33.61 μg gc−1, 432.13 μg gc−1 and 710.1 μmol (L gc)−1, respectively, which are lower than those obtained in this work under the different examined conditions.
Although our results show that NO plays a significant role in the fixation of N2, different reactions used as controls indicate that the contribution of NO to this process is indirect. For example, no nitrogen-containing compound (i.e. urea and or NH4+) was detected when the reaction was conducted for 48 h using CO2 + N2 (3 bar each without NO) as feed gas and without UV illumination. In that case, the identified reaction products (not shown) were identical to those reported for the reaction of CO2 alone.22 On the other hand, detection of the NH4+ reaction product was achieved with N2/NO alone (6 bar of N2 containing 314 ppm of NO) and 20 mL of water without UV radiation at 120 °C using p-HAp as a catalyst, yielding a higher NH4+ quantity compared to the catalytic reaction of sole N2 with the same conditions (Fig. S13†). It is worth highlighting that for the N2/NO reaction without UV radiation the NH4+ production yield reached similar values to the ones reported in the literature when using UV illumination.31 Therefore, the crucial role of NO in the activation of N2 can be corroborated.
In order to ascertain the mechanism, the reaction products formed at shorter times, lower temperatures and lower pressures were identified and quantified. The results obtained individually for the catalyst and supernatant are shown in Fig. S14–S16,† while Fig. 3 displays the sum of the yields considering the two phases. Both NH4+ and acetone were detected in the supernatant at the shortest reaction time (6 h) but disappeared at 24 h. Instead, the yield of urea clearly increases with time. At the lowest temperature (60 °C) the amount of acetic acid and acetone in the supernatant was significant after 24 h (Fig. 3b). Besides, for a reaction time of 6 h, the reaction conducted at the lower pressures (especially 1 bar) allowed the identification of significant amounts of NH4+ and acetone in the supernatant (Fig. 3c and S16†).
 | ||
| Fig. 3 Total yield of the reaction products identified by 1H NMR (i.e. sum of the yields quantified in the catalyst and the supernatant; see Fig. S14–S16†) as a function of: (a) the reaction times (6, 12, 24 and 48 h) for a mixture of 3 bar of CO2 and 3 bar of N2/NO (314 ppm of NO) at 120 °C; (b) the temperature (60, 80, 100 and 120 °C) for a mixture of 3 bar of CO2 and 3 bar of N2/NO (314 ppm of NO) and 24 h of reaction; and (c) the pressure (1, 2, 4 and 6 bar; half and half of CO2 and N2/NO) at 120 °C for 24 h. In all cases, the reactions were conducted without UV illumination, using 20 mL of de-ionized water and p-HAp as the catalyst. | ||
The results displayed in Fig. 3 suggest that NH4+ plays a crucial role in the formation of urea catalyzed by p-HAp in the absence of UV illumination. The electro-reduction of NO to NH3 was reported as a sustainable route to convert such a pollutant into a value-added chemical using nanoscale Fe on carbon black (Fe/C)41 and a nanoporous VN film supported on carbon fiber cloth42 as catalysts while applying a cathodic potential.
| NO + 6H+ + 5e− ⇆ NH4+ + H2O | (3) |
This reaction, which occurs in the absence of UV radiation, could also be proposed for p-HAp, which is known to act as an electrocatalyst able to reduce CO2 without the assistance of an external potential,21–23 due to its capacity to store charge at the surface.26,30 It is worth noting that the amount of NO in the feed mixture is too small to explain the high yield of urea by a mechanism involving this process, and therefore it is necessary to propose an additional source of NO. However, N2 in the presence of water can oxidize to nitrous oxide (N2O) that, subsequently, can oxidize to NO. The global process involved in this double oxidation is:
| N2 + 2H2O ⇆ 2NO + 2H2 | (4) |
The consumption of NO to produce NH4+ (eqn (3)) would favor NO formation, shifting the equilibrium of eqn (4) towards the right. Accordingly, the complete redox process obtained by applying the capacitive charge to p-HAp and combining the oxidation of N2 to NO and the reduction of NO into NH4+ is compatible with both: (1) the lack of NH4+ when NO is not included in the feed gas mixture and the reaction occurs without UV illumination; and (2) the high yield of urea. Subsequently, the presence of H2 and CO2 on the surface of the catalyst leads to a self-reinforcing process by means of a reverse water–gas shift reaction (RWGSR) as follows:
| CO2 + H2 ⇆ HCOOH ⇆ CO + H2O | (5) |
The presence of formic acid (HCOOH), a known intermediate of the CO2-to-CO RWGSR,43 can be observed for short reaction times (Fig. S14†). Thus, both HCOOH and CO can contribute to positive feedback towards the final urea yield. The presence of C1–C3 products (Fig. S15 and S16†) has been attributed to CO2 methanation, a side reaction of the RWGSR that is thermodynamically favorable at low temperature and pressure.44 Indeed, the presence of p-HAp favors the incorporation of ˙CH3 from methane into other C1 and C2 products obtained by the direct fixation of CO2.45 Therefore, the reduction of acetone observed at the early stages of the reaction (Fig. S14†) seems to be more plausible if assigned to CO2 methanation, which at higher temperatures has been reported to occur only at the start of the RWGSR.46
Finally, the reaction of NH4+ with CO2 and water gives ammonium salts (eqn (6)).47 Although such intermediates are not thermally stable and tend to decompose into ammonia and CO2, some catalysts promote the conversion of ammonium carbamate to urea at mild temperatures (∼100–120 °C).48,49 This process is proposed for this work (eqn (7)), and the high yield of urea suggests that ammonium carbamate is the most abundant among the formed ammonium salts:
 | (6) |
NH4+−O–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–NH2 → CO(NH2)2 O)–NH2 → CO(NH2)2 | (7) |
In order to support the proposed mechanism, a reaction consisting of 6 bar of CO2, 19 mL of water and 1 mL of NH3 was carried out at 120 °C for 24 h, obtaining a urea yield of 1.99 ± 0.26 mmol gc−1. The 1H NMR spectrum (Fig. S17†) confirmed the presence of the carbamate ion as a subproduct of the reaction (i.e. a new singlet at 3.65 ppm, corresponding to methylcarbamate). Accordingly, both the acetone and the acetic acid peaks displayed well-resolved shoulders which were attributed to the partial substitution of methyl groups by amino species.
In addition, analyses of the gas products obtained for a standard urea synthesis reaction (3 bar CO2, 3 bar N2/NO, 120 °C, with UV radiation and p-HAp for 24 h) were carried out by means of gas chromatography. No urea was detected in the gas phase suggesting the complete dissolution of this reaction product into the supernatant. Nonetheless, a non-negligible amount of N2O can be observed in the chromatographs, as displayed in Fig. S18.† Such a result strongly supports the overall proposed reaction mechanism, confirming the oxidation of N2 to NO presented in eqn (4). Because of the fact that N2O is identified as an intermediate of the reaction (i.e. very small amount observed) and that the retention time almost overlaps the CO2 signal, proper quantification cannot be performed. As it was the only compound with catalytic properties inside the reactor, p-HAp is proposed to catalyze not only the redox reactions needed to produce ammonium from NO and NO from N2, but also the conversion of ammonium carbamate into urea. Besides, the elimination of C1, C2 and C3 products is probably due to their electro-oxidation to CO2 at an advanced stage of the reaction.
As a proof of concept, the urea synthesis catalyzed by p-HAp was evaluated using engine exhaust gases coming out of the fumes of gasoline vehicle engines that do not incorporate the use of AdBlue to reduce NOx-type pollutants (see Methods). After reaction at 120 °C for 24 h without UV illumination using 1 bar of polluted air, the total amount of urea detected in the corresponding 1H NMR spectra (mainly at the supernatant) was 8.0 ± 0.4 mmol gc−1 (Fig. S19†). The migration of the product from the catalyst to the supernatant (3% vs. 97%) is expected not only to avoid poisoning of the catalyst but also to facilitate the recovery of the reaction product.
Conclusions
A nanoporous p-HAp catalyst produces urea starting from N2 and CO2 gases and liquid water using mild reaction conditions. With UV radiation, a high urea yield (1.5 ± 0.1 mmol gc−1 for 48 h) with a selectivity close to 80% is obtained, whereas no reaction is observed without UV illumination. Thus, urea forms through the reaction of NH4+, which comes from the N2 fixation catalyzed by p-HAp, with CO2 to give ammonium carbamate that subsequently decomposes into urea.When a small amount of NO (314 ppm) is introduced in the feed gas mixture, the urea yield significantly increases to 15.2 ± 0.6 mmol gc−1 under UV illumination and, in addition, the reaction is successful without UV irradiation with a yield of 4.6 ± 0.4 mmol gc−1, Furthermore, the selectivity in both cases increases to almost 100%. A mechanism based on the reduction of NO to NH4+ is proposed, while NO regenerates through the oxidation of N2. Finally, as a proof of concept, the applicability of this process is proven using engine exhaust gases with NOx-type pollutants from the fumes of gasoline vehicle engines. The reaction, which produced a high yield of urea, evidences the potential of p-HAp in the automotive sector.
In summary, this work offers a catalyst, based on a green ceramic, for converting N2 and CO2 to urea with high yield and selectivity using mild reaction conditions and UV irradiation. The addition of a small amount of NO to the feed gas mixture allows the UV illumination to be eliminated, resulting in an outstanding yield and selectivity towards urea.
Conflicts of interest
The authors declare that the preparation and application of permanently polarized hydroxyapatite as a catalyst was patented by the Universitat Politècnica de Catalunya and B. Braun Surgical S.A. (EP16382381, EP16382524, P27990EP00, PCT/EP2017/069437, P58656 EP, P59205 EP, P59091 EP, P59008 EP, and P59528 EP).Acknowledgements
The authors acknowledge the Agència de Gestió d'Ajuts Universitaris i de Recerca (2021 SGR 003879). Support for the research of C. A. was also received through the prize “ICREA Academia” for excellence in research funded by the Generalitat de Catalunya.References
- B. Comer, P. Fuentes, C. Dimkpa, Y. Liu, C. Fernandez, P. Arora, M. Realff, U. Singh, M. Hatzell and A. Medford, Joule, 2019, 3, 1578–1605 CrossRef CAS.
- Y. Abghoui, A. L. Garden, V. F. Hlynsson, S. Björgvinsdóttir, H. Ólafsdóttir and E. Skúlason, Phys. Chem. Chem. Phys., 2015, 17, 4909–4918 RSC.
- N. Yahya, in Green Urea: for Future Sustainability, Springer, Singapore, 2018 Search PubMed.
- M. K. Xia, C. L. Mao, G. Ozin, M. Xia, G. Mao, A. Gu, A. A. Tountas, C. Qiu, T. Wood, E. Thomas, Y. F. Li, U. Ulmer, Y. Xu, C. J. Viasus, J. Ye, C. Qian and G. Ozin, Angew. Chem., Int. Ed., 2022, 61, e202110158 CrossRef CAS PubMed.
- J. Li, J. Li, T. Liu, L. Chen, Y. Li, H. Wang, X. Chen, M. Gong, Z.-P. Liu and X. Yang, Angew. Chem., Int. Ed., 2021, 60, 26656–26662 CrossRef CAS PubMed.
- H. N. Sun, J. P. Liu, W. Jung, H. Sun, J. Liu, G. Chen, H. Kim, S. Kim, Z. Hu, J.-M. Chen, S.-C. Haw, F. Ciucci and W. C. Jung, Small Methods, 2022, 6, 2101017 CrossRef CAS PubMed.
- S. Ji, X. Y. Wang, H. Wang, X. Xu, S. Ji, H. Wang, X. Wang, V. Linkov and R. Wang, J. Colloid Interface Sci., 2022, 615, 163–172 CrossRef PubMed.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef CAS.
- X. Zhu, X. Dou, J. Dai, X. An, Y. Guo, L. Zhang, S. Tao, J. Zhao, W. Chu, X. C. Zeng, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2016, 55, 12465–12469 CrossRef CAS PubMed.
- S. Chen, J. Duan, A. Vasileff and S. Z. Qiao, Angew. Chem., Int. Ed., 2016, 55, 3804–3808 CrossRef CAS PubMed.
- M. Kitano, S. Kanbara, Y. Inoue, N. Kuganathan, P. Sushko, T. Yokoyama, M. Hara and H. Hosono, Nat. Commun., 2015, 6, 6731 CrossRef CAS PubMed.
- D. Kayan and F. Köleli, Appl. Catal., B, 2016, 181, 88–93 CrossRef CAS.
- C. Chen, X. Zhu, X. Wen, Y. Zahng, L. Zhou, H. Li, Q. Li, S. Du, T. Liu, D. Yan, C. Xie, Y. Zou, Y. Wang, R. Chen, J. Huo, Y. Li, J. Cheng, H. Su, X. Zhao, W. Cheng, Q. Liu, H. Lin, J. Luo, J. Chen, M. Dong, K. Cheng, C. Li and S. Wang, Nat. Chem., 2020, 12, 717–724 CrossRef CAS PubMed.
- M. Yuan, H. Zhang, Y. Xu, R. Liu, R. Wang, T. Zhao, J. Zhang, Z. Liu, H. He, C. Yang, S. Zhang and G. Zhang, Chem Catal., 2022, 2, 309–320 CrossRef CAS.
- M. Yuan, J. Chen, Y. Bai, Z. Liu, J. Zhang, T. Zhao, Q. Wang, S. Li, H. He and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 10910–10918 CrossRef CAS PubMed.
- H. Maimaiti, B. Xu, J.-Y. Sun and L.-R. Feng, ACS Sustain. Chem. Eng., 2021, 9, 6991–7002 CrossRef CAS.
- G. Bharath, G. Karthikeyan, A. Kumar, J. Prakash, D. Venkatasubbu, A. K. Nadda, V. K. Gupta, M. A. Haija and F. Banat, Appl. Energy, 2022, 318, 119244 CrossRef CAS.
- D. Li, Y. Zhao, Y. Miao, C. Zhou, L.-P. Zhang, L.-Z. Wu and T. Zhang, Adv. Mater., 2022, 34, 2207793 CrossRef CAS PubMed.
- S. Yang, J. Deng, J. Chen, Q. Tan, T. Liu, K. Chen, D. Han, Y. Ma, M. Dai and L. Niu, Catal. Sci. Technol., 2023, 13, 1855–1865 RSC.
- Y. Wang, S. Wang, J. Gan, J. Shen, Z. Zhang, H. Zheng and X. Wang, ACS Sustainable Chem. Eng., 2023, 11, 1962–1973 CrossRef CAS.
- J. Sans, M. Arnau, P. Turon and C. Alemán, Mater. Horiz., 2022, 9, 1566–1576 RSC.
- J. Sans, V. Sanz, P. Turon and C. Alemán, ChemCatChem, 2021, 13, 5025–5033 CrossRef CAS.
- J. Sans, G. Revilla-López, V. Sanz, J. Puiggalí, P. Turon and C. Alemán, Chem. Commun., 2021, 57, 5163–5166 RSC.
- J. Sans, V. Sanz, J. Puiggalí, P. Turon and C. Alemán, Cryst. Growth Des., 2021, 21, 748–756 CrossRef CAS.
- M. Miyauchi, T. Watanabe, D. Hoshi and T. Ohba, Dalton Trans., 2019, 48, 17507–17515 RSC.
- J. Sans, M. Arnau, F. Estrany, P. Turon and C. Alemán, Adv. Mater. Interfaces, 2021, 8, 2100163 CrossRef CAS.
- J. Sans, J. Llorca, V. Sans, P. Turon and C. Alemán, Langmuir, 2019, 35, 14782–14790 CrossRef CAS PubMed.
- Z. Iqbal, V. P. Tomaselli, O. Fahrenfeld, K. D. Möller, F. A. Ruszala and E. Kostiner, J. Phys. Chem. Solids, 1977, 38, 923–927 CrossRef CAS.
- F. Müller, C. A. Ferreira, D. S. Azambuja, C. Alemán and E. Armelin, J. Phys. Chem. B, 2014, 118, 1102–1112 CrossRef PubMed.
- J. Sans, M. Arnau, V. Sanz, P. Turon and C. Alemán, Adv. Mater. Interfaces, 2022, 9, 2101631 CrossRef CAS.
- J. Sans, M. Arnau, V. Sanz, P. Turon and C. Alemán, Chem. Eng. J., 2022, 446, 137440 CrossRef CAS.
- R. Y. Hodgetts, A. S. Kiryutin, P. Nichols, H.-L. Du, J. M. Bakker, D. R. Macfarlane and A. N. Simonov, ACS Energy Lett., 2020, 5, 736–741 CrossRef CAS.
- Q. Wu, F. Zhu, G. Wallace, X. Yao and J. Chen, Chem. Soc. Rev., 2024, 53, 557–565 RSC.
- J. Xian, S. Li, P. Liao, S. Wang, Y. Zhang, W. Yang, J. Yang, Y. Sun, Y. Jia, Q. Liu, Q. Liu and G. Li, Angew. Chem., Int. Ed., 2023, 62, e202304007 CrossRef CAS PubMed.
- Y. Chen, Y. Liu, S. Hu, D. Wu, M. Zhang and Z. Cheng, Sci. Total Environ., 2024, 913, 169722 CrossRef CAS PubMed.
- J. Sans, V. Sanz, L. J. del Valle, J. Puiggalí, P. Turon and C. Alemán, J. Catal., 2021, 397, 98–107 CrossRef CAS.
- G. Revilla-López, J. Sans, J. Casanovas, O. Bertran, J. Puiggalí, P. Turón and C. Alemán, Appl. Catal., A, 2020, 596, 117526 CrossRef.
- S. Qureshi, M. Mumtaz, F. K. Chong, A. Mukhtar, S. Saqib, S. Ullah, M. Mubashir, K. S. Khoo and P. L. Show, Chemosphere, 2022, 291, 132806 CrossRef CAS PubMed.
- B. H. Ko, B. Hasa, H. Shin, E. Jeng, S. Overa, W. Chen and F. Jiao, Nat. Commun., 2020, 11, 5856 CrossRef CAS PubMed.
- D. Li, Y. Zhao, Y. Miao, C. Zhou, L.-P. Zhang, L.-Z. Wu and T. Zhang, Adv. Mater., 2022, 34, 2207793 CrossRef CAS PubMed.
- S. Cheon, W. J. Kim, D. Y. Kim, Y. Kwon and J.-I. Han, ACS Energy Lett., 2022, 7, 958–965 CrossRef CAS.
- D. Qi, F. Lv, T. Wei T, M. Jin, G. Meng, S. Zhang, Q. Liu, W. Liu, D. Ma, M. S. Hamdy, J. Luo and X. Liu, Nano Research Energy, 2022, 1, e9120022 CrossRef.
- N. Ishito, K. Hara, K. Nakajima and A. Fukuoka, J. Energy Chem., 2016, 25, 306–310 CrossRef.
- X. Chen, Y. Chen, C. Song, P. Ji, N. Wang, W. Wang and L. Cui, Front. Chem., 2020, 8, 709 CrossRef CAS PubMed.
- J. Sans, M. Arnau, V. Sanz, P. Turon and C. Alemán, Chem. Eng. J., 2022, 433, 133512 CrossRef CAS.
- Y. Zhou, Y. Ma, G. Luan, H. Tang, W. Han, H. Liu and Y. Li, Chin. J. Catal., 2019, 40, 114–123 CrossRef CAS.
- F. Barzagli, F. Mani and M. Peruzzini, Green Chem., 2011, 13, 1267–1274 RSC.
- D. S. Hanson, Y. Wang, X. Zhou, E. Washburn, M. B. Ekmekci, D. Dennis, A. Paripati, D. Xiao and M. Zhou, Inorg. Chem., 2021, 60, 5573–5589 CrossRef CAS PubMed.
- C. Chen, X. Zhu, X. Wen, Y. Zhou, L. Zhou, H. Li, L. Tao, Q. Li, S. Du, T. Liu, D. Yan, C. Xie, Y. Zou, Y. Wang, R. Chen, J. Huo, Y. Liu, J. Cheng, H. Su, X. Zhao, W. Cheng, Q. Liu, H. Lin, J. Luo, J. Chen, M. Dong, K. Cheng, C. Li and S. Wang, Nat. Chem., 2020, 12, 717–724 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3se01704d |
| This journal is © The Royal Society of Chemistry 2024 |