
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The sooting behavior of lactones as sustainable fuels†
Zhanhong
Xiang
 a,
Mehmet B.
Acikel‡
a,
Collin J.
Hansen
b,
Ga-Un
Jeong
c,
Raúl
Pérez-Soto
b,
David Z.
Wang§
a,
Vivian C.
Whoriskey
a,
Seonah
Kim
b,
Charles S.
McEnally
*a,
Lisa D.
Pfefferle
a and
Yuan
Xuan
c
a,
Mehmet B.
Acikel‡
a,
Collin J.
Hansen
b,
Ga-Un
Jeong
c,
Raúl
Pérez-Soto
b,
David Z.
Wang§
a,
Vivian C.
Whoriskey
a,
Seonah
Kim
b,
Charles S.
McEnally
*a,
Lisa D.
Pfefferle
a and
Yuan
Xuan
c
aDepartment of Chemical & Environmental Engineering, Yale University, New Haven, CT 06520, USA. E-mail: charles.mcenally@yale.edu
bDepartment of Chemistry, Colorado State University, Fort Collins, CO 80523, USA
cDepartment of Mechanical Engineering, The Pennsylvania State University, State College, PA 16801, USA
First published on 8th October 2024
Abstract
Lactones are an interesting category of sustainable fuels since they have the same carbon backbones as sugars but are liquids at room temperature. Engine studies have shown that lactones can reduce soot emissions as well as net carbon dioxide emissions. In this study quantitative sooting tendencies were measured for 10 lactones with a wide range of molecular structures. They included compounds with ring sizes varying from three to six carbons, unsubstituted compounds, substituted compounds with side chain lengths ranging from one to seven carbons, and one compound with a double bond in the ring. Two alkenoic acids were also tested since they are possible isomerization products of lactones. The sooting tendencies were characterized by yield sooting index (YSI), which is based on the soot yield when a methane/air nonpremixed flame is doped with 1000 μmol mol−1 of the test fuel. The results show that the lactones have lower sooting tendencies than conventional gasoline, diesel fuel, and Jet A aviation fuel, even when accounting for their lower heats of combustion. However, the sooting tendencies depend strongly on molecular structure, so the right lactones must be chosen to maximize the emissions benefits. The measured sooting tendencies are generally larger than those predicted with a group contribution method, which indicates that the lactones have high sooting tendencies given the set of atoms they contain. To explain this observation, reactive molecular dynamics simulations and quantum chemistry calculations were performed. The results show that the lactones tend to decompose directly to CO2, so the oxygen atoms are being used inefficiently to sequester only one carbon atom.
1. Introduction
Sugars are the largest sustainable hydrocarbon resource on Earth. About 75% of biomass consists of the inedible sugar polymers cellulose and hemicellulose,1 and about 1011 kg of sugars are produced on land each year.2 As shown on the left side of Fig. 1, sugars can have a variety of structures: (1) they can be pentoses with the formula C5H10O5 or hexoses with the formula C6H12O6; (2) in either of these cases they can adopt a furanose configuration with a C4O ring or a pyranose configuration with a C5O ring; and (3) in the case of hexo-furanoses the two non-ring carbons can form two C1 side-chains or one C2 side-chain. Unfortunately—from the perspective of fuels—in all cases they are heavily functionalized with hydroxy groups, so they are solids with melting points (Tmelt) > 100 °C.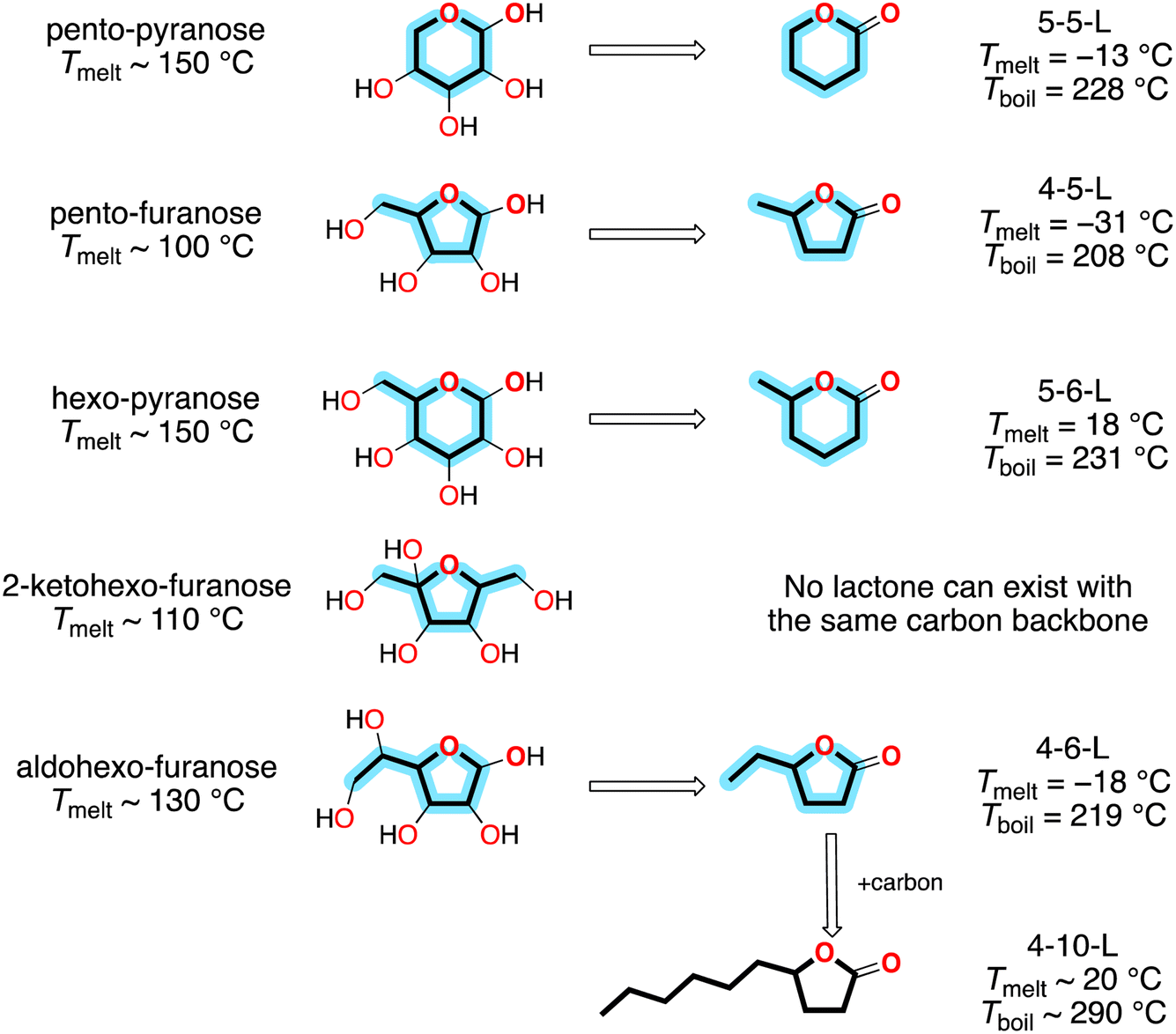 | ||
| Fig. 1 Relationship between sugars and lactones. Property data from ref. 3. | ||
However, if one of the hydroxy groups is converted to a carbonyl group and the others are removed, then the products are the liquid lactones on the right side of Fig. 1. Consequently, lactones have generated interest as potential alternative fuels. Specific processes have been developed for their production [e.g., ref. 1 and 4–6], and they have been tested in engines.7–9
In Fig. 1 and throughout this paper, we use a custom nomenclature where saturated lactones are designated by NC,ring-NC,total-L, where NC,ring is the number of carbons in the ring and NC,total is the total number of carbons. This is a compact and comprehensible alternative to the common names for lactones, where NC,ring is designated by a Greek letter offset from one (α = 2, β = 3, γ = 4, etc.) and NC,total is designated by a series of non-standard prefixes (butyro = 4, valero = 5, capro = 6, etc.). For example, 4-5-L corresponds to γ-valerolactone. Table 1 lists our abbreviations and the common names for all the lactones discussed in this paper.
| Abbreviation | Common name | Measured YSI |
|---|---|---|
| 3-3-L | β-Propiolactone | 8.8 ± 5.0 |
| 4-4-L | γ-Butyrolactone | 17.2 ± 5.0 |
| 4-5-L | γ-Valerolactone | 34.9 ± 5.0 |
| 4-6-L | γ-Caprolactone | 41.0 ± 5.0 |
| 4-7-L | γ-Heptanolactone | 45.2 ± 5.0 |
| 4-11-L | γ-Undecanolactone | 58.2 ± 5.0 |
| 5-5-L | δ-Valerolactone | 21.1 ± 5.0 |
| 5-6-L | δ-Caprolactone | 38.0 ± 5.0 |
| 6-6-L | ε-Caprolactone | 35.7 ± 5.0 |
| α-A-L | α-Angelicalactone | 18.2 ± 5.0 |
| 4PA | 4-Pentenoic acid | 37.3 ± 5.0 |
| 5HA | 5-Hexenoic acid | 37.2 ± 5.0 |
The lactones with sugar backbones have boiling points (Tboil) that are below the diesel range (∼260 to ∼340 °C). However, compounds with longer side-chains can have diesel range volatilities; for example, 4-10-L, shown at the bottom of Fig. 1, has been successfully used as a single-component diesel fuel.9 These compounds can be produced by adding carbon with C–C coupling reactions10 or directly via biological conversion.11
The objective of this study was to compare the sooting behavior of lactones to other hydrocarbons under well-defined burning conditions. Engine experiments suggest that lactones can reduce particulate emissions,8,9 so sugar-derived lactone biofuels could have the significant co-benefit of improving local air quality as well as reducing net carbon emissions. However, laboratory-scale sooting tendencies have not been reported for them. Lactones would be expected to have low sooting tendencies since they contain two oxygen atoms, but they also contain a ring, which tends to promote soot production. Furthermore, both oxygen atoms are bonded to the same carbon atom, so they could directly form CO2, which would use the oxygen atoms inefficiently to sequester only one carbon atom.12
In this work sooting tendencies were measured for 10 lactones and two alkenoic acids, which are potential isomerization products of lactones.13 The sooting tendencies were measured as yield sooting indices (YSIs),14 which allows the results to be compared with previous measurements for conventional fuels15–17 and hundreds of pure hydrocarbons.18,19Fig. 1 shows that sugars can lead to a variety of lactones, so the test compounds included a range of ring sizes and a varying number of carbon atoms in the side-chain (including some cases with no side-chain).
Simulations were also performed to interpret the experimental results and extrapolate them to other combustion conditions. These included molecular dynamics (MD) simulations using the ReaxFF force field20–25 and quantum chemistry calculations.26–29 The ReaxFF simulations consider pure pyrolysis at high temperature, which has been shown to be a good approximation to the conditions in the soot-forming regions of the nonpremixed YSI flames in previous work where we directly simulated the YSI flames by solving the governing conservation equations.30 In contrast, the quantum chemistry calculations are for bimolecular reactions of lactones with H atoms, so they indicate how the reaction pathways might differ in lower temperature or more premixed combustion environments.
2. Methods
2.1. YSI measurements
Sooting tendencies were measured using a yield-based approach we developed previously.14 The specific procedure used in this study is described in ref. 16. It consists of three steps: (1) we sequentially doped 1000 μmol mol−1 (1000 ppm) of n-heptane (H), toluene (T), and each test fuel (TF) into the fuel stream of a base methane/air flame; (2) we measured the maximum soot concentration in each flame with line-of-sight spectral radiance (L); and (3) we rescaled the results into a yield sooting index (YSI) defined as: | (1) |
This rescaling eliminates sources of systematic uncertainty such as the optical properties of the soot. Furthermore, it allows the new results to be quantitatively compared to a database that contains measured YSIs for hundreds of organic compounds.19 The parameters YSIT and YSIH are constants that define the YSI scale; their values—170.9 and 36.0—were taken from the database so that the newly measured YSIs would be on the same scale for a direct comparison. The dopants are added at a small concentration to eliminate indirect effects such as changes in the flame temperature or residence time.
ESI (SI A)† lists the sources and purities of each reactant. SI B† shows a schematic diagram of the apparatus and describes the experiments in detail. SI C† gives details of the burner.31,32 The liquid lactones were injected into the gas-phase CH4 fuel mixture with a syringe pump. SI D† lists the liquid-phase flowrates corresponding to 1000 μmol mol−1 in the gas-phase for each test fuel, and the property values that were used to calculate them.33 The fuel lines were heated to 100 °C, and the burner was heated to 170 °C. Each test fuel was injected for 600 s and L was averaged from 300 to 600 s—SI E† shows that the initial 300 s is adequate for all the test fuels to equilibrate with the walls of the fuel line and burner. SI F† experimentally confirms that the test fuels were not condensing in the fuel delivery system. Isooctane was included in each measurement set as an internal standard—SI G† shows that its measured YSIs were consistent over time and agreed with previous measurements.19
2.2. ReaxFF molecular dynamics simulations
ReaxFF is a bond-order-based force field with instantaneous connectivity for chemical bonds depending on the atomic local environment. The bond order is calculated directly from an interatomic distance and updated at every iteration, which captures bond formation and breaking. The total energy of a system consists of many-body empirical potential terms. There are bond-order-dependent energy terms such as bond, angle, and torsion, and nonbonded interactions terms such as van der Waals and Coulomb interactions. The ReaxFF method uses Morse-potential for the van der Waals interactions to properly describe the short-range interactions and uses the Electronegativity Equalization Method (EEM) for Coulomb interactions to calculate atomic charges. ReaxFF calculates the energy of each atom in the system using the following equation: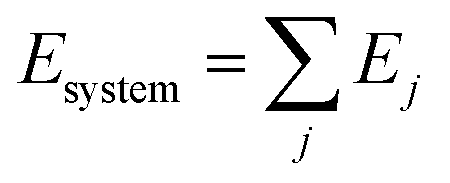 | (2) |
For each fuel investigated, we used the previously established simulation framework for pyrolysis studies using ReaxFF.24,25 First, the fuel molecular structure was energy minimized at 0 K. Then, 40 fuel molecules were randomly placed in a 3D-periodic box of appropriate size to reach the desired density of 0.2 kg dm−3. Next, the system was equilibrated at 1500 K for 2.5 ps with constant number of atoms, volume, and temperature (NVT) conditions, using a Berendsen thermostat with a temperature damping constant of 100 fs, to stabilize the randomly arranged molecules without any chemical reaction occurring. Finally, for each fuel, three statistically independent NVT simulations with different initial configurations were performed at 2000 K for 3 ns with a time step of 0.1 fs. This temperature is higher than in the soot-forming regions of the YSI flames, but prior studies have shown that while increased temperatures accelerate the process, they do not influence the types of reactions occurring.34,35 All ReaxFF results reported in this work were based on ensemble-averaged data from the three NVT-MD simulations to minimize statistical noise. The C/H/O force field that was previously developed and validated23 was used in all ReaxFF simulations. We used an in-house reaction analysis code to identify all reactions that occurred during MD simulations. This code assumes that a reaction event occurs when a species with a different chemical formula is identified.24,25
2.3. Quantum mechanical calculations
Quantum mechanical calculations were carried out using Gaussian 16.26 Geometry optimizations and frequency calculations were obtained with the G4 methodology27 which has proven to provide low errors in the calculation of formation enthalpies of both closed-shell and open-shell species.28 Intrinsic reaction coordinate (IRC) calculations were carried out on the key optimized transition state structures to ensure that they are connected to the correct minima. Free energies at 1500 K and 1 atm were obtained using the Goodvibes software.293. Results and discussion
3.1. Measured YSIs
Table 1 lists the YSIs measured in this study for 10 lactones and two alkenoic acids. Each YSI was measured three times and then averaged. There are three main sources of uncertainty: (1) random uncertainty, which is estimated to be ±2% based on the standard deviation of the internal standard measurements (see SI G†); (2) systematic uncertainty, which is estimated to be ±1%, mainly due to possible error in the ratios of the mass densities between the test compounds, and (3) the uncertainty in comparing the current measurements with the previous measurements in ref. 19, which is estimated to be ±2% based on the differences observed for the internal standard. Overall, we estimate that the uncertainty in the measured YSIs is ±5% or ±5.0, whichever is larger.Fig. 2 compares the sooting behavior of the lactones with several conventional fuels. The figure plots the data as YSI/LHV, where LHV is lower heating value, since the oxygen atoms in the lactones significantly reduce their heat of combustion. The units of LHV are molar (i.e., MJ mol−1) since YSI measures the amount of soot produced per mole of fuel consumed. The conventional fuels include several test gasolines (the TG fuels),15 a Jet A aviation fuel,16,36 and a diesel fuel.17,37 The TG fuels were produced by conventional fuel refineries and mostly meet the specifications for gasoline, but they were formulated to have enhanced concentrations of various hydrocarbon classes and they bracket the composition space of commercial gasolines. The LHVs of the conventional fuels are measured values, while the LHVs of the lactones were estimated with an empirical relationship.38 SI H† provides further details and the raw data.
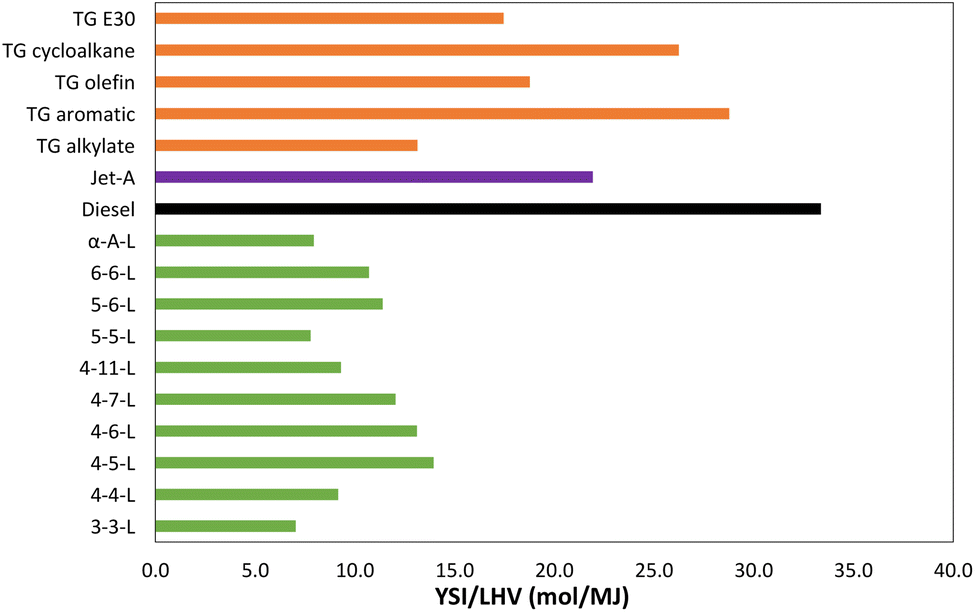 | ||
| Fig. 2 YSI/LHV for the lactones and several conventional fuels. LHV = lower heating value. See SI H† for further details and the raw data. | ||
The results show that the lactones produce less soot than conventional fuels even when accounting for the differences in heat of combustion—i.e., even though a greater quantity of the lactones must be burned to perform a given amount of work, the total soot production is lower. All the lactones have a YSI/LHV at least 50% lower than the diesel fuel and at least 30% lower than the Jet A fuel. Most of the lactones also have lower YSI/LHV than all the TG fuels—the exception is 4-5-L, which has a slightly higher value than TG alkylate, an alkane-rich gasoline. But even 4-5-L has lower values than the other TG fuels, including TG E30, which is a conventional gasoline containing 30% ethanol.
Bereczky et al. have measured the particulate matter (PM) emissions from a diesel engine when oxygenates are added to the fuel.8 The mass concentration of PM in the exhaust ΓPM was reduced by 23% when 16 mol% of a methyl ester biodiesel (BD) was added to a conventional diesel fuel (D), and by 36% when 14 mol% of the BD and 15 mol% of 4-5-L were added to the D. We estimate YSIs and LHVs for each of these fuel mixtures in SI I.†Fig. 3 shows that there is a reasonably monotonic relationship between the measured ΓPM and the estimated YSI/LHV for each fuel. This observation indicates that the differences in YSI/LHV observed in this study between the lactones and conventional fuels are likely to translate into reductions in PM emissions from engines.
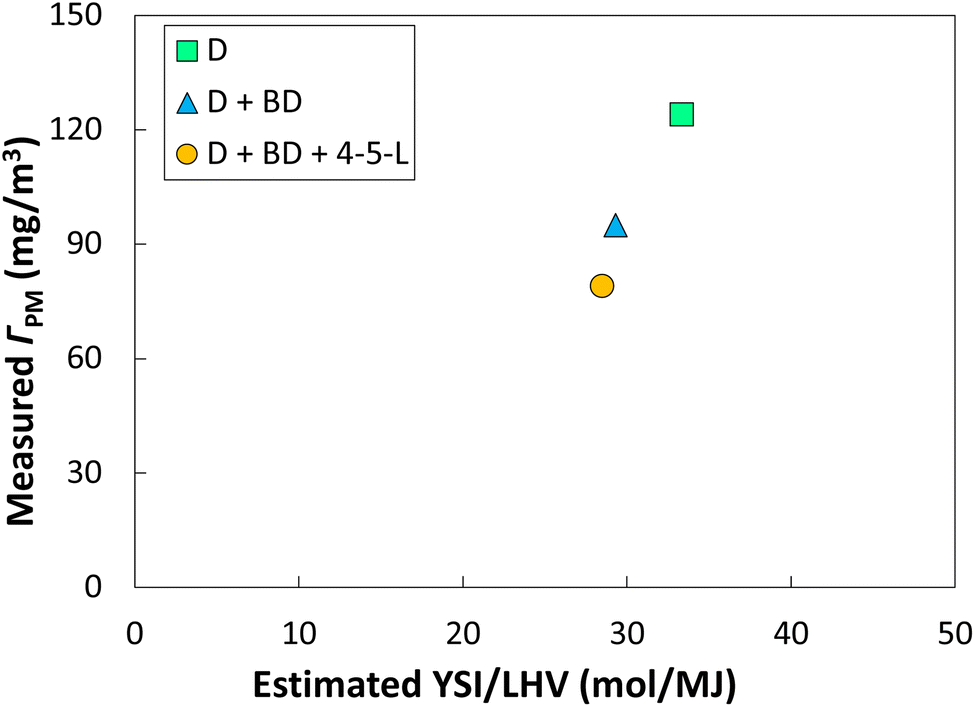 | ||
| Fig. 3 Diesel engine PM emissions measured in ref. 8vs. estimated YSI/LHVs for three fuel mixtures. D = diesel and BD = biodiesel. See SI I† for further details and the raw data. | ||
The YSIs of the lactones depend strongly on the molecular structure of the molecule. As an example, Fig. 4 shows how the YSI of 4-5-L changes when its structure is altered in various ways. Removal of the methyl side chain produces 4-4-L and reduces the sooting tendency by 17.7 YSI units (left side of the figure). Extension of the side chain to an ethyl group produces 4-6-L and increases the YSI, but only by 6.1 (right). The comparisons to the molecules in the top row are particularly interesting since they have the same number of carbon atoms as 4-5-L. Ring-opening isomerization produces 4PA and only changes the YSI slightly, by +2.4 (upper left). However, rearrangement of the side-chain carbon into the ring produces 5-5-L and reduces the YSI by 13.8 (upper middle). Most strikingly, addition of a double carbon–carbon bond to the ring produces α-A-L and reduces the YSI by 16.7 (upper right)—this is one of very few cases where a greater degree of unsaturation corresponds to a lower sooting tendency.18
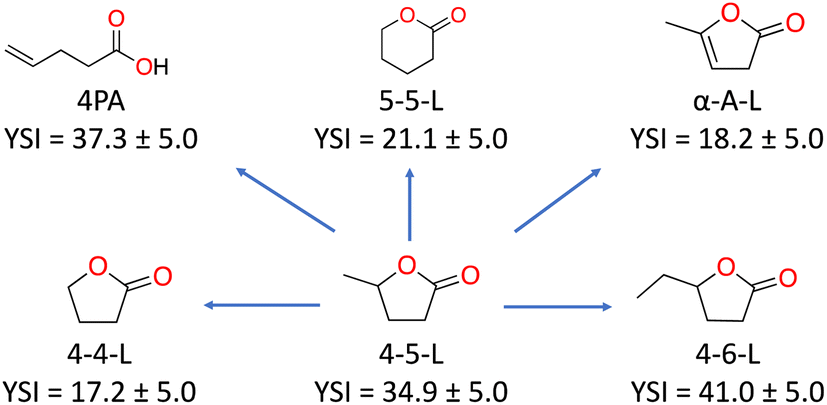 | ||
| Fig. 4 The effects of small structural changes on the YSI of 4-5-L. | ||
Overall, these results show that while all lactones have lower sooting tendencies than conventional fuels, the specific choice of lactone has a large impact on the soot reduction benefit. Of particular significance, 4-5-L is the lactone that has been discussed most as a fuel, but its isomer 5-5-L is a better choice from the viewpoint of reducing soot emissions.
As discussed in Section 1, addition of carbon to the side-chain is a potential strategy for forming diesel-range lactones. Fig. 5 plots the YSIs for 4-x-L lactones as a function of the number of carbons in the side chain (=x − 4). The YSI jumps significantly from no side chain (4-4-L) to a methyl side chain (4-5-L). As the side chain grows longer, the YSI continues to increase, but by a smaller increment. The linear fit shows that this increment is about 3.7 YSI units per carbon atom.
 | ||
| Fig. 5 The YSIs of 4-x-L lactones as a function of the number of carbons in the side chain. | ||
Fig. 6 compares the YSIs measured for the lactones to predictions with a group contribution method (GCM).18 The GCM decomposes the target molecule into a series of carbon-centered groups, where each group is defined by all the atoms directly bonded to the central carbon, the order of the bonds, and whether the carbon atom is part of a ring. Then the YSI is estimated from:
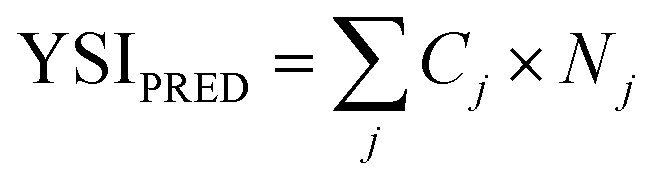 | (3) |
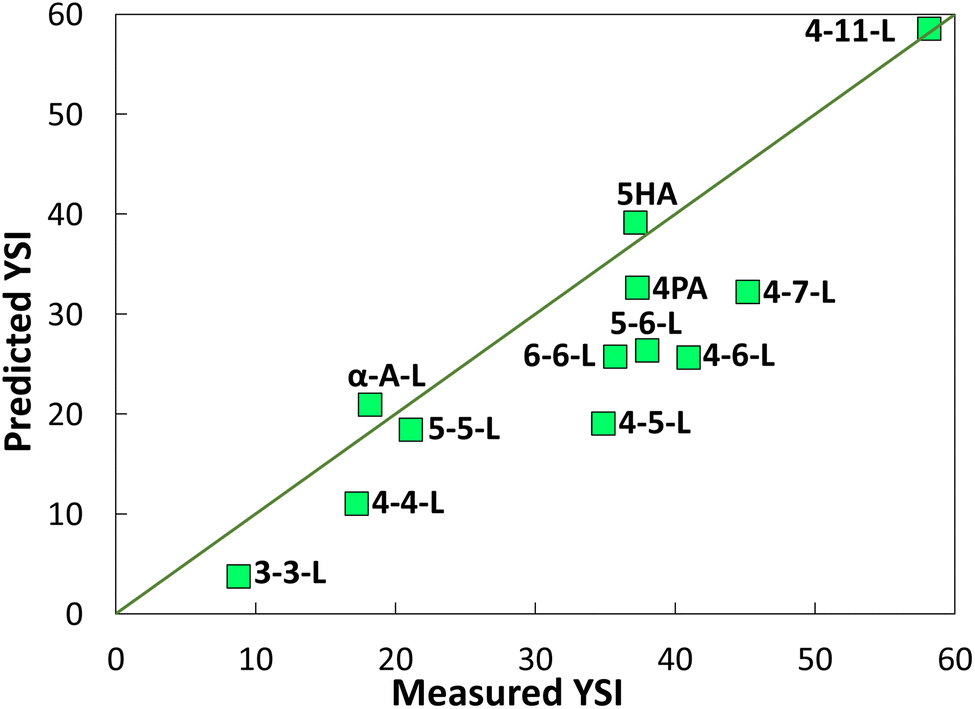 | ||
| Fig. 6 YSIs predicted with a group contribution method (GCM)18vs. measured YSIs. The line represents y = x. See SI J† for further details and the raw data. | ||
Most of the data points in Fig. 6 differ significantly from the y = x line, which shows that the YSIs are not simple functions of the atoms that are present—instead, the YSIs depend on kinetic pathways that are affected by non-nearest-neighbor interactions. Furthermore, most of the data points are below the line, which means the measured YSIs are larger than the predicted YSIs. This observation indicates that the sooting tendencies of the lactones are relatively high for compounds with two oxygen atoms.
3.2. ReaxFF simulation results
ReaxFF MD simulations were performed to understand the decomposition of the lactones in pure pyrolysis at high temperatures. These conditions are a good approximation to the soot formation region in the nonpremixed YSI flames.24,25 A key benefit of MD simulations is that they do not require a pre-existing chemical kinetic mechanism. This capability is critical since mechanisms are currently available for only one lactone, 4-5-L.39 In this study we simulated three lactones to examine the effects of ring size (4-4-Lvs.5-5-L) and the consequence of a side chain (4-5-Lvs.4-4-L).Fig. 7 presents the results for the three lactones: 4-4-L (top panel), 5-5-L (middle), and 4-5-L (bottom). Each panel shows the number of molecules of the fuel and of the six highest concentration products as a function of time during the simulation. These products account for over 98% of the oxygen and 65% of the carbon. In all three cases, the reactant lactone (grey lines) is consumed over a timescale of 0.5 to 3 ns. The major products are alkenes (butenes, C4H8; propene, C3H6; and ethylene, C2H4) and oxygen-containing species (CO2; CO; and formaldehyde, CH2O).
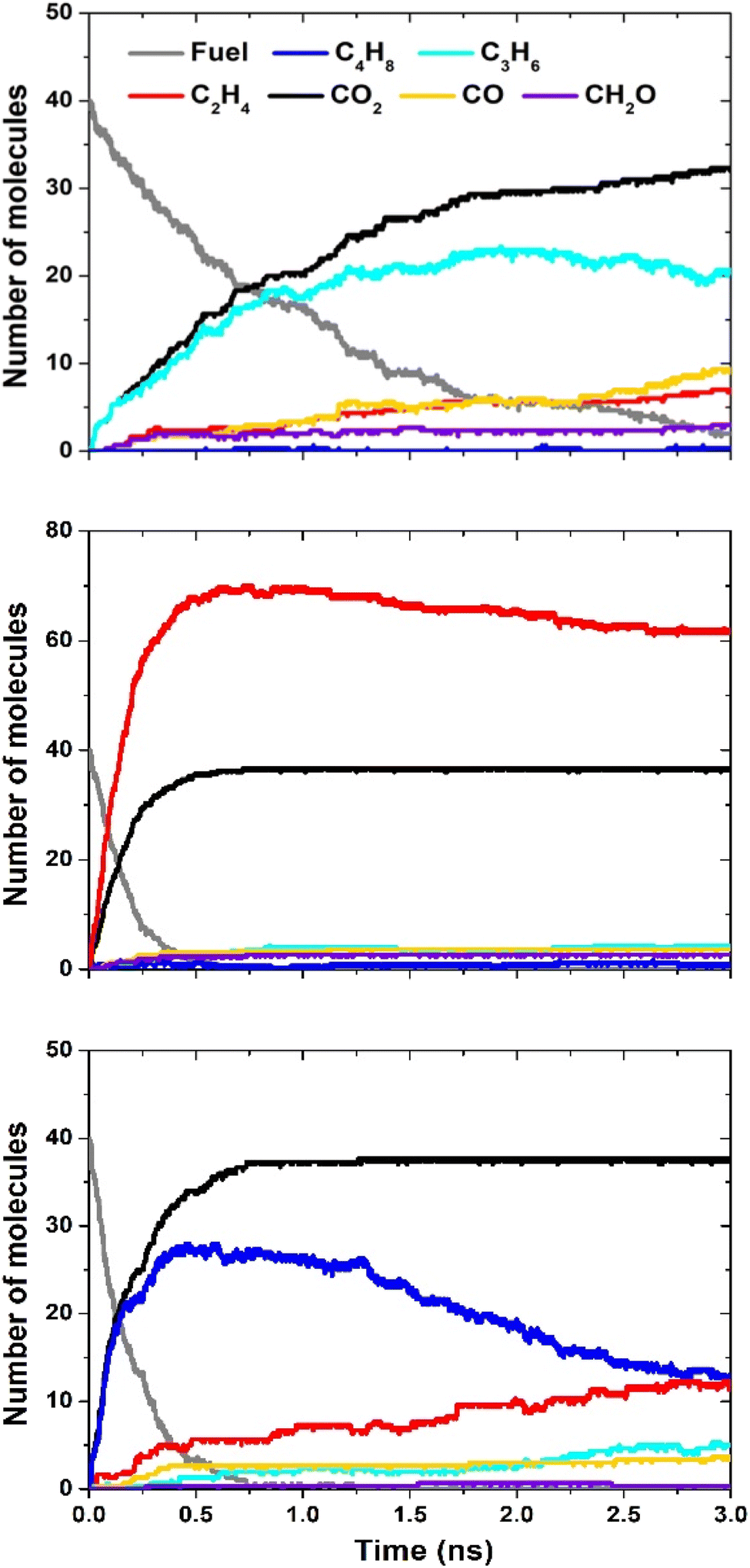 | ||
| Fig. 7 ReaxFF simulation results for 4-4-L (top), 5-5-L (middle), and 4-5-L (bottom). The legend in the top panel applies to all three panels. | ||
Significantly, CO2 (black lines) is the main oxygen-containing product for all three lactones, and its level is close to the maximum possible (40 molecules). This result explains the observation in Fig. 6 that lactones have large sooting tendencies relative to other di-oxygenated hydrocarbons. In general, the decomposition reactions of lactones can either produce CO2 and a regular hydrocarbon (R1), or two oxygenated hydrocarbons (R2):
| Lactone → CO2 + CaHb | (R1) |
| Lactone → CcHdO + CeHfO | (R2) |
If (R1) occurs, the CO2 is inert and will not lead to soot, but the other product CaHb can grow to larger hydrocarbons and eventually soot. If (R2) occurs, the oxygen atoms in the products CcHdO and CeHfO interferes with either of them growing to soot. Thus, while (R1) will reduce soot formation compared to a regular hydrocarbon fuel, (R2) uses the oxygen atoms more efficiently and will reduce soot formation even more. The ReaxFF results in Fig. 7 show that (R1) dominates for a range of lactones.
The distribution of alkenes varies among the lactones, with 4-4-L producing mostly propene (light blue lines), 5-5-L producing mostly ethylene (red lines), and 4-5-L producing mostly butenes (dark blue lines). These trends may explain some of the differences observed between the lactone YSIs. For example, molecules that decompose primarily to ethylene (e.g., n-alkanes) usually have lower sooting tendencies than molecules that decompose to larger alkenes (e.g., branched alkanes),18 so the preferential formation of ethylene from 5-5-L could explain why it has a much lower YSI than 4-5-L.
3.3. Quantum chemistry results
Quantum chemistry calculations were performed to determine the effects of different lactone structures on abstraction reaction pathways. Previous studies are limited to 4-5-L13,39–41 and 4-4-L.42Fig. 8 shows some of the results for five lactones, including three cases with different ring sizes (4-4-L, 5-5-L, 6-6-L; left column) and two of their methyl-substituted analogues (4-5-L, 5-6-L; middle column). In each case, the initial reaction considered is abstraction of a hydrogen atom from the lactone by a H radical produced elsewhere in the flame. The products are H2 and various lactone radicals (designated 44a, 44b, 55a, etc.), depending on which hydrogen atom is abstracted. The lactone radicals then undergo ring-opening unimolecular reactions to various linear intermediates (designated A, B, C, etc.). | ||
| Fig. 8 (Panel A) hydrogen atom-initiated abstraction pathways that lead to ring opening reactions of 4-4-L, 5-5-L, and 6-6-L, and their methyl-substituted analogues. The numbers in parentheses are the relative free energies in kcal mol−1. The numbers in red are activation barriers in kcal mol−1. The numbers in blue are the experimentally measured YSIs. (Panel B) overall reverse pathways from molecule C, the common intermediate of lactones 4-5-L and 5-5-L, and from molecule E, the common intermediate of 5-6-L and 6-6-L. The numbers in parenthesis are free energies in kcal mol−1 relative to the common intermediate. | ||
The red numbers in Fig. 8 are the barriers to the ring-opening reactions. They vary significantly depending on the structure of the initial lactone. For the unsubstituted lactones (left column), the barriers systematically decrease as the ring becomes larger, from 30.1 and 30.5 kcal mol−1 for 4-4-L to 19.0 and 25.2 kcal mol−1 for 6-6-L. There is also a change in the order of the ring-opening channels. For 4-4-L the lowest barrier leads to the carbonyl intermediate B, but for 5-5-L and especially 6-6-L the lowest barriers lead to the carboxylic acid intermediates C and E. In the methyl-substituted lactones (middle column), the magnitude of the barrier decreases only very slightly as the ring becomes larger. However, the range of barriers narrows from 4.5 kcal mol−1 for 4-5-L to 1.1 kcal mol−1 for 5-6-L.
In terms of sooting tendency, the most important observation is that the ring-opening pathways to carboxylic acid intermediates (A, C, E, and I) are important for all the lactones. These intermediates are likely to rapidly dissociate to CO2 + a hydrocarbon. Thus, CO2 is a major oxygenated product for abstraction-dominated regime as well as the pure pyrolysis conditions of the ReaxFF MD simulations. This observation suggests that the lactones are likely to have high sooting tendencies relative to the atoms they contain under a wide range of conditions.
4. Conclusions
The experiments and simulations in this study lead to the following conclusions:• The measured sooting tendencies of lactones are less than conventional fuels, even when accounting for their lower heats of formation, so using lactones as fuels will likely reduce particulate emissions from combustion devices.
• The measured sooting tendencies depend strongly on the molecular structure of the lactone, so careful selection of lactones is necessary to maximize the emissions benefits.
• The measured sooting tendencies are mostly larger than the values predicted by a structure–property relationship based on contributions from carbon-centered groups, which indicates that lactones are sootier than other species with two oxygen atoms, and that non-nearest-neighbor kinetic interactions are important.
• The molecular dynamics simulations suggest that the decomposition of lactones in pure pyrolysis directly produces CO2. This pathway uses the two oxygen atoms inefficiently to sequester only one carbon atom, and potentially explains the high sooting tendencies of lactones relative to other species with two oxygen atoms.
• The quantum chemistry calculations indicate that the reactions of lactones with H atoms are also likely to directly produce CO2, so this pathway may be a common feature of lactone combustion chemistry under both nonpremixed and premixed conditions.
• The kinetic mechanisms of the lactones affect their sooting behavior, so further kinetic studies are warranted, especially for a wider range of lactones than just γ-valerolactone.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization: SK, CSM, YX. Investigation: all authors. Writing—original draft: ZX, CJH, GJ, CSM, RPS. Writing—review & editing: all authors. Project administration: SK, LDP, YX.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The work at Yale was supported by the U.S. National Science Foundation (NSF) award CBET 2210894. VCW was supported by a First-Year Summer Research Fellowship from Yale College. Muhammad Oruc and Zhengrong Qian contributed to the experiments. The work at CSU was supported by the Colorado State University startup funds for the PI (Seonah Kim) and the computer time was provided by the NSF Extreme Science and Engineering Discovery Environment (XSEDE, now ACCESS) award TG-CHE210034.References
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- C. B. Field, M. J. Behrenfeld, J. T. Randerson and P. Falkowski, Science, 1998, 281, 237–240 CrossRef CAS PubMed.
- Chemspider, https://www.chemspider.com/, accessed December 2023.
- F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6, 4497–4559 RSC.
- S. Gupta, R. Arora, N. Sinha, M. I. Alam and M. A. Haider, RSC Adv., 2016, 6, 12932–12942 RSC.
- R. Xu, K. Liu, H. Du, H. Liu, X. Cao, X. Zhao, G. Qu, X. Li, B. Li and C. Si, ChemSusChem, 2020, 13, 6461–6476 CrossRef CAS.
- I. T. Horváth, H. Mehdi, V. Fábos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238–242 RSC.
- Á. Bereczky, K. Lukács, M. Farkas and S. Dóbé, Nat. Resour., 2014, 5, 177–191 Search PubMed.
- J. Frost, P. Hellier and N. Ladommatos, Energy Convers. Manage., 2023, 283, 116886 CrossRef CAS.
- G. Li, R. Wang, J. Pang, A. Wang, N. Li and T. Zhang, Chem. Rev., 2024, 124, 2889–2954 CrossRef CAS.
- R. Silva, E. Coelho, T. Q. Aguiar and L. Domingues, Appl. Sci., 2021, 11, 8500 CrossRef CAS.
- C. K. Westbrook, W. J. Pitz and H. J. Curran, J. Phys. Chem. A, 2006, 110, 6912–6922 CrossRef CAS.
- R. De Bruycker, H. H. Carstensen, M. F. Reyniers, G. B. Marin, J. M. Simmie and K. M. Van Geem, Combust. Flame, 2016, 164, 183–200 CrossRef CAS.
- C. S. McEnally and L. D. Pfefferle, Combust. Flame, 2007, 148, 210–222 CrossRef CAS.
- C. S. McEnally, Y. Xuan, P. C. S. John, D. D. Das, A. Jain, S. Kim, T. A. Kwan, L. K. Tan, J. Zhu and L. D. Pfefferle, Proc. Combust. Inst., 2019, 37, 961–968 CrossRef CAS.
- J. Zhu, J. V. Alegre-Requena, P. Cherry, D. Curtis, B. G. Harvey, M. A. Jabed, S. Kim, C. S. McEnally, L. D. Pfefferle and J.-D. Woodroffe, Proc. Combust. Inst., 2023, 39, 877–887 CrossRef.
- D. L. Bartholet, M. A. Arellano-Treviño, F. L. Chan, S. Lucas, J. Zhu, P. C. S. John, T. L. Alleman, C. S. McEnally, L. D. Pfefferle, D. A. Ruddy, B. Windom, T. D. Foust and K. F. Reardon, Fuel, 2021, 295, 120509 CrossRef CAS.
- D. D. Das, P. C. S. John, C. S. McEnally, S. Kim and L. D. Pfefferle, Combust. Flame, 2018, 190, 349–364 CrossRef CAS.
- C. S. McEnally, D. D. Das and L. D. Pfefferle, Yield Sooting Index database volume 2: sooting tendencies of a wide range of fuel compounds on a unified scale, Harvard Dataverse, accessed July 2024, DOI:10.7910/DVN/7HGFT8.
- A. C. T. Van Duin, S. Dasgupta, F. Lorant and W. A. Goddard, J. Phys. Chem. A, 2001, 105, 9396–9409 CrossRef CAS.
- K. Chenoweth, A. C. T. Van Duin and W. A. Goddard, J. Phys. Chem. A, 2008, 112, 1040–1053 CrossRef CAS.
- T. P. Senftle, S. Hong, M. M. Islam, S. B. Kylasa, Y. Zheng, Y. K. Shin, C. Junkermeier, R. Engel-Herbert, M. J. Janik, H. M. Aktulga, T. Verstraelen, A. Grama and A. C. T. van Duin, npj Comput. Mater., 2016, 2, 1–14 CrossRef.
- C. Ashraf and A. C. T. Van Duin, J. Phys. Chem. A, 2017, 121, 1051–1068 CrossRef CAS.
- H. Kwon and Y. Xuan, Fuel, 2021, 306, 121616 CrossRef CAS.
- H. Kwon, A. Lele, J. Zhu, C. S. McEnally, L. D. Pfefferle, Y. Xuan and A. C. T. van Duin, Fuel, 2020, 279, 118548 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (Revision C.01), Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- L. A. Curtiss, P. C. Redfern and K. Raghavachari, J. Chem. Phys., 2007, 126, 084108 CrossRef PubMed.
- J. M. Simmie and K. P. Somers, J. Phys. Chem. A, 2015, 119, 7235–7246 CrossRef CAS PubMed.
- G. Luchini, J. V. Alegre-Requena, I. Funes-Ardoiz and R. S. Paton, F1000Research, 2020, 9, 291 Search PubMed.
- H. Kwon, S. Lapointe, K. Zhang, S. W. Wagnon, W. J. Pitz, J. Zhu, C. S. McEnally, L. D. Pfefferle and Y. Xuan, Fuel, 2020, 276, 118059 CrossRef CAS.
- J. Gau, D. Das, C. McEnally, D. Giassi, N. Kempema and M. Long, Yale Coflow Burner Information and CAD Drawings, Figshare, accessed July 2024, DOI:10.6084/m9.figshare.5005007.v1.
- B. Franzelli, M. Roussillo, P. Scoflaire, J. Bonnety, R. Jalain, T. Dormieux, S. Candel and G. Legros, Proc. Combust. Inst., 2019, 37, 1355–1363 CrossRef CAS.
- D. Mathieu and R. Bouteloup, Ind. Eng. Chem. Res., 2016, 55, 12970–12980 CrossRef CAS.
- C. Ashraf, S. Shabnam, A. Jain, Y. Xuan and A. C. T. van Duin, Fuel, 2019, 235, 194–207 CrossRef CAS.
- H. Kwon, B. D. Etz, M. J. Montgomery, R. Messerly, S. Shabnam, S. Vyas, A. C. T. van Duin, C. S. McEnally, L. D. Pfefferle, S. Kim and Y. Xuan, J. Phys. Chem. A, 2020, 124, 4290–4304 CrossRef CAS.
- N. A. Huq, G. R. Hafenstine, X. Huo, H. Nguyen, S. M. Tifft, D. R. Conklin, D. Stück, J. Stunkel, Z. Yang, J. S. Heyne, M. R. Wiatrowski, Y. Zhang, L. Tao, J. Zhu, C. S. McEnally, E. D. Christensen, C. Hays, K. M. Van Allsburg, K. A. Unicic, H. M. Meyer III, Z. Abdullah and D. R. Vardon, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2023008118 CrossRef CAS.
- C. J. Mueller, W. J. Cannella, J. T. Bays, T. J. Bruno, K. DeFabio, H. D. Dettman, R. M. Gieleciak, M. L. Huber, C.-B. Kweon, S. M. McConnell, W. J. Pitz and M. A. Ratcliff, Energy Fuels, 2016, 30, 1445–1461 CrossRef CAS PubMed.
- K. Schmidt-Rohr, J. Chem. Educ., 2015, 92, 2094–2099 CrossRef CAS.
- A. Sudholt, R. Tripathi, D. Mayer, P.-A. Glaude, F. Battin-Leclerc and H. Pitsch, Proc. Combust. Inst., 2017, 36, 577–585 CrossRef CAS.
- L. Ye, W. Li and F. Qi, RSC Adv., 2018, 8, 12975–12983 RSC.
- D. Liu and A. Farooq, Combust. Flame, 2023, 253, 112771 CrossRef CAS.
- Z.-H. Li, W.-N. Wang, K.-N. Fan, M. W. Wong, H.-H. Huang and W. Huang, Chem. Phys. Lett., 1999, 305, 474–482 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se00883a |
| ‡ Permanent address: Newark Academy, Livingston, NJ 07039, USA. |
| § Permanent address: Choate Rosemary Hall, Wallingford, CT 06492, USA. |
| This journal is © The Royal Society of Chemistry 2024 |