
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular gels – a panorama of low-molecular-weight gelators from ancient origins to next-generation technologies
David K.
Smith

Department of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: david.smith@york.ac.uk
First published on 11th December 2023
Abstract
Supramolecular gels, self-assembled from low-molecular-weight gelators (LMWGs), have a long history and a bright future. This review provides an overview of these materials, from their use in lubrication and personal care in the ancient world, through to next-generation technologies. In academic terms, colloid scientists in the 19th and early 20th centuries first understood such gels as being physically assembled as a result of weak interactions, combining a solid-like network having a degree of crystalline order with a highly mobile liquid-like phase. During the 20th century, industrial scientists began using these materials in new applications in the polymer, oil and food industries. The advent of supramolecular chemistry in the late 20th century, with its focus on non-covalent interactions and controlled self-assembly, saw the horizons for these materials shifted significantly beyond their historic rheological applications, expanding their potential. The ability to tune the LMWG chemical structure, manipulate hierarchical assembly, develop multi-component systems, and introduce new types of responsive and interactive behaviour, has been transformative. Furthermore, the dynamics of these materials are increasingly understood, creating metastable gels and transiently-fueled systems. New approaches to shaping and patterning gels are providing a unique opportunity for more sophisticated uses. These supramolecular advances are increasingly underpinning and informing next-generation applications – from drug delivery and regenerative medicine to environmental remediation and sustainable energy. In summary, this article presents a panorama over the field of supramolecular gels, emphasising how both academic and industrial scientists are building on the past, and engaging new fundamental insights and innovative concepts to open up exciting horizons for their future use.
 David K. Smith | David Smith is Professor of Chemistry at University of York where he carries out research into self-assembling molecular materials. He was educated at the University of Oxford (BA 1993, DPhil 1996) and carried out postdoctoral research at ETH Zurich (1997–1998), before being appointed Lecturer in York in 1999, and promoted to a Professorship in 2006. In recognition of his work on supramolecular gels, he was awarded the 2022 Royal Society of Chemistry ‘Tilden Prize’. He also received the 2022 Society of Chemical Industry's ‘Science for Society Award’ in honour of his work in chemical education and outreach. Dave has written and lectured widely on wide-ranging aspects of inclusion and diversity in modern science, and was recognized by Chemical and Engineering News as an ‘LGBT+ Trailblazer’. After the death of his husband, Dave has worked part-time and is a single parent to their son. His family's love of food and cooking led him to write the award-winning cookbook/memoir, ‘Tw-Eat Together’. |
1. Introduction
Supramolecular gels based on low-molecular-weight gelators (LMWGs) self-assemble through non-covalent interactions to form nanostructured ‘solid-like’ network architectures that immobilise ‘liquid-like’ solvents (Fig. 1).1 Gels are archetypal nanomaterials, in which ‘information’ is translated from the molecular scale, via nanoscale assembly, up to the macroscopic scale, directly impacting on physical properties. In recent years, supramolecular chemists have found such gels to be a fascinating playground to test concepts of dynamic assembly, and create systems with exciting potential applications.2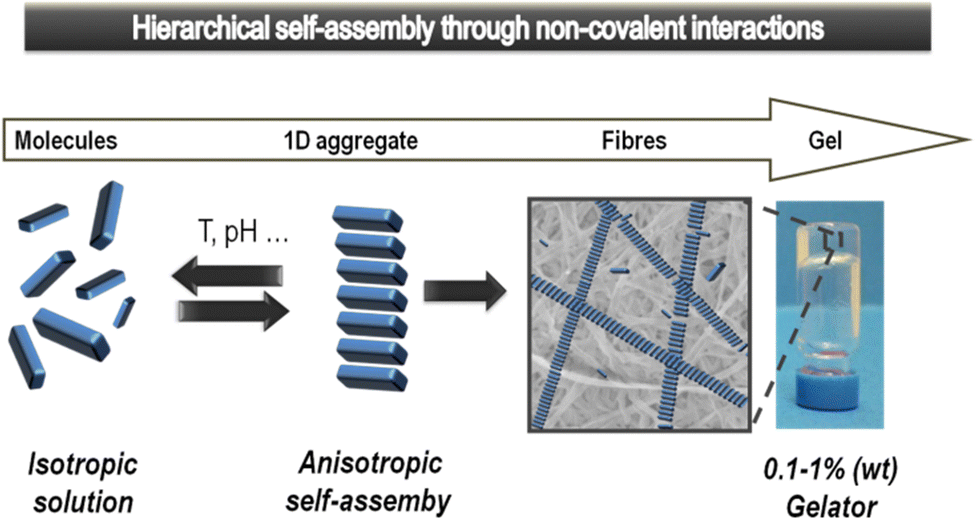 | ||
| Fig. 1 Schematic of low-molecular-weight gelator (LMWG) assembly from an isotropic solution to anisotropic self-assembled objects that hierarchically assemble further to ultimately form a gel-phase material. Reproduced from ref. 5 with permission from the Royal Society of Chemistry, © 2014. | ||
Gels surround us in everyday life, but many of them are based on polymers. Indeed, polymer-based gels far outnumber supramolecular gels.3 Polymers are well-placed to form extended networks because they have relatively large molecular sizes – they can then form extended networks either via non-covalent interactions between polymer chains (‘physical’ gels) or covalent crosslinking (‘chemical’ gels). It is worth noting that an outstanding review from Eelkema and Pich explored the pros and cons of polymer and low-molecular-weight gelators,4 but here we focus our attention specifically on LMWGs.
The initial assembly step in gel formation is often referred to as ‘supramolecular polymer’ formation.6 However, not all supramolecular polymers go on to assemble into a gel. Strictly, supramolecular gels are a subset of supramolecular polymers, in which the self-assembled fibrils can interact with one another to hierarchically form bundles, fibres and an extended sample-spanning network. It is also worth noting that in recent years, the term ‘supramolecular polymer’ has been further extended, and is increasingly used to refer to any kind of polymeric system held together by non-covalent interactions.7 Indeed, the breadth of use of the ‘supramolecular polymer’ terminology can makes effective interrogation of the gel literature difficult. For example, within the field sometimes described as ‘supramolecular polymer gels’ are polymers that have been covalently-modified with self-assembling units capable of forming non-covalent interactions that ultimately lead to polymer crosslinking.8 The responsive nature of the non-covalent interactions means they share some of the dynamic characteristics of gels based on LMWGs, and there is much to learn from their study, however, in order to clearly define the scope of this review, we focus here on gels that primarily contain assembled low-molecular-weight systems.
Although supramolecular chemistry is usually considered to have begun with the field-defining work of Pedersen, Lehn and Cram from the 1960s onwards,9 in reality, ‘supramolecular’ gels based on the self-assembly of LMWGs have much older origins.10 This review aims to tell the full history of such gels – from their ancient origins, all the way through to the ways in which supramolecular chemists are developing new concepts that will transform 21st century technologies.
In Section 2, I address the historical development of supramolecular gels from ancient times, and the way early colloid chemists began to develop understanding of gels, enabling industry to build on their insights. These early applications made use of the physical rheological properties of gels as soft solids. In Section 3, I reflect on how supramolecular chemists entered the field and introduced new fundamental concepts, propelling the field forwards. This includes understanding structural effects on assembly, manipulating dynamics and responsiveness, developing multi-component systems, and shaping/patterning the resulting gels. Finally, in Section 4, we see how these new concepts are allowing gels to approach far more sophisticated applications, in which they extend beyond their simple rheological properties and begin to show added features as a result of the precise details of their chemical programming.
It is hoped this review will allow academic researchers to see a wider scope of supramolecular gels than they are perhaps aware of, while giving industrial researchers an insight into the ways academic chemists have been expanding their potential. It is also hoped, that by placing supramolecular gels into a broader context, a wide range of chemists will come to appreciate the development of the field, and gain insight into how gels have influenced scientific progress, and the world around us. There is currently significant interest in interrogating the way supramolecular chemistry influences real-world applications.11 Furthermore, a broad panorama of a field is very rare, and it is hoped new researchers in the field will find it beneficial. This review does not intend to be comprehensive, the study of LMWGs and their applications is now a vast enterprise. Instead, key examples, principles, and personal favourites have been selected, that exemplify how the field of supramolecular gels has developed into the vibrant area it is today.
2. Early applications and insights
2.1 Gels in lubrication – from ancient origins to a modern industry
Archaeologists divide the Ancient World into epochs based on the technologies used – Stone Age, Bronze Age, Iron Age, etc. Some would say we are now living in the Polymer Age,12 and perhaps beginning to think about how we transition beyond it towards a greener and more sustainable future. However, these ‘ages’ all refer to ‘hard’ materials – the things that are left behind and survive, meaning they can be excavated and investigated millennia later. Perhaps surprisingly, we rarely think about the soft, plant- and animal-derived materials that also underpinned society and were a key part of technology – the resins, reeds, stems, oils, fibres, and indeed, gels.Knowing when a technology first emerged is always difficult to establish – often we rely on the first written records, or the places most extensively studied by archaeologists, and this can bias understanding. For supramolecular gels, one of their earliest recorded practical uses is associated with the development of the wheel. The wheel was invented in ca. 4000 BCE, but gaining speed from early wheels was a significant challenge, and for this reason, lubricating greases were developed. This invention is sometimes attributed to the Hittites, who were renowned for their fast-moving battle chariots that carried archers into battle against their enemies, such as those in Ancient Egypt.13 Ultimately, the Egyptians started using lubricant technology, and some high quality archaeological remains have been extensively investigated. Chariots from Tutankhamun's tomb (ca. 1300 BCE) had traces of grease on their axles, and the lubricants used were what we would think of today as ‘supramolecular gels’ (Fig. 2, top).14 Organogels (gels formed in organic media) are ideal for lubricating grease, because the solid-like nature means the grease remains in place when applied to the axle, while the ‘liquid-like’ characteristics allow movement and prevent wear of the moving parts, especially at high speeds.
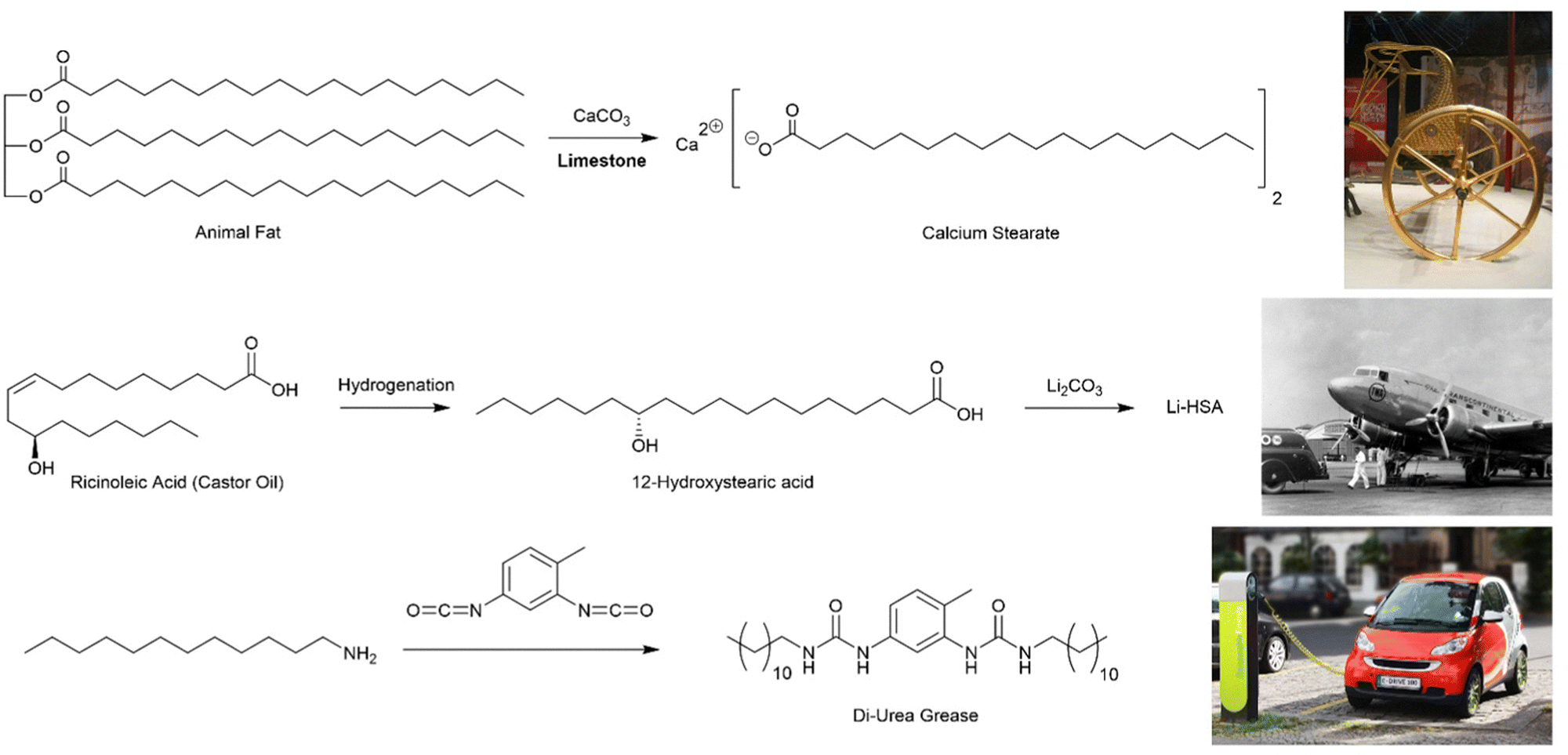 | ||
| Fig. 2 Chemical structures of lubricating greases. (top) Synthesis of calcium stearate, used as a lubricant since ancient times, from animal fat and limestone, alongside a photograph of King Tutankhamun's chariot. (centre) Synthesis of 12-hydroxstearic acid and its lithium salt from ricinoleic acid, extracted from castor oil, alongside a photograph of an early aeroplane that benefitted from this type of lubricant. (bottom) Synthesis of a di-urea grease from dodecylamine and toluene di-isocyanate, alongside a photograph of an electric car, for which this class of lubricant is well suited. Images reproduced with creative commons licences: (top) reproduced from ref. 16 with CC0 Public Domain Image. (centre) Reproduced from ref. 17 with CC BY-SA 3.0 licence, image cropped from family photo © Wikimedia user: postdlf (bottom) Reproduced from ref. 18 with CC BY-SA 3.0 licence, © M. Movchin & F. Müller. | ||
These ancient lubricating greases were gels based on calcium stearate (Fig. 2, top), created by mixing animal fat with limestone (CaCO3). Through modern eyes, it is clear that basic limestone can saponify the triglyceride esters in the fat to yield long chain carboxylic acids in the form of their calcium salts. The calcium stearate was suspended in an oily phase (essentially an organic solvent), and it will therefore self-assemble into nanostructured fibres. Specifically, the polar head groups become hidden in the interior of the self-assembled architecture, avoiding contact with the apolar ‘solvent’, while the long alkyl chains extend into the surrounding organic phase. In this way, calcium stearate assembles into an extended solid-like network that is stable in the surrounding liquid-like phase and forms a gel. The field of supramolecular gels, at least in terms of recorded history, was born.
In 1845, a British patent was filed for a lubricating grease.15 The formulation was also based on animal fat and limestone, combined with a mineral oil. Clearly, this was no great advance over the technologies known from ancient times, and stands as an example of the codification of ancient knowledge. This was the height of the Industrial Revolution. Greases and lubricants played a vital role in enabling the efficient operation of machinery. Controlling patent rights was one way in which Britain could maintain its economic (and military) advantage, alongside the ability to exploit resources and access cheap labour, provided by its global empire.
For a steam-powered revolution, ‘calcium grease’ had some disadvantages – most significantly, it did not have great stability ≥100 °C, and would regularly need replacing. In the language of modern gel chemistry, we would say that its Tgel value (its gel–sol transition temperature) was too low. Therefore, although calcium grease was effective for chariots and waterwheels, something better was needed for high-energy steam engines. One simple strategy was to change the counter-cation from calcium, and as a result, sodium grease, based on beef tallow and sodium carbonate, was developed.19 This had greater thermal stability (a higher Tgel value) and could operate at much higher temperatures. However, it suffered from higher water solubility, which in a steam-powered engine is problematic.
In the later 1800s, there was a move away from using animal fat for grease formation, and instead use purified chemical products directly. This led to significantly greater understanding of the chemistry underpinning these gels, which allowed those working in the lubrication industry to take a chemistry-led approach to the discovery of new systems with advantageous properties.20
Perhaps the biggest step in the lubrication industry was made by the rapidly-developing aviation industry in the 1930s and 40s. Their engines required high-performance lubricants, and they thus developed gels based on 12-hydroxystearic acid (12-HSA, Fig. 2, centre). This compound, a naturally-derived product made from castor oil via the hydrogenation of ricinoleic acid, had been reported as an early gelator in its own right.21 However, it was as its lithium salt that the aviation industry developed it as an outstanding lubricant.22 Lithium 12-hydroxystearate went into production as a multi-purpose grease and remains the cornerstone of the industry.20
Recent studies have demonstrated that Li-12-HSA self-assembles in organic media to create nanofibers with the polar head groups on the interior, as would be expected.23 The alcohol groups part-way down the hydrocarbon chain interact with one another through hydrogen bonds to reinforce the alignment of the assembly. Even today, 12-HSA remains a gelator of intense interest and untapped potential.24
In the early 1950s, a new class of lubricant was developed, based not on fatty acids, but ureas – initially a range of mono-substituted ureas were preferred,25 but this was soon extended to di-ureas26 (and also polyureas)27 which are now widely used (Fig. 2, bottom). Such ureas assemble as a result of self-complementary hydrogen bond interactions between the urea motifs. The carbonyl oxygen can form two hydrogen bonds to the two N–H protons on an adjacent molecule, leading to intermolecular stacked assembly. These low-molecular-weight systems were the first fully-synthetic lubricants, and are easily modified by varying amine/isocyanate units in the synthesis, giving an ability to tune performance as required.28 Much of the further development of urea greases took place in Japan, where they are now the most commonly-used lubricants. They have high thermal stability and very good resistance to shear, which means they see particular use in high-performance applications. It has been suggested that they are particularly well-suited to the lubrication of electric-powered motors such as those in electric cars.29 In recent years, the bis-urea motif has become one of the most explored gelation motifs in academic science (see Section 3.1).
Overall, the history of lubrication demonstrates that LMWGs have much earlier origins than might have been expected. Furthermore, it demonstrates one of the principles that drives research in the field to this day, that structure–activity effects can be explored by making simple modifications to a known gelator and investigating the impact on performance. The idea that molecular structure can program self-assembly, and hence modify overall materials performance, lies at the heart of modern nanotechnology, yet is clearly a concept with much older origins than often acknowledged.
2.2 Gels in personal care – from ancient origins to the modern bathroom
The origins of LMWGs in personal care also date back to ancient times.30 Soaps are based on fatty acid metal salts, which, in concentrated form, create a type of hard gel. These soaps are directly related to the softer lubricating greases described in Section 2.1, where the stearate is mixed with larger amounts of a carrier oil. A Mesopotamian tablet dating to ca. 2200 BCE describes mixing potash (K2CO3) and oils to create soaps, and in Egyptian times, the Ebers Papyrus (ca. 1550 BCE) provides a more detailed record of soap production.31 Sweet-smelling perfumes would be mixed into these soaps which, in modern terms, can be considered as ‘fragrance delivery vehicles’.Recent archaeological studies have evidenced the use of gels in Ancient Egypt for hair styling.32 Detailed analysis has indicated the presence of fats added to the hair of a number of Egyptian ‘mummies’. Given the use of fatty acids as thickeners and soaps in this period, it seems likely that a similar approach was used to form a gel to provide the hair with styling, probably during mummification. A similar approach may also have been used during life. It is likely that many other cultures developed similar products, but are not so well documented. For example, Chinese culture has, since at least the Tang dynasty in ca. 700 AD, soaked elm bark in water to release a polysaccharide polymer gel for purposes of hair styling.33 In the modern era, one of the first hair-gel products was Brylcreem, released to market in 1928.34 Brylcreem is an emulsion of water and mineral oil, thickened to a gel using beeswax. Beeswax is a complex mixture of low-molecular weight compounds – notably long chain alkanes, acids, alcohols and esters,35 exactly the type of compounds known to assemble into supramolecular gels.
Later in the 20th century, the deodorant gel stick was developed, initially based on simple sodium stearate gels, directly related to ancient soap technology.36 The role of the gel is to act as a delivery vehicle for deodorant and antiperspirant agents, with gentle rubbing onto the skin leading to gel breakdown and delivery. In the years that followed, the industry used more advanced LMWGs to formulate these materials. For example, Procter and Gamble hold an extensive series of patents on 12-HSA based deodorant gels.37
Interestingly, alongside the development of 12-HSA as an organogelator in the late 19th century, another class of industrially-important LMWG originated, which also ultimately had an impact on the development of deodorants. In 1891, Meunier reported that when sorbitol was reacted with two equivalents of benzaldehyde, it formed ‘une gelée transparente’.38 The product was later shown to be 1,3:2,4-dibenzylidene sorbitol (DBS, Fig. 3). This molecule, sometimes referred to as a ‘butterfly surfactant’, self-assembles in organic media as a result of hydrogen bond interactions between sorbitol ‘bodies’ and π–π stacking (and solvophobic interactions) between aromatic ‘wings’. In later years, Gillette developed RightGuard (now owned by Henkel) based on a DBS gel stick.39 The acid-instability of DBS as a result of its acetal functionality, limited its use with acidic aluminium-based antiperspirants, and efforts were put into formulating additives into the gel to stabilise it.40 Interestingly, additives were also used to control the size of the DBS assemblies, as this was shown to decrease the visible residue left by the deodorant.41 There was therefore significant industrial focus on reducing the size of assembled structures to 2–200 nm by adding nucleation agents that would nucleate many more gel fibres, and prevent each of them from growing too large – a clear example of supramolecular control of aspect ratio. Fascinatingly, this type of dynamic control over gel assembly is a hot topic in modern academic gel chemistry (Section 3.7) – it is interesting to reflect that some principles are already well-understood in an industrial setting.
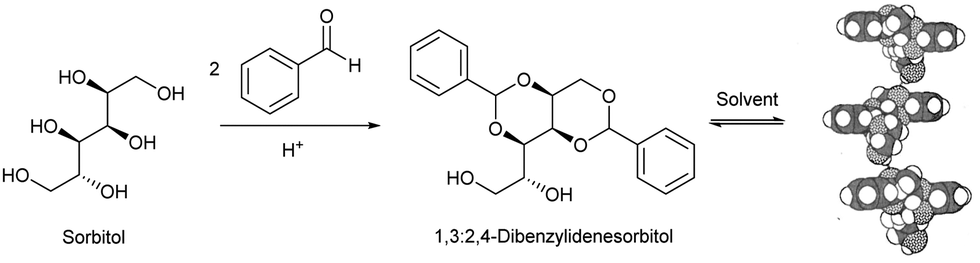 | ||
| Fig. 3 Synthesis of 1,3:2,4-dibenzylidenesorbitol (DBS), and qualitative model of self-assembly indicating intermolecular hydrogen bonds between sorbitol units, model reproduced from ref. 42 with permission from Chemical Society of Japan, © 1995. | ||
Related to personal care, stearates and DBS have also both been used in adhesives. The renowned ‘Pritt Stick’ is a stearate-based gel that releases adhesive components on rubbing.43 The Japanese competitor product uses DBS as a gelation agent.44 It was suggested in the patent that DBS enhanced performance in humid climates compared to stearic acids. As in the lubrication industry, markets were segmented between West and East based on different gelator chemistries.
2.3 Gels in the polymer industry
In the early 20th century, polymer chemistry rose to prominence. LMWGs have been used as additives in the polymer industry for some years. For example, during the melting of polyolefins, a 1,3:2,4-dibenzylidenesorbitol (DBS) additive increases clarity and reduces shrinkage in the moulded plastic on cooling.45 DBS assembles into a solid-like nanoscale ‘gel’ network in the molten polymer, which alters polymer crystallisation kinetics, hence improving transparency and rheologically-reinforcing the polymer, making for more effective moulding.In 20th century patent literature, a number of chemical modifications of DBS were reported, with pioneering work in this area from Milliken. As one example, incorporating alkyl groups onto the aromatic rings of DBS both optimises the clarifying effects of the additive on poly(propene) and overcomes problems with poor taste and smell, presumably by making the gelator more hydrophobic and less able to leach from the moulded polymer.46 This enabled the widespread use of this technology in plastic food packaging applications. From an academic perspective, this also demonstrates how small changes to a gelator structure can often be tolerated without destroying gelation ability. This is one of the main ways in which LMWG ‘chemical space’ has been expanded by supramolecular scientists (see Section 3.1).
Recently, LMWGs have been combined with polymers in dentistry. A self-assembled LMWG can assist with the UV-induced polymerisation of dental composites, increasing the rate of monomer conversion, improving the strength of the resulting polymer, and reducing shrinkage during polymerisation within the mouth.47 One 3M patent, lists many of the organogelators from recent academic literature as potential additives for the manufacture of dental composites.48
At the end of the 20th century, academic interest in combining LMWGs with polymers also blossomed, with researchers intrigued by the potential advantages of having a self-assembled LMWG skeleton within a polymer matrix.49 It was reported that this can have significant impact on the rheological performance,50 and furthermore, that the LMWG could be washed away to yield nano-imprinted materials.51 This demonstrates how industrial and academic chemistry can explore similar concepts, without influencing each other perhaps as much as they should. More recently, academics have used LMWGs to assemble higher-level functions into polymeric materials (see Sections 3 and 4).
2.4 Gels as weapons of war
Sadly, supramolecular gels have played a key role in developing weapons. Given the ability of gelators such as 12-HSA to form gels in oils, it was perhaps not surprising that attention should turn to the gelation of highly flammable fuel oils. In the 1940s, this led to the development of ‘napalm’, a sticky incendiary gel, first used by Allied forces during World War Two. The goal was to improve the ‘effectiveness’ of flammable liquids by allowing them to adhere to materials, making ‘flamethrower’ weapons significantly more destructive. Supramolecular gels undergo shear-thinning, and can be sprayed as liquids, whilst their thixotropy (see Section 3.9) gives some of them the ability to re-form a gel on contact with the target, making them ideal for this application.The original napalm formulation was published in 1946, and was revealed to be a combination of the aluminium salts of naphthenic and palmitic acids, (Fig. 4) hence the name ‘na’-‘palm’.53 This gel therefore follows the tried-and-tested formula of using a metal salt of an apolar carboxylic acid. Combining two different carboxylic acids optimised performance making this an early example of a two-component LMWG (see Section 3.4). In later years, the supramolecular gel formulation was replaced by a polymer gel.54 Although the composition had been changed, the name napalm was retained – it went on to become infamous, and much-feared in the Vietnam conflict.
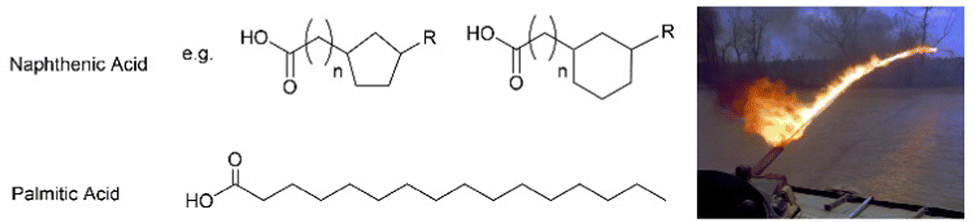 | ||
| Fig. 4 Chemical structures of naphthenic and palmitic acids, the original components of napalm, alongside a photograph of a napalm flamethrower being used during the Vietnam conflict. Image is public domain and reproduced from ref. 52 based on CC0 Creative Commons licencing. | ||
2.5 Gels in the oil industry
LMWGs that operate in fuel oils have also been of significant importance in the oil industry. For example, in oil pipelines, build-ups of gel-like deposits can occur, causing blockages.55 These are especially problematic when pipelines are at low temperatures. The gelation is caused by the self-assembly of long-chain alkanes into solid-like aggregates within the liquid-like short-chain alkane solvent. Long-chain alkanes crystallise into nano- and micro-crystalline 2D-platelets, and these platelets then yield an extended network that forms a gel (Fig. 5).56 As such, this is a relatively rare example of gelation through a 2D-platelet morphology, rather than 1D-nanofibers. Over the years, oil companies have developed additives and physical methods to break down these gels, and restore pipeline flow.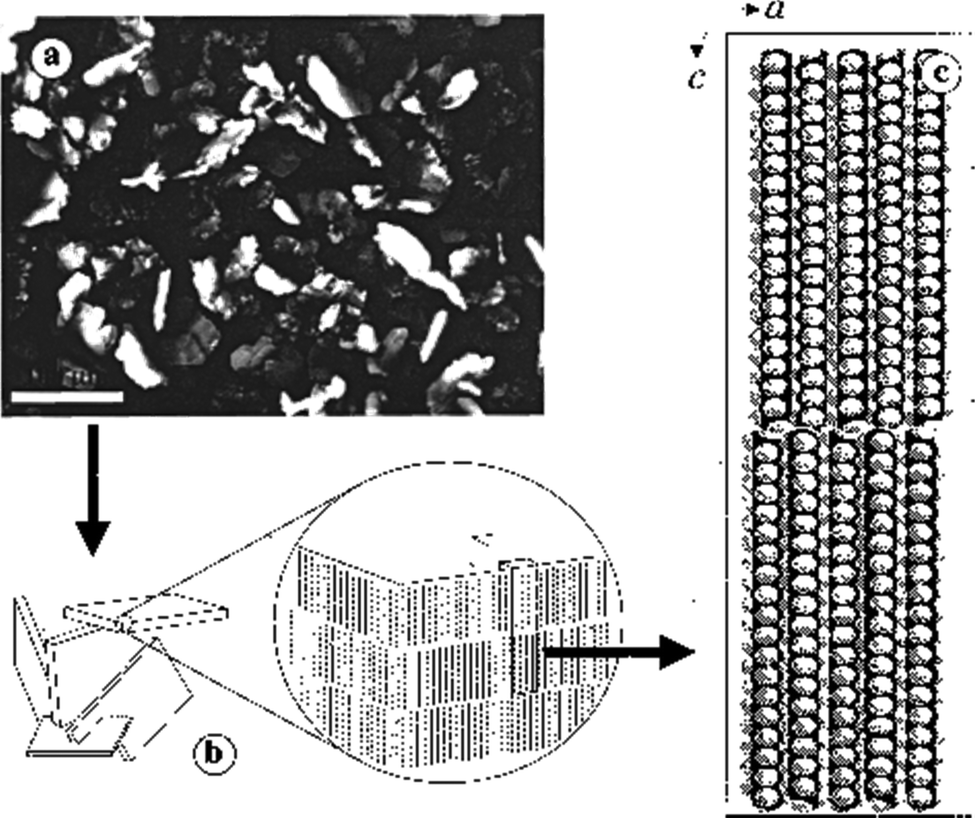 | ||
| Fig. 5 (a) Optical micrograph of a 4 wt% C36H74/1-octanol gel viewed through crossed polars (scale bar = 100 μm), (b) a cartoon representation of the micro-platelets showing the orientations of the long molecular axes of C36H74 molecules, and (c) the molecular packing of C36H74. Figure reproduced from ref. 56 with permission from the American Chemical Society, © 2000. | ||
In addition to gels causing problems, they can also offer opportunities to the oil industry. For example, fracking (hydraulic fracturing) to extract natural gas from shale deposits is facilitated by pumping solid-like materials down into a geological fracture, opening it up, and displacing the gas. Although polymer gels are most commonly used, low-molecular-weight organogels have been applied, in particular, aluminium salts of long chain fatty acids.57 Optimising such fluids to limit environmental damage is important, although ultimately (and hopefully), the move towards cleaner forms of energy (where supramolecular gels can also play a key role, see Section 4.6) will render this type of technology obsolete.
As well as extracting fossil fuel deposits, LMWGs have been explored for their potential to clean up problems caused by their uncontrolled release into the environment. Specifically, gels have been investigated for their ability to remediate oil spillages, by immobilising liquid crude oil, enabling its clean-up.58 The advantages of LMWGs in terms of their low-cost synthesis and environmental compatibility are significant. Research started as early as the 1970s, with a wide range of fatty acid metal salts and ureas being tested by the US Environmental Protection Agency.59 Their preferred formulation was a combination of amine and isocyanate that formed a urea by in situ covalent synthesis (similar to urea grease synthesis). However, it did not prove practically useful in the field. In the following years, a number of other LMWGs were tested, including an amide-derivative of glutamic acid (one of the earliest examples of an amino acid LMWG),60 and DBS.61
In 2001, this application of LMWG technology, which until that point had been largely ‘hidden’ in technical reports and patents, was introduced to an a academic audience, when Bhattacharya and Krishnan-Ghosh used an amide-modified alanine derivative to gel commercial fuels in biphasic oil–water mixtures.62 In the years since, this context has been returned to many times by different researchers, who have tested a wide range of LMWGs (e.g.Fig. 6).63
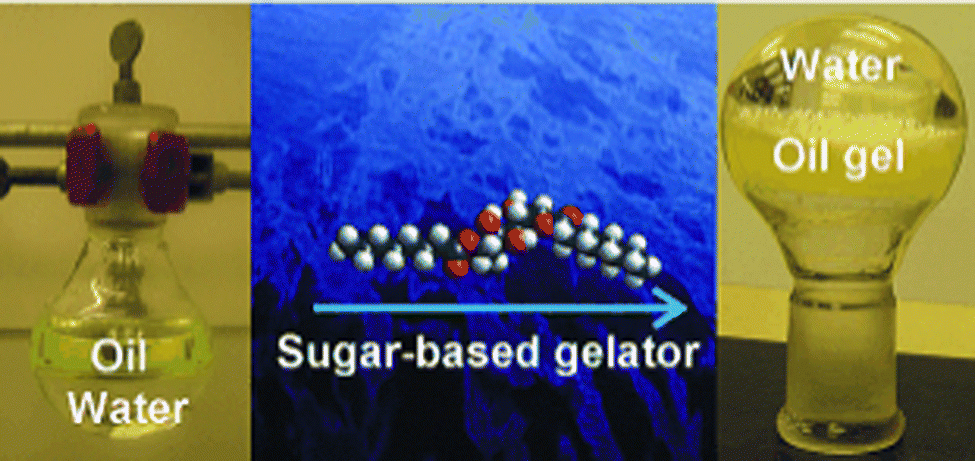 | ||
| Fig. 6 Gels for remediation of oil from a biphasic oil/water mixture – image reproduced from ref. 63 with permission from Wiley-VCH, © 2010. | ||
One of the challenges is to deliver the LMWG to the oil spillage, and achieve efficient gelation in ambient environmentally-relevant conditions. In an interesting patent, Weiss and co-workers employed 12-HSA, dissolving the LMWG in a water-miscible solvent and encapsulating it in a water-soluble bag.64 The active agent was released on environmental contact, with gelation then enabled by solvent mixing.
There is also interest in preventing oil loss from ruptured pipelines or drilling rigs. In elegant work,65 Raghavan and co-workers demonstrated DBS could be added to the organic medium in a model pipeline with a small amount of DMSO co-solvent. On pipeline breakage, the DMSO partitions into the surrounding water, lowering the polarity of the model oil, and causing the DBS to rapidly form a gel at the damage site, limiting further oil loss. This research builds elegantly on the enhanced understanding of solvent effects on LMWGs that emerged during the early 21st century (see Section 3.3).
2.6 Gels in the food industry
Hydrogels (gels formed in water) based on polymers have been used in culinary applications since ancient times. Gelatin from boiled animal bones (polypeptide),66 agar from seaweed (polysaccharide)67 and pectin from fruit (polysaccharide)68 all form physical gels assembled through non-covalent interactions between polymer chains, and assemble via a heat/cool cycle – ideal for culinary processing. Recently, calcium alginate formed when alginic acid (a polysaccharide) is added to calcium chloride solution,69 with the calcium ions crosslinking between carboxylic acids, has also been used in creating bubble-type food gels. However, all of these systems are polymer hydrogels – given they are well-established, it is not surprising that low-molecular-weight hydrogels have not been used in the food industry. In contrast, supramolecular organogels do have a significant history of use in thickening foods based on fats and oils, as described below.A key ingredient in food-based organogels is lecithin (Fig. 7) – a zwitterionic phospholipid originally isolated from egg yolk in 1846.70 More recently, soy-lecithin, extracted from soy beans, has become the dominant source of industrially-produced lecithin.71 Lecithin is well-known for its properties as an emulsifier (surfactant) helping stabilise combinations of oil and water. It can be fully metabolised, is safe for food use, and is found in a huge range of products, such as chocolate, where it stabilises fats, and prevents phase separation.72 Lecithin also provides thickening effects – consider for example mayonnaise, invented in the 18th century.73 Oil is mixed with a small amount of vinegar or lemon juice, and egg yolk is added to create a thickened water-in-oil emulsion via a gelation mechanism. The ability of eggs to act as a thickening (gelation) agent has probably been known since ancient times, even if not formally recognised.
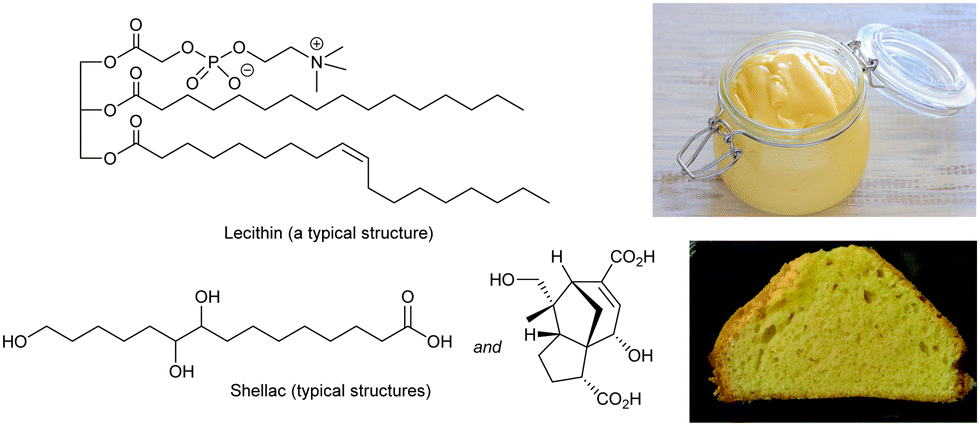 | ||
| Fig. 7 Typical chemical structures of lecithin (top) and shellac (bottom) with images of mayonnaise thickened with natural lecithin from egg yolk, and a cake baked with shellac-thickened oleogel emulsion as a replacement for margarine. Image of mayonnaise, from ref. 74 CC BY 2.0 licence, © jules:stonesoup (Flickr), image of cake reproduced from ref. 79 with permission from the Royal Society of Chemistry, © 2014. | ||
In the late 1980s, work from Luisi and co-workers formally described the ability of lecithin to act as an organogelator in organic solvents in the presence of small amounts of water.75 Combined with a rapidly growing interest in a healthy diet, this led food scientists to consider using lecithin, and other LMWGs, to create replacement fats. Polyunsaturated fats, such as those in olive oil, are healthier than saturated fats, such as those in dairy products, but the healthier fats are liquids, and lack the mouthfeel of solid saturated fats. This mouthfeel helps make saturated fats enjoyable to eat. The concept emerged that a gelated polyunsaturated fat (an ‘oleogel’) could potentially replicate the mouthfeel of a solid saturated fat, but with the health benefits of the polyunsaturated fat.76
Lecithin has a long history of culinary application,77 and has therefore been explored in oleogel formation. Alternatively, shellac is a food-safe insect byproduct that is a mixture of fatty acids with pendant alcohol groups (Fig. 7),78 similar to 12-hydroxystearic acid. It is widely used as a glazing agent, but has also been tested as an oleogelation agent and formulated into (e.g.) modified margarines, chocolates and cakes (Fig. 7).79 Shellac forms nano- or micro-crystalline platelets when it assembles, and there has been interest in whether the non-fibrillar nanoscale morphology gives rise to a different mouthfeel.80
An increasing range of LMWGs are being investigated in oleogels for the food industry.81 For example, a 12-HSA oleogel has been tested as a replacement for butter, including human trials which indicated a lowering of markers of cardiovascular disease.82 There remains a significant need for increased understanding of the impact of oleogels on flavour, texture and sensory properties. Furthermore, many potential oleogelators are not currently approved for food use, meaning further in vivo work is required. Recent attention has increasingly focussed on food-grade LMWGs such as phytosterols,83 or discovering natural extracts with oleogelation potential.
2.7 Gels in water – early low-molecular-weight hydrogels
As noted above, polymer gels in water, such as hydrogels based on gelatin and pectin, have a very long history of use, probably dating back to the Stone Age. Perhaps surprisingly, given the importance of water to life on Earth, LMWG hydrogels have, historically, been less extensively developed than LMWG organogels.The first low-molecular weight hydrogelator in the academic literature was lithium urate, reported in 1841 by Lipowitz (Fig. 8).84 Uric acid is found in high concentrations in urine, and is a breakdown product of purine nucleotides. Lipowitz noted that on heating 1 part lithium carbonate, 4 parts uric acid and 90 parts water to 90 °C, then cooling, a gelatinous mass was formed, which readily re-dissolved on heating – a thermo-responsive hydrogel. A number of researchers returned to this gelator in the early part of the 20th century, and there was considerable speculation about the bonding in the uric acid molecule, the way it may interact with lithium ions, and changes in bonding that might be induced by heating.85 This work was performed before any sort of detailed understanding of the hydrogen bonding possible in nucleotides, therefore the inaccuracies in bonding models should be forgiven. There has also been considerable interest in lithium urate solubility and gel assembly because of the importance of the crystallization of uric acid in the disease gout86 – indeed, the improved solubility of lithium urate compared to uric acid itself has therapeutic relevance in this area.
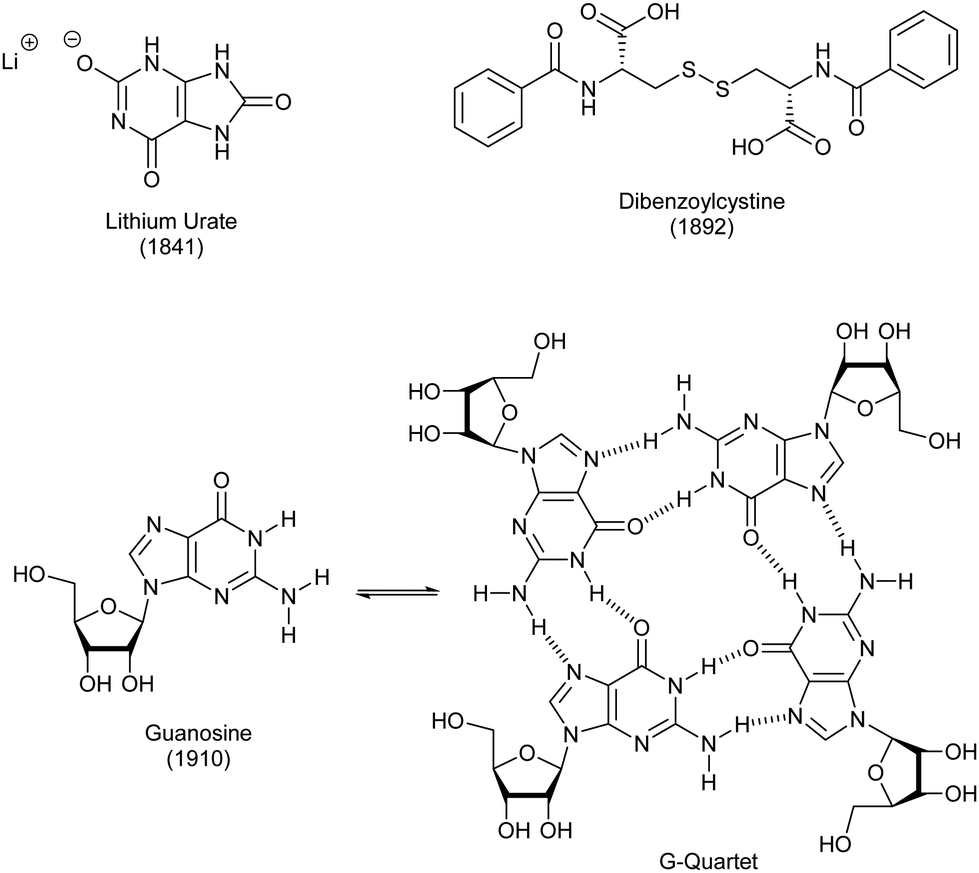 | ||
| Fig. 8 Early low-molecular-weight hydrogelators with origins dating back over 100 years. | ||
In 1910, a related hydrogel was reported when Bang investigated guanosine assembly (Fig. 8).87 It was only 50 years later, in 1962 that a seminal paper understood in detail how it assembled into gels.88 Specifically, X-ray studies, combined with spectroscopic work, including optical rotation studies, indicated that the assembly was helical, with the motif responsible for assembly being a quartet of guanosine units (Fig. 8). These ‘G-quartets’ stacked, one on top of another, to form extended 1-D objects. It has since become understood that such gels are further stabilized by metal cations, one of which binds in the centre of the G-quartet.89 This stoichiometry aligns with the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio of lithium and uric acid required for lithium urate gelation. G-quartet gels have been widely explored in academic science since 1990 – several reviews have outlined their potential and briefly explored their history.90 Guanosine can be synthetically modified in order to introduce other functionality, and the G-quartet now constitutes a versatile self-assembly ‘platform’, providing access to tailored nanostructured materials.
:4 ratio of lithium and uric acid required for lithium urate gelation. G-quartet gels have been widely explored in academic science since 1990 – several reviews have outlined their potential and briefly explored their history.90 Guanosine can be synthetically modified in order to introduce other functionality, and the G-quartet now constitutes a versatile self-assembly ‘platform’, providing access to tailored nanostructured materials.
Only one other low-molecular-weight hydrogelator dates back over 100 years, being first reported in 1892 by Brenzinger.91 Dibenzoylcystine forms remarkably effective gels in water (Fig. 8), typically, achieved by dissolving it in a small amount of ethanol, and then adding to water. This gelator came to wider attention in the 1921 report from Gortner and Hoffmann, who studied it in some detail.92 They attempted to understand how the molecules formed a gel, and proposed self-assembly based on intermolecular interactions. They studied analogues without the S–S linkage and found they did not form gels, leading them to postulate that it played a key role in assembly. Although not all suggested interactions were plausible (this work pre-dated Pauling's seminal ‘The Nature of the Chemical Bond’93), the paper indicates a good degree of conceptual understanding of the way small molecules could underpin the formation of a gel by forming non-covalent supramolecular polymers (although of course the word ‘supramolecular’ was not used at the time). This is perhaps surprising to the modern supramolecular chemist, who would imagine this type of ‘assembly mechanism’ work belonging more to the end of the 20th century than the beginning. Gortner and Hoffmann also investigated the ability of this gel to adsorb the dye methylene blue – this type of work came back into fashion in the 21st century, with interest in using gels to remediate pollutants from the environment (Section 4.3).
During the 1920s, there were a number of further academic studies of dibenzoylcystine, including its application in brine,94 and further studies of dye uptake.95 In the years that followed, it is less easy to track development, although in a 1961 patent, the abilities of dibenzoylcystine as a hydrogelator are discussed in some detail.96 The patent uses a hydrogel to stabilise pipelines by forming a flexible, corrosion-resistant gel layer between the interior plastic pipeline and the outer metal pipe, with dibenzoylcystine being suggested as the hydrogelator. With the advent of fibre-optic cables, the use of a gel to prevent water ingress, assist cable flexibility and protect from damage, became standard industrial practice, albeit with the gel having a different (polymeric) composition.97
Dibenzoylcystine reappears in the literature in 1978,98 when Menger and co-workers applied modern analytical methods to its study. In particular, NMR techniques proved that the gelator existed in two different environments – one highly mobile and ‘liquid-like’ the other effectively immobilised and ‘solid-like’. However, this work received relatively little attention and remains little-known. Menger and co-workers then went on to study dibenzoylcystine a few more times before publishing their seminal and erudite article in 2000 about the performance of this hydrogel, which helped establish LMWG hydrogels as a major field of academic research.99
2.8 Supramolecular gels in colloid and polymer science
From an academic perspective, gels were first studied as a class of material by Thomas Graham in the mid-19th Century. Indeed, Graham is often considered the founder of colloid science, and introduced the word ‘gel’ into the scientific language (shortened from gelatin). Graham separated matter into ‘crystalloids’ that would diffuse through a semi-permeable membrane (e.g. small molecules and soluble ionic salts), and ‘colloids’ which would not (e.g. larger molecules, and assemblies of molecules). On considering gels in his visionary 1861 paper, he wrote: “while the rigidity of the crystalline structure shuts out external expressions, the softness of the gelatinous colloid partakes of fluidity and enables the colloid to become a medium for liquid diffusion, like water itself.”100 It was clear, even from these very early studies, that gels were understood as materials that combined solid-like ‘crystallinity’ and liquid-like ‘diffusion’. In the same work, Graham noted the dynamic nature of colloidal gels and their importance as structuring agents in living systems – both topics of intense current interest (Sections 3.7 and 4.7). He went on to write: “the colloid possesses ‘Energia’. It may be looked upon as the probable primary source of the force appearing the phenomena of vitality.” Although concepts of ‘energia’ disappeared as scientists dispensed with the idea (common in the 19th century) that there is anything unique in chemical terms about ‘living’ matter, it remains a matter of modern-day speculation that gels may have played a role in the evolution of living systems – potentially forming the basis of early cellular lifeforms.101Further study of colloids led to their classification as materials containing one dispersed phase within a continuous phase.102 These two phases would normally be immiscible – in the case of gels, a solid dispersed within a liquid. By confining the dimensions of the dispersed phase (in this case the solid), it can extend throughout the liquid and remain compatible with it, forming a network, without sedimenting or separating. The dimensions of the dispersed phase should be in the order of 1–1000 nm. Even by the 1870s, it is evident that the role of colloidal systems in gel formation was relatively well understood – in his book on fermentation, von Nägeli talks about gels in terms of the aggregation of micelles, leading to the creation of a mesh in which solvent becomes trapped by molecular attraction but is not completely immobilized.103 This explanation of gelation is little different to that used in modern-day understanding of many supramolecular gels (Section 3.1).
In the later years of the 19th century and the early years of the 20th century, the study of colloids continued apace. Although the atomic nature of the world was broadly understood, the structure, or indeed existence, of the atom had not yet been definitively proven. Colloids have much larger particle sizes than atoms, and were therefore much more amenable to experimental study, providing early insight into the particulate nature of matter. For example, Tyndall showed that colloids interact with, and scatter, visible radiation104 – key evidence of a particulate world. In 1902, this ultimately led to the invention of the ultramicroscope by Zsigmondy to study particles as small as 4 nm.105 This early interest was reflected by the fact that by 1930, three Nobel Prizes had been awarded for work on colloids, with colloidal materials beginning to dominate a wide range of 20th century industrial applications (see above). Nonetheless, even in 1926, Dorothy Jordan Lloyd, one of the pioneers of colloid chemistry, pragmatically wrote: “the colloid condition, the gel, is easier to recognize than to define.”106
This was a key point in science – the field of colloid chemistry was well established, but polymer science was in its infancy. Indeed, it was only in 1920 that Herman Staudinger proposed polymeric materials were comprised of long chain molecules.107 In the years that followed, there was an explosive growth of interest in polymers, and it became evident they would transform science and society, ushering in a new materials age. Branched or crosslinked polymers gained importance as sample-spanning gels and Paul Flory applied statistical methods to understand their behaviour, ultimately receiving the Nobel Prize in 1974.108 As a part of this work, Flory–Stockmayer theory emerged, which helped understand the gelation of polymeric systems.109 This set limits on the amount of crosslinking required to obtain a gel and was the first theoretically rigorous way of understanding gel-phase materials.
During the 20th century, therefore, the balance of interest in materials science switched, to a large extent, away from colloids, and towards polymers, with covalently-crosslinked polymer gels transforming technologies, including personal care, drug formulation, contact lenses, etc. Indeed, polymer gels came to dominate the scientific discourse, while LMWGs became somewhat forgotten and overlooked. Only with the advent of supramolecular chemistry as first defined by Jean-Marie Lehn in 1978,110 and the development of an intense interest in self-assembly,111 did academic attention really shift back to the true potential of ‘molecular materials’.
In Section 3, I explore the ways in which supramolecular chemistry has informed, and indeed transformed, the field of self-assembled gels over the last 40–50 years.
3. Emergence of supramolecular concepts
Perhaps the most influential academic article on supramolecular gels was published in 1997 by Terech and Weiss.112 Now cited thousands of times, this early review captured the rapidly-growing field of ‘supramolecular gels’ and led to many academic chemists recognising the phenomenon of gelation and finding it interesting, rather than just seeing it as an oddity, or a synthetic annoyance. Certainly, it was a huge inspiration to my own early work, at the very beginning of my academic career, which started with the serendipitous discovery of an acid-amine two-component gel.113As supramolecular chemists have increasingly built on the early foundations (Section 2), they have begun to expand the scope of LMWGs. This has led to new concepts being developed. As a result, supramolecular gels now exhibit far more complex types of behaviour than the simple rheology modification on which their applications historically depended. Gels have moved well beyond the simple photographic image of an inverted vial, ubiquitous in the early academic literature. This section explores the most significant conceptual advances, many of which are used in the further development of high-tech gel applications, which are explored further in Section 4.
3.1 Expanding LMWG chemical space
Given supramolecular gels are multi-scale, hierarchical materials (Fig. 1), it is crucial to apply different characterisation techniques at different length-scales in order to gain a holistic understanding of their properties.114 At the molecular level, spectroscopy provides insight into the non-covalent interactions between LMWGs, and the dynamics of these materials, with IR, UV-Vis, fluorescence, and NMR all having particular value. On the nanoscale, techniques like electron microscopy and X-ray/neutron scattering provide insight into self-assembled nanostructures, while circular dichroism spectroscopy can characterise chiral assembled architectures. At the microscale, confocal or optical microscopy can characterise the wider network properties. On the macroscale, rheology and calorimetry provide insight into materials performance and thermal properties. The wide range of techniques required to properly understand gels makes their study a challenging endeavour, providing exceptional training to the next generation of scientists. Furthermore, the development of innovative methods to better characterise gels remains an area of intense activity,115 with such experiments having the potential to unlock more secrets of gels.
Historically, most gelators were discovered serendipitously rather than by design, but increasingly, simple design tools have been developed to assist in this process.116 It is often possible to develop new gelators in a semi-rational way by modifying the structures of known gelators. Liu and co-workers suggested using the terminology ‘gelatons’ (in analogy to ‘synthons’) to describe motifs that were well-known to be capable of gel assembly.117 Advances in supramolecular chemistry have provided a better understanding of self-assembly, enabling rationalisation of why certain molecules form gels. For example, ‘one dimensional’ non-covalent interactions are known to be of particular importance, and attempts have been made to relate such interactions in crystal structures to the potential for gel assembly.116c Recently, computational methods have increasingly been used in an attempt to understand and/or predict the existence of gel-forming systems.118
As an example of iterative gelator design, academic researchers realised the potential of the bis-urea motif in underpinning organogels as a result of its self-complementary hydrogen bonds (Section 2.1).119 This motif is one of the key ‘gelatons’. The oriented tape-like intermolecular interactions allow the incorporation of a range of functionalities into the remainder of the structure (Fig. 9). This expanded the potential of this class of gelator far beyond the greases for which they were being used.120 For example, by modifying the peripheral groups, Hamilton and co-workers showed it was possible to change the solvent in which they assemble to water, creating hydrogels (Fig. 9).
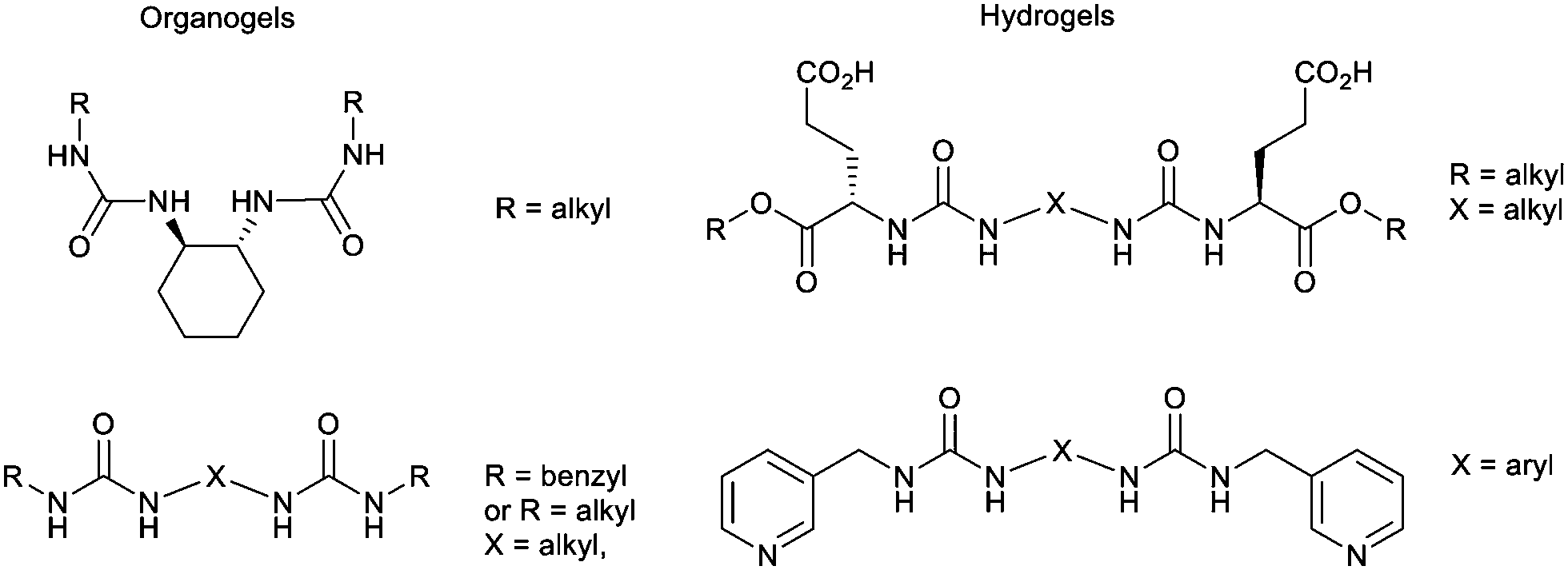 | ||
| Fig. 9 Selected examples of urea-based gels – organogels (left) have apolar peripheral groups while hydrogels (right) introduced a degree of hydrophilicity into the peripheral units. | ||
Hydrogen bonding plays a key role in underpinning the assembly of many organogelators. Functional groups such as amides, alcohols and acids are widely found in LMWG organogels.121 Other non-covalent interactions that can establish themselves in organic solvents have also been explored, including donor–acceptor interactions, π–π stacking and metal coordination, giving organogels broad chemical scope and a wide range of potential uses (see Section 4).122 Indeed, many thousands of compounds have now been reported as organogelators in one organic solvent or another, representing many classes of molecule and modes of assembly.
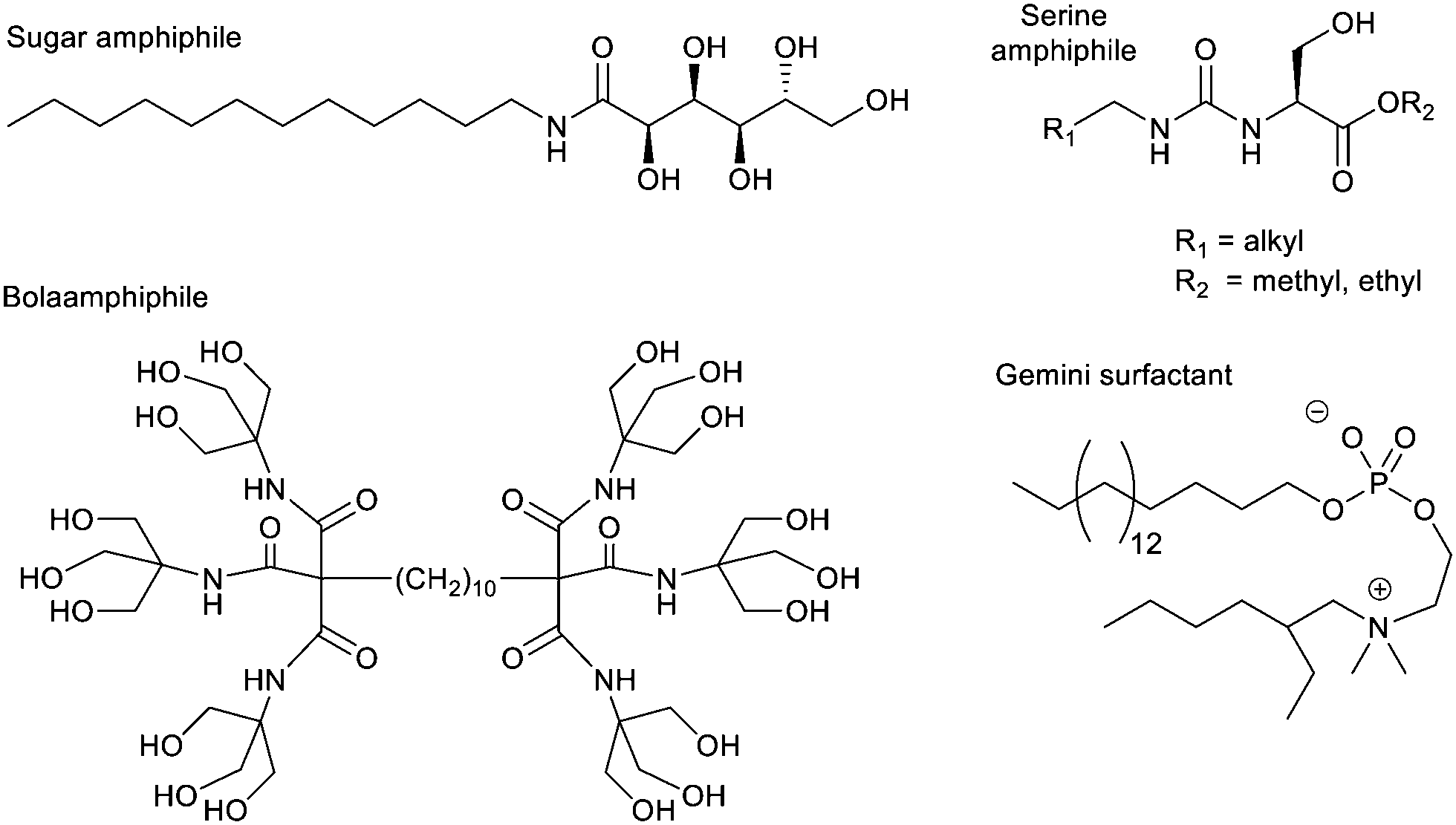 | ||
| Fig. 10 Selected examples of charge-neutral amphiphile-based gels. | ||
It is well-understood from the landmark work of Israelachvili in the 1970s, that amphiphile design controls assembly mode.125 In particular, the hydrophilic/lipophilic balance (HLB) defines the structure of the aggregates formed, with the geometry playing a key role in determining whether an amphiphile assembles into spherical micelles, cylindrical micelles, vesicle bilayers or inverted structures (Fig. 11). Although considered ‘colloid science’, and often overlooked by supramolecular chemists, these conceptual developments sit perfectly alongside the concepts of controlled assembly being concurrently pioneered by early supramolecular chemists.126 In the early 1980s, Kunitake and co-workers applied Israelachvili's approach to practical studies of lipids to understand gel fibre assembly in terms of a preference for cylindrical micelle morphologies.127 For lipids to be capable of gel formation, it became understood that the assembly of ‘1-dimensional’ cylindrical micelles is ideal, as at sufficient concentration they can assemble further into fibres, that ultimately form a sample-spanning gel network. It is interesting to reflect that this (albeit much more detailed) model of interacting micelles underpinning a gel matches that originally conceptualised as early as the 1870s by von Nägeli (Section 2.8).103
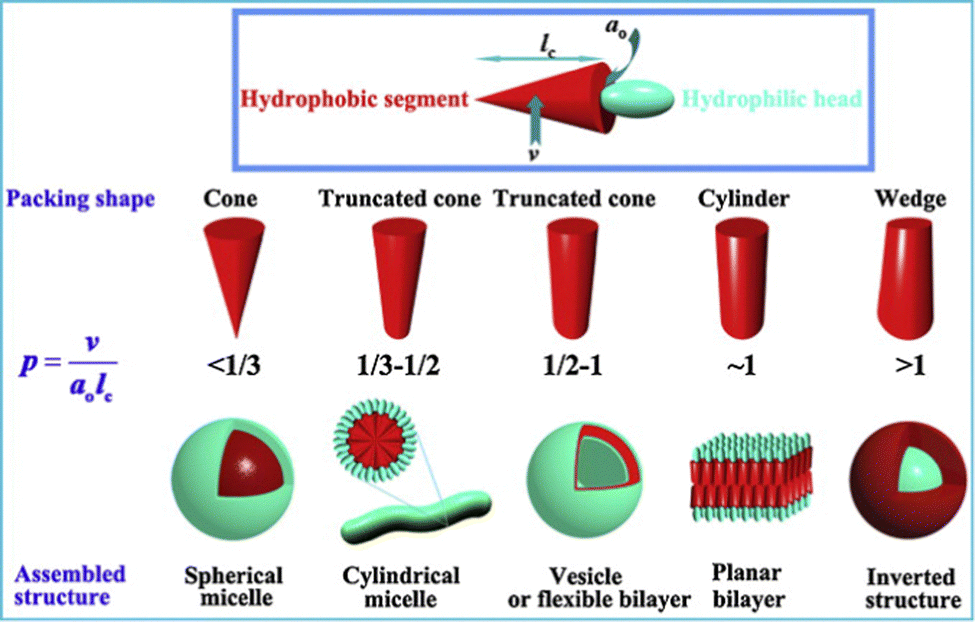 | ||
| Fig. 11 The impact of surfactant geometry, hydrophilic/lipophilic balance, and packing parameter on the structures of assemblies formed in water. For gel formation via lipid assembly, cylindrical micelle formation is usually required. Figure reproduced from ref. 128 with permission from Elsevier, © 2012. | ||
Great efforts have been made to understand neutral amphiphile assembly using surfactant science techniques.129 The assembly of the cylindrical micelles is primarily driven by the hydrophobic effect associated with the apolar units that separate themselves from the bulk water and pack into the interior of the structure. It is further supported by hydrogen bond interactions between polar head groups, and interactions between the aqueous solvent and the head groups, which therefore occupy the exterior of the assembled structure. Interactions between head groups also mediate micelle–micelle interactions and the formation of a sample spanning network. Indeed, it is vital that the micelles form constructive interactions with one another in order for a gel network to form (see below).
Bola-amphiphiles, which contain two polar neutral head groups, connected by an apolar linker, have also been used as hydrogelators, and constitute a general ‘design principle’. These molecules are compatible with a surrounding aqueous solvent, while also containing a significant hydrophobic domain to drive self-assembly. One of the earliest modern hydrogelators was the bolaamphiphile discovered by Newkome and co-workers during their pioneering work on dendritic molecules in 1986 (Fig. 10).130 A number of other researchers have since found bolaamphiphiles support hydrogelation.131 At the time of Newkome's early work, the field of ‘supramolecular gels’ had not nucleated, and therefore a number of contributions were building up from different, often disconnected, areas of chemistry, not being linked until much later. For example, colloid scientists increasingly focussed on ‘gemini surfactants’, which are dimeric surfactant structures with two linked polar head groups, each flanked by a flexible hydrophobic chain (Fig. 10) – these often assemble in such a way as to form hydrogels.132
In contrast to the non-ionic amphiphiles described above, ionic (charged) surfactants are much less effective at forming gels. Even if cylindrical micelles do assemble, the polar headgroups typically repel one another, preventing the formation of the beneficial inter-micelle interactions required to underpin a sample-spanning network. Nonetheless, in some cases, charged amphiphiles could be encouraged to self-assemble into gels, for example if additional interactions were placed into the hydrophobic units, such as amide groups capable of forming intermolecular hydrogen bonds.127
An alternative way of developing hydrogels from charged amphiphiles is to remove the charge as a trigger to gelation – i.e. the colloidal system is soluble in water when the amphiphile is charged, then assembles into a gel on becoming neutral. In early work from 1992, this was demonstrated using glutamic acid as head group (Fig. 12, top).133 On protonation, it forms gels as the cylindrical micelles begin to interact with one another. This strategy has become hugely important in LMWG science, leading to a very significant class of peptide hydrogels.
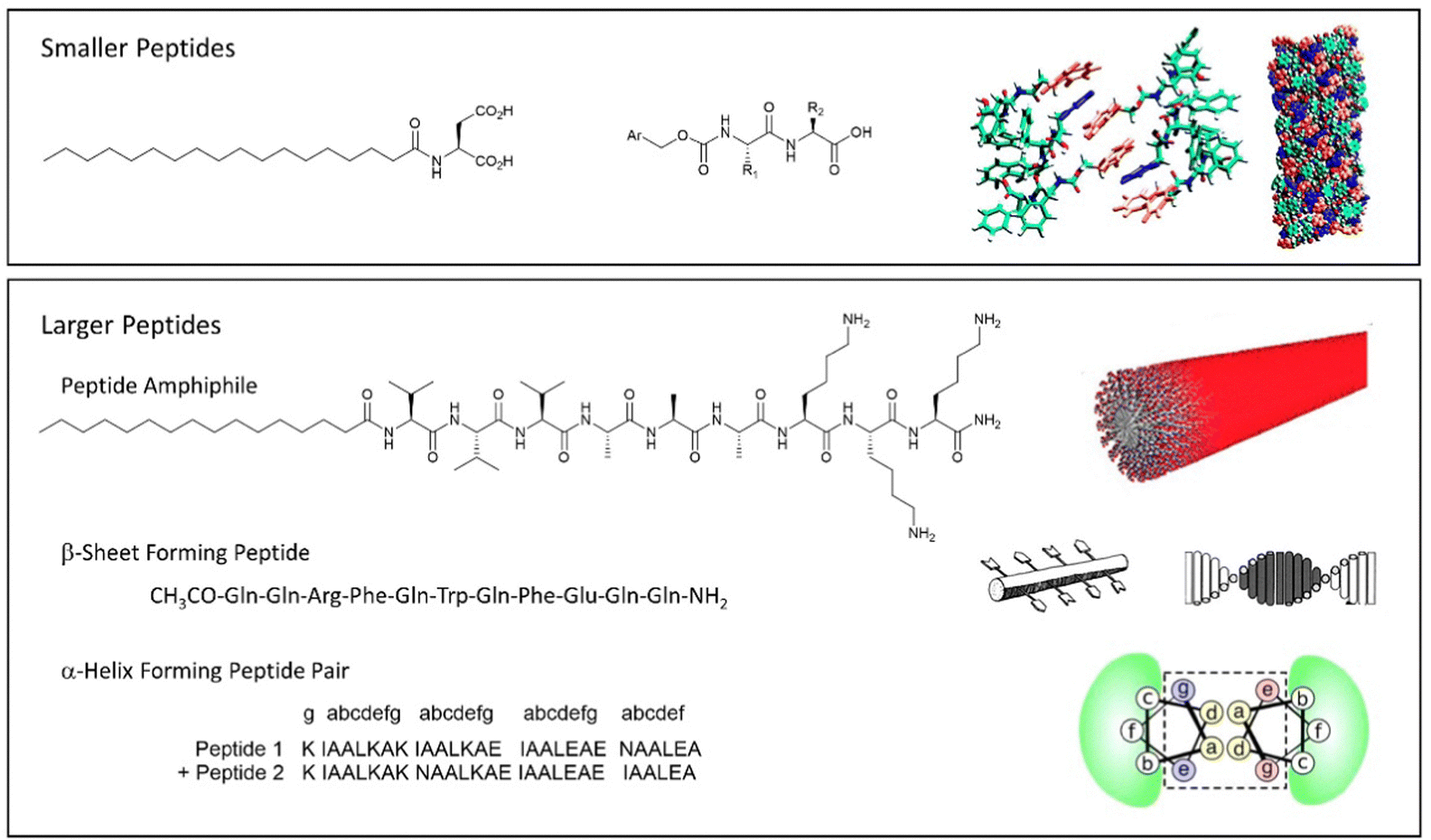 | ||
| Fig. 12 Selected examples of peptide-based gels. (top) Smaller peptides are those based on 1–3 amino acids, modelling of Fmoc-YY assembly is reproduced from ref. 135b with permission from Wiley-VCH, © 2008. (bottom) Larger peptides are based on >3 amino acids. Schematics of assembly are reproduced from ref. 141 (peptide amphiphile) with permission of the American Chemical Society, © 2017; ref. 139b (β-sheet forming peptide) with permission of National Academy of Science, © 2001; and ref. 140b (α-helix forming peptide) with permission of Wiley-VCH, © 2014. | ||
Beyond the small peptide gelators described above, larger peptides also underpin a wide range of self-assembled gels (Fig. 12, bottom). This work can be considered to originate from knowledge of gelatin assembly, and understanding of protein structure that emerged during the 20th century. Progress in this area has often been driven by bio-chemists/physicists. Larger peptide LMWGs are 5–50 amino acid units in length – as such, they span the range between low-molecular-weight gelators and something more like a polymer gelator.138
Typically, larger peptide gelators self-assemble as a result of the secondary peptide structures. For example, as pioneered by Aggeli and co-workers in the late 1990s, peptides with an extended conformation assemble through β-sheet interactions (Fig. 12, bottom).139 Alternatively, as exploited by Woolfson and co-workers, coiled peptides can form α-helices, that further assemble into networks via hydrogen bonding or hydrophobic coil-coil contacts (Fig. 12, bottom).140 It was noted that gels that formed as a result of hydrogen bonds between coils weakened on heating, whereas those based on hydrophobic contacts were stabilised by heating, indicative of the opposite role of entropy in these different non-covalent interactions.
Combining peptide assembly with amphiphile behaviour, Stupp and co-workers developed peptide amphiphiles that have extended peptide sequences, functionalised with a large hydrophobic unit (Fig. 12, bottom).141 Such gelators can be considered a scaled-up version of simple peptide LMWGs like Fmoc-YY. They benefit from peptide-peptide hydrogen bonding and a powerful hydrophobic effect during assembly. Stupp's research team have explored a wide range of applications of such gelators (see Section 4).
The ability of amino acids to code for many different types of chemical ‘behaviour’ – from hydrophobic to hydrophilic, neutral to charged, acidic to basic, interactive to reactive, means peptide gelators are highly programmable. Solid-phase peptide synthesis makes it easy to synthesise peptides in which individual amino acids have been changed, hence gaining a detailed structure–activity relationship understanding.142 Furthermore, the opportunity to incorporate bioactive ‘tags’ into self-assembling peptides, which can then interact with key target proteins and enzymes, provides an easy methodology for the embedding functions into these materials.143
In terms of understanding the impact of composition on gelation, in landmark work, Tuttle and co-workers modelled the self-assembly of all 8000 possible tripeptides to determine the sequence space capable of supporting self-assembly.118a They applied the CG MARTINI forcefield and ranked the output of simulations according to descriptors such as propensity to aggregate and hydrophilicity, hence allowing the selection of a candidate gelators. Experimental methods were then used to validate this predictive approach. In more recent work, these researchers simulated larger peptides.144 In this case, exhaustive simulation becomes impossible because the sequence space is too large, but they developed an active machine-learning method which leverages a lower-resolution dataset encompassing the whole search space and a just-in-time high-resolution dataset which further analyzes the target peptides selected by the lower-resolution model.
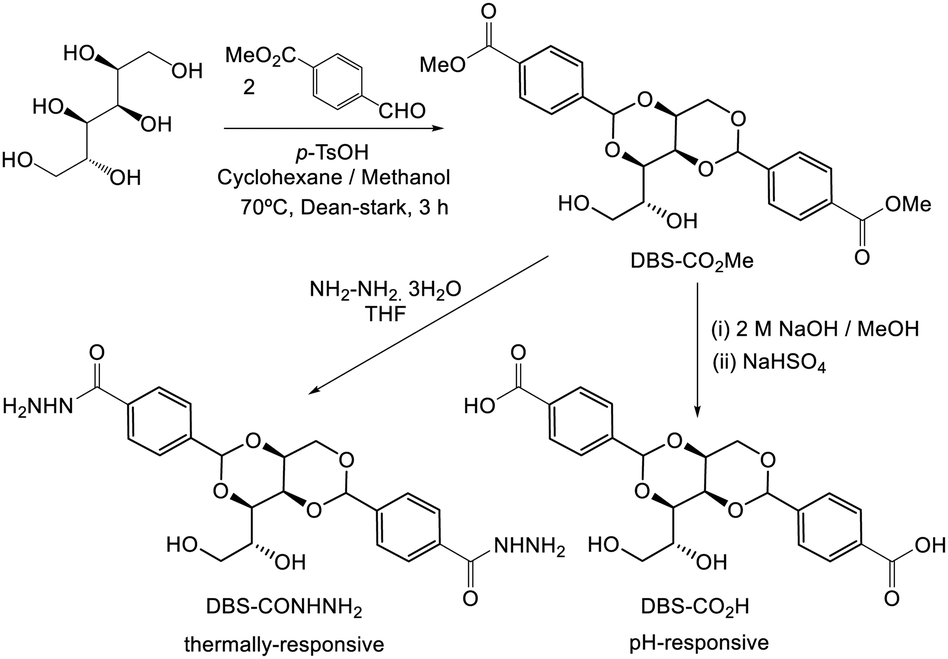 | ||
| Fig. 13 Synthesis of DBS derivatives, DBS-CONHNH2 and DBS-COOH which act as hydrogels in pure water. Gels based on DBS-CONHNH2 are thermally-responsive, while those based on DBS-COOH are pH-responsive. | ||
As might be expected, most successful examples include the types of functional group well known to have assembly potential, including surfactant-like nature, hydrophobic surfaces, oriented hydrogen bonding units, etc. John and co-workers reported early examples. For example, they developed gels based on the sugar amphiphile amygdalin, found in apricot kernels,150 and also modified hydrophilic ascorbic acid (Vitamin C) with long chain esters to create LMWGs (Fig. 14).151 These researchers also used design principles building on the knowledge of sugar amphiphiles to modify cardanol, a relatively hydrophobic molecule found in cashew nut shells, with a sugar headgroup, inducing gelation.152 The ongoing development of ‘green’ LMWGs remains a topic of considerable interest.
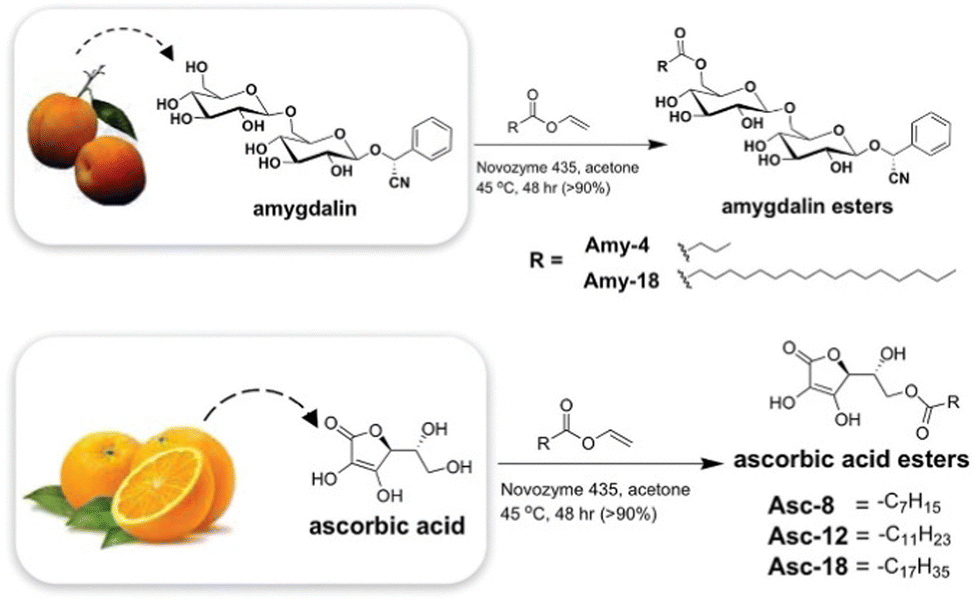 | ||
| Fig. 14 Biorefinery gels developed from natural products, amygdalin from (apricot kernels) and ascorbic acid (from oranges) by John and co-workers. Figure reproduced from ref. 148 with permission of the American Chemical Society, © 2010. | ||
The examples described above are not meant to be comprehensive in terms of the structures incorporated into LMWGs – thousands of systems are known to form gels under one set of conditions or another. Rather, the examples aim to illustrate key principles and provide rational ways of thinking about LMWG design and assembly. The effort of synthetic chemists means that many more LMWG structures are known now than 50 years ago. Although cataloguing them is an important task, thinking about what they can do in terms of behaviour and applications is increasingly important. In particular, it is important to develop and understand new LMWGs that do things that are not possible for well-established ‘historic’ LMWGs. This is explored in later sections.
3.2 Understanding hierarchical assembly
Although much progress has been made in understanding self-assembly at the molecular level using detailed experimental methods,114 understanding how the resulting one-dimensional fibres go on to form a sample-spanning network is often poorly understood – this was discussed in detail in Adams’ thoughtful personal perspective on gels.1c Clearly, there must be interactions between fibrillar assemblies as discussed above, but in many cases, it is unclear whether these result from specific non-covalent interactions, generalised solvent-based effects, or even simple fibre–fibre physical entanglement. Probing this level of hierarchical assembly experimentally remains a challenge. Although researchers are beginning to understand this kind of process, as will be presented, it remains a key area where conceptual progress, and new techniques, are needed. Computational modelling techniques will also have a role to play.Perhaps the best early progress in understanding such processes was provided by those studying larger peptide gels as described in Section 3.1 (Fig. 12, bottom). Contacts between α-helices or the hierarchical assembly of β-sheet fibrils were predicted and in some cases modelled.139b,140b This reflects the fact that peptides have well-understood, defined molecular shapes, and their non-covalent interactions across multiple length-scales had already been studied in the context of protein folding. As such, it was natural for those engaged in these systems to attempt a full structural understanding of hierarchical assembly.
Working with smaller LMWGs, attempts have been made to understand hierarchical assembly, but this remains challenging. In recent work, Steed and co-workers investigated the ability of fibrils in supramolecular gels to undergo a braiding process, with detailed analysis giving rise to an understanding of the defect points at which network branching occurred (Fig. 15).153
 | ||
| Fig. 15 (a) SEM image of a single three-stranded braid showing a defect in which the braid separates into separate fibrils. This defect results in a branch point, increasing the branching density in the gel. (b) SEM image showing that the separation of branch points in dried gel samples is highly variable. Figure reproduced from ref. 153 with permission of Royal Society of Springer Nature, © 2019. | ||
Adams and co-workers made careful use of small angle neutron scattering (SANS) to probe the temporal assembly of 2Nap-FF peptide gels, demonstrating that the hollow cylindrical micelles formed at high pH initially lose their hollow core on pH-lowering and gel assembly, then laterally associate, becoming elliptical in shape, with double the original diameter.154 In this way, fibril–fibril assembly processes were better understood. There are also seminal contributions in this area from Hamachi and co-workers – these are described in Section 3.4, as they employ multi-component systems to help understand fibre–fibre interactions at the network level.
Demonstrating the impact that network-level assembly has on materials properties, George, Maitra and co-workers recently used a seeding approach to stimulate secondary nucleation-triggered cross-linking of a metastable gel, leading to fibre bundling.155 This fibre-linking process generated a new gel state with significantly enhanced mechanical strength. They argued that their seed-induced network strengthening approach was a non-covalent crosslinking strategy analogous to classical crosslinking of polymer chains. If generalisable, this could be extremely powerful in LMWG science. Metastable and dynamic systems are discussed in more detail in Section 3.7.
In other work to influence network-level properties, van Esch and co-workers reported the first example of a strain-stiffening supramolecular gel.156 By introducing a degree of anionic charge to the periphery of their gel nanofibers, the resulting electrostatic repulsion stopped the nanofibre bundling which normally prevents stiffening. As a result of this minor molecular ‘edit’, their materials stiffened in response to strain. Intriguingly, the authors noted that this type of mechanism is used by living systems to protect against deformation.
Although LMWG structures can to some extent be predicted/rationalised (Section 3.1), it remains very difficult to predict, a priori, the macroscopic mechanical properties of the gel formed by a particular LMWG. Although mechanical properties are routinely measured by rheology,157 it is almost impossible to know whether a gel will be stiff or weak based on inspection of molecular structure. It seems logical that less-soluble gelators may form more rigid crystalline assemblies that might be more brittle, while more-soluble gelators will tend to form softer gels on the brink of disassembling into the solvent. It is also broadly understood that the nanoscale assembled architecture will direct rheological performance. However, a detailed, predictive understanding of how molecular structure translates into rheological performance via nanoscale assembly has not yet been achieved and remains a major target.
3.3 Understanding the role of solvent
Possibly the best way to consider gelation is as a balanced process where the LMWG sits on a knife-edge between the conditions where it is fully soluble (and dissolves), and is fully insoluble (and precipitates). Between these two, is the point at which the molecule starts to assemble, but in a controlled manner, giving rise to the 1-dimensional fibrils (or occasionally 2-dimensional platelets) that underpin gel networks. The gel fibres/platelets must be sufficiently soluble to disperse in the solvent, yet sufficiently insoluble that they want to form interactions with one another (network junction points), which allow the gelator to form a sample-spanning network.
The best overall description of solvent effects in LMWG assembly, however, was given by Rogers and co-workers in their seminal review from 2015.159 Specifically, they used Hansen parameters, as first introduced to the field of gel assembly by Raynal and Bouteiller in 2011.160 Hansen parameters are subdivided into interactions based on dispersion forces, dipolar interactions and hydrogen bonds, and importantly, these three parameters define ‘Hansen space’ with the coordinates of molecules within Hansen space representing their similarity to one another. By plotting the ability of an LMWG to form gels in different solvents onto the Hansen coordinates (Fig. 16), it was found that in many cases, the LMWG would be fully soluble in a small (blue) zone of Hansen coordinates, around that would be zones in which opaque or transparent gels were formed (green and red zones), and finally, beyond those limits, LMWG solubility would be so low that it would be insoluble. These plots visualise the outcomes when the gelator either interacts strongly with solvent and therefore dissolves, is totally incompatible with the solvent and therefore precipitates, or sits in between the two extremes and forms an opaque (less soluble) or transparent (more soluble) gel. By measuring the dimensions of Hansen space in which gels could be formed, it was possible to gain insight into gelator versatility – some LMWGs have limited solvent scope, while others, such as DBS (Fig. 16), have wide gelation potential.161 However, many LMWGs do not show perfect behaviour – there are clearly subtleties that are not yet fully understood.
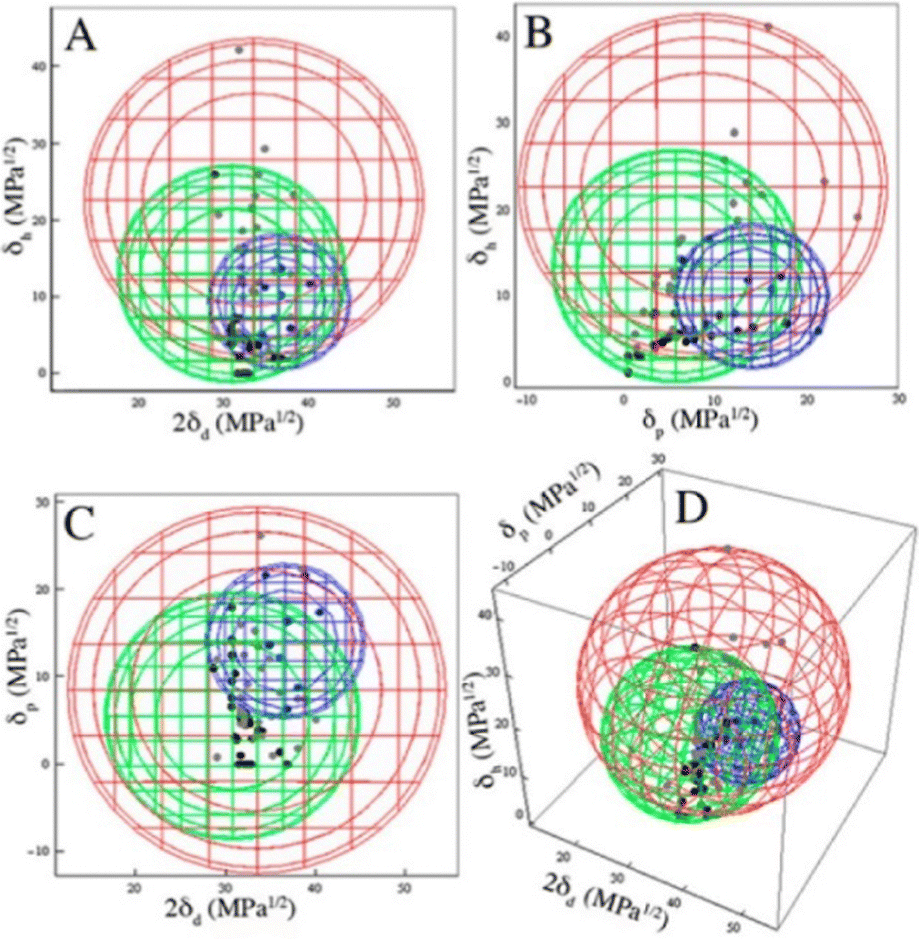 | ||
| Fig. 16 Two-dimensional projections (A)–(C) and a 3D rendering (D) of the Hansen space using minimal enclosing spheres and the appearance at the CGC. The blue sphere encloses DBS solutions, the green sphere encloses opaque gels, and the red sphere encloses clear gels. Figure reproduced from ref. 161a with permission of the American Chemical Society, © 2014. | ||
Importantly, Rogers and co-workers concluded that no LMWG can (or likely will) gel all solvents (a so-called ‘universal’ gelator).159 This emphasises the importance of considering solvent when developing and applying LMWGs. Furthermore, they demonstrated that, as would be expected, modifying LMWG structure, could change the range of solvents in which gels were formed, extending the range of well-known ‘gelatons’ into new solvent spaces.162 Although this was well-known in simply qualitative terms, plotting it onto Hansen space provides a way of quantifying this process, and offers a greater degree of predictive power in terms of future LMWG development.
Rather than thinking about general solvent effects, Meijer and co-workers explored the role of solvent on the molecular level, with key work demonstrating that solvent could play an intimate role in assembly.163 Specifically, they found that self-assembly depended on solvent structure, with an organized shell of solvent molecules playing an explicit role in rigidifying aggregates and guiding them towards further assembly into gels. Ghosh and co-workers have also reported a system which appears to exhibit shape matching between LMWG and chemically-similar but differently-shaped solvents, indicative of intimate LMWG–solvent interactions that might not have been predicted just by considering general solvent effects.164
Deep eutectic solvents, formed by mixing two solids (e.g. glycerol and choline chloride) which interact with one another non-covalently to form an ionic liquid at room temperature, have also been immobilised to form ‘eutectogels’ (Fig. 17).168 Once again, high ion mobility is retained in the liquid-like phase – these gels exhibit good conductivity and have potential in electronic applications (see Section 4.6). It has also recently been reported that an LMWG eutectogel has potential applications as an environmentally-friendly adhesive.169 The authors noted that the high viscosity, high polarity, and abundant electrostatic and hydrogen-bonding sites of deep eutectic solvents enable strong interfacial interactions with diverse substrates, which when combined with the gel-like nature, yielded effective adhesive performance. To form gels in ionic media, it is important that, just like in organogels and hydrogels, the LMWG retains a balance between solubility and aggregation.
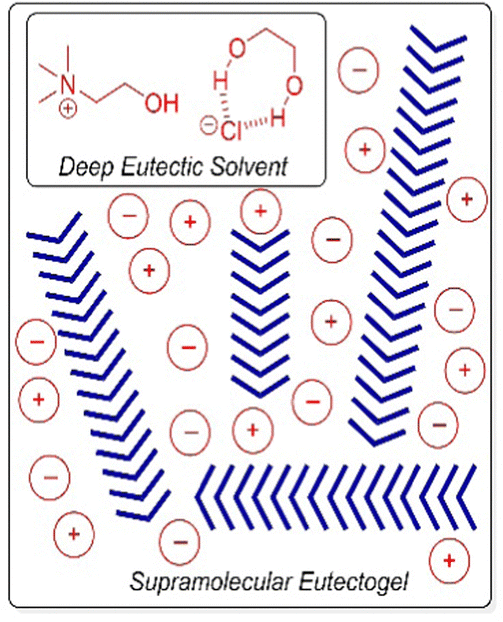 | ||
| Fig. 17 Schematic of assembly of an LMWG (blue chevrons) within a deep eutectic solvent based on a mixture of choline chloride and ethane-1,2-diol. Figure reproduced from ref. 168b with permission of Wiley-VCH, © 2019. | ||
There has also been interest in assembling supramolecular gels in thermotropic liquid crystals (LCs) – a combination pioneered by Kato and co-workers in the early 2000s.170 Such composite materials can exhibit two independent thermoreversible transitions based on (i) the gel network, and (ii) the liquid crystal phase. If the thermally reversible LMWG sol–gel transition (Tsol–gel) is higher than the isotropic–anisotropic LC transition temperature (Tiso-Ic), this was defined this as a Type I material, in which a random gel-network forms in the isotropic liquid-like LC-phase. Conversely, if Tsol–gel is lower than Tiso-Ic, this was defined as a Type II material; the ordered state of the LC acts as a template and induces the assembly of an oriented gel network (e.g. aligned fibres). Kato and co-workers also developed a photo-responsive liquid-crystalline gel by incorporating azobenzene into the LMWG.171 Reversible structural changes between LC nematic and cholesteric phases could be induced via trans–cis photoisomerisation of the gelator – hence the response of the LMWG led to a response of the LC.
The discussion above clearly demonstrate how the liquid-like phase in a supramolecular gel plays an active role in gel assembly. Furthermore, the solvent can also perform a functional role in the resulting gels. It is likely that solvent modification will continue to lead to significant developments in terms of gel tuning and the development of new high-tech applications (Section 4).
3.4 Multi-component gels
Self-assembled materials can easily be formulated from multiple components by mixing different molecules in a solvent – this is a key advantage of the supramolecular approach to materials science. As such, there has been considerable interest in multi-component gels.172 In their landmark 2012 review, Buerkle and Rowan then developed a system for classifying such gels that has benefits, both in terms of its simplicity and its conceptual use (Fig. 18).173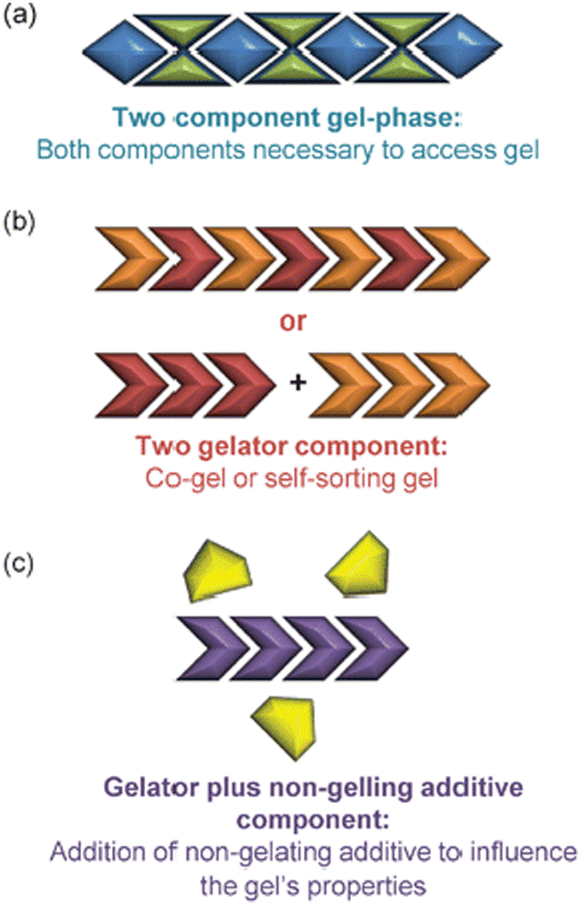 | ||
| Fig. 18 Schematic of multi-component gels according to Buerkle and Rowan: (a) Two component gel phase, (b) Two gelator components, (c) Gelator plus non-gelling additive component, figure reproduced from ref. 173 with permission of the Royal Society of Chemistry, © 2012. | ||
• Multi-component gels. Gels that require reaction or interaction between components in order to assemble.
• Multi-gelator gels. Gels formed from multiple compounds, each of which is capable of forming a gel in its own right.
• Gelator-additive gels. Gels in which an additive is formulated into the gel – it is possible that the additive has no effect on gelation, but it also possible it may interact with the LMWG and hence impact on gelation.
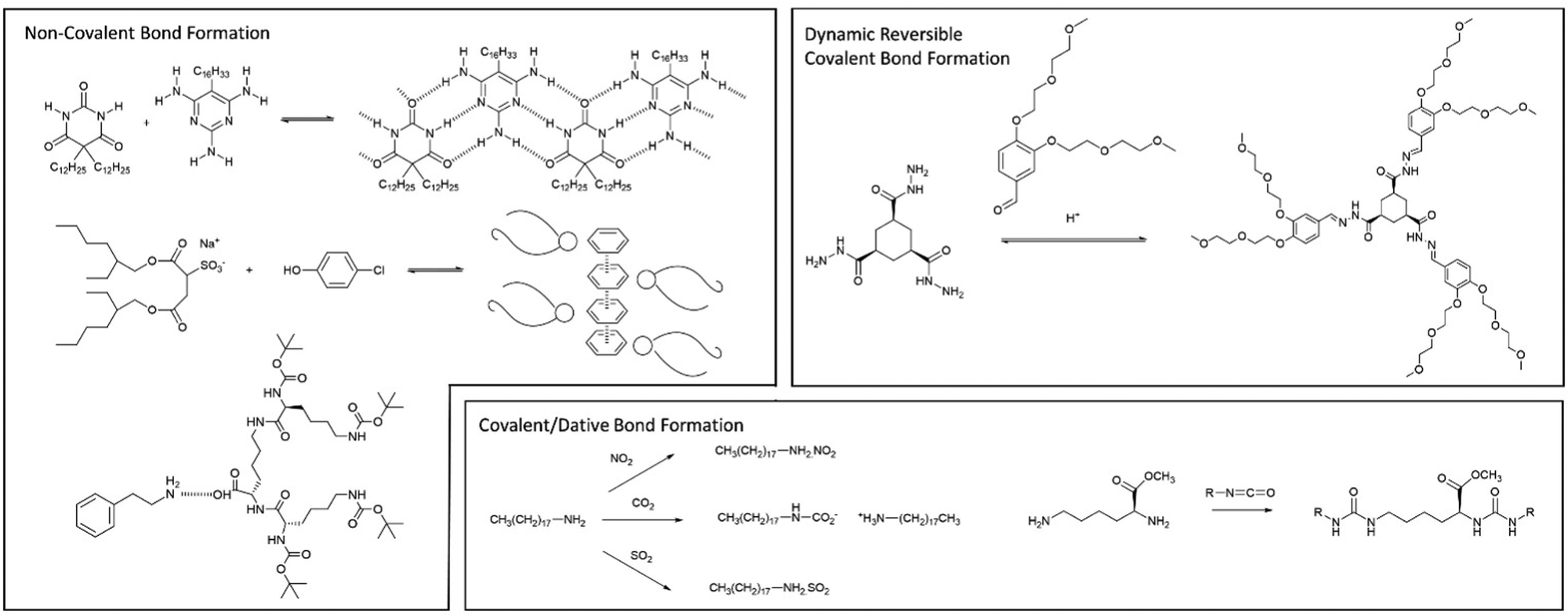 | ||
| Fig. 19 Examples of multi-component gels based on different types of interaction between the individual components. | ||
In later work, researchers moved on from considering irreversible bond forming processes to explore more dynamic reactions with a degree of reversibility. For example, van Esch and co-workers employed the acid-catalysed reaction between aldehydes and acyl hydrazides. The resulting acylhydrazone linkage(s) connected together the building blocks, forming a self-assembling LMWG (Fig. 19).176 The reversibility means this approach is particularly useful for developing highly dynamic materials with the potential for active patterning (see Section 3.9). Many small molecule aldehydes have applications in the fragrance industry, and Lehn, Herrmann and co-workers made use of reversible acylhydrazone-forming reactions to generate multi-component gels that incorporated fragrance molecules.177 Such gels have a variety of potential controlled release applications in homecare and personal care.
Also in 1993, McPherson and co-workers, coming from a colloid chemistry background, developed two-component gels based on the interaction between the twin-tailed anionic surfactant bis(2-ethylhexyl)sulfosuccinate and a variety of phenols (Fig. 19). The hydrogen bond interaction between the sulfonate group and the phenol led to a complex, that helped shield the negative charge of the surfactant, and thus mediated gelation of the cylindrical micelles in non-polar solvents.179 In this case, detailed structural studies indicated the assembly of molecular-scale strands (ca. 2.1 nm diameter), into fibres (ca. 10 nm), then into bundles (ca. 20–100 nm) – an early example in which hierarchical assembly was understood (Section 3.2).
In our own work, starting in 2001, we pioneered gels based on the simple interaction between dendritic peptide carboxylic acids and apolar amines, which give rise to a stable acid–base complex in organic solvents (Fig. 18).113 The resulting complex, has modified solubility, triggering further assembly into nanofibers as a result of hydrogen bond interactions between the peptide groups. The acid-amine motif turns out to be a simple and generalisable way of achieving self-assembly of many organogels,180 somewhat in analogy to the fatty acid metal salts widely used across organogel history (Section 2). However, compared with metal ions, the choice of amine offers more tunability with regards to structure and performance, and can introduce useful functionalities into the gelator. For example, organogels were assembled incorporating phenylethylamine (Fig. 19), a key pharmacophore in psychoactive drugs.181 Incorporating drug-like molecules into the network hints at the potential of gels in terms of drug delivery or sensing applications (Sections 4.1 and 4.5).
In the years since these early studies, a wide range of multi-component gels have been reported, based on many different interactions. This remains a simple way of assembling gels, with component mixing often acting as the only trigger required for assembly. Furthermore, it creates highly tunable gels, as any of the constituent components can be synthetically varied to program the performance of the resulting gel.
 | ||
| Fig. 20 Cartoon schematic showing how the assembly of two LMWG (red and blue) can lead to (a) random mixing; (b) alternating co-assembly; (c) self-sorting. Figure reproduced from ref. 182d with permission of the Royal Society of Chemistry, © 2014. | ||
It has been suggested that molecules with similar structures, self-assembled using the same trigger and through similar interactions, are more likely to co-assemble, whereas molecules with divergent structures, assembled via orthogonal triggers or through different interactions, are more likely to self-sort – there is significant interest in understanding and controlling these processes.183 In this way, molecular recognition pathways and gel fabrication methods program the behaviour of a multi-gelator gel.
However, the situation is actually significantly more complex than that, because in addition to whether systems self-sort, co-assemble or disrupt at the molecular level, it is important to also consider what is happening to the fibres at the network level. For example, do ‘self-sorted’ fibres also self-sort in terms of their network interactions, or will they interact with other fibres irrespective of their molecular-scale composition?
As noted by Adams,1c we don’t even really have clear terminology for these types of differences, and often simply refer to ‘self-sorting gels’ without reflecting on whether this applies across all length scales. In a key review, Adams and Draper gave guidelines for detailed characterisation of multi-component gels, which aim to help avoid some of the pitfalls of treating these complex system in a superficial manner.184
In the search for deeper understanding, Hamachi and co-workers have performed elegant studies to explore self-sorting in gels using time-resolved confocal laser scanning microscopy as a characterisation tool.185 They have exemplified the way different gelators can self-sort, gaining insight into the temporal resolution of these processes, following the assembly of two different gel networks with different kinetics in real time. They went on to control the network-level pattern of self-sorted systems by controlling the kinetics of seed formation using fast or slow oxime bond-forming reactions to generate a peptide LMWG in the presence of lipid-type nanofibers.186 When the peptide LMWG was generated rapidly it led to interpenetrated networks, but when it was generated slowly, the network was formed in parallel to the pre-formed lipid network, with the nanofibers being formed proximal to the lipid nanofibers rather than in the interstitial water space. Van Esch and co-workers also explored a multi-component system that self-sorts on the molecular-scale, then further separates into self-sorted micro-scale domains, such that hierarchical control over the self-assembled material occurs from the bottom-up.187
In addition to mixing two different LMWGs together to create multi-gelator gels, it is also possible to create multi-gelator gels in which an LMWG is mixed with a polymer gelator (PG).49 This is seen as a useful way of fusing desirable properties of LMWGs (responsiveness, programmability, sustainability) with the enhanced rheological performance often obtained from PGs. Hamachi and co-workers made a careful study of a range of LMWGs assembling with an agarose PG, and proposed four distinct assembly patterns that could result at the network level (Fig. 21) depending on network fabrication kinetics and interactions between the different components.188 We will further explore combining LMWGs with PGs to create hybrid gels with applications in later sections of this review.
 | ||
| Fig. 21 Cartoon schematic illustrating how LMWGs, when combined with agarose PG, can give rise to different types of network-level assembly pattern depending on the network fabrication kinetics and the interactions between network components. Figure reproduced from ref. 188 with permission of Springer Nature, © 2023. | ||
Beyond simple molecular additives, such as fragrances or drugs, a significant source of supramolecular interest has focussed on combining LMWGs with other self-assembling components. It is of fundamental interest to understand how different supramolecular pathways can be expressed in a multi-component smart material. In early work, van Esch and co-workers reported self-sorted assembly of an LMWG with lipid-based vesicles.189 If LMWG assembly was triggered in the supporting solution, then the vesicles became enmeshed in a gel network (Fig. 22a), whereas if LMWG assembly was triggered within the vesicle itself via a controlled pH change, it was possible to form rigid self-assembled nanofibers inside the vesicles, leading to their distortion (Fig. 22b).190 This has potential implications for the co-assembly of LMWGs with biological assemblies, such as cell membranes (see Section 4.7).
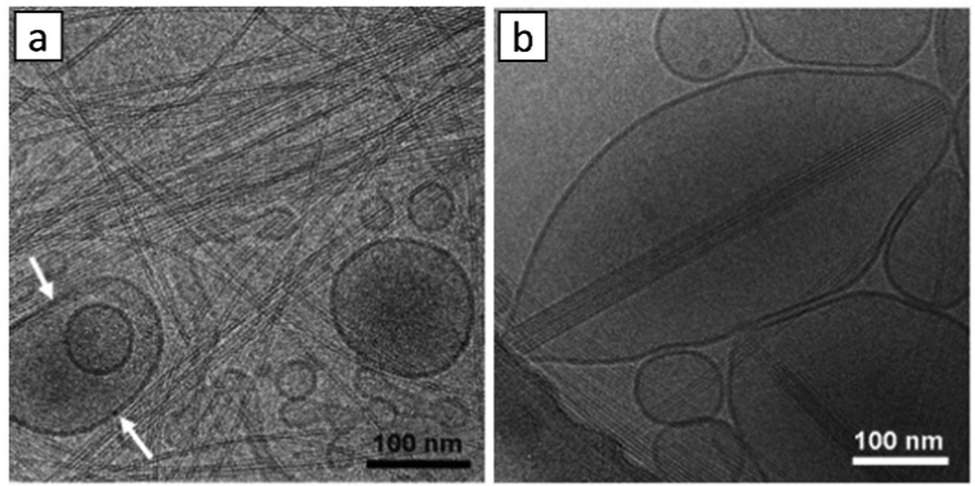 | ||
| Fig. 22 TEM images of self-sorted LMWG and surfactant structures. (a) LMWG assembled outside the vesicles. (b) LMWG assembled inside the vesicles via pH change. Image adapted and reproduced from ref. 190 with permission of Wiley-VCH, © 2008. | ||
Other researchers have combined surfactants and LMWGs with a range of different outcomes from self-sorted structures to disrupted co-assembled nanostructures.191 Stubenrauch and co-workers have studied in detail the co-assembly of LMWGs with surfactant-based lyotropic liquid crystal phases, and developed an understanding of the way different LMWGs interact with these phases, either acting as co-surfactants, or as potent gelators, depending on their structures.192 Meijer and co-workers have also explored combining gelators and surfactants with interests in controlling gel–sol transitions in a dynamic way as a function of concentration.193 Such materials are ‘complex fluids’, combining characteristics of surfactants and gels, with potential applications including in household cleaning and personal care.
As the 21st century has progressed, interest in multi-component gels has intensified. They offer advantages in terms of their ease of formation and high levels of structural variability. Blending materials together, or ‘formulation’, is a common strategy in both polymer and colloid science, which translates well to the supramolecular world of LMWGs. Developing multi-component gels also uses the expertise of supramolecular chemists in employing multiple interactions between different building blocks, and then understanding, optimising and using them to yield highly functional materials. It seems likely that such systems will underpin a range of important new developments, both in fundamental LMWG science, and its translation into new technologies.
3.5 Chirality in gels
Studies of chiral gelators date back further than might be expected. As described earlier, the helical packing of G-quartets in guanosine hydrogels was reported in 1962, using X-ray diffraction.88a The first visualisation of chiral LMWG nanofibers was reported in 1965, using transmission electron microscopy (TEM).195 As illustrated in Fig. 23, lithium 12-HSA formed fibres with opposite helicities depending on whether the chirality at the 12-position was ‘D’ or ‘L’. When a racemic mixture was used, although nanofibers still formed, they had no helical twist. Studies of 12-HSA assembly in the 1980s used circular dichroism (CD) spectroscopy, with significant CD spectra observed in the self-assembled gel-phase.196 For 1,3:2,4-dibenzylidenesorbitol, although each enantiomer is a proficient gelator in its own right, a racemic mixture of the two is not capable of forming a gel at all.197
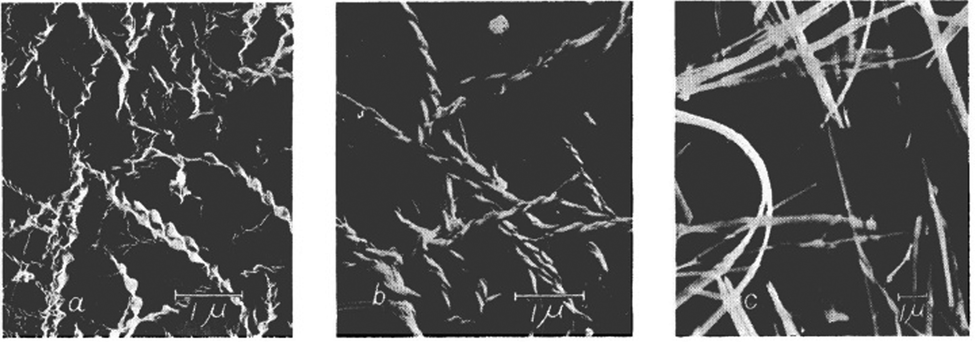 | ||
| Fig. 23 Original 1965 TEM images of lithium 12-hydroxystearate: (left) D-form, right handed twist, (centre) L-form, left-handed twist, (right) D,L-racemate, no twist. Reproduced from ref. 195 with permission of the American Chemical Society, © 1965. | ||
Clearly, the chirality of LMWGs plays a role in orienting the specific non-covalent interactions responsible for high-fidelity assembly, therefore it is not surprising it should have a significant impact on gelation. Although it is tempting to consider observations of nanostructures as part of the late 20th century ‘nanochemistry’ revolution, they were evidently quite well established before that, albeit with a lower level of prominence in the scientific discourse.
Electron microscopy and circular dichroism have remained at the heart of studies of chiral LMWGs. In particular, significant focus has been placed on using CD in more sophisticated ways. By varying the temperature at which spectra are recorded, it is possible to assemble and disassemble the gel. Although individual molecules in the disassembled state usually have small CD signals, in general, on gel assembly, the CD spectrum is enhanced as a result of the organisation of multiple chiral molecules within a helical nanostructure. This means CD spectroscopy is an effective means of quantifying gel assembly.
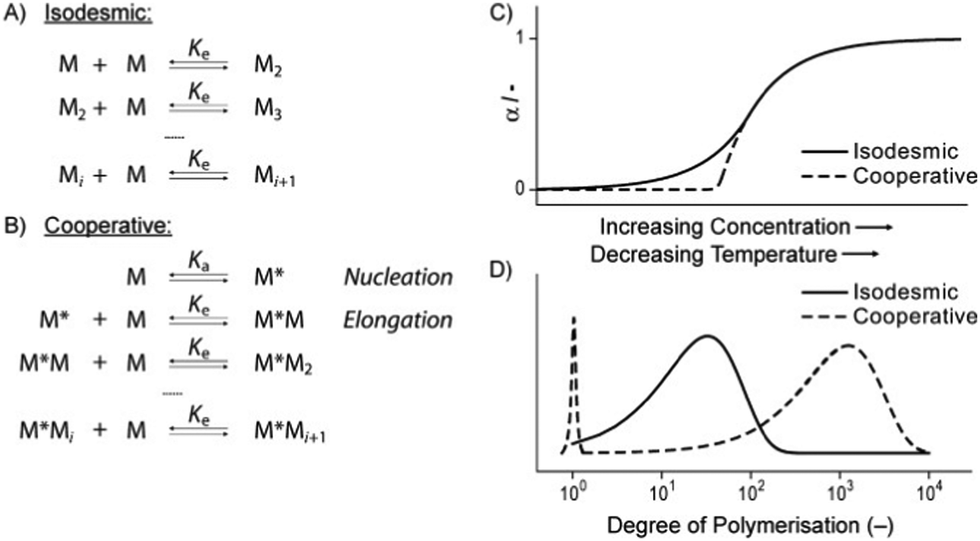 | ||
| Fig. 24 (A) Isodesmic and (B) cooperative models of self-assembly, (C) graph indicating how CD spectroscopy can differentiate between the two outcomes based on line-shape analysis, (D) differences in chain length for supramolecular polymers formed under the two assembly regimes. Figure reproduced from ref. 198 with permission from Wiley-VCH, © 2010. | ||
Thinking in more depth about how chirality influences assembly, Meijer and co-workers demonstrated that a small amount of chiral assembling ‘additive’ can bias the assembly of structurally-similar achiral building blocks into chiral nanostructures. This is described as ‘sergeants and soldiers’ assembly – one chiral ‘sergeant’ molecule is able to direct the assembly of hundreds of achiral ‘soldiers’.199 This occurs because the molecules co-assemble into mixed systems (see Section 3.4) and the chiral ‘information’ is transmitted along molecular recognition pathways between molecular-scale building blocks, with the chiral imbalance leading to an energy difference between one assembly pathway and the other. This type of process has been demonstrated to operate in a number of different gels.200 For example, just 1% of an aromatic amide gelator modified with chiral amino acid linker groups can direct the assembly of an achiral analogue into M or P helices depending on the chirality of the ‘sergeant’ molecule (Fig. 25).
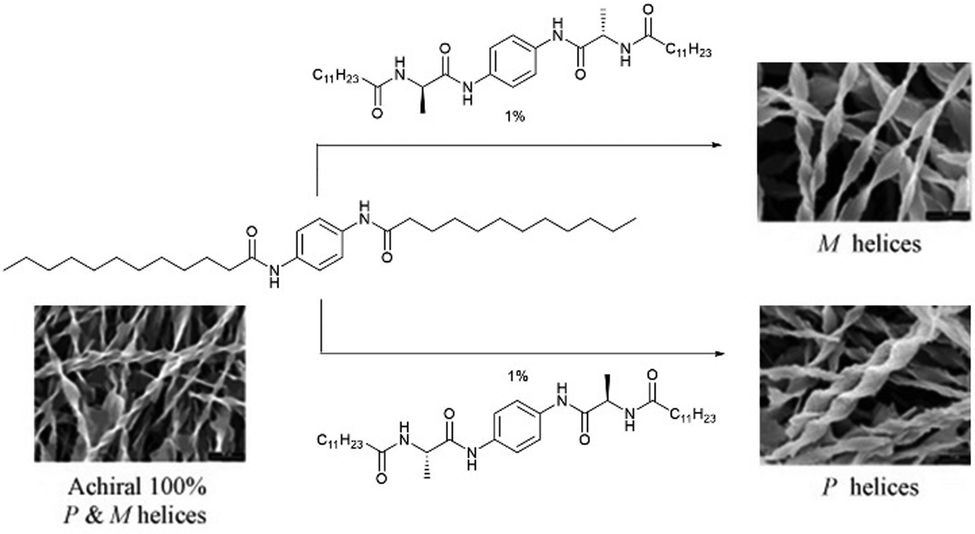 | ||
| Fig. 25 Sergeants and soldiers concept in which 1% of a modified chiral LMWG can direct the assembly of 99% of achiral gelator into a specifical chiral superstructure. Figure adapted and reproduced from ref. 200a with permission of Wiley-VCH, © 2008. | ||
• LMWG enantiomers disrupt one another's assembly creating a weaker gel than individual enantiomers, or no gel at all – this is the most common outcome and occurs when interactions between enantiomers have similar energies, meaning no/little discrimination in binding,
• LMWG enantiomers self-sort into separate chiral nanostructures – this indicates a strong preference for homochiral interactions at the molecular level,
• LMWG enantiomers co-assemble to create a more effective gel – this rare case201 suggests a preference for heterochiral assembly at the molecular level.
There is also an interest in the assembly of mixtures of enantiomers where the ratio is not 50:50. Meijer and co-workers demonstrated ‘majority rules’ systems, in which a small excess of one enantiomer can bias the assembly of the gel towards its preferred chiral nanostructure.202 As in sergeants and soldiers assembly, this indicates a difference in energies being induced by the slight excess of one enantiomer.
In addition to mixing LMWG enantiomers to create ‘multi-gelator gels’, it is also possible to mix a chiral gel with a chiral additive to form gelator-additive gels. It has been demonstrated that the chirality of the gel can influence the way it interacts with chiral additives, for example giving rise to gels with applications in enantioselective recognition or asymmetric catalysis (see Section 4.2).203 We demonstrated that the carboxylic acid at the focal point of a dendritic L-lysine preferentially formed gels with R-2-aminohexane rather than S-2-aminohexane.204 If the organogel was formed with the less-favoured S-enantiomer, and then exposed to the preferred R-enantiomer, it dynamically exchanged components, adapting and changing its composition (Fig. 26). This hints at the dynamic nature of gels, a topic returned to in Section 3.7.
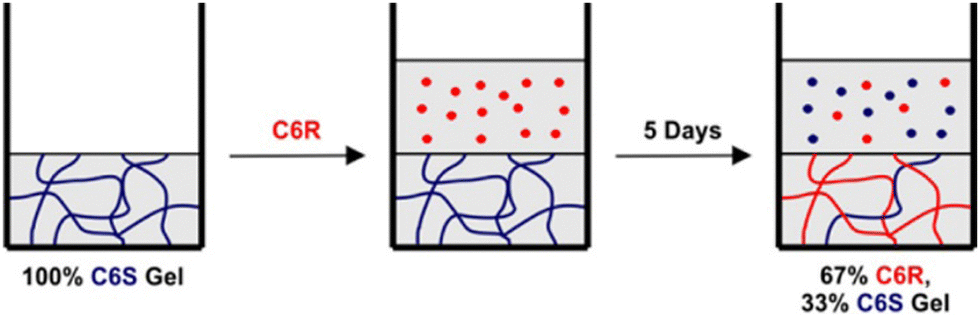 | ||
| Fig. 26 Schematic of thermodynamically controlled dynamic gel evolution on addition of R-2-hexylamine (C6R) to a gel made from a second generation L-lysine-based dendron (G2-Lys) and S-2-hexylamine (C6S). Figure reproduced from ref. 204 with permission of the American Chemical Society, © 2014. | ||
There has also been interest in determining, for two-component gels, which of two different chiral components can dominate assembly.205 Furthermore, it has been demonstrated that enantioselective recognition can differentiate between co-assembly and self-sorting, and in the case of donor and acceptor components, this can lead to control over energy transfer, with co-assembled components being in much closer proximity to one another than self-sorted architectures.206 This type of application is explored further in Section 4.6.
Feng and co-workers reported that enantiomeric forms of a peptide gelator interacted differently with cells, with cells appearing to thrive in one enantiomeric form of the gel, but not the other.207 They later demonstrated that the left-handed or right-handed helical nanofibers impacted differently on cell adhesion and growth, even if they were composed of the same naturally-occurring form of the amino acid.208 Intriguingly, this suggests that nanoscale chiral interactions can impact cell growth, not just the chirality of the amino acid building block.
Working with supramolecular polymers, Palmans, Meijer and co-workers have demonstrated that the solvent can also be considered an ‘additive’ and bias the assembly of chiral molecules.209 Specifically, a chiral solvent could overrule the stereochemical preference of the monomer. This work reinforces the importance of specific solvent-gelator interactions at the molecular level in mediating self-assembly (Section 3.3). Although unclear whether the systems reported formed gels, it seems likely such concepts will transfer to gels.
In addition to chiral building blocks giving rise to chiral nanostructures, achiral molecules can also assemble into chiral architectures. Of course, this usually results in a 50:50 mixture of mirror image nanostructures. In elegant work, however, Liu and co-workers created chiral microvortices in a microfluidics system, biasing nucleation, and assembling stereochemically-controlled supramolecular gels from achiral building blocks.210 They noted that micropores are common in rocks near volcanic vents, and went on to suggest that vortices in flow processes may plausibly have played a role in chiral amplification on the early Earth.
In summary, the fundamental nature of chirality in chemical science, and the fact many gelators are based on chiral building blocks and/or assemble into helical nanoscale architectures, mean that this remains fertile ground for investigation. Such studies give rise to new fundamental insights, but also can impact on applications in biomedical science, where chirality is ubiquitous (Section 4.7), and reaction engineering, where stereoselectivity is a prized outcome (Section 4.2).
3.6 Responsive gels
Perhaps one of the most obvious features of supramolecular gels on the macroscopic scale is that often, on heating, they undergo a transition from gel to sol.211 Indeed, this is one of the first things many researchers characterise when working with a new LMWG. The gel–sol transition occurs because as the temperature increases, the favourable entropy term associated with disassembly becomes more important and outweighs the favourable enthalpy associated with assembly, meaning the free energy of gel disassembly becomes favourable.The gel–sol transition has long been recognised as an important parameter – for example, the thermal instability of calcium stearate organogels drove developments in lubrication technology during the late 19th and early 20th centuries (Section 2.1). More recently, there has been interest in gel–sol transitions at body temperature for use in drug delivery (Section 4.1).
Typically, the gel–sol transition temperature increases with concentration until it reaches a plateau value associated with saturation. We demonstrated this concentration-dependent behaviour could be modelled by understanding of LMWG solubility thermodynamics based on the van’t Hoff equation – i.e. within a family of related gelators the solubility of the LMWG could predict both the magnitude of Tgel, and the overall lineshape of its response to LMWG concentration (Fig. 27).212
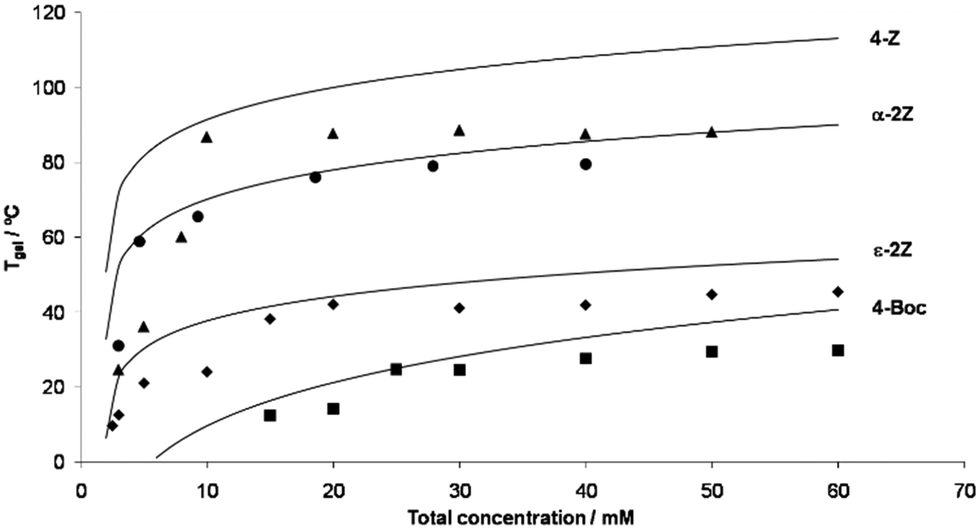 | ||
| Fig. 27 Illustration of fitting of experimental Tgel values based on tube inversion measurements for four different gelator structures, fitted to solid lines representing the Tgel values calculated based on solubility values extracted from the van’t Hoff equation, relating to complete solubilization of the networked gelator, demonstrating how solubility considerations predict the general values and lineshapes, including plateau regions. Figure reproduced from ref. 212 with permission of the American Chemical Society, © 2008. | ||
Intriguingly, a recent study reported an LMWG that only formed gels on raising the temperature above a lower critical solution temperature.213 On heating, the LMWG aggregated to form a turbid solution – this was entropically driven through desolvation, and was required prior to assembly into a gel. This demonstrates how different assembly mechanisms, with alternative thermodynamic driving forces, can modify the sol–gel transition.
Given the well-defined non-covalent interactions involved in self-assembly, there has also been increasing interest in intervening in the assembly process to trigger the gel–sol transition using different mechanisms and create a diverse range of responsive gels.214
As noted previously, many hydrogels are pH-responsive. Acid-functionalised gels assemble on protonation to the neutral form at lower pH values,133 while (more rarely) amine functionalised gels assemble on deprotonation to the neutral form at higher pH values.215 These processes decrease solubility and enhance fibre-fibre interactions. Kinetically slow pH changes, such as the hydrolysis of glucono-δ-lactone (GdL),136 are beneficial in yielding homogeneous gels. In recent work, Ravoo and co-workers introduced an innovative new chemical strategy for lowering pH in a controlled way,216 using the redox reaction between iodine and a thioether to create protons in situ, and trigger gel assembly.
Light is also a fascinating trigger. Photoacids have been used in combination with LMWGs so light can trigger protonation – a good example is diphenyliodonium nitrate, which breaks down under UV irradiation to yield iodobenzene and nitric acid.217 Alternatively, LMWGs have been designed in which photoirradiation induces a conformational switch that alters the molecule from a soluble form to a self-assembling form (or vice versa) – typical examples are based on azobenzenes which undergo light induced trans–cis interconversions.218 Thordarson and co-workers reported an ultra-low-molecular-weight photoresponsive gelator based on E/Z isomerisation of an arylazopyrazole (Mr = 258).219 This clearly indicates how synthetic supramolecular chemists can incorporate design features into LMWGs to give them behaviours significantly beyond what can be achieved by traditional LMWGs.
Magneto-responsive gels have also been reported – in general, this is achieved by incorporating magnetic nanoparticles into the gel, such that exposure to a magnetic field, results in the network being broken down as the magnetic nanoparticles get attracted towards it.220
The interest of supramolecular chemists in LMWGs, with a focus on binding events, has led to gels which undergo gel–sol transitions in the presence of an analyte.221 Multi-component gels (Section 3.4) provide some examples. Typically, the binding of a guest leads to a new structure with different solubility and assembly profiles, hence triggering gel assembly (or disassembly). In early work, Steed and co-workers recognised that urea functional groups, as well as being privileged self-assembly motifs, can also bind anions though hydrogen bonding.222 When bound to an anion, the urea groups are no longer able to form extended hydrogen bonded tapes, and it was demonstrated that urea-based gels can be responsive to the different anions, which can convert the gel to a sol.223 The degree of responsiveness depends on the strength of anion binding, and the visual nature of the gel–sol transition means such systems have potential applications as sensors – a theme we return to in Section 4.5.
An alternative way of triggering gel assembly/disassembly is to use enzyme-catalysed reactions to convert a gelator into a non-gelator (or vice versa). This concept was explored in a seminal review from Xu and co-workers.224 Indeed, the first example of enzyme-triggered gelation was reported by them, using alkaline phosphatase to dephosphorylate an Fmoc-protected amino acid, generating an active LMWG in situ (Fig. 28, top left).225 This led to an explosion of interest in using enzymes to trigger gel assembly.226 The use of enzymes to trigger gel assembly/disassembly has significant relevance in the development of next-generation applications, because it enables supramolecular gels to detect or intervene in specific biological processes (see Sections 4.1, 4.5 and 4.7).
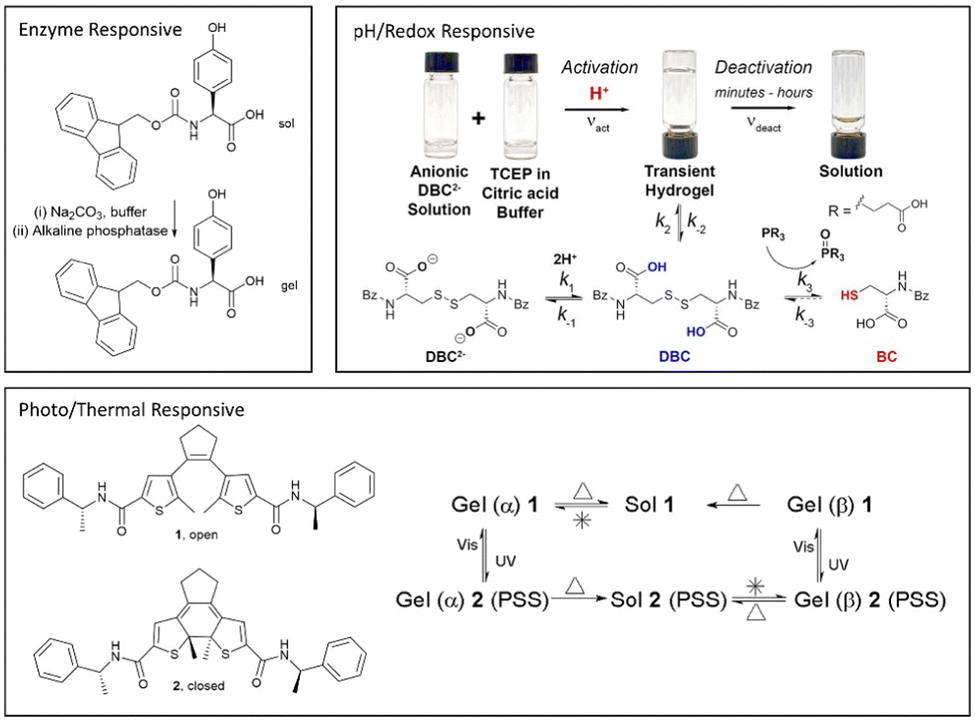 | ||
| Fig. 28 Examples of responsive gelators. (Top left) Enzyme responsive gelator first reported by Xu and co-workers. (Top right) Response of dibenzoylcystine to pH and redox chemistry, figure reproduced from ref. 229 with permission of the American Chemical Society, © 2018. (bottom) Response of diarylethene gel system to UV/Vis-radiation and variable temperature. | ||
There has also been considerable interest in using enzymes to create dynamic materials.227 In a landmark paper, Ulijn and co-workers demonstrated that varying the amount of enzyme could modify the extent of higher level supramolecular order in ways not easily possible using other assembly triggers.228 By controlling enzyme loading, it was possible to obtain a variety of different materials from the same concentration of LMWG. They postulated the presence of biocatalytic clusters that helped to enhance order at molecular, nano and micro levels.
Older gels have also been made more responsive. In elegant work, Thordarson and co-workers took dibenzoylcystine (Section 2.7) and demonstrated that it could exhibit different response mechanisms (Fig. 28, top right).229 Not only is it pH-responsive as a result of the carboxylic acids, it is also redox-responsive because the disulfide linkage can be reduced to individual thiols, switching off gelation. Fascinatingly, if both acid and reducing agent were applied at the same time, gel formation induced by acid, slowed down the gel disassembly induced by the reducing agent, leading to a transient gel. This illustrates how gel responsiveness can induce dynamic/transient behaviour (Sections 3.7 and 3.8).
Beyond focus on simple gel–sol transitions, there is increasing attention on supramolecular gels that respond by transforming from one ‘state’ to another. For example, it has been demonstrated that azobenzene isomerisation, instead of switching gelation on and off, can convert a gel from a stiffer to a softer form, modifying elasticity in a reversible manner.230 In fascinating early work, Feringa and co-workers demonstrated that a chiral diarylethene LMWG had a preference to assemble into one helical form (α), with UV irradiation then driving a ring closing reaction that, when combined with a heat–cool cycle, enabled switching of the α helical form of nanofibers to the β form (Fig. 28, bottom).231 This ability of gels to exist in different dynamic states underpins much current interest, and will likely see significant growth in the coming years. Some further examples are given in the following sections.
3.7 Dynamic gels
The difference in dynamics between the solid-like and liquid-like parts of a gel can be detected by NMR methods – molecules in the liquid-like phase can be detected using standard NMR spectroscopy, while the resonances of molecules associated with the solid-like network are broadened and not visible in the NMR spectrum.98,232 With the use of mobile, non-interacting, internal standards, this can allow detailed quantification of the ‘mobile’ and ‘immobile’ parts of a gel and provide insight into its dynamic nature. Furthermore, if something in the liquid-like phase interacts with the solid-like network it may become immobilised and hence invisible to NMR. This approach is particularly useful in characterising, in a quantitative way, multi-component gelator-additive gels. Diffusion NMR methods can also provide quantitative measures of diffusion coefficients within a gel.233
It is worth noting here that the ability of small molecules to diffuse within gels, also gives them capacity to diffuse out of gels leading to intriguing mechanisms of gel growth and adhesion – these principles are explored further in Section 3.9 on shaping and patterning gels.
LMWG assembly is dynamic, and is usually under kinetic control. During self-assembly, molecules explore an energy landscape, and can sometimes initially assemble into what is only a local minimum.234 There remains sufficient energy in the system for it to reorganise over longer timescales into a different material. As such, gels should be considered as metastable, kinetically-trapped materials. Although they may be stable on experimentally-useful timescales, they may also evolve over time. There can be multiple minima in an energy landscape, and a gelator might therefore assemble in several different forms simultaneously, or be able to be switched from one structural polymorph to another either at random, or with relatively small changes to gelation conditions.235 This demonstrates how gelation is related to crystallisation, which is well-known to exhibit this type of effect, with different crystal polymorphs resulting depending on the nucleation event that starts the assembly process.236
A good example of an LMWG that explores an energy landscape was reported by Stupp and co-workers.237 They used a peptide amphiphile (PA) which forms hydrogen bonded β-sheets between peptides, and explored its response to: (i) dilution, (ii) thermal annealing and (iii) addition of salt. They found that they could access a thermodynamic product, a metastable product, or a kinetically-trapped product depending on the order in which stimuli were applied. By navigating the energy landscape (Fig. 29) in different ways, it was found that β-sheet formation could kinetically lock molecules in conformations that are thermodynamically unfavourable, leading to the different outcomes. The PA gels formed using different approaches had different properties in terms of biological cell adhesion and survival, emphasising that nanostructure, not just molecular structure, can impact on the way in which a gel performs.
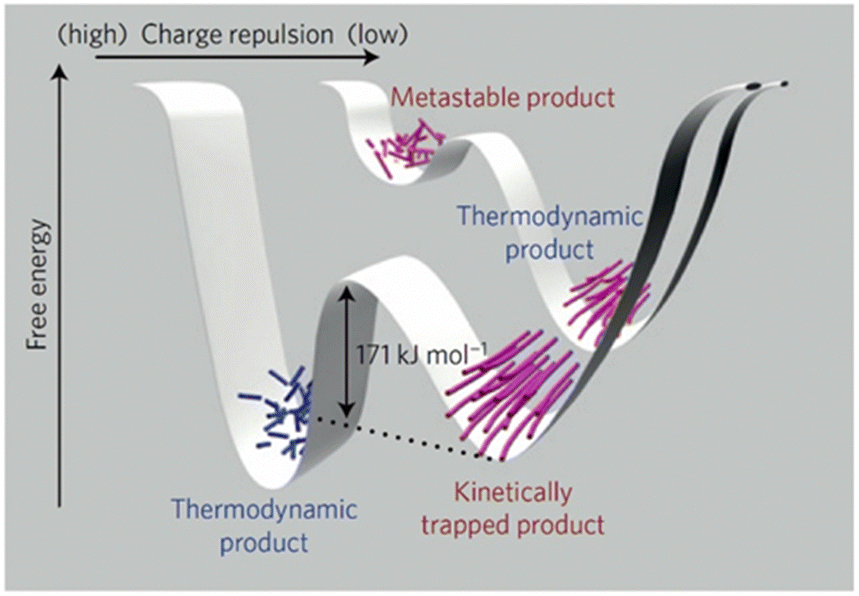 | ||
| Fig. 29 Schematic representation of the free energy landscapes of peptide amphiphile self-assembly with relatively low (back) and high (front) charge-charge repulsions. At low intermolecular repulsion, long fibres with β-sheets are favoured and monodisperse short fibres represent a metastable state. At high repulsion, a kinetically trapped assembly and a thermodynamically favoured product separated by an energy barrier (Eb) of 171 kJ mol−1 were found. Figure reproduced from ref. 237 with permission of Springer Nature, © 2016. | ||
The fact that gels occupy a balanced position between solubility and assembly means that some gels can evolve into a crystalline form, that phase separates from the solvent over time.238 This gel-crystal transition has been characterised in real-time for a pentapeptide LMWG by Gazit and co-workers.239 They applied microscopic and microrheological methods to show that the gel–crystal transition initiates sporadically and follows a sigmoidal kinetic profile, exploring the complex local dynamics of gel weakening and dissolution associated with microcrystalline growth. They also found that external factors such as temperature, solvent and mechanical perturbation, can have significant impacts on this process.
This type of metastability can limit the application of some gels, and it is therefore important to test temporal stability. Furthermore, on ageing, one gel nanostructure can evolve over time into another, for example by lateral assembly of individual gel fibres into larger assemblies, or by slow reorganisation of one form into another more stable form.240 Such considerations are relatively commonplace in polymer science, where it is well understood that processes occurring on different length-scales can have different kinetics.241 The fact that LMWGs assemble into polymer-sized objects means it is not surprising that they can also exhibit similar behaviour, although for some small molecule chemists it is initially unexpected.
Adams and co-workers have spent considerable time in seminal work exploring the assembly of LMWGs based on amino acid building blocks and have developed a detailed understanding of the way the precise conditions employed during gel preparation can control the outcome. For example, they found their dipeptide-acid gels differ depending on whether the LMWG is dissolved in an organic solvent or basic solution prior to assembly, and whether assembly is triggered by acid or salt.242 Furthermore, they demonstrated that in multi-gelator self-sorted gels, performing an annealing step on the kinetically-controlled gel could modify it at the network level of assembly.243 Elegantly, Draper and co-workers found that in their multicomponent gels, mixing the components as solids led to co-assembled gel fibres, whereas mixing them as solutions led to self-sorted nanofibers.244
The impact of ageing on seeded assembly has also been investigated by van Esch and co-workers, who developed a multi-component system in which ageing one component led to a degree of pre-assembly of ‘seeds’ which subsequently directed gel assembly down a different pathway, into a metastable state that then reorganised into a more thermodynamically stable form.245 As such, not only is ageing of the gel important, but also the way gel precursors are treated can have a significant impact on assembly mode.
The relationship of gels to concepts of crystallisation is also reflected in the fact that gels have different assembly rates, with some gels forming quickly and some slowly. Meijer and co-workers demonstrated that solvent could play an intimate role in mediating the kinetics of assembly.164,246 Efforts have been made to understand gelation kinetics in a more predictive way, for example, we reported a family of gelators in which the rate of nucleation and assembly correlates with LMWG solubility, while the thermodynamic gel stability is controlled by different factors.182
In short, it is not enough to simply characterise ‘a gel’ formed by an LMWG and expect to capture a detailed understanding of the way that system behaves. It is vital to consider how the gel has been made, the stimuli it has been exposed to, the way things have been mixed, the length of time gel has been aged, and the conditions in which the ageing itself has taken place, amongst numerous other factors. It is expected that many more studies of this type, getting to grips with the true complexity of gel assembly, will be performed in future.
In order for a gel to ‘self-heal’, it is suggested here that the energy barrier to gelation should be overcome without need for a significant triggering event. Systems that seem well-suited to exhibiting self-healing properties are ones where the LMWG has significant solubility in its own right and readily exchanges between liquid-like and solid-like phases. Alternatively, systems based on two components that interact with one another to form a gelator (e.g. Section 3.4) are also suited to self-healing, as the individual components often have high solubility in their own right and are in equilibrium with the gelator that assembles into a gel.249 As such, on forced disassembly, the individual components remain in solution, prior to reforming the gelator and reassembling into a gel.
Aida and co-workers made use of a self-healing mechanism to adhere supramolecular gel blocks together.250 The ability of supramolecular systems to assemble further through new non-covalent interactions played a key role. Freshly cut surfaces of LMWG gel blocks acted as nucleation sites for further assembly, which caused effective adhesion of one block to another at the exposed surface as new supramolecular interactions were formed. We return to this approach in Section 3.9 as a way of shaping and patterning gels.
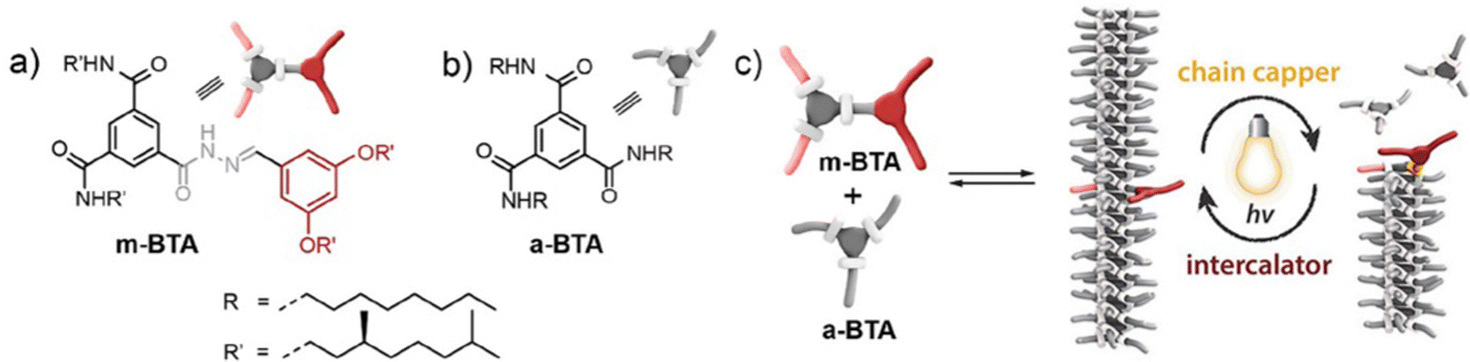 | ||
| Fig. 30 Molecular structure of (a) mono acylhydrazone functionalized BTA (m-BTA), (b) achiral octyl BTA (a-BTA) monomers, and (c) copolymer formed by intercalation of chiral m-BTA into helical stacks of a-BTA. Light induced isomerisation of the acylhydrazone linkage in m-BTA converts it into a chain capper, and shortens the length of the assembled nanofibers. Figure reproduced adapted from ref. 251 with permission of the American Chemical Society, © 2020. | ||
Thinking about molecular mobility within a gel, Zeng, Li and co-workers incorporated a rotational molecular motor, of the type popularised by Feringa, into a supramolecular gel (Fig. 31).252 They demonstrated that the motor retained its diffusional mobility and light-activated ability to rotate. Fascinatingly, rotation of the motor perturbed the gel, causing a gel-to-gel transition, modifying morphological and rheological properties. This work creates the intriguing suggestion that the energy of a molecular motor can be coupled to material outputs – a new mechanism of dynamics within these gels.
 | ||
| Fig. 31 Incorporation of molecular motor into self-assembled gel, with light-induced rotation of the motor giving rise to a gel-to-gel transition. Figure reproduced from ref. 252 with permission of Wiley-VCH, © 2023. | ||
In summary therefore, gelation is a much more complex process than some appreciate. For a full understanding, it is essential to consider the entirety of the assembly pathway/process, and carefully characterise the materials that result. Although this adds significant challenges to gel chemists in terms of characterisation compared to those working on ‘hard’ materials, it is also one of the primary advantages of supramolecular gels. They are highly dynamic, evolvable and adaptable materials – an understanding of this can unlock a variety of high-tech applications, moving them well beyond being simple ‘on–off’ rheologically-active materials, and enabling them to play more active and engaged roles in next-generation technologies (see Section 4).
3.8 Dissipative (‘fuelled’) assembly
Building on the dynamic nature of gel assembly discussed above, there has been considerable interest in using dynamic processes to ‘fuel’ gel formation in a so-called ‘dissipative assembly’ process. Key reviews describing this concept have been produced by the groups of Boekhoven and Hemans.253 This idea was pioneered by van Esch and co-workers, who developed a multi-component gel based on well-established dibenzoylcystine, that used an alkylating agent as ‘fuel’ to generate an ester LMWG from the anionic soluble carboxylate, with the process being slowly reversed over time by ambient hydrolysis (Fig. 32, top).254 The amount of fuel controlled the extent of assembly, a process that slowly reversed over time once the fuel supply was exhausted. Refuelling the system caused assembly to be triggered again. In other words, the gel can be considered transient or ‘dissipative’. | ||
| Fig. 32 Examples of fueled assembly. (top) General schematic of fueled assembly and example of fueled assembly based on esterification of dibenzoylcystine – figure adapted and reproduced from ref. 254 with permission of Wiley-VCH, © 2010. (bottom left) Tuning a gelator for fueled assembly. If there are insufficient interactions the system remains as a sol, with too many interactions it is a gel even without fueling, and if the interactions are just right, its assembly can be transiently fueled – figure adapted and reproduced from ref. 256 with permission of the American Chemical Society, © 2020. (bottom right) System capable of transient sol–gel–sol–gel–sol assembly – figure reproduced from ref. 257 with permission of Wiley-VCH, © 2023. | ||
In their review,253a Boekhoven and co-workers suggested three strategies by which a system could be triggered to self-assemble in response to a fuelled reaction:
• loss of charge leading to greater ability to aggregate,
• coupling two non-assembling units to create an assembling unit,
• conformational change into a self-assembling form.
A number of reports have built on the concept of dissipative assembly, demonstrating that hydrogels can exist in non-equilibrium states, responding to a variety of fuels, including solvent playing an active role, and having their properties reprogrammed over a number of cycles.255
In one interesting example, Boekhoven and co-workers took a rational design approach to tune the number of attractive interactions between hydrophobic self-assembling peptides in order to toggle between permanently and transiently assembled structures (Fig. 32, bottom left).256 It is also possible to create systems showing multiple transitions – for example Singh and Hermans reported a fueled acid/aldehyde/imine system that underwent sequential sol–gel–sol–gel–sol transitions (Fig. 32, bottom right).257 Furthermore, it was demonstrated by Quintard and co-workers that dissipative assembly can be used to switch a sol–sol–gel system between three different chiroptical states, with the potential to cycle multiple times.258
Fuel-driven self-assembly widely exists in biology, with functional micro- or nanostructures in living bodies often being transiently formed by biomolecular self-assembly far from equilibrium, driven by active molecules (chemical fuel), like adenosine triphosphate (ATP).259 Given this biomimetic relevance, there has been considerable interest in using enzymes to drive such processes. In relatively early work, Ulijn and co-workers developed a cycle of gelators and non-gelators that could be interconverted using chymotrypsin and thermolysin, and demonstrated that the system could be fueled by addition of an amino acid precursor – in this way, non-assembling units were reversibly connected together to give a self-assembling system capable of forming transient gels.260
In a number of cases, reactivity and self-assembly are coupled processes. Das and co-workers reported an example in which histidine catalysed an ester formation to form a gel, but then a second process, catalysed by the now-proximal histidine units in the self-assembled gel state, led to hydrolysis of the ester to break down the gel once again.261 In this way, not only can the rates of chemical processes influence transient assembly, but the assembly event itself can influence the rates of chemical processes. The effect of self-assembly on catalysis is a topic that we will return to in Section 4.2.
Recently, Hermans and co-workers demonstrated that transient assembly can be coupled with multi-component systems to yield self-sorted gels. Building on their dissipative system based on an aldehyde-containing saccharide, disassembled by conversion into its soluble hydroxysulfonate analogue,262 they created a small family of similar molecules, and demonstrated they were capable of fuel-driven self-sorting.263 On simple cooling, these molecules tended to co-assemble, but with chemical fueling, the ‘chemi-gels’ self-sorted dependent on the rates of reaction of different components with the fuel, a process which could be kinetically predicted based on an understanding of electronic effects.
The capacity to fuel the assembly of transient gels offers considerable potential in applications where adaptive, dynamic materials may be required – for example to interface with living tissue, which is itself adaptive and dynamic (Section 4.7). Currently, researchers studying dissipative assembly are exploring scope primarily from an academic point of view, but it is anticipated that innovative uses of this technology will being to emerge in the coming years, especially at the biological interface.
3.9 Shaping and patterning gels
In many of the earliest supramolecular gels, attention simply focussed on gel formation, and photographic images of upturned vials containing a gel were ubiquitous in the literature. However, for more sophisticated applications it may be necessary to provide these materials with shapes and patterns (Fig. 29). As such, in recent times, attention has begun to turn to addressing this goal – a topic we unified for the first time in a 2019 review.264 Many supramolecular gels are relatively weak in rheological terms, and this makes shaping and patterning a challenge. A number of chemical engineering approaches have been developed, and a brief overview is given here. Many of these strategies rely on the responsive nature of supramolecular gels (Section 3.6). | ||
| Fig. 33 Overview of methods for shaping and patterning gels. Moulding and healing are used to create gels in defined shapes, with self-assembly enabling repair if the gel is damaged. Photopatterning can be used to assemble (or disassemble, not shown) a gel network with spatial resolution. 3D printing is used to print self-assembled gels with spatial and temporal resolution – this can be combined with photo-patterning to achieve additional patterning. In surface patterning a catalytically active patterned surface triggers spatially resolved gel assembly. Diffusion assembly uses the diffusion of gelators and gelation triggers through a pre-formed matrix to generate an LMWG in situ leading to spatially and temporally resolved gel assembly. Figure reproduced and adapted from ref. 264 with permission of Springer Nature, © 2019. | ||
Other ways of enhancing rheological performance are to formulate LMWGs with additives, such as polymer gels. Such materials combine two different networks – one to provide function (LMWG), and one to reinforce and shape the material (PG).49 This strategy was first employed by Yang and co-workers who used agarose as a robust polymer gel supporting a LMWG.266 The fracture stresses of the hybrid hydrogels were 20 times higher than those of hydrogels in the absence of agarose. The hybrid hydrogels could be fabricated into different shapes, and allowed other components to be incorporated.
The use of polymers can also allow more sophisticated gel shaping. By adding a solution of LMWG and alginic acid drop-wise to aqueous CaCl2, we created LMWG gel beads (Fig. 34, left and centre).267 The supramolecular gel assembled as the core inside a stabilising shell of calcium alginate. Essentially, this is a LMWG moulding process using a soft PG-based mould. We then went on to demonstrate that these gel beads can be made with diameters as small as ca. 800 nm (Fig. 34, right), by fabricating them in aqueous surfactant-stabilised droplets in paraffin oil, and crosslinking with Ca2+. These nanogel beads can be injected through a needle without damage,268 opening a range of possible biological applications (see Sections 4.1 and 4.7). We have also developed methods to fabricate hybrid gel beads with other polymers like agarose and gellan gum.269
 | ||
| Fig. 34 (left) Schematic of gel beads formed using calcium alginate to encapsulate an self-assembled LMWG core. (centre) SEM image of gel bead formed with diameter of ca. 2 cm by dropping LMWG and alginic acid into aqueous calcium chloride. (right) SEM image of gel beads with diameters of ca. 800 nm formed by dropping alginic acid, LMWG and surfactant into a paraffin bath, followed by cross-linking with aqueous CaCl2. Figures adapted from ref. 267 with permission of Wiley-VCH, © 2020, and 268 with permission of the Royal Society of Chemistry, © 2021. | ||
It is also possible to adhere moulded gel blocks together in order to construct larger architectures, using them rather like Lego™ bricks. In an early study, Aida and co-workers adhered individual moulded supramolecular gel blocks (reinforced with a nanoscale clay) via a self-healing dynamic mechanism (Section 3.7).250 As noted above, some gels have greater capacity to self-heal,247 and this gives them particular potential with regard to creating interfaces that adhere to one another, and structures that can repair themselves once damaged (Fig. 33).
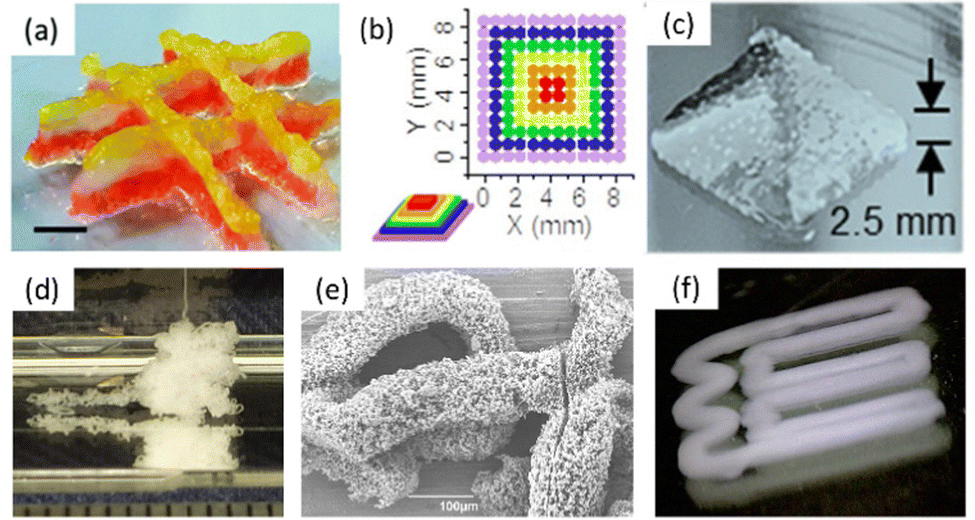 | ||
| Fig. 35 (a) Extrusion printing of an LMWG loaded with different coloured dyes in layers, figure reproduced from ref. 270 with permission of the Royal Society of Chemistry, © 2017. (b) Pattern for layer-by-layer printing of a pyramid, and (c) printed gel pyramid based on two different LMWGs with opposite charges, figures reproduced from ref. 273 with permission of the American Chemical Society, © 2019. (d) Wet-spinning of an LMWG, (e) SEM image of wet spun fibre showing the nanofibrillar assembled architecture, and (f) 3D-printed object produced by layer-by-layer wet-spinning printing method, figures (d)–(f) reproduced from ref. 278 with permission of the Royal Society of Chemistry, © 2022. | ||
An alternative approach to extruding a pre-formed gel, is to inject a dissolved gel precursor into a ‘coagulant solvent’ to initiate gel assembly. Pioneering research from Stupp and co-workers, fabricated string-shaped hydrogels by injecting a peptide amphiphile into calcium chloride, with the divalent ions inducing fibre crosslinking.275 A similar methodology was adopted by the groups of Hartgerink and Mihara.276
Perhaps more generally, Fitremann and co-workers introduced wet-spinning methods in which an LMWG is dissolved in a ‘good solvent’ (e.g. DMSO) and then injected into a ‘bad solvent’ (e.g. H2O) to trigger rapid self-assembly.277 This gives rise to thin gel filaments that can also be printed in water onto a substrate, layer-by-layer, creating 3D-printed gel objects with good resolution (Fig. 35d–f). Working with Fitremann, we demonstrated that the choice of gelator could enhance the stability of these printed objects depending on its solubility, with some patterns remaining stable for many days in water.278 We went on to demonstrate that multiple gelators could be wet-spun simultaneously, with the resulting gel filaments being co-assembled as a result of their rapid formation, and exhibiting combined responsive properties from the two gelators.279 This simple, low-tech approach to printing objects has considerable versatility, and can co-assemble different functional units to create multi-functional materials.
Rather than printing a gel, Davis and co-workers developed an innovative approach to print onto a gel.280 They used a hydroxamic acid functionalized G-quartet gelator, and on exposure to iron(III) the gel gained an orange/red colour via complexation. Using a 3D-printer to deliver the Fe(III) solution allowed them to write patterns into the gel. These patterns could be reduced using ascorbic acid (vitamin C), converting Fe(III) to Fe(II). Although the printing here was primarily visual, it is possible to imagine using this approach to deliver additives that change mechanical, or electronic/biological properties of a gel in a spatially resolved manner.
In 2012, Adams and co-workers used the photo-acid diphenyliodonium nitrate (DPIN) to activate a pH-responsive gel – using of a mask led to spatial resolution.217 We also applied DPIN-triggered assembly to pattern a pH-responsive LMWG within a pre-formed pH-stable supporting LMWG network, leading to multi-component gels with excellent spatial resolution (Fig. 36, centre).283 The light thus achieves ‘positive writing’ of one LMWG network within another. We described such materials as ‘multi-domain’ gels because they contain domains with different compositions. In later work, we combined these LMWGs with a photocrosslinkable polymer gel (PG), leading to three-component patterned gels, where the timescale of irradiation controlled whether only the PG was crosslinked (fast) or the photo-responsive LMWG was also assembled (slow).284 In this way, we patterned a gel with four different domains (Fig. 36, right). Photopatterned gels have also been created using ‘negative writing’ in which two gelators are assembled into self-sorted networks, and then one of them is disassembled by UV irradiation – in the example reported by Adams and co-workers, as a result of conversion of a trans-stilbene into a cis-stilbene.285 Once again, this gave a multi-domain gel in which either one or both networks were assembled in different domains (Fig. 36, left).
 | ||
| Fig. 36 Examples of photopatterned gels. (left) Gel in which a star image is photopatterned into a two component gel by disassembling one of the two self-sorted gel components as a result of photo-isomerisation (negative writing). Figure reproduced from ref. 285 with permission of Springer Nature, © 2015. (centre) Gel in which one LMWG is assembled in the shape of the Eye of Horus by phototriggered acid release within a pre-formed pH-stable supporting supramolecular gel. Figure reproduced from ref. 283 with permission of the American Chemical Society, © 2015. (right) Four-domain gel, in which each domain has a different assembled composition, by using three orthogonal components triggered in different ways – pH change (LMWG1), slow UV irradiation (LMWG2) and fast UV irradiation (PG). Figure reproduced from ref. 284 with permission of The Royal Society of Chemistry, © 2020. | ||
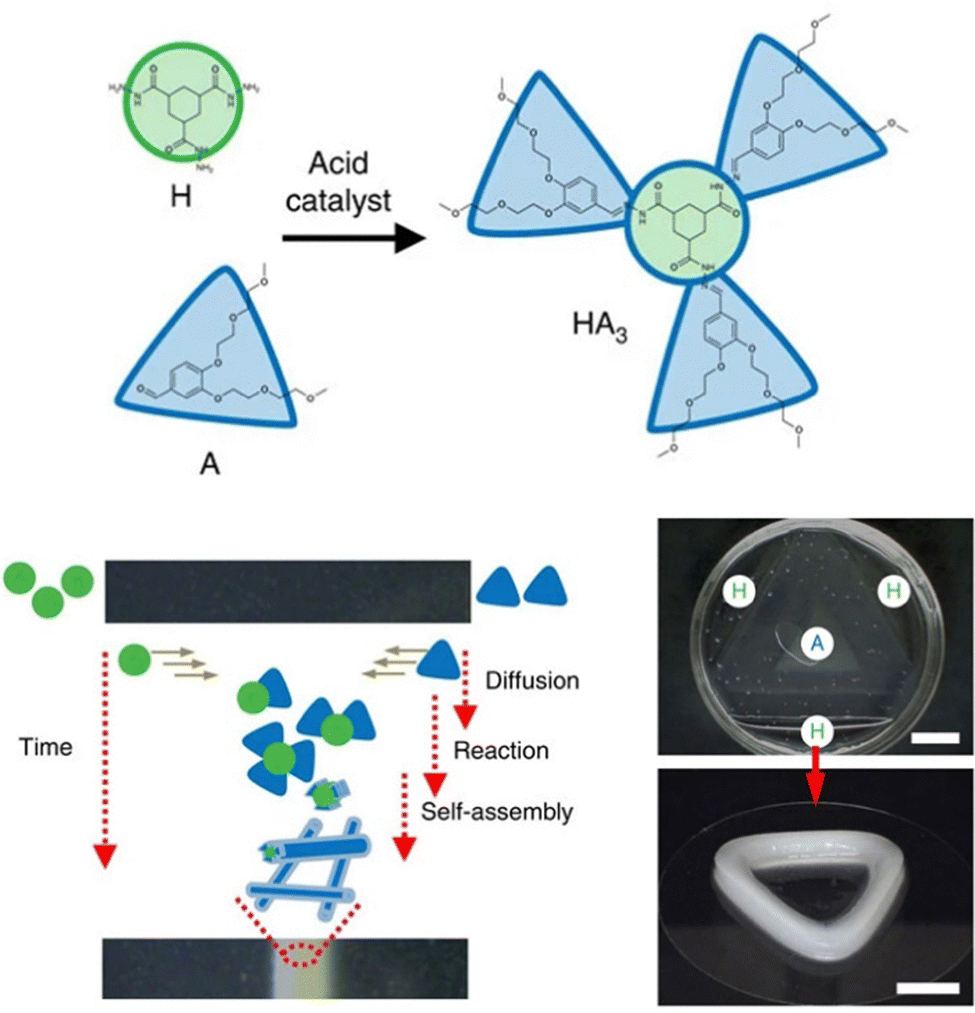 | ||
| Fig. 37 Two-component gel formed by dynamic reaction between acylhydrazide (H) and aldehyde (A). Allowing diffusion to bring the two-components together within a pre-formed polymer gel matrix causes the LMWG to assemble with spatial resolution dependent on the starting locations of the H and A components. Figure adapted and reproduced from ref. 286a with permission of Springer Nature, © 2017. | ||
This dynamic covalently bound two-component gel could also be used to adhere polymer gel blocks together. If blocks were loaded with each of the two components, then as they diffused and came into contact at the interface between blocks, they reacted to form the LMWG, and the self-assembled structures that formed gave highly effective adhesion.287
Diffusion can also create transient gels that assemble with spatial resolution and then disassemble again. By diffusing an acid through a pre-formed pH-stable supramolecular gel, we assembled a pH-responsive LMWG within it, creating a multi-domain gel with the patterning depending on acid loading.288 If the amount of acid was limited, then the surrounding basic solution eventually disassembled the growing domain of pH-responsive gel. This process of gel growth and loss could be repeated by loading more acid (Fig. 38). We went on to demonstrate that by loading acid, and deprotonated gelator into different wells, diffusion of the two components could pattern gels analogous to those described by van Esch and co-workers,289 with assembly kinetics controlling the shapes.
 | ||
| Fig. 38 Photographs of gels indicating how acid addition triggers growth of an assembled ring of DBS-COOH. Over time (+48 h), diffusion of base from the exterior then largely disassembles the transient DBS-COOH gel, which can be re-assembled by fuelling with more acid – creating a dissipative, oscillating gel system driven by the diffusion of fuel. Figure reproduced from ref. 288 with permission of the Royal Society of Chemistry, © 2021. | ||
In work using a photo-acid generator, Adams and co-workers showed that a weak gel formed on the addition of a small amount of GdL, could be converted into a stiffer gel on activation of a photoacid. When only one part of the gel was irradiated, this led to a gradient from stiffer to softer regions as photo-generated protons diffused. Over time, this gradient dissipated, indicating transient pH-mediated patterning.290
Besenius, Hermans and co-workers used diffusion of an acid out of a polymer gel cube to assemble a pH-responsive LMWG on its surface, with the choice of polymer gel controlling acid diffusion and LMWG assembly kinetics.291
Diffusion across an oil–water interface has been used by Maruyama and co-workers to assemble supramolecular hydrogels on oil droplet surfaces in an oil-in-water emulsion.292 They used a water-soluble amine and an oil-soluble aldehyde – on diffusion, they reacted to form a gel coating at the microdroplet surface, leading to stabilisation.
In an eye-catching example, Hamachi and co-workers combined diffusion-directed assembly with dissipative assembly to develop a gel system that assembled in the presence of Zn(II) and disassembled when exposed to H2O2.293 By exposing the gel to propagating waves of these orthogonal chemical stimuli, they created a propagating wave of supramolecular nanofibers that created a measurable force. This force could move fluorescent nanobeads in a directional way. Hamachi and co-workers also coupled phototriggered assembly with dynamic processes to yield out-of-equilibrium patterns of peptide nanofibers in self-sorting gels.294 This demonstrates how multiple concepts can be combined in synergistic ways to yield highly sophisticated behaviour.
In summary, a range of strategies exist for shaping and patterning gels. Given the importance of heterogeneous materials in the biological world, many of which are patterned with compositional gradients, such gels have considerable importance in terms of both biomimetics and biointervention. Importantly, patterned gels are not necessarily static – they may be dynamic, transient, adaptive and evolvable. These aspects that may sometimes be considered weaknesses of supramolecular gels in terms of their traditional use, are increasingly seen as some of their most uniquely useful forms of behaviour, which may help underpin next-generation applications. We anticipate that ‘engineering’ self-assembled gels in new ways will continue to be a vibrant area of fundamental research as a way of endowing LMWG materials with additional function. Such methods are particularly powerful when combined with multi-component materials which allow the fabrication of gels containing multiple domains, each with different properties.
4. Advanced technologies
Building on the historic applications of gels (Section 2), and the modern conceptual toolkit developed by supramolecular chemists (Section 3), there are a number of future-looking applications of LMWGs in advanced technologies. Some of these are already in everyday use, while others are more speculative. These advanced applications demonstrate the potential of supramolecular gels to play a transformative role in a wide range of technologies.Before diving into the details of individual applications, it is worth reflecting that supramolecular gels are sometimes (but not always) sustainable materials. They are assembled from low-molecular-weight systems, and can disassemble and degrade over time. Furthermore, in many cases they are derived from renewable feedstocks from the natural world (Section 3.1). In ancient times (Section 2), all materials were green and sustainable. Only in the 20th century did gel technologies move extensively towards polymer gels derived from petroleum resources, albeit often with the good intention of creating high performance materials, lowering cost, and removing pressures from other natural resources. It would seem logical, as we think more seriously about longer-term impacts of technology, that materials like supramolecular gels should dominate soft materials applications in the 21st century and beyond.
4.1 Drug delivery and formulation
There is considerable interest in multi-component gels, including gelator-additive gels (Section 3.4). In the case where the additive is an active pharmaceutical ingredient (API), such materials have potential use in drug formulation and delivery, as described in a classic review from Marlow and co-workers.298 In medicine, there is often a need to assist pharmaceuticals into the human body, or control their distribution rates. With these goals in mind, there have been considerable investigations of LMWGs as drug delivery vehicles, with some already being in everyday use (see below). In terms of formulating APIs into a gel, there are a number of possible outcomes (Fig. 39):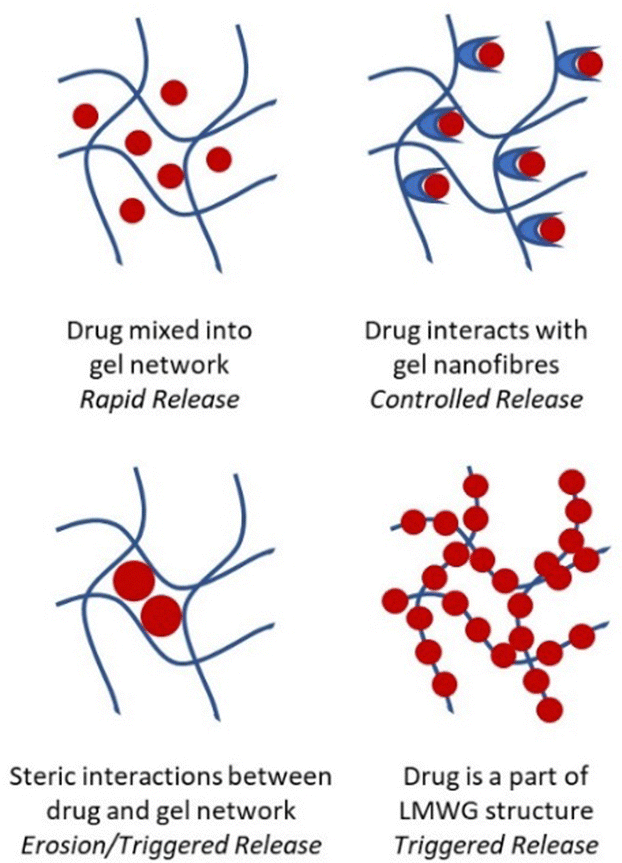 | ||
| Fig. 39 Schematic diagram of different ways in which LMWGs can be combined with APIs and the possible outcomes that these formulation modes can have on release profiles. (top left) API mixed into the gel network achieves rapid release, (top right) API interacting with gel nanofibers gives controlled/slow release, (bottom left) steric interactions between API and gel network require erosion or triggered release. (bottom right) API as an integral part of the LMWG structure requires triggered release. | ||
• the API is mixed into the gel, but remains mobile in the liquid-like phase, making rapid release likely;
• the API interacts with the solid-like network, becoming immobilised, making controlled or slow release possible;
• the API remains in the liquid-like phase but is physically trapped in the pores of the gel network, only achieving very slow release, or requiring erosion release;
• the API is an integral part of the LMWG itself and requires either activated release from the gel network, or triggered breakdown of the whole gel structure.
Given that gels can be formulated with widely differing rheological properties, as well as potentially being injected, or even shaped and patterned, it is clear that such materials may be useful for drug administration via a range of modalities, including transdermal, subcutaneous, transmucosal, intranasal, implantation, oral delivery, etc. This opens a wide range of possibilities in pharmaceutical science.
More recently, supramolecular gels were developed for transdermal delivery and subcutaneous injection. The earliest reports from Luisi and co-workers from 1989 use lecithin as a gelator.74,300 This naturally-derived surfactant is a key food industry additive (Section 2.6). It is ‘generally regarded as safe’, lowering the barrier to application in medicine. Lecithin assembles into cylindrical micelles in water, which form an organogel on mixing with an organic solvent (water-in-oil gelation). Adding a polar polymer to the aqueous phase gives very stable ‘pluronic’ lecithin hydrogels of particular use in drug formulation. These multi-component gels were originally developed by compounding pharmacists Jones and Kloesel in the 1990s.301 This is an early example of the way LMWGs can be combined with a polymer to create materials with enhanced performance (Section 3.4). Pluronic hydrogels have been applied in API formulation, including clinical use, particularly for transdermal delivery of active agents like muscle relaxants and non-steroidal anti-inflammatory drugs (NSAIDs).
Organogels have been extensively developed by Leroux and co-workers using solvents such as soybean or safflower oil combined with LMWGs based on amino acid derivatives, leading to a number of patents.302 Such gels have been investigated for potential as subcutaneous injections delivery systems,303 as well as having been explored for in situ gel formation to achieve (e.g.) controlled local release of hormones used to treat prostate cancer.304 In an in vivo model system, release was achieved for prolonged periods of 14–25 days, comparing very well against polymer-based gel delivery vehicles which poorly-retained similar drugs, leading to rapid release. Organogels based on 12-HSA have also been investigated and considered for use as injectable and implantable systems.305 Indeed, there remains considerable ongoing interest in organogels as drug delivery vehicles.306
The earliest work reporting a supramolecular hydrogel approach to API delivery is from 1995, and used a dipeptide hydrogel (Fmoc-Leu-Asp) – one of the first reports of such systems as LMWGs.308 When the gel was loaded with antiviral adamantanamine derivatives, it induced an antibody response when injected into rabbits, clearly demonstrating bioactivity. Building on this, Zhang and co-workers studied peptide hydrogels loaded with APIs and demonstrated that the release rate depended on the interactions between API and gel network, with electrostatic interactions between API and LMWG, or steric hindrance, playing key roles in lowering the drug release rate (Fig. 39).309
A wide range of supramolecular hydrogels have been explored with regards to drug delivery and tested against a range of biological targets. There are many hundreds of LMWG structures, each of which can potentially exhibit different drug release profiles and selectivities, and there are hundreds of different active APIs, each of which has different structural properties and specific drug delivery needs. As such, there has been an explosion of literature. Díaz Díaz's 2018 review did an excellent job of drawing this work together and placing it in a logical framework.310 Here, I simply select some examples which illustrate interesting drug delivery paradigms.
Nilsson and co-workers illustrated that a simple peptide hydrogel, could be loaded with diclofenac and locally injected into the ankle of a mouse model with ankle inflammation and pain (Fig. 40, top left).311 Remarkably, pain relief from the hydrogel lasted as long as 2 weeks, while standard injected diclofenac remediated pain for less than a day. This is an excellent example of the in vivo benefits that can result using a simple hydrogel drug delivery vehicle.
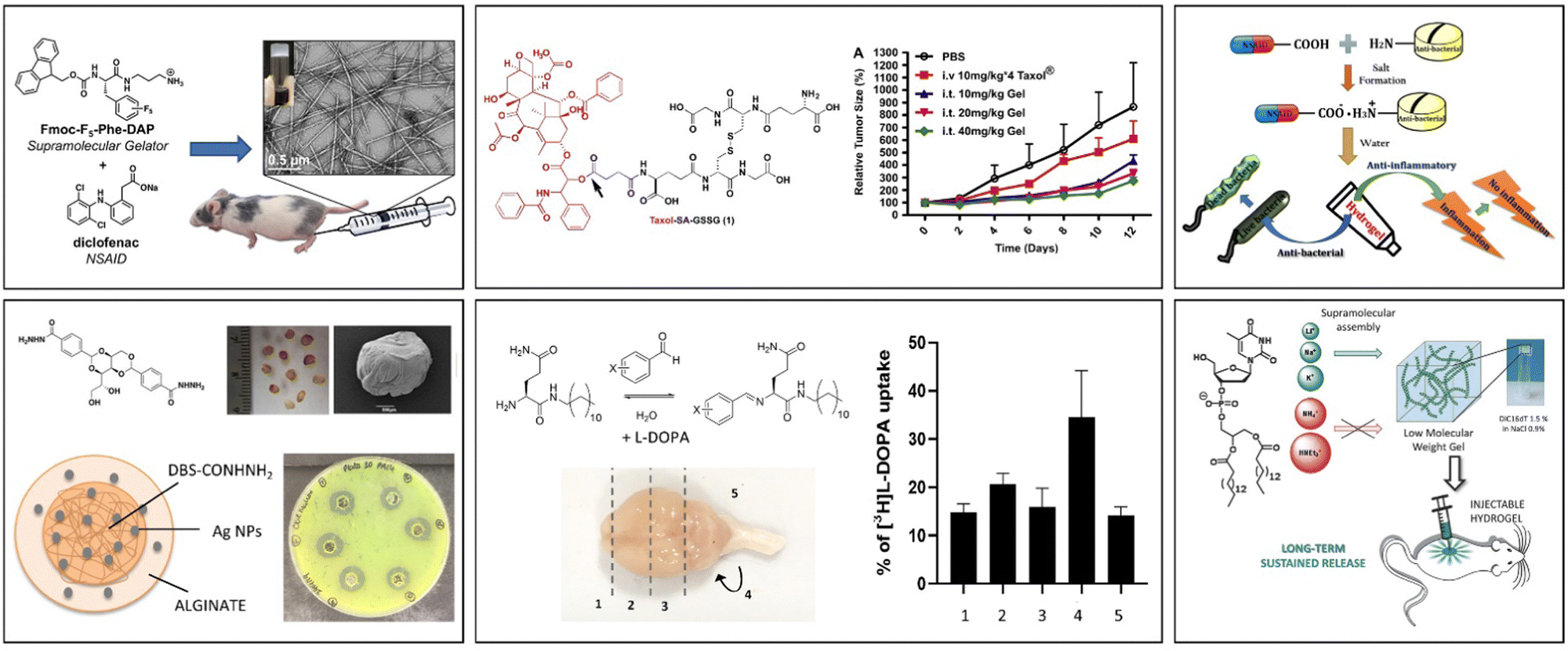 | ||
| Fig. 40 Selected examples of drug delivery based on LMWG technology. (top left) Peptide gelator loaded with diclofenac for controlled long-term release of the painkiller, as demonstrated in vivo, figure reproduced from ref. 311 with permission of the American Chemical Society, © 2019. (top centre) Modified taxol LMWG showing enhanced in vivo compared to IV taxol, figure reproduced from ref. 314a with permission of Elsevier, © 2019. (top right) Schematic of two-component LMWG combining non-steroidal anti-inflammatory drug (NSAID) and antibacterial agent for dual therapy, figure reproduced from ref. 318 with permission of the American Chemical Society, © 2022. (bottom left) DBS-CONHNH2 gelator fabricated into gel beads with calcium alginate, loaded with silver nanoparticles in situ and then tested for bacterial activity against multi-drug resistant MRSA and vancomycin-resistant VRE, figure adapted from ref. 326 with permission of Wiley-VCH © 2020. (bottom centre). Two component self-healing gel encapsulated L-DOPA used for nasal delivery and demonstrating enhanced brain uptake compared with a solution-phase control, figure adapted from ref. 329 with permission of Wiley-VCH, © 2021. (bottom right) Nucleoside gelator tuned by choice of cation and then used for long-term sustained release of a model drug in vivo, figure reproduced from ref. 323 with permission of Wiley-VCH, © 2017. | ||
The incorporation of APIs into the covalent framework of an LMWG is an important strategy for controlling API release, and was originally pioneered by Xu and co-workers in 2002 with a modified vancomycin derivative.312 This group went on to demonstrate gel formation from a paclitaxel derivative, which could be switched by an enzyme (Section 3.6) between sol and gel depending on whether it was phosphorylated.313 Others made gels that can release paclitaxel based on ester hydrolysis, achieving sustained release in vitro and demonstrating activity in vivo in a mouse model (Fig. 40, top centre).314 The ability to trigger API–gelator systems to break down and release the API in the presence of specific enzymes gives them great potential as targeted delivery vehicles (see below).
In an alternative approach to incorporating an API into the LMWG scaffold, Barthélémy and co-workers reported a disulfide-containing gelator, which could exchange with a thiol-containing protein loaded into the gel. This anchored the protein into the gel network, with the material then exhibiting sustained long term protein release via a thiol-exchange mechanism.315 Given the importance of protein therapeutics, such strategies have value, especially in treating lifelong conditions such as diabetes.
Recently, Kieltyka and co-workers explored a dynamic assembly system in which the anticancer drug doxorubicin could be incorporated non-covalently into LMWG assemblies by intercalation, or could react with the gelator scaffold via a bond forming reaction.316 These outcomes could be tuned based on the dynamics of assembly and the formulation strategy. In the case where the drug became covalently coupled to the gelator framework, a greater degree of sustained release was observed.
Dastidar and co-workers pioneered hydrogels based on converting APIs into their salt form to give them gelling potential.317 Recently, they combined a mafenide (a topical anti-burn agent) as an ammonium cation, with non-steroidal anti-inflammatory drugs as carboxylates, demonstrating the LMWG salt can achieve multi-drug delivery (Fig. 40, top right).318 The gel had dual activity to kill bacteria and reduce inflammation, making it an ideal candidate to treat burns. In a key review, these researchers presented their rational ‘supramolecular synthon’ strategy to facilitate therapeutic gel design.319
Researchers, including Gunnlaugsson and co-workers, and Kumar and co-workers reported LMWGs with antibacterial activity in their own right, without any need to load an API.320 Kumar and co-workers designed LMWGs based on a hydrophobic peptide, with a cationic ammonium or guanidinium head group. The gels were inherently toxic to bacteria, but not healthy human cells, with toxicity caused by the release of nanofibers with high surface charge-density.
Another benefit of incorporating APIs into self-assembling LMWGs is the potential to modify solubility. By conjugating triclosan to a self-assembling peptide, a dramatic increase in its solubility and bioavailability was achieved.321 The resulting self-assembled system, delivered by injection, showed excellent in vivo performance as a broad spectrum antibiotic, significantly out-performing unmodified triclosan.
There has also been interest in how gelator–API interactions can play an intimate structural role that may potentially impact on API release. For example, Perez-Garcia, Amabilino and co-workers reported a system where on drug release, the gel nanostructure was transformed.322 Barthélémy and co-workers reported an injectable nucleotide phospholipid, which exhibited controlled release of a fluorescent protein in vivo (Fig. 40, bottom right).323 LMWG assembly could be tuned by the choice of cation, with smaller hydrophilic cations giving fibrillar assemblies, while larger more lipophilic cations yielded lamellar systems – simple component modification directs assembly and impacts on API release. The release of small hydrophobic molecules required gel degradation. There is also developing interest in combining multiple LMWGs, with each component playing a different active role in API formulation.324 This builds on emergent expertise in multi-component gels (Section 3.4).
The fact LMWGs are highly-responsive to stimuli gives them potential to control API release in ways that cannot easily be achieved with polymer gels. Furthermore, being based on well-defined low-molecular-weight building blocks means regulatory hurdles to their use can be addressed in analogy to the development of small molecule drugs. It is anticipated there will be many more examples of active LMWG-based delivery systems, that will hopefully build on the market penetration of lecithin-based gels for passive release. It is expected that these next-generation gels will exhibit smart features that control delivery in more active ways to suit the needs of the patient.
:1 acid:base complex formed between citric acid and nicotine, which could form a supramolecular gel in the presence of a variety of fragrance molecules.328 It was proposed that this system had applications in e-cigarettes with the gel prolonging nicotine release and stabilising fragrance molecules. Clearly gels may have application in other inhaled therapies.
Recently, we demonstrated that a rheologically-soft, self-healing gel could be applied for nasal delivery (Fig. 40, bottom centre).329 Nasal administration has the potential for enhanced uptake in the bloodstream, and importantly also avoid the blood–brain barrier to achieve direct brain delivery as a result of direct uptake through the olfactory and trigeminal nerves. We demonstrated that a drug-loaded thixotropic gel could be delivered into the nasal cavity as a liquid and assemble in situ, hence delivering the Parkinson's drug L-DOPA. The gel enhanced residence times in the nose, giving improved uptake in the bloodstream and brain, compared to either nasal or intravenous delivery of a standard L-DOPA solution.
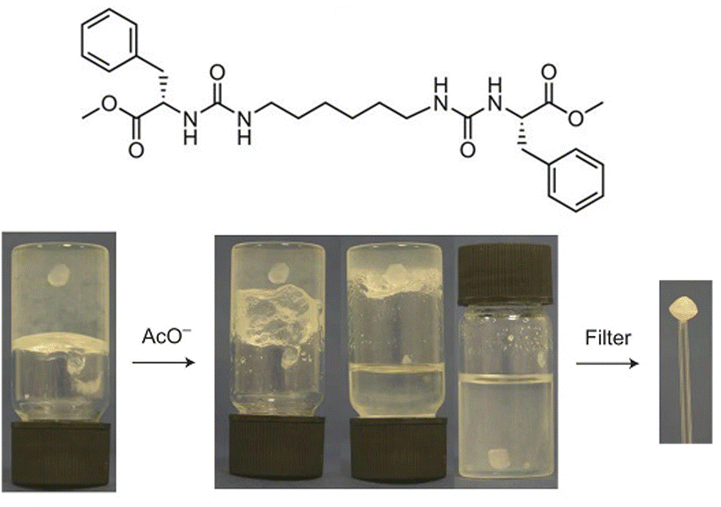 | ||
| Fig. 41 Structure of a urea-based gelator used to control API crystallisation and photographs of the growth of a crystal of carbamazepine within the gel, and the use of acetate anions to disassemble the gel and allow recovery of the crystal. Figure reproduced from ref. 331 with permission of Springer Nature, © 2010. | ||
A range of researchers have since explored API crystallisation, demonstrating it can be facilitated, or inhibited, by tunable self-assembled gels.332 In elegant work, Steed and co-workers demonstrated that matching the chemical structure of the LMWG to the API, makes it possible to intervene directly in crystallisation by providing a nano-surface to initiate crystal nucleation.333 As such, LMWGs have considerable potential in the pharmaceutical industry during drug manufacture.
4.2 Reaction engineering
The combination of solid-like and liquid-like characteristics in gels allows compatibility with a solvent phase and rapid diffusion of small molecules, but also provides the opportunity to immobilise active species – gels therefore have considerable potential in reaction engineering. It is possible to immobilise a catalyst in the solid-like gel network, generating solvent-compatible materials that promote chemical reactions, with the potential to be reused, enabling sustainable workflows. Excellent reviews of supramolecular gels in catalysis have been produced by a number of groups.334 Furthermore, by performing reactions in gel media, the confined environments can be used to achieve specific outcomes. Alternatively, it is possible to envisage a gel as a delivery vehicle for reagents or catalysts, releasing them in a controlled manner. | ||
| Fig. 42 Selected examples of supramolecular gels applied in catalytic processes. (Left) Early example of a palladium-binding LMWG which self-assembles into nanofibers and catalyses the oxidation of benzyl alcohol to benzaldehyde, figure reproduced from ref. 335 with permission of the Royal Society of Chemistry, © 2005. (Right) Hybrid hydrogel formed from DBS-CONHNH2 and agarose reduces Pd(II) waste to Pd nanoparticles in situ with the resulting PdNP-loaded gel then catalysing Suzuki–Miyaura cross coupling reactions, figure reproduced from ref. 338a with permission of the Royal Society of Chemistry, © 2018. | ||
 | ||
| Fig. 43 (top left) Self-assembled peptide amphiphile functionalized with histidine amino acids catalyses ester hydrolysis, figure reproduced from ref. 341 with permission of the American Chemical Society, © 2007. (top right) Self-assembled proline-functionalised LMWG exhibits enhanced basicity and catalyses the nitroaldol reaction more effectively when assembled than in solution, figure reproduced from ref. 342 with permission of the American Chemical Society, © 2009. (bottom left) Self-sorted hydrogel network catalyses sequential acetal deprotection and aldol reactions, with each of the networks catalysing one of the steps, figure reproduced from ref. 343 with permission of the Royal Society of Chemistry, ©2016. (bottom right) Self-assembled LMWG containing a secondary amine catalyses the aldol reaction between glycolaldehyde in water to give threose and erythrose products with diastereo- and enantio-selectivity, figure adapted and reproduced from ref. 345 with permission of the American Chemical Society, © 2020. | ||
However, this field has really been pioneered by Escuder and co-workers, who introduced a number of key principles.334a In early work, they demonstrated that placing proline on the periphery of bolaamphiphile-style organogelators meant that on self-assembly, the basicity was increased by three orders of magnitude.342 This enabled the gel to catalyse a nitroaldol reaction as a result of self-assembly, whereas individual LMWGs were inactive (Fig. 43, top right). They also explored the impact of gel polymorphism on reaction outcomes, demonstrating that specific details of self-assembled morphology can impact on catalytic performance, indicating the importance of precise organisation of catalytic groups on the molecular level.235
In visionary work, Escuder and co-workers created self-sorted gels containing two orthogonal self-assembled networks, each of which could catalyse a different reaction, demonstrating that the multi-component gel could catalyse a multi-step reaction.343 The first gelator was acidic and catalysed acetal deprotection, while the second contained basic proline and catalysed aldol condensation (Fig. 43, bottom left). These acid/base catalysts are orthogonal to one another and could not usually be used together in one-pot. Indeed, the researchers showed that if the LMWG structures were modified, such that they co-assembled, rather than self-sorted, then the gel was no longer catalytically proficient. It was thus demonstrated that multi-component gel-based catalysts, with precise control over organisation on the molecular level, can enable shorter syntheses by telescoping multiple reactions into a single vessel.
Studying hydrogels capable of aldol catalysis also led Escuder and co-workers to explore reactions of prebiotic relevance, albeit with protecting groups in place.344 Building on this, working with Clarke, we went on to demonstrate that a relatively simple LMWG, synthesised from prebiotically-relevant building blocks could catalyse the aqueous aldol reaction of unprotected glycolaldehyde to give threose and erythrose with good diastereoselectivity and some enantioselectivity (Fig. 43, bottom right).345 This sugar-forming reaction is considered a fundamental reaction in the prebiotic origins of life on the early Earth – its catalysis by a gel indicates how simple LMWGs can have the potential to create the building blocks of life. The ability of gels to play a role in the evolution of life has been postulated.101 It is possible that the physical characteristics of gels may have combined with their ability to play an active role in catalysing key processes and guiding reaction outcomes on the Early Earth.
Recently, Escuder and co-workers assembled an imidazole-functionalised gelator inside a polymerosome assembled from block co-polymers,346 using techniques pioneered by van Esch and co-workers.190 They demonstrated that the encapsulated gel nanofibers were catalytically proficient in ester hydrolysis, also highlighting the relevance of this work in prebiotic science, with regard to the assembly of minimalistic protocells.
An alternative way of encapsulating enzymes in gels is to convert a pre-gelator to the LMWG in the presence of the enzyme via triggered gel assembly – this avoids exposing enzymes to potentially stressful thermal treatment on gel formation.348 Once encapsulated, these enzymes showed enhanced (e.g.) thermal stability. Furthermore, LMWG structure can be used to tune the efficiency of an encapsulated enzyme, indicating synergy between the catalytically-active unit and the self-assembled network.349 Recently, Marr and co-workers have immobilised enzymes in supramolecular ionic liquid gels, achieving good reactivity.350 They shaped the system as gel beads to enable the reactions to be easily performed and facilitate recycling.
In addition to using gels as enzyme supports, we attempted to shape gels in order to create ‘gel reactors’ – for example a ring of enzyme-loaded gel in a tray.351 When the interior of the ring was loaded with substrate, diffusion through the ring led to products being released into the outer compartment. Although requiring further optimisation, this demonstrated how shaped gels can potentially enable new approaches in reactor design and reaction engineering. Recently supramolecular gels capable of catalysing the Knoevenagel reaction have been used in flow reactors.352
 | ||
| Fig. 44 Organolithium gel loaded with phenyllithium can be handled under ambient conditions in air, and used to perform a wide range of organic reactions, including nucleophilic additions such as that shown here. The gels are homogeneously loaded with organolithium reagents and can therefore be subdivided for dosing into reactions. Figure adapted from ref. 348 with permission of Springer Nature, © 2023. | ||
More recently, there has been interest in performing photocatalysed organic reactions within gels.358 Confining reactions in a gel network can improve photochemical reactions, for example by enhancing light absorption or intermediate lifetimes. Photodimerization reactions have been carried out in supramolecular gels with higher efficiencies and different selectivities.359 Recently, Ajayaghosh and co-workers demonstrated that ‘in-gel’ synthesis could lead to difficult-to-obtain photo-cycloaddition products, with potential applications in creating white-light emission devices, hence demonstrating how unique synthetic outcomes can ultimately be coupled to applications.360 Díaz and co-workers investigated processes such as photooxidation and photoreduction in supramolecular gels.361 The precise nanoscale morphology impacted on reaction outcomes – entangled fibrillar networks with relatively high mechanical strength were usually associated with lower reaction rates, whereas wrinkled laminated morphologies seemed to favour reaction. These researchers have also shown that air-sensitive photoredox chemistry can be performed in a self-assembled gel under aerobic conditions as a result of reactant confinement and blockage of oxygen diffusion.362
In summary, gels are fascinating media for organic reactivity. Their combination of solid-like and liquid-like characteristics, combined with their unique internal environments gives them significant potential in catalysis, reagent delivery and reaction engineering. We anticipate continued growth in the use of gels as unique materials in the synthetic toolkit. In particular, as synthetic chemistry moves towards greater automation, and the desirability of ‘kit-form’ chemistry becomes more evident, gels will have significant roles to play in enabling effective, easy-to-use reaction workflows.
4.3 Environmental remediation
As global attention focusses on environmental sustainability, there has been intense interest in using gels to remediate pollutants.363 Gels are highly adsorbent materials, compatible with solvent, and with nanoscale internal structuring which possesses very large surface-area compared to interior volume – as such, they are ideally suited for adsorption of unwanted compounds from water (Fig. 45). Gel disassembly offers a route by which the pollutant can potentially be recovered and the LMWG reused, enabling sustainable workflows. | ||
| Fig. 45 General schematic of the use of gels for environmental remediation, figure reproduced from ref. 363a with permission from the Royal Society of Chemistry, © 2016. | ||
In 2007, attention returned to this long-forgotten area of research, with the publication of Banerjee and co-workers’ key paper using a supramolecular gel based on a phenylalanine bolaamphiphile, the assembly of which was triggered by divalent metal cations. Having formed the gel, they dried it to a nanostructured xerogel, and used this to extract dyes from water (Fig. 46, top left).364 Although modest uptakes were obtained, there was specificity for remediation of ionic dyes rather than non-ionic dyes, indicating active control of uptake. Hayes and co-workers went on to report very high uptakes of methylene blue using a urea derivative of isophthalic acid.365 Selectivity studies suggested dye uptake was driven by intercalation, with a degree of selectivity for the precise nature of the aromatic dye. Yamanaka and co-workers managed to obtain >1.25 g g−1 uptake of methylene blue using a related amphiphilic tris-urea LMWG.366 Working with rhodamine-6G, fluorescence microscopy suggested uptake occurs on the gel nanofibers, then in the pores once the fibres become saturated.
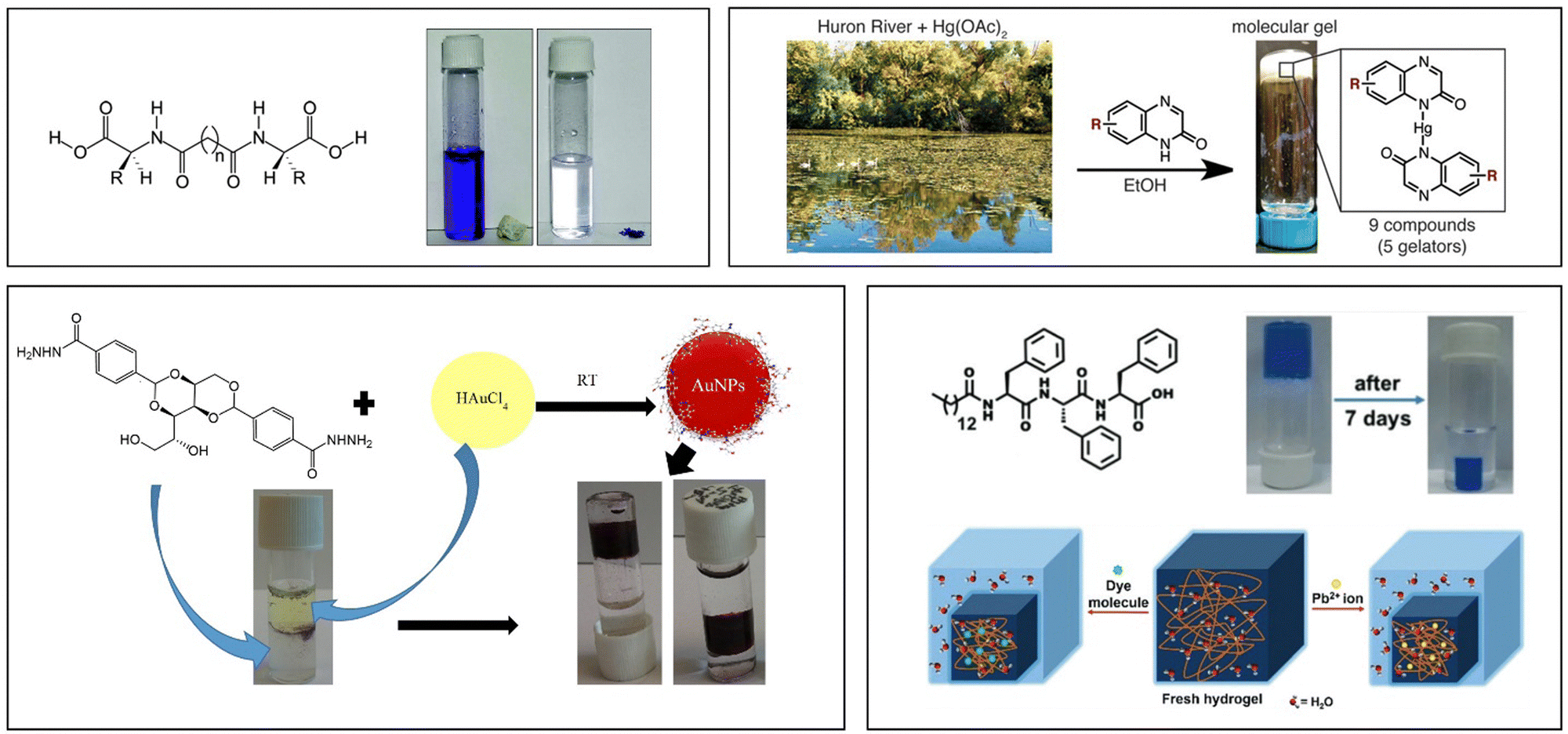 | ||
| Fig. 46 Examples of gels used for environmental remediation. (top Left) Bolaamphiphile gel formed on addition of divalent metal ions, used for remediation of dyes from water – photograph shows xerogel being used to remove methylene blue from water. Figure reproduced from ref. 359 with permission of the American Chemical Society, © 2007. (top right) Pre-gelator assembles into a gel on binding Hg(II) and was demonstrated to remediate Hg(II) from artificially-contaminated river water. Figure reproduced from ref. 368 with permission of the American Chemical Society, © 2014. (bottom left) LMWG based on DBS remediates precious metals via reductive mechanism with nanoparticles being generated in situ. Figure reproduced from ref. 369 with permission of Wiley-VCH, © 2016. (bottom right) Tripeptide gel exhibits syneresis on gel formation, with this shrinkage being used to trap pollutant dyes and Pb(II) ions. Figure reproduced from ref. 372 with permission of the Royal Society of Chemistry, © 2017. | ||
We investigated dye uptake onto our DBS-CONHNH2 hydrogels.147 We found high levels of uptake, with a preference for dyes in their least-charged form – we reasoned that more highly-charged dyes preferred to stay in the aqueous phase. Davis and co-workers, revisited the classic G-quartet gel system, and adapted it to enhance uptake of anionic dyes by stabilising assembly with divalent barium(II) cations. As a result, a preference for anionic dyes was built into the gel nanofibres, demonstrating how dye uptake can be controlled.367
One of the problems with remediating dyes in this way is that supramolecular gels are often mechanically weak, making filtration difficult. Yang and co-workers addressed this problem by combining an LMWG with agarose PG.368 The LMWG gave the resulting hybrid gel dye uptake capacity, while the agarose made it more robust, enabling the use of stirring to maximise dye uptake kinetics. This was one of the earliest examples in which a PG was combined with an LMWG to create a hybrid gel – now a widely-used strategy.49
There is also interest in using gels based on environmentally-friendly alternative solvents for remediation. D’Anna and co-workers further developed ionogels and eutectogels (Section 3.3),369 demonstrating the use of ionogels in an adsorption column where 95% dye removal could be achieved in less than 10 minutes, with regeneration and reuse for >20 cycles being possible, indicating the practical applicability of this approach.
In an elegant study demonstrating the potential of the LMWG approach to metal remediation, McNeil and co-workers used Hg(II) to trigger gelation of an LMWG by binding to a quinoaxalinone, creating a complex with two ligands and one mercury ion (Fig. 46, top right).372 They demonstrated that 3800 ppm Hg(II) in water could be reduced to ca. 290 ppm. Although this remains above acceptable Hg(II) levels, it was argued that it may also have potential as a sensor for mercury-polluted water (see Section 4.5). They further developed the system by structural modification to optimise mercury uptake, such that >98% of mercury was remediated.373 The gelator worked on river water that had been artificially contaminated with Hg(II), indicating that the system could potentially function in real-world environmental samples.
Although metal–ligand binding is the simplest way in which a pollutant metal becomes trapped in a gel, there are other strategies to achieve metal immobilisation. We used DBS-CONHNH2 to remediate precious metals from water – specifically Pd(II), Au(III), Ag(I) and Pt(II).374 In each case the metals were reduced to their level zero oxidation state by the acylhydrazide functional group on the LMWG,339 and spontaneously, on exposure to the gel, assembled into nanoparticles, which became trapped in the gel (Fig. 46, bottom left). This type of system has the potential to remediate precious metals from mining waste or e-waste. Beyond simple remediation, the nanoparticle-loaded gels created in the process would also then have their own value-added functions (e.g. catalysis [Pd],339 conductivity [Au],374 antibacterial activity [Ag]326etc.) – a so-called ‘waste-to-wealth’ strategy.375
D'Anna and co-workers employed sugar-derived gelators to remediate Cr(VI) anions from waste water at neutral pH.376 This demonstrates the way metal remediation can also rely on binding to anionic targets, depending on the speciation of the metal of interest. The sugar units played a dual role of (i) binding the chromium-based oxoanions through hydrogen bond interactions, and (ii) acting as a reducing agent to convert toxic Cr(VI) into Cr(III). Their most efficient gel achieved ca. 600 mg g−1 chromium uptake, and could be recycled up to 4 times.
In search of a different mechanism of pollutant entrapment, Banerjee, Hamley and co-workers reported a tripeptide gel based on phenylalanine repeat units (Fig. 42, bottom right), that shrinks after assembly (syneresis).377 It was suggested that the highly hydrophobic nature of the three Phe repeat units may be responsible for the shrinkage of the gel, and expulsion of water. When assembled in a solution containing Pb(II) the pollutant became almost completely entrapped in the shrunken gel, with excellent levels of uptake (1995 mg g−1). The authors noted that soaking the shrunken gel in EDTA solution led to leaching of the Pb(II) and suggested that this indicated a physical mechanism of entrapment. The LMWG could be recycled between different solvent phases, and reused several times. Others have also applied the gel syneresis approach for waste-water treatment.378 The ability to fundamentally understand syneresis in gels is of considerable current fundamental interest.379
There are now a plethora of papers in which a variety of LMWGs extract wide-ranging pollutants from water. These experiments are relatively straightforward to perform, and the wide range of LMWGs and pollutants opens up many possibilities. Increasingly it is being reported that individual LMWGs are capable of remediating multiple different species from waste water.380 Furthermore, an increasing focus on waste-to-wealth strategies tries to integrate this approach into the circular economy. It is hoped that in the coming years, the most promising systems will be developed into technologies to remediate waste from (e.g.) industrial processes. This will require supramolecular chemists to work more closely with end-users to translate results into the real-world.
4.4 Printing applications
As discussed in Section 3.9 there is developing academic interest in 3D-printing LMWGs to create shaped and patterned gels, with researchers beginning to understand how LMWG structure and dynamics impact on printability. Ground rules and for such processes are emerging. Interestingly, these concepts intersect with industrial work that has recently been incorporating LMWG technology into printing applications.For ink-jet printing, an ideal ink should be stored as a solid, converted to a liquid on heating, and then rapidly resolidify on contact with the print medium to prevent ‘bleed’ of the ink. The ability of phase-change materials like gels to control these processes makes them potentially useful. An early patent from 1989 reported phase change materials based on fatty amides for ink-jet printing.381 Although not completely clear if they were gels or waxes, these materials have found their way into Xerox solid ink products, and led to significant further work.
A key Xerox patent from 2005 suggested that organogelators were ideally compatible with a system for printing containing an ink vehicle and a dye.382 This patent protects an exceptionally large scope of LMWG chemistries from a wide range of academic literature for this application. Later patents combined LMWG chemistries with photocurable technologies, for example, by incorporating polymerisable groups into the LMWG, or adding photocrosslinkable monomers to the LMWG.383 Such systems have potential applications in 3D printing devices, where the supramolecular gel helps structure the system during initial printing, enabling rapid layer-by-layer deposition and avoiding the need for intermediate curing steps, prior to a later photocuring step on the finished object to provide full rigidity through polymerisation. This is related to the ways academic chemists have combined polymers with LMWGs (Section 3.4) and developed polymerizable gelators (Section 4.2). It also links to the concept of using gels in applications such as in photocrosslinkable composites in dentistry (Section 2.3).
It seems likely that the interests of academics and industrialists will intersect further in this area of research, especially as 3D printing increasingly becomes a mass-market technology. Thus far, most commercial 3D-printing has largely focussed on hard ‘plastic’-type materials. It seems likely that LMWG technologies will enable the fabrication of soft objects that are more dynamic and evolvable – such materials can play a key role in ‘4D printing’, where time (the fourth dimension) is an important factor, and objects are designed for dynamic properties as much as their static structure.384 Gels are uniquely suited for such materials, given their responsive, dynamic properties (Sections 3.6–3.8). 4D-printing has particular potential value in regenerative medicine (Section 4.7).
4.5 Sensor technology
The ability of gels to undergo a gel–sol transition makes them of high interest for application in sensor technologies, as this response offers a mechanism through which key analytes can be detected. Indeed, some examples of triggered gel responses that could be considered to ‘sense’ analytes or environmental changes were discussed in Section 3.6. | ||
| Fig. 47 Selected examples of sensors based on LMWG technology. (top left) Compound incorporating a β-lactam functional group, which when exposed to β-lactamase enzyme found in penicillin-resistant bacteria, is converted into an active LMWG, switching gelation on, figure reproduced from ref. 385 with permission of the American Chemical Society, © 2007. (top right) Peptide-modified antitumour agent that self-assembles at the disease site and can be used to image and treat glioma brain tumours, figure reproduced from ref. 389 with permission of the American Chemical Society, © 2017. (bottom left) Design of system which forms an LMWG on binding Pb2+ and photograph of samples of paints with or without lead which could be tested via gel formation, figure reproduced from ref. 390 with permission of the American Chemical Society, © 2016. (bottom right) Design and photographs of a simple LMWG sensor for chemical weapons agents such as soman, in which the chemical weapons simulant leads to breakdown of the gel, and as the copper coil drops from being suspended in the gel, it makes contact with a pair of electrodes, completing a circuit and turning on a red LED, Figure reproduced from ref. 391a with permission of the Royal Society of Chemistry, © 2015. | ||
There are now many enzyme-sensitive gels, ideally-placed for the detection of key biological processes.386 By triggering self-assembly of a gelator at a specific disease site, it is also possible to achieve effective bioimaging.387 For example, Liang and co-workers activated a gadolinium-containing LMWG by alkaline phosphatase enzymes present in tumours, and observed enhanced MRI contrast imaging as the gelator assembled and accumulated at the tumour site.388 Cui, Liu and co-workers reported a peptide-modified antitumour agent that self-assembled in vivo. The antitumour agent also exhibited a label-free chemical exchange saturation transfer (CEST) MRI signal. The system was used to simultaneously image and treat glioma brain tumours in a mouse model, combining sensing and drug delivery applications (Fig. 47, top right).389
Gale and co-workers pushed LMWG technology into the area of chemical weapons detection and developed a gel that underwent a gel–sol transition on exposure to the nerve agent soman.391 A copper coil was embedded in the gel, and on exposure to a chemical weapons simulant, the gel–sol transition caused it to fall. This brought it into contact with a metal plate and switched on a warning LED (Fig. 47, bottom right). Although a low-tech approach, this demonstrated how a gel–sol transition can be converted into an simple ‘warning’ response. There has also been interest in developing gel-based sensors for toxic species like cyanide anions,392 or explosives like picric acid.393
In recent work, Bhattacharya and co-workers developed a biocompatible fluorescent LMWG which underwent oxidative decomposition in the presence of hypochlorite, leading to a gel–sol transition and changing the fluorescence intensity.396 This is a reaction-based sensing mechanism, rather than one based on non-covalent interactions. Harnessing the inherent materials properties, gel-coated paper strips were developed to detect hypochlorite. Furthermore the system gave fluorescent imaging of hypochlorite in live mammalian cells. Fluorescent hydrogels have also been used to detect toxic species including phosgene via a reaction-based sensing mechanism.397
In summary, gels are dynamic, responsive materials, with rapid diffusion kinetics in the liquid-like phase, and hence very well-placed for rapid analyte sensing. The ability to use the properties of the gel itself to detect analytes, as well as coupling gel characteristics with other mechanisms of response, means the potential use of LMWGs in this field is wide-ranging.
4.6 Nanostructured electronics
Gels with electronic properties have considerable potential in electronic, energy and optical technologies. Conductive gels are of particular interest, and therefore, a number of approaches have been taken to create them (Fig. 48): | ||
| Fig. 48 Schematic of different conductivity mechanisms possible within self-assembled gel-phase materials. | ||
• using a conductive liquid-like phase;
• embedding conductive units into the gel;
• designing conductive gel nanofibers.
In academic LMWG research, this concept was pioneered in 1999 by Hanabusa and co-workers who prepared supramolecular organogels in solvents like propylene carbonate loaded with a supporting electrolyte and explored the conductivity.399 Conductivity plots indicated that the ionic mobility of the electrolyte was scarcely affected by the presence of the self-assembled LMWG network.
In 2004, Grätzel and co-workers used 1,3:2,4-dibenzylidenesorbitol (DBS) as a simple LMWG approach to electrolyte immobilisation in dye-sensitised solar cells (DSCs).400 In particular, methyl-substituted DBS derivatives had ideal compatibility with the polar organic solvents being used. DSCs incorporating the LMWG achieved energy conversion efficiencies equivalent to those using a liquid electrolyte. In 2008, Dai and co-workers reported the use of 12-HSA with the same goal in mind.401 Once again, this demonstrates how DBS and 12-HSA are often the initial ‘go-to’ organogels as a result of their commercial-relevance and ease-of-supply. In later work, it was noted that the molecular structure of the LMWG can influence self-assembled morphology and hence electron transfer/recombination kinetics and DSC performance.402 In this way, Grätzel and co-workers developed optimised gel-based DSCs with excellent solar energy conversion efficiencies, of commercial relevance.403
In recent years, multi-component gels have been used to enhance performance of gel electrolyte devices. Researchers combined bis-amide and valine-derivative organogelators and found that in the two-component system, the 3D network of the gel electrolyte became looser, accelerating electron transport, and prolonging the electron recombination lifetime.404 This illustrates how fundamental concepts of multi-component gels can transfer to an applied setting to improve device efficiency. Optimising gel–sol transition temperatures can also play a role in enhancing the safety of these devices.405
There have also been attempts to use gels to create battery technologies. For example, Raghavan and co-workers used DBS in propylene carbonate and lithium perchlorate to create a gel electrolyte of relevance in lithium ion batteries (LIBs).406 Glutamic acid based LMWGs have also been reported to directly form gels in the electrolyte used in commercial LIBs, providing an easy route to potential use.407
Super-capacitors for power storage are of key interest as society moves to more sustainable energy generation. A small-scale supercapacitor based on monobenzylidenesorbitol (MBS) hydrogels was demonstrated to have higher specific capacitance than liquid electrolyte equivalents, as well as solving problems with leakage.408
Alternative to dissolving ions in organic/aqueous solvents to create an electrolyte and achieve conductivity, it is also possible to have a fully ionic liquid phase. The use of ionic liquids and deep eutectics to create ionogels165 and eutectogels168 was described in Section 3.3. As early as 2003, Yanagida and co-workers used ionogels to fabricate DSCs.409 Ongoing development of LWMG ionogels by Grätzel and co-workers increased the power conversion efficiencies,410 and some cells were even tested outdoors at high temperatures in Jeddah, Saudi Arabia, with excellent performance under field conditions.411
Given the simplicity of formulating LMWGs in different fluids, the importance of conductive fluids in energy devices, and the ability of gels to outperform solids and liquids, it seems very likely that LMWG-stabilised conductive fluids will underpin practical technologies in the coming years, potentially playing important roles in the ‘green energy revolution’.
An LMWG was combined with poly(3-hexylthiophene) and the organisation of the two components was investigated, with both of them forming assembled nanofibres.413 Interestingly, a dried film of the gel with only ca. 20% P3HT content gave the same conductivity as a pristine P3HT sample, and it was suggested that the materials might have relevance in the development of conducting films for optoelectronic or biomedical applications.
Gazit and co-workers combined a dipeptide LMWG hydrogel with conducting polyaniline.414 The LMWG loading tuned the mechanical properties of the material and endowed it with self-healing characteristics, which meant that separated blocks could be joined together, establishing conductivity pathways between them. The gel provides a conductive interface for electrogenic cardiac cells and supported cardiomyocyte organization into a spontaneously contracting system, with potential applications in regenerative medicine (Section 4.7).
Recently, potentially-conducting poly(3,4-ethylenedioxy thiophene) (PEDOT) was combined with a supramolecular eutectogel to create an injectable, self-healing material.415 In this case, the conductivity was due to the deep eutectic solvent, with PEDOT only making negligible contributions. This demonstrates the importance of careful characterisation and highlights the fact that the bulk liquid-like phase plays a large role in mediating conductivity in gels.
In 2004, Stupp and co-workers demonstrated that an oligo(thiophene)-based gelator could create films that, on doping with iodine, became highly conducting (Fig. 49, top).417 In the absence of self-assembly, no conductivity was observed, emphasising that nanoscale organisation of the LMWGs switches on conducting pathways in the material. Oxidation with iodine is a classic way of doping organic conductors, and Ajayaghosh and co-workers also used it to dope self-assembled trithienylenevinylene LMWGs and induce conductivity.418
 | ||
| Fig. 49 Early examples of conducting nanowires. (top) Stupp and co-workers reported an oligo(thiophene) gelator that became conducting on doping only when in the self-assembled gel state, figure reproduced from ref. 417 with permission of the American Chemical Society, © 2004. (bottom) TTF-based gelator which self-assembles into nanowires that were demonstrated to become conducting on doping using current-sensing AFM, figure reproduced from ref. 420 with permission of Wiley-VCH, © 2007. | ||
Using concepts of gel shaping, Stupp and co-workers extruded gel ‘noodles’ of their oligothiophene gelators, to align the nanofibers and enhance conductivity – demonstrating the anisotropic potential of self-assembled gel nanofibers as conducting one-dimensional ‘wires’.275 Ajayaghosh and co-workers also explored alignment and organisation at the molecular scale, using innovative methods of assembly at an interface to modify the arrangement of self-assembled nanostructures, creating structures with different properties and potential applications depending on whether the molecules were assembled parallel or perpendicular to one another.419
Tetrathiafulvalenes (TTFs) have high potential for conductivity, and Amabilino and co-workers made a TTF derivative that formed a gel with π-stacked columnar structures and acted as a conductor on oxidation with iodine (Fig. 45, bottom).420 In a landmark experiment, they used current-sensing atomic force microscopy to detect metallic-type conductivity along the nanowires – the first time conductivity pathways had been visualised in this way at the molecular scale.
Although much work focuses on self-assembled π-systems,416 intriguingly, it has been reported that gels based on α-helical peptide gelators may conduct charge over long distances, even in the absence of π-stacked aromatic rings.421 Although the conductivity mechanism is not fully understood,422 this indicates exciting new prospects in the fabrication of biomimetic conducting systems.
Self-organisation through gel formation can potentially separate D and A units at the molecular level, while nonetheless allowing contract points between them, ideal in PVD design. In a key study, Shinkai and co-workers achieved this by applying concepts of self-sorting (Section 3.4) to assemble individual nanofibers based on thiophene (p-type) and perylene (n-type) LMWGs (Fig. 50, top).425 The nanoscale dimensions of the self-sorted fibres means the interfacial area between them is large, and nanoscale mixing of the gel fibres is intimate, thus creating multiple p–n heterojunction points. However, at the molecular level, there is separation between donors and acceptors. On photoirradiation, a robust and reversible photocurrent was generated, demonstrating the success of this supramolecular PVD design strategy. Ajayaghosh and co-workers also developed self-sorting D/A molecules and reported >10-fold enhancement in anisotropic photo-conductivity.426 Draper, Adams and co-workers used a self-sorting strategy to create photoconductive gels.427 The self-sorting approach was also applied by del Guerzo and co-workers, who self-sorted n-acene nanoribbons and then observed spatially-resolved electroluminescence at the p–n junction points using innovative luminescence microscopy techniques.428
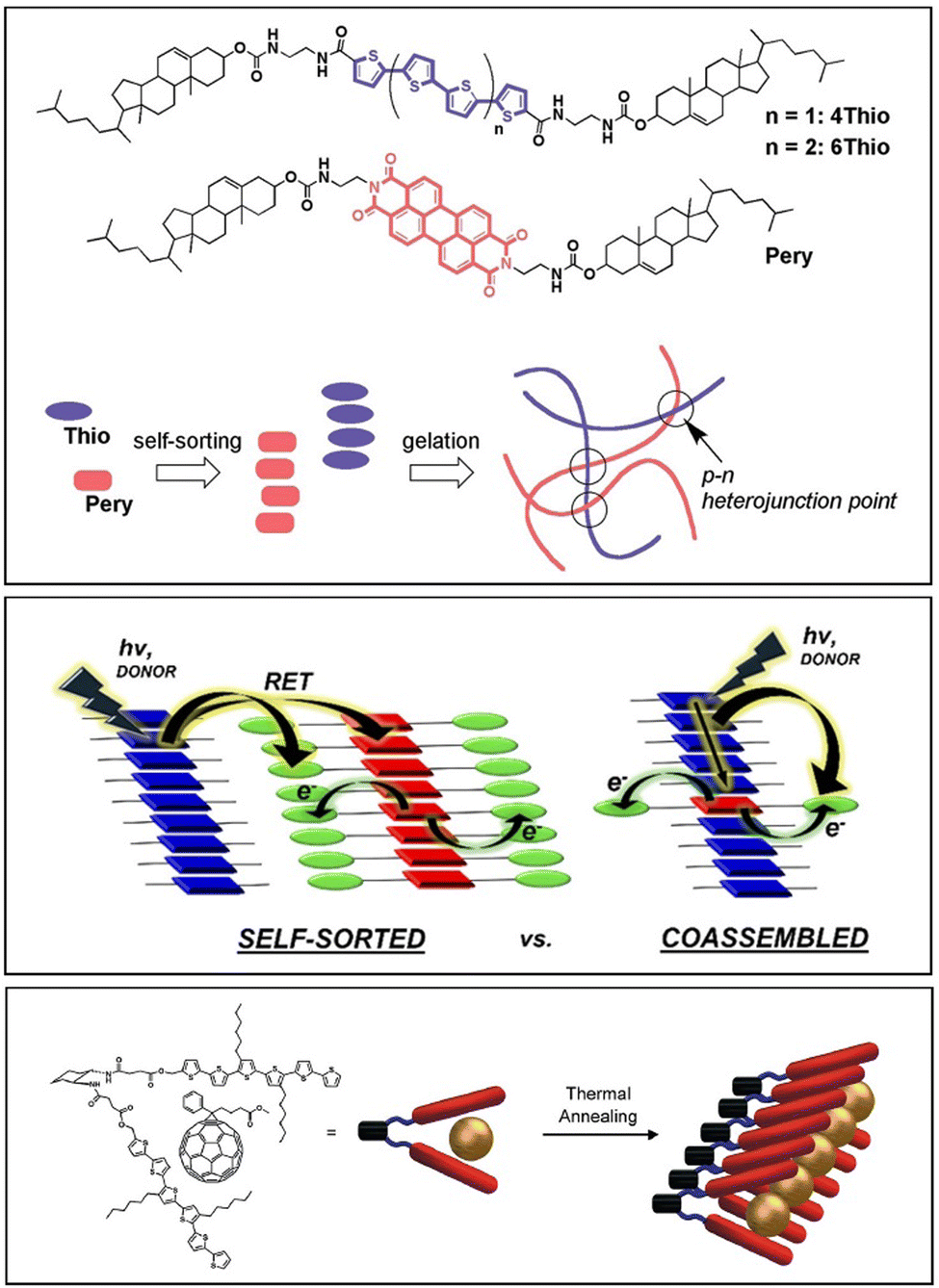 | ||
| Fig. 50 Examples of donor–acceptor self-assembled systems. (top) Donor and acceptor LMWGs that self-sort into independent nanostructures creating p–n heterojunction points between the self-assembled architectures, figure reproduced from ref. 425 with permission of the American Chemical Society, © 2008. (centre) Schematic diagram demonstrating how self-sorted or co-assembled systems can lead to different outcomes with self-sorting systems achieving better charge separation while co-assembled systems optimize energy transfer, figure reproduced from ref. 430 with permission of the American Chemical Society, © 2017. (bottom) Combination of a fullerene derivative with self-assembling oligo(thiophene) used to fabricate a bulk heterojunction photovoltaic device, figure reproduced from ref. 431 with permission of the American Chemical Society, © 2012. | ||
In addition to PVDs, donor–acceptor materials are of interest for energy transfer applications in light harvesting and molecular electronics. Adams and co-workers reported D/A hydrogels capable of energy transfer from a naphthalene dipeptide LMWG to dansyl or anthracene chromophores hosted in the gel via non-covalent interactions.429 With Tovar, they went on to compare systems in which donors and acceptors were either part of different self-sorted architectures, or randomly co-assembled (Fig. 46, centre).430 They found that self-sorting was important for establishing p–n heterojunctions that achieve the charge separation required for energy applications (see above), while random co-mixing was better for the energy transfer efficiency required in photonic applications. This work is an elegant example of how innovations in supramolecular control can be harnessed for high-tech outputs.
Combining electroactive nanofibers with other conductive units also has significant potential. For example, fullerenes are widely used as electron acceptors in solar cell PVD design, facilitating charge separation as a result of their ability to be reduced by up to six electrons. Combining fullerene derivatives with an electron donor LMWG, Stupp and co-workers used the dried gel to fabricate a bulk heterojunction PVD (Fig. 46, bottom).431 Thermal annealing encouraged domain growth and improved the power conversion efficiency, demonstrating how supramolecular materials retain a degree of dynamic and adaptive behaviour that can be harnessed to enhance performance. In related work, Lu and co-workers bound an acid-functionalised fullerene to an amine-functionalised LMWG through acid–base interactions, creating a multi-component gel, and a large stable photocurrent was generated.432
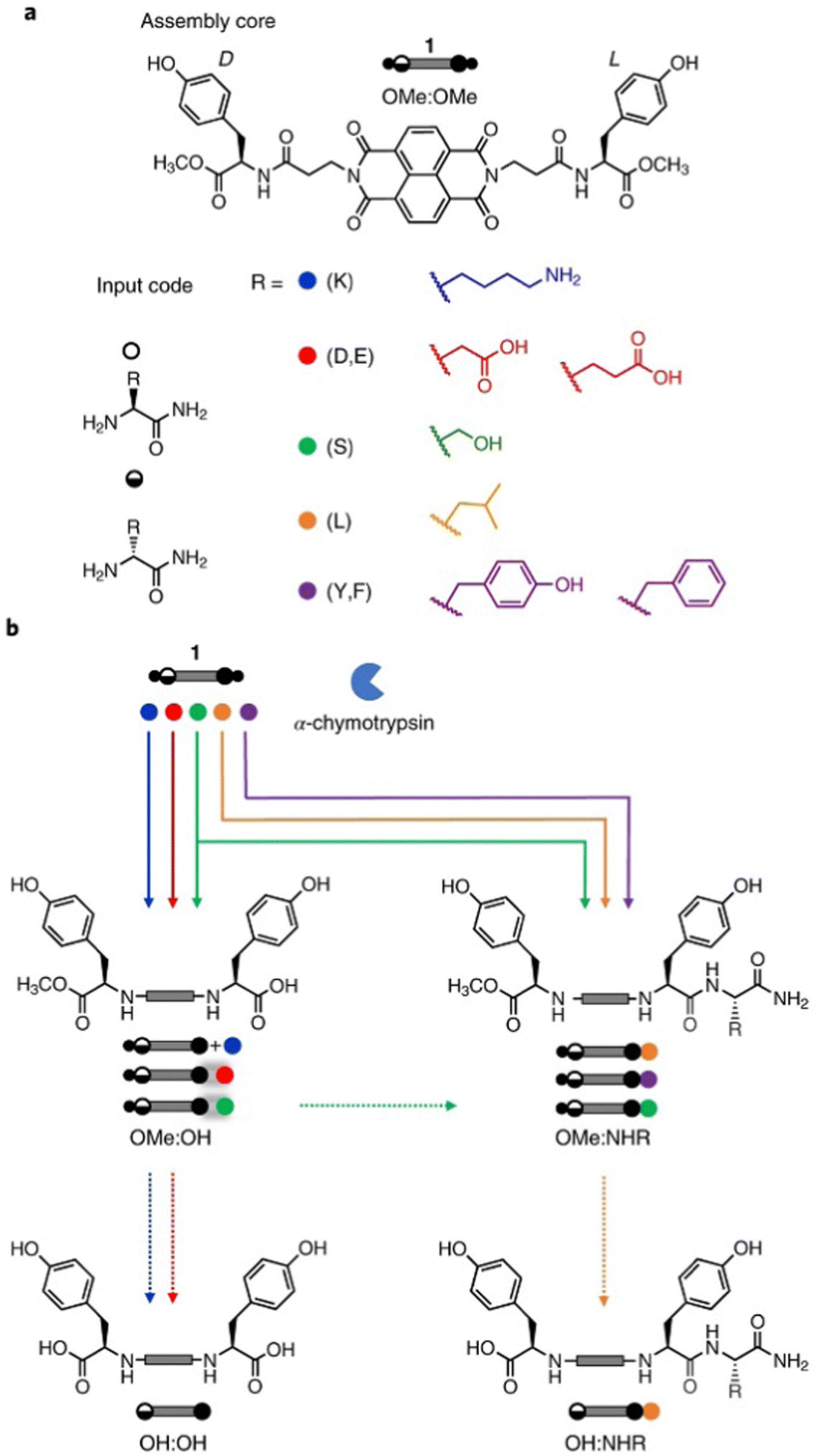 | ||
| Fig. 51 In the presence of an enzyme, (a) different amino acid modified LMWGs (b) respond with different reaction pathways and different kinetics depending on the LMWG structure. This can be used to create transient conductivity profiles that differ from one LMWG to the next, demonstrating how concepts of dynamic and dissipative assembly can achieve dynamic and dissipative physical outputs. Figure adapted and reproduced from ref. 435 with permission of Springer Nature, © 2018. | ||
These examples demonstrate how molecular design can underpin the assembly of systems for optoelectronic applications, with the combination of solid-like and liquid-like characteristics providing gels with significant advantages in device fabrication. Supramolecular principles increase the degree of control by organising molecular building blocks in precise positions with respect to one another. Furthermore, the supramolecular nature of these materials can give them dynamic behaviour. Combined with smart engineering approaches to create sophisticated patterned gels, and the potential of such materials to heal if damaged,247 it is anticipated this will increasingly lead to sophisticated new soft electronic devices.
4.7 Regenerative medicine
As early as the mid-19th century, Thomas Graham noted that gels underpin living systems (Section 2.8). They play vital roles – for example as the cytoplasm of cells, in the soft materials that make up human tissue, and in the extracellular matrix that supports and directs tissue growth. Adhesion points between cells and the gel-like extracellular matrix control and direct cell growth and differentiation.436 Given the biomedical importance of such processes, there has been considerable interest in using synthetic LMWGs to mimic these biological environments and hence control and direct cell growth – either in vivo or in vitro (Fig. 52).437 Self-assembled LMWGs are versatile biomaterials, as discussed in Xu and co-workers’ seminal review.438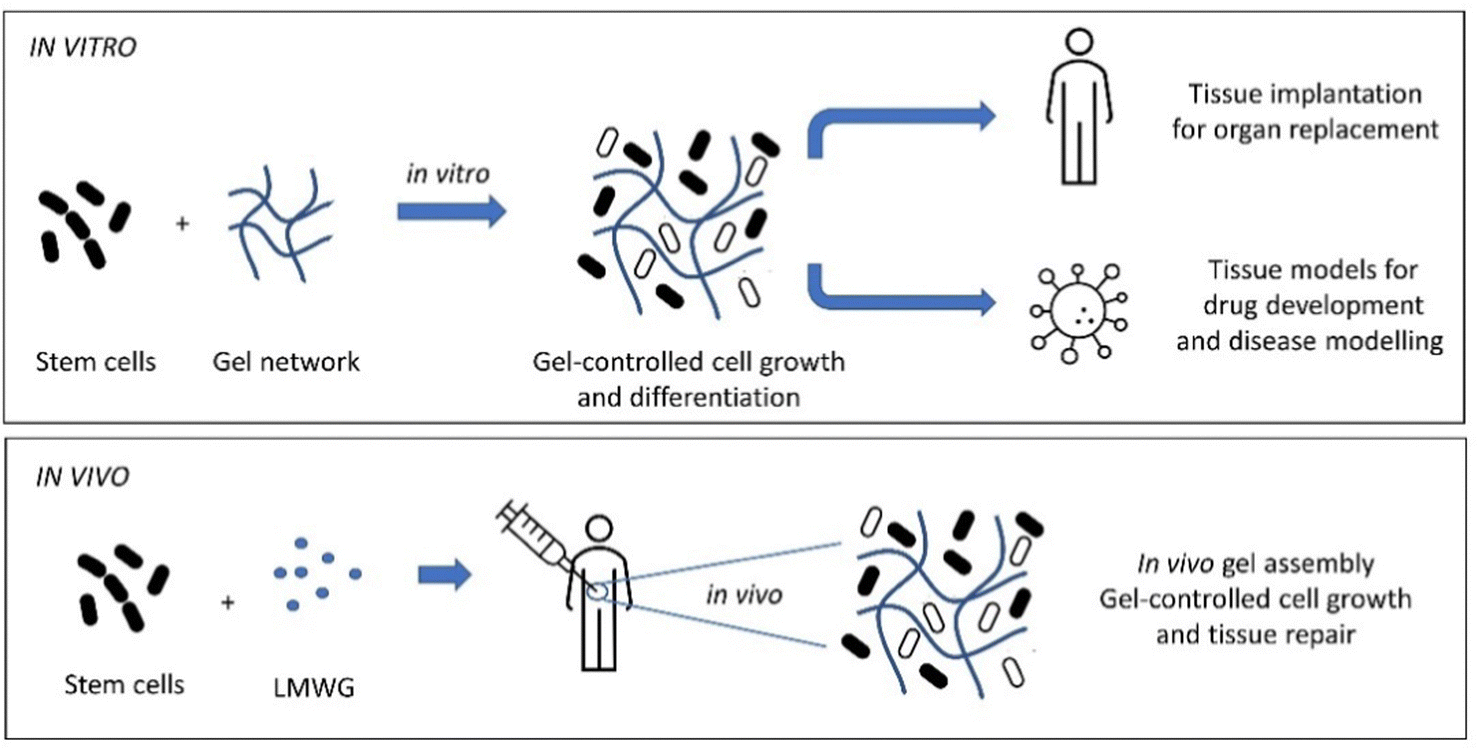 | ||
| Fig. 52 Schematic diagram illustrating in vitro and in vivo approaches to tissue engineering and regenerative medicine. | ||
During in vivo regenerative medicine, the body is encouraged to repair itself more effectively. An ex vivo approach involves the growth of new tissues and organs in vitro, followed by implantation into the patient. Over the next 100 years, regenerative medicine will play a key role in extending lifespans and helping treat a wide range of conditions. Gels are set to play a key role in this revolution in medical technology.439
It is worth noting that polymer gels and thickeners are already beginning to be clinically used in vivo in this regard – for example hyaluronic acid (HA) has been routinely injected into the hips or knees of patients suffering from osteoarthritis.440 Although there remains debate about the effectiveness of this ‘visco-supplementation’ treatment, the concept is that the gel-like nature of the HA may assist joint lubrication and mobility, extending natural joint lifetime, and lubricating replacement synthetic joint implants. Clearly there is potential for application of more advanced, and potentially beneficial, LMWGs.
In regenerative medicine, stem cells are also a source of considerable excitement.441 Stem cells have the potential to differentiate into the range of cells that make up the human body. They can be extracted from the patient themselves, and therefore the application of stem cell technologies in terms of implantation or supplementation can avoid rejection problems that can arise when using donor tissue. Furthermore, the use of stem cells potentially avoids the need to wait for a donor to become available. The use of stem cells in combination with gels offers the potential to support, shape and control tissue growth, helping direct the fabrication of tissue ex vivo, or mediate compatibility between implanted materials/tissue and the human body in vivo.
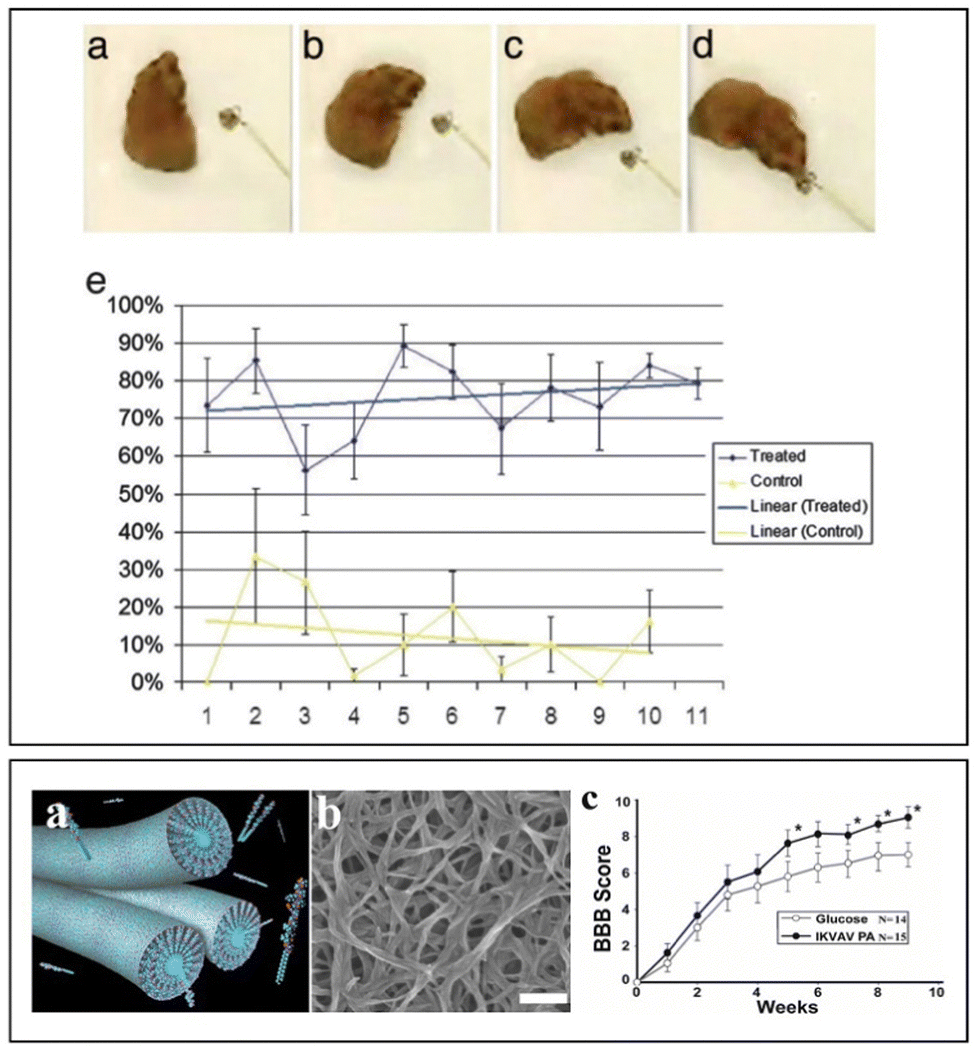 | ||
| Fig. 53 (top) Peptide hydrogelator applied to damaged optic nerve of hamsters with blinding in their right eye, regenerates vision and (a)–(d) the hamster responds to stimulus. (e) Data indicated treated hamsters regained ca. 80% of vision; untreated animals only 10%. Adapted from ref. 442a with permission of the National Academy of Sciences, © 2006. (bottom) (a) image of self-assembling peptide amphiphiles labelled with IKVAV signalling peptides, (b) SEM image of gel nanostructure in vitro (scale bar = 200 nm), (c) graph showing mean mouse BBB locomotor scores between IKVAV PA and glucose injections after spinal cord injury, demonstrating enhanced recovery of animals treated with peptide gel. Figure adapted from ref. 443c with permission of to Society for Neuroscience, © 2008. | ||
Stupp and co-workers used peptide amphiphiles functionalised with the neuroactive pentapeptide IKVAV, in combination with stem cells, to help regenerate damaged spinal cord tissue in a rat model (Fig. 53, bottom).443 Enhanced locomotor scores were observed in animals treated with LMWG rather than with a saline control. It was suggested that the assembly of a gel network in vivo provided a matrix that favoured cellular regrowth and repair. When combined with their gel extrusion nanofibre alignment strategy,276 they showed that mesenchymal stem cells began to grow in alignment with the fibres.444 It has also been demonstrated that IKVAV-functionalised self-assembled peptides can help the proliferation of human embryonic stem cells in the inner ear, with possible applications in regeneration of the spiral ganglion.445 The self-assembled nature of the gel allowed simple delivery as a liquid into the inner ear prior to in situ assembly.
Hartgerink and co-workers have also worked with peptide amphiphile gels, developing an understanding of issues such as fibrous encapsulation, unwanted immune responses and degradation by-products.446 These kind of detailed studies play a key role in translating gel-based technologies into the clinic. By avoiding some of these problems, the team observed excellent results for the in vivo treatment of ischemic tissue disease in a mouse model.447
Alongside in vivo studies, there has been considerable interest in gaining a detailed understanding the way cells behave in supramolecular gels. Working with long-chain peptides, Collier and co-workers focussed on the impact of structure on immunogenic response, finding significant variation even with minor changes.448 Aggeli and co-workers also noted similar effects on cell proliferation in their peptide gels.449 The significance of precise structural features, even in generally biocompatible building blocks like peptides, indicates that care must be taken with LMWG design for applications in regenerative medicine, where immunological responses are generally undesirable. However, the immune-adjuvant effects of some systems do enable potential applications in (e.g.) vaccine development.450
Given the challenges of working with larger peptide gels, which include relatively costly synthesis and the risk of off-target effects, there has been significant interest in very simple gels based on smaller LMWGs. In 2007, Liebmann and co-workers reported the peptide gelator Fmoc-FF as a potential scaffold for cell culture.451 Ulijn and co-workers went on to demonstrate that co-assembling it with Fmoc-RGD created materials suitable for use as 3D scaffolds with anchorage-dependent human dermal fibroblast cells (Fig. 54, top).452 The RGD peptide motif is well-known to encourage cell adhesion, as a result of its interaction with integrins – its incorporation into the self-assembled nanostructures is a good strategy to encourage cell growth. This approach was also applied by Collier and co-workers.453 The co-assembly strategy of combining Fmoc-FF with Fmoc-RGD is something that cannot readily be achieved with polymer gels, and constitutes a significant advantage of LMWGs in this type of application – molecular mixing in supramolecular gels is experimentally simple. Furthermore, the loading-level of co-assembled supramolecular systems can be varied, potentially changing the output, meaning such gels are readily tunable bioactive materials.
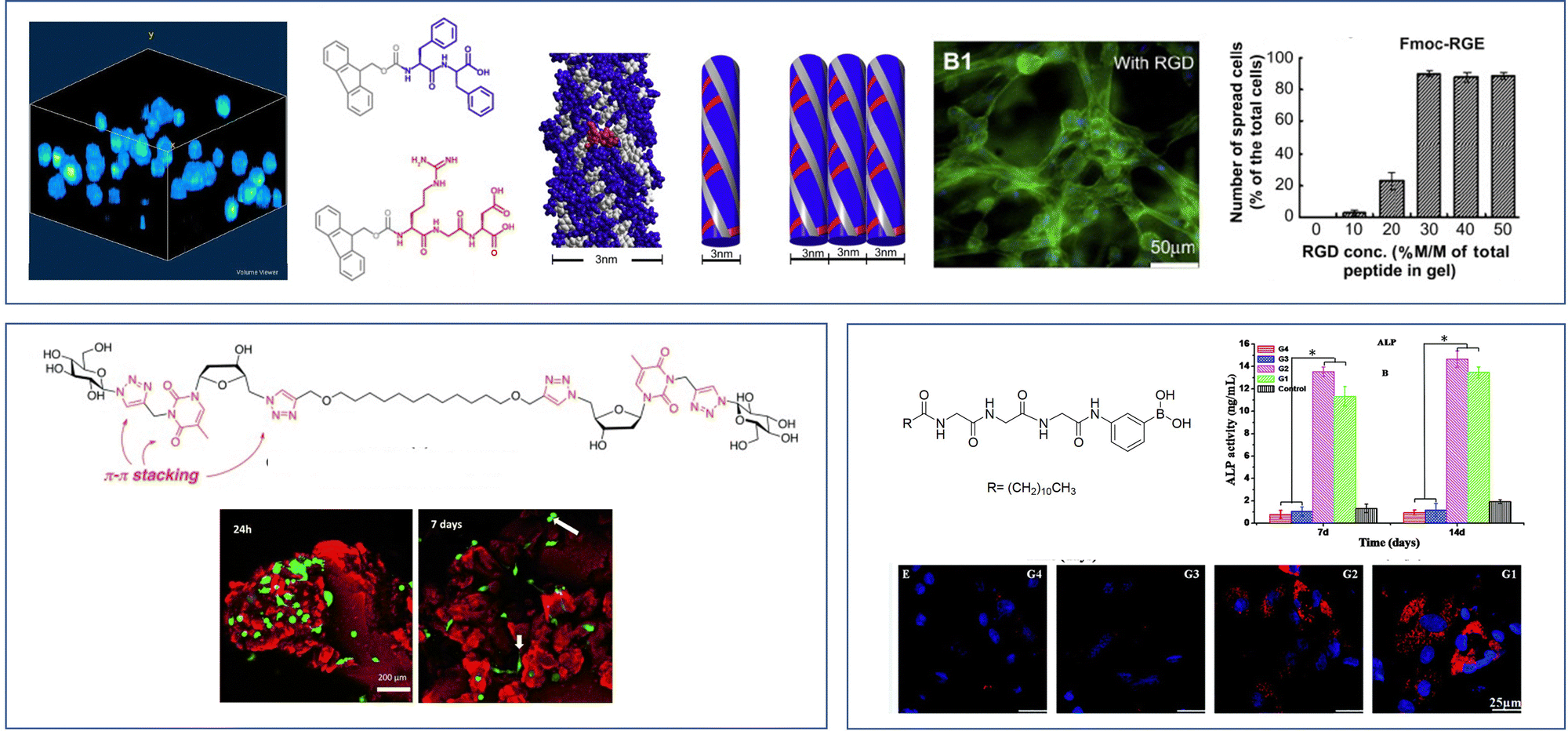 | ||
| Fig. 54 Selected examples and concepts of in vitro tissue growth. (top) Tissue growth on Fmoc-FF. Left panel is a fluorescent image of 3D tissue growth reproduced from ref. 451 with permission of Springer Nature, © 2007. Combination with Fmoc-RGD creates co-assembled nanostructures shown schematically that encourage cell spreading as shown by microscopy imaging, and graph indicating how the loading of Fmoc-RGD switches on this process. Figures reproduced from ref. 452 with permission of Elsevier, © 2008. (bottom left) Injectable nucleobase gelator which gives rise to effective cell growth. Microscopy imaging shows that over time, the cells spread from the surface throughout the gel, and that cell division takes place, figure adapted and reproduced from ref. 455 with permission of Wiley-VCH, © 2015. (bottom right) Gelator which gives rise to four different gels with increasing G′ (G1 < G2 < G3 < G4). Graph indicates greater alkaline phosphatase activity of stem cells grown on the stiffer gels, and imaging indicates greater osteocalcein secretion, both markers of osteogenic differentiation into bone tissue. Figure reproduced from ref. 458a with permission of the Royal Society of Chemistry, © 2016. | ||
Demonstrating this high degree of tunability, Martin and co-workers explored a series of peptide gels in which the LMWG contained two lysines and two phenylalanines, simply located in different positions, to gain an insight into which encouraged the growth of neurons at the nanofibre interface.454 They found significant levels of sequence control, concluding that the mobility of phenylalanine residues at the nanofibre surface was the determining factor in governing the suitability of a given peptide as a scaffold for primary neurons.
Moving beyond peptides, Barthélémy and co-workers developed nucleobase gels for use with human mesenchymal stem cells (Fig. 54, bottom left).455 These soft gels possess self-healing characteristics, meaning they are well suited to syringe injection. Furthermore, cells seeded onto the surface gradually spread throughout the gel. Stem cell survival depended on LMWG structure and gel stiffness. Clearly such systems have potential use in applications encouraging tissue growth and repair in vivo. In recent work, Lampe and co-workers used computational methods to predict a gelator based on a simple pentapeptide, and indicated that its injectability may facilitate cell delivery and protection.456
One of the ways gels can control the behaviour of stem cells is through their rheological parameters. As the G′ value of gels increases from soft to rigid, mesenchymal stem cells can be encouraged to differentiate into different types of cell, from fat, through to muscle, and ultimately bone.457 This mimics how rheology of the extracellular matrix control stem cells. Although well-known from polymer gel research, this principle has been demonstrated by He and co-workers, using LMWGs, using a system with tunable gel stiffness.458 Stiffer gels led to osteogenic differentiation into bone, as indicated by increased alkaline phosphatase activity and greater secretion of osteocalcein (Fig. 54, bottom right).
Beyond the relatively large rheological changes associated with directing stem cells towards fat/muscle/bone, more subtle rheological triggers have also been reported. For example, the loading level of a peptide gelator can provide environments that either encourage the growth of healthy breast cells (G′ < 1 kPa) or breast tumour cells (G′ > 1 kPa), with the system being used to understand the impact of different components on cell proliferation.459 LMWG modification can also lead to small changes in rheological stiffness that have a significant impact on cell survival and neurite growth.460 These observations are relevant in the central nervous system, where small changes in mechanical strength can have major impacts on cell biology.
Ulijn and co-workers went beyond gel-directed rheological control, and carefully studied the cells growing in each type of gel. The cells in each case produced different lipid metabolites.461 Feeding experiments using these lipids then demonstrated that, in their own right, these helped induce differentiation, identifying their importance for the first time. This demonstrates that in addition to being useful cell growth scaffolds, gels can be tools for learning about fundamental biological processes.
Gel composition can also play a role in engineering tissue. In 2001, in work that was well-ahead of its time, Stupp and co-workers used a gelator that incorporated both RGDS peptides and a phosphorylated serine group capable of binding to calcium ions.462 These two factors are tailored to encourage bone growth, and the resulting apatite crystals were aligned with the long axes of the gel fibres, analogous to the alignment observed between collagen fibres and hydroxyapatite crystals.
As noted earlier, multi-component systems have great potential in tissue engineering. For example, Mata and co-workers combined two different self-sorting LMWGs to optimise bulk properties and enhance cytocompatibility in comparison to either single-component.463 There has also been interest in blending LMWGs with PGs. For example, Li and co-workers combined peptide hydrogels with silk fibroin, which is otherwise only a very weak gel.464 The resulting injectable hybrid gel was loaded with vascular endothelial growth factor (VEGF) and subcutaneously injected in mice to trigger the generation of new blood capillaries in vivo.
In the future, in vitro studies will increasingly focus on achieving more complex structured outcomes from stem cell growth. In the field of polymer gels, there is much interest in fabricating patterned materials that shape and direct stem cell growth and behaviour, with the potential to create more complex organs based on multiple interacting cell types.465 With increasing focus on patterning LMWG gels (Section 3.9), it is increasingly possible to generate suitable supramolecular gels.
In early work, Hamachi and co-workers developed a light-sensitive gelator, which underwent a gel–sol transition on exposure to light.466 Using lasers, channels were fabricated in the gel and on seeding with cells, the growth of cellular tissue became spatially patterned within these channels.
Taking a multi-component approach to shaping, we have blended a stem-cell compatible LMWG (DBS-CONHNH2) with a robust but non-interactive PG. The hybrid gels combine the stem cell compatibility of the LMWG with the capacity of the PG to be shaped.278 Furthermore, the ability of the LMWG to generate gold nanoparticles in situ further enhanced stem cell growth. Alternatively, by creating silver nanoparticles in situ, we created shaped gels that enable stem cell growth and have antibacterial activity, with potential applications in orthopaedic regenerative medicine.467 We also formulated injectable nanogels (diameters ca. 800 nm) loaded with heparin, the release of which enhanced stem cell growth.268 Such microgels could potentially be injected into damaged tissue, or after surgical intervention, to encourage tissue repair.
In interesting work, He and co-workers created a multi-domain LMWG/PG system in which a peptide LMWG acted as a soft scaffold, while photopolymerised PEGDA acted as a harder photo-patterned PG domain (Fig. 55).468 This strategy, built on prior work from our own lab, photopatterning the same type of PG/LMWG combinations.469 He and co-workers found that their multidomain materials had an interpenetrated interface region between LMWG and PG domains, possessing a gradient of composition and stiffness. They went on to show that these two domains, connected via the interface, could differentiate cells towards cartilage and bone respectively, an approach that may have application in osteochondral tissue regeneration.
 | ||
| Fig. 55 Combination of LMWG and photo-crosslinkable PEGDA PG create patterned materials with gradients at interpenetrated interfaces between materials which can encourage controlled differentiation of stem cells in osteogenic/chondrogenic ways, with relevance for osteochondral tissue regeneration. Figure reproduced from ref. 468 with permission of Elsevier, © 2021. | ||
In terms of in vitro cell culture the peptide gelator RADA-16, developed by Zhang and co-workers, has been commercialised as ‘PuraMatrix’ and is sold for use in 3D tissue culture. It is a sol in acidic media and forms a gel on mixing with neutral saline, encapsulating cells in situ.470 It is necessary to mix other bioactive units into the gel to optimise cell growth, and the high cost, associated with the relatively complex peptide, suggest there is significant scope for simpler LMWGs to have an impact in this application. Nonetheless, this work Indicates the potential of LMWGs as commercially-viable in vitro tissue culture materials.
Collier and co-workers have used peptide hydrogels to create prostate cancer spheroids in vitro.471 The tissue created behaved like prostate cancer cells in terms of producing prostate specific antigen – such artificial tumour spheroids have the potential to enable the study of metastasis, migration and invasion, and enable more personalised treatments to be developed that target the specific tumour type in an individual patient. Self-assembling peptide gels have also been used to develop an effective and practical breast cancer model, suitable for the growth and study of patient-derived tumour samples, with the added benefits of being relatively cheap, fully-defined and free from the use of animals or animal products.472
In very early work, Zhang and co-workers demonstrated that a self-assembling peptide gel could achieve hemostasis in less than 15 seconds.474 The gel established a near-instant nanofibre barrier when applied directly to a wound in the brain, spinal cord, femoral artery, liver, or skin of mammals, stopping bleeding without pressure, cauterization, vasoconstriction, coagulation, or cross-linked adhesives. More recently, other researchers combined polymeric alginate with peptide amphiphile gels, creating a hybrid gel, to enhance the mechanical robustness of this type of material.475
There are many possible applications of gels in wound repair, and dentistry is an area of particular interest. In fascinating work, a monomeric nucleoside-based LMWG formed hydrogels with a remarkably high storage modulus of ca. 1 MPa, in seconds after shear-thinning injection at 37 °C.476 This LMWG was applied to rat molar extraction sockets, and performed very well in assisting bone healing.
There is increasing focus on the rational design of peptide units to facilitate desired biological outcomes.477 For example, a negatively-charged heparin-mimetic peptide amphiphile gel promotes regeneration of full thickness burn injuries, playing an active role in triggering angiogenesis as a result of its affinity for growth factors.478 Non-bioactive peptide amphiphile gels did not perform as well. In other work, modifying a fibronectin-mimetic peptide sequence with Nap-FF, introduced gel properties and increased the stability of the peptide sequence against protease hydrolysis.479 The resulting new LMWG, Nap-FFPHSRN, performed well in treatment of corneal injuries. A single daily dose of hydrogel facilitated corneal re-epithelialization with morphological and architectural recovery.
 | ||
| Fig. 56 Impact of gelation on cell viability. (Left) Gel assembly within bacterial cells triggered by enzymatic activation leads to bacterial cell inhibition, figure reproduced from ref. 480 with permission of Wiley-VCH, © 2007. (Right) Gel assembly on the cell surface of cancer cells, triggered by over-expressed enzymes on the cell surface, leads to cell death, figure reproduced from ref. 481 with permission of Wiley-VCH, © 2014. | ||
In summary, LMWGs are able to direct cell growth in a variety of ways (mechanical and chemical), and are suitable for both in vitro and in vivo use. Undoubtedly, the tunability, responsiveness, degradability and biocompatibility of supramolecular gels offers great potential in regenerative medicine. It seems likely that the adaptive and dynamic nature of these materials, currently the subject of intense academic investigation will be of significant importance in this application. Living tissue is itself dynamic and adaptive, and the potential synergies between tissue and LMWG matrix, with each being able to impact upon the other in responsive ways, will be an exciting arena for further exploration.
5. Conclusions
Supramolecular gels are materials that have a surprisingly long history, significant uses in everyday life, and a very bright future. From the soft matter technologies of the ancient world such as lubrication and personal care, through to next generation applications in sustainable energy and regenerative medicine, gels have been an ever-present part of human society. The earliest gelators depended on products extracted from the natural world, with metal salts of fatty acids playing a particularly important role.In academic terms, it was early colloid scientists who first got to grips with the physical, molecular, nature of these materials. During the 19th and 20th century, industrial chemists then made significant inroads into understanding how to manipulate these materials, particularly with regard to their rheological performance. This led to the applications of gels in a variety of industries, ranging from the polymer and oil industries through to military and food applications. During this time, it became understood that molecular modification could control performance, with structure–activity relationships being derived. Furthermore it was noted that multi-component systems were a useful way of optimising behaviour in the application of choice. A range of new gelator chemistries emerged during the early part of the 20th centuries. This included organogels based on sugar derivatives and ureas, both of which employ hydrogen bonds in their self-assembly and hydrogels based on nucleobases and amino acids. In all cases, their discovery was serendipitous.
With the advent of supramolecular chemistry, and greater focus on non-covalent interactions and control over self-assembly, the potential to understand and design gelators significantly increased. This led to a huge increase in the chemical space for gelator chemistries. However, within the field there remains an emphasis on the use of bioderived molecules, some of which are fully green and sustainable. There is now much greater understanding of the impact of solvent on gelation, with a degree of predictive capacity based on solvent parameters. Nonetheless, there remains a need to better understand all levels of the hierarchical assembly process – although assembly is quite well understood on the molecular level, network level assembly remains challenging to characterise and/or predict. It is anticipated that significant further breakthroughs await, building on some of the recent early studies in this area.
With a keen focus on interactions between molecules, supramolecular chemists have radically increased the potential of multi-component gels, with interest in gels in which each component plays an active, and predictable role, either in the assembly process, or in the properties of the resulting material. Supramolecular chemists are also increasingly exploring the dynamics of these materials and concluding that they have complex adaptive behaviour, and can often be considered as multi-state materials, rather than simple on–off systems. Careful characterisation of these complex materials, including focus on each process applied to them, is therefore vital. Greater insight into the dynamic nature of gels has enabled the introduction of new specific types of responsive and interactive behaviour as a result. Furthermore, it is increasingly being explored to create smart soft materials in which fuels can temporarily drive self-assembly, but once the fuel runs out, the gel disassembles again – such evolving systems have fascinating future potential in terms of interfacing with dynamic living tissue. Making use of advances in chemical engineering, the ability to shape and pattern supramolecular gels has radically improved in the past 10 years, providing opportunity for more tailored and sophisticated use of these materials.
As a result of these supramolecular developments, the horizons for applications these gels have been shifted very significantly beyond the rheological applications for which they have historically been used. Most modern applications aim to harness both the materials properties of the gel, and some added function programmed in by the chemical structure of the LMWG. Smart gels are designed to interact with drug molecules and hence control their release in active ways, which using their gel characteristics to facilitate delivery. In reaction engineering, carefully-designed gels incorporate catalytically-active units to create recyclable catalysts or can be used to stabilise highly reactive species facilitating their application in synthesis. Alternatively, by designing LMWGs to interact with pollutants, it is possible to use these solvent compatible porous materials to remove pollutants from the environment, with the high nanoscale surfaces providing effective pollutant. The responsive nature of gels allows them to act as sensors, while careful molecular design of gels based on π-systems opens up a wide range of uses as conductive or donor–acceptor materials in energy and photonic applications. Finally, the ability of LMWGs to mimic the extracellular environment makes them ideally placed for use in regenerative medicine, assisting with processes such as tissue growth and wound healing. Their capacity to be used ex vivo or in vivo makes them highly versatile, and once again, the ability to incorporate bioactive chemical functionalities into LMWGs enables them to play specific active roles in living systems.
In the future, it is anticipated that applications will be extended, and new concepts developed, particularly as a result of the enhanced understanding of gels as dynamic and adaptive systems, the engineering skill to pattern them in a controlled way, and the ability to combine multi-components with precise structural control within the gel network in order to endow the materials with multi-function. As selected examples: (i) the dynamic nature of transient gels would be fascinating to interface in regenerative medicine with living systems, which are themselves dynamic and adaptive, in order to achieve temporal and spatial control over cell growth; (ii) the combination of multiple components with gel shaping and patterning may enable the design of gels that can catalyse multistep processes in a single pot; (iii) controlled multi-component gels have considerable potential in smart personal care applications; (iv) enhanced control over nanoscale shaping of LMWGs may open new possibilities in drug delivery as a result of the capacity of nanogels to circulate in the bloodstream for extended periods of time; (v) the combination of innovative gel engineering methods with conductive LMWGs may enable the fabrication of soft adaptive circuitry for nanoscale electronic devices. It is therefore hoped that supramolecular advances will continue to be translated into advanced applications.
It is worth noting that there are some potential pitfalls to using LMWGs. In general, supramolecular gels are rheologically quite weak, and cannot usually reach the strengths or stiffnesses achieved by crosslinked polymer gels. This can often be addressed by blending LMWGs with additives to enhance rheological performance. Supramolecular gels are not necessarily benign/non-toxic even if formed from bio-derived molecules – this must be carefully demonstrated for the application of interest, and it should be noted that the toxicity may be different when assembled and this may be problematic given the dynamic nature of these materials. Unlike some polymer gels, they are mostly not yet approved for use in vivo, and in addition to understanding toxicity, it is also important to ensure that uncontrolled assembly of LMWG into persistent fibrils cannot occur in undesirable locations. However, unlike many areas of supramolecular science, supramolecular gels already have widespread industrial use, and this significantly lowers the barrier for the use of smart LMWGs in next generation applications. As such, the future of supramolecular gels is very promising.
It is hoped that this article has provided a unique panorama over this fascinating class of materials, giving greater appreciation of both the global historical sweep of supramolecular gels, and the way in which both academic and industrial scientists are creating and applying new and innovative fundamental insights and concepts to open up new horizons for their future use.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
DKS acknowledges funding from the Horizon Europe Marie Skłodowska-Curie Action (MSCA) MULTISMART, with local funding of this international network being provided by EPSRC (EP/X02895X/1).References
- (a) R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed; (b) E. R. Draper and D. J. Adams, Chem, 2017, 3, 390–410 CrossRef CAS; (c) D. J. Adams, J. Am. Chem. Soc., 2022, 144, 11047–11053 CrossRef CAS.
- (a) N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC; (b) A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS.
- (a) Polymer Gels: Fundamentals and Applications, ed. H. B. Bohidar, P. Dubin and Y. Osada, American Chemical Society, Washington DC, 2002 Search PubMed; (b) V. K. Thakur and M. K. Thakur, Polymer Gels: Science and Fundamentals, Springer, 2018 Search PubMed.
- R. Eelkema and A. Pich, Adv. Mater., 2020, 32, 1906012 CrossRef CAS.
- Functional Molecular Gels, ed. B. Escuder and J. F. Miravet, RSC, Cambridge, 2014 Search PubMed.
- (a) L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4097 CrossRef CAS; (b) T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed; (c) M. Wehner and F. Würthner, Nat. Rev. Chem., 2020, 4, 38–53 CrossRef CAS.
- B. Qin, Z. Yin, X. Tang, S. Zhang, Y. Wu, J.-F. Xu and X. Zhang, Prog. Polym. Sci., 2020, 100, 101167 CrossRef CAS.
- A. D. O’Donnell, S. Salimi, L. R. Hart, T. S. Babra, B. W. Greenland and W. Hayes, React. Funct. Polym., 2022, 172, 105209 CrossRef.
- (a) J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89–112 CrossRef; (b) D. J. Cram, Angew. Chem., Int. Ed. Engl., 1988, 27, 1009–1020 CrossRef; (c) C. J. Pedersen, Angew. Chem., Int. Ed. Engl., 1988, 27, 1021–1027 CrossRef.
- D. K. Smith, Applications of Supramolecular Gels, in Molecular Gels: Structure and Dynamics, ed. R. G. Weiss, RSC, Cambridge, 2018, pp. 300–371 Search PubMed.
- G. T. Williams, C. J. E. Haynes, M. Fares, C. Caltagirone, J. R. Hiscock and P. A. Gale, Chem. Soc. Rev., 2021, 50, 2737–2763 RSC.
- M. Sutton, The Birth of the Polymer Age, Chemistry World, 2020. https://www.chemistryworld.com/features/the-birth-of-the-polymer-age/4011418.article, accessed Nov 2023.
- L. A. Ruiz, The Hittites’ fast war chariots threatened mighty Egypt, National Geographic, 2020, May 4. https://www.nationalgeographic.co.uk/history-and-civilisation/2020/05/hittites-fast-war-chariots-threatened-mighty-egypt, accessed Nov 2023.
- (a) A. Rovetta, A. Emanueli, I. Nasry and A. Helmi, Ancient Egyptian Chariots Design and Functional Aspects, International Symposium on History of Machines and Mechanisms Proceedings, Springer, 2000, pp. 149–154 Search PubMed; (b) A. Rovetta, I. Nasry and A. Helmi, Mech. Mach. Theory, 2000, 35, 1013–1031 CrossRef.
- J. Lewkowitsch, Chemical technology and Analysis of Oils, Fats and Waxes, Vieweg, Braunschweig, 1904, vol. 2 Search PubMed.
- King Tutankhamun's Chariot, https://www.publicdomainpictures.net/en/view-image.php?image=113775&picture=king-tutankhamuns-chariot, accessed Nov 2023.
- TWA, 1940, https://commons.wikimedia.org/wiki/File:TWA_1940.jpg, accessed Nov 2023.
- Electric car recharging, https://commons.wikimedia.org/wiki/File:Electric_Car_recharging.jpg, accessed Nov 2023.
- W. Little, Brit. Pat., 12571, 1849 Search PubMed.
- C. J. Donahue, J. Chem. Educ., 2006, 83, 862–869 CrossRef CAS.
- R. Zsigmondy and W. Batchmann, Z. Chem. Ind. Kolloide, 1912, 11, 145–157 CrossRef.
- (a) C. E. Earle, US Pat., 2274676, 1942 Search PubMed; (b) C. E. Earle, US Pat., 2274673, 1942 Search PubMed; (c) H. M. Fraser, US Pat., 2397956, 1946 Search PubMed.
- (a) V. A. Mallia and R. G. Weiss, J. Phys. Org. Chem., 2014, 27, 310–315 CrossRef CAS; (b) R. Gordon, S. T. Stober and C. F. Abrams, J. Phys. Chem. B, 2016, 120, 7164–7173 CrossRef CAS.
- A. L. Fameau and M. A. Rogers, Curr. Opin. Colloid Interface Sci., 2020, 45, 68–82 CrossRef CAS.
- B. W. Hotten, US Pat., 2698300, 1954 Search PubMed.
- (a) H. Kinoshita, M. Sekiya and N. Makino, US Pat., 4115284, 1978 Search PubMed; (b) H. Yasui, T. Yoshida, H. Komiya, T. Oguchi and S. Okamura, US Pat., 4668411, 1987 Search PubMed; (c) H. Kinoshita, M. Mishima, M. Sekiya and K. Oyama, US Pat., 4780231, 1988 Search PubMed.
- J. L. Dreher and J. E. Goodrich, US Pat., 3242210, 1966 Search PubMed.
- A. S. Lyadov, Y. M. Maksimova, A. S. Shakhmatova, V. V. Kirillov and O. P. Parenago, Russ. J. Appl. Chem., 2018, 91, 885–894 CrossRef CAS.
- Polyurea greases – applications and properties, https://www.molygraph.com/newsroom/polyurea-greases---applications-and-properties, accessed Nov 2023.
- H. B. Routh, K. R. Bhowmik, L. C. Parish and J. A. Witkowski, Clin. Dermatol., 1996, 14, 3–6 CrossRef CAS.
- The Papyrus Ebers: The Greatest Egyptian Medical Document. T. E. Bendix, Copenhagen, Levin & Munksgarrad, 1937 Search PubMed.
- N. C. McCreeh, A. P. Gize and A. R. David, J. Archaeol. Sci., 2011, 38, 3432–3434 CrossRef.
- History of women's hairstyles in Ancient China: https://www.chinoy.tv/history-hairstyles-ancient-china/, accessed Nov 2023.
- Brylcreem: https://en.wikipedia.org/wiki/Brylcreem, accessed Nov 2023.
- A. P. Tulloch, Bee World, 1980, 61, 47–62 CrossRef CAS.
- W. K. Teller, US Pat., 2732327, 1956 Search PubMed.
- (a) D. L. Shelton, US Pat., 4120948, 1978 Search PubMed; (b) A. D. Sabatelli, US Pat., 4822602, 1989 Search PubMed; (c) B. D. Hofrichter, J. M. Gardlik, P. A. Sawin, J. P. Luebbe and B. J. Bradbury, US Pat., 5429816, 1995 Search PubMed.
- M. J. Meunier, Ann. Chim. Phys., 1891, 22, 412–432 Search PubMed.
- (a) E. L. Roehl and H. B. Tan, US Pat., 4154816, 1979 Search PubMed; (b) E. L. Roehl, US Pat., 4346079, 1982 Search PubMed.
- T. J. Schamper, M. M. Perl and J. D. Warren, US Pat., 4720381, 1988 Search PubMed.
- G. K. Guskey, C. B. Motley and G. E. Tzeghai, US Pat., 5849276, 1998 Search PubMed.
- S. Yamasaki, Y. Ohashi, H. Tsutsumi and K. Tsujii, Bull. Chem. Soc. Jpn., 1995, 68, 146–151 CrossRef CAS.
- J. A. Muszik and W. Diehrichs, US Pat., 3576776, 1971 Search PubMed.
- T. Ando and Y. Yamazaki, US Pat., 3846363, 1974 Search PubMed.
- (a) K. Hamada and H. Uchiyama, US Pat., 4016118, 1977 Search PubMed; (b) H. Uchiyama, US Pat., 4483952, 1984 Search PubMed; (c) G. R. Titus and J. L. Williams, US Pat., 4808650, 1989 Search PubMed.
- J. W. Rekers, US Pat., 5049605, 1991 Search PubMed.
- E. A. Wilder, K. S. Wilson, J. B. Quinn, D. Skrtie and J. M. Antonucci, Chem. Mater., 2005, 17, 2946–2952 CrossRef CAS.
- N. Karim, T. D. Jones, K. M. Lewandowski, B. D. Craig, S. B. Mitra and M. C. P. Alencar, US Pat., 8445558, 2013 Search PubMed.
- D. J. Cornwell and D. K. Smith, Mater. Horiz., 2015, 2, 279–293 RSC.
- (a) E. R. Zubarev, M. U. Pralle, E. D. Stone and S. I. Stupp, Adv. Mater., 2002, 14, 198–203 CrossRef; (b) J. C. Stendahl, L. Li, E. R. Zubarev, Y.-R. Chen and S. I. Stupp, Adv. Mater., 2002, 14, 1540–1543 CrossRef CAS.
- H. Gankema, M. A. Hempenius, M. Möller, G. Johansson and V. Percec, Macromol. Symp., 1996, 102, 381–390 CrossRef CAS.
- A U.S. Navy ZIPPO flamethrower is test from a patrolboat, https://nara.getarchive.net/media/a-u-s-navy-zippo-flame-thrower-is-test-from-a-patrol-boat-ae8915, accessed Nov 2023.
- (a) L. F. Fieser, G. C. Harris, E. B. Hershberg, M. Morgana, F. C. Novello and S. T. Putnam, Ind. Eng. Chem., 1946, 38, 768–773 CrossRef CAS; (b) K. J. Mysels, Ind. Eng. Chem., 1949, 41, 1435–1438 CrossRef CAS.
- Napalm-B to Use Huge Amount of Polystyrene, Chem. Eng. News, 1966, 44, 24 Search PubMed.
- S. P. Srivastava, A. K. Saxena, R. S. Tandon and V. Shekher, Fuels, 1997, 76, 625–630 CrossRef CAS.
- D. J. Abdallah, S. A. Sirchio and R. G. Weiss, Langmuir, 2000, 16, 7558–7561 CrossRef CAS.
- (a) J. B. Clark, US Pat., 2596844, 1952 Search PubMed; (b) M. A. McCabe, L. R. Norman and J. R. Stanford, US Pat., 5514645, 1996 Search PubMed; (c) R. Barati and J.-T. Liang, J. Appl. Polym. Sci., 2014, 40735 CrossRef.
- A. M. Vibhute and K. M. Sureshan, ChemSusChem, 2020, 13, 5343–5360 CrossRef CAS.
- USEPA-Water Quality Office, Gelling Crude Oils to Reduce Marine Pollution from Tanker Oil Spills, Water Pollution Control Research Series 15080DJN 1/71, US Government Printing Office, Washington DC, 1971.
- T. Saito, Y. Matsuzawa, S. Ninagawa, M. Honna, M. Takesada and M. Takehara, US Pat., 3969087, 1976 Search PubMed.
- T. Kobayashi, Y. Kawashima, M. Yoshimura, M. Sugiura, T. Nobe and S. Fujimoto, US Pat., 4502975, 1985 Search PubMed.
- S. Bhattacharya and Y. Krishnan-Ghosh, Chem. Commun., 2001, 185–186 RSC.
- S. R. Jadhav, P. K. Vemula, R. Kumar, S. R. Raghavan and G. John, Angew. Chem., Int. Ed., 2010, 49, 7695–7698 CrossRef CAS.
- V. Mallya and R. G. Weiss, Pat., WO2012047251, 2012 Search PubMed.
- H. Oh, N. Yaraghi and S. R. Raghavan, Langmuir, 2015, 31, 5259–5264 CrossRef CAS.
- J. Poppe, Gelatin, in Thickening and Gelling Agents for Food, ed. A. P. Imeson, 1997, pp. 144–168 Search PubMed.
- R. Armisén and F. Gaiatas, Agar, in Handbook of Hydrocolloids, ed. G. O. Phillips and P. A. Williams, Woodhead Publishing, 2nd edn, 2009, pp. 82–107 Search PubMed.
- H. Braconnot, Ann. Chim. Phys., 1825, 28, 173 Search PubMed.
- E. Onsøyen, Alginates, in Thickening and Gelling Agents for Food, ed. A. P. Imeson, 1997, pp. 22–44 Search PubMed.
- T. Gobley, J. Pharm. Chem., 1846, 9, 81–91 Search PubMed.
- N. Gutiérrez-Méndez, D. R. Chavez-Garay and M. Y. Leal-Ramos, J. Food Biochem., 2022, 46, e14157 CrossRef.
- J. Chevalley, J. Texture Studies, 1975, 6, 177–196 CrossRef.
- A Brief History of Mayonnaise, https://slate.com/culture/2013/12/mayonnaise-history-was-it-invented-by-the-french-or-the-spanish.html, accessed Nov 2023.
- Mayonnaise, https://commons.wikimedia.org/wiki/File:Mayonnaise_%281%29.jpg, accessed Nov 2023.
- (a) P.-L. Luisi, WO1989000077, 1989; (b) R. Scartazzini and P.-L. Luisi, J. Phys. Chem., 1988, 92, 22–31 CrossRef.
- (a) N. E. Hughes, A. G. Marangoni, A. J. Wright, M. A. Rogers and J. W. E. Rush, Int. J. Gastron. Food Sci., 2014, 2, 470–480 Search PubMed; (b) A. J. Martins, A. A. Vicente, R. L. Cunha and M. A. Cerqueira, Food Funct., 2018, 9, 758–773 RSC; (c) A. Puscas, V. Muresan, C. Socaciu and S. Muste, Foods, 2020, 9, 70 CrossRef CAS PubMed; (d) P. M. Silva, M. A. Cerqueira, A. J. Martins, L. H. Fasolin, R. L. Cunha and A. A. Vicente, J. Am. Oil. Chem. Soc., 2022, 99, 911–923 CrossRef CAS; (e) R. C. da Silva, M. J. Ferdaus, A. Foguel and T. L. T. da Silva, Gels, 2023, 9, 180 CrossRef.
- (a) C. Fresenius, Ger. Pat., 142397, 1902 Search PubMed; (b) H. Bollman, Ger. Pat., 439130, 1927 Search PubMed.
- A. R. Patel, D. Schatterman, W. H. De Vos, A. Lesaffer and K. Dewettinck, J. Colloid Interface Sci., 2013, 411, 114–121 CrossRef CAS.
- A. R. Patel, P. S. Rajarethinem, A. Gredowska, O. Turhan, A. Lesaffer, W. H. De Vos, D. Van de Walle and K. Dewettinck, Food Funct., 2014, 5, 645–652 RSC.
- C. D. Doan, I. Tavernier, P. K. Okuro and K. Dewettinck, Innov. Food Sci. Emerg. Technol., 2018, 45, 42–52 CrossRef CAS.
- (a) A. Singh, F. I. Auzanneau and M. A. Rogers, Food Res. Int., 2017, 97, 307–317 CrossRef CAS PubMed; (b) C. Park and F. Maleky, Front. Sust. Food Syst., 2020, 4, 139 CrossRef; (c) Z. Huang, B. Guo, D. Gong and G. Zhang, Food Chem., 2023, 404, 134553 CrossRef CAS PubMed.
- (a) N. E. Hughes, A. G. Marangoni, A. J. Wright, M. A. Rogers and J. W. E. Rush, Trends Food Sci. Technol., 2009, 20, 470–480 CrossRef CAS; (b) T. A. Stortz, A. K. Zetzl, S. Barbut, A. Cattaruzza and A. G. Marangoni, Lipid Technol., 2012, 24, 151–154 CrossRef CAS.
- A. Matheson, G. Dalkas, P. S. Clegg and S. R. Euston, Nutr. Bull., 2018, 43, 189–194 CrossRef CAS PubMed.
- A. Von Lipowitz, Justus Liebigs Ann. Chem., 1841, 38, 348–355 CrossRef.
- H. Henstock, Trans. Faraday Soc., 1934, 30, 403–406 RSC.
- H. Schade and E. Boden, Z. Physiol. Chem., 1913, 83, 347 CrossRef CAS.
- I. Bang, Biochem. Z., 1910, 26, 293–311 CAS.
- (a) M. Gellert, M. N. Lipsett and D. R. Davies, Proc. Natl. Acad. Sci. U. S. A., 1962, 48, 2013–2018 CrossRef CAS; (b) J. F. Chantot and W. Guschlbauer, Jerusalem Symposium on Quantum Chemistry and Biochemistry, 1972, 4, 205–214 Search PubMed.
- D. Bhattacharyya, G. M. Arachchilage and S. Basu, Front. Chem., 2016, 4, 38 CrossRef PubMed.
- (a) T. Bhattacharyya, P. Saha and J. Dash, ACS Omega, 2018, 3, 2230–2241 CrossRef CAS PubMed; (b) M. Godoy-Gallardo, M. Merino-Gómez, L. C. Matiz, M. A. Mateos-Timoneda, F. J. Gil and R. A. Perez, ACS Biomater. Sci. Eng., 2023, 9, 40–61 CrossRef CAS PubMed.
- K. Brenzinger, Z. Physiol. Chem., 1892, 16, 537 Search PubMed.
- R. A. Gortner and W. F. Hoffman, J. Am. Chem. Soc., 1921, 43, 2199–2202 CrossRef CAS.
- L. Pauling, The Nature of the Chemical Bond, Cornell University, New York, 1939 Search PubMed.
- H. Zocher and H. W. Albu, Koll. Z., 1928, 46, 27–33 CrossRef CAS.
- E. K. Rideal, J. Roy. Soc. Arts, 1924, 72, 849–854 Search PubMed.
- W. B. Buck and G. I. Freeze, US Pat., 3192955, 1961 Search PubMed.
- G. Langer, DE 2946027, 1982.
- F. M. Menger and K. S. Venkatasubban, J. Org. Chem., 1978, 43, 3413 CrossRef CAS.
- F. M. Menger and K. L. Caran, J. Am. Chem. Soc., 2000, 122, 11679–11691 CrossRef CAS.
- T. Graham, X. Phil. Trans. Roy. Soc. Ldn., 1861, 151, 183–224 Search PubMed.
- J. T. Trevors and G. H. Pollack, Prog. Biophys. Mol. Biol., 2005, 89, 1–8 CrossRef CAS PubMed.
- Colloid Science: Principles, Methods and Applications, ed. T. Cosgrove, Wiley-Blackwell, 2010 Search PubMed.
- K. von Nägeli, Theorie der Gärung, München, 1879, p. 162.
- J. Tyndall, Proc. Roy. Soc. Ldn., 1868, 17, 223–233 Search PubMed.
- H. Siedentopf and R. Zsigmondy, Ann. Phys., 1902, 315, 1–39 CrossRef.
- D. J. Lloyd, Colloid Chemistry: Theoretical and Applied, ed. J. Alexander, The Chemical Catalog Co., New York, vol 1, 1926, p. 7 Search PubMed.
- H. Staudinger, Ber. Dtsch. Chem. Ges., 1920, 53, 1073–1085 CrossRef.
- P. J. Flory, Angew. Chem., 1975, 87, 787–797 CrossRef CAS.
- (a) P. J. Flory, J. Am. Chem. Soc., 1941, 63, 3083–3090 CrossRef CAS; (b) W. H. Stockmayer, J. Chem. Phys., 1944, 12, 125 CrossRef CAS.
- J.-M. Lehn, Pure Appl. Chem, 1978, 50, 871–892 CrossRef CAS.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS.
- P. L. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3160 CrossRef CAS PubMed.
- (a) K. S. Partridge, D. K. Smith, G. M. Dykes and P. T. McGrail, Chem. Commun., 2001, 319–320 RSC; (b) A. R. Hirst, D. K. Smith, M. C. Feiters, H. P. M. Geurts and A. C. Wright, Two-component Dendritic Gels: Easily Tunable Materials, J. Am. Chem. Soc., 2003, 125, 9010–9011 CrossRef CAS.
- (a) G. Yu, X. Yan, C. Han and F. Huang, Chem. Soc. Rev., 2013, 42, 6697–6722 RSC; (b) V. J. Nebot and D. K. Smith, Techniques for the Characterization of Molecular Gels, in Functional Molecular Gels, ed. B. Escuder and J. F. Miravet, Royal Society of Chemistry, Cambridge, 2013, pp. 30–66 Search PubMed; (c) B. R. Denzer, R. J. Kulchar, R. B. Huang and J. Patterson, Gels, 2021, 7, 158 CrossRef CAS.
- (a) A. Caragheorgheopol, W. Edwards, J. G. Hardy, D. K. Smith and V. Chechik, Langmuir, 2014, 30, 9210–9218 CrossRef CAS PubMed; (b) A. M. F. Fuentes-Caparrós, B. Dietrich, L. Thomson, C. Chaveau and D. J. Adams, Soft Matter, 2019, 15, 6340–6347 RSC; (c) D. McDowall, D. J. Adams and A. M. Seddon, Chem. Soc. Rev., 2022, 18, 1577–1590 CAS; (d) S. Bianco, S. Panja and D. J. Adams, Gels, 2022, 8, 132 CrossRef CAS PubMed; (e) R. Chevigny, E. D. Sitsanidis, J. Schirmer, E. Hulkko, P. Myllyperkio, M. Nissinen and M. Petterson, Chem. – Eur. J., 2023, 29, e202300155 CrossRef CAS PubMed.
- (a) J. H. van Esch, F. Schoonbeek, M. De Loos, E. M. Veen, R. M. Kellogg and B. L. Feringa, Low molecular weight gelators for organic solvents – From serendipity towards design, Supramolecular science: where it is and where it is going, Springer, Dordrecht, 1999, pp. 233–259 Search PubMed; (b) D. M. Zurcher and A. J. McNeil, J. Org. Chem., 2015, 80, 2473–2478 CrossRef CAS; (c) P. Dastidar, Gels, 2019, 8, 15 CrossRef PubMed.
- M. Liu, G. Ouyang, D. Niu and Y. Sang, Org. Chem. Front., 2018, 5, 2885–2900 RSC.
- (a) P. W. J. M. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7, 30–37 CrossRef CAS PubMed; (b) J. K. Gupta, D. J. Adams and N. G. Berry, Chem. Sci., 2016, 7, 4713–4719 RSC; (c) R. van Lommel, J. Zhao, W. M. De Borggraeve, F. De Proft and M. Alonso, Chem. Sci., 2020, 11, 4226–4238 RSC.
- (a) F. Fages, F. Vögtle and M. Žinić, Top. Curr. Chem., 2005, 256, 77–131 CrossRef CAS; (b) R. van Lommel, L. A. J. Rutgeerts, W. M. De Borggraeve, F. De Proft and M. Alonso, ChemPlusChem, 2020, 85, 267–276 CrossRef CAS PubMed; (c) M. Yokoya, S. Kimura and M. Yamanaka, Chem. – Eur. J., 2021, 27, 5601–5614 CrossRef CAS.
- (a) K. Hanabusa, K. Shimura, K. Hirose, M. Kimura and H. Shirai, Chem. Lett., 1996, 885–886 CrossRef CAS; (b) J. van Esch, S. De Feyter, R. M. Kellogg, F. De Schryver and B. L. Feringa, Self-Chem. Eur. J., 1997, 3, 1238–1243 CrossRef CAS; (c) L. A. Estroff and A. D. Hamilton, Angew. Chem., Int. Ed., 2000, 39, 3447–3450 CrossRef CAS.
- T. Yi, X. Yu and L. Chen, Hydrogen Bonding for the Self-assembly of Organogels and Hydrogels, Hydrogen Bonded Supramolecular Materials, Springer, 2015, pp. 69–100 Search PubMed.
- M. A. Kuzina, D. D. Kartsev, A. V. Stratonovich and P. A. Levkin, Adv. Funct. Mater., 2023, 33, 2301421 CrossRef CAS.
- (a) J. H. Fuhrhop, P. Schnieder, J. Rosenberg and E. Boekema, J. Am. Chem. Soc., 1987, 109, 3387–3390 CrossRef CAS; (b) J. H. Fuhrhop, S. Svenson, C. Boettcher, E. Rossler and H. M. Vieth, J. Am. Chem. Soc., 1990, 112, 4307–4312 CrossRef CAS.
- (a) C. Boettcher, B. Schade and J. H. Fuhrhop, Langmuir, 2001, 17, 873–877 CrossRef CAS; (b) G. Wang and A. D. Hamilton, Chem. Commun., 2003, 310–311 RSC.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc. Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- D. K. Smith, Soft Matter Science – A Historical Overview with a Supramolecular Perspective, in Supramolecular Chemistry: From Molecules to Nanomaterials, ed. P. A. Gale and J. W. Steed, Wiley-VCH, 2012, vol. 7, pp. 3169–3182 Search PubMed.
- T. Kunitake, Y. Okahata, M. Shimomura, S. Yasunami and K. Takarabe, J. Am. Chem. Soc., 1981, 103, 5401–5413 CrossRef CAS.
- J. Zhang, X. Li and X. Li, Prog. Polym. Sci., 2012, 37, 1130–1176 CrossRef CAS.
- S. Datta and S. Bhattacharya, Chem. Soc. Rev., 2015, 44, 5596–5637 RSC.
- G. R. Newkome, G. R. Baker, M. J. Saunders, P. S. Russo, V. K. Gupta, Z.-Q. Yao, J. E. Miller and K. Bouillion, J. Chem. Soc., Chem. Commun., 1986, 752–753 RSC.
- J.-H. Fuhrhop and T. Wang, Chem. Rev., 2004, 104, 2901–2938 CrossRef CAS PubMed.
- (a) F. M. Menger, H. Zhang, K. L. Caran, V. A. Seredyuk and R. P. Apkarian, J. Am. Chem. Soc., 2002, 124, 1140 CrossRef CAS; (b) F. M. Menger, V. A. Seredyuk, R. P. Apkarian and E. R. Wright, J. Am. Chem. Soc., 2002, 124, 12408 CrossRef CAS.
- T. Imae, Y. Takahashi and H. Muramatsu, J. Am. Chem. Soc., 1992, 114, 3414–3419 CrossRef CAS.
- (a) S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC; (b) L. Li, L. Xie, R. Zheng and R. Sun, Front. Chem., 2021, 9, 739791 CrossRef CAS.
- (a) M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS; (b) A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC.
- (a) K. McAulay, B. Dietrich, H. Su, M. T. Scott, S. Rogers, Y. K. Al-Hilaly, H. Cui, L. C. Serpell, A. M. Seddon, E. R. Draper and D. J. Adams, Chem. Sci., 2019, 10, 7801–7806 RSC; (b) E. R. Draper, B. Dietrich, K. McAulay, C. Brasnett, H. Abdizadeh, I. Patmanidis, S. J. Marrink, H. Su, H. Cui, R. Schweins, A. Seddon and D. J. Adams, Matter, 2020, 2, 764–778 CrossRef.
- (a) C. Tomasini and N. Castellucci, Chem. Soc. Rev., 2013, 42, 156–172 RSC; (b) R. Schweitzer-Stenner and N. J. Alvarez, J. Phys. Chem. B, 2021, 125, 6760–6775 CrossRef CAS.
- (a) A. Aggeli, M. Bell, N. Boden, J. N. Keen, P. F. Knowles, T. C. McLeish, M. Pitkeathly and S. E. Radford, Nature, 1997, 386, 259–262 CrossRef CAS; (b) A. Aggeli, I. A. Nyrkova, M. Bell, R. Harding, L. Carrick, T. C. B. McKeish, A. N. Semenov and N. Boden, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11857 CrossRef CAS PubMed.
- (a) E. F. Banwell, E. S. Abelardo, D. J. Adams, M. A. Birchall, A. Corrigan, A. M. Donald, M. Kirkland, L. C. Serpell, M. F. Butler and D. N. Woolfson, Nat. Mater., 2009, 8, 596–600 CrossRef CAS PubMed; (b) N. Mehrban, E. Abelardo, A. Wasmuth, K. L. Hudson, L. M. Mullen, A. R. Thomson, M. A. Birchall and D. N. Woolfson, Adv. Healthcare Mater., 2014, 3, 1387–1391 CrossRef CAS PubMed.
- M. P. Hendricks, K. Sato, L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2017, 50, 2440–2448 CrossRef CAS PubMed.
- C. W. G. Fishwick, A. J. Beevers, L. M. Carrick, C. D. Whitehouse, A. Aggeli and N. Boden, Nano Lett., 2003, 3, 1475–1479 CrossRef CAS.
- N. Mehrban, B. Zhu, F. Tamagnini, F. I. Young, A. Wasmuth, K. L. Hudson, A. R. Thomson, M. A. Birchall, A. D. Randall, B. Song and D. N. Woolfson, ACS Biomater. Sci. Eng., 2015, 1, 431–439 CrossRef CAS PubMed.
- A. van Teijlingen and T. Tuttle, J. Chem. Theory Comput., 2021, 17, 3221–3232 CrossRef CAS.
- B. O. Okesola, V. M. P. Vieira, D. J. Cornwell, N. K. Whitelaw and D. K. Smith, Soft Matter, 2015, 11, 4767–4787 RSC.
- D. J. Cornwell, B. O. Okesola and D. K. Smith, Soft Matter, 2013, 9, 8730–8736 RSC.
- B. O. Okesola and D. K. Smith, Chem. Commun., 2013, 49, 11164–11166 RSC.
- G. John, B. V. Shankar, S. R. Jadhav and P. K. Vemula, Langmuir, 2010, 26, 17843–17851 CrossRef CAS.
- UN DESA, The Sustainable Development Goals Report 2023: Special Edition – July 2023, New York, USA, 2023.
- P. K. Vemula, J. Li and G. John, J. Am. Chem. Soc., 2006, 128, 8932–8938 CrossRef CAS.
- P. K. Vemula, U. Aslam, V. A. Mallia and G. John, Chem. Mater., 2007, 19, 138–140 CrossRef CAS.
- G. John, J. H. Hung, M. Masuda and T. Shimizu, Langmuir, 2004, 20, 2060–2065 CrossRef CAS.
- C. D. Jones, H. T. D. Simmons, K. E. Horner, K. Liu, R. L. Thompson and J. W. Steed, Nat. Chem., 2019, 11, 375–381 CrossRef CAS PubMed.
- E. R. Draper, B. Dietrich, K. McAulay, C. Brasnett, H. Abdizadeh, I. Patmanidis, S. J. Marrink, H. Su, H. Cui, R. Schweins, A. Seddon and D. J. Adams, Matter, 2020, 2, 764–778 CrossRef.
- R. Laishram, S. Sarkar, I. Seth, N. Khatun, V. K. Aswal, U. Maitra and S. J. George, J. Am. Chem. Soc., 2022, 144, 11306–11315 CrossRef CAS.
- Y. Wang, Z. Xu, M. Lovrak, V. A. A. le Sage, K. Zhang, X. Guo, R. Eelkema, E. Mendes and J. H. van Esch, Angew. Chem., Int. Ed., 2020, 132, 4860–4864 CrossRef.
- A. Dawn and H. Kumari, Chem. – Eur. J., 2018, 24, 762–776 CrossRef CAS.
- (a) K. Hanabusa, M. Matsumoto, M. Kimura, A. Kakehi and H. Shirai, J. Colloid Interface Sci., 2000, 224, 231–244 CrossRef CAS PubMed; (b) J. Makarevic, M. Jokic, M. Peric, V. Tomisic, B. Kojic-Prodic and M. Zinic, Chem. – Eur. J., 2001, 7, 3328–3341 CrossRef CAS; (c) A. R. Hirst and D. K. Smith, Langmuir, 2004, 20, 10851–10857 CrossRef CAS; (d) G. Zhu and J. S. Dordick, Chem. Mater., 2006, 18, 5988–5995 CrossRef CAS.
- Y. Lan, M. G. Corradini, R. G. Weiss, S. R. Raghavan and M. A. Rogers, Chem. Soc. Rev., 2015, 44, 6035–6058 RSC.
- M. Raynal and L. Bouteiller, Chem. Commun., 2011, 47, 8271–8273 RSC.
- (a) Y. Lan, M. G. Corradini, X. Liu, T. E. May, F. Borondics, R. G. Weiss and M. A. Rogers, Langmuir, 2014, 30, 14128–14142 CrossRef CAS PubMed; (b) A. Singh, F.-I. Auzanneau, M. G. Corradini, G. Grover, R. G. Weiss and M. A. Rogers, Langmuir, 2017, 33, 10907–10916 CrossRef CAS PubMed.
- D. R. Nunes, M. Reche-Tamayo, E. Ressouche, M. Raynal, B. Isare, P. Foury-Leylekian, P.-A. Albouy, P. Brocorens, R. Lazzaroni and L. Bouteiller, Langmuir, 2019, 35, 7970–7977 CrossRef.
- P. Jonkheijm, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Science, 2006, 313, 80–83 CrossRef CAS.
- G. Ghosh, A. Chakraborty, P. Pal, B. Jana and S. Ghosh, Chem. – Eur. J., 2022, 28, e202201082 CrossRef CAS.
- (a) J. Le Bideau, L. Viau and A. Vioux, Chem. Soc. Rev., 2011, 40, 907–925 RSC; (b) P. C. Marr and A. C. Marr, Green Chem., 2016, 18, 105–128 RSC.
- N. Kimizuka and T. Nakashima, Langmuir, 2001, 17, 6759–6761 CrossRef CAS.
- K. Hanabusa, H. Fukui, M. Suzuki and H. Shirai, Langmuir, 2005, 21, 10383–10390 CrossRef CAS.
- (a) S. Marullo, A. Meli, F. Giannici and F. D’Anna, ACS Sustainable Chem. Eng., 2018, 6, 12598–12602 CrossRef CAS; (b) J. Ruiz-Olles, P. Slavik, N. K. Whitelaw and D. K. Smith, Angew. Chem., Int. Ed., 2019, 58, 4173–4178 CrossRef CAS PubMed.
- Y. Liang, K. Wang, J. Li, Y. Zhang, J. Liu, K. Zhang, Y. Cui, M. Wang and C.-S. Liu, Mater. Horiz., 2022, 9, 1700–1707 RSC.
- (a) T. Kato, N. Mizoshita and K. Kishimoto, Angew. Chem., Int. Ed., 2006, 45, 38–68 CrossRef CAS; (b) N. Mizoshita, T. Kutsuna, T. Kato and K. Hanabusa, Chem. Commun., 1999, 781–782 RSC; (c) T. Kato, T. Kutsuna, K. Yabuuchi and N. Mizoshita, Langmuir, 2002, 18, 7086–7088 CrossRef CAS; (d) N. Mizoshita, K. Hanabusa and T. Kato, Adv. Funct. Mater., 2003, 13, 313–317 CrossRef CAS.
- M. Moriyama, N. Mizoshita, T. Yokota, K. Kishimoto and T. Kato, Adv. Mater., 2003, 15, 1335–1338 CrossRef CAS.
- A. R. Hirst and D. K. Smith, Chem. – Eur. J., 2005, 11, 5496–5508 CrossRef CAS PubMed.
- L. E. Buerkle and S. J. Rowan, Chem. Soc. Rev., 2012, 41, 6089–6102 RSC.
- M. Suzuki, Y. Nakajima, M. Yumoto, M. Kimura, H. Shirai and K. Hanabusa, Langmuir, 2003, 19, 8622–8624 CrossRef CAS.
- (a) M. George and R. D. Weiss, J. Am. Chem. Soc., 2001, 123, 10393–10394 CrossRef CAS; (b) M. George and R. D. Weiss, Langmuir, 2003, 19, 1017–1025 CrossRef CAS.
- J. Boekhoven, J. M. Poolman, C. Maity, F. Li, L. van der Mee, C. B. Minkenberg, E. Mendes, J. H. van Esch and R. Eelkema, Nat. Chem., 2013, 5, 433–437 CrossRef CAS.
- B. Buchs, W. Fieber, F. Vigouroux-Elie, N. Sreenivasachary, J.-M. Lehn and A. Herrmann, Org. Biomol. Chem., 2011, 9, 2906–2919 RSC.
- K. Hanabusa, T. Miki, Y. Taguchi, T. Koyama and H. Shirai, J. Chem. Soc., Chem. Commun., 1993, 1382–1384 RSC.
- (a) X. Xu, M. Ayyagari, M. Tata, V. T. John and G. L. McPherson, J. Phys. Chem., 1993, 97, 11350–11353 CrossRef CAS; (b) M. Tata, V. T. John, Y. Y. Waguespack and G. L. McPherson, J. Am. Chem. Soc., 1994, 116, 9464–9470 CrossRef CAS; (c) B. A. Simmons, C. E. Taylor, F. A. Landis, V. T. John, G. L. McPherson, D. K. Schwartz and R. Moore, J. Am. Chem. Soc., 2001, 123, 2414–2421 CrossRef CAS.
- (a) M. Ayabe, T. Kishida, N. Fujita, K. Sada and S. Shinkai, Org. Biomol. Chem., 2003, 1, 2744–2747 RSC; (b) A. Ballabh, D. R. Trivedi and P. Dastidar, Chem. Mater., 2003, 15, 2136–2140 CrossRef CAS.
- S. S. Rohner, J. Ruiz-Olles and D. K. Smith, RSC Adv., 2015, 5, 27190–27196 RSC.
- (a) A. R. Hirst, B. Huang, V. Castelletto, I. W. Hamley and D. K. Smith, Chem. – Eur. J., 2007, 13, 2180–2188 CrossRef CAS; (b) C. Colquhoun, E. R. Draper, E. G. B. Eden, B. N. Cattoz, K. L. Morris, L. Chen, T. O. McDonald, A. E. Terry, P. C. Griffiths, L. C. Serpell and D. J. Adams, Nanoscale, 2014, 6, 13719–13725 RSC; (c) S. Fleming, S. Debnath, P. W. J. M. Frederix, N. T. Hunt and R. V. Ulijn, Biomacromolecules, 2014, 15, 1171–1184 CrossRef CAS PubMed; (d) J. Raeburn and D. J. Adams, Chem. Commun., 2014, 51, 5170–5180 RSC.
- (a) C. C. Piras and D. K. Smith, Chem. – Eur. J., 2019, 25, 11318–11326 CrossRef CAS; (b) S. Panja, B. Dietrich, A. J. Smith, A. Seddon and D. J. Adams, ChemSystChem, 2022, 4, e202200008 CrossRef CAS.
- E. R. Draper and D. J. Adams, Chem. Soc. Rev., 2018, 47, 3395–3405 RSC.
- (a) S. Onogi, H. Shigemitsu, T. Yoshii, T. Tanida, M. Ikeda, R. Kubota and I. Hamachi, Nat. Chem., 2016, 8, 743–752 CrossRef CAS; (b) R. Kubota, S. Liu, H. Shigemitsu, K. Nakamura, W. Tanaka, M. Ikeda and I. Hamachi, Bioconj. Chem., 2018, 29, 2058–2067 CrossRef CAS.
- R. Kubota, K. Nagao, W. Tanaka, R. Matsumura, T. Aoyama, K. Urayama and I. Hamachi, Nat. Commun., 2020, 11, 4100 CrossRef CAS.
- Y. Wang, M. Lovrak, Q. Liu, C. Maity, V. A. A. le Sage, X. Guo, R. Eelkema and J. H. van Esch, J. Am. Chem. Soc., 2019, 141, 2847–2851 CrossRef CAS PubMed.
- K. Nakamura, R. Kubota, T. Aoyama, K. Urayama and I. Hamachi, Nat. Commun., 2023, 14, 1696 CrossRef CAS.
- A. Heeres, C. Van der Pol, M. Stuart, A. Friggeri, B. L. Feringa and J. Van Esch, J. Am. Chem. Soc., 2003, 125, 14252–14253 CrossRef CAS PubMed.
- A. Brizard, M. Stuart, K. van Bommel, A. Friggeri, M. de Jong and J. Van Esch, Angew. Chem., Int. Ed., 2008, 47, 2063–2066 CrossRef CAS.
- (a) V. J. Nebot, B. Escuder, J. F. Miravet, J. Smets and S. Fernández-Prieto, Langmuir, 2013, 29, 9544–9550 CrossRef CAS PubMed; (b) V. M. P. Vieira, L. L. Hay and D. K. Smith, Chem. Sci., 2017, 8, 6981–6990 RSC; (c) A. Torres-Martinez, C. A. Angulo-Pachón, F. Galindo and J. F. Miravet, Langmuir, 2019, 35, 13375–13381 CrossRef CAS.
- (a) K. Steck, J. H. Van Esch, D. K. Smith and C. Stubenrauch, Soft Matter, 2019, 15, 3111–3121 RSC; (b) K. Steck and C. Stubenrauch, Langmuir, 2019, 35, 17132–17141 CrossRef CAS; (c) K. Steck, N. Preisig and C. Stubenrauch, Langmuir, 2020, 35, 17142–17149 CrossRef; (d) K. Steck, S. Dieterich, C. Stubenrauch and F. Giesselmann, J. Mater. Chem. C, 2020, 8, 5335–5348 RSC.
- L. Su, J. Mosquera, M. F. J. Mabesoone, S. M. C. Schoenmakers, C. Muller, M. E. J. Vleugels, S. Dhiman, S. Wijker, A. R. A. Palmans and E. W. Meijer, Science, 2022, 377, 213–218 CrossRef CAS PubMed.
- (a) D. K. Smith, Chem. Soc. Rev., 2009, 38, 684–694 RSC; (b) P. Duan, H. Cao, L. Zhang and M. Liu, Soft Matter, 2014, 10, 5428–5448 RSC; (c) L. Zhang, Q. Jin and M. Liu, Chem. – Asian J., 2016, 11, 2642–2649 CrossRef CAS PubMed; (d) S. Huang, H. F. Yu and Q. Li, Adv. Sci., 2021, 8, 2002132 CrossRef PubMed; (e) Y. Sang and M. Liu, Chem. Sci., 2022, 13, 633–656 RSC.
- T. Tachibana and H. Kambara, J. Am. Chem. Soc., 1965, 87, 3015–3016 CrossRef CAS.
- T. Taro, M. Tomoko and H. Kayako, Bull. Chem. Soc. Jpn., 1980, 53, 1714–1719 CrossRef.
- S. Yamasaki, Y. Ohashi, H. Tsutsumi and K. Tsujii, Bull. Chem. Soc. Jpn., 1995, 68, 146–151 CrossRef CAS.
- M. M. J. Smulders, M. M. L. Nieuwenhuizen, T. F. A. de Greef, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Chem. – Eur. J., 2010, 16, 362–367 CrossRef CAS.
- A. R. A. Palmans, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Angew. Chem., Int. Ed. Engl., 1997, 36, 2648–2651 CrossRef CAS.
- (a) S. R. Nam, H. Lee and J.-I. Hong, Chem. – Eur. J., 2008, 14, 6040–6043 CrossRef CAS PubMed; (b) W. Cai, G.-T. Wang, P. Du., R.-X. Wang, X.-K. Jiang and Z.-T. Li, J. Am. Chem. Soc., 2008, 130, 13450–13459 CrossRef CAS PubMed; (c) F. Rodriguez-Llansola, D. Hermida-Merino, B. Nieto-Ortega, F. J. Ramirez, J. T. Navarrete, C. Lopez, J. Casado, I. W. Hamley, B. Escuder, W. Hayes and J. F. Miravet, Chem. – Eur. J., 2012, 18, 14725–14731 CrossRef CAS PubMed.
- (a) J. Makarević, M. Jokić, Z. Raza, Z. Štefanić, B. Kojić-Prodić and M. Žinić, Chem. – Eur. J., 2003, 9, 5567–5580 CrossRef; (b) K. J. Nagy, M. C. Giano, A. Jin, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2011, 133, 14975–14977 CrossRef CAS.
- J. van Gestel, A. R. A. Palmans, B. Titulaer, J. A. J. M. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2005, 127, 5490–5494 CrossRef CAS.
- (a) D. Gambhir, S. Kumar, G. Dey, V. Krishnan and R. R. Koner, Chem. Commun., 2018, 54, 11407–11410 RSC; (b) X. Xu, L. Qu, J. Song, D. Wu, X. Zhou and H. Xiang, Chem. Commun., 2019, 55, 9873–9876 RSC; (c) A. K. Patterson, L. H. El-Qarra and D. K. Smith, Chem. Commun., 2022, 58, 3941–3944 RSC.
- W. Edwards and D. K. Smith, J. Am. Chem. Soc., 2014, 136, 1116–1124 CrossRef CAS PubMed.
- (a) Y. Liu, C. Chen, T. Wang and M. Liu, Langmuir, 2016, 32, 322–328 CrossRef; (b) W. Edwards and D. K. Smith, Gels, 2018, 4, 31 CrossRef PubMed.
- D. Yang, J. Han, Y. Sang, T. Zhao, M. Liu and P. Duan, J. Am. Chem. Soc., 2021, 143, 13259–13265 CrossRef CAS.
- G. F. Liu, D. Zhang and C. L. Feng, Angew. Chem., Int. Ed., 2014, 53, 7789–7793 CrossRef CAS.
- (a) J. Liu, F. Yuan, X. Ma, D.-I. Y. Auphedeous, C. Zhao, C. Liu, C. Shen and C. Feng, Angew. Chem., Int. Ed., 2018, 57, 6475–6479 CrossRef CAS PubMed; (b) S. He, Y. Zhang, C. Zhao, X. Wang, S. Baddi, B. Wu, X. Dou and C. Feng, Chem. – Eur. J., 2022, 29, e202202735 CrossRef.
- M. L. Sleczkowski, M. F. J. Mabesoone, P. Sleczkowski, A. R. A. Palmans and E. W. Meijer, Nat. Chem., 2021, 13, 200–207 CrossRef CAS.
- J. Sun, Y. Li, F. Yan, C. Liu, Y. Sang, F. Tian, Q. Feng, P. Duan, L. Zhang, X. Shi, B. Ding and M. Liu, Nat. Commun., 2018, 9, 2599 CrossRef PubMed.
- C. D. Jones and J. W. Steed, Chem. Soc. Rev., 2016, 45, 6546–6596 RSC.
- A. R. Hirst, I. A. Coates, T. R. Boucheteau, J. F. Miravet, B. Escuder, V. Castelletto, I. W. Hamley and D. K. Smith, J. Am. Chem. Soc., 2008, 130, 9113–9121 CrossRef CAS.
- S. Wu, Q. Zhang, Y. Deng, X. Li, Z. Luo, B. Zheng and S. Dong, J. Am. Chem. Soc., 2020, 142, 448–455 CrossRef CAS PubMed.
- S. Panja and D. J. Adams, Chem. Soc. Rev., 2021, 50, 5165–5200 RSC.
- M. de Loos, A. Friggeri, J. van Esch, R. M. Kellogg and B. L. Feringa, Org. Biomol. Chem., 2005, 3, 1631–1639 RSC.
- B. P. Nowak, L. Schlichter and B. J. Ravoo, Angew. Chem., Int. Ed., 2022, 61, e202201791 CrossRef CAS PubMed.
- J. Raeburn, T. O. McDonald and D. J. Adams, Chem. Commun., 2012, 48, 9355–9357 RSC.
- (a) Y. Wu, S. Wu, X. Tian, X. Wang, W. Wu, G. Zou and Q. Zhang, Soft Matter, 2011, 7, 716–721 RSC; (b) S. Lee, S. Oh, J. Lee, Y. Malpani, Y.-S. Jung, B. Kang, J. Y. Lee, K. Ozasa, T. Isoshima, S. Y. Lee, M. Hara, D. Hashizume and J.-M. Kim, Langmuir, 2013, 29, 5869–5877 CrossRef CAS PubMed.
- F. A. Larik, L. L. Fillbrook, S. S. Nurttila, A. D. Martin, R. P. Kuchell, K. A. Taief, M. Bhadbhade, J. E. Beves and P. Thordarson, Angew. Chem., Int. Ed., 2021, 60, 6764–6770 CrossRef CAS.
- (a) Z. Yang, H. Gu, J. Du, J. Gao, B. Zhang, X. Zhang and B. Xu, Tetrahedron, 2007, 63, 7349–7357 CrossRef CAS; (b) B. P. Nowak, M. Niehues and B. J. Ravoo, Soft Matter, 2021, 17, 2857–2864 RSC.
- M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960–2004 CrossRef CAS PubMed.
- M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Commun., 2008, 2644–2646 RSC.
- G. O. Lloyd and J. W. Steed, Nat. Chem., 2009, 1, 437–442 CrossRef CAS PubMed.
- H. He, W. Tan, J. Guo, M. Yi, A. N. Shy and B. Xu, Chem. Rev., 2020, 120, 9994–10078 CrossRef CAS PubMed.
- Z. Yang, H. Gu, D. Fu, P. Gao, J. K. Lam and B. Xu, Adv. Mater., 2004, 16, 1440–1444 CrossRef CAS.
- (a) S. Toledano, R. J. Williams, V. Jayawarna and R. V. Ulijn, J. Am. Chem. Soc., 2006, 128, 1070–1071 CrossRef CAS PubMed; (b) D. Koda, T. Maruyama, N. Minakuchi, K. Nakashima and M. Goto, Chem. Commun., 2010, 46, 979–981 RSC; (c) S. C. Bremmer, A. J. McNeil and M. B. Soellner, Chem. Commun., 2014, 50, 1691–1693 RSC; (d) J. Baillet, A. Gaubert, J. Verget, L. Latxague and P. Barthélémy, Soft Matter, 2020, 16, 7648–7651 RSC.
- (a) A. K. Das, A. R. Hirst and R. V. Ulijn, Faraday Discuss., 2009, 143, 293–303 RSC; (b) J. R. Fores, M. Criado-Gonzalez, M. Schmutz, C. Blanck, P. Schaaf, F. Boulmedais and L. Jierry, Chem. Sci., 2019, 10, 4761–4766 RSC.
- A. R. Hirst, S. Roy, M. Arora, A. K. Das, N. Hodson, P. Murray, S. Marshall, N. Javid, J. Sefcik, J. Boekhoven, J. H. van Esch, S. Santabarbera, N. T. Hunt and R. V. Ulijn, Nat. Chem., 2010, 2, 1089–1094 CrossRef CAS.
- J. P. Wojciechowski, A. D. Martin and P. Thordarson, J. Am. Chem. Soc., 2018, 140, 2869–2874 CrossRef CAS.
- (a) F. Zhao, A. Bonasera, U. Nöchel, M. Behl and D. Bléger, Macromol. Rapid Commun., 2018, 39, 1700527 CrossRef; (b) C.-W. Chu, L. Stricker, T. M. Kirse, M. Hayduk and B. J. Ravoo, Chem. – Eur. J., 2019, 25, 6131–6140 CrossRef CAS.
- J. J. D. de Jong, L. N. Lucas, R. M. Kellogg, J. H. van Esch and B. L. Feringa, Science, 2004, 304, 278–281 CrossRef CAS PubMed.
- (a) B. Escuder, M. Llusar and J. F. Miravet, J. Org. Chem., 2006, 71, 7747–7752 CrossRef CAS; (b) A. R. Hirst, J. F. Miravet, B. Escuder, L. Noirez, V. Castelletto, I. W. Hamley and D. K. Smith, Chem. – Eur. J., 2009, 15, 372–379 CrossRef CAS.
- Y. E. Shapiro, Prog. Polym. Sci., 2011, 36, 1184–1253 CrossRef CAS.
- S. Yilmazer, D. Schwaller and P. J. Mésini, Gels, 2023, 9, 273 CrossRef CAS.
- S. Díaz-Oltra, C. Berdugo, J. F. Miravet and B. Escuder, New J. Chem., 2015, 39, 3785–3791 RSC.
- A. J. Cruz-Cabeza, S. M. Reutzel-Edens and J. Bernstein, Chem. Soc. Rev., 2015, 44, 8619–8635 RSC.
- F. Tantakitti, J. Boekhoven, X. Wang, R. Kazantsev, T. Yu, J. Li, E. Zhuang, R. Zandi, J. H. Ortony, C. J. Newcomb, L. C. Palmer, G. S. Shekhawat, M. Olvera de la Cruz, G. C. Schatz and S. I. Stupp, Nat. Mater., 2016, 15, 469–476 CrossRef CAS.
- (a) V. J. Anderson and H. N. W. Lekkerkerker, Nature, 2002, 416, 811–815 CrossRef CAS; (b) J. R. Moffat and D. K. Smith, Chem. Commun., 2008, 2248–2250 RSC; (c) J. L. Andrews, E. Pearson, D. S. Yufit, J. W. Steed and K. Edkins, Cryst. Growth Des., 2018, 18, 7690–7700 CrossRef CAS; (d) D. Giuri, L. J. Marshall, C. Wilson, A. Seddon and D. J. Adams, Soft Matter, 2021, 17, 7221–7226 RSC.
- T. Guterman, M. Levin, S. Kolusheva, D. Levy, N. Noor, Y. Roichman and E. Gazit, Angew. Chem., Int. Ed., 2019, 58, 15869–15875 CrossRef CAS.
- (a) F. Rodriguez-Llansola, J. F. Miravet and B. Escuder, Chem. Commun., 2009, 209–211 RSC; (b) M. M. Smith and D. K. Smith, Soft Matter, 2011, 7, 4856–4860 RSC; (c) E. R. Draper, T. O. McDonald and D. J. Adams, Chem. Commun., 2015, 51, 6595–6597 RSC; (d) K. Gayen, N. Nandi, K. S. Das, D. Hermida-Merino, I. W. Hamley and A. Banerjee, Soft Matter, 2020, 16, 10106–10114 RSC; (e) M. Mirzamami, A. Dawn, C. J. Garvey, L. He, H. Koerner and H. Kumari, Phys. Chem. Chem. Phys., 2023, 25, 131–141 RSC.
- J. R. White, C. R. Chimie, 2006, 9, 1396–1408 CrossRef CAS.
- C. Colquhoun, E. R. Draper, R. Schweins, M. Marcello, D. Vadukul, L. C. Serpell and D. J. Adams, Soft Matter, 2017, 13, 1914–1919 RSC.
- A. M. Fuentes-Caparrós, F. de Paula Gómez-Franco, B. Dietrich, C. Wilson, C. Brasnett, A. Seddon and D. J. Adams, Nanoscale, 2019, 11, 3275–3280 RSC.
- R. I. Randle, R. E. Ginesi, O. Matsarskaia, R. Schwiens and E. R. Draper, Macromol. Rapid Commun., 2023, 44, 2200709 CrossRef CAS.
- Y. Wang, T. K. Piskorz, M. Lovrak, E. Mendes, X. Guo, R. Eelkema and J. H. van Esch, Adv. Sci., 2020, 7, 1902487 CrossRef CAS.
- S. Cantekin, Y. Nakano, J. C. Everts, P. van der Schoot, E. W. Meijer and A. R. A. Palmans, Chem. Commun., 2012, 48, 3803–3805 RSC.
- (a) X. Yu, L. Chen, M. Zhang and T. Yi, Chem. Soc. Rev., 2014, 43, 5346–5371 RSC; (b) J. Li, L. Geng, G. Wang, H. Chu and H. Wei, Chem. Mater., 2017, 29, 8932–8952 CrossRef CAS.
- R. van Lommel, J. van Hooste, J. Vandaele, G. Steurs, T. Van der Donck, F. De Proft, S. Rocha, D. Sakellariou, M. Alonso and W. M. De Borggraeve, Gels, 2022, 8, 813 CrossRef CAS PubMed.
- L. Yan, G. Li, Z. Ye, F. Tian and S. Zhang, Chem. Commun., 2014, 50, 14839–14842 RSC.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS.
- E. Weyandt, G. M. ter Huurne, G. Vantomme, A. J. Markvoort, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2020, 142, 6295–6303 CrossRef CAS.
- Y. Qin, Y. Wang, J. Xiong, Q. Li and M.-H. Zeng, Small, 2023, 2207785 CrossRef CAS.
- (a) B. Rieß, R. K. Grötsch and J. Boekhoven, Chem, 2020, 6, 552–578 CrossRef; (b) A. Sharko, D. Livitz, S. De Piccoli, K. J. M. Bishop and T. M. Hermans, Chem. Rev., 2022, 122, 11759–11777 CrossRef CAS.
- J. Boekhoven, A. M. Brizard, K. N. K. Kowlgi, G. J. M. Koper, R. Eelkema and J. H. van Esch, Angew. Chem., Int. Ed., 2010, 49, 4825–4828 CrossRef CAS.
- (a) T. Heuser, E. Weyandt and A. Walther, Angew. Chem., Int. Ed., 2015, 54, 13258–13262 CrossRef CAS; (b) M. Tena-Solsona, B. Rieß, R. K. Grötsch, F. C. Löhrer, C. Wanzke, B. Käsdorf, A. R. Bausch, P. Müller-Buschbaum, O. Lieleg and J. Boekhoven, Nat. Commun., 2017, 8, 15895 CrossRef CAS; (c) R. Chevigny, J. Schirmer, C. C. Piras, A. Johansson, E. Kalenius, D. K. Smith, M. Petterson, E. D. Sitsanidis and M. Nissinen, Chem. Commun., 2021, 57, 10375–10378 RSC.
- K. Dai, J. Rodon Fores, C. Wanzke, B. Winkeljann, A. M. Bergmann, O. Lieleg and J. Boekhoven, J. Am. Chem. Soc., 2020, 142, 14142–14149 CrossRef CAS.
- T. M. Hermans and N. Singh, Angew. Chem., Int. Ed., 2023, 62, e202301529 CrossRef CAS.
- E. Olivieri, G. Quintard, J.-V. Naubron and A. Quintard, J. Am. Chem. Soc., 2021, 143, 12650–12657 CrossRef CAS.
- G. Wang and S. Lie, ChemSystChem, 2020, 2, e1900046 CrossRef CAS.
- (a) S. Debnath, S. Roy and R. V. Ulijn, J. Am. Chem. Soc., 2013, 135, 16789–16792 CrossRef CAS; (b) C. G. Pappas, I. R. Sasselli and R. V. Ulijn, Angew. Chem., Int. Ed., 2015, 54, 8119–8123 CrossRef CAS.
- (a) S. Bal, K. Das, S. Ahmed and D. Das, Angew. Chem., Int. Ed., 2019, 58, 244–247 CrossRef CAS; (b) S. Afrose, S. Bal, A. Chatterjee, K. Das and D. Das, Angew. Chem., Int. Ed., 2019, 58, 15783–15787 CrossRef CAS PubMed.
- N. Singh, B. Lainer, G. J. M. Formon, S. De Piccoli and T. M. Hermans, J. Am. Chem. Soc., 2020, 142, 4083–4087 CrossRef CAS PubMed.
- N. Singh, A. Lopez-Acosta, G. J. M. Formon and T. M. Hermans, J. Am. Chem. Soc., 2022, 144, 410–415 CrossRef CAS.
- P. R. A. Chivers and D. K. Smith, Nat. Rev. Chem., 2019, 4, 463–478 CAS.
- (a) P. Sahoo, R. Sankolli, H.-Y. Lee, S. R. Raghavan and P. Dastidar, Chem. – Eur. J., 2012, 18, 8057–8063 CrossRef CAS PubMed; (b) Z. Xu, J. Peng, N. Yan, H. Yu, S. Zhang, K. Liu and Y. Fang, Soft Matter, 2013, 9, 1091–1099 RSC.
- J. Wang, Z. Wang, J. Gao, L. Wang, Z. Yang, D. Kong and Z. Yang, J. Mater. Chem., 2009, 19, 7892–7896 RSC.
- C. C. Piras, P. Slavik and D. K. Smith, Angew. Chem., Int. Ed., 2020, 59, 853–859 CrossRef CAS.
- C. C. Piras, A. G. Kay, P. G. Genever and D. K. Smith, Chem. Sci., 2021, 12, 3958–3965 RSC.
- (a) C. C. Piras and D. K. Smith, Chem. – Eur. J., 2021, 27, 14527–14534 CrossRef CAS; (b) C. C. Piras, P. G. Genever and D. K. Smith, Mater. Adv., 2022, 3, 7966–7975 RSC.
- M. C. Nolan, A. M. Fuentes Caparros, B. Dietrich, M. Barrow, E. R. Cross, M. Bleuel, S. M. King and D. J. Adams, Soft Matter, 2017, 13, 8426–8432 RSC.
- A. M. Fuentes-Caparrós, Z. Canales-Galarza, M. Barrow, B. Dietrich, J. Läuger, M. Nemeth, E. R. Draper and D. J. Adams, Biomacromolecules, 2021, 22, 1625–1638 CrossRef PubMed.
- M. J. S. Hill and D. J. Adams, Soft Matter, 2022, 18, 5960–5965 RSC.
- H. Jian, M. Wang, Q. Dong, J. Li, A. Wang, X. Li, P. Ren and S. Bai, ACS Appl. Mater. Interfaces, 2019, 11, 46419–46426 CrossRef CAS.
- A. C. Farsheed, A. J. Thomas, B. H. Pogostin and J. D. Hartgerink, Adv. Mater., 2023, 35, 2210378 CrossRef CAS.
- S. M. Zhang, M. A. Greenfield, A. Mata, L. C. Palmer, R. Bitton, J. R. Mantei, C. Aparicio, M. O. de la Cruz and S. I. Stupp, Nat. Mater., 2010, 9, 594–601 CrossRef CAS.
- (a) T. Sawada, M. Tsuchiya, T. Takahashi, H. Tsutsumi and H. Mihara, Polym. J., 2012, 44, 651–657 CrossRef CAS; (b) K. Fukunaga, H. Tsutsumi and H. Mihara, Biopolymers, 2016, 106, 476–483 CrossRef CAS; (c) I.-C. Li and J. D. Hartgerink, J. Am. Chem. Soc., 2017, 139, 8044–8050 CrossRef CAS PubMed.
- (a) A. Chalard, P. Joseph, S. Souleille, B. Lonetti, N. Saffon-Merceron, I. Loubinoux, L. Vaysse, L. Malaquin and J. Fitremann, Nanoscale, 2019, 11, 15043–15056 RSC; (b) A. Chalard, M. Mauduit, S. Souleille, P. Joseph, L. Malaquin and J. Fitremann, Addit. Manuf., 2020, 33, 101162 CAS.
- C. C. Piras, A. G. Kay, P. G. Genever, J. Fitremann and D. K. Smith, Chem. Sci., 2022, 13, 1972–1981 RSC.
- E. N. Drew, C. C. Piras, J. Fitremann and D. K. Smith, Chem. Commun., 2022, 58, 11115–11118 RSC.
- S. Xiao, P. J. Paukstelis, R. D. Ash, P. Y. Zavalij and J. T. Davis, Angew. Chem., Int. Ed., 2019, 58, 18434–18437 CrossRef CAS.
- J. Eastoe, M. Sanchez-Dominguez, P. Wyatt and R. K. Heenan, Chem. Commun., 2004, 2608–2609 RSC.
- J. J. D. de Jong, P. R. Hania, A. Pugžlys, L. N. Lucas, M. de Loos, R. M. Kellogg, B. L. Feringa, K. Duppen and J. H. van Esch, Angew. Chem., Int. Ed., 2005, 44, 2373–2376 CrossRef CAS PubMed.
- D. J. Cornwell, O. J. Daubney and D. K. Smith, J. Am. Chem. Soc., 2015, 137, 15486–15492 CrossRef CAS.
- D. J. Cornwell and D. K. Smith, Chem. Commun., 2020, 56, 7029–7032 RSC.
- E. R. Draper, E. G. B. Eden, T. O. McDonald and D. J. Adams, Nat. Chem., 2015, 7, 848–852 CrossRef CAS PubMed.
- (a) M. Lovrak, W. E. J. Hendriksen, C. Maity, S. Mytnyk, V. van Steijn, R. Eelkema and J. H. van Esch, Nat. Commun., 2017, 8, 15317 CrossRef CAS; (b) M. Lovrak, W. E. Hendriksen, M. T. Kreutzer, V. van Steijn, R. Eelkema and J. H. van Esch, Soft Matter, 2019, 15, 4276–4283 RSC.
- M. Lovrak, S. J. Picken, R. Eelkema and J. H. van Esch, ChemNanoMat, 2018, 4, 772–775 CrossRef CAS.
- L. Schlichter, C. C. Piras and D. K. Smith, Chem. Sci., 2021, 12, 4162–4172 RSC.
- H. S. Cooke, L. Schlichter, C. C. Piras and D. K. Smith, Chem. Sci., 2021, 12, 12156–12164 RSC.
- L. Thomson, R. Schweins, E. R. Draper and D. J. Adams, Macromol. Rapid Commun., 2020, 2000093 CrossRef CAS.
- D. Spitzer, V. Marichez, G. J. M. Formon, P. Besenius and T. M. Hermans, Angew. Chem., Int. Ed., 2018, 57, 11349–11353 CrossRef CAS.
- Y. Nishida, A. Tanaka, S. Yamamoto, Y. Tominaga, N. Kunikata, M. Mizuhata and T. Maruyama, Angew. Chem., Int. Ed., 2017, 56, 9410–9414 CrossRef CAS.
- R. Kubota, M. Makuta, R. Suzuki, M. Ichikawa, M. Tanaka and I. Hamachi, Nat. Commun., 2020, 11, 3541 CrossRef CAS PubMed.
- K. Nakamura, W. Tanaka, K. Sada, R. Kubota, T. Aoyama, K. Urayama and I. Hamachi, J. Am. Chem. Soc., 2021, 143, 19532–19541 CrossRef CAS.
- (a) J. Raeburn, B. Alston, J. Kroeger, T. O. McDonald, J. R. Howse, P. J. Cameron and D. J. Adams, Mater. Horiz., 2014, 1, 241–246 RSC; (b) C. Patterson, B. Dietrich, C. Wilson, A. R. Mount and D. J. Adams, Soft Matter, 2022, 18, 1064–1070 RSC.
- (a) R. J. Williams, A. M. Smith, R. Collins, N. Hodson, A. K. Das and R. V. Ulijn, Nat. Nanotechnol., 2009, 4, 19–24 CrossRef CAS; (b) M. P. Conte, K. H. A. Lau and R. V. Ulijn, ACS Appl. Mater. Interfaces, 2017, 9, 3266–3271 CrossRef CAS.
- B. Yang, M. Lledos, R. Akhtar, G. Ciccone, L. Jiang, E. Russo, S. Rajput, C. Jin, M. G. F. Angelereou, T. Arnold, J. Rawle, M. Vassalli, M. Marlow, D. J. Adams and M. Zelzer, Chem. Sci., 2021, 12, 14260–14269 RSC.
- K. J. Skilling, F. Citossi, T. D. Bradshaw, M. Ashford, B. Kellam and M. Marlow, Soft Matter, 2014, 10, 237–256 RSC.
- Ed. P. J. Sheskey, B. C. Hancock, G. P. Moss and D. J. Goldfarb. Handbook of Pharmaceutical Excipients: Edition 9, Pharmaceutical Press, 2020 Search PubMed.
- (a) H. Willimann, P. Walde, P. L. Luisi, A. Gazzaniga and F. Stroppolo, J. Pharm. Sci., 1992, 81, 871–874 CrossRef CAS PubMed; (b) R. Tarantino, 1994.
- (a) L. V. Allen, Int. J. Pharm. Compd., 2003, 7, 180–183 Search PubMed; (b) J. O. Trimble and C. M. Brisco. US Pat., 20090017120, 2009 Search PubMed; (c) H. Alsaab, S. P. Bonam, D. Bahl, P. Chowdury, K. Alexander and S. H. S. Boddu, J. Pharm. Pharm. Sci., 2016, 19, 252–273 CrossRef CAS.
- (a) J.-C. Leroux and A.-C. Couffin-Hoarau, US Pat., 7691408, 2010 Search PubMed; (b) J.-C. Leroux and G. Bastiat, US Pat., 8815944, 2014 Search PubMed.
- (a) A. C. Couffin-Hoarau, A. Motulsky, P. Delmas and J.-C. Leroux, Pharm. Res., 2004, 21, 454–457 CrossRef CAS PubMed; (b) A. Motulsky, M. Lafleur, A. C. Couffin-Hoarau, D. Hoarau, F. Boury, J. P. Benoit and J.-C. Leroux, Biomaterials, 2005, 26, 6242–6253 CrossRef CAS.
- F. Plourde, A. Motulsky, A. C. Couffin-Hoarau, D. Hoarau, H. Ong and J.-C. Leroux, J. Controlled Release, 2005, 108, 433–441 CrossRef CAS.
- (a) K. Iwanaga, T. Sumizawa, M. Miyazaki and M. Kakemi, Int. J. Pharm., 2010, 388, 123–128 CrossRef CAS; (b) S. R. P. Camelo, S. Franceschi, E. Perez., S. G. Fullana and M. I. Ré, Drug Dev. Ind. Pharm., 2016, 42, 985–997 CrossRef; (c) C. L. Esposito, V. Tardif, M. Sarrazin, P. Kirilov and V. G. Roullin, Mater. Sci. End. C, 2020, 114, 110999 CrossRef CAS PubMed.
- C. L. Esposito, P. Kirilov and V. Gaëlle Roullin, J. Controlled Release, 2018, 271, 1–20 CrossRef CAS.
- (a) C. B. P. Olivieira, V. Gomes, P. M. T. Ferreira, J. A. Martins and P. J. Jervis, Gels, 2022, 8, 706 CrossRef; (b) M. Godoy-Gallardo, M. Merino-Gomez, L. C. Matiz, M. A. Mateos-Timoneda, F. J. Gil and R. A. Perez, ACS Biomater. Sci. Eng., 2023, 9, 40–61 CrossRef CAS PubMed.
- R. Vegners, I. Shestakova, I. Kalvinish, R. M. Ezzell and P. A. Kanmey, J. Pept. Sci., 1995, 1, 371–378 CrossRef CAS PubMed.
- (a) Y. Nagai, L. D. Unsworth, S. Koutsopoulos and S. Zhang, J. Controlled Release, 2006, 115, 18–25 CrossRef CAS PubMed; (b) S. Koutsopolous, L. D. Unsworth, Y. Nagaia and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4623–4628 CrossRef.
- J. Mayr, C. Saldías and D. Díaz Díaz, Chem. Soc. Rev., 2018, 47, 1484–1515 RSC.
- D. M. Raymond, B. L. Abraham, T. Fujita, M. J. Watrous, E. S. Toriki, T. Takano and B. L. Nilsson, ACS Appl. Biomater., 2019, 2, 2116–2124 CrossRef CAS.
- B. Xing, C.-W. Yu, K.-H. Chow, P.-L. Ho, D. Fu and B. Xu, J. Am. Chem. Soc., 2002, 124, 14846–14847 CrossRef CAS PubMed.
- Y. Gao, Y. Kuang, Z.-F. Guo, Z. Guo, I. J. Krauss and B. Xu, J. Am. Chem. Soc., 2009, 131, 13576–13577 CrossRef CAS.
- (a) H. Wang, J. Wei, C. Yang, H. Zhao, D. Li, Z. Yin and Y. Zang, Biomaterials, 2012, 33, 5848–5853 CrossRef CAS; (b) K. Zhang, L. Zhou, F. Chen, Y. Chen and X. Luo, J. Controlled Release, 2019, 315, 197–205 CrossRef CAS.
- N. D. Bansode, K. R. Sindhu, C. Morel, M. Rémy, J. Verget, C. Boiziau and P. Barthélémy, Biomater. Sci., 2020, 8, 3186–3192 RSC.
- W. E. M. Noteborn, S. K. Vittala, M. B. Torredemer, C. Maity, F. Versluis, R. Eelkema and R. E. Kieltyka, Biomacromolecules, 2023, 24, 377–386 CrossRef CAS.
- P. Dastidar, R. Roy, R. Parveen and K. Sarkar, Adv. Ther., 2019, 2, 1800061 CrossRef.
- R. Roy, J. Majumder, H. K. Datta, R. Parveen and P. Dastidar, ACS Appl. Bio Mater., 2022, 5, 610–621 CrossRef CAS PubMed.
- P. Dastidar, R. Roy, R. Parveen and K. Sarkar, Adv. Ther., 2019, 2, 1800061 CrossRef.
- (a) K. Pandurangan, J. A. Kitchen, S. Blasco, F. Paradisi and T. Gunnlaugsson, Chem. Commun., 2014, 50, 10819–10822 RSC; (b) V. R. Aldilla, R. Chen, R. Kuppusamy, S. Chakraborty, M. D. P. Willcox, D. StC. Black, P. Thordarson, A. D. Martin and N. Kumar, Sci. Rep., 2022, 12, 22259 CrossRef PubMed.
- L. Yang, C. Zhang, F. Huang, J. Liu, Y. Zhang, C. Yang, C. Ren, L. Chu, B. Liu and J. Liu, J. Controlled Release, 2020, 324, 354–365 CrossRef CAS.
- D. Limón, C. Jiménez-Newman, A. C. Calpena, A. González-Campo, D. B. Amabilino and L. Pérez-García, Chem. Commun., 2017, 53, 4509–4512 RSC.
- M. A. Ramin, K. R. Sindhu, A. Appavoo, K. Oumzil, M. W. Grinstaff, O. Chassande and P. Barthélémy, Adv. Mater., 2017, 29, 1605227 CrossRef.
- (a) A. K. Patterson and D. K. Smith, Chem. Commun., 2020, 56, 11046–11049 RSC; (b) R. Martí-Centelles, I. Dolz-Pérez, J. De la O, I. Ontoria-Oviedo, P. Sepúlveda, V. J. Nebot, M. J. Vicent and B. Escuder, ACS Appl. Bio Mater., 2021, 4, 935–944 CrossRef.
- B. Martin, F. Brouillet, S. Franchesci and E. Perez, AAPS PharmSciTech, 2017, 18, 1261–1269 CrossRef CAS.
- C. C. Piras, C. S. Mahon and D. K. Smith, Chem. – Eur. J., 2020, 26, 8452–8457 CrossRef CAS.
- S. A. Stewart, J. Doniguez-Robles, R. F. Donnelly and E. Larraneta, Polymers, 2018, 10, 1379 CrossRef.
- H. Yi, G. Xiaowei, Z. Wei, Z. Donglai, Y. Liu, Q. Yunhua, C. Yongkuan, L. Shoubo, L. Tinghua, L. Qian, H. Liu and T. Yongfeng, Chinese Pat., CN113208156A, 2021.
- J. T.-W. Wang, A. C. Rodrigo, A. K. Patterson, K. Hawkins, M. M. S. Aly, J. Sun, K. T. Al Jamal and D. K. Smith, Adv. Sci., 2021, 8, 2101058 CrossRef CAS PubMed.
- H. K. Henisch, Crystal Growth in Gels, Dover Publications, New York, 1970 Search PubMed.
- J. A. Foster, M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke, J. A. K. Howard and J. W. Steed, Nat. Chem., 2010, 2, 1037–1043 CrossRef CAS.
- (a) F. Aparicio, E. Matesanz and L. Sánchez, Chem. Commun., 2012, 48, 5757–5759 RSC; (b) J. Buendia, E. Matesanz, D. K. Smith and L. Sánchez, CrystEngComm., 2015, 17, 8146–8152 RSC; (c) L. Kaufmann, S. R. Kennedy, C. D. Jones and J. W. Steed, Chem. Commun., 2016, 52, 10113–10116 RSC.
- (a) B. Saikia, M. T. Mulvee, I. Torres-Moya, B. Sarma and J. W. Steed, Cryst. Growth Des., 2020, 20, 7989–7996 CrossRef CAS; (b) S. S. Jayabhavan, J. W. Steed and K. K. Damodaran, Cryst. Growth Des., 2021, 21, 5383–5393 CrossRef CAS; (c) J. L. Andrews, S. R. Kennedy, D. S. Yufit, J. F. McCabe and J. W. Steed, Cryst. Growth Des., 2022, 22, 6775–6785 CrossRef CAS PubMed.
- (a) B. Escuder, F. Rodriguez-Llansola and J. F. Miravet, New J. Chem, 2010, 34, 1044–1054 RSC; (b) C. Rizzo, S. Marullo, F. Billeci and F. D’Anna, Eur. J. Org. Chem., 2021, 3148–3169 CrossRef CAS; (c) B. Escuder, Catalytic Supramolecular Gels, in: Supramolecular Catalysis: New Directions and Developments, ed. P. W. M. N. Van Leeuwen and M. Raynal, Wiley-VCH, 2022, pp. 81–92 Search PubMed; (d) N. Das and C. Maity, ACS Catal., 2023, 13, 5544–5570 CrossRef CAS.
- J. F. Miravet and B. Escuder, Chem. Commun., 2005, 5796–5798 RSC.
- Y.-R. Liu, L. He, J. Zhang, X. Wang and C.-Y. Su, Chem. Mater., 2009, 21, 557–563 CrossRef CAS.
- M. Maity and U. Maitra, J. Mater. Chem. A, 2014, 2, 18952–18958 RSC.
- (a) P. Slavik, D. W. Kurka and D. K. Smith, Chem. Sci., 2018, 9, 8673–8681 RSC; (b) P. Slavik and D. K. Smith, Tetrahedron, 2020, 76, 131344 CrossRef CAS.
- M. Albino, T. J. Burden, C. C. Piras, A. C. Whitwood, I. J. S. Fairlamb and D. K. Smith, ACS Sustainable Chem. Eng., 2023, 11, 1678–1689 CrossRef CAS.
- K. Tanaka, A. Mori and S. Inoue, J. Org. Chem., 1990, 55, 181–185 CrossRef CAS.
- M. O. Guler and S. I. Stupp, J. Am. Chem. Soc., 2007, 129, 12082–12083 CrossRef CAS PubMed.
- F. Rodriguez-Llansola, B. Escuder and J. F. Miravet, J. Am. Chem. Soc., 2009, 131, 11478–11484 CrossRef CAS.
- N. Singh, K. Zhang, C. A. Angulo-Pachon, E. Mendes, J. H. van Esch and B. Escuder, Chem. Sci., 2016, 7, 5568–5572 RSC.
- M. Tena-Solsona, J. Nanda, S. Díaz-Oltra, A. Chotera, G. Ashkenasy and B. Escuder, Chem. – Eur. J., 2016, 22, 6687–6694 CrossRef CAS PubMed.
- K. Hawkins, A. K. Patterson, P. A. Clarke and D. K. Smith, J. Am. Chem. Soc., 2020, 142, 4379–4389 CrossRef CAS.
- R. Martí-Centelles, J. Rubio-Magnieto and B. Escuder, Chem. Commun., 2020, 56, 14487–14490 RSC.
- Q. Wang, Z. Yang, L. Wang, M. Ma and B. Xu, Chem. Commun., 2007, 1032–1034 RSC.
- Q. Wang, Z. Yang, Y. Gao, W. Ge, L. Wang and B. Xu, Soft Matter, 2008, 4, 550–553 RSC.
- (a) Q. Wang, Z. Yang, X. Zhang, X. Xiao, C. K. Chang and B. Xu, Angew. Chem., Int. Ed., 2007, 46, 4285–4289 CrossRef CAS; (b) L. A. Solomon, J. B. Kronenberg and H. C. Fry, J. Am. Chem. Soc., 2017, 139, 8497–8507 CrossRef CAS PubMed.
- H. T. Imama, K. Hill, A. Reid, S. Mix, P. C. Marr and A. C. Marr, ACS Sustainable Chem. Eng., 2023, 11, 6829–6837 CrossRef.
- P. R. A. Chivers, J. A. Kelly, M. J. S. Hill and D. K. Smith, React Chem. Eng., 2020, 5, 1112–1117 RSC.
- S. C. Zacharias, M. Kamlar and H. Sunden, Ind. Eng. Chem. Res., 2021, 60, 10056–10063 CrossRef CAS.
- (a) P. Slavik, B. R. Trowse, P. O’Brien and D. K. Smith, Nat. Chem., 2023, 15, 319–325 CrossRef CAS PubMed; K. Smith, P. O’Brien and P. Slavik, WO2021214453, 2021.
- P. Visser and B. L. Feringa, Chem. Commun., 2023, 59, 5539–5542 RSC.
- (a) M. de Loos, J. van Esch, I. Stokroos, R. M. Kellogg and B. L. Feringa, J. Am. Chem. Soc., 1997, 119, 12675–12676 CrossRef CAS; (b) M. George and R. G. Weiss, Chem. Mater., 2003, 15, 2879–2888 CrossRef CAS; (c) M. Shirakawa, N. Fujita and N. Tamaoki, J. Am. Chem. Soc., 2005, 127, 4164–4165 CrossRef CAS PubMed.
- (a) D. D. Díaz, K. Rajagopal, E. Strable, J. Schneider and M. G. Finn, J. Am. Chem. Soc., 2006, 128, 6056–6057 CrossRef; (b) D. D. Díaz, J. J. Cid, P. Vázquez and T. Torres, Chem. – Eur. J., 2008, 14, 9261–9273 CrossRef.
- J. R. Moffat, I. A. Coates, F. J. Leng and D. K. Smith, Langmuir, 2009, 25, 8786–8793 CrossRef CAS PubMed.
- B. Maiti, A. Abramov, R. Pérez-Ruiz and D. D. Díaz, Acc. Chem. Res., 2019, 52, 1865–1876 CrossRef CAS PubMed.
- (a) S. Bhat and U. Maitra, Molecules, 2007, 12, 2181 CrossRef CAS; (b) A. Dawn, N. Fujita, S. Haraguchi, K. Sada and S. Shinkai, Chem. Commun., 2009, 2100–2102 RSC.
- S. Das, N. Okamura, S. Yagi and A. Ajayaghosh, J. Am. Chem. Soc., 2019, 141, 5635–5639 CrossRef CAS.
- (a) J. Bachl, A. Hohenleutner, B. B. Dhar, C. Cativiela, U. Maitra, B. König and D. D. Díaz, J. Mater. Chem. A, 2013, 1, 4577–4588 RSC; (b) M. Häring, R. P. Pérez-Ruiz, A. Jacobi von Wangelin and D. D. Díaz, Chem. Commun., 2015, 51, 16848–16851 RSC.
- M. Häring, A. Abramov, K. Okumura, I. Ghosh, B. König, N. Yanai, N. Kimizuka and D. D. Díaz, J. Org. Chem., 2018, 83, 7928–7938 CrossRef.
- (a) B. O. Okesola and D. K. Smith, Chem. Soc. Rev., 2016, 45, 4226–4251 RSC; (b) J. Y. C. Lim, S. S. Goh, S. S. Liow, K. Xue and X. J. Loh, J. Mater. Chem. A, 2019, 7, 18759–18791 RSC.
- S. Ray, A. K. Das and A. Banerjee, Chem. Mater., 2007, 19, 1633–1639 CrossRef CAS.
- (a) F. Rodriguez-Llansola, B. Escuder, J. F. Miravet, D. Hermida-Merino, I. W. Hamley, C. J. Cardin and W. Hayes, Chem. Commun., 2010, 46, 7960–7962 RSC; (b) D. M. Wood, B. W. Greenland, A. L. Acton, F. Rodriguez-Llansola, C. A. Murray, C. J. Cardin, J. F. Miravet, B. Escuder, I. W. Hamley and W. Hayes, Chem. – Eur. J., 2012, 18, 2692–2699 CrossRef CAS.
- J. Takeshita, Y. Hasegawa, K. Yanai, A. Yanamoto, A. Ishii, M. Hashegawa and M. Yamanaka, Chem. – Asian J., 2017, 12, 2029–2032 CrossRef CAS PubMed.
- T. N. Plank, L. P. Skala and J. T. Davis, Chem. Commun., 2017, 53, 6235–6238 RSC.
- J. Yang, H. Wang, Z. Song, D. Kong, X. Chen and Y. Zang, Colloids Surf., B, 2010, 80, 155–160 CrossRef PubMed.
- (a) C. Rizzo, S. Marullo, P. R. Campodonico, I. Pibiri, N. T. Dintcheva, R. Noto, D. Millan and F. D’Anna, ACS Sustainable Chem. Eng., 2018, 6, 12453–12462 CrossRef CAS; (b) S. Marullo, A. Meli, N. T. Dnitcheva, G. Infurna, C. Rizzo and F. D’Anna, ChemPlusChem, 2020, 85, 301–311 CrossRef CAS PubMed.
- (a) A. Y.-Y. Tam and V. W.-W. Tam, Chem. Soc. Rev., 2013, 42, 1540–1567 RSC; (b) Z. Liu, X. Zhao, Q. Chu and Y. Feng, Molecules, 2023, 28, 2274 CrossRef CAS PubMed.
- P. Sutar and T. K. Maji, Dalton Trans., 2020, 49, 7658–7672 RSC.
- K. N. King and A. J. McNeil, Chem. Commun., 2010, 46, 3511–3513 RSC.
- K. K. Carter, H. B. Rycenga and A. J. McNeil, Langmuir, 2014, 30, 3522–3527 CrossRef CAS PubMed.
- B. O. Okesola, S. K. Suravaram, A. Parkin and D. K. Smith, Angew. Chem., Int. Ed., 2016, 55, 183–187 CrossRef CAS PubMed.
- J. R. Dodson, H. L. Parker, A. M. Garcia, A. Hicken, K. Asemave, T. J. Farmer, H. He, J. H. Clark and A. J. Hunt, Green Chem., 2015, 17, 1951–1965 RSC.
- C. Rizzo, J. L. Andrews, J. W. Steed and F. D’Anna, J. Coll. Interface Sci., 2019, 548, 184–196 CrossRef CAS PubMed.
- S. Basak, N. Nandi, S. Paul, I. W. Hamley and A. Banerjee, Chem. Commun., 2017, 53, 5910–5913 RSC.
- B. K. Das, B. Pramanik, S. Chodhuri, O. A. Scherman and D. Das, Chem. Commun., 2020, 56, 3393–3396 RSC.
- S. Panja, B. Dietrich and D. J. Adams, Angew. Chem., Int. Ed., 2022, 61, e202115021 CrossRef CAS PubMed.
- B. Mondal, D. Bairagi, N. Nandi, B. Hansda, K. Sundar Das, C. J. C. Edwards-Gayle, V. Castelletto, I. W. Hamley and A. Banerjee, Langmuir, 2020, 36, 12942–12953 CrossRef CAS PubMed.
- C. W. Jaeger, D. R. Titterington, H. P. Le and J. J. Sopko, US Pat., 4889560, 1989 Search PubMed.
- M. P. Breton, D. C. Boils-Boissier, D. R. Titterington, J. W. Thomas Jr., J. H. Banning, C. E. Bedford and J. D. Wuest, US Pat., 6872243, 2005 Search PubMed.
- (a) R. Carlini, E. Toma, P. G. Odell and J. H. Banning, US Pat., 7153349, 2004 Search PubMed; (b) N. Chopra, M. N. Chretien, B. Keoshkerian, J. Eliyahu, D. W. Vanbiesen and A. Godedama, US Pat., 9328248, 2016 Search PubMed; (c) N. Chopra, M. N. Chretien and J. L. Belelie, US Pat., 10696857, 2020 Search PubMed.
- M. Champeau, D. A. Heinze, T. N. Viana, E. Rodrigues de Souza, A. C. Chinellato and S. Titotto, Adv. Funct. Mater., 2020, 30, 1910606 CrossRef CAS.
- Z. Yang, P.-L. Ho, G. Liang, K. H. Chow, Q. Wang, Y. Cao, Z. Guo and B. Xu, J. Am. Chem. Soc., 2007, 129, 266–267 CrossRef CAS PubMed.
- H. Shigemitsu and I. Hamachi, Acc. Chem. Res., 2017, 50, 740–750 CrossRef CAS PubMed.
- X. Liu, X. Sun and G. Liang, Biomater. Sci., 2021, 9, 315–327 RSC.
- L. Dong, J. Qian, Z. Hai, J. Xu, W. Du, K. Zhong and G. Liang, Anal. Chem., 2017, 89, 6922–6925 CrossRef CAS PubMed.
- L. L. Lock, Y. Li, X. Mao, V. Staedtke, R. Bai, W. Ma, R. Lin, Y. Li, G. Liu and H. Cui, ACS Nano, 2017, 11, 797–805 CrossRef CAS PubMed.
- G. K. Veits, K. K. Carter, S. K. Cox and A. J. McNeil, J. Am. Chem. Soc., 2016, 138, 12228–12233 CrossRef CAS PubMed.
- (a) J. R. Hiscock, M. R. Sambrook, J. A. Ede, N. J. Wells and P. A. Gale, J. Mater. Chem. A, 2015, 3, 1230–1234 RSC; (b) J. R. Hiscock, M. R. Sambrook, N. J. Wells and P. A. Gale, Chem. Sci., 2015, 6, 5680–5684 RSC.
- S. Panja, A. Panja and K. Ghosh, Mater. Chem. Front., 2021, 5, 584–602 RSC.
- (a) S. Mondal, P. Bairi, S. Das and A. K. Nandi, Chem. – Eur. J., 2018, 24, 5591–5600 CrossRef CAS PubMed; (b) B. Pramanik, N. Singha and D. Das, ACS Appl. Polym. Mater., 2019, 1, 833–843 CrossRef CAS.
- X. Cao, A. Gao, J.-T. Hou and T. Yi, Coord. Chem. Rev., 2021, 434, 213792 CrossRef CAS.
- (a) Q. Lin, T.-T. Lu, X. Zhu, B. Sun, Q.-P. Yang, T.-B. Wei and Y.-M. Zhang, Chem. Commun., 2015, 51, 1635–1638 RSC; (b) Q. Lin, T.-T. Lau, X. Zhu, T.-B. Wie, H. Li and Y.-M. Zhang, Chem. Sci., 2016, 7, 5341–5346 RSC; (c) H. Yao, Y.-B. Niu, Y.-P. Hu, X.-W. Sun, Q.-P. Zhang, Y.-M. Zhang, T.-B. Wei and Q. Lin, New J. Chem., 2022, 46, 17251–17259 RSC.
- D. Biswakarma, N. Dey and S. Bhattacharya, Chem. Sci., 2022, 13, 2286–2295 RSC.
- Q. Han, Q. Wang, A. Gao, X.-P. Chang, L. Zeng and X. Cao, ACS Sustainable Chem. Eng., 2023, 11, 2139–2150 CrossRef CAS.
- (a) C. Zhong, Y. Deng, W. Hu, J. Qiao, L. Zhang and J. Zhang, Chem. Soc. Rev., 2015, 44, 7484–7539 RSC; (b) J. Wu, Z. Lan, J. Lin, M. Huang, Y. Huang, L. Fan and G. Luo, Chem. Rev., 2015, 115, 2136–2173 CrossRef CAS PubMed; (c) A. Thamizhanban, K. Lalitha and S. Nagarajan, in Self-Assembled Soft Materials for Energy and Environmental Applications, in Emerging Nanostructured Materials for Energy and Environmental Science. Environmental Chemistry for a Sustainable World., ed. S. Rajendran, M. Naushad, K. Raju and R. Boukherroub, Spring, 2019, vol. 23 Search PubMed.
- K. Hanabusa, K. Hiratsuka, M. Kimura and H. Shirai, Chem. Mater., 1999, 11, 649–655 CrossRef CAS.
- N. Mohmeyer, P. Wang, H.-W. Schmidt, S. M. Zakeruddin and M. Grätzel, J. Mater. Chem., 2004, 14, 1905–1909 RSC.
- Z. Huo, S. Dai, C. Zhang, F. Kong, X. Fang, L. Guo, W. Liu, L. Hu, X. Pan and K. Wang, J. Phys. Chem. B, 2008, 112, 12927–12933 CrossRef CAS PubMed.
- L. Tao, Z. Huo, Y. Ding, L. Wang, J. Zhu, C. Zhang, X. Pan, M. K. Nazeruddin, S. Dai and M. Grätzel, J. Mater. Chem. A, 2014, 2, 15921–15930 RSC.
- L. Tao, Z. Huo, Y. Ding, Y. Li, S. Dai, L. Wang, J. Zhu, X. Pan, B. Zhang, J. Yao, M. K. Nazeeruddin and M. Grätzel, J. Mater. Chem. A, 2015, 3, 2344–2352 RSC.
- L. Tao, W. Zhang, Z. Wang, H. Wang, J. Zhang, Z. Huo, S. Dai, T. Hayat and N. S. Alharbi, Org. Electron., 2019, 65, 179–184 CrossRef CAS.
- D. Yu, X. Li and K. Xu, Sci. China Mater., 2019, 62, 1556–1573 CrossRef CAS.
- V. R. Basrur, J. Guo, C. Wang and S. R. Raghavan, ACS Appl. Mater. Interfaces, 2013, 5, 262–267 CrossRef CAS PubMed.
- L. Li, Q. Zhang, H. Huo, J. Zhou and L. Li, RSC Adv., 2016, 6, 88820–88825 RSC.
- S. Sun, J. Song, Z. Shan and R. Feng, J. Electroanal. Chem., 2012, 676, 1–5 CrossRef CAS.
- W. Kobo, S. Kambe, S. Nakade, T. Kitamura, K. Hanabusa, Y. Wada and S. Yanagida, J. Phys. Chem. B, 2003, 107, 4374–4381 CrossRef.
- N. Mohmeyer, D. Kuang, P. Wang, H.-W. Schmidt, S. M. Zakeeruddin and M. Grätzel, J. Mater. Chem., 2006, 16, 2978–2983 RSC.
- J.-D. Decoppet, T. Moehi, S. S. Babkair, R. A. Alzubaydi, A. A. Ansari, S. S. Habib, S. M. Zakeeruddin, H.-W. Schmidt and M. Grätzel, J. Mater. Chem. A, 2014, 2, 15972–15977 RSC.
- (a) S. Bhattacharya and S. K. Samanta, Chem. Rev., 2016, 116, 11967–12028 CrossRef CAS PubMed; (b) P. Choudhury, S. Dinda and P. K. Das, Soft Matter, 2020, 16, 27–53 RSC.
- M. Lakdusinghe, M. Abbaszadeh, S. Mishra, D. Sengottuvelu, R. Wijayapala, S. Zhang, A. R. Benasco, X. Gu, S. E. Morgan, D. O. Wipf and S. Kundu, ACS Appl. Nano Mater., 2021, 4, 8003–8014 CrossRef CAS.
- P. Chakraborty, T. Guterman, N. Adadi, M. Yadid, T. Brosh, L. Adler-Abaramovich, T. Dvir and E. Gazit, ACS Nano, 2019, 13, 163–175 CrossRef CAS PubMed.
- M. Criado-Gonzalez, N. Alegret, A. M. Fracaroli, D. Mantione, G. Guzman-González, R. Del Olmo, K. Tashiro, L. C. Tomé, M. L. Picchio and D. Mecerreyes, Angew. Chem., Int. Ed., 2023, 62, e202301489 CrossRef CAS PubMed.
- (a) S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed; (b) O. Dumele, J. Chen, J. V. Passarelli and S. I. Stupp, Adv. Mater., 2020, 32, 1907247 CrossRef CAS PubMed.
- B. W. Messmore, J. F. Hulvat, E. D. Sone and S. I. Stupp, J. Am. Chem. Soc., 2004, 126, 14452–14458 CrossRef CAS PubMed.
- S. Prasanthkumar, A. Saeki, S. Seki and A. Ajayaghosh, J. Am. Chem. Soc., 2010, 132, 8866–8867 CrossRef CAS PubMed.
- K. Sakakibara, P. Chithra, B. Das, T. mori, M. Akada, J. Labuta, T. Tsuruoka, S. Maji, S. Furumi, L. K. Shrestha, J. P. Hill, S. Acharya, K. Ariga and A. Ajayaghosh, J. Am. Chem. Soc., 2014, 136, 8548–8551 CrossRef CAS PubMed.
- J. Puigmartí-Luis, V. Laukhin, A. P. del Pino, J. Vidal-Gancedo, C. Rovira, E. Laukhina and D. B. Amabilino, Angew. Chem., Int. Ed., 2007, 46, 238–241 CrossRef PubMed.
- N. L. Ing, R. K. Spencer, S. H. Luong, H. D. Nguyen and A. I. Hochbaum, ACS Nano, 2018, 12, 2652–2661 CrossRef CAS PubMed.
- N. L. Ing, M. Y. El-Naggar and A. I. Hochbaum, J. Phys. Chem. B, 2018, 122, 10403–10423 CrossRef CAS PubMed.
- A. del Guerzo, A. G. L. Olive, J. Reichwagen, H. Hopf and J.-P. Desvergne, J. Am. Chem. Soc., 2005, 127, 17984–17985 CrossRef CAS PubMed.
- P. Heremans, D. Cheyns and B. P. Rand, Acc. Chem. Res., 2009, 42, 1740–1747 CrossRef CAS PubMed.
- K. Sugiyasu, S.-i Kawano, N. Fujita and S. Shinkai, Chem. Mater., 2008, 9, 2863–2865 CrossRef.
- S. Prasanthkumar, S. Ghosh, V. C. Nair, A. Saeki, S. Seki and A. Ajayaghosh, Angew. Chem., Int. Ed., 2015, 54, 946–950 CrossRef CAS PubMed.
- E. R. Draper, J. R. Lee, M. Wallace, F. Jackel, A. J. Cowan and D. J. Adams, Chem. Sci., 2016, 7, 6499–6505 RSC.
- P. Schäfer, C. de Vet, L. Gartzia-Rivero, G. Raffy, M.-T. Kao, C. Schäfer, L. J. Romasanta, B. Pavageau, Y.-T. Tsai, L. Hirsch, D. M. Bassani and A. del Guerzo, Nanoscale, 2022, 14, 8951–8958 RSC.
- L. Chen, S. Revel, K. Morris and D. J. Adams, Chem. Commun., 2010, 46, 4267–4269 RSC.
- H. A. M. Ardona, E. R. Draper, F. Citossi, M. Wallace, L. C. Serpell, D. J. Adams and J. D. Tovar, J. Am. Chem. Soc., 2017, 139, 8685–8692 CrossRef CAS PubMed.
- I. D. Tevis, W.-W. Tsai, L. C. Palmer, T. Aytun and S. I. Stupp, ACS Nano, 2012, 6, 2032–2040 CrossRef CAS PubMed.
- P. Xue, P. Wang, B. Yao, J. Sun, P. Gong, Z. Zhang and R. Lu, RSC Adv., 2015, 5, 75425–75433 RSC.
- S. K. M. Malluri and R. V. Ulijn, Chem. Sci., 2013, 4, 3699–3705 RSC.
- S. K. M. Nalluri, C. Berdugo, N. Javid, P. W. J. M. Frederix and R. V. Ulijn, Angew. Chem., Int. Ed., 2014, 53, 5882–5887 CrossRef CAS PubMed.
- M. Kumar, N. L. Ing, V. Narang, N. K. Wijerathne, A. I. Hochbaum and R. V. Ulijn, Nat. Chem., 2018, 10, 696–703 CrossRef CAS PubMed.
- J. K. Mouw, G. Ou and V. M. Weaver, Nat. Rev. Mol. Cell Biol., 2014, 15, 771–785 CrossRef CAS PubMed.
- (a) J. Hoque, N. Sangaj and S. Varghese, Macromol. Biosci., 2019, 19, e1800259 CrossRef PubMed; (b) L. Saunders and P. X. Ma, Macromol. Biosci., 2019, 19, e1800313 CrossRef PubMed; (c) J. Omar, D. Ponsford, C. A. Dreiss, T.-C. Lee and X. J. Loh, Chem. – Asian J., 2022, 17, e202200081 CrossRef CAS PubMed.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- (a) B. V. Slaughter, S. S. Khurshid, O. Z. Fisher, A. Khademhosseini and N. A. Peppas, Adv. Mater., 2009, 21, 3307–3329 CrossRef CAS PubMed; (b) S. Mantha, S. Pillai, P. Khayambashi, A. Upadhyay, Y. Zhang, O. Tao, H. M. Pham and S. D. Tran, Materials, 2019, 12, 3323 CrossRef CAS PubMed.
- R. C. Gupta, R. Lall, A. Srivastava and A. Sinha, Front. Vet. Sci., 2019, 6, 192 CrossRef PubMed.
- (a) W. Zakrzewski, M. Dobrzynski, M. Szymonowicz and Z. Rybak, Stem Cell Res. Ther., 2019, 10, 68 CrossRef CAS PubMed; (b) E. H. Ntege, H. Sunami and Y. Shimizu, Regen. Ther., 2020, 14, 136–153 CrossRef PubMed.
- (a) R. G. Ellis-Behnke, Y.-X. Liang, S.-W. You, D. K. C. Tay, S. Zhang, K.-F. So and G. E. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5054–5059 CrossRef CAS PubMed; (b) R. G. Ellis-Behnke, G. Schneider and S. Zhang, US Pat., 7846891, 2010 Search PubMed.
- (a) G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352–1355 CrossRef CAS PubMed; (b) J. D. Hartgerink, K. L. Niece and S. I. Stupp, 2004; (c) V. M. Tysseling-Mattiace, V. Sahni, K. L. Niece, D. Birch, C. Czeisler, M. G. Fehlings, S. I. Stupp and J. A. Kessler, J. Neurosci., 2008, 28, 3814–3823 CrossRef CAS PubMed.
- E. J. Berns, S. Sur, L. L. Pan, J. E. Goldberger, S. Suresh, S. M. Zhang, J. A. Kessler and S. I. Stupp, Biomaterials, 2014, 35, 185–195 CrossRef CAS PubMed.
- A. J. Matsuoka, Z. A. Sayed, N. Stephanopoulos, E. J. Berns, A. R. Wadhwani, Z. D. Morrissey, D. M. Chadly, S. Kobayashi, A. N. Edelbrock, T. Mashimo, C. A. Miller, T. L. McGuire, S. I. Stupp and J. A. Kessler, PLoS One, 2017, 12, e0190150 CrossRef PubMed.
- V. A. Kumar, N. L. Taylor, S. Shi, B. K. Wang, A. A. Jalan, M. K. Kang, N. C. Wickremasinghe and J. D. Hartgerink, ACS Nano, 2015, 9, 860–868 CrossRef CAS PubMed.
- V. A. Kumar, Q. Liu, N. C. Wickremasinghe, S. Shi, T. T. Cornwright, Y. Deng, A. Azares, A. N. Moore, A. M. Acevedo-Jake, N. R. Agudo, S. Pan, D. G. Woodside, P. Vanderslice, J. T. Willerson, R. A. Dixon and J. D. Hartgerink, Biomaterials, 2016, 98, 113–119 CrossRef CAS PubMed.
- (a) J. S. Rudra, Y. F. Tian, J. P. Jung and J. H. Collier, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 622–627 CrossRef CAS PubMed; (b) J. S. Rudra, T. Sun, K. C. Bird, M. D. Daniels, J. Z. Gasiorowski, A. S. Chong and J. H. Collier, ACS Nano, 2012, 6, 1557–1564 CrossRef CAS PubMed.
- S. Kyle, S. H. Felton, M. J. McPherson, A. Aggeli and E. Ingham, Adv. Healthcare Mater., 2012, 1, 640–645 CrossRef CAS PubMed.
- E. Froimchuk, S. T. Carey, C. Edwards and C. M. Jewell, Acc. Chem. Res., 2020, 53, 2534–2545 CrossRef CAS PubMed.
- T. Liebmann, S. Rydholm, V. Akpe and H. Brismar, BMC Biotechnol., 2007, 7, 88 CrossRef PubMed.
- M. Zhou, A. M. Smith, A. K. Das, N. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30, 2523–2530 CrossRef CAS PubMed.
- J. P. Jung, A. K. Nagaraj, E. K. Fox, J. S. Rudra, J. M. Devgun and J. H. Collier, Biomaterials, 2009, 30, 2400–2410 CrossRef CAS PubMed.
- A. D. Martin, J. P. Wojciechowski, E. Y. Du, A. Rawal, H. Stefen, C. G. Au, L. Hou, C. G. Cranfield, T. Fath, L. M. Ittner and P. Thordarson, Chem. Sci., 2020, 11, 1375–1382 RSC.
- L. Latxague, M. A. Ramin, A. Appavoo, P. Berto, M. Maisani, C. Ehret, O. Chassande and P. Barthélémy, Angew. Chem., Int. Ed., 2015, 54, 4517–4521 CrossRef CAS PubMed.
- J. D. Tang, C. Mura and K. J. Lampe, J. Am. Chem. Soc., 2019, 141, 4886–4899 CrossRef CAS PubMed.
- (a) A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed; (b) N. Huebsch, P. R. Arany, A. S. Mao, D. Shvartsman, O. A. Ali, S. A. Bencherif, J. Rivera-Feliciano and D. J. Mooney, Nat. Mater., 2010, 9, 515–526 CrossRef PubMed; (c) Y. S. Pek, A. C. A. Wan and J. Y. Ying, Biomaterials, 2010, 31, 385–391 CrossRef CAS PubMed.
- (a) Y. Hu, W. Gao, F. Wu, H. Wu, B. He and J. He, J. Mater. Chem. B, 2016, 4, 3504–3508 RSC; (b) J. He, Y. Hu, F. Wu, B. He and W. Gao, J. Bionic Eng., 2018, 15, 682–692 CrossRef.
- J. C. Ashworth, J. L. Thompson, J. R. James, C. E. Slater, S. Pijuan-Galitó, K. Lis-Slimak, R. J. Holley, K. A. Meade, A. Thompson, K. P. Arkill, M. Tassieri, A. J. Wright, G. Farnie and C. L. R. Merry, Matrix Biol., 2020, 85–86, 15–33 CrossRef CAS PubMed.
- J. M. Godbe, R. Freeman, L. F. Burbulla, J. Lewis, D. Krainc and S. I. Stupp, ACS Biomater. Sci. Eng., 2020, 6, 1196–1207 CrossRef CAS PubMed.
- E. V. Alakpa, V. Jayawarna, A. Lampel, K. V. Burgess, C. C. West, S. C. J. Bakker, S. Roy, N. Javid, S. Fleming, D. A. Lamprou, J. Yang, A. Miller, A. J. Urquhart, P. W. J. M. Frederix, N. T. Hunt, B. Péault, R. V. Ulijn and M. J. Dalby, Chem, 2016, 1, 298–319 CAS.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- B. O. Okesola, Y. Wu, B. Derkus, S. Gani, D. Wu, D. Knani, D. K. Smith, D. J. Adams and A. Mata, Chem. Mater., 2019, 31, 7883–7897 CrossRef CAS PubMed.
- B. Cheng, Y. Yan, J. Qi, L. Deng, Z.-W. Shao, K.-Q. Zhang, B. Li, Z. Sun and X. Li, ACS Appl. Mater. Interfaces, 2018, 10, 12474–12484 CrossRef CAS PubMed.
- S. Kühn, J. Sievers, A. Stoppa, N. Träber, R. Zimmermann, P. B. Welzel and C. Werner, Adv. Funct. Mater., 2020, 30, 1908857 CrossRef.
- H. Komatsu, S. Tsukiji, M. Ikeda and I. Hamachi, Chem. – Asian J., 2011, 6, 2368–2375 CrossRef CAS PubMed.
- C. C. Piras, C. S. Mahon, P. G. Genever and D. K. Smith, ACS Biomater. Sci. Eng., 2022, 8, 1829–1840 CrossRef PubMed.
- N. Zhang, Y. Wang, J. Guo and J. He, Acta Biomater., 2021, 135, 304–317 CrossRef CAS PubMed.
- P. R. A. Chivers and D. K. Smith, Chem. Sci., 2017, 8, 7218–7227 RSC.
- (a) S. Zhang, T. Holmes, C. Lockshin and A. Rich, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 3334–3338 CrossRef CAS PubMed; (b) PuraMatrixTM Self-Assembling: https://www.3d-matrix.co.jp/en/technology/tec2.html, accessed Nov 2023.
- K. M. Hainline, F. Gu, J. F. Handley, Y. F. Tian, Y. Wu, L. de Wet, D. J. Vander Griend and J. H. Collier, Macromol. Biosci., 2019, 19, 1800249 CrossRef PubMed.
- S. Jones, J. C. Ashworth, M. Meakin, P. Collier, C. Probert, A. A. Ritchie, C. L. R. Merry and A. M. Grabowska, In Vitro Models, 2023, 2, 99–111 CrossRef PubMed.
- T. Guan, J. Li, C. Chen and Y. Liu, Adv. Sci., 2022, 9, 2104165 CrossRef CAS PubMed.
- R. G. Ellis-Behnke, Y.-X. Liang, D. K. C. Tray, P. W. F. Kau, G. E. Schneider, S. Zhang, W. Wu and K.-F. So, Nanomedicine: Nanotech. Biol. Med., 2006, 2, 207–215 CrossRef CAS PubMed.
- Z. Zhai, K. Xu, L. Mei, C. Wu, J. Liu, Z. Liu, L. Wan and W. Zhong, Soft Matter, 2019, 15, 8603–8610 RSC.
- Z. Wang, Y. Zhang, Y. Yin, J. Liu, P. Li, Y. Zhao, D. Bai, H. Zhao, X. Han and Q. Chen, Adv. Mater., 2022, 34, 2108300 CrossRef CAS PubMed.
- Y. Loo, M. Goktas, A. B. Tekinay, M. O. Guler, C. A. E. Hauser and A. Mitraki, Adv. Healthcare Mater., 2015, 4, 2557–2586 CrossRef CAS PubMed.
- F. Yergoz, N. Hastar, C. E. Cimenci, A. D. Ozkan, T. Tekinay, M. O. Guler and A. B. Tekinay, Biomaterials, 2017, 134, 117–127 CrossRef CAS PubMed.
- Y. Hu, H. Shi, X. Ma, T. Xia, Y. Wu, L. Chen, Z. Ren, L. Lei, J. Jiang, J. Wang and X. Li, Acta Biomaterialia, 2023, 159, 128–139 CrossRef CAS PubMed.
- Z. Yang, G. Liang, Z. Guo, Z. Guo and B. Xu, Angew. Chem., Int. Ed., 2007, 46, 8216–8219 CrossRef CAS PubMed.
- Y. Kuang, J. Shi, J. Li, D. Yuan, K. A. Alberti, Q. Xu and B. Xu, Angew. Chem., Int. Ed., 2014, 53, 8104–8107 CrossRef CAS PubMed.
- (a) A. Tanaka, Y. Fukuoka, Y. Morimoto, T. Honjo, D. Koda, M. Goto and T. Maruyama, J. Am. Chem. Soc., 2015, 137, 770–775 CrossRef CAS PubMed; (b) T. Maruyama and W. K. Restu, Polym. J., 2020, 52, 883–889 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |