
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigating the effect of the micelle structures of block and random copolymers on dye solubilization†
Masahiko
Asada
*ab,
Airi
Wakai
b,
Hisakazu
Tanaka
a,
Yukie
Suwa
c,
Yuuji
Tamura
a,
Mariko
Kouyama
a,
Shigehito
Osawa‡
 d and
Hidenori
Otsuka
*bd
d and
Hidenori
Otsuka
*bd
aCentral Research Laboratories, DIC Corporation, 631, Sakado, Sakura, Chiba 285-8668, Japan. E-mail: masahiko-asada@ma.dic.co.jp
bDepartment of Chemistry, Graduate School of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku, Tokyo 162-8601, Japan. E-mail: h.otsuka@rs.tus.ac.jp
cSakai Plant, DIC Corporation, 3, Takasago 1-chome, Takaishi, Osaka 592-0001, Japan
dDepartment of Applied Chemistry, Faculty of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku, Tokyo 162-8601, Japan
First published on 2nd May 2024
Abstract
To elucidate the correlation between dye solubilization into micelles and their core–shell aggregated structure, the structures of block and random copolymer micelles were characterized. The block copolymer micelles exhibited a higher dye solubilization capacity which correlated with their core volume, clear core–shell contrast and slow solubilization rate.
Owing to their unique and excellent associating behavior with well-defined hydrophobic/hydrophilic molecular architectures, block copolymer micelles have been thoroughly investigated as functional vehicles capable of effectively loading hydrophobic species such as drugs and dyes.1–5 Consequently, studies have focused on understanding the self-assembly of block copolymers.6–11 However, their synthesis can be tedious and time-consuming, as it involves the sequential addition of monomers via controlled polymerization techniques in addition to post-polymerization treatments such as grafting, substitution, hydrolysis and “click” chemistries.12,13 On the other hand, random copolymers can be easily synthesized via the one-step copolymerization of two (or more) different monomers, and their structures can be adjusted by modifying the types of comonomers and their properties.14
Polymeric micelles, which are aggregation colloids formed in solution by the self-assembly of amphiphilic polymers, have been employed in dye solubilization and their micellar structures are closely related to the pigment dispersion for water-based color inks. Although block copolymers are often used in the pharmaceutical industry, random copolymers with acrylic monomers are generally used in the ink industry owing to their low manufacturing cost and suitability for large-scale production. However, there exists a tradeoff between utilizing random copolymers as dispersants for ink production and their inadequate dispersion performance. This study thoroughly investigated the phase-separated structures of block copolymer micelles and compared them with those of random copolymers to determine the micelle structure required for dye solubilization.
The amphiphilic block copolymers (BL01, BL02, BL03, BL04, and BL05) were synthesized in a microreactor15 from styrene (St), n-butyl methacrylate (BMA), and methacrylic acid (MA) (Fig. S1, ESI† and Table 1). BL01, BL03, BL04, and BL05 were prepared using St as the hydrophobic monomer and MA as the hydrophilic monomer. On the other hand, BL02 was prepared using St as the hydrophobic monomer, while the hydrophilic region consisted of random chains of the hydrophilic MA and hydrophobic BMA. BL01, BL02, BL03, and BL05 were changed in their molecular weights with almost the same acid values to observe the effect of molecular weight on pigment dispersion. BL04 was changed in the acid values different from other BL01/02/03/05 to observe the effect of hydrophilic–hydrophobic balance on pigment dispersion. BL02 randomly formulated BMA into MA in the hydrophilic block to estimate the effect of the charge density of the tail. We further compared the application of polymer dispersant hard to be soluble in water between block and random copolymers in which hydrophobic side chains based on styrene were introduced into polyacrylic acid. As reference random copolymers, RD01, RD02, and RD03 composed of St and either MA or acrylic acid (AA) were purchased. The composition was estimated from the quantitative 13C NMR and 1H NMR measurement results, and molecular weights were measured by GPC using polystyrene standard for calibration. The acid values were also listed. Furthermore, RD04 was synthesized as a high-acid-value polymer of St and MA (Fig. S1 and S2, ESI†).
| Polymers | Monomers | Mole ratio | M w | Acid value |
|---|---|---|---|---|
| a Weight average molecular weight determined by GPC. Polystyrene standards were used for calibration. b In the hydrophilic part, MA was randomly mixed with BMA. c Commercially available styrene acrylic random copolymer dispersant. The compositions were determined by quantitative 13C NMR and 1H NMR. | ||||
| BL01 | St, DPE, MA | 13/1/8 | 3107 | 129 |
| BL02b | St, DPE, BMA, MA | 3.9/1/9.1/8 | 4077 | 146 |
| BL03 | St, αMeSt, MA | 6.8/2/5 | 1737 | 143 |
| BL04 | St, αMeSt, MA | 8/2/13 | 3165 | 271 |
| BL05 | St, αMeSt, MA | 24/2/14 | 4752 | 152 |
| RD01c | St, MMA, BA, MA | 10/12.4/6.2/1.5 | 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 407 407 |
110 |
| RD02c | St, αMeSt, MA | 10/trace/5.2 | 7051 | 153 |
| RD03c | St, BA, AA | 10/6.7/16.3 | 3348 | 213 |
| RD04 | St, MA | 10/10.3 | 1571 | 301 |
Small angle X ray scattering (SAXS) was performed to obtain information on the micelle structure as well as the core and the shell size. The five block copolymers and four random copolymers were prepared as aqueous solutions (0.5 wt%). The SAXS curves of the samples (Fig. 1) revealed that all block copolymers exhibited either 1 or 2 maxima at q values between 0.1 and 1 nm−1, and the intensity tended to significantly decrease with an increase in the q value. It was thus concluded that these copolymers exhibited a typical spherical scattering pattern with a clear boundary at the interface. However, the random copolymers did not exhibit a clear maximum, and the decrease in the slope for q values above 1 was smaller than that of block copolymers. These polymers exhibited a typical random-coil scattering pattern with a continuous interface. The obtained scattering curves were subjected to curve fitting using the optimal theoretical equations. For the block copolymers, the poly-core model was the most suitable (Fig. S3a–c, ESI†). Since the random copolymers exhibited monotonic curves, any fitting model could be matched. RD01 was fitted with the polydisperse Gaussian coil model (Fig. S3d, ESI†), representing a random coil since the contrast difference was low. The shell thickness was approximately zero in the poly-core model, thus indicating a random-coil structure (Fig. S3e, ESI†). The same pattern was observed for the other random copolymers. The fitting results thus indicated that the block copolymers had a core–shell structure,16–20 whereas the random copolymers had a random coil structure. However, the radius and polydispersity values from each curve fitting (Table 2) indicated that the random copolymers exhibited high polydispersities with ambiguous and undetectable core–shell structure boundaries.
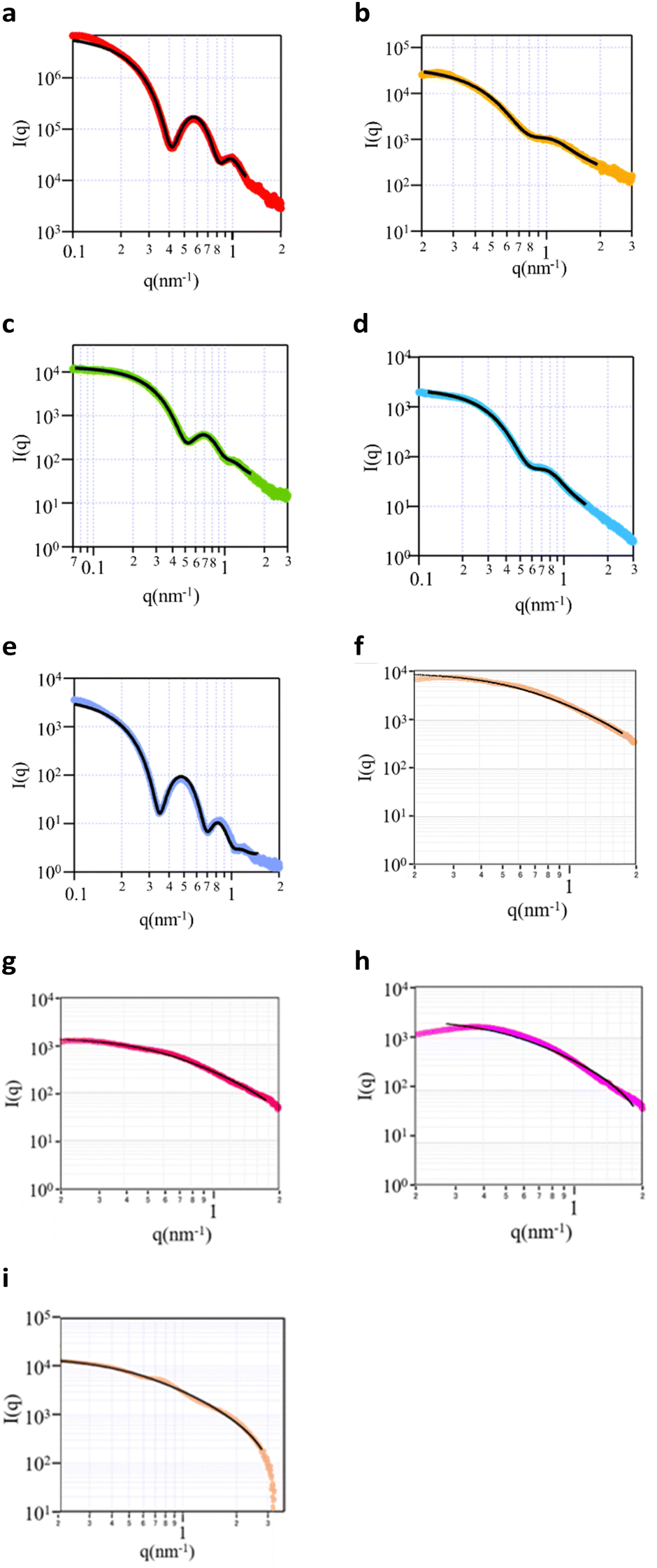 | ||
| Fig. 1 SAXS curves of (a) BL01, (b) BL02, (c) BL03, (d) BL04, (e) BL05, (f) RD01, (g) RD02, (h) RD03, and (i) RD04. Curve fitting was performed by the (a)–(e) poly-core model and (f)–(i) polydisperse Gaussian coil model. | ||
| Polymers | D H/nma | M agg | N agg | R g/nmb | Zeta potential/mV | ||
|---|---|---|---|---|---|---|---|
| Core/nme | Shell/nmf | PDI | |||||
| a Hydrodynamic diameters (DH) determined by DLS with cumulant analyses. b Radius of gyration (Rg) determined by SAXS. c Aggregate molecular weight from SLS. d Aggregation number. e Radius of core. f Thickness of the shell. g Considering the DLS value, the main particle size of RD04 is outside the measurement range of SAXS (q < 0.1), and this value may reflect small particles mixed in. h Considering the DLS value of DH, Magg of RD04 is outside the measurement of Debye plot, and thus both Magg and Nagg is not obtained. | |||||||
| BL01 | 15.7 | 103 K | 33.3 | 5.2 | 4 | 0.1 | −48.1 |
| BL02 | 14.1 | 50 K | 12.1 | 2.2 | 2.1 | 0.4 | −88.1 |
| BL03 | 26 | 116 K | 66.5 | 4.5 | 2.5 | 0.2 | −56.8 |
| BL04 | 20 | 140 K | 44.2 | 3.2 | 2.9 | 0.3 | −69.5 |
| BL05 | 21.6 | 291 K | 61.3 | 6.3 | 4.9 | 0.1 | −43.3 |
| RD01 | 6.9 | 23 K | 1.8 | 2.7 | 1.0 | −53.1 | |
| RD02 | 14.6 | 238 K | 33.8 | 3.0 | 0.97 | −55.8 | |
| RD03 | 5.9 | 10 K | 3.1 | 1.8 | 0.95 | −46.7 | |
| RD04 | 152 | NDh | NDh | 5.2g | 0.98 | ND | |
The critical micelle concentration (CMC) was determined by a fluorescent probe of pyrene.21–23 The fluorescence spectrum of pyrene monomer possesses a vibrational band structure which, due to the Ham effect, exhibits a strong sensitivity to the polarity of the microenvironment in the micelle.24 Consequently, the intensity ratio of the (0, 0) band (I1, 375 nm) to the (0, 2) band (I3, 385 nm), with a local polarity and which is highly sensitive to the polarity of the medium surrounding the pyrene molecules, was used to determine the CMC.25 The fluorescence intensity ratios range from 1.9 for water and 1.04 for toluene to 0.6 for aliphatic hydrocarbons. Plots of the I1/I3 peak height ratio from the emission spectra as a function of the copolymer concentration are shown in Fig. 2. Below the CMC, micelles were not formed and the pyrene fluorescence spectrum corresponded to that in water, with an I1/I3 ratio of approximately 1.6–1.8. As the copolymer concentration increased above the CMC (0.0001 wt%), pyrene was progressively solubilized in the hydrophobic interior, as illustrated by the drastic decrease in the I1/I3 ratio in the intermediate region of the copolymer concentration. For copolymer concentrations exceeding 0.01 wt%, the block and random copolymers exhibited constant ratios of values of 1.0 and 1.25, respectively. These values indicated that the locus of pyrene in the block micelle was a polystyrene environment more compact than that in the random micelles in which the pyrenes experienced some polarity associated with the water interface. This may be attributed to the core–shell structure of the block copolymers unlike the random copolymers which did not have a clear core. All block copolymers exhibited almost the same degree of hydrophobicity of the micelle core. Notably, the concentration at which the I1/I3 ratio of BL04 started to decrease was higher than that of the other block copolymers, which can be attributed to its high acid value. RD04, which had a higher acid value than BL04, showed a further upper shift in the CMC. Block copolymers have a core–shell structure and higher hydrophobicity in the micelle core, thus indicating their higher ability to solubilize hydrophobic compounds compared to random copolymers.
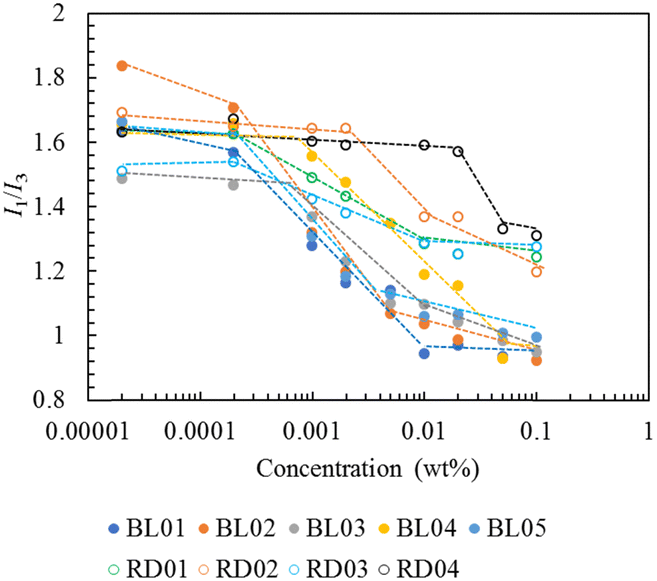 | ||
| Fig. 2 I 1/I3 ratios of the polymer solutions. | ||
The λmax of the oil orange SS in the micelles was determined to be 494 nm.26,27 The results revealed that the amount of dye solubilized in the block copolymer micelles was higher than that in the random copolymers (Fig. 3a and b/Fig. S5a and b, ESI†). This result is consistent with the differences in the polar environments probed by pyrene between the block and random copolymers. Furthermore, the random copolymer micelle was nearly saturated within 10 h, while the amount of dye solubilized in the BL01, BL03, and BL05 continued to slowly increase over two days. To compare the solubilization kinetics, the rate constants were calculated using the Noyes-Whitney equation28 (Fig. S5c, Table S1, ESI†). This result is closely related to the micelle structure. BL01, BL03 and BL05, which showed many fringes in the SAXS chart, had the smaller solubilization rate constants. This means clear core–shell contrast prevented the dye from penetrating into the micelle core of the block copolymers. On the other hand, the poor core–shell properties of BL02 and BL04 (Fig. 1, Table 2) enabled the dye to penetrate relatively easily. Similarly, the dye more readily penetrated the micelles of the random copolymers exhibiting random coil structure with ambiguous core–shell contrast (Fig. S5c, Table S1, ESI†). The block copolymers showed a large difference in the amount of dye solubilized although the hydrophobic environment was not significantly different (Fig. 2). BL03 showed the highest solubilization ability, followed by BL01 and BL05. Accordingly, we focused on the differences in the block copolymers rather than those in the hydrophobic environments.
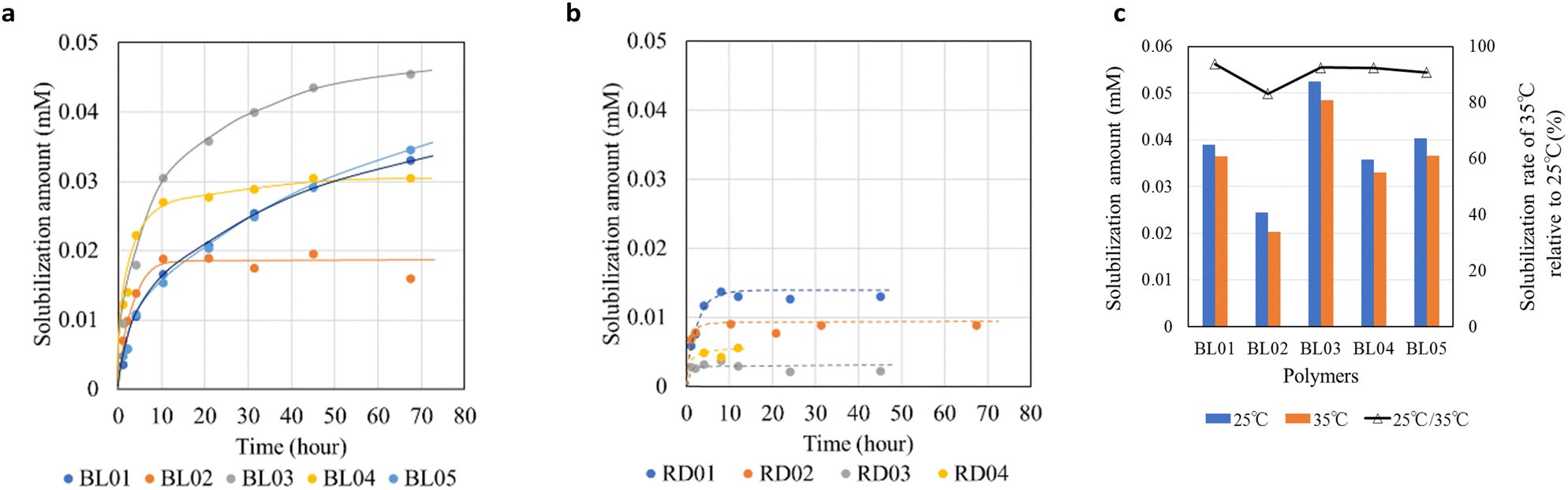 | ||
| Fig. 3 Variation of the solubilized amount of Oil Orange SS as a function of time for the polymer micelles of (a) Block copolymers and (b) Random copolymers. (c) Solubilization amount of dye in polymer micelles under temperature change (25 and 35 °C, left-axis) and solubilization rate (%, right-axis). | ||
The aggregate molecular weights (Magg) of the block copolymers in the micelles were determined by static light scattering (SLS, Fig. S6, ESI† and Table 2). To calculate the aggregation number (Nagg), we used the Mw values obtained by GPC (Table 1).29,30 The size of the block copolymer micelles determined by DLS and SAXS generally correlated with the Magg as well as Nagg. DLS diameter have been reported as hydrodynamic size based on the diffusion of the particles, mostly above the diameter of that measured with SAXS which probes the electron rich part of the particle. In fact, Rg determined by SAXS (Table 2) was smaller than RH determined by DLS, while TEM diameters were measured using freeze-fracture transmission electron microscopy (FF-TEM) (Fig. S4, ESI†) and were in close agreement with RH (DLS). Note that FF-TEM is known to facilitate the study of structure in such hydration swelling micelles. The principles behind FF-TEM could be easily extended to study the morphological details of various structures in complex liquids. Vitrified specimens were fractured at low temperature and under high vacuum to produce surfaces which were replicated and examined. BL02 had a smaller micelle size and lower Magg and Nagg values compared to BL05, which had a larger micelle size. The extent of dye solubilization was thus directly linked to Nagg and the core radius, as demonstrated in BL01, 03 and 05, where a larger core radius was positively correlated with an increase in the amount of dye solubilization, even though the polarity of the core estimated by pyrene was almost the same as those of BL02 and BL04 with lower solubilization capacities. This trend was also observed in the random copolymer systems, where small particle sizes without a clear core–shell phase separation were observed by SAXS (1.8–3.0 nm radius, Table 2), which can attribute to the low amount of dye solubilized. The micelle concentration of the block copolymers can be divided by Nagg to determine the number of micelles present, thus allowing an estimation of the number of dye molecules solubilized within each micelle. The calculations indicated that the larger the core volume and number of aggregates (Nagg) resulted in a higher number of dye molecules solubilized per micelle (Table S2, ESI†). Approximately 0.2–2 dye molecules were solubilized per micelle. Micelle size was measured by DLS before and after dye solubilization. A significant increase in the size was observed after dye solubilization in BL01, BL02, and BL04 (Table S3, ESI†). Especially in BL01 and BL02 with a small amount of dye solubilization, it might be due to small particle size, low Nagg number with weak molecular interaction, leading to size increase after dye solubilization. In light of the monomer composition, BL02 has a relatively low St ratio compared to the other block copolymers, which is probably the additional reason for the reduced solubilization. In contrast, BL03 and BL05, which have larger micelle particle diameters with higher number of Nagg, probably have a slower solubilization rate but greater solubilization potential (more solubilized molecules per micelle) due to better hydrophobic aggregation and molecular packing. As a result, the change in micelle particle size after solubilization would be small. On the other hand, the Nagg of the random copolymers were even smaller than that of the block copolymers. This result may be contributed from high polydispersities with ambiguous and undetectable core–shell structure boundaries, resulting in reduced dye solubilization.
As dye solubilization is significantly influenced by its interaction with the hydrophobic core of the micelle, the mechanism was investigated by measuring the solubilization of the dye at different temperatures. After confirming that dye solubilization in the micelles reached saturation at 25 °C (88 h), the micelles were maintained at 35 °C for 1 h, and the amount of dye solubilized was measured again (Fig. 3c). The changes in the CMC as a function of the temperature changes were also monitored using a pyrene-based fluorescent probe (Fig. S7, ESI†). The results (Fig. 3c) revealed that for all studied block copolymers the amount of dye solubilized tended to decrease with an increase in temperature. At this temperature increase, the CMC change, estimated from the I1/I3 ratio of pyrene, could not be determined for any of the block polymers. Our results followed an opposite trend to that of Chang et al. who reported that drug solubilization in pluronic micelles increased with an increase in temperature31 which was attributed to the decrease in CMC, directly leading to an increase in the number of micelles and distribution of the dye to the respective micelles. Since the CMC of the block copolymers did not change with a change in temperature in our case (Fig. S7, ESI†), the number of micelles was constant; therefore, the decrease in dye solubilization with an increase in temperature may be attributed to the exothermic nature of solubilization. When the binding of dye molecules to surfactant micelles decreases with an increase in temperature, the binding and partitioning processes are exothermic and occur spontaneously.32
Notably, BL02 showed a high dye solubilization rate, which can be attributed to its shell characteristics. BL02 was identified as a micelle with a core–shell structure but had the greatest polydispersity, and its fringe was smaller than that of the other studied block copolymers, suggesting that the boundary was somewhat ambiguous at the core–shell interface or shell–bulk solvent interface (Fig. 1). The dye dissolved poorly in the core because of the micellar structure of BL02, which caused it to dissolve rapidly (Fig. 3a). These features were attributed to the specific shell structure of BL02, in which the hydrophilic block consisted of a random mixture of MA and BMA. Combined with the weak molecular packing of the BL02 micelles, which can be judged from the Nagg, the dye could easily penetrate the micelle core and at the same time be easily excluded (Fig. 3a). A similar trend to BL02 was observed in BL04 between rapid dye solubilization (Fig. 3a) and an ambiguous boundary (Fig. 1d).
This study investigated the properties of block copolymer micelles to elucidate the correlation between the capacity of the hydrophobic substances to be incorporated into micelles and their core–shell aggregated structure by comparison with random copolymer micelles having the same composition as block copolymer. The micelle structure was characterized by fluorescence, DLS, SLS, and SAXS. All block copolymers formed core–shell micelles, whereas random copolymers showed high polydispersity values with ambiguous and undetectable core–shell structural boundaries. These structural features enabled the block copolymers to solubilize higher amounts of a hydrophobic dye compared to random copolymers. Kinetically, the random copolymers reached solubilization saturation rapidly, while the block copolymers reached saturation slowly. Notably, the weak molecular packing and ambiguous core–shell interface of BL02, resulted from the hydrophilic block of a random mixture of MA and BMA, accelerated the distribution of dyes to the micelle core.
Author contributions
M. A. carried out the all experiments and wrote the paper. A. W., H. T., Y. S., Y. T., and M. K. partly carried out the experiments. S. O. reviewed the manuscript. H. O. supervised, reviewed, and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Iketani Science and Technology Foundation of Japan. This research was financially supported by the Japan Agency for Medical Research and Development (AMED) under the Grant Number 22ym0126812j0001. The authors thank Mr Satoshi Hashimoto, Mr Iwao Hattori, Mr Takahiro Nio and Mr Shinya Takeno (DIC) for helpful discussions and supports. The authors also thank Dr Koji Tsuchiya and Prof. Hideki Sakai for taking TEM images.References
- A. W. York, S. E. Kirkland and C. L. McCormick, Adv. Drug Delivery Rev., 2008, 60(9), 1018–1036 CrossRef CAS PubMed.
- Y. Xue, J. Sun, S. Xiong, H. Chai, X. Xin, G. Xu and T. Liua, J. Mol. Liq., 2019, 278, 320–328 CrossRef CAS.
- C. A. Lorenzo, J. G. Lopez, M. F. Tarrio, I. S. Macho and A. Concheiro, Eur. J. Pharm Biopharm., 2007, 66, 244–252 CrossRef PubMed.
- S. A. Pillai, U. Sheth, A. Bahadur, V. K. Aswal and P. Bahadur, J. Mol. Liq., 2016, 224, 303–310 CrossRef CAS.
- T. Ohtake, H. Ito and N. Toyoda, Langmuir, 2022, 38, 7618–7627 CrossRef CAS PubMed.
- M. Hibino, K. Tanaka, M. Ouchi and T. Terashima, Macromolecules, 2022, 55(1), 178–189 CrossRef CAS.
- D. Chang, C. Du, J. Liu, W. Sun, Y. Su, D. Zang and T. Liu, J. Mol. Liq., 2022, 368, 120635 CrossRef CAS.
- M. Karayianni and S. Pispas, J. Polym. Sci., 2021, 59, 1874–1898 CrossRef CAS.
- Z. Ge and S. Liu, Chem. Soc. Rev., 2013, 42, 7289–7325 RSC.
- H. C. Kim, S. M. Park and W. D. Hinsberg, Chem. Rev., 2009, 110, 146–177 CrossRef PubMed.
- J. T. Early, A. Block, K. G. Yager and T. P. Lodge, J. Am. Chem. Soc., 2021, 143(20), 7748–7758 CrossRef CAS PubMed.
- D. Quémener, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Chem. Commun., 2006, 5051–5053 RSC.
- J. A. Johnson, Y. Y. Lu, A. O. Burts, Y.-H. Lim, M. G. Finn, J. T. Koberstein, N. J. Turro, D. A. Tirrell and R. H. Grubbs, J. Am. Chem. Soc., 2011, 133, 559–566 CrossRef CAS PubMed.
- Y. Q. Yang, X. D. Guo, W. J. Lin, L. J. Zhang, C. Y. Zhang and Y. Qian, Soft Matter, 2012, 8, 454–464 RSC.
- M. Asada, H. Tanaka, Y. Suwa, S. Irifune, S. Osawa and H. Otsuka, Appl. Sci., 2023, 13(3), 1834 CrossRef CAS.
- A. E. Marras, T. R. Campagna, J. R. Vieregg and M. V. Tirrell, Macromolecules, 2021, 54(13), 6585–6594 CrossRef CAS.
- C. Chakrabarti, S. K. Pathan, V. D. Punetha and S. A. Pillai, J. Mol. Liq., 2022, 366, 120289 CrossRef CAS.
- K. C. Castro, J. C. Coco, E. M. Santos, R. M. Martinez, M. H. M. Nascimento, J. Parata, P. R. M. L. Fonte, P. Severino, P. G. Mazzola, A. R. Baby, E. B. Souto, D. R. Araujo and A. M. Lopes, J. Controlled Release, 2023, 353, 802–822 CrossRef PubMed.
- Z. Chen, S. Deng and L. Tian, Proc. SPIE-Int. Soc. Opt. Eng., 2023, 12598 Search PubMed.
- M. Chountoulesi, D. Selianitis, S. Pispas and N. Pippa, Materials, 2023, 16(6), 2298 CrossRef CAS PubMed.
- K. P. Ananthapadmanabhan, E. D. Goddard, N. J. Turro and P. L. Kuo, Langmuir, 1985, 1, 352–355 CrossRef PubMed.
- T. Satomi, K. Ueno, Y. Fujita, H. Kobayashi, T. Tateishi and H. Otsuka, J. Jpn. Soc. Colour Mater., 2006, 79(11), 475–482 CrossRef CAS.
- H. Otsuka, T. Sanbai, D. Matsukuma and Y. Ikenakga, Colloid Polym. Sci., 2014, 292, 291–300 CrossRef CAS.
- J. S. Ham, J. Chem. Phys., 1953, 21, 756–758 CrossRef CAS.
- M. Wilihelm, C.-L. Zhao, Y. Wang, R. Xu and M. A. Winnik, Macromolecules, 1991, 24(5), 1033–1040 CrossRef.
- K. Fujio, T. Mitsui, H. Kurumizawa, H. Tanaka and Y. Uzu, Colloid Polym. Sci., 2004, 282(3), 223–229 CrossRef CAS.
- N. Tripathi, D. Ray, V. K. Aswal, K. Kuperkar and P. Baharur, Soft Matter, 2023, 19, 7227–7244 RSC.
- A. Dokoumetzidis and P. Macheras, Int. J. Pharm., 2006, 321, 1–11 CrossRef CAS PubMed.
- X. Li, K. Y. Mya, X. Ni, C. He, K. W. Leong and J. Li, J. Phys. Chem. B, 2006, 110, 5920–5926 CrossRef CAS PubMed.
- I. LaRue, M. Adam, E. B. Zhulina, M. Rubinstein, M. Pitsikalis, N. Hadjichristidis, D. A. Ivanov, R. I. Gearba, D. V. Anokhin and S. S. Sheiko, Macromolecules, 2008, 41(17), 6555–6563 CrossRef CAS.
- D. Chang, C. Du, J. Liu, W. Sun, Y. Su, D. Zang and T. Liu, J. Mol. Liq., 2022, 368, 120635 CrossRef CAS.
- S. Tunc, O. Duman and B. Kanci, Dyes Pigm., 2012, 94(2), 233–238 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm00009a |
| ‡ Current affiliation: Department of Biomedical Engineering, Faculty of Life Science, Toyo University, Japan. |
| This journal is © The Royal Society of Chemistry 2024 |