
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of polymerization on free water in polyacrylamide hydrogels observed with Brillouin spectroscopy
Britta R.
Gorman
and
L. E.
McNeil
 *
*
Department of Physics and Astronomy, University of North Carolina, Chapel Hill, North Carolina 27599, USA. E-mail: mcneil@physics.unc.edu
First published on 18th June 2024
Abstract
Brillouin spectroscopy is used to determine the effects of polymer concentration, crosslinking density, and polymerization on the longitudinal storage and loss moduli of polyacrylamide hydrogels. The model established by Chiarelli et al. is implemented to calculate the speed of sound in the free water [Chiarelli et al., The Journal of the Acoustical Society of America, 2010, 127(3), 1197–1207]. The polymer concentration has the greatest effect on the moduli of the polymer matrix. We determined that the crosslink density has no measurable effect on the logitudinal storage or loss modulus of polyacrylamide hydrogels when measurements are made at GHz frequencies, in contrast to measurements made at kHz frequencies as documented by other studies. However, the moduli are independent of monomer concentration if the acrylamide is not polymerized. We show at the GHz frequency that the incorporation of acrylamide polymer chains affects the mechanical properties of the free water. The speed of sound in the free water is reduced by the introduction of polymerized acrylamide. The long polymer chains and their interactions with the bounded water disrupt the bonding organization of the unbound water, causing a reduction of the average hydrogen bond strength between free water molecules. This results in a decreased speed of sound in the free water and an increase in the longitudinal storage modulus of the hydrogel.
1 Introduction
A hydrogel is a material containing a porous solid and a large amount of interstitial fluid, typically water. There are many applications for such a material due to the many possibilities of the chemical and mechanical properties. The bulk properties of a hydrogel are affected by all constituents with contributions from the solid, the fluid, and the interactions between these two components. Polyacrylamide gels are widely used in various industries. Their tunable mechanical properties are used in the oil industry to increase the viscosity of the water used to extract oil.1 Hydrogels can also be used as proton conductors in electrochemical devices.2 Because of their high water content, hydrogels are used to mimic living tissue for studies of the viscoelastic nature of tissue and for cell-growth studies. Many types of hydrogels are used in this way, including polyacrylamide.3,4The longer-timescale processes (hours to microseconds) and the very-short timescale of bond vibrations (femtoseconds) in hydrogels are well understood, but there is clearly a gap between the two. Also, mechanical characteristics are typically studied using techniques that require contact, such as rheology and atomic-force microscopy. It is therefore beneficial to the field to introduce a non-contact and non-destructive technique that can be used in situ to determine certain viscoelastic properties of tissue. For this reason, Brillouin spectroscopy has garnered attention in the field in the last decade.
Brillouin spectroscopy, commonly used in condensed matter physics, is a non-contact low-power technique that probes the viscoelastic properties of a material at the gigahertz frequency (hundreds of picoseconds timescale). While there are several more-established techniques in the soft-matter field, Brillouin spectroscopy yields information that the others do not. It is yet unclear how the information that can be extracted using this technique compares to and augments that derived from the more-established long-timescale techniques. It is therefore the goal of this project to ascertain the exact type of information gathered when using Brillouin spectroscopy to study hydrogels.
1.1 Motivation
Recently, polymer systems have been used in proton exchange membrane fuel cells, systems that alter chemical energy into electrical energy.5 For such use, potential polymer systems need to be well understood and the organization of the solvent molecules needs to be studied and tuned. It is known that water greatly increases proton conduction in solid-state material and is the main proton-conducting component.6 A highly-organized hydrogen bond network is essential for good proton conductivity in water.6,7 To understand and quantify proton transport in a non-destructive way, one can study the hydrogen bond network of the transport medium. Brillouin light spectroscopy (BLS) is a good candidate for such studies. BLS has been used to study protic conductivity and polyacrylamide gels have been incorporated into proton exchange membrane fuel cells.8,9In protein- and polymer-based electrolyte cells, the polymers are responsible for mediating water transport and thus proton conduction. This is realized by the proteins themselves, or by their interactions with the surrounding water.10 It has been shown that the macromolecule's ability to bind to surrounding water affects the degree of proton conduction.10 It then follows that the mobility and hydrogen-bonding characteristics of the water within a hydrogel affect the proton conductivity. It is therefore essential to understand the interactions between polymers and water within a specific hydrogel in order to maximize the effectiveness of proton exchange.
Certain artificial hydrogels are also used to model different tissues in the body, necessitating the tuning of the viscoelastic properties of the material such that they match physiological conditions. It is therefore essential to understand the mechanical properties of materials both at the macroscopic and microscopic level so that appropriate materials can be prepared. Unlike many industry standards, Brillouin spectroscopy is a non-contact, non-invasive, bulk measurement of viscoelastic properties. It does not require the excision or destruction of a sample, and has been used simultaneously with Raman scattering to obtain a fuller picture of the properties of materials from the GHz–THz region.11
Brillouin spectroscopy probes the sample at the length scale of an acoustic phonon, on the order of hundreds of nanometers, which corresponds to coherent molecular motion within the sample. This small length scale has allowed researchers to probe subcellular levels, and indeed use Brillouin microscopy to map certain cells.12 A better understanding of the mechanical and viscoelastic properties at the microscopic level would allow researchers a better baseline from which to build functional models for living cells and their environments, and the establishment of a non-contact technique would help ease the difficulties of in situ electrolyte studies that involve contact. The rate of microscale and nanoscale processes, such as cell transport and proton conduction, is largely affected by the viscosity of the local microenvironment. With the rising interest in Brillouin spectroscopy as a tool in biomechanics and hydration dynamics, an aim of this work is to show the efficacy of Brillouin spectroscopy in detecting changes to mechanical characteristics and its use in understanding the viscoelastic properties and relaxation processes of hydrogels.
2 Background
2.1 Viscoelasticity
Polymer networks are by nature inhomogeneous at different length and timescales. At the angstrom lengthscale they are composed of several different types of atoms. At the slightly larger lengthscale, monomers are covalently bound together along the length of the polymer, and interact with surrounding non-bound atoms in the other two directions. Along the radial direction of the polymer, it may be associated strongly with adjacent macromolecules. Each level of interactions (at the various lengthscales) has its own dominating forces and relaxation times. Every level of association between atoms, monomers, polymers, etc. has a different rate at which energy is transferred. This is referred to as temporal heterogeneity.Due to the inhomogeneity at different lengths and timescales caused by structural hierarchies and interactions with multiple types of molecules and molecular suprastructures, polymer networks are dispersive for vibrational waves. This temporal heterogeneity causes dispersion and is the basis for viscoelastic behavior in polymer materials.
Viscoelasticity is the confluence of viscosity and elasticity. In a viscoelastic material, the strain and stress rates affect the stiffness. In polymer systems this results from contributions of the various molecular rearrangement processes.13 If the polymers have enough time to rearrange under the applied stress, then they will do so. If the stress is applied over a shorter time period than the viscous reorganization time, then the polymer will react somewhat elastically and stretch and then return to its unexcited state after releasing the same amount of energy used to excite it. The energy needed to slide two independent polymers past each other is much lower than the energy needed to stretch an individual polymer; therefore, longer-timescale interactions cause the polymer network to be less stiff than do shorter-timescale interactions and permanently reorganize the material in a process called creep.13 Biologically-relevant polymer systems as well as proton-conducting polymer systems have a large amount of water and display viscoelastic properties. Therefore, a system that models these conditions will either be a solution or a more permanent network with water bound within, such as a hydrogel.
One might assume that the viscoelastic properties of isotropic and highly homogeneous hydrogels are fairly straightforward, with the network structure and trapped water working cohesively to produce bulk properties. This is not necessarily the case at all timescales. The interplay among the mechanical properties of the individual polymers, the polymer matrix, the dynamic interaction of the polymer with the surrounding water, and the complex water relaxation processes causes the viscoelastic characteristics of homogeneous hydrogels to be complex and nonuniform across all polymer types.
2.2 Water content
While matrix structure and energy dissipation play a role, the high water content in hydrogels necessitates a deep understanding of hydration dynamics to understand their mechanical properties and relaxation processes. The fluid component of the hydrogel experiences very short relaxation times, typically on the order of picoseconds, while the solid component contributes relaxation processes on a much longer timescale (microseconds to hours, depending on the concentration, crosslinking density, and the loading rate of the applied force).14The role of water in the viscoelastic properties of hydrogels is highly significant; the dynamics are largely affected by the contributions of water. Whether water is trapped in pores within the network due to surface tension, or interacts strongly with the polymer network due to hydrophilic functional chains or moieties, affects the bulk properties of the material. Polymer chains that contain amphiphilic molecules may integrate into the water network in a less disruptive way than will those that are largely hydrophobic in nature. This effect is also compounded by chain length and rotational degrees of freedom of the macromolecules. If the molecule is free to rotate and is largely hydrophilic, the polymer unit might orient itself in such a way that the somewhat-tetrahedral water network surrounding the polymer is largely unaffected by the polymer network. However, if the chain length of the base polymers is long enough that there is little rotational freedom due to physical entanglements, and the polymer interacts with the water in a largely disruptive manner, then the water network will be affected both in compressional properties and in relaxation.15 Similarly, it has been shown that as a polymer becomes hydrated (increasing the number of water molecules hydrogen bound to the polymer) the stiffness increases and the compressibility decreases.16
In hydrogels, water can be classified as bounded water or free water. The former is water bound to the polymer and inflates the perimeter of the polymer itself; it is not free to move radially away from the polymer. The free water interacts with other free water molecules or with the bounded water; it does not interact directly with the polymer.17 Compared to other types of solutions which contain smaller molecules, the molecular internal degrees of freedom in polymer solutions play a larger role in their dynamics.18 The internal motions of smaller molecules occur at frequencies between 1 GHz and 100 THz, while changes in conformation of the polymer chains occur at much lower frequencies and are therefore more prominent in low-frequency viscoelastic studies. It has been shown that there exist relaxation processes of water molecules at the GHz timescale.19 Because Brillouin spectroscopy probes in the GHz frequency range, the interactions of the small water molecules are the subject of this study.
The hydrogen-bonded network of the free water molecules governs their relaxation processes. There are two main relaxation processes of free water at high frequencies, rotational and translational processes. In reorientation, a water molecule will exchange hydrogen bond locations while it re-orients. This happens in concurrence with the rotation of other molecules around it; the hydrogen-bond network simultaneously rearranges.20 There is still much debate about the translational dynamics, though we do know that they too are governed by the hydrogen bond network and affected by the strength and number of binding sites at each water molecule.21 The rate at which these relaxation processes occur is affected by the characteristics of the hydrogen bonding of the matrix. In the presence of other molecules, this hydrogen-bonding network is perturbed and the relaxation dynamics are affected. Typically, this perturbative effect in polymers is limited to the interfacial water surrounding the polymer with the free water unaffected. However, the data presented here indicate that this is not the case for polyacrylamide gels.
2.3 Polyacrylamide hydrogels
Polyacrylamide (PA) gels are chemically-crosslinked matrices fabricated by the copolymerization of acrylamide and a crosslinking monomer, typically N,N-methylenebis(acrylamide) known as bisacrylamide. These two monomers copolymerize in the presence of free radicals to form long-chain acrylamide polymers that are crosslinked by the bisacrylamide monomers. The networks formed are highly absorbent and isotropic. These gels are commonly used in gel electrophoresis, as the pore size of the resulting matrix is highly adjustable and dependent on the overall monomer concentration (T) | (1) |
 | (2) |
Nominally, C is the crosslinking density.
It is widely accepted in the field of proton conduction that in most polymeric systems an increase in crosslinks can increase the efficiency of proton conducting membranes.22,23 This mostly depends on the increased ability to absorb water (swell) with highly crosslinked systems, as water is an effective medium for proton conduction. However, there is still some debate about the trade-offs between mechanical robustness and ion conductivity.24 There is a decrease in polymer molecule mobility when crosslinking density is increased, however, this increase is also associated with the stabilization of the hydration regions within a hydrogel and a reduction in competitive hydration. With that being said, it is an aim of this study to understand the effects of crosslinking density on the organization and average hydrogen bond strength of the free water to better understand the effects of crosslinking density.
Polyacrylamide gels have garnered attention in bioscaffolding and cell growth studies due to their flexibility, strength, and the tuneability of their mechanical properties. The concentration of monomers in the matrix and the crosslinking density have been shown to affect the stiffness of these hydrogels at low timescales.25 The final mechanical properties of these gels depend on T and C in a cooperative manner and independently, with one common industry standard of C = 3.33% used because it produces a self-supporting and optically-transparent gel across a wide range of T. We therefore paid particular attention to hydrogels with C near this value.
While it is known that pore size plays an integral role in the mechanical properties of hydrogels and polymeric networks at various timescales, the effect of crosslinking density of PA gels on their behavior at shorter timescales has yet to garner consensus. As previously stated, the shorter-timescale viscoelastic properties affect cell migration, fluid flow dynamics, and energy transfer throughout the matrix. Of particular importance to proton conduction is fluid flow and fast hydrogen bond dynamics. To develop a better understanding of the full frequency range of hydrogel's viscoelastic response, it is necessary to expand the existing data into the GHz range.
2.4 Brillouin spectroscopy
Brillouin light scattering is the inelastic scattering of a photon by a thermal acoustic phonon in a material. Non-zero temperatures cause the existence of thermal vibrations within a material; the frequency and propagation speed of these vibrations depend on the mechanical properties of the material. In the classical picture, propagating sound waves create periodic variations in mass density which produces changes to the refractive index in the material. The periodicity of these variations creates a diffraction grating, from which incident photons are scattered. Because these phonons travel at some speed, the scattered photons experience a Doppler shift proportional to the speed of sound in the material.In the quantum picture, the Stokes shift occurs when the photon energy is transferred to create a phonon in the material and an outgoing photon of lower energy; an anti-Stokes shift corresponds to an increase in photon energy due to the annihilation of a phonon. This symmetric energy difference is dependent on the elastic properties in a material. The output of this type of spectroscopy is a spectrum of intensity versus frequency shift from the incident laser line. Example spectra are shown in Fig. 1. Note that while the gel is almost entirely made of water, the Brillouin shift is visibly different from that of pure water.
 | ||
| Fig. 1 Two Brillouin spectra. The blue curve is that of water and the red curve is that of a polyacrylamide hydrogel. The Brillouin shift, Ω, is the position of the two peaks. The central peak (extending upward out of the graph) results from elastic scattering. | ||
In the simplest elastic regime, it is assumed that there is a linear relationship between stress and strain, there is no frequency or time dependence, and no viscous creep. In this regime, the elastic modulus of a material is related to the speed of sound (c) by
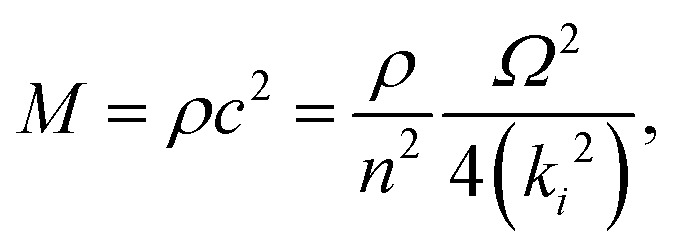 | (3) |
The different relaxation timescales of the individual molecules and macromolecular interactions necessitate the higher-order viscoelastic treatment. To do this, the longitudinal modulus is promoted to the complex form
| M*(ω) = M′(ω) + iM′′(ω), | (4) |
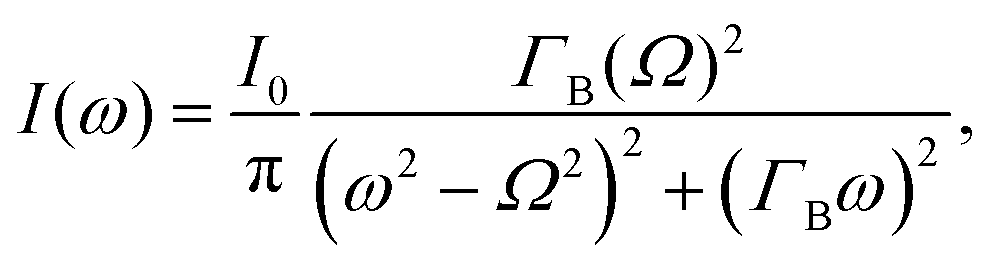 | (5) |
 | (6) |
 | (7) |
2.5 Poroelastic waves in hydrogels
A common model for the interpretation of BLS data is the viscoelastic model. This model describes the matrix as being in a single phase with an elastic component modeled by a spring, and a viscous component modeled by a dashpot. The two components are not distinguished from one another and their contributions are not delineated. In contrast, the poroelastic model describes a material as containing an elastic solid matrix with pores through which a viscous fluid is allowed to flow.26 Both models are appropriate when describing a sample that contains a polymer matrix surrounded by a fluid that is capable of flowing with respect to the solid matrix. While the viscoelastic model is useful in determining the bulk properties of the material, the poroelastic model can be utilized to understand the contributions of the two different components of the sample (solid and fluid). Both of these models have been used to describe hydrogels, and the benefits to using each model lie in what information one wants to extract. If the fluid flow is important, one might use the poroelastic model. If only the viscoelastic response is important than one might use the simpler viscoelastic model. For the present study, it is important to interpret the results of polyacrylamide data in terms of the contributions from the polymer network and the fluid portion individually.The technique used in this work, BLS, probes thermal acoustic phonons. As seen above, the speed of sound in a material can be calculated from the longitudinal modulus, as measured by BLS, using the relation
 | (8) |
The model we will use is that of poroelastic waves in hydrogels, as presented by Chiarelli et al.27 That work builds on the established relations presented by Biot in 1956, with a semi-empirical adaptation.28 The work by Chiarelli et al. confirms the present model using ultrasonic (US) data.
In Biot's theory of poroelastic waves, a hydrogel is modeled as a porous material that has an elastic component due to the polymer matrix and a fluid component. The fluid component moves through the pores of the solid according to Poiseuille's law. This theory mainly attributes the relaxations for ultrasonic (US) excitation to the dissipation of energy caused by the friction involved in fluid moving against the solid. To better match experimental data at high frequencies, Chiarelli et al. additionally consider bounded water (that which is attached to the matrix). This bounded water viscoelastically interacts with the solid and the bounded water itself interacts viscously with the remaining water.27 These are the two main components added to the Biot model.
The expression, developed by Chiarelli et al., for the speed of sound in a poroelastic material is
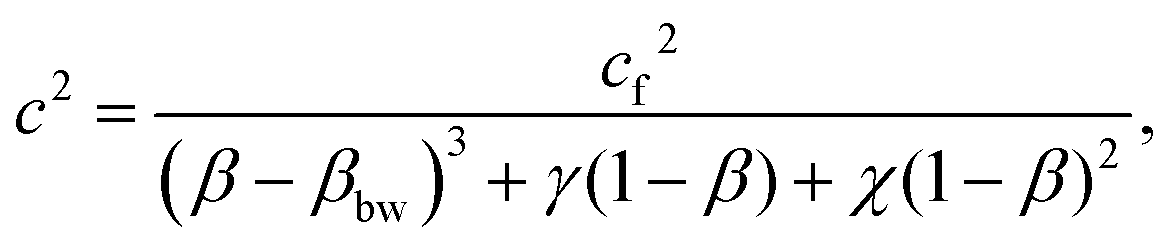 | (9) |
3 Methods
In the present study, three sets of polyacrylamide samples are studied using Brillouin spectroscopy:(1) Varying total polymer concentration with crosslinked and non-crosslinked samples
(2) Varying crosslink density while maintaining total polymer concentration
(3) Varying the total polymer concentration while maintaining crosslink density
For each sample, the Brillouin shift (Ω) and the FWHM of the Brillouin peaks (ΓB) are used to study the stiffness and relaxation rates at the GHz frequency.
3.1 Brillouin spectrometer
Our Brillouin light spectroscopy (BLS) apparatus uses a scanning multi-pass Fabry–Pérot interferometer to produce a spectrum of intensity of the scattered light vs. frequency (energy), as shown in Fig. 1. A schematic of the spectrometer is shown in Fig. 2. | ||
| Fig. 2 A schematic representation of our Brillouin spectrometer. | ||
Our Brillouin spectrometer includes a cobolt 06 diode-pumped laser (561 nm) from Hübner photonics. This is a 50 mW continuous-wave laser with single-frequency lasing and very narrow linewidth. The interferometer is a tandem three-pass scanning Fabry–Perót interferometer from JRS Scientific Instruments with accompanying control unit, and the detector is a Hamamatsu H10862 photon counter.
3.2 Polyacrylamide hydrogel fabrication
Polyacrylamide gels are synthesized at room temperature using a catalyst of tetramethylethylenediamine (TEMED) and ammonium persulfate (APS) to copolymerize acrylamide and bisacrylamide into a three-dimensional crosslinked network, where bisacrylamide acts as the chemical crosslink between polymer chains composed of acrylamide monomers. The acrylamide is a 40% (weight/volume) solution, suitable for electrophoresis and sterile-filtered; the bisacrylamide is a powder, for molecular biology and suitable for electrophoresis, ≥99.5% purity.To synthesize the samples, bis powder is dissolved in water to 1.5% w/v (0.015 g in 1 mL water); APS is dissolved in water to 10% w/v. The desired volume of acrylamide, bisacrylamide, and water are added to the sample container. This mixture is then degassed under ∼60 mTorr vacuum for 20 minutes to remove any oxygen from the solution. After degassing, the test tubes are inverted to mix, then the APS and TEMED are added in rapid succession and 2 mL is pipetted into a square cuvette, compatible with the spectrometer sample holder. Samples are topped with water-saturated butanol and covered with parafilm and allowed to gel fully at room temperature, over a full day.
3.3 Error calculations and hydrogel uniformity
The error bars in the presented data are calculated using the partial derivative error propagation method. The experimental uncertainties are the uncertainties associated with fitting the Brillouin spectrum to the convolution of the DHO and the instrument response function. There are three parameters used to fit each peak: the intensity, the FWHM, and the peak center. The peak centers are used to calculate the Brillouin shift and subsequently longitudinal storage modulus, the FWHM of the Brillouin peaks are used to calculate the longitudinal loss modulus. The uncertainty in the Brillouin shift is the sum of the maximum error of the two Brillouin shifts in the spectrum (left peak and the central peak; the right peak and the central peak). The uncertainty in the Brillouin shift (Ω) is then propagated using the partial derivative of eqn (6). Similarly, the square-root of the sum of the squares of the uncertainty in the FWHM measurements is used with the partial derivative of eqn (7).Hydrogel uniformity can affect certain light scattering results. For polyacrylamide hydrogels, increasing total polymer or crosslink concentration can increase nonuniformity.29 If the nonuniformities in the hydrogel are on the same length scale as the probing wave, mass density fluctuations can affect the consistency of the measurements. However, the length scale of the acoustic phonon for an excitation wavelength of 532 nm is approximately 0.5 μm and documented inhomogeneities in polyacrylamide hydrogels are on the order of tens of nanometers.30 While the inhomogeneity in the hydrogel may affect certain light scattering measurements, such a varying was not detected in this work. Measurements were made at various positions within the hydrogel and no measurable difference was detected.
4 Results and discussion
4.1 Crosslinked vs. not crosslinked
As explained in the previous section, we performed three experiments on polyacrylamide gel. In the first experiment, samples are fabricated with and without crosslinks to evaluate the effect of crosslinks. In this experiment, the total monomer concentration that is varied consists of acrylamide alone with no bisacrylamide. These data are shown in Fig. 3; note that the horizontal error bars are too small to see on this and many future graphs. The results indicate that there is no effect of crosslinks on the longitudinal storage modulus of PA gels as measured by Brillouin spectroscopy. Both series, crosslinked and not crosslinked, show an increase in modulus as T increases. On the whole, there is no difference between the moduli measured on crosslinked gels and those samples which are not crosslinked. While it is known that crosslinks increase the stiffness of the polymer matrix, there is no observable effect on the stiffness of the hydrogel on the whole. Similarly, merely the increased concentration of acrylamide is not responsible for this increase, as see in Fig. 4. These results are mirrored in the results of the second experiment. | ||
| Fig. 3 Longitudinal storage modulus versus total monomer concentration (T) for PA gels of crosslinker concentration (C) = 3.33% (red) and C = 0% (blue). Guide line added for clarity. | ||
 | ||
| Fig. 4 Longitudinal storage modulus versus total monomer concentration (T) for PA gels of crosslinker concentration (C) = 3.33% fully polymerized and cross-linked (black) and monomer solutions (red). Guide lines added for clarity. | ||
4.2 Varying crosslinking concentration
In the second experiment, total crosslinking concentration is varied while the total polymer concentration is held constant. The aim of the second experiment is to determine the effects of crosslinking density on the longitudinal modulus of the sample. To do this,we keep the overall monomer concentration (T) constant and vary the crosslink density (C). For this experiment, T = 12%, and C is varied from 0–5%. The results of this experiment are shown in Fig. 5. These results shows no effect of crosslinks on the longitudinal modulus of the hydrogel in this range of C at GHz frequency. This result is consistent with the results of the previous experiment in that the matrix itself would be less stiff if there are no crosslinks. If the matrix stiffness is what is causing the changes seen in the the previous experiment, then increasing crosslinking density should increase the stiffness of the bulk. However, we observe no increase in the bulk stiffness with increasing crosslinking density. Therefore, the stiffness of the matrix itself is not responsible for the increase seen in Fig. 3. | ||
| Fig. 5 Longitudinal storage modulus versus crosslink density for PA gels of total monomer concentration (T) = 12%. Guide line added for clarity. | ||
4.3 Varying overall monomer concentration
For all samples in this experiment, C = 3.33% (the ratio of acrylamide![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :bis monomer is 29:1) which is a common industry standard. PA samples are made with a range of T between 3% and 18%. The resulting data are shown in Fig. 6. The longitudinal storage modulus, M′, increased linearly with an increase in the total monomer concentration. Note that M′ for water is 2.15 GPa; all of the samples have a M′ larger than that of water even though they are all over 80% water (weight by volume). There are two procedures to quantitatively analyze these data. The first is to use a rule-of-mixtures argument (Voigt model), as has been done before with BLS in hydrogels, to extract the longitudinal modulus of the matrix itself.31 This process decomposes the contributions of the matrix and the contributions of the water from the total modulus linearly by making use of volumetric arguments of each component, treating it as a composite. The second quantitative analysis uses poroelastic wave propagation theory from ultrasonic studies of hydrogels (described above) to show that the speed of sound (and therefore the longitudinal modulus) in the water in the PA hydrogel is decreased from that of bulk water.
:bis monomer is 29:1) which is a common industry standard. PA samples are made with a range of T between 3% and 18%. The resulting data are shown in Fig. 6. The longitudinal storage modulus, M′, increased linearly with an increase in the total monomer concentration. Note that M′ for water is 2.15 GPa; all of the samples have a M′ larger than that of water even though they are all over 80% water (weight by volume). There are two procedures to quantitatively analyze these data. The first is to use a rule-of-mixtures argument (Voigt model), as has been done before with BLS in hydrogels, to extract the longitudinal modulus of the matrix itself.31 This process decomposes the contributions of the matrix and the contributions of the water from the total modulus linearly by making use of volumetric arguments of each component, treating it as a composite. The second quantitative analysis uses poroelastic wave propagation theory from ultrasonic studies of hydrogels (described above) to show that the speed of sound (and therefore the longitudinal modulus) in the water in the PA hydrogel is decreased from that of bulk water.
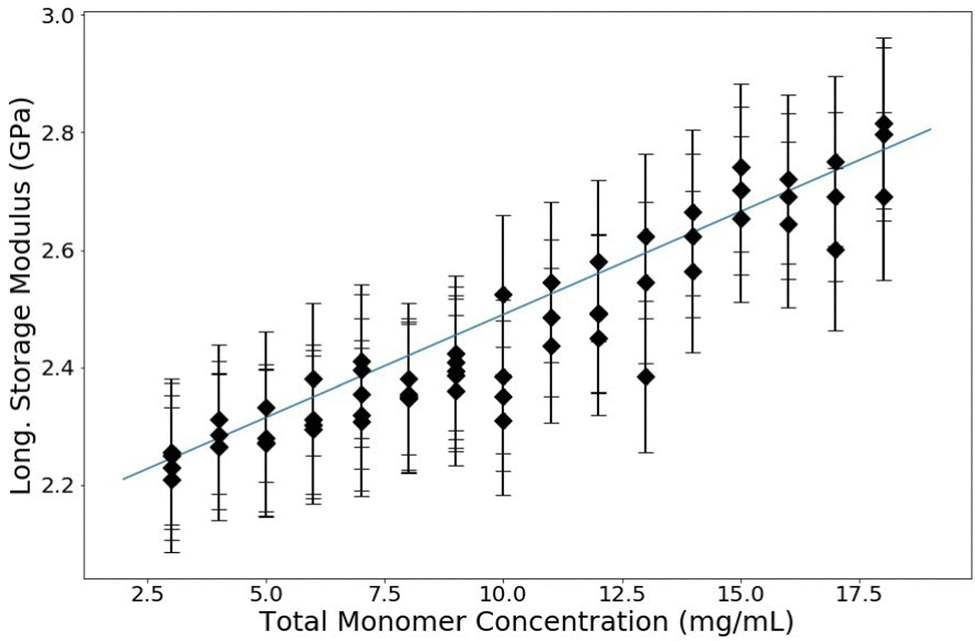 | ||
| Fig. 6 Longitudinal modulus versus overall monomer concentration (T) of PA gels. All samples in this experiment have a crosslink concentration of C = 3.33%. Guide line added for clarity. | ||
A linear regression on these raw data in Fig. 6 can be used to extrapolate the total modulus at 0% polyacrylamide, corresponding to a sample containing only water. This extrapolation yields a bulk modulus of 2.07 GPa, which is noticably lower than that of bulk water. This piece of information, along with the results of the next analysis, is necessary to elucidate the whole picture of what is changing in these polyacrylamide samples as the polymer concentration is changed. These analyses also show that specifically for polyacrylamide hydrogels, the two components (water and polymer matrix) cannot be treated as independent constituents; there is some interaction between the two that affects the bulk and thus a simple Voigt analysis is inadequate to analyze BLS data of polyacrylamide hydrogels.
As explained previously, eqn (9) can be used to calculate the high-frequency poroelastic wave speed in a hydrogel in the intermediate hydration regime (5–25% water by volume). Fig. 7 is a plot of the speed of sound in the PA gels as a function of the polymer concentration volumetric water fraction; these are the same measurements presented in Fig. 6 converted to speed from longitudinal modulus.
 | ||
| Fig. 7 The speed of sound of the hydrogel versus the volumetric water fraction within the sample. The poroelastic wave speed fit for PA gels in the intermediate hydration regime is indicated with a dashed line. Fit parameters are listed. | ||
The monomer concentration is used in the calculation of the volumetric fraction of all water in the sample, β. The corresponding plot of speed versus β in the intermediate hydration regime is shown in Fig. 7. The parameter βbw is a volumetric ratio of the water which is bound to the polymer to the total volume. It has been shown that as water content in polymeric systems increases, the number of water molecules bound to the polymer saturates.17 In polyacrylamide, each monomer of acrylamide has 2 bonding sites for water.32 Using the molar mass of acrylamide and the approximate molar density of water, the parameter βbw is calculated for each monomer concentration.
The poroelastic wave speed equation is fit to the data with βbw dependent on T; cf, γ, and χ are fitting parameters. The result of this fit is shown in Fig. 7. The wave speed in bulk (pure) water is measured to be 1468 m s−1. The inclusion of PA in the gels reduces the speed of sound in the free water in the sample, cf, to 1429 m s−1. These measurements show that the presence of polymerized acrylamide causes a decrease in the tetrahedral ordering of the free water and therefore perturbs the bulk properties of the water in the hydrogel. As discussed previously, bulk water orients itself in a generally tetrahedral matrix, with each water molecule forming hydrogen bonds with four other water molecules. When this tetrahedral organization is disrupted, the average strength of the hydrogen bonds between water molecules is decreased causing a decrease in the speed of sound in the water.33 A decrease in the hydrogen bond strength has been observed using BLS by Harley et al. who studied rat tail tendons at various levels of hydration.34 This decrease in hydrogen bond strength manifests in a decrease in the time it takes for the water molecules to reorient after excitation due to a decreased restoring force. This change in the global reorientation dynamics of water in PA gels has been recently reported by several researchers.15,20,35 Note that in all of these cases, researchers measure the reorientation time of water molecules in PA samples; while the measurements show no distinction between bounded and bulk water reorientation dynamics, there still exist populations of water molecules that are bound to the polymer and those that are not.
4.4 Loss modulus and relaxation processes
The loss modulus measured by BLS corresponds to the relaxation rates of vibrations within the sample. In this study, we found that when total monomer concentration is varied, the loss modulus of polymerized acrylamide gels is affected while samples that contain the same concentration of acrylamide in monomer form are unaffected, as shown in Fig. 8. From the data, it is clear that the loss modulus is less affected by the monomers than it is by the polymers. The dependence of the loss modulus on the polymerization and the overall monomer concentration, T, is consistent with the explanation that the reorganization dynamics of water are slowed in the presence of the long polymer chains. The monomers have little perturbative effect on the reorganization rates of the water molecules, as the water matrix is largely not disrupted. However, the presence of the long polymer chains affects the relaxation rates of the water itself and thus increases the loss modulus of the samples. | ||
| Fig. 8 Loss modulus versus T for PA gels of C = 3.33% (black) and monomer solutions (red). Guide lines added for clarity. | ||
The same trend is measured for samples with and without crosslinks, as seen in Fig. 9. Clearly from these data, there is no effect of crosslinking on the loss modulus and therefore the lifetime of these vibrations. The lifetime of the phonons is only affected by polymerization (not crosslinking) and total monomer concentration, T. This indicates that the relaxation processes responsible for thermal phonons in PA samples are dependent on the interactions of the bounded water, the free water, and the localization effects of the physically entangled polymers. If the polymer matrix itself were responsible for the relaxation dynamics, there would be a change in the loss modulus as a function of crosslinking density, as the crosslinks cause a more rigid matrix. The only measurable effect on the loss modulus is caused by polymerization and a change in overall monomer concentration. This bolsters the argument made in the analysis of the storage modulus. At this GHz timescale and hydration level, matrix rigidity has no measurable effect on the viscoelastic properties of PA gels. The phonon speeds and lifetimes are dominated by the changes to the properties of water surrounding polymerized acrylamide; at this short timescale the polymer chains do not have enough time to reorient or slide past one another and the addition of crosslinks has no effect. The bounded and free water in the samples, which are both affected by the presence of the polymers, are wholly responsible for the viscoelastic properties of polyacrylamide gels at the GHz frequency in the intermediate-high hydration regime.
 | ||
| Fig. 9 Loss modulus versus T for PA gels of C = 3.33% (black) and C = 0% (red). Guide line added for clarity. | ||
The resulting conclusion from the present experiments is that at GHz frequency and at the intermediate-high hydration regime in polyacrylamide gels, BLS is measuring a change to the fluid properties with little to no contribution from the actual matrix itself. There is a significant increase in M′ when T is increased. One might think that this is an obvious conclusion due to the inclusion of arguably stiffer material (the polymer). However, this increase in M′ is only indirectly caused by the inclusion of the polymer. The presence of the polymer, which readily forms hydrogen bonds with the surrounding water, disrupts the water network and affects the mechanical properties of the fluid component of the hydrogel. This conclusion is aligned well with the findings from Yan et al., Roget et al., and Garrett et al., though they all used techniques other than BLS.15,20,35
5 Conclusion
From the present experiments, it is shown that in this hydration and frequency regime, the interactions between the bounded and free water determine the viscoelastic properties. Polymerized acrylamide localizes the bounded water and disrupts the hydrogen bond strength of the free water matrix. The interface between the bounded water and the free water causes frictional loss and is the largest source of energy dissipation at GHz frequency. The disruption of the free water matrix results in a decrease in the average hydrogen bond strength and thus reduces the speed of sound in the free water and increases the storage modulus. It has been shown in other polymers, such as ethylene glycol, that polymer length as well as temperature affect the hydrogen bond strength of the interstitial water.36 Our BLS detects changes to longitudinal storage moduli of pure water between the temperatures of 2 °C and 84 °C of 5% lower and 5% higher than that of room temperature water, respectively. Future work on this topic should focus on the temperature dependence of the observed phenomena presented here.One of the main transport mechanisms of protons in water involves a proton hopping from molecule to molecule in a hydrogen-bound network. This involves the breaking and reforming of hydrogen bonds. As proton transport in water is largely affected by the average hydrogen bond strength, BLS and the use of the poroelastic wave model to determine the speed of sound in the free water of the hydrogel can be used to evaluate trends in the average hydrogen bond strength, and thus proton transport, in a material in a non-contact way for in situ applications.
These results show the uniqueness of Brillouin spectroscopy, and open up questions about the use of polyacrylamide and BLS in electrochemical devices as the attenuation and organization of solvent molecules is related to proton conductivity.8 In particular, what is the role of an increase in bounded water and a decrease in average hydrogen bond strength on the conduction efficiency?
Increasing the total polymer concentration of polyacrylamide hydrogels increases the storage and loss moduli of the hydrogel, as measured by Brillouin spectroscopy. Varying the crosslinking density, or indeed simply the inclusion of crosslinks, has no effect on the storage and loss moduli. Both effects are only seen when the acrylamide is polymerized; tests involving monomer solutions show minimal-to-no effect on the storage and loss moduli with varied total monomer concentration. This is the result of polymerization changing the properties of the water in the hydrogel.
These experiments show that the elasticity of the polymer matrix is not responsible for the increase in longitudinal storage modulus at this GHz timescale; rather, the decrease in the amount of free water, β − βbw, and the interactions between the bounded and free water are responsible for the increase in longitudinal storage modulus as the total monomer concentration is increased. The smaller pores may play a role; we do not know how the effects of C on pore size compare to the effects of T. It is possible that in this range of C, the pore size is not changing as drastically with C as it does with T. However, a sample with no crosslinks has the same longitudinal storage modulus as one with 5% crosslink density within experimental error as measured by BLS. Therefore, the total amount of bounded water, which is related to monomer concentration, is the determining factor in the longitudinal modulus at GHz frequency.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Freeman group of the University of North Carolina at Chapel Hill for their guidance in sample preparation, subject knowledge, and facilities. We thank Dr Kengo Nishi, Kyle Riker, and Dr Ronit Freeman.Notes and references
- S. Al-Kindi, S. Al-Bahry, Y. Al-Wahaibi, U. Taura and S. Joshi, Environ. Monit. Assess., 2022, 194, 875 CrossRef CAS.
- J. Przyłuski, Z. Połtarzewski and W. Wieczorek, Polymer, 1998, 39, 4343–4347 CrossRef.
- A. Dobreikina, T. Shklyar, A. Safronov and F. Blyakhman, Polym. Int., 2018, 67, 1330–1334 CrossRef CAS.
- W. Wieczorek, Z. Florjanczyk and J. Stevens, Electrochim. Acta, 1995, 40, 2327–2330 CrossRef CAS.
- M. Ünlü, J. Zhou and P. Kohl, Angew. Chem., Int. Ed., 2010, 49, 1299–1301 CrossRef.
- D. Lim, M. Sadakiyo and H. Kitagawa, Chem. Sci., 2019, 10, 16–33 RSC.
- N. Sakashita, H. Ishikita and K. Saito, Phys. Chem. Chem. Phys., 2020, 22, 15831–15841 RSC.
- M. Pochylski, J. Gapiński, Z. Wojnarowska, M. Paluch and A. Patkowski, J. Mol. Liq., 2019, 282, 51–56 CrossRef CAS.
- S. Yuan, Q. Tang, B. He, H. Chen, Q. Li, C. Ma, S. Jin and Z. Liu, J. Power Sources, 2014, 249, 277–284 CrossRef CAS.
- S. Mondal, Y. Agam, R. Nandi and N. Amdursky, Chem. Sci., 2020, 11, 3547–3556 RSC.
- A. J. Traverso, J. V. Thompson, Z. A. Steelman, Z. Meng, M. O. Scully and V. V. Yakovlev, Anal. Chem., 2015, 87, 7519–7523 CrossRef CAS.
- M. Nikolić and G. Scarcelli, Biomed. Opt. Express, 2019, 10, 1567–1580 CrossRef.
- S. Matsuoka, Relaxation Phenomena in Polymers, Hanser, 1992 Search PubMed.
- F. Palombo and D. Fioretto, Chem. Rev., 2019, 119, 7833–7847 CrossRef CAS.
- P. Garrett and C. B. Baiz, Soft Matter, 2022, 18, 1793–1800 RSC.
- S. Magazù, J. Mol. Struct., 2000, 523, 47–59 CrossRef.
- M. T. Caccamo and S. Magazù, Spectrosc. Lett., 2017, 50, 130–136 CrossRef CAS.
- I. Teraoka, Polymer Solutions: An Introduction to Physical Properties, Wiley, 2002 Search PubMed.
- S. Magazu, G. Maisano, D. Majolino, F. Mallamace, P. Migliardo, F. Aliotta and C. Vasi, J. Phys. Chem., 1989, 93, 942–947 CrossRef CAS.
- C. Yan, P. L. Kramer, R. Yuan and M. D. Fayer, J. Am. Chem. Soc., 2018, 140, 9466–9477 CrossRef CAS.
- A. Gomez, Z. A. Piskulich, W. H. Thompson and D. Laage, J. Phys. Chem. Lett., 2022, 13, 4660–4666 CrossRef CAS.
- H. Zhu, M. Jia, Q. Li and C. Zhang, Solid State Ionics, 2020, 351, 115325 CrossRef CAS.
- S. Tasarin, C. Panawong, J. Sumranjit and S. Budsombat, Int. J. Hydrogen Energy, 2021, 46, 36969–36981 CrossRef CAS.
- G. Lu, Y. Zhang, J. Zhang, X. Du and Z. Lv, Carbon Energy, 2022, 5, e287 CrossRef.
- A. K. Denisin and B. L. Pruitt, ACS Appl. Mater. Interfaces, 2016, 8, 21893–21902 CrossRef CAS.
- M. D. J. McGarry, C. L. Johnson, B. P. Sutton, J. G. Georgiadis, E. E. W. Van Houten, A. J. Pattison, J. B. Weaver and K. D. Paulsen, Med. Phys., 2015, 42, 947–957 CrossRef CAS.
- P. Chiarelli, A. Lanatá, M. Carbone and C. Domenici, J. Acoust. Soc. Am., 2010, 127, 1197–1207 CrossRef CAS.
- M. A. Biot, J. Acoust. Soc. Am., 1956, 28, 168–178 CrossRef.
- S. Khandai, R. A. Siegel and S. S. Jena, Colloids Surf., A, 2020, 593, 124618 CrossRef CAS.
- N. Orakdogen and O. Okay, Polym. Bull., 2006, 57, 631–641 CrossRef CAS.
- M. Bailey, M. Alunni-Cardinali, N. Correa, S. Caponi, T. Holsgrove, H. Barr, N. Stone, C. P. Winlove, D. Fioretto and F. Palombo, Sci. Adv., 2020, 6, eabc1937 CrossRef CAS.
- D. Gurina, O. Surov, M. Voronova and A. Zakharov, Nanomaterials, 2020, 10, 1256 CrossRef CAS.
- T. V. Lokotosh, S. Magazù, G. Maisano and N. P. Malomuzh, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 62, 3572–3580 CrossRef CAS.
- R. Harley, D. James, A. Miller and J. W. White, Nature, 1977, 267, 285–287 CrossRef CAS.
- S. A. Roget, Z. A. Piskulich, W. H. Thompson and M. D. Fayer, J. Am. Chem. Soc., 2021, 143, 14855–14868 CrossRef CAS.
- V. Crupi, M. Jannelli, S. Magazu, G. Maisano, D. Majolino, P. Migliardo and D. Sirna, Mol. Phys., 2006, 84, 645–652 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |