
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Engineering poly(dehydroalanine)-based gels via droplet-based microfluidics: from bulk to microspheres†
Hannah F.
Mathews
 ab,
Tolga
Çeper
cde,
Tobias
Speen
ab,
Céline
Bastard
ab,
Selin
Bulut
ab,
Maria I.
Pieper
ab,
Felix H.
Schacher
cdef,
Laura
De Laporte
abg and
Andrij
Pich
*abh
ab,
Tolga
Çeper
cde,
Tobias
Speen
ab,
Céline
Bastard
ab,
Selin
Bulut
ab,
Maria I.
Pieper
ab,
Felix H.
Schacher
cdef,
Laura
De Laporte
abg and
Andrij
Pich
*abh
aDWI – Leibniz Institute for Interactive Materials, Forckenbeckstr. 50, 52074 Aachen, Germany. E-mail: pich@dwi.rwth-aachen.de
bInstitute of Technical and Macromolecular Chemistry, RWTH Aachen University, Worringer Weg 2, 52074 Aachen, Germany
cInstitute of Organic Chemistry and Macromolecular Chemistry, Friedrich-Schiller-University Jena, Humboldtstr. 10, 07743 Jena, Germany
dJena Center for Soft Matter (JCSM), Friedrich-Schiller-University Jena, Philosophenweg 7, 07743 Jena, Germany
eCenter for Energy and Environmental Chemistry Jena (CEEC), Friedrich-Schiller-University Jena, Philosophenweg 7a, 07743 Jena, Germany
fCluster of Excellence Balance of the Microverse, Friedrich-Schiller-University Jena, Grüne Aue, 07754 Jena, Germany
gInstitute of Applied Medical Engineering (AME), Department of Advanced Materials for Biomedicine (AMB), University Hospital RWTH Aachen, Center for Biohybrid Medical Systems (CMBS), Forckenbeckstr. 55, 52074 Aachen, Germany
hAachen Maastricht Institute for Biobased Materials (AMIBM), Brightland Chemelot Campus, Maastricht University, 6167 RD Geleen, The Netherlands
First published on 25th July 2024
Abstract
Biomedical applications such as drug delivery, tissue engineering, and functional surface coating rely on switchable adsorption and desorption of specialized guest molecules. Poly(dehydroalanine), a polyzwitterion containing pH-dependent positive and negative charges, shows promise for such reversible loading, especially when integrated into a gel network. Herein, we present the fabrication of poly(dehydroalanine)-derived gels of different size scales and evaluate them with respect to their practical use in biomedicine. Already existing protocols for bulk gelation were remodeled to derive suitable reaction conditions for droplet-based microfluidic synthesis. Depending on the layout of the microfluidic chip, microgels with a size of approximately 30 μm or 200 μm were obtained, whose crosslinking density can be increased by implementing a multi-arm crosslinker. We analyzed the effects of the crosslinker species on composition, permeability, and softness and show that the microgels exhibit advantageous properties inherent to zwitterionic polymer systems, including high hydrophilicity as well as pH- and ionic strength-sensitivity. We demonstrate pH-regulated uptake and release of fluorescent model dyes before testing the adsorption of a small antimicrobial peptide, LL-37. Quantification of the peptide accommodated within the microgels reveals the impact of size and crosslinking density of the microgels. Biocompatibility of the microgels was validated by cell tests.
Introduction
In natural biomolecules, oppositely charged moieties often occur in close spatial proximity. Well-known representatives of natural zwitterions are proteins,1 small osmolytes,2,3 and phospholipids forming cell membranes.4–6 The recurring pattern of vicinity between anionic and cationic groups has been hypothesized to be largely responsible for many exceptional properties of biomaterials, one example being the adhesive behaviour of mussel proteins.7 It is hardly surprising that there have been many attempts to produce synthetic alternatives or to biomimetically optimize artificial materials, respectively.8–10Polyzwitterions, i.e. polymers that carry both positively and negatively ionizable moieties within each monomer unit,11,12 have hence been extensively investigated. Due to their high charge density, they form strong intra- and intermolecular ion pairs, which result in charge compensation at intermediate pH values and an effective net charge of zero at the isoelectric point.13,14 The strong ion complexation within the polyzwitterions also limits their overall solubility, so that the use of polar protic solvents, the addition of salt, or the administration of heat is necessary to disrupt the attractive ionic interactions and solubilize the polymer.13,15,16 Because of the structural similarity between synthetic and natural polyzwitterions, it is often assumed that the synthetic polymers are biocompatible and exhibit resistance to biofouling17,18 even though corresponding studies have only been carried out for a few of the substances known from literature.16
Recently, the focus of research has shifted from synthetic polybetaines12,17,19 to polyzwitterions derived from natural building blocks. Li et al. introduced polyzwitterions adapted from trimethylamine N-oxide (TMAO), an osmolyte found in saltwater fish. The polymers (PTMAO) were prepared by oxidation of N,N-dimethylaminopropyl acrylamide followed by photopolymerization and exhibited antifouling properties as well as low immunogenicity.20 Feng and coworkers expanded on these findings and grafted PTMAO onto commercial polyamide filtration membranes by use of controlled atom transfer radical polymerization (ATRP). In this way, the membranes were endowed with enhanced fouling resistance whilst maintaining a comparatively low flux decline and sufficient water permeability.21
In addition to osmolytes, amino acids have been used as natural building block for polyzwitterions. Wang et al. used L-carnitine, formed through metabolism of methionine and lysine, and converted it with acryloyl chloride to the respective methacrylate, which they in turn grafted onto a gold surface by ATRP. Subsequent protein binding assays with fibrinogen and lysosome demonstrated that the proteins did not adsorb to the surface, and bacterial adhesion tests with E. coli confirmed an antifouling performance similar to that of polybetaines.22 Instead of L-carnitine, Leiske and coworkers modified glutamic acid to prepare zwitterionic monomers with different vinyl end groups. They then utilized reversible addition-fragmentation chain-transfer (RAFT) polymerization whilst protecting the carboxyl and amino groups to obtain defined polymers. The authors were particularly interested in the influence of the polymerizable group on the interaction of the polyzwitterions with cells, and proved that the association of the polymers with different cell types depends on the polymer backbone.23
Starting from the amino acid serine, the synthesis of the polyzwitterion poly(dehydroalanine) (PDha) is possible.24 In contrast to the common geometry of polybetaines, the oppositely charged groups in PDha are not located within the same pendant chain but in two separate side chains connected to the same backbone atom.12,24 The concurrent presence of both carboxylic acid and amine in every monomer unit provides PDha with pH-tunable characteristics: at low and high pH values, it behaves as polyelectrolyte with either polycationic or polyanionic traits, whereas it acts as polyzwitterion in the intermediate pH range.24,25 Though PDha was initially obtained by free radical polymerization of N-acetyl dehydroalanine followed by removal of the acetoxy group,26 it is nowadays mostly synthesized via the prepolymer species poly(tert-butoxycarbonylaminomethyl acrylate) (PtBAMA).24,27 Co-polymers of PDha have either been prepared by ATRP of tBAMA with polymeric halogenides and subsequent deprotection of the ionizable moieites27–30 or by post-modification of a fraction of the amino functions in the homopolymer.31–33
In the last years, the Schacher group has focussed on exploiting the co-existence of positive and negative charges within PDha for various applications. Magnetic nanoparticles (MNPs) coated with PDha34 were shown to adsorb and desorb polyelectrolyte model cargo by electrostatic interaction depending on the pH value of the surrounding medium.35,36 While the non-specific adsorption of proteins was less pronounced for PDha-treated nanoparticles than for pristine or polyanion-coated nanoparticles, they still displayed a protein corona.37 Çeper et al. produced organogels by crosslinking the prepolymer PtBAMA with N,N′-methylenebisacrylamide before hydrolysing the groups protecting the carboxylates and amines.38 Analogous to previous experiments with PDha-coated MNPs,35,36 pH-dependent uptake and release of anionic and cationic model dyes was verified.38 Hydrogel networks were synthesized by crosslinking PDha with bifunctional poly(ethylene glycol) diglycidylether (PEG-DGE).39,40 The ionic groups within the gel network allowed for the electrostatic adsorption of various catalytic species and charged photosensitizers enabling light-driven hydrogen and oxygen evolution reactions.39–41 In contrast to that, Kowalczuk et al. first synthesized asymmetric star-shaped block copolymers from PEG and PDha, which they later incorporated into PEG-based hydrogels via their terminal thiol functions using a photoinduced thiol–ene click reaction. In addition to the adsorption of model dyes, they demonstrated that the peptide sequence RGDS could be immobilized within the hydrogels. The authors interpreted this as an indication that PDha-based hydrogels have a high potential as scaffolds for tissue engineering.
Compared to traditional bulk hydrogels, microgels offer distinct advantages in the biomedical field.42–45 Especially polyzwitterionic microgels have been considered for drug delivery, tissue engineering and antibacterial or antifouling coatings.46,47 However, neither have corresponding materials from PDha been fabricated yet nor have the existing bulk hydrogel networks been tested concerning biocompatibility. Herein, we suggest a simple engineering approach, in which amine–epoxy crosslinking of PDha with PEG-epoxides31,39,40 is employed in droplet-based microfluidics to form microgels of adaptable size (Fig. 1). After microfluidic fabrication, we analyse the composition of the microgels and test the availability of residual amino groups by a fluorescamine assay. The behaviour of the microgels is evaluated and compared to the bulk hydrogel, especially concerning their potential in biomedical applications. In this regard, we examine permeability and determine the softness of the microgels by both nanoindentation and micropipette aspiration. By measuring the electrophoretic mobility and swelling degree of the microgels, we verify that the polyzwitterionic character of PDha endows the microgels with pH- and ionic strength-responsivity. Our experiments with model dyes and the antibacterial peptide LL-37 show that the electrostatic charges within the microgels also permit the accommodation of guest molecules within the network. Cell culture confirms that the microgels affect neither viability nor proliferation and are thus, biocompatible.
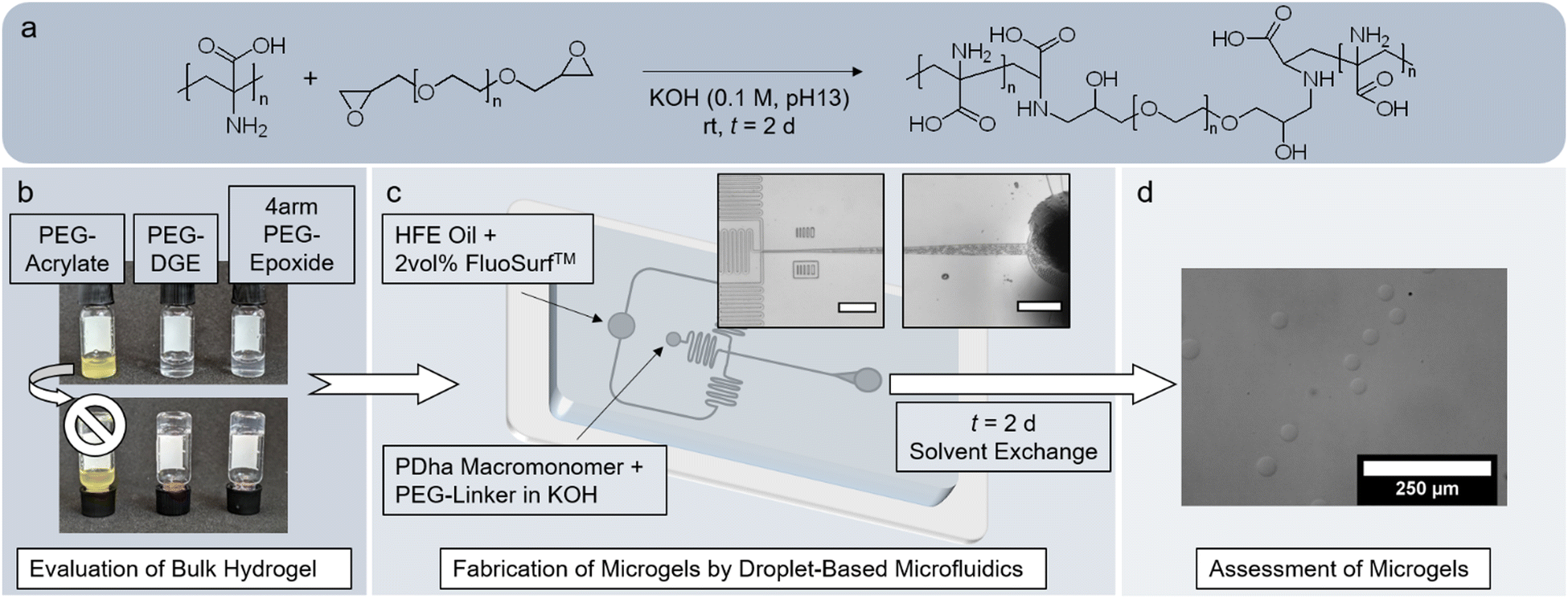 | ||
| Fig. 1 Schematic representation of our experimental concept. The underlying amine–epoxy crosslinking reaction is depicted in (a). First, gelation was tested in bulk with different types of PEG-based crosslinkers (b). Successful reaction conditions were then applied to a droplet-based microfluidics system (c). The channel design was varied to obtain differently sized microgels. Inset images show the crossflow junction and outlet of a microfluidic device with hchannel = 20 μm (scale bars: 200 μm). After t = 2 d, the product emulsion was purified by solvent exchange. The stable microspheres obtained (d) were analyzed in terms of comparability to the bulk hydrogels and with regard to their potential for biomedical applications (scale bar: 250 μm). | ||
Materials and methods
Materials
Poly(ethylene glycol) diacrylate (PEG-DA, Merck/Sigma Aldrich, Mn = 2 kDa), poly(ethylene glycol) diglycidylether (PEG-DGE, Merck/Sigma Aldrich, Mn = 2 kDa) and 4-arm poly(ethylene) glycol-epoxide (4arm-PEG-Ep, Biozol/Abbexa, Mn = 2 kDa, 95%) were used as received. PDha with an average degree of polymerization of Pi = 180 was obtained according to the procedure stated in literature.24 Briefly, tert-butoxycarbonylaminomethylacrylate (tBAMA) was first synthesized to prepare the prepolymer PtBAMA, which was subsequently deprotected to yield PDha (Fig. S1, ESI†). Prior to its use in gelation experiments, PDha was refluxed in methanol for t ≈ 2 h to remove any remaining impurities.If not indicated otherwise, HPLC-grade water (VWR, filtered at 0.2 μm) was used for the experiments. Potassium hydroxide solution (KOH, ChemSolute, 0.1 M, ≥ 85.0%) was used as received. Formic acid buffers (pH 3.0–4.4), succinic acid buffers (pH 5.0–6.0), phosphoric acid buffers (pH 6.5–8.0), tris(hydroxymethyl)aminomethane (TRIS) buffers (pH 8.5–8.9), and N-cyclohexyl-3-aminopropanesulfonic acid (CAPS) buffers (pH 9.6–11.0) were prepared according to literature48 and adjusted to an ionic strength of I = 10 mM.
For the fabrication of the microfluidic chip, a polydimethylsiloxane (PDMS) silicone elastomer kit (SYLGARD 184, Downsil) was used. As oil phase in droplet-based microfluidics, a mixture of hydrofluoroether (HFE) oil and neutral surfactant (FluoSurf™ 2 w/w% in Novec7500, Emulseo) was employed.
Sulforhodamine B (sRB, Merck/Sigma Aldrich, 75%, 1 mg) and rhodamine 6G (R-6G, Merck/Sigma Aldrich, 1 mg) were solved in V = 1 mL of either formic acid buffer (pH 3.0, I = 10 mM), water (pH ≈ 6.5), or CAPS buffer (pH 11.0, I = 10 mM) to yield solutions of c = 1 mg mL−1. Fluorescein isothiocyanate-labelled dextrans (FITC-dextran, Merck/Sigma Aldrich) were employed without modification. Fluorescamine (Merck/Sigma Aldrich, ≥98%) was solved in acetone to obtain a solution with c = 3 mg mL−1. The fluorescein-conjugated cathelicidin antimicrobial peptide LL-37 (FITC-LL-37, Biozol/Rockland Immunochemicals, lyophilized, 1 mg) was solved in V = 1 mL water before its use in experiments.
Synthesis of PDha-based bulk hydrogels
PDha was mixed with 0.1 M potassium hydroxide (KOH) solution and shaken overnight to obtain a solution with c(PDha) = 25 mg mL−1. PEG-DGE, PEGDA, and 4arm-PEG-Ep solutions, respectively, were prepared by solvation in 0.1 M KOH(aq) as specified in Table 1 and promptly mixed with PDha solution at a volume ratio of V(PDha)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :V(PEG crosslinker) = 3:1. After extensive mixing, the samples were allowed to rest for t = 2 d. The samples were purified by addition of water and exchange of the supernatant against fresh water at least thrice.
:V(PEG crosslinker) = 3:1. After extensive mixing, the samples were allowed to rest for t = 2 d. The samples were purified by addition of water and exchange of the supernatant against fresh water at least thrice.
| Samplea | c(PDha) [mg mL−1] | Crosslinker | c(PEG-epoxide) [mg mL−1] | V(PDha)/V(PEG-epoxide) | m%theo(PDha) [wt%] |
|---|---|---|---|---|---|
| a HG indicates bulk hydrogel samples. Superscript signs indicate which PEG epoxy species was used (2: bifunctional PEG-DGE, 4: tetrafunctional 4arm-PEG-Ep, DA: bifunctional PEGDA). The brackets specify the theoretical weight percentage of PDha in the hydrogel. | |||||
| HG2(13) | 25 | PEG-DGE (bifunctional) | 300 | 3:1 |
≈13 |
| HG2(16) | 25 | PEG-DGE (bifunctional) | 400 | 3:1 |
≈16 |
| HG2(20) | 25 | PEG-DGE (bifunctional) | 500 | 3:1 |
≈20 |
| HG4(16) | 25 | 4arm-PEG-Ep (tetrafunctional) | 400 | 3:1 |
≈16 |
| HGDA(16) | 25 | PEGDA (bifunctional) | 400 | 3:1 |
≈16 |
Fabrication of flow-focusing microfluidic device
PDMS-based microfluidic devices were fabricated by means of replica molding. Previously, a template design generated with the AutoCAD software (Autodesk) was printed onto a dark-field photomask (25000 dpi), which allowed for soft photolithography of the pattern onto an epoxy-based photoresist (SU-8) joined to a silicon wafer.49,50 After mixing PDMS with a curing agent (SYLGARD 184 elastomer kit) in a ratio of 10:1 and subsequent degassing, the mixture was poured into the master micromold and cured overnight at T = 60 °C. The fully crosslinked PDMS device was removed from the mold and punched with a biopsy puncher (MicrotoNano, dneedle = 0.75 mm) to generate the respective in- and outlets for the tubing. Both the PDMS device and a glass slide were thoroughly washed with water and isopropanol and then dried for t = 60 min at T = 60 °C in the oven before fixing the device onto the glass slide by plasma oxidation (Plasma TePla 100: Q = 30 mL min−1, P = 100 W, t = 40 s). Flushing with a silane solution (Aquapel®) rendered the microfluidic channels hydrophobic.
Fabrication of PDha-based microgels
Microgels were prepared in different microfluidic devices, the designs of which are depicted in Fig. S5 (ESI†). The microchannels all provided a rectangular crossflow junction with a uniform height of either 20 μm or 80 μm. The entire microfluidic station comprised PHD Ultra syringe pumps (Harvard Apparatus) and an inverted microscope (Motic AE2000, TED PELLA Inc.) equipped with a camera (Flea3, Point Grey). For the experiments, the hardened PDMS device was outfitted with fine bore polyethylene tubing (dinside = 0.38 mm, doutside = 1.10 mm) and connected to gastight Hamilton syringes (Hamilton) clamped in the syringe pumps. The continuous phase, hydrofluoroether oil, contained 2 w/w% of the neutral surfactant FluoSurf™, consisting of block copolymers from perfluoropolyether, poly(propylene oxide) and poly(ethylene oxide). For the dispersed phase, PDha solutions were first prepared in 0.1 M KOH(aq) by mixing overnight (c = 25 mg mL−1). The respective PEG epoxide solution (Table 2, V(PDha):V(PEG crosslinker) = 3:1) was either used as separate aqueous phase or mixed with the PDha solution before it was transferred to the microfluidic station. In the case of two separate aqueous solutions, the volumetric flow rate of each was set to Qwater = 75 μL min−1 and the oil phase was adjusted to Qoil = 300 μL min−1. In the case of a singular aqueous phase, the volumetric flow rate was set to Qwater = 200 μL min−1 for the dispersed phase and Qoil = 400 μL min−1 for the continuous phase. The product emulsion was collected in 2 mL vials and allowed to rest for at least t = 2 d to ensure complete gelation of the microgels.
| Samplea | Crosslinker | c(Crosslinker) [mg mL−1] | m%theo(PDha) [wt%] | m%1187(PDha)b [wt%] | m%1290(PDha)c [wt%] | No. of aqueous phases | h channel [μm] | D h [μm] |
|---|---|---|---|---|---|---|---|---|
| a MG indicates microgel samples fabricated with a microchannel height of 80 μm; mg indicates microgel samples fabricated with a microchannel height of 20 μm. Superscript signs indicate which PEG epoxy species was used (2: bifunctional PEG-DGE, 4: tetrafunctional 4arm-PEG-Ep). The brackets specify the theoretical weight percentage of PDha in the respective microgel. b Weight concentration of PDha within the respective microgel calculated by FT-IR spectroscopy exploiting the ratio between the C–N stretching vibration (1187 cm−1) of PDha and the C–O–C vibration (1100 cm−1) of the crosslink. c Weight concentration of PDha within the respective microgel calculated by FT-IR spectroscopy exploiting the ratio between the C–N valence vibration (1290 cm−1) of PDha and the C–O–C vibration (1100 cm−1) of the crosslink. d Hydrodynamic diameter of the respective microgel species as determined by optical microscopy in HPLC-grade water (average of 100 microgels measured). | ||||||||
| MG2(13) | PEG-DGE | 500 | 13 | — | — | 1 | 80 | — |
| MG2(16) | PEG-DGE | 400 | 16 | 28 | 30 | 1 | 80 | 297.4 ± 10.3 |
| MG2(20) | PEG-DGE | 300 | 20 | 40 | 40 | 1 | 80 | — |
| MG2(16)2w | PEG-DGE | 400 | 16 | 23 | 20 | 2 | 80 | 198.8 ± 63.4 |
| mg2(16) | PEG-DGE | 400 | 16 | 34 | 34 | 1 | 20 | 35.9 ± 2.5 |
| MG4(13) | 4arm-PEG-Ep | 500 | 13 | 17 | 30 | 1 | 80 | — |
| MG4(16) | 4arm-PEG-Ep | 400 | 16 | 23 | 33 | 1 | 80 | 207.9 ± 7.8 |
| MG4(20) | 4arm-PEG-Ep | 300 | 20 | 25 | 36 | 1 | 80 | — |
| mg4(16) | 4arm-PEG-Ep | 400 | 16 | 37 | 23 | 1 | 20 | 28.5 ± 1.6 |
| mg4(20) | 4arm-PEG-Ep | 300 | 20 | 34 | 30 | 1 | 20 | 33.2 ± 1.4 |
Purification of the formed microgels was achieved by successive solvent exchange. For that, the samples were allowed to sediment, the supernatant decanted and replaced with fresh solvent. In that way, the samples were washed thrice with HFE oil to remove the surfactant, twice with hexane to withdraw residual oil and at least five times with water to remove side products and unreacted polymer. As the microgels did not sediment as readily in water as they did in HFE oil and hexane, they were centrifuged (t = 30–60 s, 7500 rpm) prior to decantation and addition of fresh water.
Uptake experiments
To investigate the interaction between the microgels and charged compounds, V = 150 μL of the respective microgel dispersion was mixed with V = 100 μL of the particular dye solution before adding V = 750 μL of corresponding solvent (either formic acid buffer, water or CAPS buffer). The samples were allowed to equilibrate whilst shaking in the dark. After t = 1 d, the samples were purified by centrifugation and exchange of the solvent, wherein the corresponding solvent did not change to maintain appropriate pH value and ionic strength. To obtain a colourless supernatant, approximately 7–10 purification cycles were necessary.Before using the microgel samples to load them with FITC-LL-37 peptide, they were centrifuged (t = 30–60 s, 7500 rpm) so the supernatant could be removed and a concentrated microgel sediment was obtained. The volume of the sediment (Vsediment) was necessary to approximate the number of microgels (Nmicrogel) per sample using the respective hydrodynamic diameter (Dh) as stated in (1).
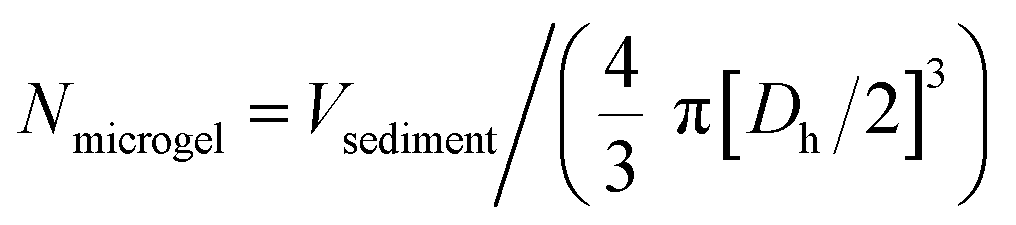 | (1) |
To each microgel sample, V = 50 μL of aqueous FITC-LL-37 solution (c = 1 mg mL−1) and V = 200 μL of water were added. The mixture was then left to shake in the dark for t = 1–2 d. Afterwards, the samples were filled with fresh water to V = 0.5 mL and purified by centrifugation (t = 1 min, 10000 rpm) before exchanging the supernatant. This procedure was repeated four times until the supernatant remained colourless and clear.
Size exclusion chromatography (SEC)
SEC traces of the prepolymer PtBAMA were measured in THF with an eluent flow rate of 1 mL min−1 using the Agilent 1260 Infinity system combined with a 1260 IsoPump and ALS autosampler (G1310B) and three consecutive PSS SDV columns (5 μm, 8 × 300 mm). The temperature of the column oven was adjusted to T = 40 °C. For calibration, PMMA standards (Mp = 2.2 M–800 Da) were used. Signals were detected with a 1260 DAD VL (G1329B) detector and a 1260 RID (G1315D) detector. The data was evaluated with the PSS WinGPC® software.1H nuclear magnetic resonance (NMR) spectroscopy
The 1H NMR spectrum of the reactive polymer PDha was measured with a Bruker Avance 400 (Bruker) instrument. The sample was solved in a D2O/NaOD mixture (7:3) and measured at 400 MHz. The spectrum was analysed with the software MestReNova.
Rheology
The viscoelasticity of the bulk hydrogels formed by amine–epoxy reaction was assessed using a discovery HR-3 hybrid rheometer (TA Instruments) equipped with a parallel plate geometry (20 mm). All experiments were carried out at 25 °C and parameters were adjusted to the linear viscoelastic region (LVR) of the hydrogels. To prevent water evaporation and consequent desiccation of the hydrogels, a solvent trap containing water was employed and the setup sealed with a lid. In general, bulk hydrogels were prepared as described and left to gelate for at least t = 2 d before transferring them onto the rheology plate. Hydrogels prepared with the bifunctional crosslinker PEG-DGE (HG2(X)) were measured as follows: After a time-dependent measurement at a fixed frequency of ω ≈ 6.28 rad s−1 (1 Hz) and 0.5% strain to ascertain complete gelation, a frequency-dependent measurement at a fixed strain of 0.5% was conducted, in which the frequency was increased from ω = 0.05 rad s−1 to ω = 100 rad s−1. In a subsequent measurement, the frequency was fixed to ω ≈ 6.28 rad s−1 (1 Hz) and the strain increased from 0.1–1000%. The sample HG4(16) was analysed analogously but due to the smaller LVR, the frequency was fixed in time- and strain-dependent measurements to ω ≈ 3.14 rad s−1 (0.5 Hz) and the strain was fixed in time- and frequency-dependent measurements to 0.2%. As the electrostatic interactions between amine groups and carboxylic acid groups were assumed to contribute to the network elasticity, the self-regeneration of the hydrogel HG2(16) was tested by repeatedly shearing the sample at high strain outside the LVR (t = 2 min, ω ≈ 6.28 rad s−1, 500%), followed by relaxation within LVR (t = 2 min, ω ≈ 6.28 rad s−1, 0.5%). To establish the duration of the gelation, a time-dependent measurement at a fixed frequency of ω = 6.28 rad s−1 (1 Hz) and 0.5% strain was conducted using a 40 mm cone plate geometry (2°). For that, V = 0.45 mL of PDha solution (c = 25 mg mL−1 in KOH, 0.1 M) was mixed with V = 0.15 mL of the respective PEG solution (c = 400 mg mL−1 in KOH, 0.1 M) on plate to form HG2(16) and HG4(16) in situ. After a pre-shear period to thoroughly mix the polymers, the measurement was performed for t = 6 h at T = 25 °C. The data was evaluated using the TRIOS software (TA Instruments).Optical microscopy
Morphology, size and responsivity were analysed either with the Olympus CKX53 inverted microscope (Olympus Corp.) or with the Motic AE2000 (TED PELLA Inc.) outfitted with a camera. To image the size and morphology of the microgels, V = 20 μL of the dilute dispersion were placed onto a glass slide and placed in the microscope. The test series concerning pH- or ionic strength-responsivity were conducted by pipetting the concentrated microgel sediment into a 96-well plate (V = 1 μL per well for the small microgels (mg), V = 5 μL per well for the large microgels (MG)) and adding either buffer solutions of varying pH value or sodium chloride salt solutions of varying salt concentration to the wells (V = 100 μL per well). Microscopic imaging was conducted after at least t = 2 h of equilibrating. For each data point, the hydrodynamic diameter of at least 50–100 microgels was measured and averaged. The diameters of the individual microgels were measured by use of the software ImageJ.Fourier-transform infrared (FT-IR) spectroscopy
Prior to FT-IR analysis, both microgel and hydrogel samples were lyophilized to obtain solid flakes. FT-IR spectra of dry samples were recorded with the Spectrum 3 FT-IR (PerkinElmer) equipped with a GladiATR diamond frontier. The measurements were conducted at room temperature in a spectral range of 4000–400 cm−1 with a spectral resolution of 4 cm−1 and a sample scan time of 4 scans. The data was baseline-corrected with the related software Spectrum (version 10.7.1, PerkinElmer) and normalized by the highest ordinate value.Confocal laser scanning microscopy (CLSM)
To acquire brightfield and fluorescence images, a Leica TCS SP8 (Leica) confocal laser scanning microscope was used. The images were processed with the software LAS X and evaluated using the ImageJ software.For the fluorometric assay of primary amine groups, V = 200 μL of the respective microgel dispersion were placed onto an object slide and thoroughly mixed with V = 10 μL of a fluorescamine solution (c = 3 mg mL−1 in acetone). Microscopic analysis was performed using a diode 405 UV laser (3% intensity, λexc = 405 nm) with a HyD detector (gain: 300%, λem = 455–500 nm, pinhole: 1.00) for fluorescence and a PMT trans detector for brightfield images.
The uptake of fluorescent dyes was investigated by applying V = 20 μL of the respective microgel sample onto an object slide and recording fluorescence and brightfield images. The fluorophore sRB was excited with a diode pumped solid state (DPSS) laser (3–4% intensity, λexc = 561 nm) and the resulting fluorescence detected with a HyD detector (gain: 400%, λem = 575–620 nm, pinhole: 1.00), while the fluorophore R-6G was excited with the argon laser (10%, 1% intensity, λexc = 514 nm) and its fluorescence detected with a HyD detector (gain: 50%, λem = 530–570 nm, pinhole: 1.00). Brightfield images were taken with the PMT trans detector in both cases.
The diffusion of FITC-dextrans of varying molecular weight into the microgels was assessed by redispersing V = 50 μL of concentrated microgel sediment in the respective FITC-dextran solutions (c = 1 mg mL−1; Mw = 4 kDa, 40 kDa and 150 kDa). The samples were investigated with an argon laser (10%, 1% intensity, λexc = 488 nm) combined with a HyD detector (λem = 500–550 nm, pinhole: 1.00) and PMT trans detector. For the permeability studies, the gain was adjusted so that the fluorescence intensity of the surrounding medium was approximately 100 RFU for all samples.
The adsorption of FITC-labelled LL-37 peptide into the microgels was verified using analogous instrument settings and a HyD gain of 10–40%.
Nanoindentation
The effective Young's modulus (E-modulus) of the large microgels (MG) was analysed with a Pavone mechanical screening platform (Optics11Life). To guarantee adequate adherence of the microgel samples, a 12-well plate was first treated with poly(L-lysine) (PLL) solution (c = 0.01% in H2O, V = 200 μL per well). After manual shaking to ensure coverage of the entire surface of each well, the solution was allowed to evaporate and dry overnight. Afterwards, V = 20 μL of the respective microgel dispersion were added to the PLL-treated wells and allowed to adhere to the surface for t = 10 min. The wells were subsequently rinsed with HPLC-grade water to remove any free-floating microgels before filling the wells with V = 1 mL HPLC-grade water and placing them into the device. The nanoindentation experiments were carried out at room temperature using a cantilever probe with a spherical indenter tip (tip radius: 10.0 μm, stiffness: 0.470 N m−1, indentation depth: 1.6 μm). For each sample, 10–15 different microgels were measured. The data was evaluated with the software Dataviewer2.0 by fitting the Hertzian contact model to each indentation curve and thus, deriving the effective Young's modulus.51Micropipette aspiration (MPA)
For MPA experiments, micropipettes from borosilicate glass capillaries (doutside = 1.50 mm, dinside = 1.17 mm) were drawn out with the P-1000 micropipette puller (Sutter Instruments) before cutting them with a heating wire under an optical microscope (MicroData Instruments Inc.). Utilizing an axopatch holder with suction port and wire (A–M systems), the pipette was linked to the Patchserver Main Unit (Multi channel systems). The PatchServer and PatchMaster software provided pressure regulation during measurements. Microgels subjected to aspiration were visualized using a LEICA DMi8 microscope, with subsequent image analysis conducted in the LAS X software. For each sample, five individual microgels were aspirated. Ten different pressure values were applied ranging from −5 mbar to −50 mbar.To determine the effective Young's modulus (E-modulus, E), homogeneity of the sample was assumed and a simple continuum-medium model (2) used.52 The force F exerted on the microgel was calculated from the applied pressure Δp and the pipette radius (RP = 32.5 μm) according to eqn (3) before plotting it against the normalized aspirated length Δx of the microgel. The term ϕP, which exhibits a partial dependency on the thickness of the pipette wall, is customarily assumed to be ϕP = 2.1. Consequently, the effective Young's modulus was calculated using the slope s of the linear fit (4).
| Δp/E = (2πΔx)/(3ϕPRP) | (2) |
| F = Δp·π·RP2 | (3) |
| E = s(3ϕP)/(2π2RP) | (4) |
Electrophoretic light scattering (ELS)
To determine the surface charge of the microgels, electrophoretic light scattering measurements were performed with a Zetasizer Ultra (Malvern Panalytical Ltd) using a capillary DTS1070 cell. For these experiments, V = 500 μL of the concentrated microgel sediment of MG2(16) were diluted with V = 500 μL of formic acid buffer (pH 3.0, I = 10 mM), phosphate buffer (pH 7.0, I = 10 mM) or CAPS buffer (pH 11.0, I = 10 mM), respectively. Measurements were conducted in folded capillary zeta cell cuvettes (DTS1070) at 22.5 °C with a scattering angle of 12.5° (forward scattering) and an effective voltage of 150 V. For the data analysis, the refractive index of the microgels was assumed to be 1.46 due to the high fraction of PEG within the microgels.53 The data was assessed with the corresponding software ZS Xplorer (Malvern Panalytical).Fluorescence intensity measurements
A Tecan Infinite M1000 PRO microplate reader (Tecan Group AG) was employed to measure the fluorescence intensity of the samples. To generate a standard fluorescence curve, the aqueous FITC-LL-37 solution (c = 1 mg mL−1) was first diluted with water to a concentration of c = 0.12 mg mL−1. This stock solution was then used as the starting point for a serial dilution using a 96-well plate with a sample volume of V = 100 μL per well. Water was used as a reference. The average fluorescence per well was subsequently measured with the microplate reader (λexc = 485 nm with bandwidth: 5 nm, gain: 75%, λem = 535 nm with bandwidth: 5 nm) and compared to the fluorescence of the loaded microgels and the fluorescence of the supernatant solution. Each dilution and sample, respectively, was measured in triplicate and averaged.Cell culture experiments
Normal human dermal fibroblasts (NHDFs) were cultured in tissue culture flasks with Dulbecco's modified eagle's medium (DMEM; Gibco) enriched with 10% fetal calf serum (FCS; Biowest) and 1% antibiotics micotic solution (ABM; Gibco). The NHDF culture proceeded in humidified environment at T = 37 °C and 5% CO2. The NHDFs were employed in passage 7–8 to determine cell viability and metabolic activity.The respective PDha-based microgel samples were washed twice with DMEM containing 10% FCS and 1% ABM. Subsequently, the microgel dispersions were diluted to c ≈ 300 microgels per μL for the small microgels (mg) or c ≈ 20 microgels per μL for the large microgels (MG). Prior to their use in cell experiments, all microgel dispersions were sterilized by UV irradiation (t = 30 min).
Cell viability tests
Biocompatibility tests were performed by adding the microgels (V = 100 μL per sample) to 96-well plates before seeding the NHDFs on top. As positive control, NHDFs were cultivated in an identical volume of DMEM containing 10% FCS and 1% ABM (without microgels). After t = 1 d and t = 7 d of culture, respectively, live/dead staining was performed by means of the LIVE/DEAD™ Cell Imaging Kit (Invitrogen). As specified by the manufacturer, a 1× working solution of the live (488/515 nm)/dead (570/602 nm) reagent was prepared by dilution in DMEM enriched with 10% FCS and 1% ABM. For the negative control experiments, NHDF samples without microgels were deliberately killed by exposure to ethanol (t = 30 s). Each sample was incubated with the reagent solution for t = 15 min at room temperature before imaging with a PerkinElmer Opera Phenix Plus microscope equipped with a 10×/N.A. 0.3 air objective. Cell viability was evaluated using the corresponding software of the microscope. All experiments were performed in triplicate.Metabolic cell activity assay
To measure the metabolic cell activity, samples were prepared by incubating the microgels (V = 100 μL per sample) with NHDFs. For the positive control experiments, NHDFs were cultured in an identical volume of DMEM enriched with 10% FCS and 1% ABM. After t = 1 d, 3 d, and 7 d in culture, a resazurin-based alamarBlue assay (Invitrogen) was carried out. According to the manufacturer's specifications, the resazurin-reagent was diluted 1:10 with DMEM containing 10% FCS and 1% ABM, and incubated with the respective samples for t = 2 h. The fluorescence intensity was measured with an Infinite M Nano+ plate reader from Tecan applying an excitation wavelength of 530 nm and an emission wavelength of 590 nm.
Statistical analysis
For the graphs, mean values were plotted with error bars indicating standard deviation. A one-way analysis of variance (ANOVA) was carried out with OriginPro 2022b software. For the comparison of mean value pairs by Tukey's range test, a p-value of < 0.05 was considered significant (p* < 0.05, p** < 0.01, p*** < 0.001).Results and discussion
Our investigation was inspired by bulk hydrogels based on PDha and PEG, which are crosslinked by the ring-opening SN reaction of the terminal epoxy groups of functionalized PEG with the primary amines in the PDha chain. Çeper et al. optimized the reaction conditions to synthesize such PDhA-based bulk hydrogels and investigated their properties in detail.39,40 The objective of our work was the fabrication of analogous PDha-based microgels to study their properties and evaluate their potential for biomedical applications.First, we tested the bulk gelation under reaction conditions suitable for use in PDMS-based droplet-based microfluidics. Due to the poor solubility of PDha (Fig. S2, ESI†),24,25 the reaction was carried out in aqueous KOH (0.1 M, pH 13). As Çeper et al. had found the bulk hydrogels to be self-supporting only when PEG-DGE with Mn = 2000 Da was used and claimed that gelation proceeded best with Pi(PDha) = 180,40 we directly employed the corresponding reactive polymers. In addition to the sample with PEG-DGE (HG2(16)), we also conducted bulk gelation trials with PEGDA (HGDA(16)) and 4arm-PEG-Ep (HG4(16)), the former to open up an alternative reaction route via aza-Michael addition, the latter to create a more finely meshed gel. To facilitate the realization of the reaction in droplet-based microfluidics, the gelation had to proceed at room temperature (rt). In contrast to previous research,39,40 gelation therefore took t = 2 d before the inverted vial test indicated successful gel formation (Fig. 1(b)). While the sample HGDA(16) did not yield a stable gel and was thus discarded for follow-up experiments, both samples containing epoxide-based PEG crosslinker formed pliable transparent bulk hydrogels.
Rheological analysis was performed for PEG-DGE crosslinked bulk hydrogels with varying amounts of crosslinker and a 4arm-PEG-Ep crosslinked bulk hydrogel (Table 1) 2 d after initial mixing. In all samples, the elastic modulus (G′) is larger than the viscous modulus (G′′) in the linear viscoelastic region (LVR) (Fig. S3(a), ESI†). Outside of the LVR, the hydrogels display a distinct G′′ maximum. The breakdown of the gel is hence characterized by the rupture of an increasing number of individual bonds (micro cracks) until a macro crack forms, which results in viscous flow of the sample. In HG2 samples, the crossover point shifts to larger strains with decreasing amounts of PEG-DGE, which suggests that not the crosslinker but PDha is reliable for the mitigation of the micro cracks. Possibly, the amine and carboxylic acid functions form dynamic intra- and intermolecular hydrogen bonds, limiting the movement of the broken chains.40
In time sweeps, a constant and parallel progression of the G′ and G′′ plots with G′ > G′′ implies that the hydrogels are fully cured and in kinetic equilibrium (Fig. S3(b), ESI†). Frequency sweeps show that G′ exceeds G′′ even towards low frequencies (Fig. S3(c), ESI†). As tan(δ) < 0.1 for all samples, the elastic, more solid-like characteristics of the hydrogels predominate. Interestingly, G′ of our PEG-DGE crosslinked bulk hydrogels is significantly lower compared to G′ of previously synthesized PDha-based hydrogels.39,40 The lower reaction temperature (rt vs. T = 60 °C) and thus, longer reaction time probably lead to increased hydrolysis of the terminal epoxy groups of the crosslinker and thus, to a lower yield of the crosslinking reaction.54 Increasing the crosslinker content in PEG-DGE crosslinked hydrogels did not necessarily lead to a higher elastic modulus. The reason for this may be that the amine groups in PDha contribute significantly to elasticity through hydrogen bonding and that PEG microdomains impair overall stability. Repeated application of high strain (outside of LVR) followed by relaxation at low strain (within the LVR) demonstrated complete regeneration in HG2(16) (Fig. S3(d), ESI†). Therefore, it is likely that the mechanical properties of the hydrogel are dominated by non-covalent dynamic interactions, such as electrostatic interaction and hydrogen bonding. This hypothesis could be verified in the future by varying the degree of crosslinking: if non-covalent interactions are indeed decisive, the behaviour of the hydrogels should remain mostly unaffected.
Carrying out post-polymerization crosslinking reactions in emulsified droplets has generally been shown to generate microgels.55–58 However, batch emulsion polymerization techniques often suffer from a broad size distribution of the microgels produced, which in turn renders characterization difficult. Therefore, we opted for a lithographic droplet-based microfluidic approach. Fluorinated oil mixed with the neutral surfactant FluoSurf™ was used as continuous phase, as it does not introduce additional charges to the system and ensures long-term stability of the product emulsion. As rheological analysis of the bulk hydrogels showed that the network strength did not improve significantly below m%(PDha) = 16 wt%, we focussed on the compositions with m%(PDha) ≥ 16 wt% and adopted the reaction conditions of the bulk hydrogel synthesis in the microfluidic approach as well. Analogously, the product emulsions were left to gel for 2 d after microfluidic droplet generation before they were purified by solvent exchange (Fig. 1(c) and (d)). The ratio between the flow rate of the oil and the flow rate of the aqueous phase(s) was set to 2:1 to avoid jetting of the dispersed phase and still achieve a sufficient flow of the more viscous water phase(s). To fabricate microgels of varying size, two different microfluidic devices with a channel height of 20 μm and 80 μm, respectively, were employed (Fig. S5, ESI†). Droplet formation occurred at the crossflow junction, where the shear force between oil and water phase prompted the generation of dispersed aqueous droplets (Fig. 1(c)).
The use of two separate water phases (one with the PEG-based crosslinker, one with the reactive polymer PDha) proved disadvantageous because the viscous aqueous solutions did not mix well within the channel. This resulted in a low PDha content and thus, structural instability of the microgels (Table 2 and Fig. S6, ESI†). Instead, the aqueous solutions were mixed prior to their transfer into the respective syringe and employed as a singular dispersed phase, since the gelation process took several hours (Fig. S4, ESI†).
Stable microgels were obtained that had a spherical morphology and appeared almost transparent in aqueous solution. Compared to the original height of the channel, the microgels had a significantly larger hydrodynamic diameter Dh, though this effect was more pronounced in the larger MG2(16) and MG4(16) samples than in the microgels prepared within the 20 μm channel. Since the aqueous droplets inside the microfluidic channel are elongated and become round only when exiting the microfluidic device, a comparatively larger hydrodynamic diameter of the microgels is to be expected. The size of the PDha-based microgels, however, corresponds to ≥150% of the initial height of the channel. On the one hand, the large hydrodynamic diameter can be attributed to the strong water affinity of PDha and thus, the high degree of swelling of the gel networks.40 On the other hand, a larger initial size of the microgels seems to further the swelling, presumably, because the pores within the MG2(16) and MG4(16) networks are larger and hence, promote the diffusion of water. Microgels containing lesser amounts of PEG-based crosslinker, e.g. mg4(20) (Table 2), are larger in size compared to microgels including more crosslinker, e.g. mg4(16) (Table 2). A higher fraction of terminal epoxy groups results in more crosslinking sites, so that the entire network becomes more tightly meshed and can therefore no longer swell as much as a less crosslinked system. The microgels with a lower overall crosslinker content are, however, structurally less stable than the more highly crosslinked samples, and suffer more strongly from droplet coalescence during the 2-day resting phase. The implementation of tetrafunctional 4arm-PEG-Ep as opposed to bifunctional PEG-DGE also leads to a denser, more crosslinked network which in turn results in smaller microgels with a narrower size distribution (Table 2 and Fig. S7, ESI†).
After purification and lyophilisation, the composition of the microgels was examined by FT-IR spectroscopy (Fig. 2). The successful integration of the PEG-based crosslinker is clearly confirmed by the signal of the C–O–C vibration (1100 cm−1) and additionally verified by the C–H valence vibration (2870 cm−1).39,59 The sharp epoxy signal at 1059 cm−1, clearly visible in both PEG-DGE and 4arm-PEG-Ep reference, disappears in the gel samples.60 While the acidic functions of PDha are clearly visible due to their C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration at 1685 cm−1, the amine groups are hidden as shoulder signals at 1600 cm−1 (N–H deformation), 1555 cm−1 (R–NH3+ deformation), and 1290 cm−1 (C–N valence vibration).39,59 Only the weak signal at 1187 cm−1 attributed to C–N stretching motions manifests as a stand-alone peak.61 By comparing the C–O–C signal (1100 cm−1) with the C–N valence signal (1290 cm−1) and the C–N stretching signal (1187 cm−1), respectively, a rough estimation of the weight fraction of PDha incorporated in the microgels was provided (Table 2 and Fig. S8, ESI†). Similar to the observations made for PDha-based hydrogels,40 the experimentally determined amount of PDha within the microgels is significantly higher than the theoretical value (Table 2). This may be explained by the fact that some of the epoxy groups of the respective PEG-based crosslinker are hydrolysed and therefore do not participate in the crosslinking reaction. Instead, the polymer strands are removed during purification, at least if they are not retained in the microgels due to chain entanglements.
O vibration at 1685 cm−1, the amine groups are hidden as shoulder signals at 1600 cm−1 (N–H deformation), 1555 cm−1 (R–NH3+ deformation), and 1290 cm−1 (C–N valence vibration).39,59 Only the weak signal at 1187 cm−1 attributed to C–N stretching motions manifests as a stand-alone peak.61 By comparing the C–O–C signal (1100 cm−1) with the C–N valence signal (1290 cm−1) and the C–N stretching signal (1187 cm−1), respectively, a rough estimation of the weight fraction of PDha incorporated in the microgels was provided (Table 2 and Fig. S8, ESI†). Similar to the observations made for PDha-based hydrogels,40 the experimentally determined amount of PDha within the microgels is significantly higher than the theoretical value (Table 2). This may be explained by the fact that some of the epoxy groups of the respective PEG-based crosslinker are hydrolysed and therefore do not participate in the crosslinking reaction. Instead, the polymer strands are removed during purification, at least if they are not retained in the microgels due to chain entanglements.
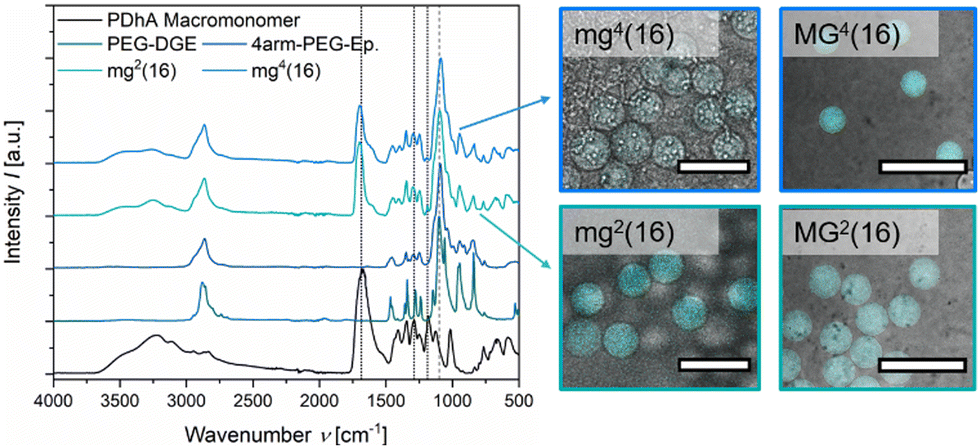 | ||
| Fig. 2 FT-IR spectra of reference materials (PDha, PEG-DGE, 4arm-PEG-Ep) and microgel samples mg2(16) and mg4(16). The corresponding fluorometric assays with fluorescamine are depicted as CLSM overlay images on the right. Scale bars for mg2(16) and mg4(16) indicate 50 μm, scale bars for MG2(16) and MG4(16) indicate 500 μm. | ||
In addition to FT-IR spectroscopy, we performed a proof-of-principle fluorometric assay to verify the presence of residual primary amine groups in the microgels. The organic spiro compound fluorescamine reacts with primary amine moieties to form a fluorescent diaryl-2-hydroxypyrrolinone derivative.62,63 Since fluorescamine only emits fluorescence after derivatization, a fluorescent signal within microgel samples after addition of fluorescamine can be considered as evidence of primary amine functions. Fig. 2 demonstrates that mg2(16), mg4(16), MG2(16), and MG4(16) all display fluorescence in the CLSM overlay images (λem = 455–500 nm). Decreased intensity in mg4(16) indicates a lower concentration of the fluorophore within the respective microgel. A possible reason for this might be that the smaller size of the initial droplet and the multiple arms of the crosslinker lead to a higher conversion of the amine–epoxy reaction, resulting in lower numbers of primary amine functions. However, smaller pores within the gel network may also limit the diffusion of fluorescamine, either because of the high hydrophilicity within the microgel network or as a consequence of spatial limitations.
For the larger microgel samples MG2(16) and MG4(16), we were able to evaluate the spatial limitations within the microgels by exploiting the correlation between different molecular weights of FITC-dextran and their corresponding hydrodynamic radius as published by Ambati et al.64 After equilibration of the respective microgel sample in an aqueous FITC-dextran solution of either 4 kDa, 40 kDa, or 150 kDa, the mixtures were subjected to CLSM. Relating the fluorescence intensity outside the microgels to the intensity within the microgels allows for an assessment regarding the ability of the respective FITC-dextran to permeate the network. Our results show that only FITC-dextran with M = 4 kDa (estimated Rh = 1.3 nm64) partially diffuses into the microgels whereas FITC-dextrans of higher molecular weights barely penetrate the microgels (Fig. S10, ESI†). While ca. 40% of 4 kDa FITC-dextran diffuse into MG2(16), the permeability is decreased for MG4(16), with only 26% of 4 kDa FITC-dextran passing into the network. This substantiates our claim that the multiple arms of the crosslinker lead to a more densely crosslinked system. Given that the mesh size of the microgels dictates their diffusivity, the permeability of FITC-dextran is poorer in MG4(16) than in MG2(16). On the basis of these experiments, it is, however, not possible to determine the exact pore size of the microgels, since electrostatic and hydrophobic interactions also influence diffusivity.
Regarding a potential biomedical use case, the mechanical properties of the microgels are decisive for their applicability. In tissue engineering, stem cell lineage specification may be governed by the softness of the surrounding matrix.65 When considering a use in tissue engineering or in vivo, the characteristics of the target organ are often mimicked by the malleability of the gel network.66,67 Therefore, we exemplarily investigated the mechanical properties of the microgel samples MG2(16) and MG4(16) by nanoindentation and MPA. Smaller microgel samples could, unfortunately, not be evaluated by these methods as the small size caused the cantilever tip to slide off the microgels in nanoindentation and made it difficult to visualize the microgel–water interface during MPA experiments. The microscopic images of the aspirated MG2(16) and MG4(16) microgels as well as the resulting linear fits are summarized in Fig. S9 (ESI†).
Fig. 3 graphically shows the effective Young's modulus of the respective microgels as measured by nanoindentation and MPA analysis. As was already discovered for the bulk hydrogels in rheology, the microgels appear to be very soft with E < 10 kPa, which is comparative to the E-modulus of muscle tissue.65 Generally, a high softness correlates with large mesh sizes, which is at odds with the previous observations concerning permeability. A potential explanation for this phenomenon may be that crosslinking within the microgels is only partially based on covalent bonds. Intra-colloidal electrostatic attraction and synergistic hydrogen bonds might allow the microgels to adapt dynamically to mechanical stress, thus rationalizing the observed softness. Both in nanoindentation and MPA experiments, MG4(16) demonstrates a higher E-modulus than MG2(16). MPA analysis provided slightly larger moduli, most likely due to adhesive forces between the capillary wall and the microgels tested. Overall, the results from nanoindentation and MPA measurements are in good agreement.
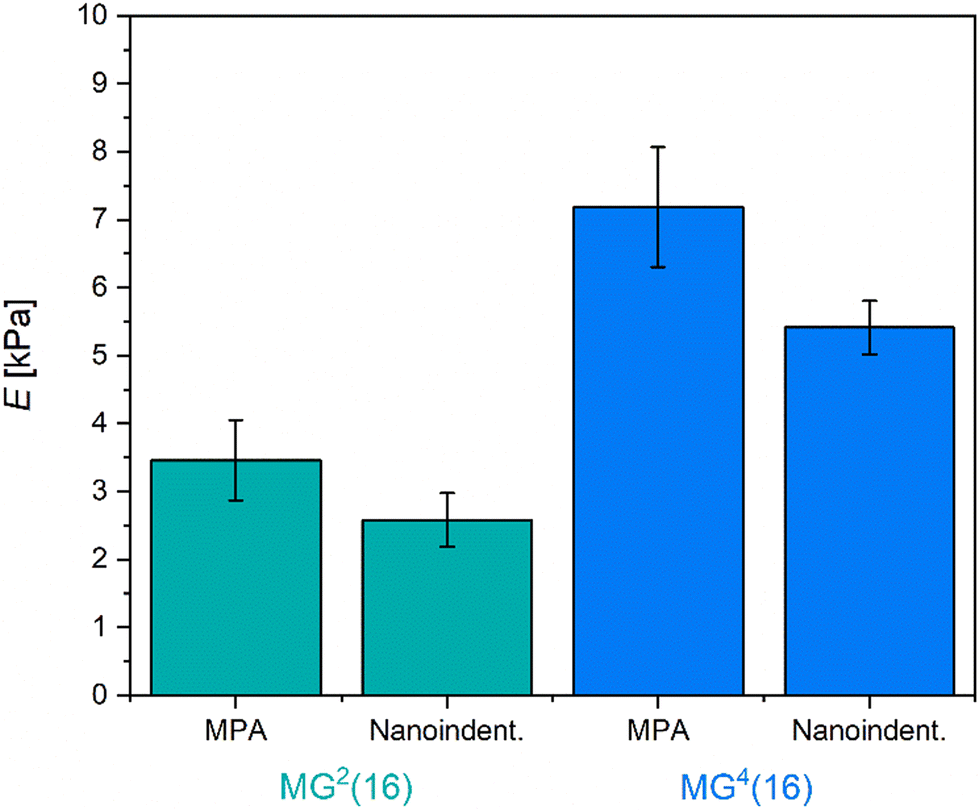 | ||
| Fig. 3 Effective Young's modulus E as determined for large microgels MG2(16) and MG4(16) by MPA and nanoindentation. All microgels are relatively soft, comparative to the E-modulus of muscle tissue. | ||
Possibly the most interesting feature of the PDha-based microgels is their ability to switch their overall net charge depending on the surrounding pH-value. We therefore studied the response of the microgels to changes in pH value and ionic strength (Fig. 4). For pH-dependent measurements, the microgels were redispersed in buffer solutions with a fixed ionic strength of I = 10 mM to keep the effects caused by the salt concentration as low as possible whilst maintaining a constant pH value. As MG2(16) was expected to exhibit the most prominent charge alteration, we examined the respective microgels by electrophoretic light scattering even though in these measurements a measurement error caused by the gravity-induced sedimentation of the microgels must be taken into account. Still, the results in Fig. 4(a) demonstrate that the surface charge of the microgels is slightly positive in acidic environment (pH 3) and becomes gradually more negative with increasing pH value. At pH 3, i.e. around the pKA value of the carboxylic acid functions,68 both acidic and basic moieties are protonated so that the microgels are predominantly positively charged. Above the pKA value of the carboxylic acid, the respective functions get deprotonated, resulting in a negative charge that compensates the positive charges from the R–NH3+ groups. As a certain percentage of the amino moieties are sacrificed to fabricate the network, the ratio between amino and carboxyl groups is not equimolar but shifted in favour of the carboxyl functions. Thus, the isoelectric point in the microgels is also shifted towards lower pH values when compared to the reactive polymer PDha.24 At higher pH values, the amino groups start to deprotonate so that the overall charge decreases even further.
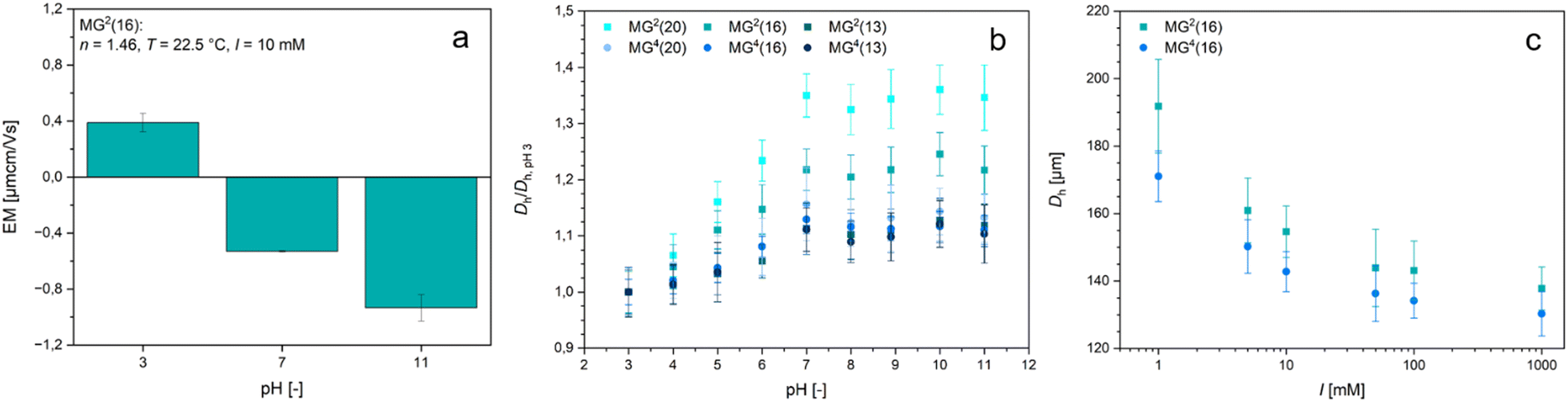 | ||
| Fig. 4 Investigation of the responsivity of the PDha-based microgels. The electrophoretic mobility of MG2(16) was measured depending on the pH-value of the environment (a). The swelling ratio of the hydrodynamic diameter of the microgel samples was evaluated as a function of pH-value (b), and the hydrodynamic diameter of MG2(16) and MG4(16) was measured as a function of ionic strength (c). | ||
Following our verification of positive and negative charges within the microgel, we studied the swelling behaviour of the samples depending on the pH value (Fig. 4(b) and Fig. S11, ESI†). All samples show an initial increase in size up to a pH value of approximately pH 7 before the hydrodynamic diameter plateaus. Usually, this curve progression is typical for polyelectrolyte microgels with weakly acidic functions.69,70 As the ratio between amino and carboxyl groups is not equimolar, we hypothesize that the influence of the protonated, i.e. positively charged amino groups is compensated for by charge- and partial-charge dependent interactions. Similar considerations have already been disclosed in the context of the published bulk hydrogels from PDha.40 Compared to their size at pH 3, the size of the microgels fabricated with 4arm-PEG-Ep crosslinker only increased by approximately 10%, whereas the microgels containing PEG-DGE grew larger (Fig. 4(b) and Fig. S11, ESI†). Although this observation is valid for both the larger (MG) and the smaller microgels (mg), the trend is more apparent in the MG samples (Fig. 4(b)), as the comparatively large standard deviation in mg2(16) and mg4(16) makes the interpretation of the data more difficult (Fig. S11(c), ESI†). This observation is congruent with the fact that the tighter the crosslinking is, the lesser the ability of the gel to swell. As evident in Fig. 4(b), a decrease in crosslinker in the PEG-DGE-crosslinked microgels leads to a more pronounced swelling (35% increase in MG2(20) vis-à-vis 22% increase in MG2(16) and 12% increase in MG2(13)) but for samples crosslinked with 4arm-PEG-Ep, the amount of crosslinker does not lead to a significant change in swelling ratio. This suggests that maximum crosslinking is already achieved in the sample MG4(20) (and potentially, MG2(13)). Most likely, the degree of crosslinking is limited by the mobility of the PEG-based crosslinker within the network, which is more strongly impaired for the multi-arm compound than for the bifunctional molecule.
Since the microgels prepared with intermediate amounts of PEG-based crosslinker (mgx(16) and MGx(16)) proved the most robust against mechanical forces exerted during centrifugation while simultaneously exhibiting responsivity indicative of the respective microgel series, they were selected as representative samples for all following experiments. To evaluate the effect of ionic strength on the size of the PDha-based microgels, we redispersed the microgels in aqueous sodium chloride solutions of varying NaCl concentration. With an increase in ionic strength, the hydrodynamic diameter of the microgels decreased (Fig. 4(c) and Fig. S11(d), ESI†). In contrast to the pH-responsive swelling, this ionic strength-induced decrease in size is independent of the crosslinker species used. The reduction of size with rising salt concentration is usually typical for weak polyelectrolyte microgels in their charged state: here, the addition of salt leads to the screening of like-charges that induce electrostatic repulsion and thus, swelling. Consequently, the swelling degree of polyelectrolyte networks decreases with an increase in screening ions.71–73 Based on our electrophoretic mobility measurements of the surface charge (Fig. 4(a)) and the pH value of HPLC-grade water (≈6.5), we assume that in pure water, the PDha-based microgels are predominantly negatively charged. Internal salt bridges with carboxylic acid functions and hydrogen bond formation may compensate for the influence of the basic amino groups. Thus, the addition of salt results in a decrease in electrostatic repulsion by screening the carboxylate functions, which in turn leads to deswelling of the microgels.
The polyzwitterionic character of PDha and the polyampholyte properties of the gel network allow for the pH-regulated uptake and release of charged (bio-) molecules. Corresponding findings have already been reported with other, chemically different PDha-based bulk hydrogel systems by demonstrating the ad- and desorption of positively charged methylene blue and negatively charged methyl blue or acid orange 7, respectively.30,38 For our studies on PDha-based microgels, we decided to perform similar experiments with fluorescent dyes instead, since the visualization of conventional dyes is challenging due to the low concentration of dye within individual microgels. As model for positively charged compounds, we used rhodamine 6G (R-6G), and as model for negatively charged compounds, sulforhodamine B (sRB). These fluorescent dyes were selected because their fluorescence was shown to be independent of the pH value in a range from pH 3 to pH 10, and because both R-6G and sRB are highly water-soluble.74,75 The fluorescent dyes were dissolved in either formic acid buffer (pH 3), water or CAPS buffer (pH 11) and mixed with microgels to enable electrostatically-driven uptake. Subsequently, excess dye was removed by allowing the microgels to sediment and replacing the supernatant several times with the respective fresh solvent.
The samples were then subjected to CLSM to verify pH-dependent adsorption. Fig. 5 exemplarily shows the resulting images for MG2(16) and a scheme of the assumed underlying mechanism. In formic acid buffer, i.e. pH 3, MG2(16) is slightly positively charged (Fig. 4(a)) due to the protonation of the amino moieties. Thus, negatively charged sRB is adsorbed into the microgels and not retrieved even by extensive washing, whereas positively charged R-6G is completely removed from the sample by solvent exchange (Fig. 5, left). In water, MG2(16) presumably has a polyampholyte character, with carboxylate groups outweighing the positive charges. Under these conditions, a significant uptake of R-6G can be observed, whereas hardly any sRB remains in the network (Fig. 5, middle). The remaining amount of sRB could either stem from interaction with individual amino groups, which remain available due to the low ionic strength of the environment, or could be caused by other interactions, such as dipole–dipole or hydrophobic interactions. In CAPS buffer, i.e. pH 11, the adsorption of sRB into the microgels is minimal (Fig. 5, lower right). Experiments with R-6G in highly basic environments remained inconclusive as the R-6G dye formed aggregates and could not be removed from the supernatant even after numerous washing cycles (Fig. 5, upper right).
 | ||
| Fig. 5 Investigation into the electrostatically-driven adsorption of model dyes R-6G (+) and sRB (−) into PDha-based microgels and scheme of the proposed mechanism. CLSM images show that R-6G is not retained in the microgels at pH 3 but readily taken up in pure water, whereas sRB is remains within the microgel at pH 3 but not in pure water or at pH 11. For R-6G, solvation in basic buffers induced aggregation so that corresponding samples could not be analysed. Scale bars indicate 100 μm. | ||
Addition of formic acid buffer (pH 3) to MG2(16) loaded with R-6G in water induced a significant decrease in fluorescence intensity, whereas only a slight decrease in fluorescence intensity could be observed, when MG2(16), previously loaded with sRB under acidic conditions, was treated with basic CAPS buffer (pH 11) (Fig. S12, ESI†). The desorption of cationic R-6G was fuelled by a change both in pH value and ionic strength: the acidic pH value of the buffer was below the pKA of the carboxylic acid, thus inducing protonation of the carboxylate groups, while the higher salt concentration (Ibuffer > Iwater) caused charge screening and osmotic pressure effects. In contrast, the addition of CAPS buffer to the formerly acidic microgel dispersion containing sRB did not change ionic strength, and the resultant pH value was only raised to intermediate values as formic acid and basic buffer were mixed. Nevertheless, the reduction of fluorescence intensity also suggests a release of the model dye in this sample.
To give a more specific example of the biomedical potential of the microgels, their loading with a bioactive substance was tested. We decided to examine targeted peptide transport with the PDha-based microgels as the concept is studied not only in the context of therapy but also in surface modification.76–78 As model substance, FITC-labelled LL-37 (FITC-LL-37), a human antimicrobial peptide derived from the cathelicidin hCAP-18,79 was selected. LL-37 comprises 37 amino acids, with multiple lysine and arginine residues contributing to the overall cationic charge of the peptide. While it adopts a random coil conformation in water, it occurs as α-helix in specific salt solutions.80 We decided to use already labelled LL-37 for our experiments to easily visualize the uptake by CLSM. As with the model dyes, microgel samples and peptide were mixed to enable uptake by electrostatic attraction before excess FITC-LL-37 was separated by decanting and replacing the supernatant.
Subsequently, the fluorescence intensity was measured both in the samples and in the total collected supernatant solution. A fluorescence calibration curve determined for FITC-LL-37 (Fig. S13, ESI†) then allowed for the deduction of the respective concentration of FITC-LL-37 in the microgels and in the corresponding supernatant. Comparison of the resulting total concentration with the amount of peptide originally implemented showed reasonable consistency of the values (Fig. 6(a)). By approximating a perfectly spherical morphology of the microgels, we specified their volume and thus, could calculate the number of microgels per sample using their respective average hydrodynamic diameter (1). This allowed for the deduction that mg2(16) adsorbed approximately 1.0 pg FITC-LL-37 per microgel, whereas mg4(16) took up about 0.5 pg (Fig. 6(b)). For the larger microgels, the loading was increased (Fig. 6(c)) to 166.0 pg for MG2(16) and 102.9 pg for MG4(16). The difference in uptake between the smaller (mg) and the larger (MG) samples corresponds roughly to their volume ratio. As was to be expected from the permeability assay (Fig. S9, ESI†), the microgels containing bifunctional PEG-DGE crosslinker adsorb substantially higher amounts of FITC-LL-37 than the microgels containing tetrafunctional 4arm-PEG-Ep. Interestingly, the amount of peptide accommodated is about one and a half to two times higher in mg2(16)/MG2(16) than in mg4(16)/MG4(16). This matches the observation in the pH-responsive swelling, where the PEG-DGE crosslinked samples mg2(16)/MG2(16) demonstrate a swelling of 20% whereas 4arm-PEG-Ep crosslinked samples mg4(16)/MG4(16) exhibit an increase in size of only 10%. Follow-up experiments are intended to fine-tune the crosslinking within the microgels and optimize uptake. Additionally, future research should determine whether the LL-37 integrated into the gels presents in its active, α-helix conformation (as it has in previous studies)77 or remains in a random coil.
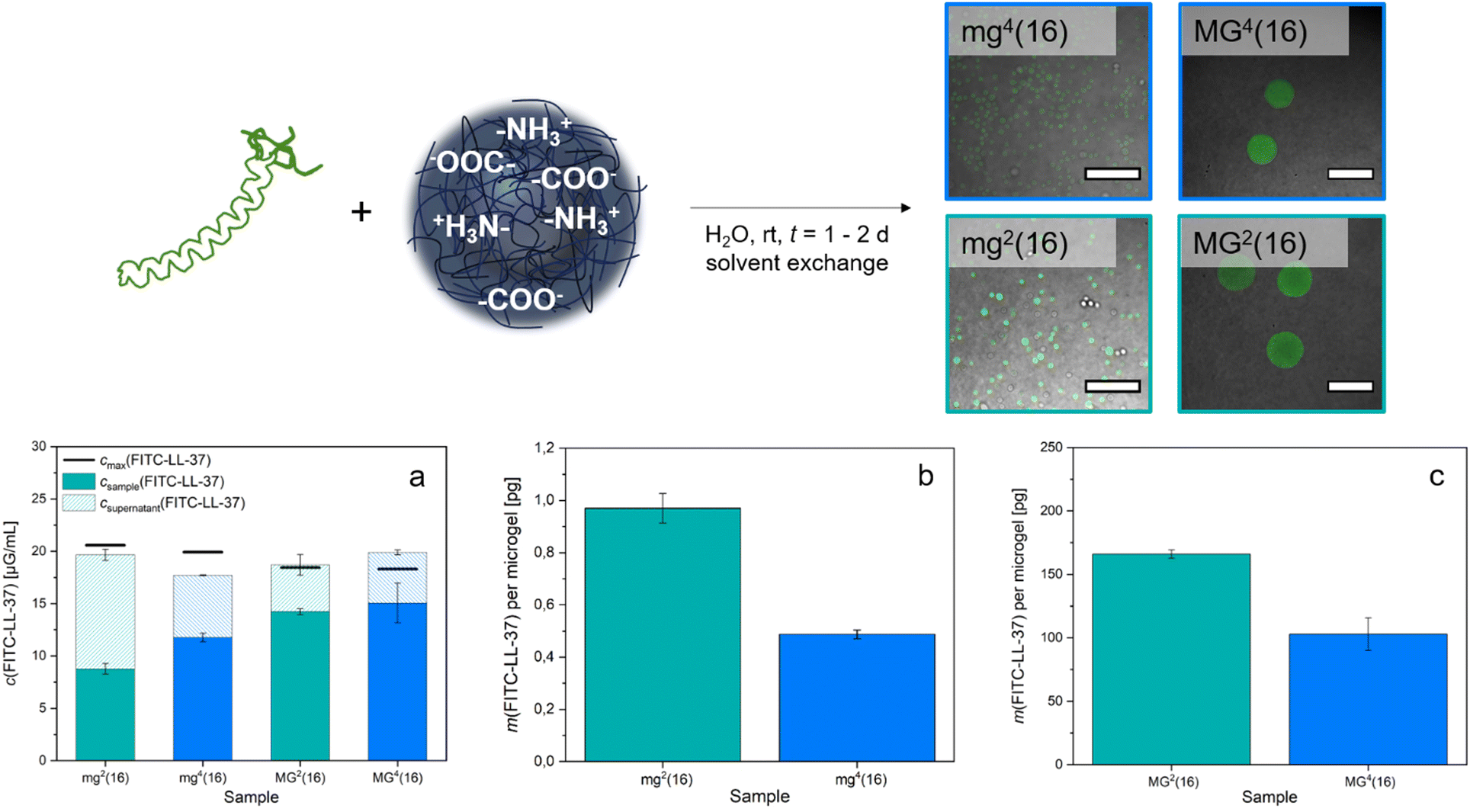 | ||
| Fig. 6 Insights into the uptake of FITC-LL-37 into PDha-based microgels. Peptide and microgels were mixed in water at rt for t = 2 d before the supernatant was decanted and replaced four times to remove any excess FITC-LL-37 (top). CLSM overlay images verify the successful adsorption of the labelled peptide into microgels of various size and crosslinker type (top). Fluorescence intensity measurements performed with a plate reader show adequate agreement between the initial concentration in the samples (cmax) and the total concentration of FITC-LL-37 measured in supernatant and in microgel dispersion (a). The amount of FITC-LL-37 adsorbed into the microgels depends on both the crosslinker species and the size of the gel network ((b), (c)). The use of the bifunctional crosslinker species almost doubles the amount of adsorbed FITC-LL-37 compared to 4arm-PEG-Ep ((b) and (c)). The difference between the FITC-LL-37 uptake within the small (b) and the large (c) samples corresponds approximately to the difference in volume. | ||
A biomedical application of the presented PDha-based gel networks is only feasible if the samples are highly biocompatible. Previous cell tests conducted with linear PDha or PDha-coated nanoparticles and human brain microvascular endothelial cells showed no apparent short-term cytotoxicity.34,37 We assessed biocompatibility by cultivating the respective microgel samples with normal human dermal fibroblasts (NHDFs) for t = 24 h and t = 7 d, respectively, before performing a live/dead assay to monitor cell viability (Fig. 7). Although the microgels took up the dead reagent (BOBO™-3 iodide), likely due to electrostatic attraction to the carboxylate groups, the fluorescence images do not show a substantial effect on cell viability induced by the microgels. Extending the duration of the experiment (t = 7 d) led to similar results as t = 1 d of culture (Fig. 7(a) and (b)). Fluorescence-based evaluation of cell proliferation indicated that cell growth in the samples with microgels progressed identically to the positive control (Fig. 7(c)). Our in vitro experiments suggest that the PDha-based microgels are fully biotolerant and non-cytotoxic.
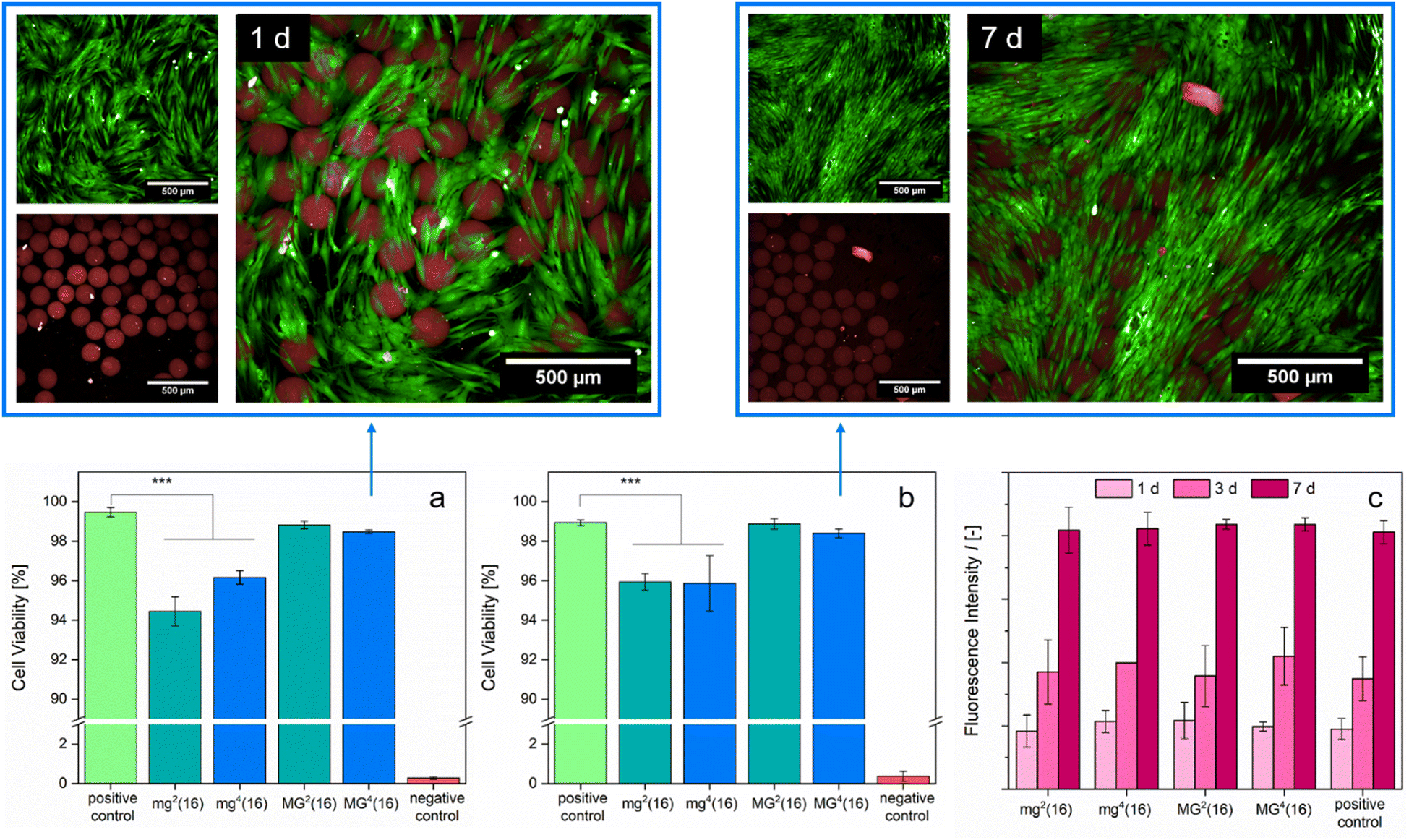 | ||
| Fig. 7 Biocompatibility assays for PDha-based microgels. Cell viability was tested by live/dead staining after NHDFs were cultivated with different microgel samples for t = 1 d (a) or t = 7 d (b). CLSM fluorescence images show live/dead staining of MG4(16) after t = 1 d (upper left) and t = 7 d (upper right), with green indicating alive and red indicating dead cells and microgels. Scale bars denote 500 μm. Cell proliferation was evaluated with a fluorescence assay after t = 1 d, t = 3 d and t = 7 d (c). All data points were obtained by performing triplicates (p* < 0.05, p** < 0.01, p*** < 0.001), with error bars denoting standard deviations. The PDha-based microgels did not exert any harmful effect onto the cells. | ||
Conclusions
PDha-based microgels were successfully fabricated by adapting the reaction conditions of bulk gelation to a droplet-based microfluidic method, where shear forces between immiscible organic and aqueous phase led to the formation of templating droplets. The high hydrophilicity of the polyzwitterionic PDha and the PEG-based crosslinker caused pronounced swelling of the microgels in water. We found that a larger amount of crosslinker and a higher number of crosslinking functions per crosslinker, respectively, led to a decrease in size in the swollen state as well as a narrower size distribution within the samples. FT-IR spectroscopy verified gelation and allowed for a rough assessment of the quantitative composition of the gels. A fluorometric assay with fluorescamine provided evidence for primary amine groups within the microgels, showing that the respective moieties are available for covalent coupling reactions. The microgels were comparatively soft, with the bifunctional crosslinker PEG-DGE leading to a slightly higher permeability and a slightly lower E-modulus than the tetrafunctional 4arm-PEG-Ep crosslinker. Based on the mechanical properties and the measurements concerning pH- and ionic strength-responsive swelling, we deduced that—in addition to covalent bonds and salt bridging—further interactions, such as dipole–dipole interactions caused dynamic crosslinking within the polyampholyte microgels. This allows for the pH-regulated adsorption and desorption of model compounds, such as R-6G, sRB and FITC-LL-37. The amount of peptide accommodated depends both on the crosslinker species and on the size of the microgels. These preliminary experiments may prove valuable for more in-depth research concerning antimicrobial surface coatings. The biocompatibility of the PDha-PEG microgels was substantiated by cell culture with NHDFs showing no cytotoxicity in either cell viability or cell proliferation assays in a period from 1 d to 7 d.As of yet, the microgels presented in this study are too large for drug delivery systems, particularly in human medicine. However, as shown, the microfluidic approach allows for facile optimization of the morphology and size, so that oral administration of drug-loaded microgels is conceivable in the future. The discovery that the E-modulus of the PDha-based microgels is in the myogenic range may also prove interesting for potential muscle tissue engineering approaches. The established electrostatic and dynamic interactions could be exploited to form switchable microporous annealed particle hydrogels, which have been found advantageous for cell spreading and proliferation.81
Author contributions
Hannah F. Mathews: conceptualization, methodology, validation, formal analysis, data curation, investigation, writing – original draft, writing – review & editing, visualization. Tolga Çeper: conceptualization, resources (PDha), writing – review & editing. Tobias Speen: investigation, formal analysis, writing – review & editing. Céline Bastard: investigation, formal analysis, validation, writing – review & editing. Selin Bulut: investigation, formal analysis, validation, writing – review & editing. Maria I. Pieper: investigation, formal analysis, validation, writing – review & editing. Felix H. Schacher: conceptualization, supervision, resources, funding acquisition, writing – review & editing. Laura De Laporte: supervision, resources, funding acquisition. Andrij Pich: conceptualization, supervision, resources, funding acquisition, writing – review & editing. All authors contributed to this manuscript and have approved of the final version.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts or competing financial interests.Acknowledgements
The authors acknowledge funding from the Deutsche Forschungsgemeinschaft (DFG) within the collaborative research center (CRC) SFB 985 “Functional Microgels and Microgel Systems” (project ID: 191948804, project A03). The DFG is further gratefully acknowledged for financial support within TRR 234 “CataLight” (project ID: 364549901, projects B03 and Z02). T. C. thanks the Deutscher Akademischer Austausch Dienst (DAAD) for a doctoral research fellowship.Notes and references
- H. Ye, L. Wang, R. Huang, R. Su, B. Liu, W. Qi and Z. He, ACS Appl. Mater. Interfaces, 2015, 7(40), 22448–22457 CrossRef CAS PubMed.
- S. Sarkar, A. Guha, T. N. Narayanan and J. Mondal, J. Phys. Chem. Lett., 2022, 13(24), 5660–5668 CrossRef CAS PubMed.
- A. Ilyas, Y. S. Wijayasinghe, I. Khan, N. M. El Samaloty, M. Adnan, T. A. Dar, N. K. Poddar, L. R. Singh, H. Sharma and S. Khan, Front. Mol. Biosci., 2022, 9, 1–21 CrossRef PubMed.
- M. S. Bretscher and M. C. Raff, Nature, 1975, 258, 43–49 CrossRef CAS PubMed.
- E. Deplazes, B. D. Tafalla, C. G. Cranfield and A. Garcia, J. Phys. Chem. Lett., 2020, 11(15), 6353–6358 CrossRef CAS PubMed.
- A. Ali, T. Dou, A. P. Holman, A. Hung, L. Osborne, D. Pickett, A. Rodriguez, K. Zhaliazka and D. Kurouski, Biophys. Chem., 2024, 306, 107174 CrossRef CAS PubMed.
- W. Wei, J. Yu, C. Broomell, J. N. Israelachvili and J. H. Waite, J. Am. Chem. Soc., 2013, 135(1), 377–383 CrossRef CAS PubMed.
- H. Erguven, L. Wang, B. Gutierrez, A. H. Beaven, A. J. Sodt and E. C. Izgu, JACS Au, 2024, 4(5), 1841–1853 CrossRef CAS PubMed.
- Z. Huang, J. Chen, R. Li, S. Shi and Y.-K. Gong, Prog. Org. Coat., 2023, 182, 107636 CrossRef CAS.
- S. Zhang, S.-H. Qian, G. Zhao, Q.-C. Pan, R. Song, T. Zhang, S. Zhang, Z. Geng, J. Huang, L. Wang and B. Zhu, Chem. Eng. J., 2024, 483, 148980 CrossRef CAS.
- P. K. Singh, V. K. Singh and M. Singh, e-Polymers, 2007, 7(1), 1–34 Search PubMed.
- A. Laschewsky, Polymers, 2014, 6(5), 1544–1601 CrossRef.
- A. B. Lowe and C. L. McCormick, Chem. Rev., 2002, 102(11), 4177–4190 CrossRef CAS PubMed.
- J. B. Schlenoff, Langmuir, 2014, 30(32), 9625–9636 CrossRef CAS PubMed.
- P. Mary, D. D. Bendejacq, M.-P. Labeau and P. Dupuis, J. Phys. Chem. B, 2007, 111(27), 7767–7777 CrossRef CAS PubMed.
- A. E. Neitzel, G. X. De Hoe and M. V. Tirrell, Curr. Opin. Solid State Mater. Sci., 2021, 25(2), 100897 CrossRef CAS.
- L. D. Blackman, P. A. Gunatillake, P. Cass and K. E. S. Locock, Chem. Soc. Rev., 2019, 48(3), 757–770 RSC.
- S. Paschke and K. Lienkamp, ACS Appl. Polym. Mater., 2020, 2(2), 129–151 CrossRef CAS.
- S. Racovita, M.-A. Trofin, D. F. Loghin, M.-M. Zaharia, F. Bucatariu, M. Mihai and S. Vasiliu, Int. J. Mol. Sci., 2021, 22(17), 9321 CrossRef CAS PubMed.
- B. Li, P. Jain, J. Ma, J. K. Smith, Z. Yuan, H.-C. Hung, Y. He, X. Lin, K. Wu, J. Pfaendtner and S. Jiang, Sci. Adv., 2019, 5(6), eaaw9562 CrossRef CAS PubMed.
- Z. Feng, X. Feng and X. Lu, Environ. Sci. Technol., 2023, 57(18), 7298–7308 CrossRef CAS PubMed.
- W. Wang, J. Yang, E. Zhang, Y. Lu and Z. Cao, J. Mater. Chem. B, 2017, 5(44), 8676–8680 RSC.
- M. N. Leiske, B. G. De Geest and R. Hoogenboom, Bioact. Mater., 2023, 24, 524–534 CAS.
- U. Günther, L. V. Sigolaeva, D. V. Pergushov and F. H. Schacher, Macromol. Chem. Phys., 2013, 214(19), 2202–2212 CrossRef.
- J. Kruse, P. Biehl and F. H. Schacher, Macromol. Rapid Commun., 2019, 40(10), 1800857 CrossRef PubMed.
- R. S. Asquith, K. L. Gardner and K. W. Yeung, J. Polym. Sci., Polym. Chem. Ed., 1978, 16(12), 3275–3280 CrossRef CAS.
- M. Billing and F. H. Schacher, Macromolecules, 2016, 49(10), 3696–3705 CrossRef CAS.
- M. Billing, G. Festag, P. Bellstedt and F. H. Schacher, Polym. Chem., 2017, 8(5), 936–945 RSC.
- P. J. Mons, N. Fribiczer, K. Kowalczuk, P. Poudel, S. Seiffert and F. H. Schacher, Polym. Chem., 2022, 13(29), 4298–4308 RSC.
- K. Kowalczuk, P. J. Mons, H. F. Ulrich, V. D. Wegner, J. C. Brendel, A. S. Mosig and F. H. Schacher, Macromol. Biosci., 2024, 24(2), 2300230 CrossRef CAS PubMed.
- J. B. Max, D. V. Pergushov, L. V. Sigolaeva and F. H. Schacher, Polym. Chem., 2019, 10, 3006–3019 RSC.
- J. B. Max, K. Kowalczuk, M. Köhler, C. Neumann, F. Pielenz, L. V. Sigolaeva, D. V. Pergushov, A. Turchanin, F. Langenhorst and F. H. Schacher, Macromolecules, 2020, 53(11), 4511–4523 CrossRef CAS.
- F. Nagler, C. Schiller, C. Kropf and F. H. Schacher, ACS Appl. Mater. Interfaces, 2022, 14(50), 56087–56096 CrossRef CAS PubMed.
- M. von der Lühe, U. Günther, A. Weidner, C. Gräfe, J. H. Clement, S. Dutz and F. H. Schacher, RSC Adv., 2015, 5, 31920–31929 RSC.
- P. Biehl, M. von der Lühe, S. Dutz and F. Schacher, Polymers, 2018, 10(1), 91 CrossRef PubMed.
- M. von der Lühe, A. Weidner, S. Dutz and F. H. Schacher, ACS Appl. Nano Mater., 2018, 1(1), 232–244 CrossRef.
- C. Gräfe, M. von der Lühe, A. Weidner, P. Globig, J. H. Clement, S. Dutz and F. H. Schacher, Nanotechnology, 2019, 30(26), 265707 CrossRef PubMed.
- T. Çeper, S. W. Mohotti, L. X. Lange and F. H. Schacher, Org. Mater., 2024, 06(01), 1–11 CrossRef.
- T. Çeper, A. Nabiyan, C. Neumann, A. Turchanin and F. H. Schacher, ACS Appl. Polym. Mater., 2023, 5(8), 6493–6503 CrossRef.
- T. Çeper, M. Langer, N. Vashistha, B. Dietzek-Ivanšić, C. Streb, S. Rau and F. H. Schacher, Macromol. Rapid Commun., 2024, 45(5), 2300448 CrossRef PubMed.
- T. Ceper, D. Costabel, D. Kowalczyk, K. Peneva and F. H. Schacher, ACS Appl. Mater. Interfaces, 2024, 16(19), 24796–24805 CrossRef CAS PubMed.
- G. Agrawal and R. Agrawal, Polymers, 2018, 10(4), 418 CrossRef PubMed.
- J. P. Newsom, K. A. Payne and M. D. Krebs, Acta Biomater., 2019, 88, 32–41 CrossRef CAS PubMed.
- Y. Kittel, A. J. C. Kuehne and L. De Laporte, Adv. Healthcare Mater., 2022, 11(6), 2101989 CrossRef CAS PubMed.
- H. Qian, K. Wang, M. Lv, C. Zhao, H. Wang, S. Wen, D. Huang, W. Chen and Y. Zhong, J. Controlled Release, 2022, 343, 492–505 CrossRef CAS PubMed.
- A. Sinclair, M. B. O’Kelly, T. Bai, H. Hung, P. Jain and S. Jiang, Adv. Mater., 2018, 30(39), 1803087 CrossRef PubMed.
- P. Saha, R. Ganguly, X. Li, R. Das, N. K. Singha and A. Pich, Macromol. Rapid Commun., 2021, 42(13), 2100112 CrossRef CAS PubMed.
- D. D. Perrin, Aust. J. Chem., 1963, 16(4), 572–578 CrossRef CAS.
- L. P. B. Guerzoni, J. C. Rose, D. B. Gehlen, A. Jans, T. Haraszti, M. Wessling, A. J. C. Kuehne and L. De Laporte, Small, 2019, 15(20), 1900692 CrossRef PubMed.
- S. H. Jung, S. Bulut, L. P. B. Busca Guerzoni, D. Günther, S. Braun, L. De Laporte and A. Pich, J. Colloid Interface Sci., 2022, 617, 409–421 CrossRef CAS PubMed.
- H. Hertz, J. fur Reine Angew. Math., 1882, 92, 156–171 CrossRef.
- B. González-Bermúdez, G. V. Guinea and G. R. Plaza, Biophys. J., 2019, 116(4), 587–594 CrossRef PubMed.
- M. Choi, J. W. Choi, S. Kim, S. Nizamoglu, S. K. Hahn and S. H. Yun, Nat. Photonics, 2013, 7(12), 987–994 CrossRef CAS PubMed.
- M. Thirumalaikumar, Org. Prep. Proced. Int., 2022, 54(1), 1–39 CrossRef CAS.
- B. Schulte, A. Walther, H. Keul and M. Möller, Macromolecules, 2014, 47(5), 1633–1645 CrossRef CAS.
- B. Schulte, K. Rahimi, H. Keul, D. E. Demco, A. Walther and M. Möller, Soft Matter, 2015, 11(5), 943–953 RSC.
- A. Gruber, L. Navarro and D. Klinger, Adv. Mater. Interfaces, 2020, 7(5), 1901676 CrossRef CAS.
- C. Hu, W. Xu, C. M. Conrads, J. Wu and A. Pich, J. Colloid Interface Sci., 2021, 582(B), 1075–1084 CrossRef CAS PubMed.
- M. Hesse, H. Meier, B. Zeeh, S. Bienz, L. Bigler and T. Fox, Spektroskopische Methoden in der organischen Chemie, Georg Thieme Verlag KG, Stuttgart, 8th edn, 2012 Search PubMed.
- H.-L. Guo, X.-F. Wang, Q.-Y. Qian, F.-B. Wang and X.-H. Xia, ACS Nano, 2009, 3(9), 2653–2659 CrossRef CAS PubMed.
- F. Navarro-Pardo, G. Martínez-Barrera, A. Martínez-Hernández, V. Castaño, J. Rivera-Armenta, F. Medellín-Rodríguez and C. Velasco-Santos, Materials, 2013, 6(8), 3494–3513 CrossRef PubMed.
- S. De Bernardo, M. Weigele, V. Toome, K. Manhart, W. Leimgruber, P. Böhlen, S. Stein and S. Udenfriend, Arch. Biochem. Biophys., 1974, 163(1), 390–399 CrossRef CAS PubMed.
- S. M. Derayea and E. Samir, Microchem. J., 2020, 156, 104835 CrossRef CAS.
- J. Ambati, C. S. Canakis, J. W. Miller, E. S. Gragoudas, A. Edwards, D. J. Weissgold, I. Kim, F. C. Delori and A. P. Adamis, Invest. Ophthalmol. Visual Sci., 2000, 41(5), 1181–1185 CAS.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126(4), 677–689 CrossRef CAS PubMed.
- D. L. Butler, S. A. Goldstein and F. Guilak, J. Biomech. Eng., 2000, 122(6), 570–575 CrossRef CAS PubMed.
- K. Xue, X. Wang, P. W. Yong, D. J. Young, Y. Wu, Z. Li and X. J. Loh, Adv. Ther., 2019, 2(1), 1800088 CrossRef.
- R. Lunkad, P. Biehl, A. Murmiliuk, P. M. Blanco, P. Mons, M. Štěpánek, F. H. Schacher and P. Košovan, Macromolecules, 2022, 55(17), 7775–7784 CrossRef CAS.
- S. Zhou and B. Chu, J. Phys. Chem. B, 1998, 102(8), 1364–1371 CrossRef CAS.
- M. Das and E. Kumacheva, Colloid Polym. Sci., 2006, 284(10), 1073–1084 CrossRef CAS.
- E. Y. Kramarenko, A. R. Khokhlov and K. Yoshikawa, Macromolecules, 1997, 30(11), 3383–3388 CrossRef CAS.
- J. B. Sokoloff, J. Chem. Phys., 2015, 142, 234903 CrossRef CAS PubMed.
- D. Sean, J. Landsgesell and C. Holm, Gels, 2017, 4(1), 2 CrossRef PubMed.
- J. Coppeta and C. Rogers, Exp. Fluids, 1998, 25, 1–15 CrossRef CAS.
- W. B. Swanson, M. Durdan, M. Eberle, S. Woodbury, A. Mauser, J. Gregory, B. Zhang, D. Niemann, J. Herremans, P. X. Ma, J. Lahann, M. Weivoda, Y. Mishina and C. F. Greineder, RSC Chem. Biol., 2022, 3(6), 748–764 RSC.
- H. Bysell, R. Månsson, P. Hansson and M. Malmsten, Adv. Drug Delivery Rev., 2011, 63(13), 1172–1185 CrossRef CAS PubMed.
- L. Nyström, A. A. Strömstedt, A. Schmidtchen and M. Malmsten, Biomacromolecules, 2018, 19(8), 3456–3466 CrossRef PubMed.
- B. C. Borro, R. Nordström and M. Malmsten, Colloids Surf., B, 2020, 187, 110835 CrossRef CAS PubMed.
- G. H. Gudmundsson, B. Agerberth, J. Odeberg, T. Bergman, B. Olsson and R. Salcedo, Eur. J. Biochem., 1996, 238(2), 325–332 CrossRef CAS PubMed.
- J. Johansson, G. H. Gudmundsson, M. E. Rottenberg, K. D. Berndt and B. Agerberth, J. Biol. Chem., 1998, 273(6), 3718–3724 CrossRef CAS PubMed.
- L. Riley, L. Schirmer and T. Segura, Curr. Opin. Biotechnol, 2019, 60, 1–8 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm00676c |
| This journal is © The Royal Society of Chemistry 2024 |