
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceShape transformations in peptide–DNA coacervates driven by enzyme-catalyzed deacetylation†
Merlijn H. I.
van Haren
 ,
Nienke S.
Helmers
,
Luuk
Verploegen
,
Viveca A. C.
Beckers
and
Evan
Spruijt
*
,
Nienke S.
Helmers
,
Luuk
Verploegen
,
Viveca A. C.
Beckers
and
Evan
Spruijt
*
Institute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6523 AJ Nijmegen, The Netherlands. E-mail: e.spruijt@science.ru.nl
First published on 11th November 2024
Abstract
Biomolecular condensates formed by liquid–liquid phase separation (LLPS) are important organizers of biochemistry in living cells. Condensate formation can be dynamically regulated, for example, by protein binding or enzymatic processes. However, how enzymatic reactions can influence condensate shape and control shape transformations is less well understood. Here, we design a model condensate that can be formed by the enzymatic deacetylation of a small peptide by sirtuin-3 in the presence of DNA. Interestingly, upon nucleation condensates initially form gel-like aggregates that gradually transform into spherical droplets, displaying fusion and wetting. This process is governed by sirtuin-3 concentration, as more enzyme results in a faster aggregate-to-liquid transformation of the condensates. The counterintuitive transformation of gel-like to liquid-like condensates with increasing interaction strength between the peptide and DNA is recapitulated by forming condensates with different peptides and nucleic acids at increasing salt concentrations. Close to the critical point where coacervates dissolve, gel-like aggregates are formed with short double stranded DNA, but not with single stranded DNA or weakly binding peptides, even though the coacervate salt resistance is similar. At lower salt concentrations the interaction strength increases, and spherical, liquid-like condensates are formed. We attribute this behavior to bending of the DNA by oppositely charged peptides, which becomes stronger as the system moves further into the two-phase region. Overall, this work shows that enzymes can induce shape transformations of condensates and that condensate material properties do not necessarily reveal their stability.
Introduction
Life requires the presence of physically separated compartments that isolate molecules and reactions. Cells achieve this with organelles that are separated from their environment by lipid bilayers, or by formation of phase separated biomolecular condensates (BMCs).1 Examples of these condensates include stress granules, Cajal bodies and the nucleolus, which are often formed by proteins and nucleic acids.2 BMCs offer advantages compared to membrane-bound organelles: they are dynamic and their formation and dissolution can be regulated by enzymes or local changes under environmental conditions, which could be beneficial for cellular processes, such as the sequestration of proteins or substrates.3 Enzymes are key regulators of BMC formation, as they adjust post-translational modifications (PTMs) of the proteins forming BMCs.4Coacervates are frequently used as in vitro models that mimic specific aspects of BMCs found in living cells, as the liquid–liquid phase separation (LLPS) underlying coacervate formation is also responsible for the formation of BMCs.5,6 Coacervates can be formed by proteins and nucleic acids based on associative and segregative interactions. By studying enzymatic reactions in coacervates, processes such as nucleation or growth of BMCs can be characterized.7 Various enzymatic reactions have been used to induce coacervation, such as phosphorylation of peptides and nucleotides or the polymerization of RNA.8–12 These enzymatic reactions increase the associative interactions between the positively and negatively charged biomolecules, leading to a more stable coacervate.3,13
Previously, it has been shown how phosphorylation and dephosphorylation of serine residues can dissolve and induce formation of peptide/RNA coacervates.8 The phosphorylation of ADP has also been used to induce phase separation with a lysine-rich elastin-like protein and create actively growing droplets, in which the growth speed could be tuned by the enzyme concentration.10 In this case, coacervate droplets were spherical from the moment they could be observed and remained spherical and liquid-like throughout the reaction. Another enzymatic reaction that gives rise to phase separation, the polymerization of RNA, was found to give rise to transient nonspherical coacervates that slowly relaxed to spherical coacervates.12 It was shown that the enzyme activity was correlated with the extent to which coacervates were driven out of equilibrium and the deviation from sphericity. However, the unknown coupling between enzymatic activity and the viscoelastic material properties of the coacervates precluded a simple explanation for this observation.12
In addition to phosphorylation, another important PTM in biochemistry that can potentially impact the phase separation propensity of biomolecules is lysine acetylation. Acetylation of lysine side chains, which neutralizes the positively charged amine group, results in weaker associative interactions with negatively charged biomolecules and an inhibitory effect on complex coacervation.14 The acetylation state of certain proteins has been shown to have an effect on LLPS, as the presence of acetylated lysine residues disfavors phase separation of Tau and DDX3.15,16 For synthetic complex coacervates like chitosan/hyaluronic acid, an increasing degree of deacetylation for chitosan promotes its ability to form coacervates with hyaluronic acid.17 Most research on the acetylation state of lysine residues has been performed in the field of chromatin research, in which the acetylation state of histone tails determines the conformation of the DNA wrapping chromatin. When histone tails are acetylated, the DNA is in an open state and can be transcribed, while deacetylated tails result in more condensed chromatin. It has been speculated that condensate formation is involved in the recruitment of certain transcription factors to the accessible DNA, possibly linking the acetylation state of histone tails to phase separation. Regarding this, it has been shown that histone H1 can form condensates in vitro and in vivo with single and double stranded DNA.18,19 The N-terminal histone tail H3 can also undergo LLPS with DNA, depending on which lysine residues are acetylated.20 Moreover, interactions of H3 with DNA can stabilize the double stranded duplex.20 Together, this research shows that the acetylation state of histones and other proteins can be important for phase separation. However, active deacetylation of histone tail peptides has not been investigated in detail, and it remains unknown how condensates emerge and develop from this process.
Here, we investigate the effect of enzyme-catalyzed deacetylation of model histone tail peptides on their phase separation with double stranded DNA (dsDNA). We use sirtuin-3 (SIRT3), which is an NAD+-dependent deacetylase, to cleave the acetyl groups of designer peptide substrates and recover the positively charged lysine side chain. We found that the peptides without acetyl groups formed more stable coacervates with dsDNA because of their stronger associative interactions. When observing the deacetylation process using microscopy, we found that nucleation of condensates started with the formation of gel-like aggregates. Surprisingly, these aggregate-like structures gradually liquefied during the reaction and turned into spherical coacervates. We investigated these shape transformations and found that counterintuitively, aggregate-like structures form close to the coacervate salt resistance as a result of screening of the associative interactions between the positively charged peptides and dsDNA.
Experimental
Materials and methods
All chemicals and reagents were used as received from commercial suppliers unless stated otherwise. Milli-Q water (i.e., ultrapure deionized water, 18.2 MΩ cm) from Millipore Corporation was used. The following chemicals were purchased from Sigma-Aldrich: volumetric 1.0 M sodium hydroxide, HEPES, low molecular weight DNA from salmon sperm, NAD+, DTT, NaCl, poly-uridylic acid potassium salt, PDDA, protamine sulfate (grade X), and complete EDTA-free Protease Inhibitor Cocktail. The following oligopeptides were purchased from Alamanda Polymers: poly-L-lysine hydrobromide (MW = 4200 Da, 20-mer), poly-L-lysine hydrobromide (MW = 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da, 100-mer), poly-L-aspartic acid sodium salt (MW = 1400 Da, 10-mer), poly-L-aspartic acid sodium salt (MW = 14000 Da, 100-mer), poly-L-arginine hydrochloride (MW = 1900 Da, 10-mer), and poly-L-arginine hydrochloride (MW = 5700 Da, 30-mer). Peptides GRKAcRG, GRKAcRGRKAcRG, FAM-GRKAcRGRKAcRG, and (KKAcASL)3 and complementary DNA sequences ACTGACTGACTGACTGACTGACTG and CAGTCAGTCAGTCAGTCAGTCAGT-Cy5 were obtained from GenScript. Peptide SRRRR was ordered from CASLO ApS as a trifluoroacetate salt. SIRT3L-12 was a gift from John Denu (Addgene plasmid # 13736; https://n2t.net/addgene:13736; RRID:Addgene_13736) Adenosine 5′-triphosphate disodium salt was purchased from Bio-Connect. pLL-g[3.5]-PEG was purchased from SuSoS. Alexa FluorTM 555 NHS ester was bought from Thermo Fisher. ELP-K72, which is a fusion protein of GFP and an elastin-like sequence [VPGKG]72, was expressed and purified as described in previous work.10
000 Da, 100-mer), poly-L-aspartic acid sodium salt (MW = 1400 Da, 10-mer), poly-L-aspartic acid sodium salt (MW = 14000 Da, 100-mer), poly-L-arginine hydrochloride (MW = 1900 Da, 10-mer), and poly-L-arginine hydrochloride (MW = 5700 Da, 30-mer). Peptides GRKAcRG, GRKAcRGRKAcRG, FAM-GRKAcRGRKAcRG, and (KKAcASL)3 and complementary DNA sequences ACTGACTGACTGACTGACTGACTG and CAGTCAGTCAGTCAGTCAGTCAGT-Cy5 were obtained from GenScript. Peptide SRRRR was ordered from CASLO ApS as a trifluoroacetate salt. SIRT3L-12 was a gift from John Denu (Addgene plasmid # 13736; https://n2t.net/addgene:13736; RRID:Addgene_13736) Adenosine 5′-triphosphate disodium salt was purchased from Bio-Connect. pLL-g[3.5]-PEG was purchased from SuSoS. Alexa FluorTM 555 NHS ester was bought from Thermo Fisher. ELP-K72, which is a fusion protein of GFP and an elastin-like sequence [VPGKG]72, was expressed and purified as described in previous work.10
SIRT3 expression and labeling
A procedure previously described by Hallows et al. was adapted for SIRT3 expression. E. coli BL21 (DE3) cells were transformed with a pQE-80 SIRT3L-12 plasmid containing ampicillin resistance using a standard heat-shock protocol.21 Overnight cultures were used to inoculate large flasks containing 2× YT medium (32 g of tryptone, 10 g of yeast extract, 5 g of NaCl in 1 L of MilliQ water, and pH 7.0) supplemented with 0.10 mg mL−1 ampicillin. Cells were grown at 37 °C till an optical density OD600 of 0.7 was reached. IPTG was added to a final concentration of 1.0 mM and the cultures were shaken at 250 rpm at 25 °C for 8 hours and cooled to 4 °C overnight, after which the cells were harvested by centrifugation. The pellet was resuspended in lysis buffer (50 mM Tris–HCl pH 7.5, 250 mM NaCl, 20 mM imidazole, 1.0 mM β-mercaptoethanol) and 1 tablet of complete EDTA-free Protease Inhibitor Cocktail (Roche) per 10 g of cells. The resuspended cells were lysed using a homogenizer. The lysate was centrifuged (45000 × g, 30 min, 4 °C) and the clarified supernatant was loaded onto a 5 mL HisTrap FF column (Cytiva, Marlborough, Ma, USA). After loading, the column was washed with 50 mL of lysis buffer, and SIRT3 was eluted using elution buffer (50 mM Tris–HCl, pH 7.5, 250 mM NaCl, 1.0 mM β-mercaptoethanol, and 500 mM imidazole). The eluted protein was dialyzed against storage buffer (25 mM Tris–HCl pH 8.0, 100 mM NaCl and 1.0 mM β-mercaptoethanol) and concentrated to 25.4 μM using a centrifugal filter (MWCO 10 kDa, 15 mL, Amicon, Merck) at 500 × g at 4 °C. Aliquots were frozen at −80 °C until further use.
The purified SIRT3 was labeled using AlexaFluorTM 555 NHS Ester (Thermo Fisher Scientific) following a previously established protocol.22 Excess dye was removed using a centrifugal filter (MWCO 10 kDa, 2 mL, Amicon, Merck) at 500 × g at 4 °C and washed with storage buffer until the flow-through contained no more free dye. The final concentration of the protein was measured using a NanoDrop OneC. A labeled SIRT3 stock was then mixed with an unlabeled protein in a 1:9 ratio of labeled to unlabeled protein, which was used for localization experiments.
Deacetylation analysis of peptides
Samples with varying amounts of SIRT3 (100 nM–2.5 μM SIRT3), acetylated peptide (100 μM–2.0 mM), NAD+ (0 or 10 mM) and dsDNA (0 or 1.0 mg mL−1) were incubated at room temperature or 37 °C and aliquots were taken at various timepoints. The aliquots were diluted 10×, filtered and characterized using analytic LC–MS using a Thermo LCQ Fleet with electrospray ionization in combination with a Shimadzu Biflex MALDI-TOF (Bruker Daltonik, Bremen, Germany) and an α-cyano-4-hydroxycinnamic acid matrix.Coacervate formation
All experiments were performed in HEPES buffer (final concentration 50 mM, pH 7.4) containing 1.0 mM DTT. Coacervates were formed by subsequently adding NaCl, NAD+, SIRT3, polyanion and polycation to the buffer. NaCl, NAD+ and SIRT3 were only added when applicable and in various concentrations. Mixing was done by vortexing for 10 seconds at 2800 rpm, except for samples containing SIRT3, in which case mixing was done by pipetting up and down three times.Coacervate salt resistance
The salt resistance of coacervates was assessed using a commonly used turbidity assay, combined with microscopy. 50 μL coacervate suspensions containing 2.0 mM peptide and a varying concentration of dsDNA in HEPES buffer (pH 7.4, final concentration 50 mM) with 10 mM NAD+ and 1.0 mM DTT were added to 384-well plates (Greiner Bio-one, clear flat bottom) and absorbance at 600 nm was measured using a microplate reader (Tecan Spark M10) equipped with an automated injector. After every readout, 2.0 μL of NaCl solution (500 mM) was added to the coacervate suspensions and the samples were mixed by shaking for 5 s. A total of 20 readouts were made, and all measurements were made in triplicate. Turbidity was calculated as 100 − T%, with T% being the fraction of transmitted light at 600 nm, calculated as T% = I/I0 × 100% = 10−Abs × 100%, where Abs is the measured absorbance at 600 nm of the sample minus the absorbance at 600 nm of a solution containing 50 μL of HEPES buffer. Turbidity curves as a function of the NaCl concentration were fitted to a Hill equation, following previous reports by Keating and co-workers:23,24T([Na+]) = Tmax(1/(([Na+]/CSR)n + 1)) using a custom MATLAB script, where T is the turbidity, Tmax is the maximum recorded turbidity at 0 mM NaCl, CSR is the NaCl resistance of the coacervate, which was defined as the concentration at the one-half y-maximum and n is the fitting parameter ranging from 2 to 20, similar to previous reports.23,24 The mean salt resistance and standard deviation were calculated by taking an average of the three CSR values resulting from the fit.Image and video acquisition
Coacervate samples were imaged on μ-Slide 18 well slides (glass bottom, Ibidi) and reaction samples on μ-Slide VI 0.4 slides (polymer coverslip, Ibidi), all of which were cleaned with an oxygen plasma cleaner, incubated for 24 h with 0.10 mg mL−1 pLL-g[3.5]-PEG (SuSoS, Dübendorf, Switzerland) dissolved in 10 mM HEPES, pH 8.0, and then washed and dried with Milli-Q water and pressurized air, respectively. Before image acquisition, 50 μL coacervate samples were incubated for 1 h to allow droplets to coalesce and settle on the glass surface. 100 μL of enzyme reaction samples were imaged immediately after the addition of the peptide. The samples were imaged using an Olympus IX83 inverted fluorescence microscope equipped with a motorized stage (TANGO, Märzhäuser) and LED light source (pE-4000 CoolLED). The images were recorded using a 40× universal plan fluorite objective (WD 0.51 mm, NA 0.75, Olympus) with a temperature controlled CMOS camera (Hamamatsu Orca-Flash 4.0).Circularity calculations
Microscopy images and videos were processed and analysed using the MATLAB 2021 Image Processing Toolbox. Objects smaller than 1000 pixels were excluded from the analysis to avoid false positives caused by dust grains, and the area was extracted for every object in each frame. For circularity calculations, the perimeter of each object was calculated by taking the sum of all pixels that were directly adjacent to the dilute phase plus half the sum of all pixels that were only diagonally adjacent to the dilute phase. The circularity of the object cross section was then calculated as circularity = 4πA/P2, where A is the area of the object and P is the perimeter. Subsequently, a mean circularity and a standard deviation for each frame were calculated.Localization and FRAP measurements
To determine the partition coefficients of coacervate constituents, a Leica SP8x confocal inverted microscope (Leica Microsystems, Germany) equipped with a DMi8 CS motorized stage, a pulsed white light laser, 2 PMT detectors, HyD SP GaAsP detectors and a Leica DRC7000 GT monochrome camera was used. All images were recorded using an HC PLAPO 63× oil immersion objective (WD 0.14 mm, NA 1.4) and a HyD detector in the emission range of the used fluorophore. Samples were prepared with a final concentration of 2 mM GRK2, 1 mg mL−1 dsDNA, 10 mM NAD+, 1 mM DTT, 1 μM SIRT3 and 50 mM HEPES pH 7.4. For each probed molecule, individual samples were prepared, one using 5% FAM-GRK2, one using an additional 10 μM [ACTG]6 and 10 μM [CAGT]6-Cy5, and one using 10% SIRT3-Alexa555. For each sample, three images were recorded and three background images were taken using a sample containing the described mixture, without any fluorescent molecules while using the same laser intensities used for recording the labeled molecules. Partition coefficients for each droplet were calculated using the following formula: KP = (Ic − Ibg)/(Id − Ibg), where Ic is the mean fluorescence intensity of a single coacervate, Id is the mean fluorescence intensity of the image with all of the coacervates removed, and Ibg is the mean fluorescence intensity of the background image, which did not contain labeled molecules. The mean KP and standard deviation were calculated by taking the average KP of all condensates from the three recorded images.For FRAP analysis, time-lapse videos were recorded using a CSU X-1 Yokogawa spinning disk confocal unit attached to an Olympus IX81 inverted microscope. Imaging was conducted with a ×100 piezo-driven oil immersion objective (NA 1.3) and 488 nm laser line. Emission was captured using an Andor iXon EMCCD camera, acquiring 120 frames per minute. Photobleaching was performed by selecting a small region of interest in the center of a condensed droplet, using the 488 nm laser line set to 100% power for 100 pulses of 100 μs each. Recovery was subsequently imaged at 10% laser intensity. Recovery profiles were analyzed using a custom MATLAB script, in which fluorescence intensities were normalized and fitted to an exponential decay function f(t) = A(1 − e−bt) + C, following the previous literature.25,26 The recovery half-life was calculated as ln(2)/b, and the maximum recovered fraction fluorescence intensity was (A + C) × 100%.
Results and discussion
To investigate whether coacervation could be driven by deacetylation, we selected the enzyme sirtuin-3 (SIRT3), which is an NAD+-dependent deacetylase originally found in the mitochondrial matrix of mammalian cells. SIRT3 acts on multiple acetylated substrates in vivo, though it can also use small synthetic acetylated peptides as substrates, using NAD+ as a cofactor, and yielding a deacetylated peptide, nicotinamide and O-acetyl-ADP-ribose (OAADPr) as products.27,28 Deacetylation of acetylated lysine residues of a peptide substrate will increase its charge, which promotes phase separation with negatively charged DNA (Fig. 1a). Three positively charged acetylated peptides had been selected for the deacetylation assay: GRKAcRG (GRK1Ac), which is a known substrate of SIRT3,29 an elongated version GRKAcRGRKAcRG (GRK2Ac), and a lysine-rich polypeptide [KKAcASL]3 (KKASL3Ac) (Fig. 1b). The protein was expressed using BL(21)DE3 E. coli transformed with a plasmid containing the SIRT3 gene using IPTG induction, and purified with a Ni-NTA affinity column to yield a 83% pure protein based on gel intensity (Fig. 1c).21 The ability of SIRT3 to deacetylate these peptides was investigated by incubating varying concentrations of the enzyme with the peptide substrate and NAD+. For KKASL3 and GRK2, mono-, di- and tri-deacetylated (when applicable) forms were found after incubation for 7 hours (Fig. S1, ESI†). For GRK1, the deacetylated peptide could not be detected, as its m/z value was lower than the detection limit. Next, the reaction was performed in the presence of short double stranded DNA (dsDNA), and again deacetylated forms of KKASL3 and GRK2 were detected, indicating that our selected substrates are recognized by SIRT3 and dsDNA does not substantially inhibit the deacetylation reaction (Fig. 1d).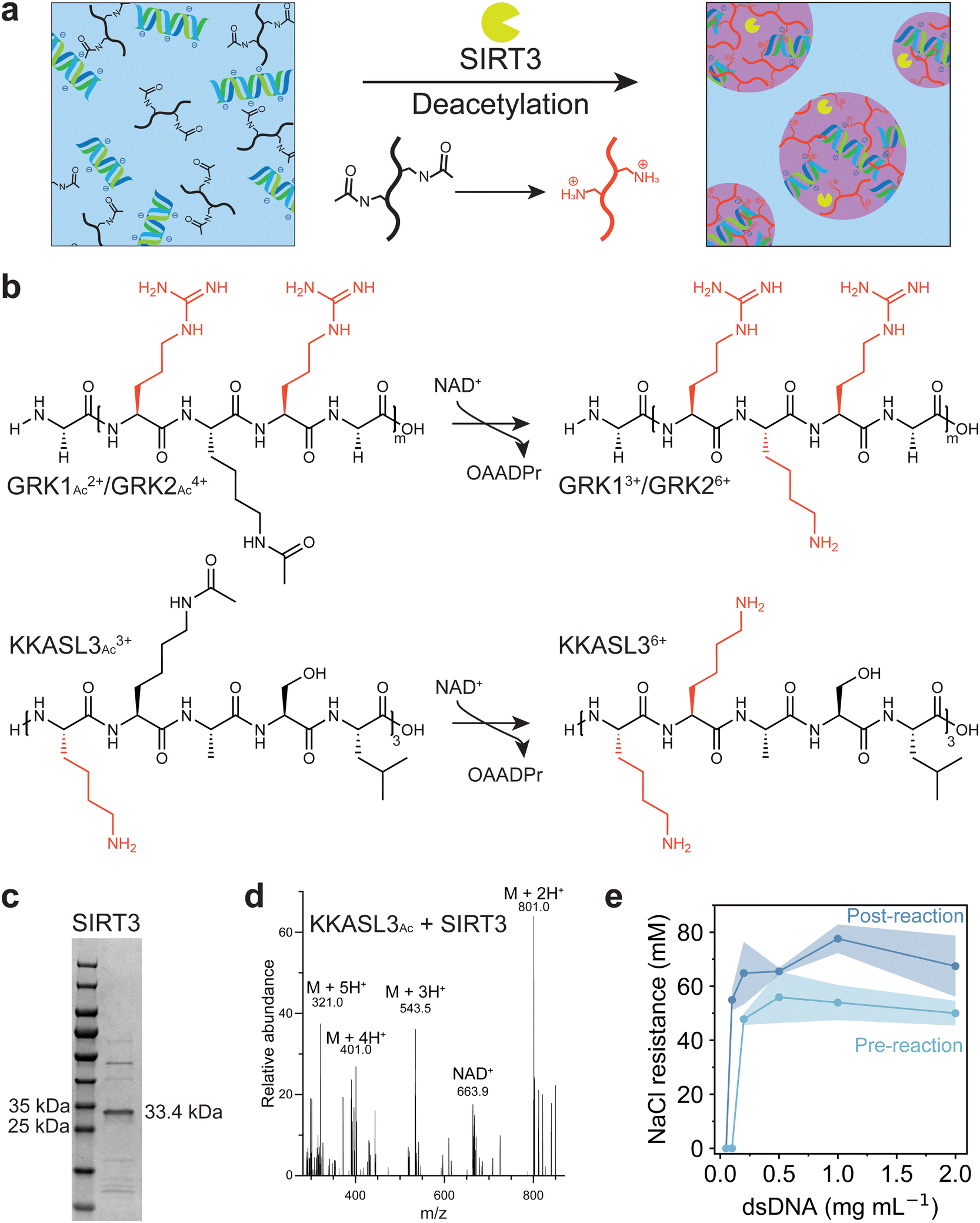 | ||
| Fig. 1 (a) Schematic design of coacervation induced by deacetylation, SIRT3 (yellow) deacetylates peptide substrates (black), which results in a more positively charged peptide (red) that phase separates with dsDNA (blue, green) to form condensates. (b) Peptides GRK1 (top, m = 1, sequence: GRKRG), GRK2 (top, m = 2, sequence: GRKRGRKRG) and KKASL3 (bottom, sequence: KKASLKKASLKKASL) used to assess the activity of SIRT3. Incubation of the peptide with the enzyme and NAD+ leads to deacetylation of the acetylated lysine side chains and formation of O-AADPR. Positively charged residues are colored red. (c) SIRT3 purified by Ni-NTA affinity chromatography resolved by SDS/PAGE and detected by Coomassie, the calculated weight for SIRT3 is 33.4 kDa. (d) Mass spectrum of deacetylated KKASL3 (M + H+ = 1601.9) after 8 h of incubation with SIRT3 and NAD+. (e) Phase diagram of GRK2Ac before (light blue) and after (dark blue) the deacetylation reaction with SIRT3. Shaded regions indicate standard deviation (n = 3). Mixture contains 2 mM GRK2, 10 mM NAD+, 1 mM DTT and 2.5 μM SIRT3 in 50 mM HEPES pH 7.4. For the pre-reaction sample, SIRT3 was incubated at 95 °C to ensure denaturation of the enzyme. | ||
To investigate the ability of these peptides to form complex coacervates, KKASL3Ac and GRK2Ac were mixed with dsDNA at different ratios. Additionally, the reaction mixtures of both peptides after incubation with the enzyme were mixed at the same ratios with dsDNA. GRK2 was able to phase separate with dsDNA before and after incubation with SIRT3, whereas KKASL3 did not phase separate (Fig. S2, ESI†). It has been shown that arginine residues that are not present in KKASL3 interact stronger with nucleobases than lysine residues and that arginine has a higher stabilizing effect on complex coacervate formation.30,31 Additionally, KKASL3Ac needs to have three acetyl groups removed before it reaches a +6 charge, while GRK2Ac only contains two acetyl groups. Subsequently, the coacervate salt resistance (CSR) of each sample was determined by measuring the turbidity at 600 nm during a sodium chloride titration. The turbidity data were fitted with a Hill equation, and the parameter indicating the half-maximum was taken as the CSR (Fig. S3, ESI†). GRK2 formed coacervates with dsDNA, and samples that were incubated with SIRT3 had a higher CSR, likely resulting from the positively charged lysine residues after the deacetylation reaction. A phase diagram for GRK2 and dsDNA was constructed, which clearly depicts the stability difference between the coacervates before and after the enzymatic reaction (Fig. 1e).
Having established that the deacetylation reaction can lead to GRK2/dsDNA coacervates with a higher salt resistance, we decided to follow the reaction using microscopy. A reaction composition at 60 mM NaCl was selected, where dsDNA forms coacervates with the deacetylated peptide but not with the acetylated form according to Fig. 1e. At the start of the reaction, a dilute solution containing aggregate-like structures was observed. Over the course of the reaction, these aggregates grew in size, became more rounded and finally transformed into spherical droplets that displayed fusion and wetting (Fig. 2a). This process is remarkable, as a regular LLPS process involves nucleation of spherical droplets, and fusion of these droplets into larger spherical droplets.32,33 As the deacetylation reaction increases the associative interactions between GRK2 and dsDNA, the CSR becomes higher and LLPS of GRK2 and dsDNA occurs. Non-active model systems have shown that increasing the interaction strength between two attracted species by increasing multivalent interaction sites can lead to aggregate formation.34 For example, poly-L-lysine and poly-L-arginine with a length of 30 (K30 and R30, respectively) form liquid droplets with ATP, while K100 and R100 form solid aggregates, presumably because the associative interactions are so strong that irreversible bonds are formed.23 However, here the opposite happens, as the deacetylation reaction increases the multivalent interactions and amorphous gel-like aggregates slowly transform into spherical liquid droplets over the course of hours (Fig. 2a and Movie S1, ESI†). This process is driven by the enzymatic reaction, as samples without cofactor NAD+ or inactive SIRT3 show no shape transformation of the amorphous gel-like structures (Fig. S4, ESI†).
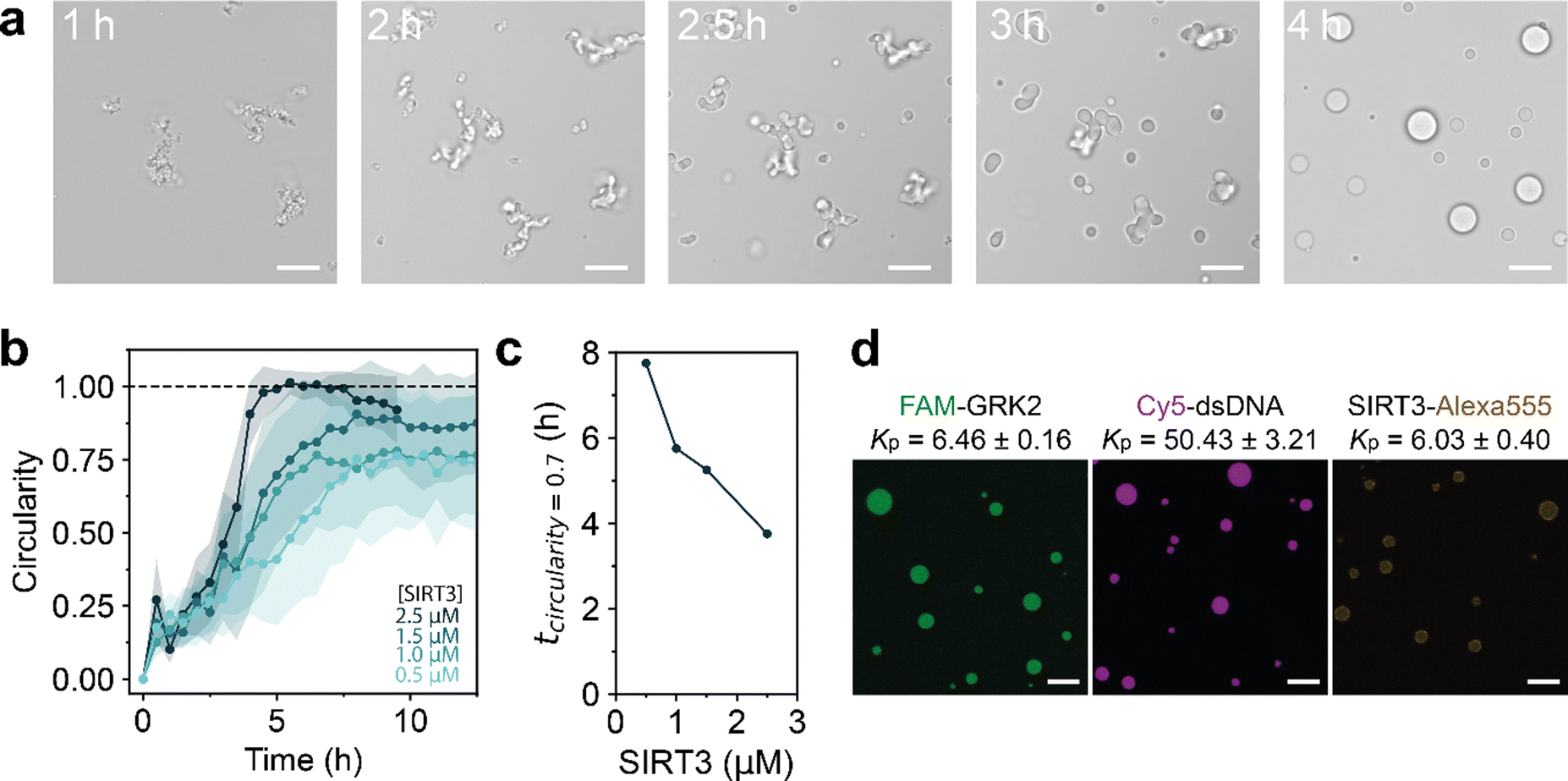 | ||
| Fig. 2 (a) Formation of amorphous gel-like condensates of GRK2Ac and dsDNA resulting from the deacetylation reaction. Over time, the gels relax and get a spherical shape, indicative of liquid droplets. (b) Circularity of condensates formed during the reaction at different SIRT3 concentrations. Points are mean circularities and shaded areas indicate the standard deviation. (c) The time it takes to reach spherical droplets decreases with a higher SIRT3 concentration. (d) GRK2, SIRT3 and dsDNA all localize to the coacervate phase. Molecules were labeled and their partition coefficient (KP) was calculated by their relative fluorescence intensity inside the coacervate phase compared to the dilute phase. Mixtures contain 2 mM GRK2, 10 mM NAD+, 1 mM DTT, 60 mM NaCl and varying SIRT3 concentrations in 50 mM HEPES pH 7.4. Scale bars are 10 μm. | ||
To study whether this shape transformation was dependent on the deacetylation reaction, we performed the reaction with varying concentration of SIRT3 (Fig. S5, ESI†). By calculating the circularity of the structures (circularity = 4πA/P2), the speed at which shape transformations occur can be quantified.12 A higher concentration of SIRT3 increases the speed at which structures form and how they liquefy to spherical droplets (circularity = 1), indicating that the enzyme drives this transformation process (Fig. 2b). Here, the deacetylation reaction driven by SIRT3 increases the interactions between GRK2 and dsDNA by increasing multivalent positive stickers in the peptide.
Nonspherical enzyme-driven coacervates have been reported before by Spoelstra et al., where they reported a system that polymerizes short polyU RNA into long RNA.12 This reaction starts off with structures looking similar to the Fig. 2a, which liquefy during the polymerization reaction. At high PNPase concentrations, coacervates were less spherical at the start of the reaction but reached a round shape in a similar time as low PNPase concentrations. Additionally, when the substrate concentration of UDP increased, condensates started out less spherical than low concentrations and took longer to reach a circular shape.12 The transformation we observe in the SIRT3 system is different, as coacervates with 2.5 μM SIRT3 reach a circular droplet state within 4 hours, while it takes around 7.5 hours for droplets with a lower enzyme concentration of 1.5 μM (Fig. 2c). Moreover, when we used inactivated enzymes or no NAD+, droplets remained nonspherical for 48 hours (Fig. S4, ESI†).
To determine which molecules are enriched in the condensates with respect to the aqueous phase, we calculated the partition coefficient (KP) of GRK2, dsDNA and SIRT3 by labeling them with fluorescent probes. Using confocal microscopy, FAM-GRK2 and a 24-mer dsDNA labeled with Cy5 at the 3′ end were homogeneously localized to the coacervate phase with a KP of 6.46 ± 0.16 and 50.43 ± 3.21, respectively (Fig. 2d). SIRT3 was labeled with an Alexa555-NHS ester, and localized to the coacervate phase with a KP of 6.03 ± 0.40, albeit slightly less homogeneous. Brightened spots were found around the interface of the droplets, indicating a slight preference of the protein for the coacervate interface. However, it is also possible that the bright spots are aggregated clusters of SIRT3, as we observed that the protein has a tendency to stay in insoluble fractions during purification. These results show that the droplets we form consist mostly of GRK2 and dsDNA and that the enzyme is present inside the coacervates.
As the GRK2/dsDNA shape transformations are different than the ones observed in the PNPase reaction, we have to consider what happens to molecules constituting the coacervate during the deacetylation reaction. As the reaction progresses, the system phase separates at its CSR and transitions to a composition at which coacervates are significantly more stable (Fig. 1e). Because of this, we investigated GRK2Ac/dsDNA condensates at increasing salt concentrations. As the NaCl concentration increases, droplets seem to become more gel-like and slowly fuse (Fig. 3a), indicating that the gel-to-condensate transition is an inherent property of GRK2Ac/dsDNA coacervates and not caused by the presence of an enzymatic deacetylation reaction. Additionally, the circularity of coacervates becomes lower, which indicates that these droplets have a higher viscosity (Fig. 3b).
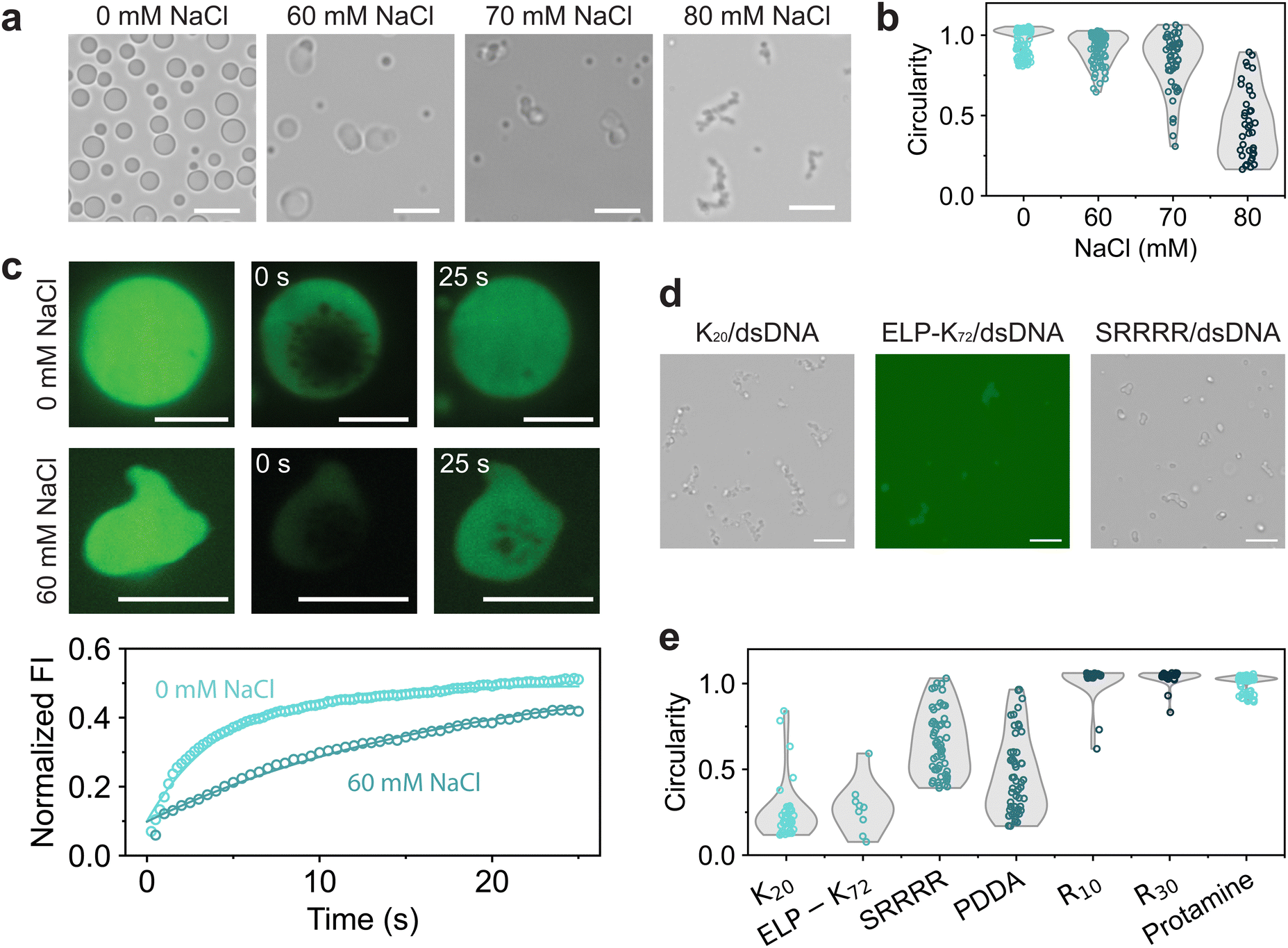 | ||
| Fig. 3 (a) GRK2Ac/dsDNA coacervates form gel-like structures near their coacervate salt resistance. (b) Circularities of GRK2/dsDNA coacervates at increasing NaCl concentrations. (c) FRAP measurements of GRK2Ac/dsDNA coacervates at no salt and 60 mM NaCl. The recovery half-life at 0 mM NaCl was 3.07 seconds, while at 60 mM, it took 12.5 seconds to reach half-maximum recovery. Data were fitted with an exponential decay function f(t) = A(1 – e−bt) + C. (d) dsDNA forms nonspherical coacervates with K20, ELP-K72 and SRRRR near their CSR. (e) Circularities of dsDNA coacervates near their CSR. R10 and protamine form circular droplets, while other polycations form nonspherical droplets. When interactions between polycation and dsDNA are relatively strong, no gel-like aggregates are found. Scale bars are 10 μm. | ||
To probe the fluidity of these coacervates, we used fluorescence recovery after photobleaching (FRAP) and measured the diffusion of FAM-GRK2Ac at low and high NaCl concentrations.25 For both samples, vacuoles formed the moment the condensates were bleached (Fig. 3c). These vacuoles did not hinder the recovery analysis, and dissipated away by fusing, shrinking, or moving to the edge of the coacervate. At 0 mM NaCl, coacervates were more spherical and recovered faster, with a recovery half-life of 3.07 seconds compared to 60 mM NaCl, where the coacervates had a recovery half-life of 12.5 seconds. Under both conditions, the droplets only recovered to approximately 50% of their original fluorescence intensity as the bleached area was relatively large compared to the droplet area, indicating that exchange from the dilute phase to the condensate phase is substantially slower than diffusion within the condensate phase. The lower circularity and diffusion coefficient of FAM-GRK2Ac show that the droplets at higher salt are more gel-like. This is the opposite of what is expected in complex coacervates: increasing salt concentrations leads to electrostatic screening, resulting in weaker associative interactions and a lower viscosity.12,35,36 GRK2 and dsDNA form complex coacervates based on electrostatic interactions and cation–pi stacking, both of which would be reduced by the increasing salt concentration.
To investigate whether the structures were caused by the presence of GRK2, dsDNA, or both, we replaced them with other polycations and polyanions, respectively. When dsDNA was replaced by polyU, polyA, D100 or E100, spherical coacervates formed by complexation with GRK2 and no gel-like structures were found when the salt concentration was increased until the droplets dissolved (Fig. S6, ESI†). As none of the tested GRK2/polyanion combinations formed nonspherical coacervates, we conclude that the nonspherical structures are not caused by the presence of GRK2. We then investigated the role of dsDNA by exploring its phase separating behavior with a variety of positively charged macromolecules at increasing NaCl concentrations. For several polycations, such as K20, ELP-K72, a positively charged elastin-like polypeptide containing 72 repeats of VPGKG, the peptide SRRRR, and polydiallyldimethylammonium chloride (PDDA) we found nonspherical structures just below the coacervate salt resistance (Fig. 3d and Fig. S7, ESI†). However, other polycations with higher charge densities, such as protamine, R10, and R30, never formed nonspherical coacervates with dsDNA and only reduced in size with increasing salt concentrations (Fig. 3e). Comparing the differences between the polycations that are able to form nonspherical coacervates and ones that are not, the polycations that only form spherical coacervates have stronger interactions with dsDNA. Protamine, R10 and R30 consist of mainly arginine residues, which have been shown to form strong bonds with nucleotides.30 SRRRR also consists of mainly arginine residues, but is shorter and can sustain fewer multivalent interactions with the dsDNA.34 These results point to a mechanism in which the polycation-dsDNA interaction strength determines the morphology of the coacervates that form. During the SIRT3-induced shape transformation shown in Fig. 2, the same mechanism can explain the observed shape transformation. SIRT3 slowly converts GRK2Ac, which forms gel-like structures with dsDNA, to GRK2, which interacts more strongly with dsDNA and is able to induce sufficient DNA bending to liquefy the peptide/dsDNA condensates, resulting in spherical liquid droplets.
Previously, it has been shown how associative LLPS can induce solubilization of dsDNA precipitates.37 This process can be considered by the charge neutralization that occurs when complex coacervation is induced. Polyelectrolytes constituting the coacervate may need to bend to maximize charge neutralization, which requires sufficiently strong associative interactions. When dsDNA is complexed with positively charged peptides, the rigid molecules can bend in order to achieve maximal charge neutralization, which leads to a liquid phase.38 These observations could explain the gel-like structures near the CSR of the condensates, as salt screens the electrostatic interactions and weakens the peptide complexation with dsDNA, leading to non-bended nucleic acid, which tends to form aggregate-like structures. At low salt concentrations, the interactions are not screened and the associative interactions are strong enough to induce complete charge neutralization. Polycations such as protamine, R10 and R30 have very strong interactions with dsDNA, as the arginine residues can form cation–pi bonds with the nucleobases and ionic bonds with the phosphate backbone. Even at NaCl concentrations near the CSR, these peptides still have enough interactions to bend the dsDNA and keep it condensed (Fig. 4a). The other amine-based polycations (K20, PDDA and ELP-K72) all have weaker interactions with dsDNA and at a high enough NaCl concentration, the associative interactions weaken enough to not fully neutralize all the dsDNA, leading to gelation. The peptides SRRRR and GRK2 do contain arginine residues, however are either too short or have a low charge density, which leads to weaker interactions. These weaker polycations are not able to fully bend dsDNA near the CSR, leading to the formation of gel-like aggregates (Fig. 4b).
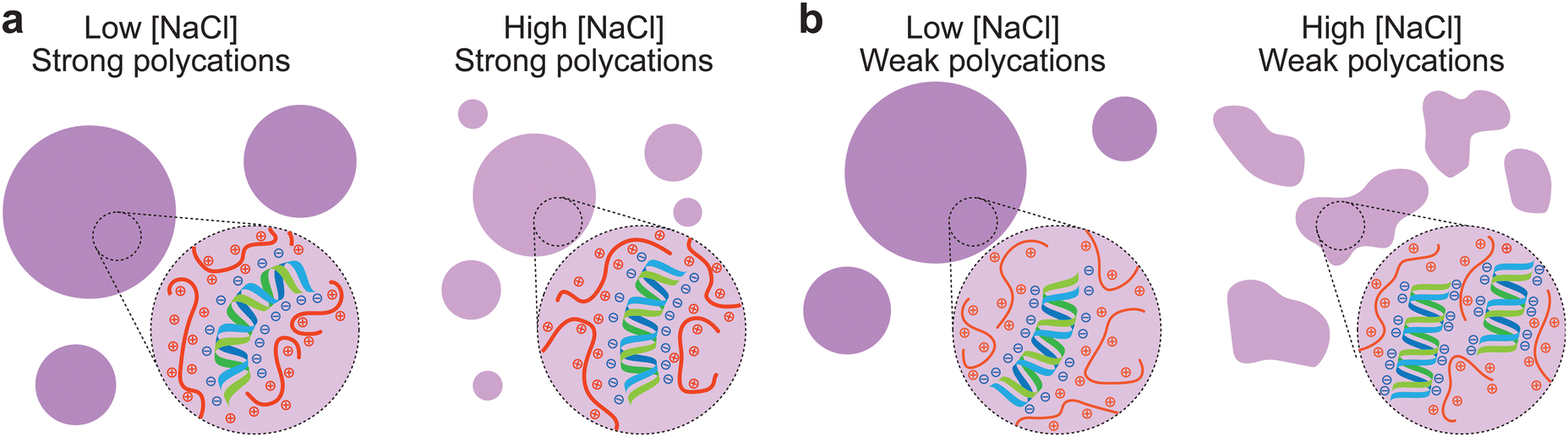 | ||
| Fig. 4 Schematic overview of complex dsDNA coacervates. (a) Strong polycations, such as R10 and protamine can bend dsDNA at high and low salt concentrations to maximize charge neutralization, resulting in spherical liquid condensates. (b) Weak polycations, such as GRK2Ac, K20 and PDDA cannot bend dsDNA at high salt concentrations, resulting in spherical condensates with low salt and gel-like aggregates at high salt concentrations. | ||
Additionally, dsDNA has been shown to form liquid crystal phases in complex coacervates.37,39 As the gel-like aggregates found are reminiscent of the liquid crystalline structures found, we studied GRK2/dsDNA coacervates at different NaCl concentrations using polarized light microscopy. No combination we tested showed any birefringent signals. A reason for this is that the liquid crystal phases observed by Fraccia et al. were mostly present at low salt concentrations, whereas high NaCl concentrations resulted in liquid droplets.39
In a biological context, our findings on deacetylation of lysine residues inducing phase separation with dsDNA could have implications on histone dynamics. It has been shown that several actively transcribed genes have hyperacetylated histones, which indicates a role for histone acetylation facilitating transcription.40 It would be interesting to compare the RNA transcription in DNA coacervates containing acetylated and deacetylated histones. It has been shown that coacervation of cell lysate can enhance the transcription rate of T7 RNA polymerase, though these coacervates consist mostly of polyethylene glycol.41 In complex coacervates, it has been shown that ELP-K72/DNA coacervates can support in vitro transcription and a translation reaction, though it was the only working system out of the eight tested.42 The histone H1 protein can form coacervates with dsDNA and the phosphorylation state of the histone peptides could influence DNA partitioning.18 These model systems could aid in the design of bottom-up phase-separated transcription hubs, where peptide–DNA interactions are regulated by enzymatically driven PTMs.
In a material science context, our results indicate that shape transformations and concomitant changes in material properties from gel-like to liquid-like can be controlled by a balance between intermolecular interaction strength (governed by charge density, salt concentration, pH and temperature), and the energy required to induce bending of the condensate components, which depend mostly on the molecular structure. Besides double stranded DNA, other polyanions or supramolecular polymers with comparable persistence lengths, such as dsRNA, noncrosslinked diacetylene filaments, intermediate protein filaments (IF), and FtsZ filaments, may also display the same behavior. It would be interesting to investigate the required degree of bending of DNA or other polymers for a gel-to-liquid transformation, for instance, by molecular dynamics simulations.
Conclusions
We have shown that enzymatically driven deacetylation can be used to induce complex coacervation of DNA with a small acetylated peptide substrate. The GRK2/dsDNA coacervates appeared gel-like close to their coacervate salt resistance and turned more liquid-like and spherical during the deacetylation reaction. This process could be controlled by the enzyme concentration, as a high SIRT3 concentration led to coacervate formation in 4 h, while a low concentration required 10 h. FRAP measurements confirmed that GRK2/dsDNA coacervates were gel-like at high salt concentration, as the time to half-maximum fluorescence recovery was four times as long for coacervates at high salt concentrations compared to low salt concentrations. The gel-like droplets were a result of the dsDNA, as GRK2 always formed spherical condensates when dsDNA was substituted with other polyanions. However, when coacervates were prepared with other polycations and dsDNA, gel-like structures similar to GRK2/dsDNA were found near the critical point. These shape transformations can be explained by the bending and charge neutralization of dsDNA, as polycations containing mostly arginine residues that can have strong associative interactions with dsDNA restrain from forming aggregates. At high salt concentrations, the electrostatic interactions between polycations and dsDNA are screened, leading to insufficient bending of the dsDNA and the formation of more gel-like droplets or aggregates. Overall, this work shows that enzymatic deacetylation can be used in vitro to actively form coacervates and that complexation of polycations and dsDNA can lead to liquid and gel-like coacervates, depending on the ionic strength of the solution.Author contributions
E. S. and M. H. I. V. H. designed and supervised the project. M. H. I. V. H. performed and analyzed the microscopy recordings. N. S. H. expressed and purified SIRT3 and analyzed peptide deacetylation. L. V. performed the FRAP experiments. V. A. C. B. investigated dsDNA phase separation. M. H. I. V. H. and E. S. wrote the manuscript, with input from all authors.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program under grant agreement number 851963.Notes and references
- S. Boeynaems, S. Alberti, N. L. Fawzi, T. Mittag, M. Polymenidou, F. Rousseau, J. Schymkowitz, J. Shorter, B. Wolozin, L. Van Den Bosch, P. Tompa and M. Fuxreiter, Trends Cell Biol., 2018, 28, 420–435 CrossRef CAS PubMed.
- S. F. Banani, H. O. Lee, A. A. Hyman and M. K. Rosen, Nat. Rev. Mol. Cell Biol., 2017, 18, 285–298 CrossRef CAS PubMed.
- K. K. Nakashima, M. A. Vibhute and E. Spruijt, Front. Mol. Biosci., 2019, 6, 21 CrossRef CAS PubMed.
- J. Söding, D. Zwicker, S. Sohrabi-Jahromi, M. Boehning and J. Kirschbaum, Trends Cell Biol., 2020, 30, 4–14 CrossRef PubMed.
- N. A. Yewdall, A. A. André, T. Lu and E. Spruijt, Curr. Opin. Colloid Interface Sci., 2021, 52, 101416 CrossRef CAS.
- C. P. Brangwynne, C. R. Eckmann, D. S. Courson, A. Rybarska, C. Hoege, J. Gharakhani, F. Jülicher and A. A. Hyman, Science, 2009, 324, 1729–1732 CrossRef CAS PubMed.
- J. Kirschbaum and D. Zwicker, J. R. Soc., Interface, 2021, 18, 20210255 CrossRef CAS PubMed.
- W. M. Aumiller, Jr. and C. D. Keating, Nat. Chem., 2016, 8, 129–137 CrossRef PubMed.
- K. K. Nakashima, J. F. Baaij and E. Spruijt, Soft Matter, 2018, 14, 361–367 RSC.
- K. K. Nakashima, M. H. I. van Haren, A. A. M. André, I. Robu and E. Spruijt, Nat. Commun., 2021, 12, 3819 CrossRef CAS PubMed.
- S. Deshpande, F. Brandenburg, A. Lau, M. G. F. Last, W. K. Spoelstra, L. Reese, S. Wunnava, M. Dogterom and C. Dekker, Nat. Commun., 2019, 10, 1800 CrossRef PubMed.
- W. K. Spoelstra, E. O. van der Sluis, M. Dogterom and L. Reese, Langmuir, 2020, 36, 1956–1964 CrossRef CAS PubMed.
- A. D. Slootbeek, M. H. I. van Haren, I. B. A. Smokers and E. Spruijt, Chem. Commun., 2022, 58, 11183–11200 RSC.
- J. Li, M. Zhang, W. Ma, B. Yang, H. Lu, F. Zhou and L. Zhang, Mol. Biomed., 2022, 3, 13 CrossRef PubMed.
- J. C. Ferreon, A. Jain, K. J. Choi, P. S. Tsoi, K. R. MacKenzie, S. Y. Jung and A. C. Ferreon, Int. J. Mol. Sci., 2018, 19, 1360 CrossRef PubMed.
- M. Saito, D. Hess, J. Eglinger, A. W. Fritsch, M. Kreysing, B. T. Weinert, C. Choudhary and P. Matthias, Nat. Chem. Biol., 2019, 15, 51–61 CrossRef CAS PubMed.
- A. B. Kayitmazer, A. F. Koksal and E. Kilic Iyilik, Soft Matter, 2015, 11, 8605–8612 RSC.
- A. L. Turner, M. Watson, O. G. Wilkins, L. Cato, A. Travers, J. O. Thomas and K. Stott, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 11964–11969 CrossRef CAS PubMed.
- R. Leicher, A. Osunsade, G. N. L. Chua, S. C. Faulkner, A. P. Latham, J. W. Watters, T. Nguyen, E. C. Beckwitt, S. Christodoulou-Rubalcava, P. G. Young, B. Zhang, Y. David and S. Liu, Nat. Struct. Mol. Biol., 2022, 29, 463–471 CrossRef CAS PubMed.
- M. Mimura, H. Sugai, T. Kameda, R. Kitahara, S. Kitazawa, Y. Shinkai, R. Kurita and S. Tomita, ChemRxiv, 2023, 1–19, DOI:10.26434/chemrxiv-2023-1zplz.
- W. C. Hallows, S. Lee and J. M. Denu, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10230–10235 CrossRef CAS PubMed.
- K. K. Nakashima, A. A. M. André and E. Spruijt, Methods Enzymol., 2021, 646, 353–389 CAS.
- F. P. Cakmak, S. Choi, M. O. Meyer, P. C. Bevilacqua and C. D. Keating, Nat. Commun., 2020, 11, 5949 CrossRef CAS PubMed.
- S. Choi, A. R. Knoerdel, C. E. Sing and C. D. Keating, J. Phys. Chem. B, 2023, 127, 5978–5991 CrossRef CAS PubMed.
- N. A. Yewdall, A. A. M. André, M. H. I. van Haren, F. H. T. Nelissen, A. Jonker and E. Spruijt, Biophys. J., 2022, 121, 3962–3974 CrossRef CAS PubMed.
- L. Hubatsch, L. M. Jawerth, C. Love, J. Bauermann, T. Y. D. Tang, S. Bo, A. A. Hyman and C. A. Weber, eLife, 2021, 10, e68620 CrossRef CAS PubMed.
- B. C. Smith, B. Settles, W. C. Hallows, M. W. Craven and J. M. Denu, ACS Chem. Biol., 2011, 6, 146–157 CrossRef CAS PubMed.
- J. M. Denu, Curr. Opin. Chem. Biol., 2005, 9, 431–440 CrossRef CAS PubMed.
- K. Patel, J. Sherrill, M. Mrksich and M. D. Scholle, J. Biomol. Screening, 2015, 20, 842–848 CrossRef CAS PubMed.
- I. Alshareedah, T. Kaur, J. Ngo, H. Seppala, L. D. Kounatse, W. Wang, M. M. Moosa and P. R. Banerjee, J. Am. Chem. Soc., 2019, 141, 14593–14602 CrossRef CAS PubMed.
- I. B. A. Smokers, M. H. I. van Haren, T. Lu and E. Spruijt, ChemSystemsChem, 2022, 4, e202200004 CrossRef CAS.
- A. A. Hyman, C. A. Weber and F. Jülicher, Annu. Rev. Cell Dev. Biol., 2014, 30, 39–58 CrossRef CAS PubMed.
- P. Zhang and Z.-G. Wang, Macromolecules, 2022, 55, 3910–3923 CrossRef CAS.
- A. Sathyavageeswaran, J. Bonesso Sabadini and S. L. Perry, Acc. Chem. Res., 2024, 57, 386–398 CrossRef CAS PubMed.
- Y. Liu, H. H. Winter and S. L. Perry, Adv. Colloid Interface Sci., 2017, 239, 46–60 CrossRef CAS PubMed.
- Q. Wang and J. B. Schlenoff, Macromolecules, 2014, 47, 3108–3116 CrossRef CAS.
- A. Shakya and J. T. King, Biophys. J., 2018, 115, 1840–1847 CrossRef CAS PubMed.
- A. A. M. André and E. Spruijt, Biophys. J., 2018, 115, 1837–1839 CrossRef PubMed.
- T. P. Fraccia and T. Z. Jia, ACS Nano, 2020, 14, 15071–15082 CrossRef CAS PubMed.
- V. G. Allfrey, R. Faulkner and A. E. Mirsky, Proc. Natl. Acad. Sci. U. S. A., 1964, 51, 786–794 CrossRef CAS PubMed.
- E. Sokolova, E. Spruijt, M. M. Hansen, E. Dubuc, J. Groen, V. Chokkalingam, A. Piruska, H. A. Heus and W. T. Huck, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11692–11697 CrossRef CAS PubMed.
- L. L. J. Schoenmakers, N. A. Yewdall, T. Lu, A. A. M. André, F. H. T. Nelissen, E. Spruijt and W. T. S. Huck, ACS Synth. Biol., 2023, 12, 2004–2014 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm01091d |
| This journal is © The Royal Society of Chemistry 2024 |