
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHexafluorophosphate ionic liquid-modified silica sorbent for selective separation and preconcentration of Pb2+, Cd2+, and Cr3+ in water samples†
Linh Dieu
Nguyen
ab,
The Thai
Nguyen
ab,
Nhi Hoang
Nguyen
ab,
Chi Thien
Gia Hua
ab,
Tan Hoang
Le Doan
 bc,
Linh Thuy
Ho Nguyen
bc and
Phuong Hoang
Tran
*ab
bc,
Linh Thuy
Ho Nguyen
bc and
Phuong Hoang
Tran
*ab
aDepartment of Organic Chemistry, Faculty of Chemistry, University of Science, Ho Chi Minh City, Vietnam. E-mail: thphuong@hcmus.edu.vn
bVietnam National University, Ho Chi Minh City, Vietnam
cCenter for Innovative Materials and Architectures (INOMAR), Ho Chi Minh City, Vietnam
First published on 10th November 2023
Abstract
A sorbent based on ionic liquid-modified silica has been developed for the determination of Cd2+, Pb2+, and Cr3+ in water. Five materials, namely DL1@SiO2 to DL5@SiO2, were prepared from five ionic liquids, including N,N,N-trimethyl-N-hexadecylammonium and N-alkylpyridinium hexafluorophosphate, which were grafted onto silica gel. These materials were used to enrich the trace concentration of three metallic cations (Cd2+, Pb2+, and Cr3+) in water samples, followed by the ICP-MS analysis. Among these sorbents, DL1@SiO2 showed highly efficient sorption. The detection limit of this method for Cd2+, Pb2+, and Cr3+ was 0.173 μg L−1, 0.046 μg L−1, and 0.185 μg L−1, and the relative standard deviation (RSD) was 4.9%, 3.4%, and 1.2%, respectively. The procedure was applied to determine Cd2+, Pb2+, and Cr3+ contents in a spiked tap water sample (Ho Chi Minh City) with satisfactory recovery from 74 to 100.8%.
Sustainability spotlightSilica gel grafted with ionic liquids has emerged as a promising and cost-effective material for various environmental applications. One critical aspect is the detection and preconcentration of heavy metal concentrations in tap water, which plays a crucial role in monitoring water quality and safeguarding human health. In line with this objective, we synthesized N,N,N-trimethyl-N-hexadecylammonium and N-alkylpyridinium hexafluorophosphate ionic liquids immobilized on silica gel, which were subsequently employed as stationary phases in the solid phase extraction method for preconcentrating trace heavy metals. The utilization of these materials significantly contributed to the enhancement of a simple and affordable solid-phase extraction method for the precise analysis and determination of trace heavy metals in water samples. |
1. Introduction
Industrial development, agricultural practices, and human activities are causing major environmental issues, which significantly increase toxic pollutants in water, food, and soil.1,2 Over the past few years, water pollution by heavy metals with an atomic density higher than or equal to 4.5 kg dm−3, such as cadmium (Cd), lead (Pb), mercury (Hg), arsenic (As), chromium (Cr), etc. caused severe problems to human health due to the high level of toxicity.3–6 While they prove resistant to natural degradation, they readily accumulate in living organisms, thereby presenting a potential hazard to human health, even at low concentrations.7 Water contamination with heavy metals is a global issue that affects human health directly and indirectly.8 Cadmium at trace levels accumulates in the human body, affecting numerous organs such as liver, lung, brain, etc. Cadmium is the reason for hepatic, hematological, and immunological effects on human health. Lead potentially harms every organ and system in the body.9 Adults who have been exposed for an extended period may experience lower performance in various tests that assess nervous system functions, weakness in their fingers, wrists, or ankles, minor increases in blood pressure, and anemia.10 Heavy metals exist in ultra-trace level concentrations and in complicated matrices, in which there are many other substances that interfere with the analysis of the target heavy metal ions.4 Consequently, it is a great challenge to design and construct a reliable and effective technique for analytical determinations of heavy metals.11,12Traditionally, there are several methods for extraction of heavy metals, including liquid–liquid extraction (LLE),13 chemical precipitation,14 and ion exchange.15 These methods, however, have several issues, including requirements for large volumes of high-purity organic solvents, expensive glassware and equipment, and low quantitative recoveries. Besides, these problems can be mitigated by using solid-phase extraction (SPE) techniques,16 which are easily automated and rapid extractions and provide better efficiency and an eco-friendly environment.17 Compared with other extraction methods, SPE has the characteristics of high sensitivity, high enrichment factors, and less organic solvent usage. SPE is also an efficient and selective method for the preconcentration of analytes.18–21 SPE is a cost-effective and reliable technique as an alternative to LLE in sample preparation.19,22
To extract and enrich trace metal ions from environmental samples, a range of ligands or functional groups are immobilized onto a solid support matrix which are called solid phase extractants. Silica gel is extremely significant as an ideal form of support since it has numerous different benefits. Silica gel is chosen as a support because of its high surface area, and high mechanical and thermal stability.23 In addition, silica gel is also effortlessly modified by interacting with silane-coupling agents via the silanol group on the surface.24,25 Immobilization of organic compounds with specific functional groups on the surface of silica gel has found widespread application in research and industrial domains.25–28 While organic compound-modified silica gel for chromatography was popularly used, sol–gel silicates were studied and used as efficient adsorbent materials for the removal of these pollutants from water.29,30
Ionic liquids (ILs) are composed entirely of cations and anions that melt below 100 °C.31 ILs are mainly organic compounds with asymmetric alkyl chains that avoid forming lattice structures, which ensures that the ionic liquids exist in the liquid phase at below 100 °C. Currently, ILs are widely considered green materials and applied in various fields due to their diversity in chemical structures. ILs can be easily tuned by substituting cations or anions according to the particular purpose. Their success comes mainly from their unique and fascinating characteristics, including low or negligible vapor pressure associated with high thermal stability, tunable viscosity, and miscibility with water and organic solvents.18,32,33 ILs have been known as green solvents in various fields, including organic synthesis, extraction, polymers, and nanomaterials.18,34,35 These stationary phases can be used as sorbents for sample preparation through a variety of sorption mechanisms, including anion exchange, hydrogen bonding, and hydrophobic interactions. The immobilization of ILs to silica materials and the diversity of IL structures provide a new application direction in analytical chemistry. Immobilization of ILs to silica materials and the diversity of IL structures offer a new approach to application in analytical chemistry.36–38 Liu and co-workers first introduced ionic liquid-immobilized silica for headspace solid-phase microextraction of benzene, xylene, toluene, and ethylbenzene.39 In the last decade, the ILs supported onto silica gel used as efficient sorbents in SPE have been studied intensively.40–52
In 2010, Liang and co-workers prepared 1-butyl-3-methylimidazolium hexafluorophosphate-modified silica via physical modification and applied it for the determination of trace amount of Cd2+ in water samples with adequate outcomes.53 1-Methyl-3-butyl-imidazolium bromide ionic liquid was a moiety physically attached to silica gel by Ayata and co-workers.54 The separation and determination of Pb2+ ion in environmental samples was developed with an SPE column containing ionic liquid-modified silica. Gharehbaghi and co-workers prepared a 1-butyl-3-methylimidazolium hexafluorophosphate ionic liquid-modified silica sorbent for simultaneous preconcentration of Cd2+, Co2+, Mn2+, Ni2+, and Pb2+ ions from water samples.55 In order to increase the adsorption, the complex of metal ions and 1-(2-pyridylazo)-2-naphthol ligand was formed at a suitable pH. Saleem and co-workers reported extraction of Pb2+ from water samples using ionic liquid-modified silica sorbents.56 In their research, both 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl) imide and 1-hexyl-3-methylimidazolium bis(trifluoromethylsulfonyl) imide were coated onto the surface of silica which was demonstrated to be a suitable stationary phase for the SPE process. In the same year, 1-(2-aminoethyl)-3-butylimidazolium hexafluorophosphate was prepared and modified onto silica gel by Shengping Wen and co-workers.57 This material was used in the direct retention of Cd2+via the complex formation of a metal ion and amino group of silica-supported ionic liquid. Although the modification of an ionic liquid onto the surface of a silica hinge on physical interaction is easy and simple, they were corroborated as a novel stationary phase for the SPE route in the qualitative analysis of metallic elements.
In this paper, we develop a sorbent using ionic liquids grafted onto silica gels for SPE ionic liquids. Five materials, including N,N,N-trimethyl-N-hexadecylammonium and N-alkylpyridinium hexafluorophosphates grafted onto silica gel were synthesized and characterized by FTIR, TGA, BET, SEM, and EDX. It is the first time these materials were applied as solid-phase extractants to extract Pb2+, Cd2+, and Cr3+ from an aqueous solution. To examine the effect of these materials, extractions were carried out in a standard solution and real water samples. The optimized conditions for sorption and enrichment of Pb2+, Cd2+, and Cr3+ were studied. The extraction performance of the material can be compared to that of the previous literature.
2. Experimental section
2.1. Materials, equipment, and analytical methods
N,N,N-trimethyl-N-hexadecylammonium bromide (≥99%), potassium hexafluorophosphate (≥99%), Triton X-100, and nitric acid (70% in H2O) were purchased from Sigma-Aldrich. The standards of Pb2+, Cd2+, and Cr3+ ions were purchased from Fisher-Acros. 1-(2-Pyridylazo)-2-naphthol (PAN), toluene, acetonitrile (99.8%), and methanol were obtained from Merck. Silica gel was obtained from Himedia (India) (60–200 μm). Deionized water was prepared using a Milli-Q system. Molded Frits (20 μm) were used for a 3 mL empty tube.The pH analysis was performed with the Mettler–Toledo parameter. FT-IR analyses were performed with a Bruker E400 using KBr pellets. N2 isotherm measurements were recorded on a Quantachrome NOVA 3200e. TG analysis was measured using a Q-500 instrument. Scanning electron microscopy (SEM) was performed on a Hitachi S-4800. EDX analysis was performed on an EMAX energy EX-400. Elemental analysis was performed on an Agilent 7500X with an Octopole Reaction System Inductively Coupled Plasma Mass Spectrometer.
2.2. Preparation of ionic liquid immobilized onto silica
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, w/w) at 80 °C for 4 h. The purpose of activation is not only to upgrade the content of silanol groups on the surface but also to ease metal oxides. Next, silica gel was stirred with hydrochloric acid (1:1, w/w) for a day at room temperature. Finally, silica gel was washed twice with distilled water until pH 7.0 was reached and dried under vacuum at 120 °C for 24 h.
:1, w/w) at 80 °C for 4 h. The purpose of activation is not only to upgrade the content of silanol groups on the surface but also to ease metal oxides. Next, silica gel was stirred with hydrochloric acid (1:1, w/w) for a day at room temperature. Finally, silica gel was washed twice with distilled water until pH 7.0 was reached and dried under vacuum at 120 °C for 24 h.
 | ||
| Fig. 1 The preparation of DL1. | ||
FT-IR (4000–400 cm−1, KBr): 3376 cm−1, 2917 cm−1, 2850 cm−1, 1385 cm−1, 1055 cm−1, 788 cm−1.
1H NMR (500 MHz, MeOD) δ 3.41–3.33 (m, 2H), 3.31 (dt, J = 3.2, 1.6 Hz, 1H), 3.14 (s, 9H), 1.82–1.75 (m, 2H), 1.44–1.23 (m, 29H), 0.90 (t, J = 7.0 Hz, 3H) (Fig. S1†).
13C NMR (125 MHz, MeOD) δ 138.50 (s), 123.31 (s), 121.81 (s), 51.91 (s), 50.84 (s), 36.75 (s), 24.24 (s), 6.07 (s) (Fig. S2†).
N,N,N-trimethyl-N-hexadecylammonium hexafluorophosphate [Me3NC16H33]PF6 (3.5 mmol, 1.503 g) was heated with activated silica gel (1:3, w/w) in 30 mL acetone in a round-bottomed flask for 4 h. Upon completion, acetone was evaporated using a vacuum rotavap to provide a solid white product (DL1@SiO2).
:1 in a round-bottomed flask. The reaction mixtures were heated at 100 °C for 4 h. The crude product was obtained in yellow color with high viscosity and washed with diethyl ether (3 × 10 mL) to remove excess substrates. Next, N-butylpyridinium bromide (2 mmol, 0.432 g) and potassium hexafluorophosphate (2 mmol, 0.368 g) were heated for 4 h for anion metathesis (Fig. 2). The mixture was filtered through Celite to remove KBr. N-butylpyridinium hexafluorophosphate (DL2) was obtained after evaporating the methanol. The same procedure was adopted for other ionic liquids (DL3, DL4, and DL5) with the replacement of butyl bromide with octyl bromide, decyl bromide, and octadecyl bromide, respectively.
 | ||
| Fig. 2 The preparation of DL2 to DL5 ionic liquids. | ||
N-alkylpyridinium hexafluorophosphate (0.5 g) was entirely dissolved in 50 mL acetone in a 250 mL round-bottomed flask. After that, activated silica was added to the solution and stirred for 4 h. The acetone was removed under a vacuum rotavap to provide white products (DL2@SiO2 to DL5@SiO2).
DL2, DL3, DL4, and DL5 ionic liquids were characterized by FTIR, 1H and 13C NMR.
2.2.3.1 DL2 ionic liquid. FT-IR (4000–400 cm−1, KBr): 3428 cm−1, 2970 cm−1, 2880 cm−1, 1637 cm−1, 1057 cm−1, 792 cm−1 (Fig. S11†).
1H NMR (500 MHz, MeOD): δ 9.06 (d, J = 5.9 Hz, 2H), 8.62 (t, J = 7.8 Hz, 1H), 8.14 (t, J = 6.5 Hz, 2H), 4.69 (t, J = 7.6 Hz, 2H), 2.08–1.97 (p, J = 7.4 Hz, 2H), 1.47–1.39 (m, 2H), 1.01 (t, J = 7.4 Hz, 3H) (Fig. S3†).
13C NMR (125 MHz, MeOD) δ 145.6, 144.7, 128.3, 61.8, 25.8, 22.3, 13.2 (Fig. S4†).
2.2.3.2 DL3 ionic liquid. FT-IR (4000–400 cm−1, KBr): 3424 cm−1, 2954 cm−1, 2856 cm−1, 1638 cm−1, 1051 cm−1, 821 cm−1 (Fig. S12†).
1H NMR (500 MHz, MeOD) δ 9.05 (d, J = 5.8 Hz, 2H), 8.62 (t, J = 7.8 Hz, 1H), 8.14 (t, J = 6.8 Hz, 2H), 4.71–4.65 (t, J = 7.6 Hz, 2H), 2.07–2.00 (p, J = 7.4 Hz, 2H), 1.39 (d, J = 3.8 Hz, 4H), 1.32 (dd, J = 12.7, 5.9 Hz, 6H), 0.90 (t, J = 6.9 Hz, 3H) (Fig. S5†).
13C NMR (125 MHz, MeOD) δ 146.9, 145.9, 129.5, 63.2, 32.8, 32.4, 30.4, 27.1, 23.6, 14.3 (Fig. S6†).
2.2.3.3 DL4 ionic liquid. FT-IR (4000–400 cm−1, KBr): 3441 cm−1, 2954 cm−1, 2853 cm−1, 1639 cm−1, 1054 cm−1, 822 cm−1 (Fig. S13†).
1H NMR (500 MHz, MeOD): δ 9.19–9.16 (m, 2H), 8.73 (t, J = 7.8 Hz, 1H), 8.28–8.23 (m, 2H), 4.81–4.77 (t, J = 7.6 Hz, 2H), 2.15 (p, J = 7.4 Hz, 2H), 1.50 (d, J = 3.9 Hz, 4H), 1.42–1.37 (m, 9H), 0.99 (t, J = 7.0 Hz, 3H) (Fig. S7†).
13C NMR (125 MHz, MeOD) δ 145.5, 144.6, 128.2, 61.8, 31.6, 31.1, 29.3–28.8, 28.7, 25.8, 22.3, 13.0 (Fig. S8†).
2.2.3.4 DL5 ionic liquid. FT-IR (4000–400 cm−1, KBr): 3990 cm−1, 2953 cm−1, 2849 cm−1, 1639 cm−1, 1051 cm−1, 826 cm−1 (Fig. S14†).
1H NMR (500 MHz, MeOD) δ 9.05 (d, J = 5.5 Hz, 9H), 8.61 (t, J = 7.8 Hz, 4H), 8.13 (t, J = 6.9 Hz, 7H), 4.71–4.62 (m, 10H), 2.09–2.01 (m, 8H), 1.28 (s, 30H), 0.89 (t, J = 7.0 Hz, 3H) (Fig. S9†).
13C NMR (125 MHz, MeOD) δ 146.9, 145.9, 129.5, 63.1, 32.9, 32.4, 30.9–30.2, 30.0, 27.1, 23.6, 14.4 (Fig. S10†).
2.3. Preparation of the column
To prepare an SPE column, 200 mg of ionic liquid immobilized on activated silica gel was precisely weighed and placed between two 20 μm frits in the empty cartridge with 6.0 cm length and 0.9 cm internal diameter. The columns were labeled and stored at room temperature for sample preparation.2.4. General procedure for the solid-phase extraction
For DL1 to DL5, the working solution, including 50 μg L−1 of each metal and PAN 2.5 × 10−3 M, which is an orange-colored dye, acts as a tridentate ligand and can form stable chelate complexes between metal ions with its oxygen atom and azo group. It was freshly prepared in distilled water from 1000 mg L−1 stock solution of three metal ions (Pb2+, Cd2+, Cr3+). The pH of the working solution was adjusted to 8.5 using borax buffer to form metal–PAN complexes. The SPE columns were cleaned and conditioned with distilled water (5 × 3 mL). The working solution (30 mL, 50 μg L−1) and PAN using borax buffer (pH 8.5) were loaded on the column. The molar ratio of metal ions and PAN (1:6) was applied for the present method, followed by our previous report.58 A small amount of KCl was added to enhance the ionic force, and Triton-X 100 played the role of a surfactant to dissolve all the precipitate to generate a homogeneous solution.
2.5. Adsorption experiment
The adsorption experiment was performed by adding 100 mg of DL1-SiO2 adsorbent into 20 mL of the metallic solution, including Cr3+, Cd2+, and Pb2+, at room temperature to evaluate the adsorption capacity. The metallic solution consisting of three metallic ions, Cr3+, Cd2+, and Pb2+, was prepared at different concentrations 10, 30, 50, and 100 μg L−1 with PAN and the pH was adjusted to 8.5. After adsorption, DL1-SiO2 was removed from the working solution using a 0.45 μm syringe and the concentration of the metallic solution was determined by ICP-MS. The adsorption process was explained by Langmuir (1) and Freundlich models (2). These models can be expressed as an equation where a and amax are the equilibrium and monolayer sorption capacities of the sorbents (μg mg−1), respectively, C is the equilibrium concentration (mass per unit volume) (μmol L−1), and k is the rate constant related to adsorption.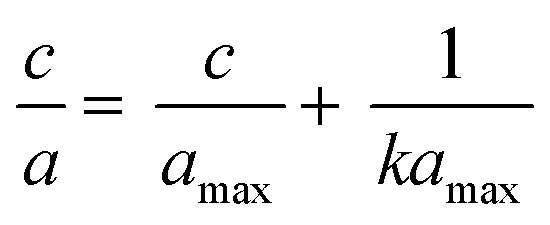 | (1) |
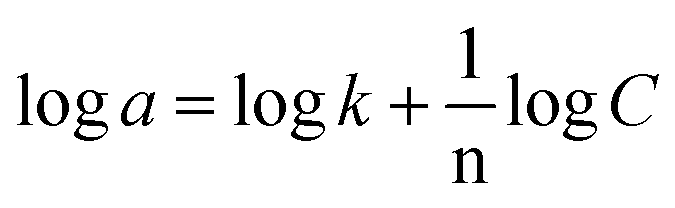 | (2) |
3. Results and discussion
3.1. Characteristics of materials
DL1 ionic liquid was synthesized following the procedure described in the previous literature. The structure of DL1 was confirmed by FTIR, 1H, and 13C NMR analyses. The as-synthesized DL1 was grafted onto silica to provide DL1-SiO2. The structure of DL1-SiO2 was determined by FTIR spectroscopy. The FTIR spectra of DL1, silica, and DL1-SiO2 are presented in Fig. 3. The peak at 1050 cm−1 was related to asymmetric Si–O–Si vibration. The variation in the width of the Si–O–Si vibration at 1050 cm−1 suggested that free silanols were involved in the silylation process, as indicated by the increase in the silanol band in the ionic liquid-modified silica spectrum.59 As depicted in Fig. 3, there was a typical broad peak, which was referred to as O–H stretching vibration. FT-IR spectra of the IL showed characteristic peaks at 2917, 2850, and 1385 cm−1, which were associated with the stretching vibration of C–H groups and C–N bonding.60 The peak at 819 cm−1 can be attributed to P–F stretching vibration.59 These signals of FT-IR spectra can prove that the ionic liquid N,N,N-trimethyl-N-hexadecylammonium (DL1) was successfully grafted onto a silica gel surface.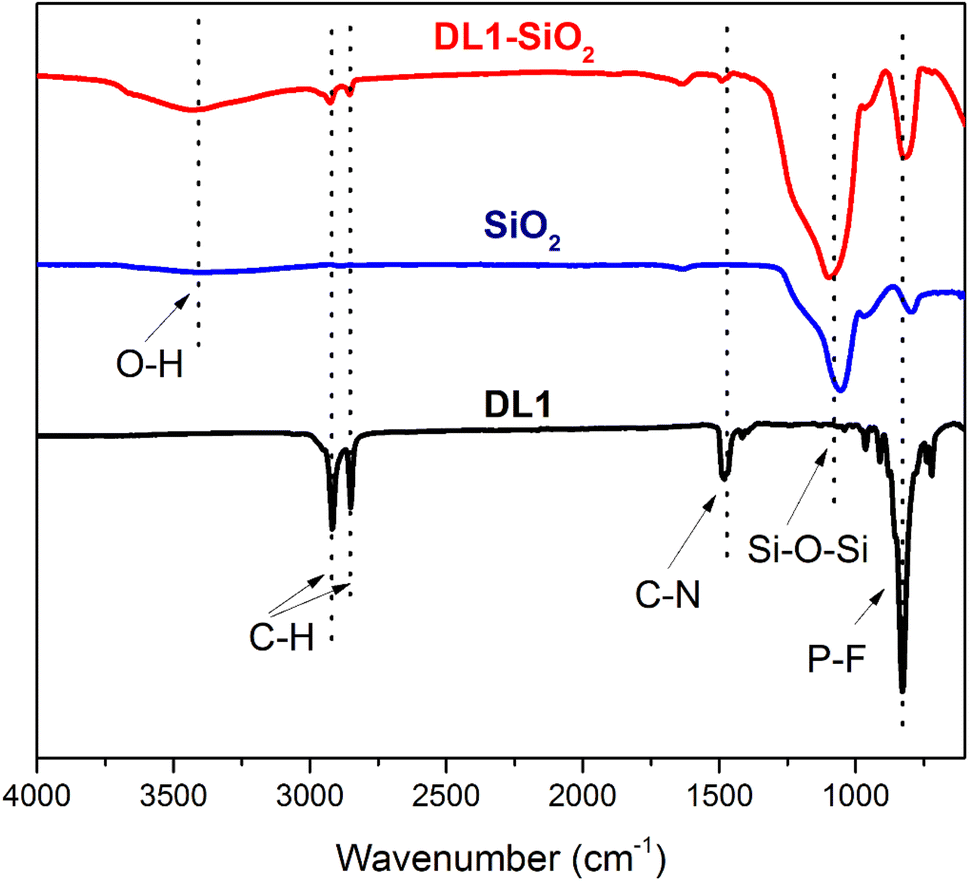 | ||
| Fig. 3 FT-IR spectra of DL1-SiO2, SiO2, and DL1. | ||
Thermal gravimetric analysis (TGA) was used to determine the stability and the mass of the ion liquid grafted on the silica gel (Fig. 4). TGA was conducted in the temperature range from 25 to 800 °C with a 5 °C min−1 heating rate in an air atmosphere. N,N,N-trimethyl-N-hexadecylammonium hexafluorophosphate showed a decline in weight (∼4%) at below 250 °C, which could be attributed to the solvent and H2O. From 250 to 350 °C, there was a significant weight loss (10%) that was due to an organic molecule that corresponded to the ionic liquid. The result showed that the ionic liquid immobilized silica gel was successfully synthesized.
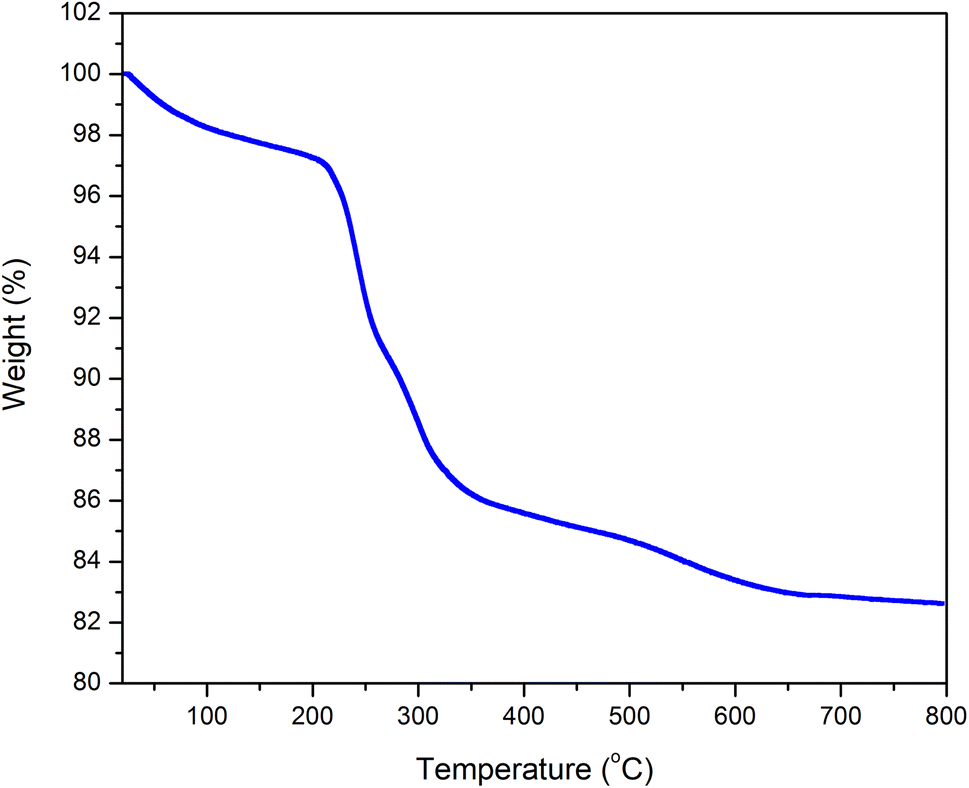 | ||
| Fig. 4 Thermal gravimetric analysis diagram of DL1-SiO2. | ||
The N2 sorption isotherm was recorded at 77 K and the specific surface area of the DL1-SiO2 sorbent was 166.595 m2 g−1 with an average pore width of 36 Å. Fig. S15† demonstrates that immobilizing the ionic liquid on silica reduced the surface area and pore width of silica gel, at about 166.595 m2 g−1 and 0.469 cm3 g−1, respectively. The results indicated that the immobilization process for the ionic liquid on silica was successfully achieved.
Energy-dispersive X-ray spectroscopy was used to assess the elemental composition of the ionic liquid-functionalized silica gel surface (EDX). EDX mapping was utilized to estimate the distribution of components derived via random scanning of particular locations. As depicted in Fig. 5, the majority of DL1-SiO2 was composed of silica and oxygen. The EDX spectrum of the synthesized material exhibits distinctive peaks at energy levels of 0.28 and 0.39 keV, indicating the likely presence of carbon (C) and nitrogen (N) on the material's surface. Furthermore, the appearance of elements F and P found in the spectrum at 0.70 keV and 2.06 keV showed that the PF6 anion was successfully altered and grafted onto the silica gel surface. In conclusion, the EDX results revealed the contribution of carbon, nitrogen, oxygen, silica, and phosphonium in the composition, showing that ionic liquids were successfully coated onto the silica.
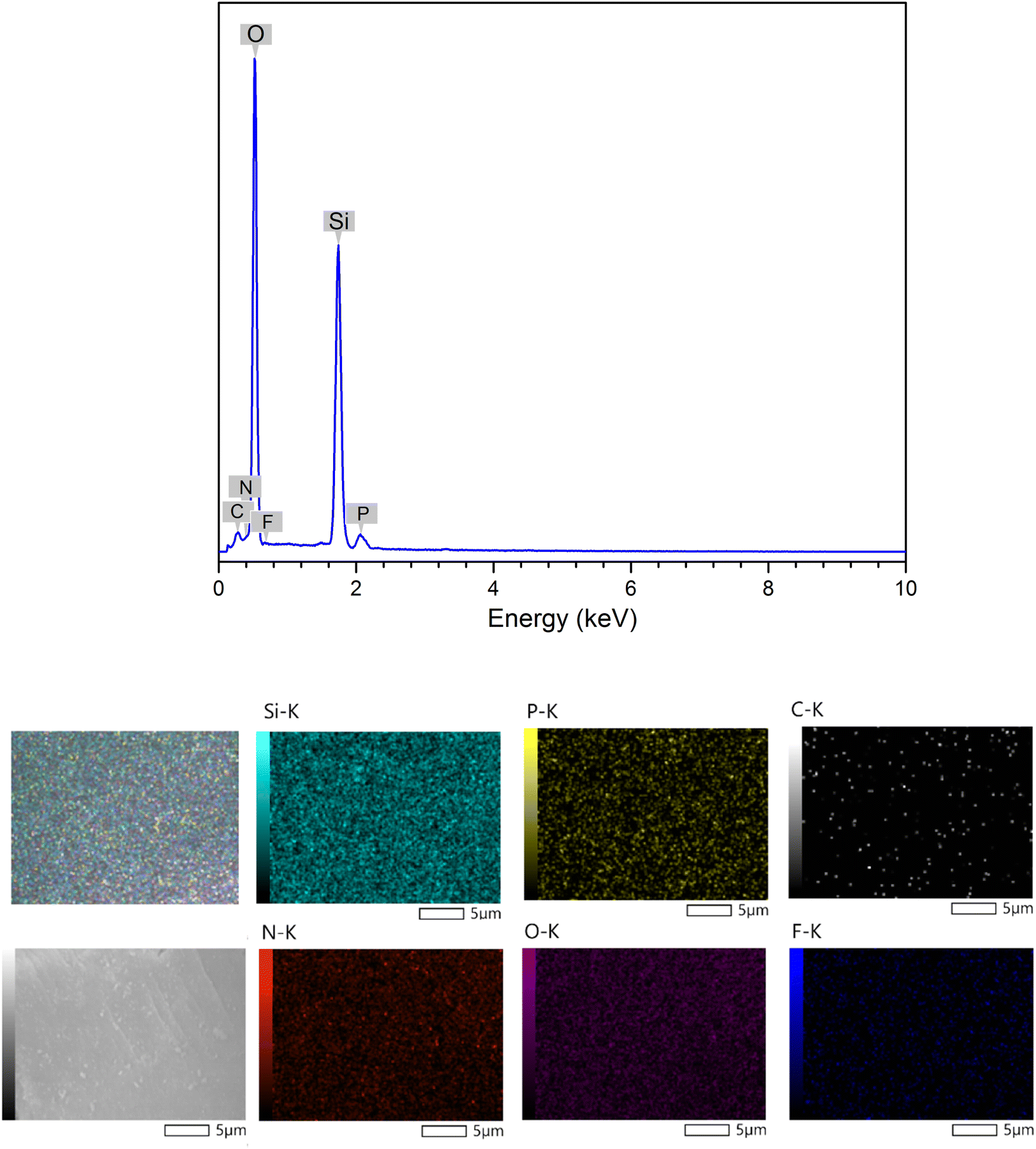 | ||
| Fig. 5 EDX spectrum and SEM mapping of elemental distribution in DL1-SiO2. | ||
Scanning electron microscopy was used to examine the morphological properties of the synthesized material DL1-SiO2 at various magnifications. As seen in Fig. 6, after immobilizing the ionic liquid DL1 onto silica gel, the material has a lamellar fracture and a rough surface, the same as the shape of regular silica gel. There is no substantial change in particle size between commercial silica gel and DL1 immobilized silica gel. This suggests that the silica gel particles were mechanically stable during immobilization. Besides, at magnification 2.000 and 10.000, several macro channels were evenly distributed on the material's surface as opposed to thin surface morphologies of pure silica gel, indicating the increase in sorption efficiency toward metal ions. Overall, the results have confirmed that the grafting process has a significant influence on the material's morphology which determines the metal adsorption capacity.
 | ||
| Fig. 6 Scanning electron microscopy images of (a) commercial silica gel and (b) ionic liquid DL1 grafted onto silica gel at different magnifications. | ||
3.2. The ability of retention
Five types of sorbents were tested to find the best sorbent for the preconcentration of metal ions (Fig. 7). All the sorbents that were placed into the SPE columns were preconditioned with 5 × 3.0 mL of distilled water. 30 mL of a mixed standard working solution including three metallic ions (50 μg L−1 for each) and the PAN indicator with the ratio 1:6 was prepared and adjusted to pH 8.5. Next, the working solution was loaded through the SPE column at a flow rate of 1.0 mL min−1. Five materials were tested, including silica modified with imidazolium ionic liquids and pyridinium ionic liquids with different alkyl chains. After loading through the column, the leaching solution from 5 cartridges was determined by ICP-MS to analyze the concentration of each metallic ion. The results demonstrated that the pyridinium hexafluorophosphate provided low effectiveness in the sorption of Cr3+, Cd2+, and Pb2+. The study revealed that the alkyl chain of the pyridinium ionic liquids did not affect the sorption of metal ions. The DL1-SiO2 sorbent with ammonium hexafluorophosphate provided the best sorption efficiency toward metal ions.
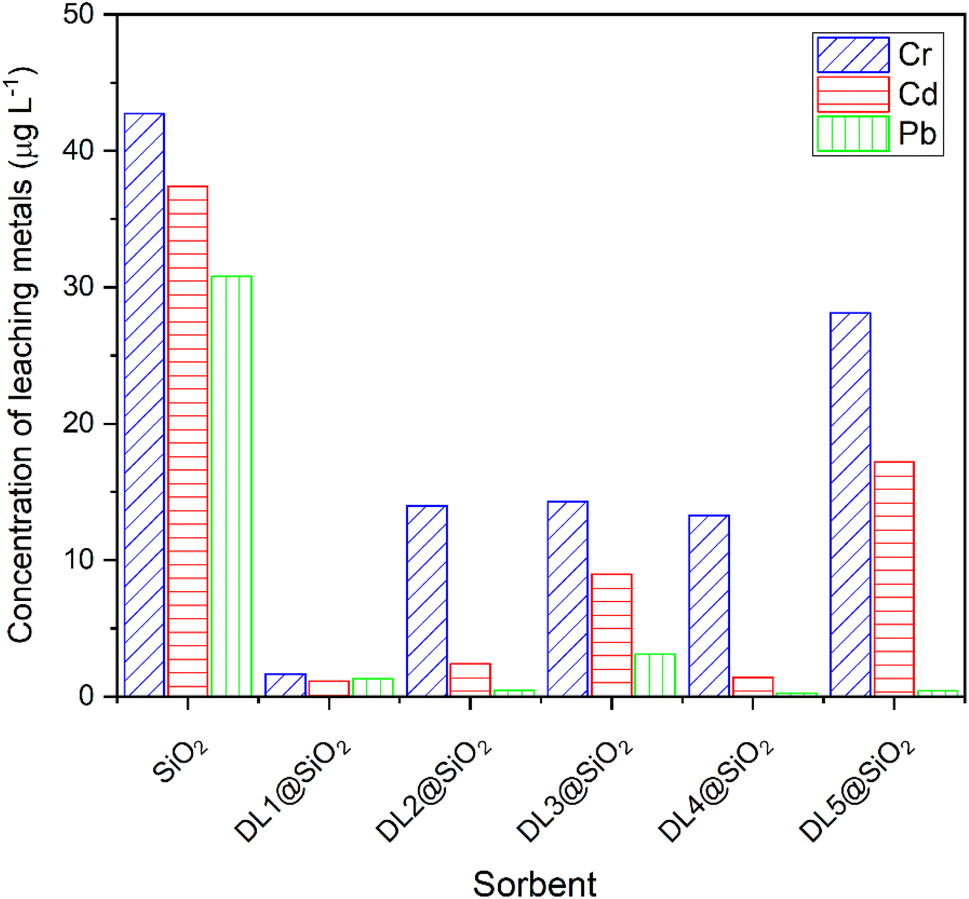 | ||
| Fig. 7 The concentration of leaching metals applying different materials. | ||
3.3. Effect of sample volume
The sample volume plays a key role in obtaining a high preconcentration. It was determined that combining a high-loading volume with a low concentration decreased retention. After choosing DL1@SiO2 as the best adsorbent, two working solutions, including 30 mL, 50 μg L−1 and 150 mL, 10 μg L−1 of the three investigated metallic ions were loaded through the column containing DL1@SiO2. Both of them were adjusted to pH 8.5 and when the loading process was finished, the ions retained on the materials were eluted using HNO3/ethanol (1 mol L−1). The content of the eluted solution was determined by ICP-MS to obtain the recovery. The recovery of a material is the amount of a metallic ion that is present in the solution after the cartridge is eluted by the suitable solvent. The results were compared with the total amount of the metallic ions found in both leaching solution and eluent solution. The recovery was calculated according to the following equations:where C0 is the initial concentration of each metallic ion in the solution (μg L−1); Ce is the concentration of each metallic ion in the eluted solution (μg L−1).
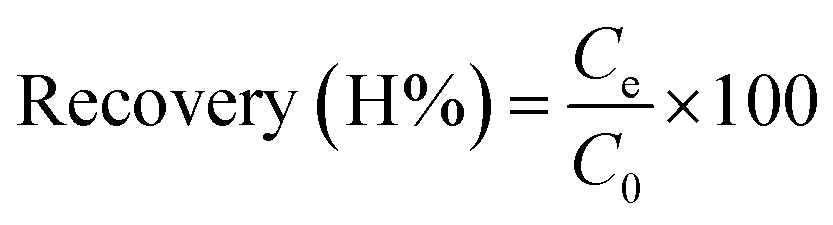
As depicted in Fig. 8, at a concentration of 50 μg L−1 and a loading volume of 30 mL, the retention of Cr2+ and Pb2+ was over 95%, while the retention of Cd2+ was below 60%. With the same concentration of metal ions, the use of 10 μg L−1 and a loading volume of 150 mL provided lower recovery than 50 μg L−1 and a loading volume of 30 mL, presumably because of self-elution.
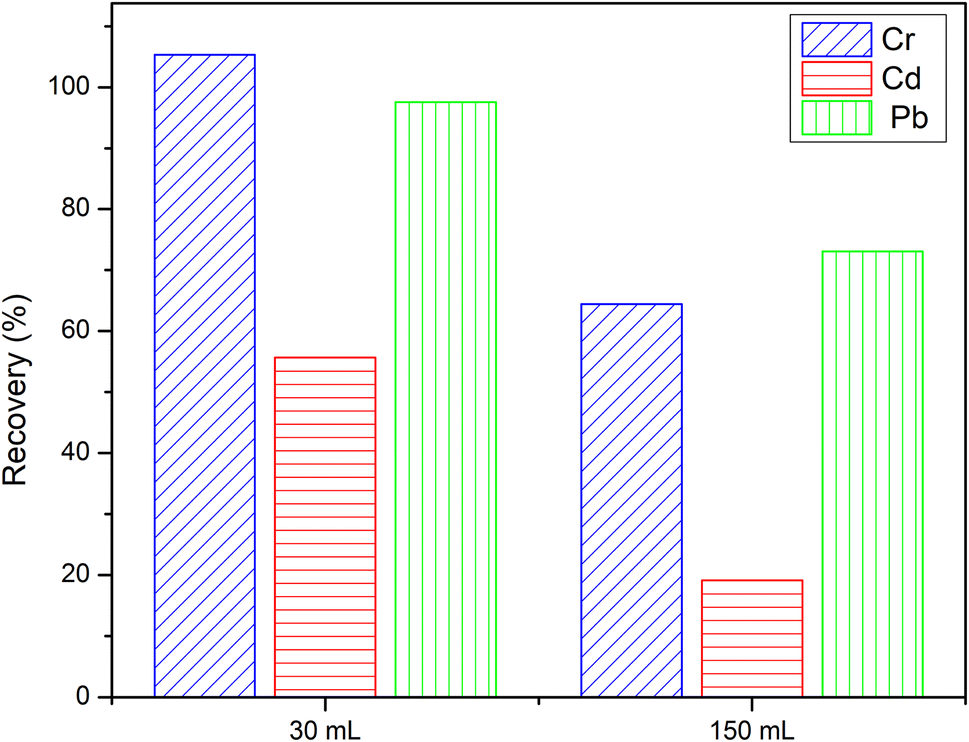 | ||
| Fig. 8 Effect of sample volume. | ||
3.4. Effect of pH
The sorbent did not retain the pure metal ions, so the popular reagent PAN was chosen to complex with metal ions. The pH of sample solutions, which has a significant impact on the stability of the metal–PAN complexes, is one of the essential factors for efficient retention of the metal ions on the sorbent. To find the optimal pH for simultaneous extraction of the metal ions from aqueous samples, pH was varied in the range of 6.5–9.5 using buffers prepared from borax and sodium hydroxide or hydrochloric acid (0.1 mol L−1). The sorption percentage of metal ions was calculated based on the difference between the concentration of metal ions in the initial solution and the solution leaching from the SPE column. It was evident that the sorption of all metal ions on the DL1@SiO2 increased with the rise of pH and reached the maximum at pH 8.5. The low efficiency of the sorption was observed at pH < 7.5 due to the unstable metal–PAN complex. All subsequent work was, therefore, carried out at the optimum pH of 8.5 (Fig. 9).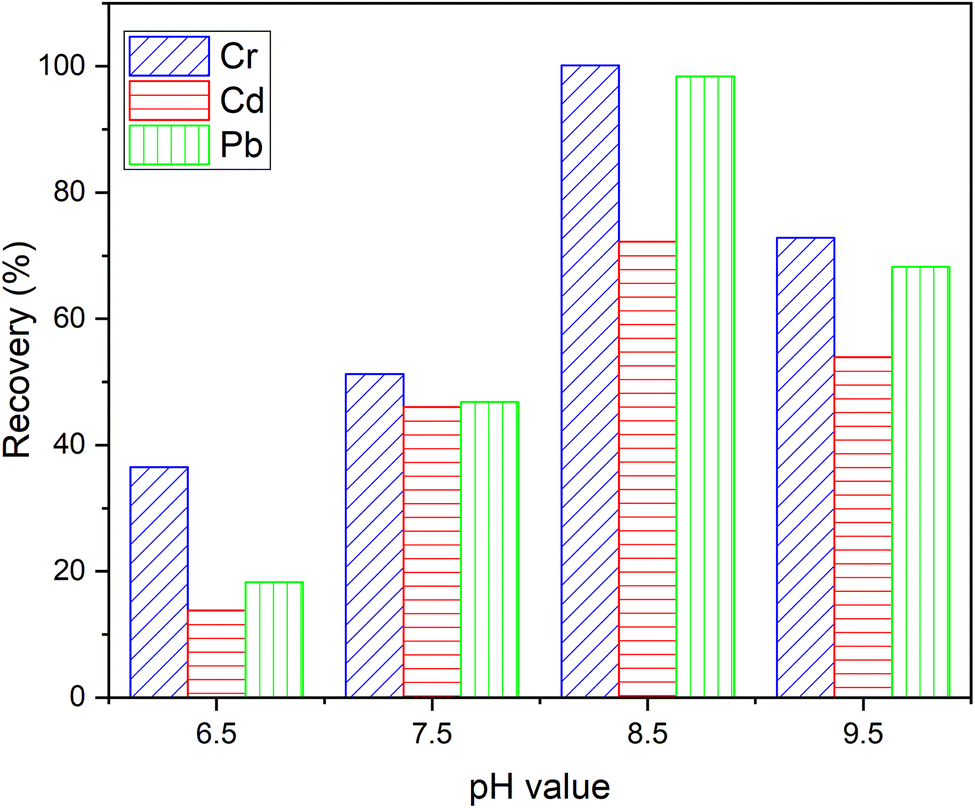 | ||
| Fig. 9 Effect of pH on the recovery of three metallic ions. | ||
3.5. Effect of elution conditions on recovery
The eluent is an important parameter for SPE investigation. According to preliminary studies and experiments, HNO3/ethanol (1.0 mol L−1) was used as a good eluent for column elution to cleave metal complexes' bonds.37 As shown in Fig. 10, quantitative recoveries were afforded with Cr3+ and Pb2+ using 1 or 2 mL of HNO3/ethanol (1.0 mol L−1). A low recovery of Cd2+ (∼60%) was obtained with 2 mL of HNO3/ethanol. Actually, there is a slight discrepancy in the recovery of three metallic cations between eluting with 1 and 2 mL of HNO3/ethanol. Therefore, the small amount of elution will contribute to minimizing the consumption of acid and reducing the amount of acid released into the environment.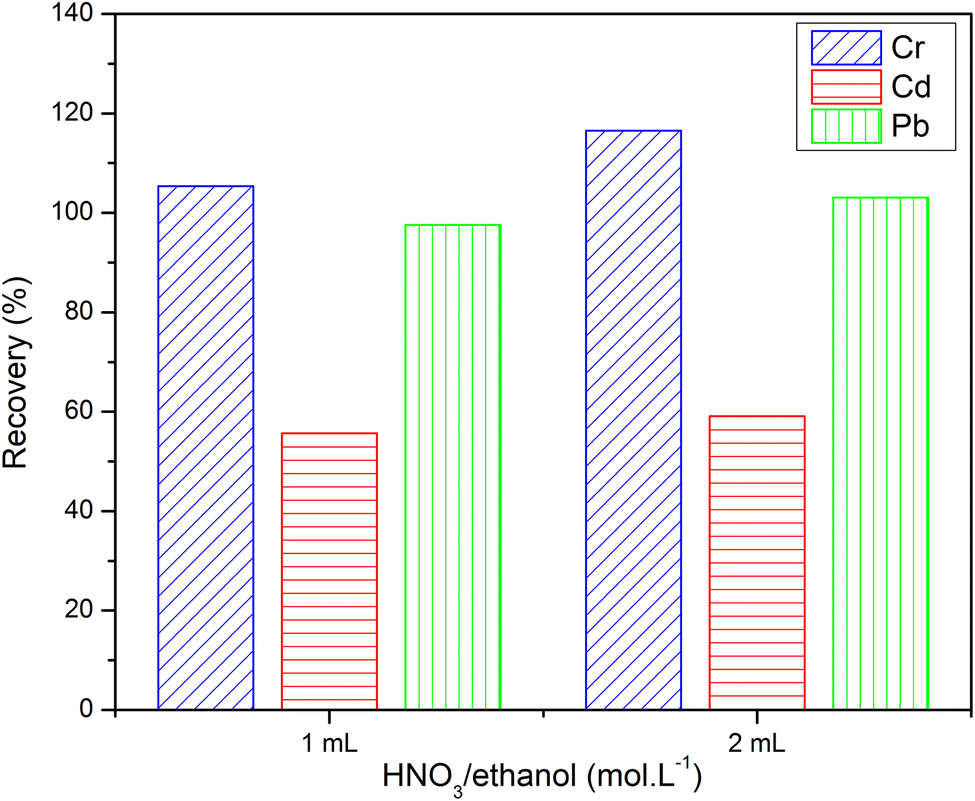 | ||
| Fig. 10 Effect of nitric acid volume on the elution recovery. | ||
3.6. Interference study
Water matrices are complicated in which several interfering ions can affect the result of analysis. After loading the mixed standard solution (50 μg L−1 for each metallic ion) containing foreign ions with high concentration into the column, the recovery of the three ions, Cr3+, Cd2+, and Pb2+, was achieved by eluting the mixed standard solution with 1.0 mL of HNO3/ethanol (1 mol L−1) solution. Then the content in the effluents was determined using ICP-MS and the recoveries were calculated. As shown in Table 1, the results indicated that most of the tested ions had no notable influence on the determination of the solution consisting of Cr3+, Cd2+, and Pb2+ species. The following tolerance amount ratios were obtained: 500-fold excess of Na+, K+, Ca2+ and Mg2+, 450-fold excess of Cl−, 2000-fold excess of SO42− and 650-fold excess of CO32−. The results showed that the interferences of most foreign ions for the preconcentration of Cr3+, Cd2+, and Pb2+ species in real samples were negligible. This can be explained by the fact that interfering cations in this experiment cannot form coordinate covalent bonds with the colored dye PAN. Besides, the complexation between the three metallic cations and PAN occurred without the interference of the coexisting anions, namely, Cl−, SO42−, and CO32− in the matrices. The method was also selective for preconcentrating and extracting Cr3+, Cd2+, and Pb2+.| Ion | Added salt | Ratio [interfering ion]/[metallic ion] | Recovery (%) | ||
|---|---|---|---|---|---|
| Cr | Cd | Pb | |||
| Na+ | Na2CO3 | 500 | 100.01 | 74.56 | 97.23 |
| K+ | KCl | 500 | 100.12 | 78.32 | 98.11 |
| Ca2+ | CaCl2 | 500 | 100.67 | 73.24 | 98.20 |
| Mg2+ | MgSO4 | 500 | 98.39 | 74.67 | 97.96 |
| Cl− | KCl | 450 | 99.37 | 74.39 | 93.11 |
| SO42− | MgSO4 | 2000 | 98.26 | 75.21 | 95.96 |
| CO32− | Na2CO3 | 650 | 100.24 | 74.42 | 96.23 |
3.7. Adsorption isotherm
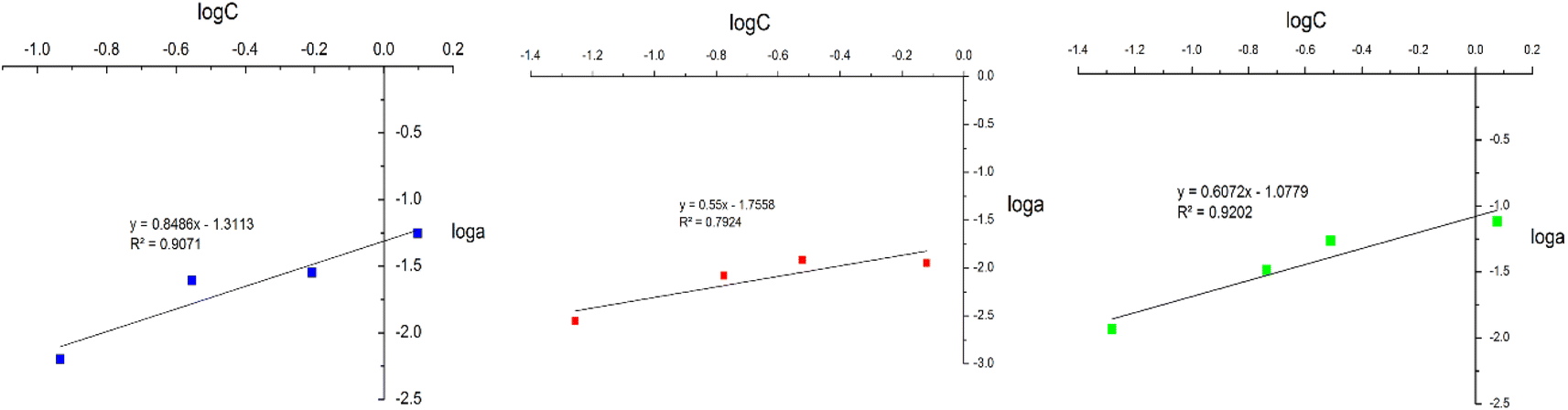
In this study, adsorption isotherms gave information about the concentration of metallic ions between the standard solution and adsorbent, the number of metals adsorbed by DL1@SiO2 and the mechanism of the PAN-metal complexes adsorbed by DL1@SiO2. The Langmuir and Freundlich isotherm models were used to analyze the adsorption data, and the results are shown in Fig. 11 and 12. Herein, the Langmuir isotherm model assumes that there are a fixed number of active sites in a single layer, that each active site can only adsorb an ion, and there is no interaction between the adsorbed ions. Besides, the Freundlich isotherm model shows that the energy and behavior of active sites on the absorbent vary depending on the adsorbent. As depicted in Fig. 11 and 12, the R2 values from the Langmuir model of the three metallic ions were 0.9826, 0.8199, and 0.9805, which were higher than the Freundlich values. Thus, the Langmuir model is suitable for the adsorption process with the amax of 14.58, 10.89, and 20.63 μg mg−1 (Fig. 11).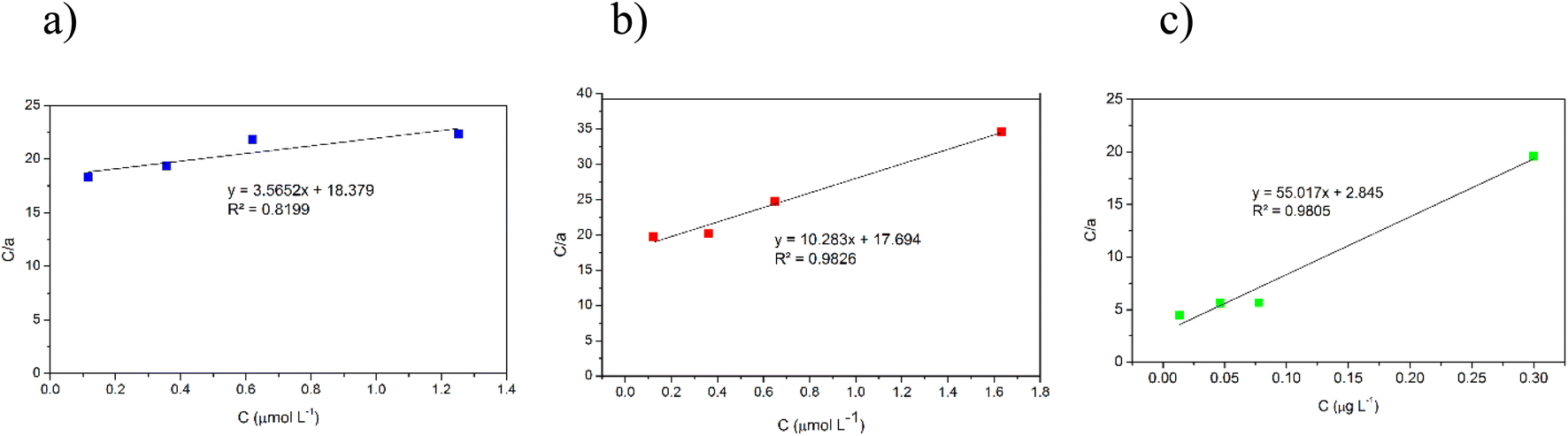 | ||
| Fig. 11 Langmuir model of (a) Cr3+, (b) Cd2+, (c) Pb2+. | ||
 | ||
| Fig. 12 Freundlich model of (a) Cr3+, (b) Cd2+, (c) Pb2+. | ||
The Freundlich isotherm assumes that each adsorption site has different energy and interacts differently with different molecules. The 1/n value indicates the type of isotherm as follows: irreversible (1/n = 0), favorable (0 < 1/n < 1), or unfavorable (1/n > 1). As shown in Fig. 12, the slope values of 1/n for Cr3+, Cd2+ and Pb2+ being lower than 1 indicate that complexes of PAN and the three metallic ions Cr3+, Cd2+, and Pb2+ were adsorbed favorably onto the DL1@SiO2 surface. It was obvious from the correlation coefficient value (R2) that the Langmuir model fitted the experimental data better than the Freundlich model, which indicated the monolayer adsorption of Cr3+, Cd2+, and Pb2+ onto the homogeneous surface of DL1@SiO2 (Fig. 12).
3.8. Reusability of DL1@SiO2
The advantage of adsorbent reuse is greatly related to the cost of practical application of the material. The reusability of DL1@SiO2 was repeatedly investigated for all three metallic ions. The difference in metal removal efficiency of the first and fifth adsorption–desorption cycles for the ions Cr3+, Cd2+, and Pb2+ was 4.8%, 5.7% and 4.5%, respectively, indicating that DL1@SiO2 can be reused at least five times without a significant decrease of the sorption capacity.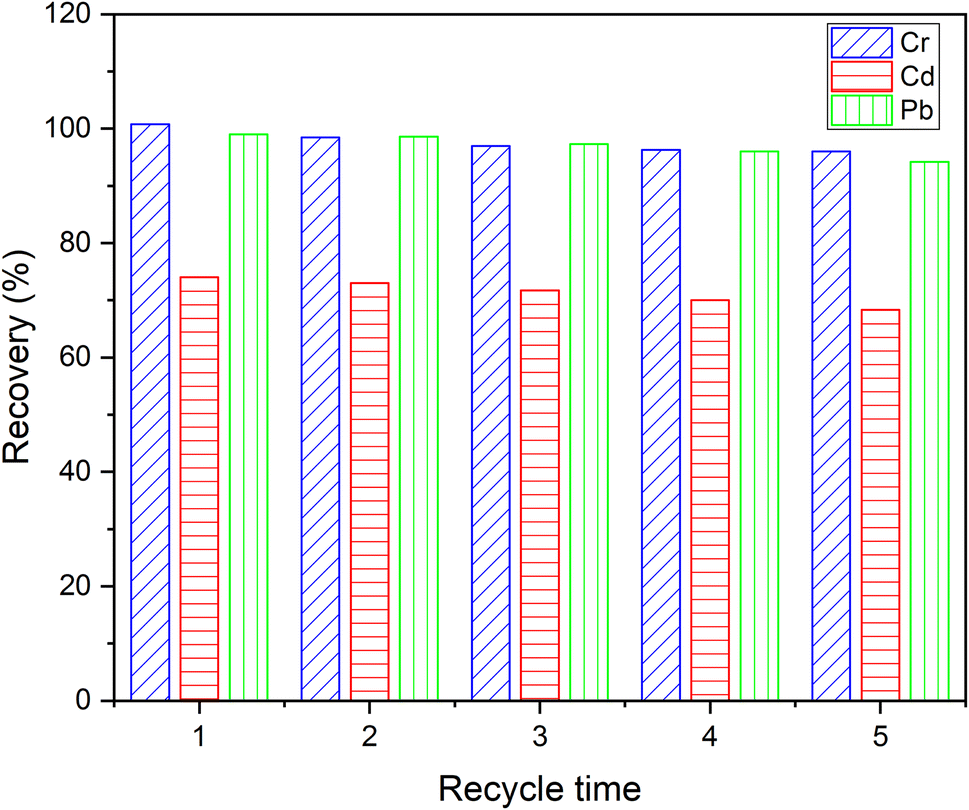
3.9. Validation of the method
The current method's quantitative characteristics, including LOD, LOQ, and RSD, were investigated by determining the metallic cations in tap water, and the results are presented in Table 2. Tap water was taken in Ho Chi Minh City and was added to six volumetric flasks with the same volume (25 mL). Next, 0.2 μg L−1 standard solution and PAN were spiked into the tap water with the molar ratio 1:6. The real sample solution (100 mL) was loaded through the column at a flow rate of 0.7–0.8 mL min−1. The elution step started when the solution HNO3/ethanol 1 mol L−1 (1 mL) was applied. Six replicates were performed to assess the method's precision, and the RSDs were calculated. The LOD and LOQ values are estimated by analyzing 6 duplicates of spiked tap water samples with low analyte concentration. As indicated in Table 2, the results yielded the average concentration value (![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) ), relative standard deviation (SD), LOD, and LOQ.
), relative standard deviation (SD), LOD, and LOQ.| LOD = 3SD, LOQ = 3LOD |
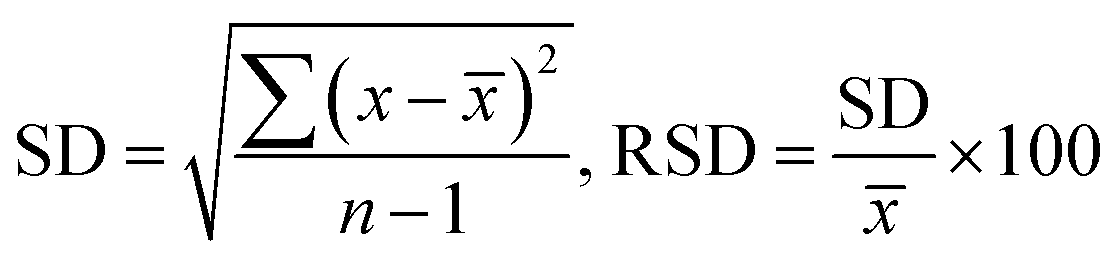
| Analyte | LOD (μg L−1) | LOQ (μg L−1) | RSD (%) |
|---|---|---|---|
| Cr | 0.173 | 0.347 | 1.2 |
| Cd | 0.046 | 0.093 | 4.9 |
| Pb | 0.185 | 0.370 | 3.4 |
The RSD values of the current method were approximately 1.2–4.9%.
The current method's analysis performance was compared with that of the previous literature (Table 3). Our method can be attributed to the novel N,N,N-trimethyl-N-hexadecylammonium ionic liquid immobilizing on silica sorbent, which exhibited high extraction of Cr3+, Cd2+, and Pb2+.
| Method | Metal ions | Enrichment factor | LOD (μg L−1) | RSD (%) | Reference |
|---|---|---|---|---|---|
| 1-Methyl-3-butyl-imidazolium bromide grafted silica gel | Pb2+ | 185 | 0.7 | 4.2 | 54 |
| Graphene oxide (GO)–titanium dioxide (TiO2) | Cu2+, Pb2+, La3+, Ce3+, Eu3+, Dy3+ and Yb3+ | 10 | 0.13–2.64 | 3.2–9.8 | 61 |
| Aminothioamidoanthraquinone grafted silica gel | Pb2+, Cu2+, Ni2+, Co2+ and Cd2+ | 100 | 1.0–22.5 | ≤9 | 62 |
| Iminodiacetic acid (IDA) grafted MWCNTs | V5+, Cr6+, Pb2+, Cd2+, Co2+, Cu2+ and As3+ | 66–101 | 0.4–3.4 | 1–4 | 63 |
| Activated carbon | Cd2+, Co2+, Cu2+, Ni2+, Pb2+, and Zn2+ | 80 | 0.17–2.6 | 1.3–3.7 | 64 |
| This work | Cr3+, Cd2+, Pb2+ | 30–150 | 0.046–0.185 | 1.2–4.9 |
3.10. Real sample analysis
To test the efficiency of the present method, the tap water in Ho Chi Minh City was analyzed using the procedure. The sample was spiked with 0.2 μg L−1. As depicted in Table 4, the recovery values for Cr3+, Cd2+, and Pb2+ were 100.8%, 74% and 97%, respectively. The result showed that the present method was accurate.| Sample | Tap water | Recovery (%) | ||
|---|---|---|---|---|
| Added (μg L−1) | 0 | 0.2 | ||
| Found | Cr | 0.032 ± 0.009 | 0.248 ± 0.015 | 100.8 |
| Cd | 0.153 ± 0.010 | 0.301 ± 0.070 | 74 | |
| Pb | 0.118 ± 0.044 | 0.312 ± 0.062 | 97 | |
4. Conclusion
We have developed ionic liquid-modified silica, which is an effective sorbent for Cr3+, Cd2+, and Pb2+. The ionic liquid-immobilized silica sorbent was characterized by FTIR, TGA, BET, SEM, and EDX. The material provided high efficiency toward Cr3+ and Pb2+. However, Cd2+ was not suitable for the SPE process using this sorbent. The current method is very simple, inexpensive, and environmentally benign. The results demonstrated that the ionic liquid-modified silica could be suitable for further potential applications of SPE technology for analytical chemistry.Author contributions
Linh Dieu Nguyen: conceptualization, methodology, investigation, data curation, writing – original draft, writing – review & editing. The Thai Nguyen: methodology, investigation, data curation. Nhi Hoang Nguyen: methodology, investigation, data curation. Tan Hoang Le Doan: methodology, investigation, data curation. Linh Thuy Ho Nguyen: methodology, investigation, data curation. Phuong Hoang Tran: conceptualization, methodology, writing – review & editing, supervision.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
We thank INOMAR Center (VNU-HCM) for supporting the FTIR and TGA analysis.References
- S. Bolisetty, M. Peydayesh and R. Mezzenga, Chem. Soc. Rev., 2019, 48, 463–487 RSC.
- D. Ramutshatsha-Makhwedzha, R. Mbaya, M. L. Mavhungu and P. N. Nomngongo, J. Environ. Chem. Eng., 2022, 10, 108187 CrossRef CAS.
- C. V. Mohod and J. Dhote, Int. J. Innov. Res. Sci. Eng., 2013, 2, 2992–2996 Search PubMed.
- M. L. Sall, A. K. D. Diaw, D. Gningue-Sall, S. Efremova Aaron and J.-J. Aaron, Environ. Sci. Pollut. Res., 2020, 27, 29927–29942 CrossRef CAS.
- H. Sahebi, S. Massoud Bahrololoomi Fard, F. Rahimi, B. Jannat and N. Sadeghi, Food Chem., 2022, 396, 133637 CrossRef CAS PubMed.
- L. Zhu, L. Guo, Z. Zhang, J. Chen and S. Zhang, Sci. China: Chem., 2012, 55, 1479–1487 CrossRef CAS.
- C. Mohod and J. Dhote, Int. J. Innov. Res. Sci. Eng., 2013, 2, 2992–2996 Search PubMed.
- F. Bucatariu, C.-A. Ghiorghita, M.-M. Zaharia, S. Schwarz, F. Simon and M. Mihai, ACS Appl. Mater. Interfaces, 2020, 12, 37585–37596 CrossRef CAS PubMed.
- S. Morais, F. G. Costa and M. d. L. Pereira, Environ. Health, 2012, 10, 227–245 Search PubMed.
- M. S. Sankhla, M. Kumari, M. Nandan, R. Kumar and P. Agrawal, Int. J. Curr. Microbiol. Appl. Sci., 2016, 5, 759–766 CrossRef CAS.
- R. Verma and P. Dwivedi, Res. Sci. Technol., 2013, 5, 98–99 Search PubMed.
- H. A. Shaheen, H. M. Marwani and E. M. Soliman, J. Mol. Liq., 2017, 232, 139–146 CrossRef CAS.
- M. Mirzaei, M. Behzadi, N. M. Abadi and A. Beizaei, J. Hazard. Mater., 2011, 186, 1739–1743 CrossRef CAS PubMed.
- N. Jiang, X. Chang, H. Zheng, Q. He and Z. Hu, Anal. Chim. Acta, 2006, 577, 225–231 CrossRef CAS PubMed.
- S. Abo-Farha, A. Abdel-Aal, I. Ashour and S. Garamon, J. Hazard. Mater., 2009, 169, 190–194 CrossRef CAS PubMed.
- S. Büyüktiryaki, R. Keçili and C. M. Hussain, TrAC, Trends Anal. Chem., 2020, 127, 115893 CrossRef.
- I. López-Martin, E. Burello, P. N. Davey, K. R. Seddon and G. Rothenberg, Chem. Phys. Chem., 2007, 8, 690–695 CrossRef PubMed.
- L. Vidal, M.-L. Riekkola and A. Canals, Anal. Chim. Acta, 2012, 715, 19–41 CrossRef CAS PubMed.
- A. Andrade-Eiroa, M. Canle, V. Leroy-Cancellieri and V. Cerdà, TrAC, Trends Anal. Chem., 2016, 80, 641–654 CrossRef CAS.
- J. Haginaka, TrAC, Trends Anal. Chem., 2005, 24, 407–415 CrossRef CAS.
- X. Cai, J. Li, Z. Zhang, F. Yang, R. Dong and L. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 305–313 CrossRef CAS.
- B. Hashemi and S. Rezania, Microchim. Acta, 2019, 186, 1–20 CrossRef.
- A. Sarkar, P. Datta and M. Sarkar, Talanta, 1996, 43, 1857–1862 CrossRef CAS PubMed.
- J. López-Darias, V. Pino, Y. Meng, J. L. Anderson and A. M. Afonso, J. Chromatogr. A, 2010, 1217, 7189–7197 CrossRef PubMed.
- M. Tian, W. Bi and K. H. Row, J. Sep. Sci., 2009, 32, 4033–4039 CrossRef CAS PubMed.
- S.-B. Qin, Y.-H. Fan, X.-X. Mou, X.-S. Li and S.-H. Qi, J. Chromatogr. A, 2018, 1568, 29–37 CrossRef CAS PubMed.
- F. Aydin, R. Çakmak, A. Levent and M. Soylak, Appl. Organomet. Chem., 2020, 34, e5481 CrossRef CAS.
- S. A. Salami, X. Siwe-Noundou and R. W. Krause, Molecules, 2022, 27, 4213 CrossRef CAS PubMed.
- D. Chandra, S. K. Das and A. Bhaumik, Microporous Mesoporous Mater., 2010, 128, 34–40 CrossRef CAS.
- S. Das, S. Chatterjee, S. Mondal, A. Modak, B. K. Chandra, S. Das, G. D. Nessim, A. Majee and A. Bhaumik, Chem. Commun., 2020, 56, 3963–3966 RSC.
- A. Hajipour and F. Rafiee, J. Iran. Chem. Soc., 2009, 6, 647–678 CrossRef CAS.
- H. Niedermeyer, J. P. Hallett, I. J. Villar-Garcia, P. A. Hunt and T. Welton, Chem. Soc. Rev., 2012, 41, 7780–7802 RSC.
- T. D. Ho, A. J. Canestraro and J. L. Anderson, Anal. Chim. Acta, 2011, 695, 18–43 CrossRef CAS PubMed.
- N. Fontanals, F. Borrull and R. M. Marcé, TrAC, Trends Anal. Chem., 2012, 41, 15–26 CrossRef CAS.
- M. Sajid, M. K. Nazal and I. Ihsanullah, Anal. Chim. Acta, 2021, 1141, 246–262 CrossRef CAS PubMed.
- L. Vidal, M.-L. Riekkola and A. Canals, Anal. Chim. Acta, 2012, 715, 19–41 CrossRef CAS PubMed.
- T. T. Nguyen, T.-H. Duy Nguyen, T. T. Thi Huynh, M.-H. Dinh Dang, L. H. Thuy Nguyen, T. Le Hoang Doan, T. P. Nguyen, M. A. Nguyen and P. H. Tran, RSC Adv., 2022, 12, 19741–19750 RSC.
- N. Wang and B. Cui, TrAC, Trends Anal. Chem., 2022, 146, 116496 CrossRef CAS.
- J.-f. Liu, N. Li, G.-b. Jiang, J.-m. Liu, J. Å. Jönsson and M.-j. Wen, J. Chromatogr. A, 2005, 1066, 27–32 CrossRef CAS.
- F. Zhao, Y. Meng and J. L. Anderson, J. Chromatogr. A, 2008, 1208, 1–9 CrossRef CAS.
- N. Mohammadnezhad, A. A. Matin, N. Samadi, A. Shomali and H. Valizadeh, J. AOAC Int., 2017, 100, 218–223 CrossRef PubMed.
- G. Fang, J. Chen, J. Wang, J. He and S. Wang, J. Chromatogr. A, 2010, 1217, 1567–1574 CrossRef CAS PubMed.
- P. Zhang, S. Dong, G. Gu and T. Huang, Bull. Korean Chem. Soc., 2010, 31, 2949–2954 CrossRef CAS.
- S. Ayata, S. S. Bozkurt and K. Ocakoglu, Talanta, 2011, 84, 212–215 CrossRef CAS.
- H. M. Al-bishri, T. M. Abdel-Fattah and M. E. Mahmoud, J. Ind. Eng. Chem., 2012, 18, 1252–1257 CrossRef CAS.
- M. Gharehbaghi and F. Shemirani, Anal. Methods, 2012, 4, 2879 RSC.
- F. Galán-Cano, R. Lucena, S. Cárdenas and M. Valcárcel, Microchem. J., 2013, 106, 311–317 CrossRef.
- H. M. Marwani, J. Dispersion Sci. Technol., 2013, 34, 117–124 CrossRef CAS.
- S. Sadeghi and A. Z. Moghaddam, Anal. Methods, 2014, 6, 4867 RSC.
- S. Saleem, A. N. S. Saqib, A. Mujahid, M. Hanif, G. Mustafa, T. Mahmood, A. Waseem and A. R. Khan, Desalin. Water Treat., 2014, 52, 7915–7924 CrossRef CAS.
- H. Zhao, B. Dai, L. Xu, X. Wang, X. Qiao and Z. Xu, J. Sci. Food Agric., 2014, 94, 1787–1793 CrossRef CAS.
- K. Dasthaiah, B. Robert Selvan, A. S. Suneesh, K. A. Venkatesan, M. P. Antony and R. L. Gardas, J. Radioanal. Nucl. Chem., 2017, 313, 515–521 CrossRef CAS.
- P. Liang and L. Peng, Talanta, 2010, 81, 673–677 CrossRef CAS PubMed.
- S. Ayata, S. S. Bozkurt and K. Ocakoglu, Talanta, 2011, 84, 212–215 CrossRef CAS PubMed.
- M. Gharehbaghi and F. Shemirani, Anal. Methods, 2012, 4, 2879–2886 RSC.
- S. Saleem, A. N. S. Saqib, A. Mujahid, M. Hanif, G. Mustafa, T. Mahmood, A. Waseem and A. R. Khan, Desalin. Water Treat., 2014, 52, 7915–7924 CrossRef CAS.
- S. Wen, X. Zhu, Q. Huang, H. Wang, W. Xu and N. Zhou, Microchim. Acta, 2014, 181, 1041–1047 CrossRef CAS.
- T.-H. D. Nguyen, T. T. T. Huynh, M.-H. D. Dang, L. H. T. Nguyen, T. L. H. Doan, T. P. Nguyen, M. A. Nguyen and P. H. Tran, RSC Adv., 2022, 12, 19741–19750 RSC.
- L.-r. Nie, J. Lu, W. Zhang, A. He and S. Yao, Sep. Purif. Technol., 2015, 155, 2–12 CrossRef CAS.
- J. Cheng, X. Ma and Y. Wu, Food Anal. Methods, 2014, 7, 1083–1089 CrossRef.
- Y. Zhang, C. Zhong, Q. Zhang, B. Chen, M. He and B. Hu, RSC Adv., 2015, 5, 5996–6005 RSC.
- W. Ngeontae, W. Aeungmaitrepirom and T. Tuntulani, Talanta, 2007, 71, 1075–1082 CrossRef CAS PubMed.
- J. Wang, X. Ma, G. Fang, M. Pan, X. Ye and S. Wang, J. Hazard. Mater., 2011, 186, 1985–1992 CrossRef CAS PubMed.
- B. Feist and B. Mikula, Food Chem., 2014, 147, 302–306 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00247k |
| This journal is © The Royal Society of Chemistry 2024 |