
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium catalyzed transformation of levulinic acid to γ-valerolactone in water†
Bhanu
Priya‡
,
Vinod K.
Sahu‡
and
Sanjay K.
Singh
 *
*
Catalysis Group, Department of Chemistry, Indian Institute of Technology Indore, Simrol, Indore 453552, India. E-mail: sksingh@iiti.ac.in
First published on 1st December 2023
Abstract
High catalytic activity for selective and efficient transformation of levulinic acid (LA) to γ-valerolactone (GVL) in water was achieved over (η6-p-cymene)Ru(II)-pyridylamine, [(η6-C10H14)RuCl(κ2-L)]+ (L = Namine-substituted pyridylamine ligands) based molecular catalysts. A series of complexes with pyridylamine ligands having different electronic and steric properties were synthesized and characterized. A significant influence of the Namine-substituents of the pyridylamine ligand on the catalytic activity was observed where the [(η6-p-cymene)RuCl(κ2-pyNHnpr)]+ catalyst ([Ru]-2) outperformed others with 87% yield and >99% selectivity for GVL at 80 °C in water. Advantageously, the activity of [Ru]-2 was also scaled up to gram scale transformation of LA to GVL. Control experiments, pH dependent NMR and mass studies revealed the involvement of crucial reaction intermediates and catalytic species in the transformation of LA to GVL.
Sustainability spotlightWith the increase in global population, industrialization, and civilization, there is a rapid growth in energy consumption and environmental pollution. To address these issues, we need to find alternative renewable energy resources and advanced cost-effective technologies. Abundantly available lignocellulosic biomass can serve as an alternative energy resource, as it can be transformed into a variety of bio-platform chemicals including furfural, 5-hydroxymethyl furfural, levulinic acid, and γ-valerolactone (GVL). Herein, we demonstrated low temperature hydrogenation of levulinic acid to GVL using N-pyridylamine based Ru catalysts in water. Notably, GVL has wide application in the production of biofuels, fuel additives, and as a green solvent. This work addresses the UN SDG 7 (Affordable and Clean Energy) and SDG 13 (Climate Action). |
Introduction
In the current scenario of depleting fossil fuel resources, abundantly available biomass is being extensively explored as alternative sustainable feedstock for the production of a variety of value-added chemicals and fuel components.1,2 Among several biomass-derived chemicals, levulinic acid (LA) can be readily produced by the acid catalyzed hydrolysis of sugars and is one of the top ten biomass-derived platform chemicals.3,4 The valorization of LA can produce a wide variety of value-added chemicals and fuel blenders such as γ-valerolactone (GVL), valeric acid, 2-methyl tetrahydrofuran, and valeric esters.5 GVL is of interest as it offers tremendous flexibility in downstream applications and upgradation into a variety of value-added chemicals.6–9 Due to its chemical and physical properties, it can be used in various ways, for example as a gasoline blender, as a natural aromatic substance,10 as an additive in cosmetics and in domestic products.11 Moreover, enantio-pure GVL can be utilized as a chiral building block for the synthesis of various pharmaceutical products.12Various homogeneous as well as heterogeneous catalytic systems have been extensively explored for the transformation of LA to GVL. A number of heterogeneous catalysts based on various noble and non-noble metals (such as Pt,13 Ru,14 Au,15 Fe,16 Pd,17 Ni,18 and Cu,19,20) were employed and found efficient for the transformation of LA to GVL. However, reaction conditions were relatively harsher (higher temperature, 130–150 °C and higher pressure of H2 gas, 12–60 bar) while using these catalysts. A number of review articles listing the various heterogeneous catalytic systems for this transformation are published in recent years.10,21 On the other hand, in the search for efficient homogeneous catalytic systems for the transformation of LA to GVL, several Pd (1 in Scheme 1),22 Pt (2 in Scheme 1),23 and Ir (3, 4 in Scheme 1)24,25 based molecular catalysts were explored. For instance, Zhou et al. reported an iridium trihydride complex with the PNP pincer ligand (5 in Scheme 1) for LA to GVL conversion at 100 °C and 50 bar H2 and achieved a TOF of nearly 1480 h−1.26 Fu et al. reported a half sandwich Ir-cp*-bipyridine based complex (6 in Scheme 1) for the transformation of LA to GVL in water at 10 bar H2 or with formic acid at 120 °C.27 Jang et al. also reported an Ir(triscarbene) catalyst (7 in Scheme 1) for transfer hydrogenation of LA to GVL in water at 150 °C using glycerol as a hydrogen source in the presence of Ba(OH)2 and observed an appreciably high TON of 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.28 Recently, Tu et al. used the self-supporting strategy and reported a solid molecular catalyst based on bis-N-heterocyclic-carbene-iridium complexes for LA to GVL conversion at 100 °C and 1 bar H2.29 Although, Ir based molecular catalysts are reported to be more active and display a higher TOF than Ru based catalysts, the Ru catalysts have relatively low cost and easy availability and thus used more widely at laboratory and industrial scales. Among them, most of the Ru catalysts explored were associated with the use of phosphine-based ligands (Table S1†). For instance, Horvath et al. reported an in situ generated catalyst by using Ru(acac)3 and the TPPTS (triphenylphosphine trisulfonate) ligand at 140 °C and 70 bar H2 pressure for the hydrogenation of LA to GVL and achieved complete conversion of LA with a turnover frequency (TOF) of 50 h−1.30 Furthermore, Guo et al. used RuCl3 with PPh3 for the hydrogenation of LA to GVL using HCOOH as the H2 source (or 40 bar H2 with 40 bar CO2) in the presence of pyridine at 150 °C and observed a TOF of 155 h−1.31 Beller et al., Mika et al., and Leitner et al. also used triphos/biphos ligands with Ru(II) precursors at 140–160 °C and obtained a TOF of 452 h−1, 3540 h−1, and 144 h−1 respectively.32–34 Furthermore, Horvath et al. explored a ruthenium based Shvo catalyst (8 in Scheme 1), while using formic acid as a hydrogen source at 100 °C under solventless conditions to achieve a TOF of 177 h−1.35 Darkwa et al. reported solvent-free transformation of LA to GVL using pyrazolylphosphite and pyrazolylphosphinite ruthenium(II) complexes (9 in Scheme 1), using both formic acid (TOF 60 h−1) and molecular hydrogen (15 bar) as hydrogen sources at 100–120 °C.11 Makhubela et al. replaced the phosphine ligands with N,N donor iminopyridine ligands for Ru(II) (10 in Scheme 1) catalyzed LA to GVL transformation and the reaction was performed under solvent free conditions using formic acid as a hydrogen source to achieve a TOF of 81.6 h−1 at 150 °C.36 Miller et al. reported triphos complexes of ruthenium (11 in Scheme 1) for the transformation of LA to GVL at 130 °C and 50 bar H2 (TOF 48 h−1).37 Makhubela et al. reported a dinuclear Ru complex having multidentate pyridyl-aminophosphinite and pyridyl-phosphoramidite ligands (12 in Scheme 1) for the transfer hydrogenation of LA using formic acid as a hydrogen source at 120 °C under neat conditions and observed complete conversion of LA in 8 h with a TOF of 123 h−1.38 Wildeman et al. used a η4-(2,3,4,5-tetraphenylcyclopentadienone)-Ru(CO)3 complex (13 in Scheme 1) for LA to GVL transformation under solvent free conditions at 100 °C and 50 bar H2 pressure and observed a TOF of 144 h−1.39 More recently, Hou et al. tested a ruthenium hydride complex (14 in Scheme 1) for solvent assisted hydrogenation of methyl levulinate to GVL at 100 °C and 10 bar H2 pressure.40 They used tetrabutylammonium acetate (TBAOAc) as the solvent, which not only acted as a reaction medium but also coordinated to Ru resulting in the formation of Ru-OAc species, which further helped in the activation of H2. Some iron complexes (15 and 16 in Scheme 1) were also reported for the transfer hydrogenation41 and direct hydrogenation42 of LA and alkyl levulinates. LA to GVL transformation was also explored using in situ generated Fe-nanoparticles from the Fe3(CO)12 precursor in the presence of imidozyl, pyridine, triethylamine or KOH at 180 °C yielding GVL in good yield (92%).43 Similarly, cobalt based catalysts using Co(BF4)2·6H2O and a tetradentate phosphine ligand P(CH2CH2PPh2)3 at 100 °C and 1 bar H2 in 1,3-dimethyl-2-imidazolidinone44 and Ni based catalysts using Ni(OAc)2·4H2O and the triphos ligand at 1 atm H2 pressure45 were also utilized for the hydrogenation of LA to GVL. Recently, Gracia et al. reported a [(dippe)Ni(COD)] catalyst (17 in Scheme 1) for transfer hydrogenation of LA in a biphasic solvent system (H2O:THF (5:1)) at 120 °C using formic acid as the hydrogen source.46 More recently, the same group reported a homogeneous Cu(I) based catalytic system comprising [(PPh3)2Cu(NO3)] and the dippe ligand for the hydrogenation of LA at 140 °C and 20.6 bar H2 pressure in hexane and yielded >99% GVL.47 A titanium based catalyst [Cp2TiCl2] (18 in Scheme 1) is also reported for the hydrogenation of LA at 100 °C and 6.8 bar H2.48
000.28 Recently, Tu et al. used the self-supporting strategy and reported a solid molecular catalyst based on bis-N-heterocyclic-carbene-iridium complexes for LA to GVL conversion at 100 °C and 1 bar H2.29 Although, Ir based molecular catalysts are reported to be more active and display a higher TOF than Ru based catalysts, the Ru catalysts have relatively low cost and easy availability and thus used more widely at laboratory and industrial scales. Among them, most of the Ru catalysts explored were associated with the use of phosphine-based ligands (Table S1†). For instance, Horvath et al. reported an in situ generated catalyst by using Ru(acac)3 and the TPPTS (triphenylphosphine trisulfonate) ligand at 140 °C and 70 bar H2 pressure for the hydrogenation of LA to GVL and achieved complete conversion of LA with a turnover frequency (TOF) of 50 h−1.30 Furthermore, Guo et al. used RuCl3 with PPh3 for the hydrogenation of LA to GVL using HCOOH as the H2 source (or 40 bar H2 with 40 bar CO2) in the presence of pyridine at 150 °C and observed a TOF of 155 h−1.31 Beller et al., Mika et al., and Leitner et al. also used triphos/biphos ligands with Ru(II) precursors at 140–160 °C and obtained a TOF of 452 h−1, 3540 h−1, and 144 h−1 respectively.32–34 Furthermore, Horvath et al. explored a ruthenium based Shvo catalyst (8 in Scheme 1), while using formic acid as a hydrogen source at 100 °C under solventless conditions to achieve a TOF of 177 h−1.35 Darkwa et al. reported solvent-free transformation of LA to GVL using pyrazolylphosphite and pyrazolylphosphinite ruthenium(II) complexes (9 in Scheme 1), using both formic acid (TOF 60 h−1) and molecular hydrogen (15 bar) as hydrogen sources at 100–120 °C.11 Makhubela et al. replaced the phosphine ligands with N,N donor iminopyridine ligands for Ru(II) (10 in Scheme 1) catalyzed LA to GVL transformation and the reaction was performed under solvent free conditions using formic acid as a hydrogen source to achieve a TOF of 81.6 h−1 at 150 °C.36 Miller et al. reported triphos complexes of ruthenium (11 in Scheme 1) for the transformation of LA to GVL at 130 °C and 50 bar H2 (TOF 48 h−1).37 Makhubela et al. reported a dinuclear Ru complex having multidentate pyridyl-aminophosphinite and pyridyl-phosphoramidite ligands (12 in Scheme 1) for the transfer hydrogenation of LA using formic acid as a hydrogen source at 120 °C under neat conditions and observed complete conversion of LA in 8 h with a TOF of 123 h−1.38 Wildeman et al. used a η4-(2,3,4,5-tetraphenylcyclopentadienone)-Ru(CO)3 complex (13 in Scheme 1) for LA to GVL transformation under solvent free conditions at 100 °C and 50 bar H2 pressure and observed a TOF of 144 h−1.39 More recently, Hou et al. tested a ruthenium hydride complex (14 in Scheme 1) for solvent assisted hydrogenation of methyl levulinate to GVL at 100 °C and 10 bar H2 pressure.40 They used tetrabutylammonium acetate (TBAOAc) as the solvent, which not only acted as a reaction medium but also coordinated to Ru resulting in the formation of Ru-OAc species, which further helped in the activation of H2. Some iron complexes (15 and 16 in Scheme 1) were also reported for the transfer hydrogenation41 and direct hydrogenation42 of LA and alkyl levulinates. LA to GVL transformation was also explored using in situ generated Fe-nanoparticles from the Fe3(CO)12 precursor in the presence of imidozyl, pyridine, triethylamine or KOH at 180 °C yielding GVL in good yield (92%).43 Similarly, cobalt based catalysts using Co(BF4)2·6H2O and a tetradentate phosphine ligand P(CH2CH2PPh2)3 at 100 °C and 1 bar H2 in 1,3-dimethyl-2-imidazolidinone44 and Ni based catalysts using Ni(OAc)2·4H2O and the triphos ligand at 1 atm H2 pressure45 were also utilized for the hydrogenation of LA to GVL. Recently, Gracia et al. reported a [(dippe)Ni(COD)] catalyst (17 in Scheme 1) for transfer hydrogenation of LA in a biphasic solvent system (H2O:THF (5:1)) at 120 °C using formic acid as the hydrogen source.46 More recently, the same group reported a homogeneous Cu(I) based catalytic system comprising [(PPh3)2Cu(NO3)] and the dippe ligand for the hydrogenation of LA at 140 °C and 20.6 bar H2 pressure in hexane and yielded >99% GVL.47 A titanium based catalyst [Cp2TiCl2] (18 in Scheme 1) is also reported for the hydrogenation of LA at 100 °C and 6.8 bar H2.48
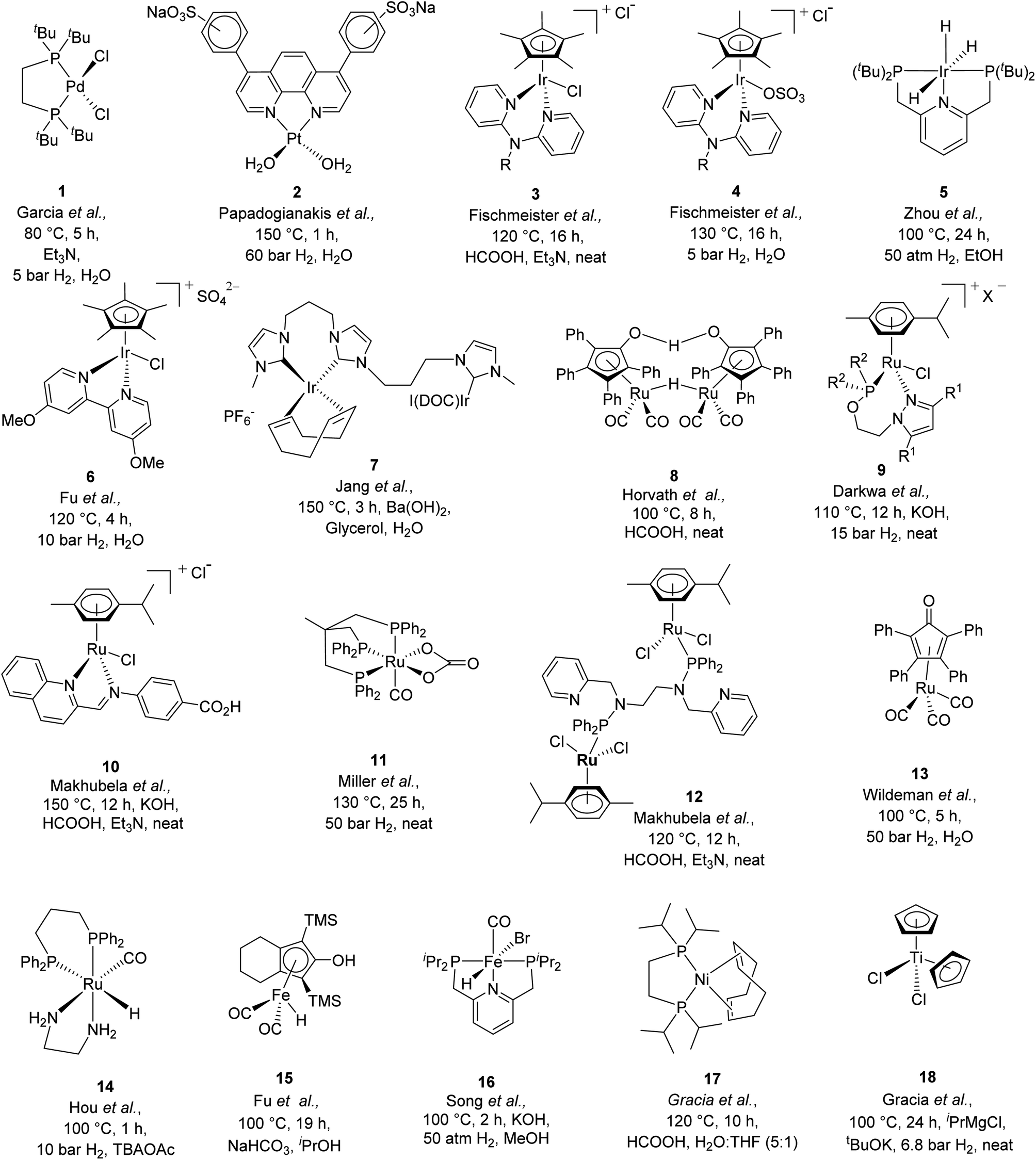 | ||
| Scheme 1 Molecular catalysts explored for the transformation of LA to GVL. | ||
It is thus evident from the rich literature reports that most of the catalytic systems explored for LA to GVL transformation either use external H2 gas (as high as 100 bar) or organic hydrogen donors such as formic acid, isopropanol, methanol, ethanol, or glycerol (Scheme 1 and Table S1†). Utilizing organic hydrogen donors eliminates the need for pressurized hydrogen gas in pressure reactors. Notably, Guo et al. demonstrated that formic acid produced during the transformation of 5-hydroxymethylfurfural (5-HMF) to LA can serve as a hydrogen source for further hydrogenation of LA to GVL.31 However, a wide range of molecular catalysts explored for the transformation of LA to GVL using HCOOH require high reaction temperatures (as high as 150 °C) (Scheme 1 and Table S1†). Therefore, it is desirable to explore molecular catalysts which can avoid the use of elevated reaction temperatures and higher H2 pressures for the transformation of LA to GVL. In this regard, herein, we report an efficient transformation of LA to GVL at a lower reaction temperature over water-soluble (η6-p-cymene)Ru(II)-pyridylamine-based molecular catalysts. A series of water-soluble (η6-p-cymene)Ru(II) complexes containing Namine substituted pyridylamine ligands are synthesized and characterized, and the X-ray molecular structure of the complexes [Ru]-4, [Ru]-5, and [Ru]-8 were determined. The synthesized complexes catalyzed the hydrogenation of biomass-derived LA to GVL using HCOOH as a hydrogen source under milder reaction conditions (80 °C) in water. Advantageously, using pyridylamine ligands with substituted Namine enabled us to effectively explore and tune the steric bulkiness and basicity of Namine of the synthesized Ru complexes and hence the catalytic activity for the transformation of LA to GVL. Mechanistic investigations using mass and NMR studies were performed to identify the crucial catalytic intermediate species involved in the transformation of LA to GVL. Moreover, the optimized methodology for LA to GVL was scaled up to the gram-scale.
Results and discussion
Synthesis and characterization of (η6-p-cymene)Ru(II)-pyridylamine complexes ([Ru]-1–[Ru]-8)
Cationic (η6-p-cymene)Ru(II)-pyridylamine complexes [Ru]-1–[Ru]-8 having N-pyridylamine ligands L1–L8 were synthesized by treating [{(η6-p-cymene)RuCl2}2] with the respective Namine substituted pyridylamine ligand in acetonitrile at room temperature (Scheme 2), following our previously reported process.49 The synthesized (η6-p-cymene)Ru(II)-pyridylamine complexes vary in terms of the substituents at Namine ([Ru]-1: NamineH2; [Ru]-2: Namine-n-propyl; [Ru]-3: Namine-i-propyl; [Ru]-4: Namine-n-butyl; [Ru]-5: Namine-i-butyl; [Ru]-6: Namine-n-pentyl; [Ru]-7: Namine-i-pentyl; [Ru]-8: Namine-t-pentyl). Varying the substituents at the Namine group may play a crucial role in tuning the electronic and steric properties and hence further tune the catalytic performance of the synthesized complexes [Ru]-1–[Ru]-8 for the transformation of LA to GVL.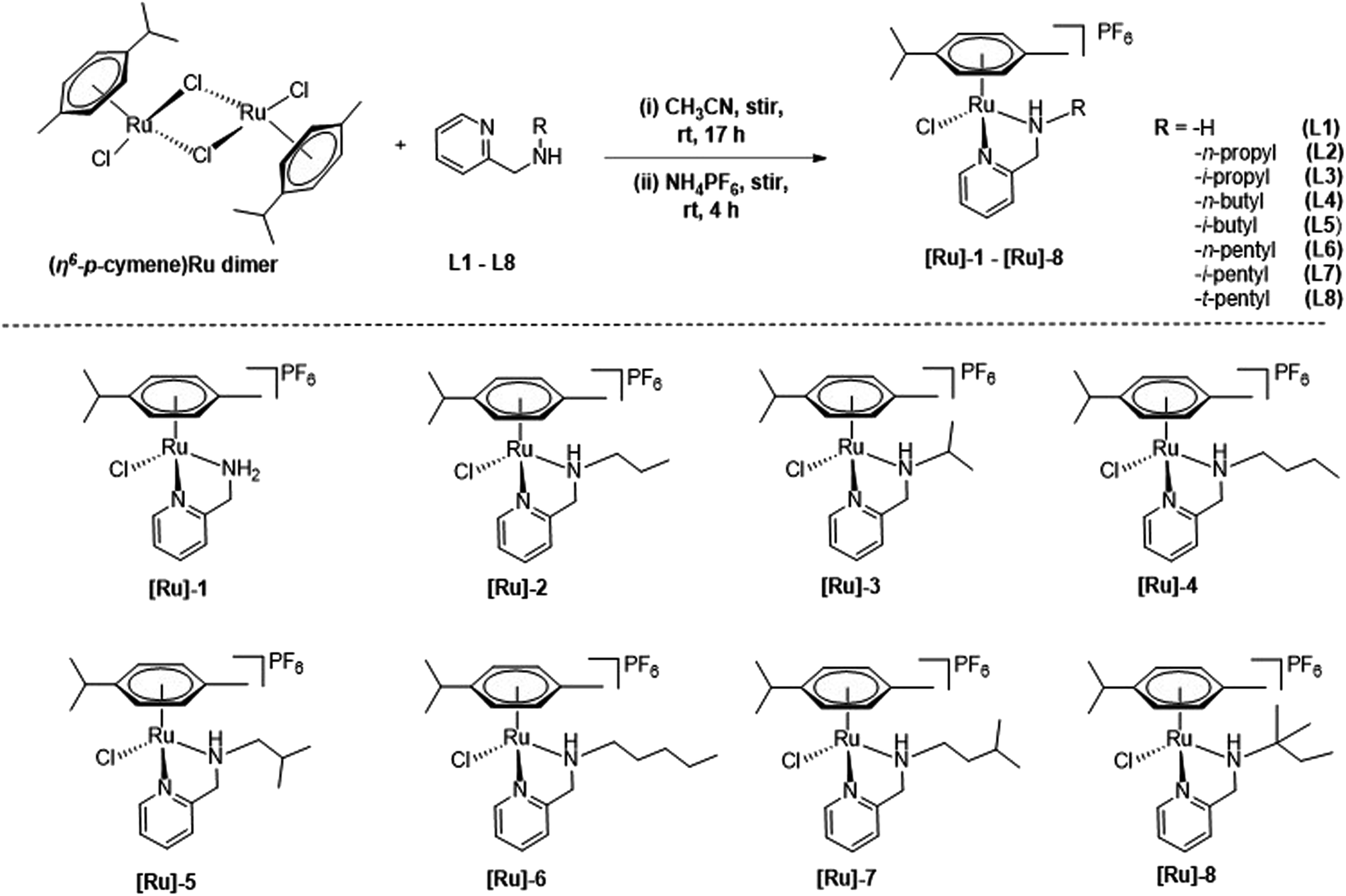 | ||
| Scheme 2 Synthesis of (η6-p-cymene)Ru(II) complexes [Ru]-1–[Ru]-8, containing pyridylamine ligands L1–L8. | ||
The spectro-analytical analysis of the synthesized complexes corroborated well with the proposed structures. The 1H NMR of the [Ru]-1–[Ru]-8 displayed a similar trend of downfield chemical shift (8.93–9.12 ppm) as compared to that of the free pyridylamine ligands (8.42–8.48 ppm) for the ortho –CH protons.49 The significant downfield shifts for the –CH2 protons indicated the ligand coordination to the ruthenium metal center. Upon introducing an electron rich alkyl substituent at the aminic nitrogen (Namine), a significant upfield shift of resonance is observed for the ortho –CH protons of the pyridine ring from 9.12 ppm in [Ru]-1 to 8.93–9.03 ppm in [Ru]-2–[Ru]-8. The coordination of (η6-p-cymene) to the Ru(II) center is also confirmed by the presence of peaks for aromatic protons in the range of 5.65–5.92 ppm, the methyl proton in the field of 1.88–1.98 ppm, and the methine and methyl protons of the isopropyl group, respectively in the ranges of 2.59–3.65 and 1.00–1.12 ppm.49 The counter anion PF6− resonated at −144 ppm in the 31P NMR spectrum for [Ru]-1–[Ru]-8. The characteristic isotopic pattern of ruthenium was observed with a significantly intense base peak at [M]+ for all the synthesized complexes [Ru]-1–[Ru]-8.
Furthermore, to confirm the molecular identity of (η6-p-cymene)Ru(II)-pyridylamine complexes, we tried to obtain single crystals using various methods, including the slow evaporation of methanol-dichloromethane (1:1) solution of the respective complexes, but we were able to grow the crystals only for the complexes [Ru]-4, [Ru]-5, and [Ru]-8. The single crystal X-ray diffraction studies inferred that the complexes [Ru]-4, [Ru]-5 and [Ru]-8, respectively crystallize in the triclinic (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group), monoclinic (P21/c space group) and orthorhombic (Pbca space group) crystal systems. The solid-state structure clearly demonstrates the pseudo-octahedral geometry for all the complexes which is characteristic of half-sandwich arene–ruthenium complexes. In typical piano-stool geometry around the ruthenium metal center, the three legs are occupied by the bis-chelating N-pyridylamine ligand coordinated to the ruthenium metal center in a κ2-bidentate fashion through pyridine (Npy) and amine nitrogen (Namine), and the chloro ligand (–Cl) while the arene ring is placed at the apex of the structure in a η6 manner. The displacement of the (η6-p-cymene) ring from the ruthenium metal center for the synthesized complexes lies in the range of 1.67–1.69 Å. The Ru–Npy and Ru–Namine bond distances are also in the range of 2.07–2.19 Å and are comparable to those for analogous N-pyridylamine arene–ruthenium complexes.49,50 Ru–Cl bond distances are observed in the range of 2.39–2.40 Å. The Npy–Ru–Namine bond angles lie in the range of 76.1–78.1°, while Npy–Ru–Cl and Namine–Ru–Cl bond angles are observed between 83.8–85.6° and 82.5–84.6°, respectively. The bond angle between Npy/Namine and ruthenium metal with the centroid of the η6-arene ring (Cct) is in the range of 129.2–137.3° for complexes [Ru]-4, [Ru]-5 and [Ru]-8, which further supports the piano-stool geometry of the (η6-p-cymene)Ru(II)-pyridylamine complexes (Fig. 1). The crystal refinement data and some other important bond lengths and bond angles are summarized in Tables S2–S4† for the complexes [Ru]-4, [Ru]-5 and [Ru]-8.
space group), monoclinic (P21/c space group) and orthorhombic (Pbca space group) crystal systems. The solid-state structure clearly demonstrates the pseudo-octahedral geometry for all the complexes which is characteristic of half-sandwich arene–ruthenium complexes. In typical piano-stool geometry around the ruthenium metal center, the three legs are occupied by the bis-chelating N-pyridylamine ligand coordinated to the ruthenium metal center in a κ2-bidentate fashion through pyridine (Npy) and amine nitrogen (Namine), and the chloro ligand (–Cl) while the arene ring is placed at the apex of the structure in a η6 manner. The displacement of the (η6-p-cymene) ring from the ruthenium metal center for the synthesized complexes lies in the range of 1.67–1.69 Å. The Ru–Npy and Ru–Namine bond distances are also in the range of 2.07–2.19 Å and are comparable to those for analogous N-pyridylamine arene–ruthenium complexes.49,50 Ru–Cl bond distances are observed in the range of 2.39–2.40 Å. The Npy–Ru–Namine bond angles lie in the range of 76.1–78.1°, while Npy–Ru–Cl and Namine–Ru–Cl bond angles are observed between 83.8–85.6° and 82.5–84.6°, respectively. The bond angle between Npy/Namine and ruthenium metal with the centroid of the η6-arene ring (Cct) is in the range of 129.2–137.3° for complexes [Ru]-4, [Ru]-5 and [Ru]-8, which further supports the piano-stool geometry of the (η6-p-cymene)Ru(II)-pyridylamine complexes (Fig. 1). The crystal refinement data and some other important bond lengths and bond angles are summarized in Tables S2–S4† for the complexes [Ru]-4, [Ru]-5 and [Ru]-8.
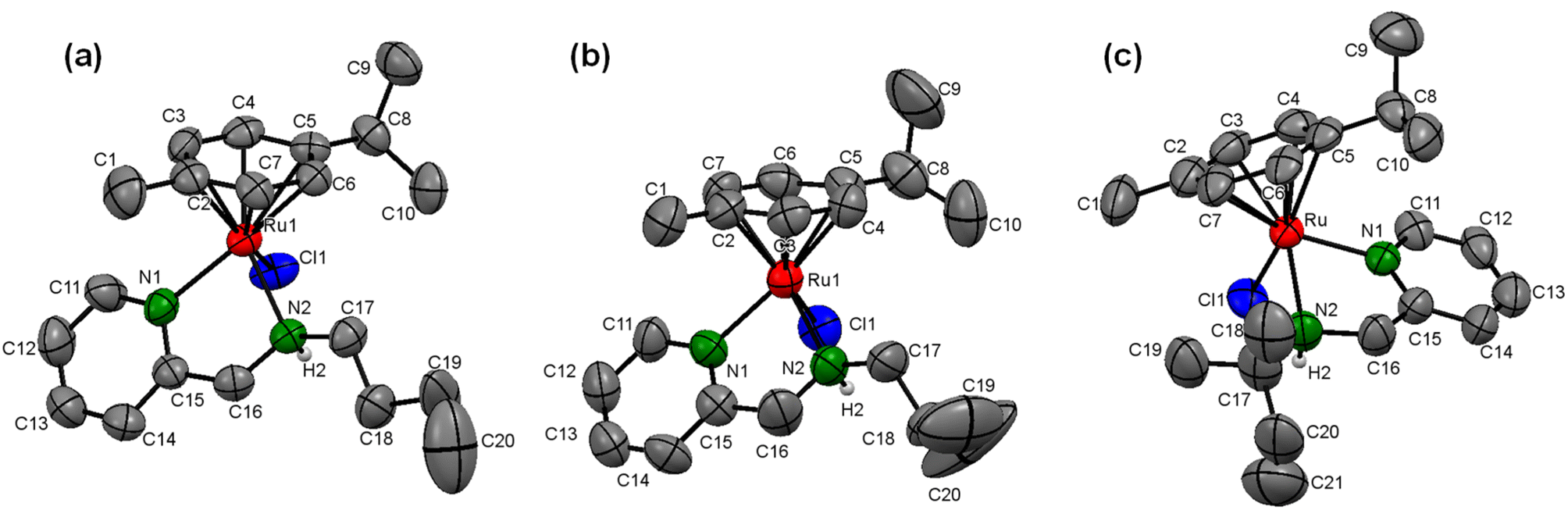 | ||
| Fig. 1 X-ray crystal structures of complexes (a) [Ru]-4, (b) [Ru]-5, and (c) [Ru]-8 with 30% ellipsoid probability. Counteranions (PF6−) and H atoms (except for those on N) are omitted for the sake of clarity. | ||
Catalytic transformation of LA to GVL
At an outset, the catalytic activity of the synthesized (η6-p-cymene)Ru(II)-pyridylamine complexes ([Ru]-1–[Ru]-8) for the transformation of biomass derived LA to GVL was investigated using formic acid as a hydrogen source along with triethylamine as a base at 80 °C in water (Table 1).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | T (°C) | t (h) | Convb. (%) | Selb. (%) GVL/HPA | TOF (h−1) |
| a Reaction conditions: Ru catalyst (2.5 mol%), LA (1 mmol), HCOOH (6 mmol), Et3N (1 mmol), and water (5 mL). b Conversion and selectivity determined by 1H NMR. c Isolated yield, TOF – turnover frequency, and n.d. – not determined. | ||||||
| 1 | [η6-p-cymene)RuCl(κ2-pyNH2)]+[Ru]-1 | 80 | 2 | 14 | 80/20 | 2.8 |
| 2 | [η6-p-cymene)RuCl(κ2-pyNHnpr)]+[Ru]-2 | 80 | 2 | >99 | >99(87%)/- | 20 |
| 3 | [η6-p-cymene)RuCl(κ2-pyNHipr)]+[Ru]-3 | 80 | 2 | 12 | 96/4 | 2.4 |
| 4 | [η6-p-cymene)RuCl(κ2-pyNHnbu)]+[Ru]-4 | 80 | 2 | 97 | 93/7 | 19.4 |
| 5 | [η6-p-cymene)RuCl(κ2-pyNHibu)]+[Ru]-5 | 80 | 2 | 75 | 77/23 | 15 |
| 6 | [η6-p-cymene)RuCl(κ2-pyNHnpen)]+[Ru]-6 | 80 | 2 | 95 | 97/3 | 19 |
| 7 | [η6-p-cymene)RuCl(κ2-pyNHipen)]+[Ru]-7 | 80 | 2 | 93 | 92/8 | 18.6 |
| 8 | [η6-p-cymene)RuCl(κ2-pyNHtpen)]+[Ru]-8 | 80 | 2 | 95 | 95/5 | 19 |
| 9 | [η6-p-cymene)RuCl2]2[Ru]-PC | 80 | 2 | 4 | n.d. | 0.8 |
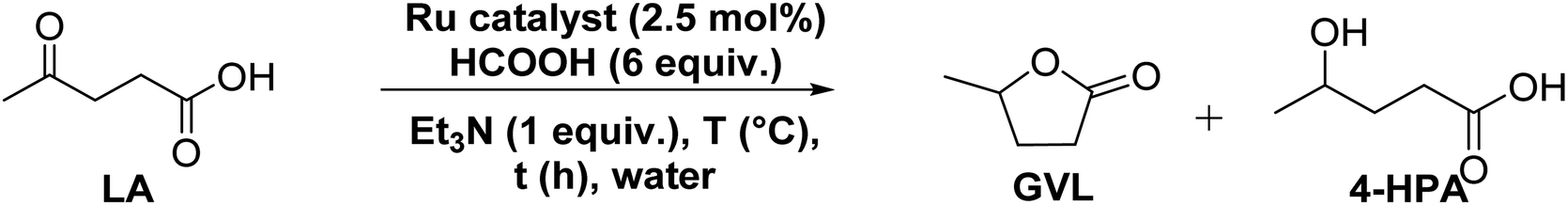
Results inferred that (η6-p-cymene)Ru(II)-pyridylmethyl amine ([Ru]-1) exhibited only 14% conversion of LA to GVL with 80% selectivity for GVL and 20% for 4-hydroxypentanoic acid (HPA) (Table 1, entry 1). A significantly enhanced conversion (>99%) of LA selectively to GVL (yield 87%) with a TOF of 20 h−1 was achieved over the [Ru]-2 containing Namine-n-propyl substituted pyridyl methyl amine ligand in 2 h (Table 1, entry 2). In contrast to [Ru]-2, the [Ru]-3 having Namine-i-propyl substituted pyridyl methyl amine displayed a very poor conversion (TOF 2.4 h−1) (Table 1, entry 3). Notably, enhancement in the catalytic activity (75–95% conversion of LA, 77–97% selectivity for GVL and a TOF of 15–19.4 h−1) was observed while performing the catalytic reaction over (η6-p-cymene)Ru(II) complexes having other N-substituted pyridyl methyl amine ligands ([Ru]-4: Namine-n-butyl; [Ru]-5: Namine-i-butyl; [Ru]-6: Namine-n-pentyl; [Ru]-7: Namine-i-pentyl; [Ru]-8: Namine-t-pentyl) (Table 1, entries 4–8). In contrast to the high activity observed for (η6-p-cymene)Ru(II)-pyridylamine, the precursor [(η6-p-cymene)RuCl2]2 ([Ru]-PC) exhibited almost negligible activity (Table 1, entry 9). The results show that the basicity of Namine as well as steric crowding played an important role in tuning the catalytic activity of these (η6-p-cymene)Ru(II)-pyridylamine complexes.
The strong interaction of the NH2 group with the solvent molecule (H2O) in [Ru]-1 permanently retarded the availability of the –NH group for the reaction, leading to lower activity. Notably, the basicity of Namine increases with increase in the alkyl chain length or branching in the ligands.40,49–52 However, with increase in the carbon chain length or branching the steric hindrance at Namine may also increase. For instance, in the case of [Ru]-3 having the Namine-i-propyl substituted pyridylmethyl amine ligand, the methyl groups are placed closer to Namine, which increased the steric crowding resulting in the lower activity of [Ru]-3 as compared to [Ru]-2. Furthermore, the increase in the N-substituted alkyl chain length or branching causes increase in the steric crowding around the –NH group, which is expected to play a crucial role in the observed trend in the catalytic activity of [Ru]-4–[Ru]-8 for LA to GVL transformation (Fig. S1†).40,49,50 From single crystal X-ray crystallography, the Ru-Namine bond length in [Ru]-4 (Py-NHnbu) is shorter than that of [Ru]-5 (Py-NHibu) and [Ru]-8 (Py-NHtpen). Moreover, the Namine–βC and Namine–γC bond lengths in [Ru]-4, [Ru]-5, and [Ru]-8 suggested more crowding around Namine for [Ru]-5 (Namine–βC 2.540 Å and Namine–γC 3.181 Å) and [Ru]-8 (Namine–βC 2.521 Å and Namine–γC 3.048 Å) as compared to [Ru]-4 (Namine–βC 2.544 Å and Namine–γC 3.895 Å). Also, it was reported in the literature that when more electron rich substituents are attached to the metal center, transfer hydrogenation using formic acid is facilitated.53 Hence, in accordance with these reports and our findings, it is clear that the increased basicity due to different steric and electronic properties of different ligands attached to Namine catalyzed the dehydrogenation of formic acid which in turn increased the rate of hydrogenation of LA to GVL.
Further optimization of the reaction conditions to get the best suited reaction conditions for LA to GVL was performed with [Ru]-2. Though the reaction performed with 2.5 mol% catalyst loading yields the best result with a TOF value of 20 h−1, reactions with lower catalyst loadings of 1 and 0.25 mol% were also performed and we achieved 98 and 92% conversion of LA to GVL in a longer duration (Table S5†). The results showed that upon performing the reaction at lower temperatures (below 80 °C), lower conversion was observed and while increasing the reaction temperature to 90 and 100 °C, the conversion of LA increased (Table S6†). Over [Ru]-2, the estimated activation energy was found to be 46.89 kJ mol−1 (Fig. S2†). The presence of a base is crucial for the activation and deprotonation of HCOOH to form formate that can coordinate to the metal centre and forms the Ru-formato species, which is an important intermediate to initiate the transfer hydrogenation reactions. Unlike other inorganic bases, transfer hydrogenation is more facile with Et3N, presumably due to its ability to facilitate the reversible proton abstraction/release from HCOOH.54 Therefore, change in the base from Et3N to inorganic bases such as K3PO4, NaOH, NaHCO3 and HCOONa could not improve the conversion of LA to GVL significantly, while in the absence of the base (Et3N) reaction could not occur (Table S7†). The reaction performed for various time intervals showed that the reaction proceeds via the formation of 4-hydroxypentanoic acid (4-HPA) as an intermediate with continuous consumption of LA (Table S8† and Fig. 2).
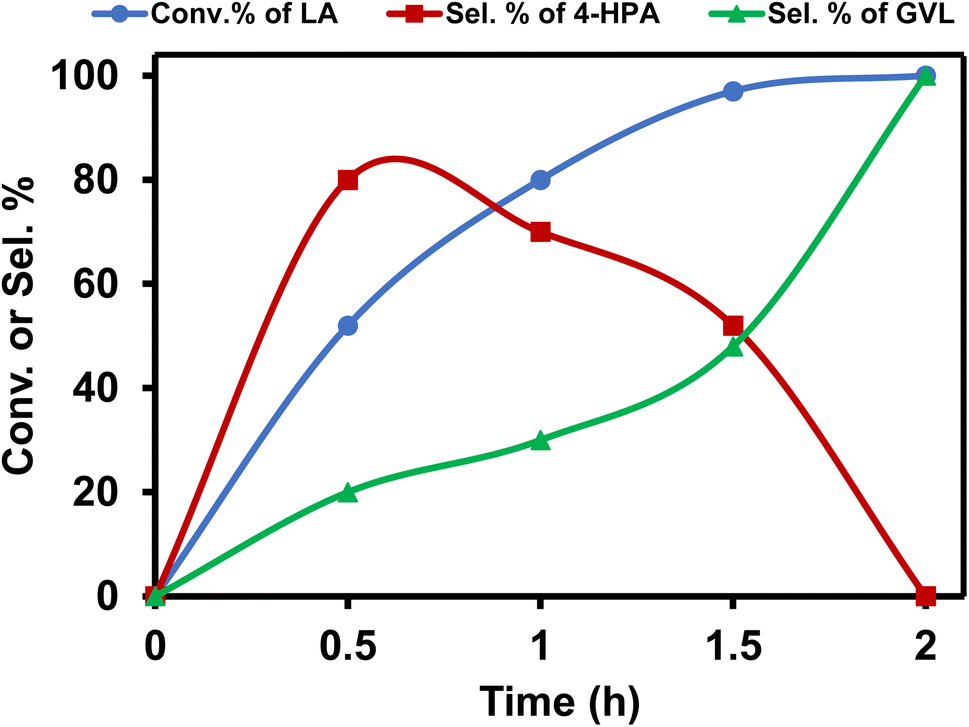 | ||
| Fig. 2 Time dependent reaction profile for catalytic transformation of LA to GVL over the [Ru]-2 catalyst. Reaction conditions: [Ru]-2 (2.5 mol%), LA (1 mmol), HCOOH (6 mmol), Et3N (1 mmol), water (5 mL), 80 °C, 0–2 h. | ||
Furthermore, pH dependent 1H NMR studies showed that lower pH is favorable for the high selectivity of GVL. It was observed that at pH 6.1, (HCOOH:Et3N – 6:1), 4-HPA which is the hydrogenated form of LA, was observed. With the further decrease in the pH to 4.7, 3.6, and 2.7, the intensity of peaks corresponding to 4-HPA decreased, while those corresponding to GVL became more prominent (Fig. 3 and Table S9†). We observed complete conversion of LA with complete selectivity for GVL in 2 h in the pH range of 2.5–2.7. The identification of 1H NMR peaks corresponding to 4-HPA, further reinforced that the transformation of LA to GVL proceeded through the intermediate 4-HPA.
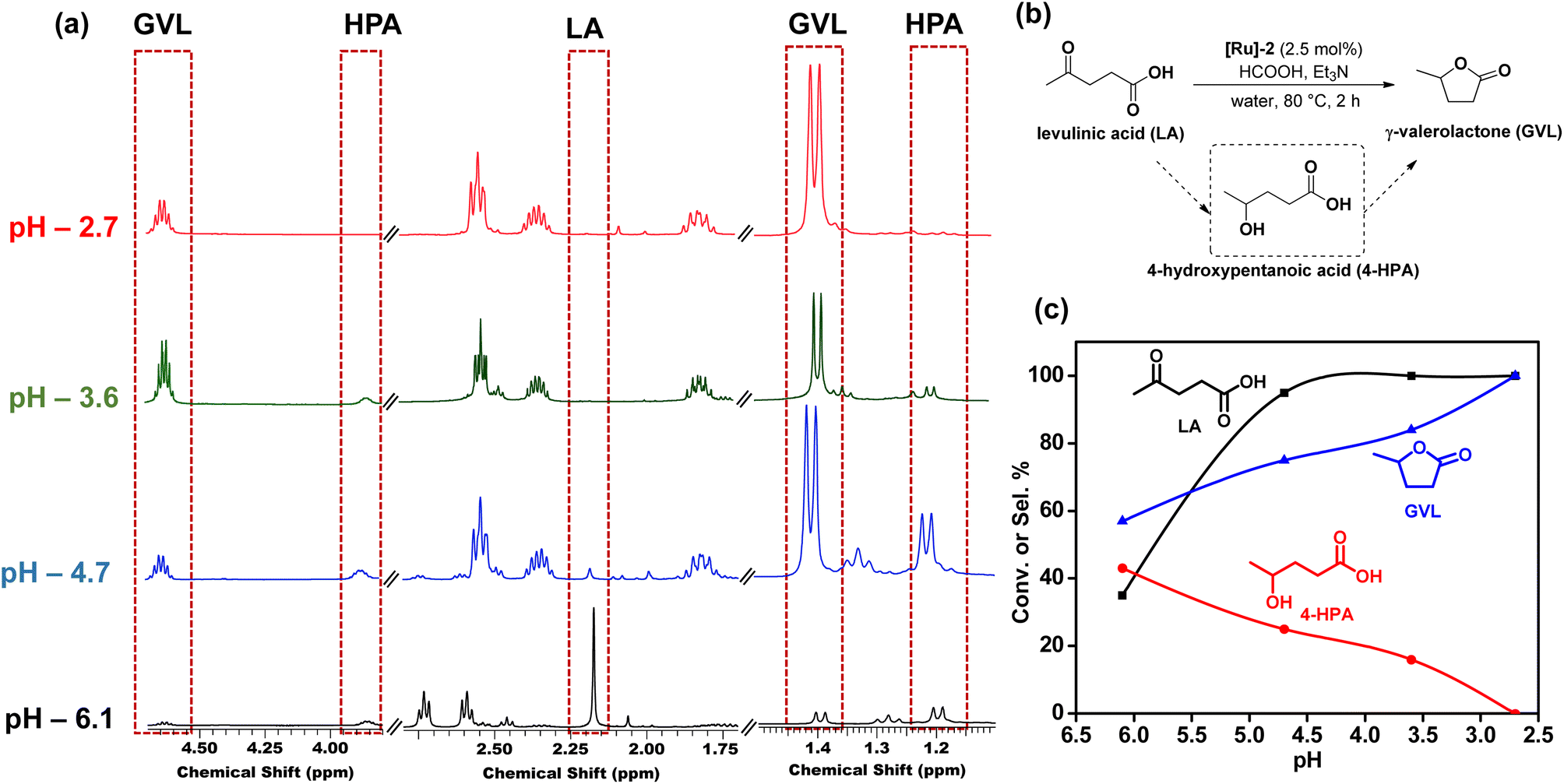 | ||
| Fig. 3 (a) 1H NMR for pH dependent transformation in CDCl3, (b) schematic representation, and (c) pH dependent transformation of biomass derived LA to GVL through the formation of intermediate 4-HPA over the [Ru]-2 catalyst. Reaction conditions: [Ru]-2 (2.5 mol%), LA (1 mmol), HCOOH (1–6 mmol), Et3N (1 mmol), water (5 mL), 80 °C, 2 h. | ||
To further investigate a plausible mechanistic pathway for the transformation of LA to GVL over the [Ru]-2 catalyst, we performed extensive mass studies to identify several intermediate species involved during the course of reaction (Fig. 4). In a control experiment, the [Ru]-2 catalyst was stirred for 5–10 min in water where a prominent peak at m/z = 385.1 corresponding to the species [(η6-p-cymene)Ru(κ2-PyNpr)]+ ([Ru]-2A) having a vacant coordination site on Ru was observed. We further noticed that the formation of species [Ru]-2A was independent of the presence of HCOOH and Et3N. In the subsequent step, the addition of 6 mmol of HCOOH, 1 mmol of Et3N and 1 mmol of LA followed by stirring at 60 °C for 30 minutes resulted in the appearance of new peaks at m/z = 431.1 and m/z = 501.2 corresponding to the [(η6-p-cymene)Ru(κ2-PyNHpr)(HCOO)]+ ([Ru]-2B) and [(η6-p-cymene)Ru(κ2-PyNHpr)(levulinate)]+ ([Ru]-2C) coordinated species.
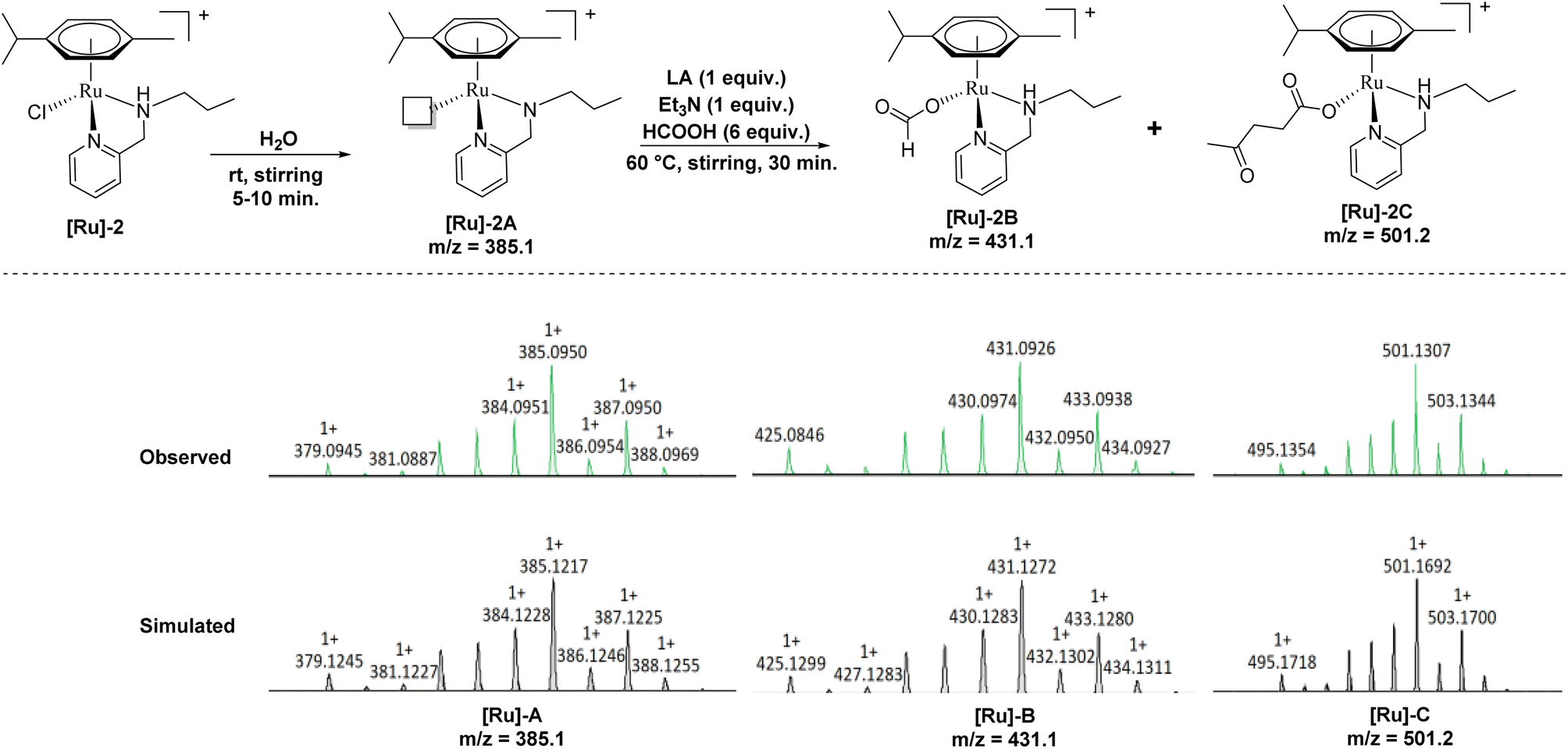 | ||
| Fig. 4 Ru-formato and Ru-levulinate species observed during mass investigation of the catalytic reaction aliquots. | ||
Based on extensive mass studies, we anticipated that the generation of active catalytic species [Ru]-2A from [Ru]-2 is the first step in the mechanistic cycle for the transformation of LA to GVL. Furthermore, the addition of formic acid, Et3N, and LA resulted in the formation of the Ru-formato ([Ru]-2B) and Ru-levulinate ([Ru]-2C) species. The [Ru]-2B species then undergoes decarboxylation to form the Ru-hydrido species ([Ru]-2D), which is responsible for the transfer hydrogenation of LA to 4-HPA (identified as an intermediate during the catalytic reaction, Fig. 2) and the product GVL was finally formed by dehydration and cyclization of 4-HPA (Scheme 3). Upon further investigation, we noticed that the formation of the [Ru]-2C species depends on the ratio of HCOOH:Et3N. Notably, mass studies inferred that high intensity mass peaks of the [Ru]-2C species appeared for a HCOOH:Et3N ratio of 3:1, while the intensity of this species greatly reduced with a 6:1 ratio of HCOOH:Et3N (Fig. S3†). Therefore, presumably the [Ru]-2C species is not directly involved in the LA hydrogenation to GVL.
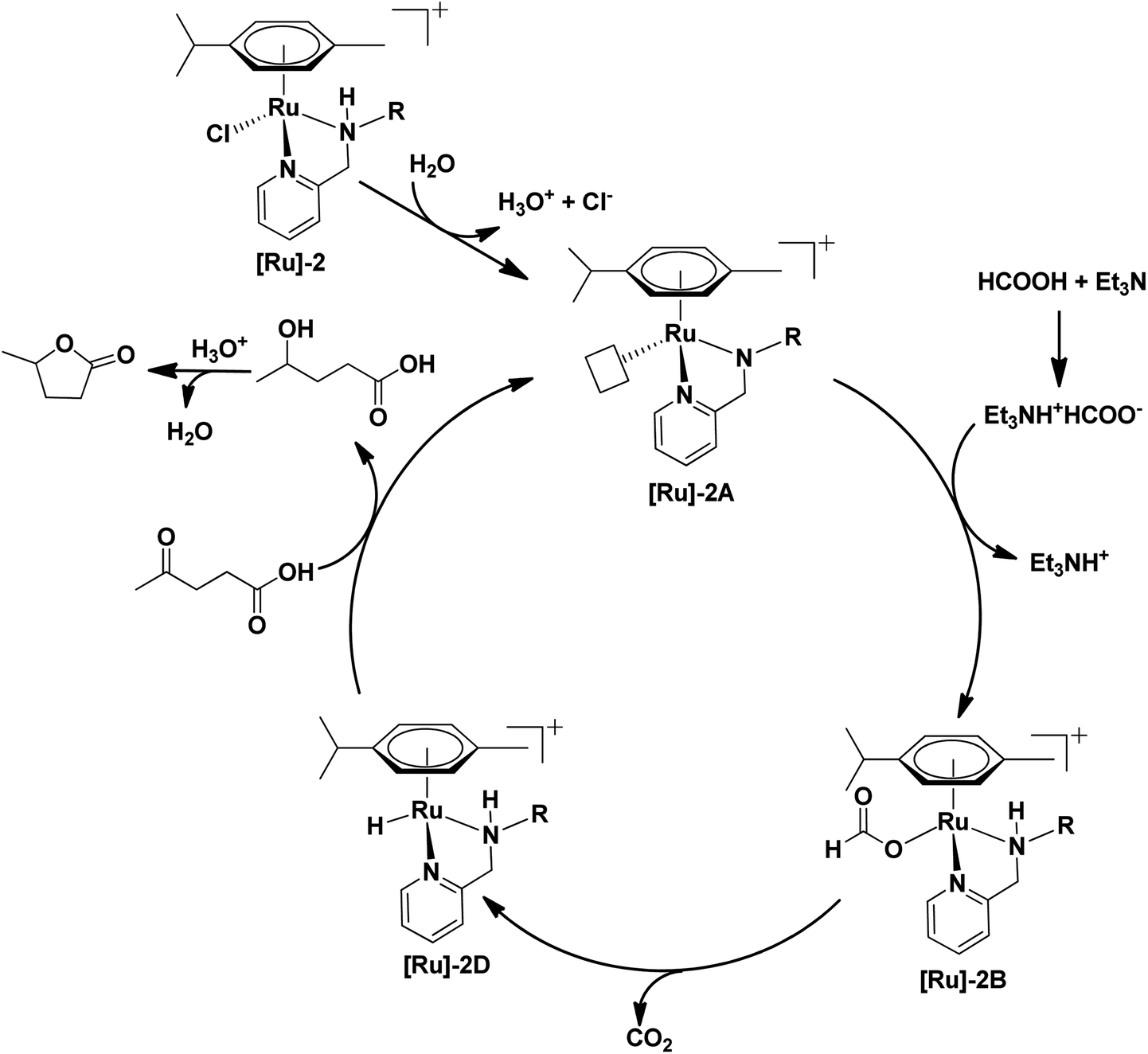 | ||
| Scheme 3 Plausible mechanism for the transformation of biomass derived LA to GVL. | ||
Catalyst recyclability and gram-Scale reaction for the transformation of LA to GVL
Notably, the [Ru]-2 catalyst also displayed good recyclability for five consecutive catalytic runs, where complete conversion was achieved for the initial three cycles with some decline in the conversion for the fourth and fifth catalytic runs to 82 and 74%, respectively (Table S10 and Fig. S4†). We performed the ICP-AES analysis of the organic and aqueous phases of the reaction mixture. The ICP-AES analysis of the organic portion inferred a loss of 10–12 ppm Ru in each catalytic run, which may account for the observed loss in catalytic activity during recyclability experiments. Furthermore, the aqueous layer inferred a prominent mass peak at m/z 385 (corresponding to [Ru]-2A) (Fig. S5†), suggesting the high stability of the catalyst under catalytic reaction conditions. Advantageously, the studied catalytic system can be scaled up to the gram-scale transformation of LA to GVL over the [Ru]-2 (0.025 mmol) catalyst to yield 95% GVL (TON 380) with complete conversion of LA in 24 h at 80 °C (Scheme 4). | ||
| Scheme 4 Gram scale catalytic transformation of LA to GVL over the [Ru]-2 catalyst. | ||
Catalytic one-pot two step transformation of furfural (FAL) to GVL
We also explored the catalytic one-pot transformation of furfural (FAL) to GVL over the [Ru]-2 catalyst (Fig. 5a). For this, to an aqueous suspension of [Ru]-2 (2.5 mol% in 5 mL water) in a graduated 10 mL test tube, 1 mmol FAL was added and heated at 80 °C in an oil bath for 12 h in the presence of 12 equivalents of formic acid. FAL is first hydrogenated to furfuryl alcohol (FA) and then acid catalyzed ring opening of FA resulted in the formation of LA, which is further confirmed by the 1H NMR analysis of the reaction aliquot (Fig. 5b). HCOOH served as both a hydrogenating agent and an acid source during the progress of reaction. To the same reaction vessel, 4 equiv. of HCOOH and 1 equiv. of Et3N were further added and the reaction mixture was allowed to stir at 80 °C for 4 h. 1H NMR analysis confirmed the formation of GVL as the product. Thus, we achieved complete conversion of FAL with 100% selectivity for GVL (yield 47%). The observed lower yield of GVL can be attributed to the poor stability of FAL in aqueous medium. These results demonstrated that the [Ru]-2 catalyst can be used for the production of GVL from the biomass platform chemical FAL with the aid of HCOOH in one pot without any need for the isolation of LA.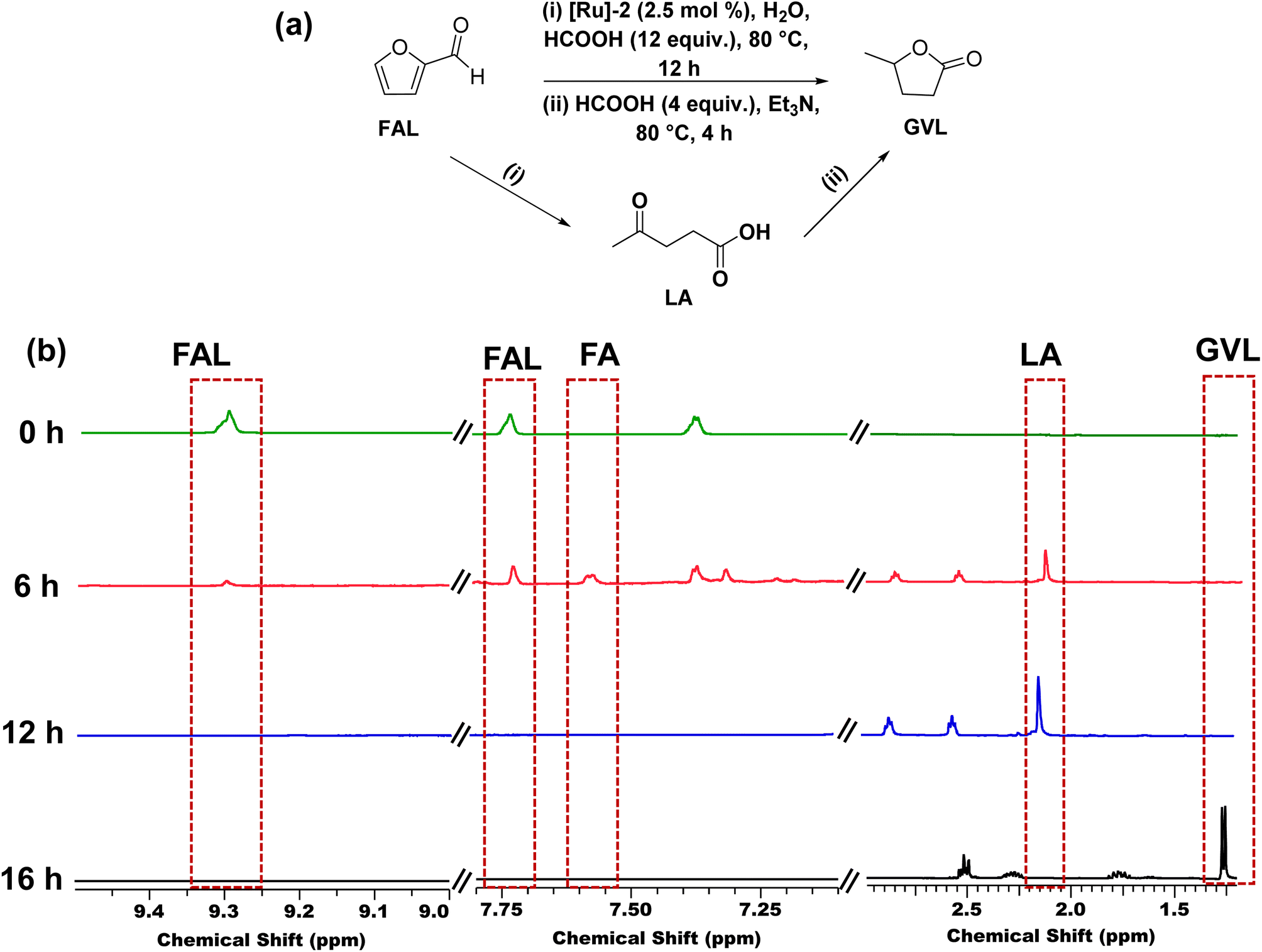 | ||
| Fig. 5 (a) Schematic representation and (b) time-dependent 1H NMR (in D2O) of one pot-two step catalytic transformation of FAL to GVL over the [Ru]-2 catalyst. Reaction conditions: FAL (1 mmol), [Ru]-2 (2.5 mol%), HCOOH (12 mmol), H2O (5 mL), 80 °C, and 12 h, and after 12 h Et3N (1 mmol) and HCOOH (4 mmol) were added, and the reaction continued for another 4 h. | ||
Therefore, our findings inferred that the [Ru]-2 catalyst demonstrated high catalytic activity for the complete and selective transformation of LA to GVL in water under much milder reaction conditions (80 °C) using HCOOH as a H2 source, as compared to previously reported Ru based catalytic systems (Table S1†). For instance, the literature revealed the use of Ru-phosphine-based catalysts that are active only at high H2 pressure (as high as 100 bar)37,39 and high reaction temperature (as high as 160 °C) to achieve the hydrogenation of LA to GVL (Table S1†). Advantageously, we could achieve one-pot transformation of furfural to GVL over the [Ru]-2 catalyst, which clearly demonstrated the advancement of our catalytic system for future applications.
Conclusions
We demonstrated herein a series of (η6-p-cymene)Ru(II)-pyridylamine based complexes for the efficient catalytic transformation of biomass-derived LA to GVL in water using formic acid at 80 °C. The water soluble (η6-p-cymene)Ru(II)-pyridylamine complexes with varying substituents at Namine were synthesized and their molecular structure was established using various characterization techniques including NMR, mass spectrometry and single crystal X-ray diffraction. The substituents at Namine of the pyridyl methyl amine ligands exerted a significant effect on the catalytic activity by tuning the steric crowding and the basicity at Namine. Among the studied complexes, [(η6-p-cymene)RuCl(κ2-pyNHnpr)]+ ([Ru]-2) outperformed with complete conversion of LA selectively to GVL (87% yield) at 80 °C in 2 h. Detailed mass investigations and control experiments provided a detailed mechanistic insight by revealing the possible involvement of [Ru]-2A and [Ru]-2B (Ru-formato) species in the process of LA hydrogenation, where the ratio of HCOOH:Et3N was found to be crucial to generate the Ru-formato ([Ru]-2B) and subsequently the Ru-hydrido ([Ru]-2D) species to facilitate the transformation of LA to GVL in water. Furthermore, time dependent studies revealed that 4-hydroxypentanoic acid (4-HPA) is formed as an intermediate during the catalytic transformation of LA to GVL. [Ru]-2 also exhibited high recyclability up to 4 cycles for LA to GVL transformation. Moreover, the developed methodology was also scaled up to gram-scale transformation of LA to GVL, highlighting the practical application of the system.
Experimental section
Synthesis of complexes [Ru]-1–[Ru]-8
Complexes [Ru]-1–[Ru]-4 and [Ru]-7 are known in the literature and synthesized by our previously reported procedures.49 Complexes [Ru]-5, [Ru]-6 and [Ru]-8 are not known and synthesized using our previously reported procedures.49 Typically, for the synthesis of [Ru]-1–[Ru]-8 complexes, [(η6-p-cymene)RuCl2]2 was suspended in acetonitrile (25 mL), stirred for 30 min at room temperature, and N-pyridylamine ligands (L1–L8) were added. After stirring the reaction mixture for 17 h at room temperature, NH4PF6 (3 equiv.) was added, and the solution was stirred for another 4 h at room temperature. Then, the solvent was removed under vacuum and the obtained residue was dissolved in a minimum amount of dichloromethane, filtered to remove unreacted NH4PF6 salt, and an excess of diethyl ether was poured into the above filtered solution to precipitate out the desired complexes as yellow solids. Precipitates were washed with diethyl ether (3 × 10 mL) and dried in air. Single crystals of [Ru]-4, [Ru]-5 and [Ru]-8 were obtained by diffusion of diethyl ether into methanolic solution of respective complexes. The CCDC deposition numbers of the complexes [Ru]-4, [Ru]-5, and [Ru]-8 are 1971292, 1971295 and 1971294, respectively.Synthesis of [(η6-p-cymene)RuCl(κ2-pyNH2)]PF6 ([Ru]-1)
Complex [Ru]-1 was synthesized by stirring 2-(aminomethyl)-pyridine (L1) (0.083 g, 0.8 mmol) and [(η6-p-cymene)RuCl2]2 (0.153 g, 0.25 mmol) in acetonitrile (25 mL) at room temperature. NH4PF6 (0.244 g, 1.5 mmol) was added and stirred for another 4 h. Yellow-brown solid, yield 0.165 g (63%). 1H NMR (400 MHz, DMSO-d6): δ (ppm) 9.11 (d, 1H, J = 4.00 Hz), 7.96 (t, 1H, J = 8.00 Hz), 7.53 (t, 1H, J = 8.00 Hz), 7.51 (d, 1H, J = 4.00 Hz), 5.91 (d, 1H, J = 8.00 Hz), 5.85 (d, 1H, J = 8.00 Hz), 5.72 (d, 1H, J = 8.00 Hz), 5.66 (d, 1H, J = 8 Hz), 4.45–4.39 (m, 1H), 4.21–4.07 (m, 1H), 2.74–2.66 (m, 1H), 1.95 (s, 3H), 1.11 (d, 6H, J = 4 Hz). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 161.88, 155.19, 139.78, 125.51, 121.79, 103.69, 98.66, 85.47, 83.71, 82.92, 82.35, 52.68, 30.87, 22.97, 22.07, 18.28. 31P NMR (161.97 MHz, DMSO-d6): δ (ppm) −144.19 (sep, PF6). MS (ESI): m/z calculated: 379.0150 [M]+, found: [M]+379.0776.Synthesis of [(η6-p-cymene)RuCl(κ2-pyNHnpr)]PF6 ([Ru]-2)
Complex [Ru]-2 was synthesized by stirring N-(pyridine-2-ylmethyl)-propan-1-amine (L2) (0.083 g, 0.55 mmol) and [(η6-p-cymene)RuCl2]2 (0.153 g, 0.25 mmol) in acetonitrile (25 mL). NH4PF6 (0.244 g, 1.5 mmol) was added and stirred for another 4 h. Pale yellow solid, yield 0.255 g (90%). 1H NMR (400 MHz, DMSO-d6): δ (ppm) 9.03 (d, 1H, J = 4.00 Hz), 8.02–7.98 (t, 1H, J = 8.00 Hz), 7.62–7.60 (d, 1H, J = 8 Hz), 7.58–7.54 (t, 1H, J = 8 Hz), 5.88–5.85 (t, 2H, J1 = 4 Hz, J2 = 8 Hz), 5.83–5.82 (d, 1H, J = 4 Hz), 5.75–5.73 (d, 1H, J = 8 Hz), 4.35–4.18 (m, 2H), 3.43–3.40 (m, 1H), 3.29–3.27 (m, 1H), 2.62–2.56 (m, 1H), 1.90 (s, 3H), 1.74–1.64 (m, 2H), 1.09–1.04 (m, 6H), 0.98–0.94 (t, 3H, J = 8 Hz). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 159.45, 154.88, 139.22, 125.13, 121.62, 105.14, 96.20, 85.54, 83.85, 82.45, 81.60, 61.12, 59.00, 30.53, 22.15, 21.40, 21.13, 17.40, 11.25. 31P NMR (161.97 MHz, DMSO-d6): δ (ppm) −144.42 (sep, PF6). MS (ESI): m/z calculated: 421.0981 [M]+, found: 421.0976 [M]+.Synthesis of [(η6-p-cymene)RuCl(κ2-pyNHipr)]PF6 ([Ru]-3)
Complex [Ru]-3 was synthesized by stirring N-(pyridine-2-ylmethyl)-propan-2-amine (L3) (0.083 g, 0.55 mmol) and [(η6-p-cymene)RuCl2]2 (0.153 g, 0.25 mmol) in acetonitrile (25 mL). NH4PF6 (0.244 g, 1.5 mmol) was added and stirred for another 4 h. Pale yellow solid, yield 0.179 g (63%). 1H NMR (400 MHz, DMSO-d6): δ (ppm) 9.00 (d, 1H, J = 4.00 Hz), 8.02–7.98 (t, 1H, J = 8.00 Hz), 7.65 (d, 1H, J = 8.00 Hz), 7.58–7.55 (t, 1H, J1 = 8.00 Hz, J2 = 4.00 Hz), 5.91 (d, 1H, J = 4.00 Hz), 5.82 (s, 2H), 5.71 (d, 1H, J = 4.00 Hz), 4.32–4.20 (m, 2H), 3.85–3.82 (m, 1H), 2.11–2.07 (m, 1H), 1.98 (s, 3H), 1.38 (d, 3H, J = 4.00 Hz), 1.31 (d, 3H, J = 4.00 Hz), 1.09 (d, 3H, J = 8.00 Hz), 1.00 (d, 3H, J = 8.00 Hz). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 159.57, 154.86, 139.17, 125.09, 121.79, 105.28, 96.58, 86.19, 84.92, 82.41, 80.38, 58.60, 56.18, 30.45, 23.42, 21.75, 21.64, 20.26, 17.38. 31P NMR (161.97 MHz, DMSO-d6): δ (ppm) −144.19 (sep, PF6). MS (ESI): m/z calculated: 421.0981 [M]+, found: 421.0977 [M]+.Synthesis of [(η6-p-cymene)RuCl(κ2-pyNHnbu)]PF6 ([Ru]-4)
Complex [Ru]-4 was synthesized by stirring N-(pyridin-2-ylmethyl)-butan-1-amine (L4) (0.090 g, 0.55 mmol) and [(η6-p-cymene)RuCl2]2 (0.153 g, 0.25 mmol) in acetonitrile (25 mL). NH4PF6 (0.244 g, 1.5 mmol) was added and stirred for another 4 h. Pale yellow solid, yield 0.263 g (91%). 1H NMR (400 MHz, DMSO-d6): δ (ppm) 9.03–9.01 (d, 1H, J = 8 Hz), 8.02–7.98 (t, 1H, J = 8 Hz), 7.62–7.60 (d, 1H, J = 8 Hz), 7.58–7.55 (t, 1H, J1 = 8 Hz, J2 = 4 Hz), 5.88–5.86 (d, 2H, J = 8 Hz), 5.83–5.81 (d, 1H, J = 8 Hz), 5.75–5.73 (d, 1H, J = 8 Hz), 4.35–4.18 (m, 2H), 3.46–3.44 (m, 2H), 2.60–2.55 (m, 1H), 1.89 (s, 3H), 1.69–1.63 (m, 2H), 1.39 (m, 2H), 1.09–1.05 (t, 6H, J = 8 Hz), 0.95–0.92 (t, 3H, J1 = 8 Hz, J2 = 4 Hz).). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 159.43, 154.87, 139.21, 125.13, 121.62, 105.25, 95.96, 85.71, 83.84, 82.41, 81.57, 61.22, 57.00, 30.55, 30.12, 22.14, 21.09, 19.62, 17.35, 13.79. 31P NMR (161.97 MHz, DMSO-d6): δ (ppm) −144.20 (sep, PF6). MS (ESI): m/z calculated: 435.1138 [M]+, found: 435.1112 [M]+.Synthesis of [(η6-p-cymene)RuCl(κ2-pyNHibu)]PF6 ([Ru]-5)
Complex [Ru]-5 was synthesized by stirring 2-methyl-N-(pyridine-2-ylmethyl)-propan-1-amine (L5) (0.090 g, 0.55 mmol) and [(η6-p-cymene)RuCl2]2 (0.153 g, 0.25 mmol) in acetonitrile (25 mL). NH4PF6 (0.244 g, 1.5 mmol) was added and stirred for another 4 h. Pale yellow solid, yield 0.168 g (58%). 1H NMR (400 MHz, methanol-d4): δ (ppm) 8.95–8.93 (d, 1H, J = 8 Hz), 7.95–7.91 (t, 1H, J = 8 Hz), 7.51–7.45 (m, 2H), 5.79–5.75 (t, 2H, J = 8 Hz), 5.68–5.66 (d, 1H, J = 8 Hz), 5.59–5.58 (d, 1H, J = 4 Hz), 4.40–4.35 (dd, 1H, J = 4 Hz), 4.25–4.18 (t, 1H, J1 = 12 Hz, J2 = 16 Hz), 3.38–3.28 (m, 2H), 2.66–2.56 (m, 1H), 2.18–2.11 (m, 1H), 1.97 (s, 3H), 1.13–1.08 (m, 6H), 0.99–0.97 (m, 6H). 13C NMR (100 MHz, methanol-d4): δ (ppm) 160.45, 159.19, 140.63, 126.78, 123.08, 107.73, 98.44, 87.19, 85.30, 84.02, 83.12, 66.92, 62.37, 32.49, 27.94, 22.85, 21.62, 20.96, 20.15, 18.18. 31P NMR (161.97 MHz, methanol-d4): δ (ppm) −144.58 (sep, PF6). MS (ESI): m/z calculated: 435.1138 [M]+, found: 435.1141 [M]+.Synthesis of [(η6-p-cymene)RuCl(κ2-pyNHnpen)]PF6 ([Ru]-6)
Complex [Ru]-6 was synthesized by stirring N-(pyridine-2-ylmethyl)-pentan-1-amine (L6) (0.098 g, 0.55 mmol) and [(η6-p-cymene)RuCl2]2 (0.153 g, 0.25 mmol) in acetonitrile (25 mL). NH4PF6 (0.244 g, 1.5 mmol) was added and stirred for another 4 h. Pale yellow solid, yield 0.241 g (81%). 1H NMR (400 MHz, DMSO-d6): δ (ppm) 9.03–9.01 (d, 1H, J = 8 Hz), 8.02–7.98 (t, 1H, J = 8 Hz), 7.62–7.60 (d, 1H, J = 8 Hz), 7.58–7.54 (t, 1H, J = 8 Hz), 5.88–5.86 (d, 2H, J = 8 Hz), 5.83–5.81 (d, 1H, J = 8 Hz), 5.74–5.73 (d, 1H, J = 4 Hz), 4.35–4.18 (m, 2H), 3.45–3.41 (m, 2H), 2.62–2.57 (m, 1H), 1.89 (s, 3H), 1.69–1.64 (m, 2H), 1.35–1.30 (m, 4H), 1.09–1.05 (t, 6H, J = 8 Hz), 0.90 (t, 3H). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 159.45, 154.87, 139.20, 125.12, 121.62, 105.21, 95.96, 85.71, 83.84, 82.43, 81.57, 61.19, 57.24, 30.55, 28.53, 27.75, 22.14, 21.96, 21.10, 17.35, 13.89. 31P NMR (161.97 MHz, DMSO-d6): δ (ppm) −144.18 (sep, PF6). MS (ESI): m/z calculated: 449.1294 [M]+, found: 449.1266 [M]+.Synthesis of [(η6-p-cymene)RuCl(κ2-pyNHipen)]PF6 ([Ru]-7)
Complex [Ru]-7 was synthesized by stirring 3-methyl-N-(pyridine-2-ylmethyl)-butan-1-amine (L7) (0.098 g, 0.75 mmol) and [(η6-p-cymene)RuCl2]2 (0.153 g, 0.25 mmol) in acetonitrile (25 mL). NH4PF6 (0.244 g, 1.5 mmol) was added and stirred for another 4 h. Pale yellow solid, yield 0.187 g (63%). 1H NMR (400 MHz, DMSO-d6): δ (ppm) 9.02–9.01 (d, 1H, J = 4 Hz), 8.00–7.98 (t, 1H, J = 8 Hz), 7.62–7.50 (m, 2H), 5.88 (m, 2H), 5.83–5.81 (d, 1H, J = 8 Hz), 5.74–5.73 (d, 1H, J = 4 Hz), 4.36–4.20 (m, 2H), 3.48–3.47 (m, 2H), 2.65–2.58 (m, 1H), 1.88 (s, 3H), 1.70–1.55 (m, 3H), 1.10–1.08 (t, 6H, J = 8 Hz), 0.94–0.92 (t, 6H, J = 8 Hz). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 159.40, 154.84, 139.20, 125.13, 121.63, 105.34, 95.78, 85.86, 83.81, 82.37, 81.61, 61.31, 55.67, 36.78, 30.58, 25.60, 22.57, 22.36, 22.17, 21.02, 17.30. 31P NMR (161.97 MHz, DMSO-d6): δ (ppm) −144.08 (sep, PF6). MS (ESI): m/z calculated: 449.1294 [M]+, found: 449.1294 [M]+.Synthesis of [(η6-p-cymene)RuCl(κ2-pyNHtpen)]PF6 ([Ru]-8)
Complex [Ru]-8 was synthesized by stirring 2-methyl-N-(pyridine-2-ylmethyl)-butan-2-amine (L8) (0.098 g, 0.75 mmol) and [(η6-p-cymene)RuCl2]2 (0.153 g, 0.25 mmol) in acetonitrile (25 mL). NH4PF6 (0.244 g, 1.5 mmol) was added and stirred for another 4 h. Pale yellow solid, yield 0.258 g (86%). 1H NMR (400 MHz, DMSO-d6): δ (ppm) 9.03–9.02 (d, 1H, J = 4 Hz), 8.77–8.66 (d, 1H, J1 = 40 Hz, J2 = 4 Hz), 8.02–8.00 (t, 1H, 8 Hz), 7.65–7.55 (m, 1H), 6.01–5.90 (m, 4H), 4.35–4.29 (m, 2H), 3.65–3.57 (m, 1H), 1.92 (s, 3H), 1.72–1.65 (m, 2H), 1.36–1.30 (d, 6H, J = 24 Hz), 1.06–1.00 (dd, 6H, J = 4 Hz), 0.90–0.86 (t, 3H, J = 8 Hz). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 159.50, 155.02, 139.23, 125.12, 122.08, 105.40, 96.41, 85.69, 84.55, 82.52, 79.79, 60.70, 55.40, 32.53, 30.53, 24.75, 24.57, 21.88, 17.18, 8.66. 31P NMR (161.97 MHz, DMSO-d6): δ (ppm) −144.20 (sep, PF6). MS (ESI): m/z calculated: 449.1294 [M]+, found: 449.1232 [M]+.General procedure for the catalytic transformation of LA to GVL in water
All the reactions were performed in a 10 mL graduated reaction test tube using water as solvent. Typically for the catalytic reaction, 1.0 mmol of LA, 6 mmol of HCOOH, and 1 mmol of Et3N were added to an aqueous suspension of the catalyst (2.5 mol% in 5 mL water). The reaction test tube was fitted to a condenser, and the reaction mixture was stirred at 80 °C in an oil bath for a specified time. All the catalytic reactions were performed using formic acid as the source of hydrogen without using any external hydrogen gas pressure. After reaction completion, the reaction mixture was extracted with diethyl ether (12 × 10 mL) and then acidified with 1 M HCl solution till pH 1. Then it was again extracted with ethyl acetate (6 × 10 mL). The combined organic portions were dried under reduced pressure. Conversion and selectivity were determined by 1H NMR.Control experiments for mechanistic study
The [Ru]-2 (0.025 mmol) catalyst was dissolved in 2.5 mL of water and stirred for 5–10 minutes, and the analysis of the reaction aliquot by mass spectrometry revealed the formation of [Ru]-A after dissociation of one Cl. Furthermore, the addition of LA (1 mmol), Et3N (1 mmol) and HCOOH (6 mmol) followed by stirring for 30 min at 60 °C and the subsequent analysis of the reaction aliquot by mass spectrometry inferred the presence of the Ru-formato ([Ru]-B) and Ru-levulinate ([Ru]-C) species. Reaction aliquots were taken out at definite time intervals and analyzed by mass spectrometry to detect the active catalytic species involved in the transformation of LA to GVL. The pH dependent studies were carried out by varying the ratios of HCOOH:Et3N to obtain a specific pH and the reaction aliquots were analyzed by 1H NMR to know the presence of intermediates involved in the transformation of LA to GVL every 30 minutes.
General procedure for the catalytic one-pot two-step transformation of FAL to GVL in water
To an aqueous suspension of the catalyst (2.5 mol% in 5 mL water) in a 10 mL graduated reaction test tube fitted with a condenser, 1.0 mmol of FAL and 12 mmol of HCOOH were added and the reaction mixture was stirred at 80 °C for a specified time in an oil bath. Reaction progress was monitored by TLC and after complete conversion of FAL, 4 mmol of formic acid and 1 mmol of triethylamine were added to the same reaction vessel and the reaction mixture was continued to stir at 80 °C for another 4 h until complete conversion of LA to GVL was achieved.Gram-scale and recyclability experiments
For the catalytic gram-scale reaction, to an aqueous suspension of [Ru]-2 catalyst (2.5 mol% in 5 mL water), 10 mmol (1.15 g) of LA, 60 mmol of HCOOH, and 10 mmol of Et3N were added, and the reaction mixture was stirred for 24 h at 80 °C in an oil bath to achieve complete conversion of LA. For the recyclability experiment, to an aqueous suspension of the catalyst (2.5 mol% in 5 mL water), 1.0 mmol of LA, 6 mmol of HCOOH and 1 mmol of Et3N were added and the reaction mixture was stirred at 80 °C in an oil bath for a specified time. After each run, the reaction mixture was extracted with diethyl ether and ethyl acetate, while the catalyst remains in the aqueous phase. Before performing the consecutive run, the required amount of HCOOH and Et3N was added to adjust pH to 2.7 and then the hydrogenation reaction of LA was carried out.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank IIT Indore, SERB DST (EMR/2016/005783), and SICI-SICRG grant, for the financial support. The Sophisticated Instrumentation Centre (SIC), IIT Indore, DST-FIST 500 NMR facility of the Department of Chemistry, IIT Indore, ICP-AES facility of SAIF IIT Bombay, India are gratefully acknowledged. B. P. and V. K. S. thank DST and UGC, New Delhi, for their fellowship.References
- L. Yan, Q. Yao and Y. Fu, Green Chem., 2017, 19, 5527–5547 RSC.
- F. Valentini, V. Kozell, C. Petrucci, A. Marrocchi, Y. Gu, D. Gelman and L. Vaccaro, Energy Environ. Sci., 2019, 12, 2646–2664 RSC.
- A. T. Adeleye, H. Louis, O. U. Akakuru, I. Joseph, O. C. Enudi and D. P. Michael, AIMS Energy, 2019, 7, 165–185 CAS.
- V. S. Shende, A. B. Raut, P. Raghav, A. A. Kelkar and B. M. Bhanage, ACS Omega, 2019, 4, 19491–19498 CrossRef CAS.
- A. S. Amarasekara and Y. M. Lawrence, Tetrahedron Lett., 2018, 59, 1832–1835 CrossRef CAS.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584–595 RSC.
- X. Tang, X. Zeng, Z. Li, L. Hu, Y. Sun and S. Liu, Renew. Sustain. Energy Rev., 2014, 40, 608–620 CrossRef CAS.
- G. Morales, J. A. Melero, J. Iglesias, M. Paniagua and C. López-Aguado, React. Chem. Eng., 2019, 4, 1834–1843 RSC.
- M. Ma, H. Liu, J. Cao, P. Hou, J. Huang, X. Xu, H. Yue, G. Tian and S. A. Feng, Mol. Catal., 2019, 467, 52–60 CrossRef CAS.
- K. Yan, Y. Yang, J. Chai and Y. Lu, Appl. Catal., B, 2015, 179, 292–304 CrossRef CAS.
- G. Amenuvor, B. C. E. Makhubela and J. Darkwa, ACS Sustain. Chem. Eng., 2016, 4, 6010–6018 CrossRef CAS.
- L. Wu, T. Moteki, A. A. Gokhale, D. W. Flaherty and F. D. Toste, Chem, 2016, 1, 32–58 CAS.
- M. Nemanashi, J. Noh and R. Meijboom, Appl. Catal., A, 2018, 550, 77–89 CrossRef CAS.
- M. Chen, Q. Dong, W. Ni, X. Zhao, Q. Gu, G. Tang, D. Li, W. Ma and Z. Hou, ChemistrySelect, 2017, 2, 10537–10545 CrossRef CAS.
- X. L. Du, Q. Y. Bi, Y. M. Liu, Y. Cao and K. N. Fan, ChemSusChem, 2011, 4, 1838–1843 CrossRef CAS PubMed.
- H. Wang, C. Chen, H. Zhang, G. Wang and H. Zhao, Chin. J. Catal., 2018, 39, 1599–1607 CrossRef CAS.
- B. Banerjee, R. Singuru, S. K. Kundu, K. Dhanalaxmi, L. Bai, Y. Zhao, B. M. Reddy, A. Bhaumik and J. Mondal, Catal. Sci. Technol., 2016, 6, 5102–5115 RSC.
- D. Liu, L. Zhang, W. Han, M. Tang, L. Zhou, Y. Zhang, X. Li, Z. Qin and H. Yang, Chem. Eng. J., 2019, 369, 386–393 CrossRef CAS.
- S. Lomate, A. Sultana and T. Fujitani, Catal. Sci. Technol., 2017, 7, 3073–3083 RSC.
- P. Balla, V. Perupogu, P. K. Vanama and V. R. C. Komandur, J. Chem. Technol. Biotechnol., 2016, 91, 769–776 CrossRef CAS.
- S. Dutta, I. K. M. Yu, D. C. W. Tsang, Y. H. Ng, Y. S. Ok, J. Sherwood and J. H. Clark, Chem. Eng. J., 2019, 372, 992–1006 CrossRef CAS.
- C. Ortiz-Cervantes, M. Flores-Alamo and J. J. García, ACS Catal., 2015, 5, 1424–1431 CrossRef CAS.
- K. Krommyda, C. Panopoulou, C. Moustani, E. Anagnostopoulou, K. Makripidi and G. A. Papadogianakis, Catal. Lett., 2019, 149, 1250–1265 CrossRef CAS.
- S. Wang, V. Dorcet, T. Roisnel, C. Bruneau and C. Fischmeister, Organometallics, 2017, 36, 708–713 CrossRef CAS.
- S. Wang, H. Huang, V. Dorcet, T. Roisnel, C. Bruneau and C. Fischmeister, Organometallics, 2017, 36, 3152–3162 CrossRef CAS.
- W. Li, J. H. Xie, H. Lin and Q. L. Zhou, Green Chem., 2012, 14, 2388–2390 RSC.
- J. Deng, Y. Wang, T. Pan, Q. Xu, Q. X. Guo and Y. Fu, ChemSusChem, 2013, 6, 1163–1167 CrossRef CAS.
- K. Sung, M. Lee, Y. J. Cheong and H. Y. Jang, Appl. Organomet. Chem., 2021, 35, e6102 CrossRef.
- L. Shen, Q. Zheng, Y. Liu, J. Wu, Z. Lua and T. Tu, Green Chem., 2021, 23, 5037–5042 RSC.
- H. Mehdi, V. Fabos, R. Tuba, A. Bodor, L. T. Mika and I. T. Horvath, Top. Catal., 2008, 48, 49–54 CrossRef CAS.
- L. Deng, J. Li, D.-M. Lai, Y. Fu and Q.-X. Guo, Angew. Chem., Int. Ed., 2009, 48, 6529–6532 CrossRef CAS PubMed.
- A. Dutta Chowdhury, R. Jackstell and M. Beller, ChemCatChem, 2014, 6, 3360–3365 CrossRef CAS.
- J. M. Tukacs, D. Király, A. Strádi, G. Novodarszki, Z. Eke, G. Dibó, T. Kégl and L. T. Mika, Green Chem., 2012, 14, 2057–2065 RSC.
- F. M. A. Geilen, B. Engendahl, M. Holscher, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2011, 133, 14349–14358 CrossRef CAS.
- V. Fábos, L. T. Mika and I. T. Horváth, Organometallics, 2014, 33, 181–187 CrossRef.
- N. K. Oklu and B. C. E. Makhubela, Inorg. Chim. Acta, 2018, 482, 460–468 CrossRef CAS.
- U. Omoruyi, S. J. Page, S. L. Apps, A. J. P. White, N. J. Long and P. W. Miller, J. Organomet. Chem., 2021, 935, 121650–121662 CrossRef CAS.
- G. Amenuvor, C. K. Rono, J. Darkwa and B. C. E. Makhubela, Eur. J. Inorg. Chem., 2019, 36, 3942–3953 CrossRef.
- C. A. M. R. Slagmaat, M. A. F. Delgove, J. Stouten, L. Morick, Y. Meer, K. V. Bernaerts and S. M. A. D. Wildeman, Green Chem., 2020, 22, 2443–2458 RSC.
- H. Liao, M. Chen, Y. Ma, Q. Peng, X. Wei and Z. Hou, Ind. Eng. Chem. Res., 2022, 61, 15156–15168 CrossRef CAS.
- N. Dai, R. Shang, M. Fu and Y. Fu, Chin. J. Chem., 2015, 33, 405–408 CrossRef CAS.
- Y. Yi, H. Liu, L. P. Xiao, B. Wang and G. Song, ChemSusChem, 2018, 11, 1474–1478 CrossRef CAS PubMed.
- G. Metzker and A. C. B. Burtoloso, Chem. Commun., 2015, 51, 14199–14202 RSC.
- Z. Liu, Z. Yang, P. Wang, X. Yu, Y. Wu, H. Wang and Z. Liu, ACS Sustain. Chem. Eng., 2019, 7, 18236–18241 CrossRef CAS.
- B. Zada, R. Zhu, B. Wang, J. Liu, J. Deng and Y. A. Fu, Green Chem., 2020, 22, 3427–3432 RSC.
- T. J. Vázquez, A. Arévalo and J. J. Garcia, Eur. J. Inorg. Chem., 2021, 2021, 445–450 CrossRef.
- T. Jurado-Vázquez, E. Rosaldo, A. Arévalo and J. García, ChemCatChem, 2022, 14, e202200628 CrossRef.
- D. A. Roa and J. J. Garcia, J. Catal., 2022, 413, 1028–1033 CrossRef CAS.
- M. K. Awasthi and S. K. Singh, Inorg. Chem., 2019, 58, 14912–14923 CrossRef CAS.
- A. D. Dwivedi, V. K. Sahu, S. M. Mobin and S. K. Singh, Inorg. Chem., 2018, 57, 4777–4787 CrossRef CAS.
- A. K. Chakraborty, K. B. Bischoff, G. Astarita and J. R. Damewood, J. Am. Chem. Soc., 1988, 110, 6947–6954 CrossRef CAS.
- J. Graton, M. Berthelot and C. Laurence, J. Chem. Soc., 2001, 2, 2130–2135 Search PubMed.
- L. Ouyang, Y. Xia, J. Liao and R. Luo, Eur. J. Org Chem., 2020, 2020, 6387–6391 CrossRef CAS.
- G. Amenuvor, J. Darkwa and B. C. E. Makhubela, Catal. Sci. Technol., 2018, 8, 2370–2380 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1971292, 1971295 and 1971294. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3su00285c |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |