
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanocrystalline iron hydroxide lignocellulose filters for arsenate remediation†
Steven A.
Soini
 ,
Sofia M.
Feliciano
,
Bobby G.
Duersch
and
Vivian M.
Merk
*
,
Sofia M.
Feliciano
,
Bobby G.
Duersch
and
Vivian M.
Merk
*
Department of Chemistry and Biochemistry, Department of Ocean and Mechanical Engineering, Florida Atlantic University, Boca Raton, FL 33431, USA. E-mail: vmerk@fau.edu
First published on 15th January 2024
Abstract
Harmful levels of environmental contaminants, such as arsenic (As), persist readily in the environment, threatening safe drinking water supplies in many parts of the world. In this paper, we present a straightforward and cost-effective filtration technology for the removal of arsenate from potable water. Biocomposite filters comprised of nanocrystalline iron oxides or oxyhydroxides mineralized within lignocellulose scaffolds constitute a promising low cost, low-tech avenue for the removal of these contaminants. Two types of iron oxide mineral phases, 2-line ferrihydrite (Fh) and magnetite (Mt), were synthesized within highly porous balsa wood using an environmentally benign modification process and studied in view of their effective removal of As from contaminated water. The mineral deposition pattern, minerology, as well as crystallinity, were assessed using scanning electron microscopy, transmission electron microscopy, micro-computed X-ray tomography, confocal Raman microscopy, infrared spectroscopy, and X-ray powder diffraction. Our results indicate a preferential distribution of the Fh mineral phase within the micro-porous cell wall and radial parenchyma cells of rays, while Mt is formed primarily at the cell wall/lumen interface of vessels and fibers. Water samples of known As concentrations were subjected to composite filters in batch incubation and gravity-driven flow-through adsorption tests. Eluents were analyzed using microwave plasma optical emission spectroscopy (MP-AES) and inductively coupled plasma mass spectrometry (ICP-MS). By subjecting the filters to a flow of contaminated water, the time for As uptake was reduced to minutes rather than hours, while immobilizing the same amount of As. The retention of As within the composite filter was further confirmed through energy-dispersive X-ray mappings. Apart from addressing dangerously high levels of arsenate in potable water, these versatile iron oxide lignocellulosic filters harbor tremendous potential for addressing current and emerging environmental contaminants that are known to adsorb on iron oxide mineral phases, such as phosphate, polycyclic aromatic hydrocarbons or heavy metals.
Sustainability spotlightSafe and readily available drinking water is vital for public health, yet financial barriers limit access in some parts of the world. As a confirmed carcinogen, the World Health Organization classified inorganic arsenic as the most significant chemical contaminant in potable water around the globe. This work focuses on developing, characterizing, and testing arsenate filters based on sustainable lignocellulosic material and immobilized iron oxide mineral phases. The chemical modification of the wood filters fulfills green chemistry guidelines by using non-toxic chemicals and water as solvent while harnessing the largest source of renewable biomass. Apart from arsenate, our technology may address a variety of current and emerging environmental contaminants that adsorb on iron oxide mineral phases, including phosphates, polycyclic aromatic hydrocarbons, perfluorocarbons, or heavy metals. |
Introduction
Drinking water contamination with arsenic is a common problem in many parts of the world.1,2 Heavy metals, such as arsenic, can leach into potable water supplies or groundwater from household plumbing and service lines, mining operations, fertilizers, petroleum refineries, electronics manufacturers, municipal waste disposal, cement plants, and natural mineral deposits.1 The high toxicity and group-1 carcinogenicity of arsenic prompted the World Health Organization (WHO)3 and the U.S. Environmental Protection Agency (EPA)1 to limit the maximum tolerable level of arsenic in drinking water to 10 μg L−1 (=10 ppb).4 Yet, some water sources in densely populated developing countries, such as Bangladesh or India, often exceed this threshold by ten times and more,1,2 presenting a long-term threat to human health. Around 35 to 77 million people in Bangladesh are susceptible to lung, bladder and skin cancer due to unsafe levels of arsenic in drinking water.1,2 In first-world countries, arsenic is released into the environment through natural geochemical processes, such as mineral dissolution by weathering, microbial activity, or complexation with natural organic materials.5 As a result, many domestic wells across the United States exceed the maximum contaminant level of arsenic in drinking water (>10 μg L−1),1 thereby threatening local drinking water supplies. Depending on the chemical form and oxidation state, arsenic compounds vary in toxicity, with the trivalent form being generally more hazardous than the pentavalent form.6 Under oxidizing conditions, As(V) is the predominant species occurring as oxyanions of arsenic acid (H3AsO4, H2AsO4−, HAsO42−and AsO43−).5One method for the removal of As from drinking water is reverse osmosis.7 This approach, however, can involve significant costs that hamper a broader application in many parts of the world.8,9 In addition, these systems may require pre-treatment systems to remove suspended matter to prevent clogging. In the past decade, there has been a large push towards functional materials from sustainable resources. The raw materials necessary for devising green composite materials are found in abundance in the biosphere, for instance in form of plant or marine waste.10 Lignocellulosic materials are readily available and exhibit excellent mechanical stability at low specific density.11 Wood presents a hierarchical interconnected pore network that can be functionalized by thermal or chemical modification.12–14 Due to its excellent biocompatibility and low cost, chemically modified wood has found application as versatile filter material for copper,15 crude oil16 and other organic pollutants.17 Previous research demonstrated the innate filtering capacity of the plant xylem for nanoscopic particulate matter, including waterborne pathogens.18 By utilizing intact lignocellulose, further pretreatment or activation steps required for chemically modifying waste biomass can be avoided, substantially lowering the cost of production and the environmental impact.19
In this study, we pursued the precipitation of nanocrystalline ferrihydrite and magnetite inside porous lignocellulosic supports using a straightforward in situ chemical reaction of iron salts and potassium hydroxide. With an average density of only ρ ≈ 160 kg m−3, balsa wood (Ochroma lagopus) provides a void volume up to 97%,4,11 resulting in a large wettable pore surface area that is amenable to chemical functionalization.
By varying the reaction chemistry (Fig. 1A and B), we altered the spatial distribution, mineralogy, grain size, nanostructure, and aggregation state of the iron oxide mineral phase and optimized interactions with the environmental contaminant.20 The ultralow solubility of the iron oxides21 ensures a complete immobilization of the mineral phase within the scaffold, thereby minimizing leaching over time.20 Both iron oxide mineral phases, ferrihydrite (Fe10O14(OH)2)22 and magnetite (Fe3O4),23 are known to exhibit high adsorption capacity for As(III) and As(V) at neutral pH values and very similar inner-sphere complexation constants for arsenic adsorption.5 Apart from using wood as a solid support, we explored synthetic protocols that specifically target the cell wall (sub)micropores. In doing so, we aimed to stabilize the metastable two-line ferrihydrite mineral through nanoscale confinement, thereby preserving its high surface reactivity.24 Previous research has shown that a cell-wall specific treatment can be achieved by adjusting the reaction chemistry25,26 or by removing a part of the wood constituents through delignification.27
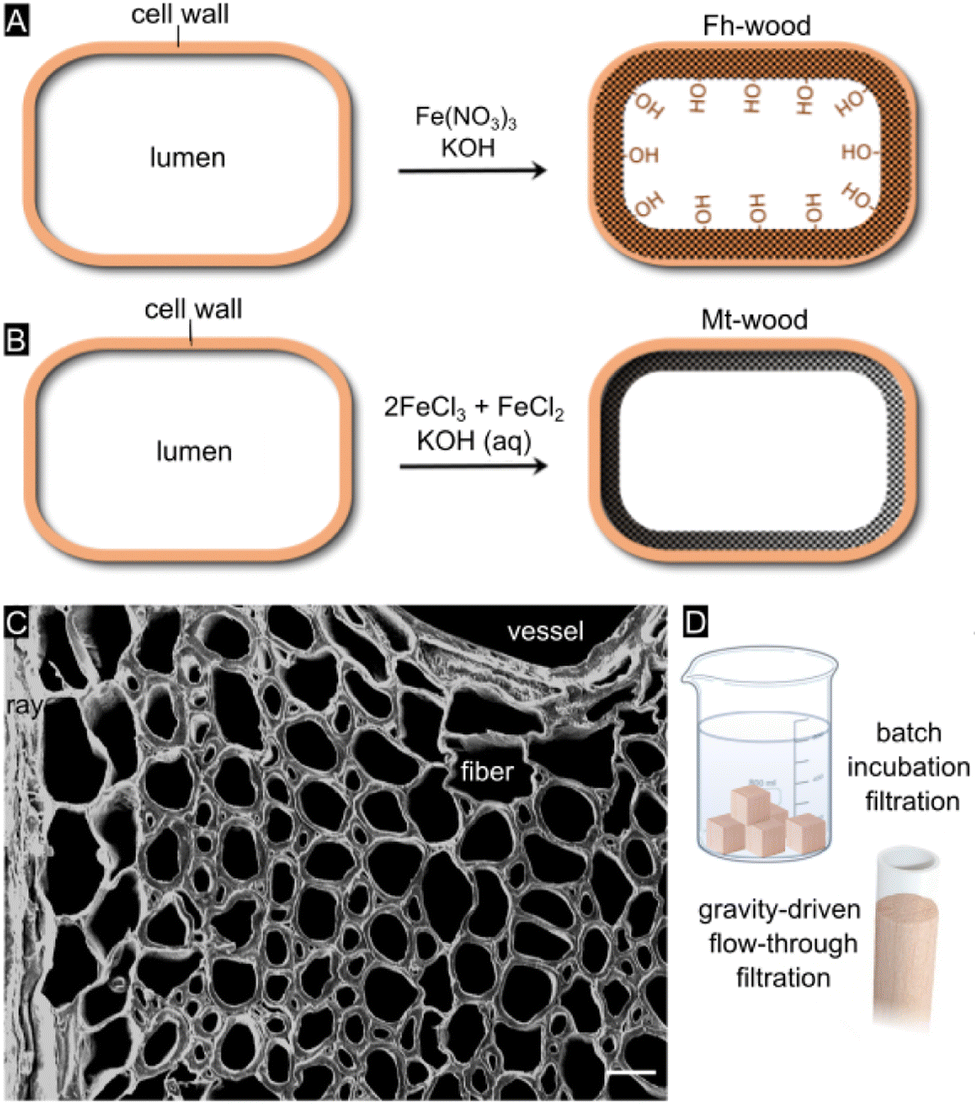 | ||
| Fig. 1 (A) Schematic of in situ synthesis of ferrihydrite (Fh) mineral within the cell wall of thin-walled balsa wood. (B) Schematic of in situ synthesis of magnetite at the cell wall/lumen interface. (C) Field-emission SEM image of ferrihydrite-balsa cross section. Scale bar equals 10 μm. (D) Schematic of batch incubation filtration and gravity-driven flow-through filtration of As-contaminated drinking water. | ||
Our overall goal was to develop a straightforward, low-cost, and rapid filter technology for generating clean drinking water in resource-limited settings. We were motivated to devise adsorbent media that can be operated either in a simple batch mode or under gravity-driven flow without a vacuum pump (Fig. 1D). As wood is available in different types, arbitrary shapes and sizes, our filtration technology can be adjusted to the user's needs and be upscaled to large-scale operation. A common drawback of conventional adsorbents based on nanocrystalline iron oxides is that substantial periods of time (24–48+ hours) are required to reduce arsenic to tolerable levels,5 plus, they are not easy or convenient to use for consumers. A benefit of our water treatment device is that mineralized wood filter cartridges can be easily separated from water, and subsequently discarded or burnt in hazardous waste incinerators after As immobilization.
Materials and methods
Materials
Balsa wood (Ochroma pyramidale) was purchased from the company Specialized Balsa Wood and cut to size before mineralization. Ferric nitrate nonahydrate (98%+, Fe(NO3)3·9H2O), ferrous chloride tetrahydrate (FeCl2·4H2O), ferric chloride hexahydrate (FeCl3·6H2O), sodium hydrogen arsenate heptahydrate (98%, Na2HAsO4·7H2O), potassium hydroxide (KOH) were purchased from Alfa Aesar. Reagents were not subjected to any further purification before use.Mineralization
A precursor solution for Fh precipitation was made by dissolving 8.08 g of Fe(NO3)3·9H2O in 50 mL Milli-Q® ultrapure water (resistivity 18.2 MΩ cm at 25 °C; total organic carbon (TOC) ≤ 5 ppb) under stirring. Balsa wood blocks of 20 mm × 20 mm × 25 mm were vacuum-impregnated in an aqueous Fe(NO3)3 solution for 24 h to ensure complete saturation of the blocks. To precipitate the mineral phase, a solution of 1.0 M KOH was slowly added under constant stirring until the pH of the solution was 7–8. The samples were again vacuum-impregnated and left to soak in solution over 24 hours before thoroughly washing in deionized water until reaching a neutral pH. Mt mineralization was achieved using 0.2 M ferric chloride and 0.4 M ferrous chloride and subsequent pH adjustment to 10–11 with 1.0 M KOH solution.Microstructural and chemical imaging
Cylindrical balsa samples (h = 12.5 mm, ⌀ = 10 mm) were mineralized with Fh. X-ray micro-computed tomographies were collected using a Bruker Skyscan 1173 X-ray microCT scanner utilizing a voltage of 60 kV, current of 133 μA, pixel size of 7.917 μm and rotation between sequential captured images of 0.16°.Smooth blockfaces and thin sections were prepared from the mineralized filters using a Leica RM2125 RTS rotary microtome. Light microscopy images were collected using a Leica M125 C stereoscope paired with a Leica DMC 6200 digital camera. For the Raman measurements, 35 μm cross sections were placed on a microscopic glass slide with a few drops of Milli-Q® water and sealed with a coverslip and nail polish to avoid evaporation. Confocal Raman mappings were collected with a Horiba XploRa confocal Raman microscope equipped with a 532 nm laser, 600 grooves mm−1 grating and 100× oil objective (NA = 1.3), a step width of 300–500 nm and two 30 s exposures per pixel for the automated removal of cosmic rays in the LabSpec6 data acquisition software. The laser power was always kept <1.5 mW to prevent thermal transformation of the iron oxide phases.28,29 Raman spectra were baseline-corrected using the adaptive baseline subtraction tool in the optical spectroscopy software SpectraGryph 1.2. Single spectra were plotted using OriginPro. Additionally, for the extraction of hyperspectral data, Vertex Component Analysis (VCA) was performed in the spectral range from 200–2000 cm−1 with the Matlab-based software Cytospec v. 2.00.07 assuming 4 endmembers.
Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDS) microanalysis were carried out on an FEI Quanta 200 SEM driven by an accelerating voltage of 15 kV in high-vacuum mode using an Everhard Thornley secondary electron detector and an UltimMax65 large-area analytical silicon drift EDS detector. Prior to SEM/EDS analysis, 35 μm sections were mounted onto Al stubs using carbon tape and sputter-coated with a thin layer of Pt under an argon atmosphere at 8 mA for 30 s using a MicroNanoTools MNT-JS1600 plasma sputtering coater. Tru-Q® EDS mappings were collected using the AZtec software package, which includes an algorithm for real-time peak deconvolution and background removal. Additional SEM images were collected with a JEOL 6330F field-emission SEM at 5 kV acceleration voltage. A TA Instruments Thermal Gravimetric Analyzer (TGA) was used to measure weight loss and the rate of weight change as a function of temperature. Fourier-Transform Infrared Spectra (ATR-FT-IR) were obtained with a Thermo Scientific Nicolet iS10 Infrared spectrometer equipped with attenuated total reflection. FT-IR spectra were acquired with 160 scans at 2 cm−1 resolution using the OMNIC Series Software and the data were displayed and analyzed using the Origin 2020b software. A background spectrum was collected prior to data collection and subtracted from the spectra. Further atmospheric corrections were not performed, but ATR correction was included. Samples were powdered using the rotary microtome set to 1 mm increments. X-ray powder diffraction patterns were collected from ground composites and mineral formed outside the wood samples using a Siemens D5000 Diffractometer equipped with a Cu X-ray source (λ = 0.149 nm) over the 2Θ range of 10–80°. A PerkinElmer STA 6000 Thermogravimetric Analyzer (TGA) was used to measure weight loss and rate of weight change as a function of temperature. Approximately 0.01 g of powdered sample was used for each analysis and each analysis was repeated 3 times. The temperature was scanned from 50 to 995 °C at 10 °C min−1.
A 120 kV JEOL JEM-1400 transmission electron microscope (TEM) was utilized to study the nanoscale Fh distribution in the cell wall. Samples were dehydrated using an ethanol series (EtOH/H2O: 25/75, 50/50, 75/25, 90/10, 100/0, 100/0) and embedded in araldite resin (Embed 812, Electron Microscopy Sciences). 100 nm slices were prepared using a Leica Ultracut EM UC7RT ultramicrotome with a 35° DiATOME® ultra diamond knife and placed on formvar-coated 200 mesh Cu grids (PELCO®). Nanoparticle sizes were measured using ImageJ (Version 1.54 g).
Arsenate adsorption studies
The adsorption capacity of the mineralized filters for arsenate species (AsO4) was probed through soaking and flow-through tests. Results for Fh filters were compared with pristine wood filters and those functionalized with a different iron oxide mineral phase, magnetite (Mt). To remove residual metal ions, glassware was thoroughly washed with hydrochloric acid. Solutions of 1 mg L−1 (1 ppm) AsO4 concentration were prepared from Na2HAsO4·7H2O in nanopure water. For soaking trials, filters were exposed to the AsO4 solution for 24 hours to allow for the solution to be taken up by the filter and allow for sufficient interactions with the mineral phase(s). After 24 hours, the filters were removed from the solution and the remaining solutions were collected for analysis.Two flow-through analysis tests for AsO4 removal were conducted using cylindrical filters (⌀ = 20 mm, h = 25 mm height). Filters were confined within a section of flexible PVC piping to create a tight seal with the axial direction of the structure parallel to the direction of water flow. For the first test. 100 mL of solution with a known starting arsenate concentration (1 mg L−1 = 1 ppm) was passed gravimetrically (20–25 mL min−1) through each of the 3 filter types with 10 mL of eluent being collected after each pass through the filters.
After AsO4 trials, filters were air dried and stored at 4 °C for subsequent elemental analysis using SEM-EDS. The pH of the resulting eluents was measured and recorded to be 7.04 prior to the addition of concentrated HNO3 (15.8 M) to result in 2% HNO3 final concentration in each. The solutions were then analyzed via Microwave Plasma Atomic Emission Spectroscopy (MP-AES) at a wavelength of 188.979 nm using an Agilent 4210 MP-AES instrument to determine the As(V) concentrations remaining in each solution. Concentrations reported are the average of 3 replicate measurements for each solution. Between each set of measurements, 2 samples of black coffee and 1 blank were run through the instrument to remove any adsorbed As from the sample chamber and ensure an accurate concentration reading for the measured samples.
The second set of analyses for As were conducted using a starting solution of 100 μg L−1 (100 ppb) AsO4 to more accurately represent contaminated environmental levels. Eluents were collected from each of the 3 filter types at the 10th and 15th passes in the flow-through and after 24 hours for batch incubation. Samples were diluted with an equal volume of 1% nitric acid before analysis. Inductively Coupled Plasma Mass Spectrometry (ICP-MS) was conducted using an Agilent 8900 triple quad with an SPS auto sampler, radio frequency power of 1550W, single-element As standard (Spex CertiPrep CLAS2-2Y), and argon plasma gas flow of 15 L min−1. Data were quantified using weighed, serial dilutions of a multi-element standard (CEM 2, VHG labs, VHG-SM70B-100) for Mn, Fe, Cu, and Zn, as well as a single-element standard for As (Spex CertiPrep CLAS2-2Y). Data were quantified using a 12-point calibration curve. A NIST SRM 1683f was prepared at 8× dilution (3.5 mL of 1% HNO3 + 500 μl of NIST SRM 1683f) to ensure accuracy of the calibration curve. For each sample, data were acquired in triplicate and averaged. A coefficient of variance (CoV) was determined from frequent measurements of a sample containing 10 ppb of the elements. An internal standard (Sc, Ge, Bi) continuously introduced with the sample was used to correct for detector fluctuations and to monitor plasma stability.
Results and discussion
Wood mineralization
As the arsenate sorption capacity crucially depends on the exposed specific surface area as well as the mineral nanostructure, we set out to explore two synthesis routes to examine different iron oxide mineral phases and their effectiveness in terms of contaminant removal. By varying the experimental conditions, two types of iron oxide mineral phases commonly used as adsorbents for the removal of arsenate, ferrihydrite and magnetite, were formed within the complex organic scaffold. The synthesis protocols for ferrihydrite and magnetite were adapted from Schwertmann and Cornell.30 The fast basic hydrolysis of Fe(NO3)3 as the iron source yields disordered, poorly crystalline two-line ferrihydrite (Fe10O14(OH)2), a Fe(III) oxyhydroxide with the simplified stoichiometry Fe(OH)3, whereas Mt was synthesized through the co-precipitation of mixed valent ferric and ferrous chloride salts at a more alkaline pH value.31 The reaction conditions, particularly the pH value and the speed of hydroxide addition,31 affect the mineralogy and therefore the adsorption capability of the reaction product. The high reactivity of ferrihydrite stems from its high surface area, poor crystallinity and defect structure.32–36 According to literature data, the underlying molecular–level interactions for adsorption of arsenic on ferrihydrite are predominantly based upon ligand exchange with surface –OH groups.19 Previous research showed that the surface reactivity is highly dependent on the crystal size, even after normalizing by specific surface area.37 Various oxyanions, such as nitrate,11,38 bicarbonate,11,38 phosphate,39 arsenite32 and arsenate,32,40 as well as heavy metal cations, such as Pb2+, Cd2+, Cu2+ and Zn2+41 are known to adsorb on Fe-(hydr)oxide surfaces.Mineral distribution in hierarchical scaffold
The mineral deposition pattern, minerology, along with the crystallinity of the mineral phases, were assessed using micro-computed X-ray tomography, scanning electron microscopy together with energy-dispersive X-ray spectroscopy, confocal Raman microscopy, transmission electron microscopy, Fourier-transform infrared spectroscopy, and X-ray powder diffraction. The synthesis protocol, specifically the pH at which the iron oxide minerals precipitated, determines which mineral phase is being formed, but it also affects the mineralization pattern within the complex and highly directional cell architecture.In the present study, micro-computed X-ray tomography (MicroCT) provided deeper insights into the bulk mineral distribution and possible solute transport pathways within the three-dimensional structure (Fig. 2A, S1†). Hardwood species such as Ochroma pyramidale contain three types of cellular structures, namely vessels, rays, and fibers11 (Fig. 1C), whereby vessels are widely regarded as the main water conduits in angiosperm trees.42,43 In hardwood species such as balsa, most of the axial fluid transport occurs through the interconnected vessel network.43–47 Fluid passage through the transverse vessels results in an overall homogeneous distribution of solutes, while rays mediate the radial fluid transport across growth rings.45 Based on the analysis of the X-ray MicroCT, both rays and vessels display high degrees of mineral deposition, likely due to their prominent roles in fluid transport, allowing for a thorough infiltration of the solutions used for mineralization. The mineral distribution throughout the smaller wood fibers, which make up the bulk volume of the composite material and provide mechanical strength, appears to be homogenous across the examined cross sections (Fig. 2B–D).
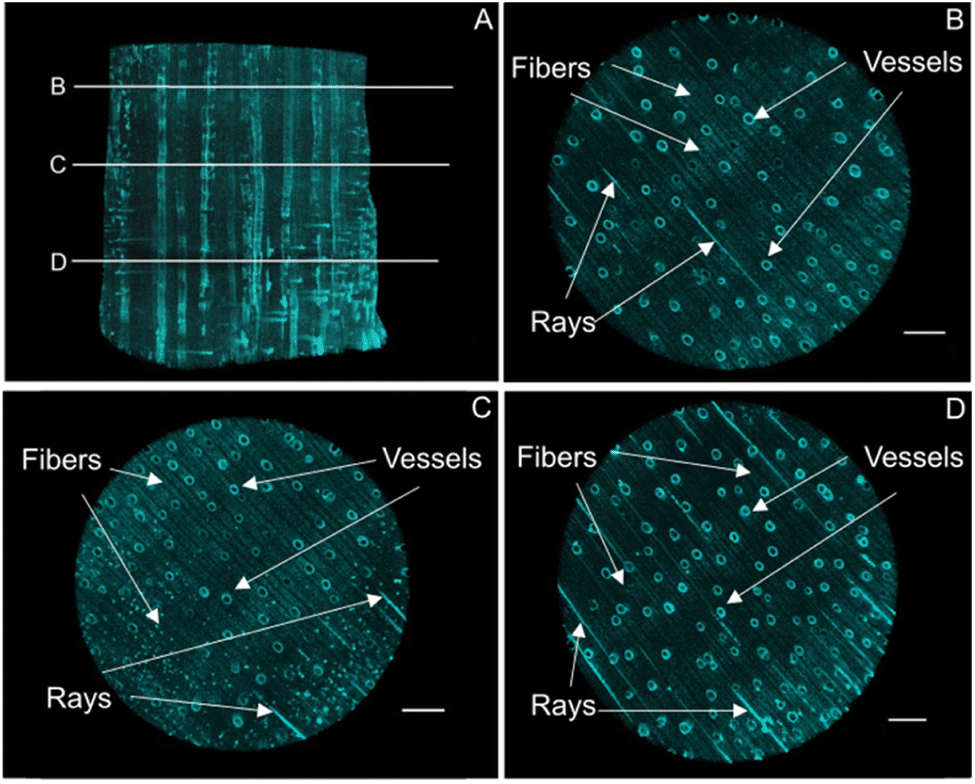 | ||
| Fig. 2 X-ray microtomography of Fh mineralized balsa scaffolds. X-ray absorbent regions pertaining to the Fh mineral phase were false coloured in turquoise to visualize their three-dimensional distribution. (A) 3D volumetric rendering of the mineralized composite highlighting the extensive mineralization of vessels and rays throughout the sample. The positions of 2D transverse projections are marked. (B–D) Cross-sectional projections showing mineral deposition at the vessel/lumen interfaces, in rays and within fibre cell walls. Scale bars correspond to 1 mm. | ||
Confocal Raman spectroscopy and scanning electron microscopy with energy dispersive X-ray spectroscopy (SEM-EDS) were utilized to examine the distribution of the mineral phase within the lignocellulose scaffold at the micron level. SEM-EDS microanalysis of mineralized balsa cross sections revealed a relatively homogenous distribution of Fh within the cell wall (Fig. 3B), while mineral deposition within the compound middle lamellae of adjacent fibers and radial parenchyma cells appears to occur more sporadically and clustered. As previous research suggested, fine-grained Mt is primarily formed at the cell wall/lumen interface of vessels and fibres.20 EDS analysis of the Fe content in the scaffolds gave between 15 and 20% Fe in all examined samples, while the measured weight gain amounted to 21% for Fh and 18% for Mt (Table S1†).
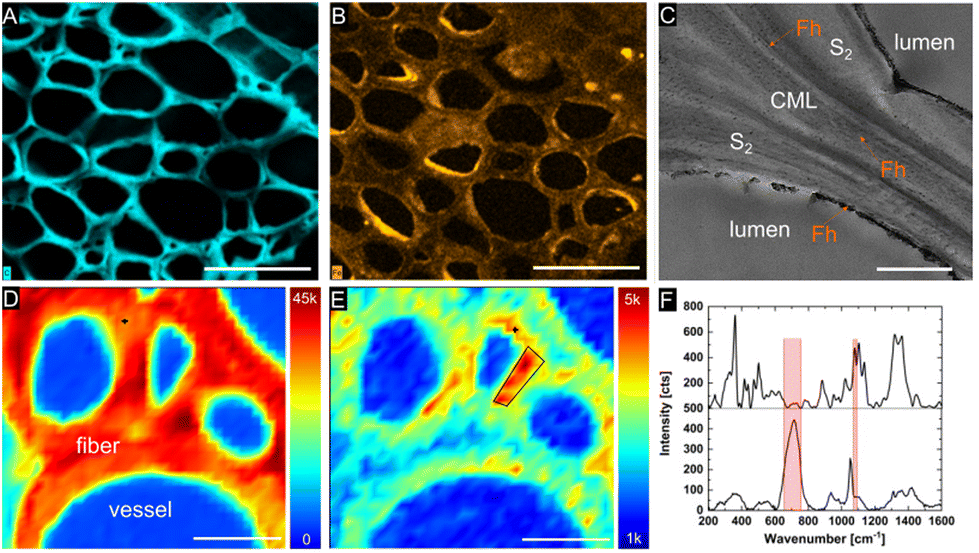 | ||
| Fig. 3 SEM/EDS mappings and microanalysis. Elemental mappings of Fh composite showing carbon (A) and iron (B) distribution. Scale bar corresponds to 50 μm. (C) TEM image shows formation of nanocrystalline Fh particles throughout compound middle lamella (CML), S1, and S2 cell wall layers and at S3 cell wall/lumen interface. Scale bar corresponds to 1 μm. Confocal Raman microscopy: chemical mapping illustrating the distribution of cellulose |1068–1190 cm−1| (D) and ferrihydrite |650–750 cm−1| (E) across Fh mineralized wood cells. Scale bars correspond to 10 μm. (F) Average Raman spectrum from region of interest marked in E (upper curve) and pure Fh powder (lower curve). | ||
According to SEM/EDS microanalysis (Table S1†), K+ ions are widely and homogeneously distributed across the wood cell walls, suggesting that the alkaline KOH solution fully saturated the entire wood structure. With regards to the chemical modification of the cell wall, a limiting factor is the slow diffusion of precursor solutes from the bulk solution into the water-saturated cell wall, with the diffusion depth being proportional to the square root of the diffusion time.48,49 Experimental evidence suggested that monovalent ions, such as K+, diffuse faster than bivalent or trivalent ions through the negatively charged capillaries.50 The precursor concentration in the wood tissue is proportional to the bulk concentration of the electrolyte, but prior to equilibrium, a concentration gradient across the cell wall with the highest retention in surface layers is common.48
We characterized the mineralogy and crystallinity of iron oxyhydroxide and iron oxide mineral phases using powder XRD, FT-IR spectroscopy and confocal Raman microscopy. Using powder XRD, we confirmed that two-line ferrihydrite and magnetite were formed under the reaction conditions (Fig. 4, S2†). To further confirm the nanocrystalline nature of the magnetite, the average particle size was calculated using the Scherrer equation and the peak of highest intensity in the XRD pattern (Fig. 4A), the (311) reflection, which resulted in an average grain size of 13.6 nm. Due to the amorphous nature of the ferrihydrite mineral phase, a particle size analysis using XRD was not possible. In Fh-mineralized balsa, we further detected infrared bands attributed Fe–OH bending (825 cm−1) and Fe–O stretching (1558 cm−1),41 which are characteristic of 2-line ferrihydrite (Fig. S3†). To localize the mineral phase at the micron scale, spectroscopic Raman mappings were obtained from the mineralized cell wall (Fig. 3D–F, S4†) using extended acquisition times, since iron oxide minerals are prone to phase transformation when irradiated at high laser powers.28,29 The vibrational spectra exhibit one prominent Raman band at 710 cm−1 (Fig. 4F), which is consistent with the formation of poorly crystalline 2-line ferrihydrite,29,41 whereas six-line ferrihydrite would be characterized by three Raman bands, 370 cm−1, 510 cm−1 and 710 cm−1.29 Due to the high fluorescence background of rays and vessels stemming from aromatic cell wall constituents,51 our Raman analysis primarily focused on the cell walls of mineralized fibers. To further decipher the Raman spectrum from the mineralized wood cell wall of fibers and an adjacent vessel, the hyperspectral datasets were processed using Vertex Component Analysis (VCA) to separate them into their most distinct spectral endmembers, with both methods resulting in close agreement with regards to the mineral distribution pattern. Using these two methods, we found that while a part of the Fh is deposited within the middle lamellae and lumina, significant amounts of Fh formed within the sub-microporous cell wall. To further confirm the deposition of Fh within the cell wall, TEM imaging of the Fh composites revealed the presence of clustered nanoscopic Fe crystallites at the S3-lumen interface, as well as finely dispersed Fh nanoparticles with an average size of 27 ± 6 nm throughout the S2 cell wall and compound middle lamella, CML (Fig. 3C, S4†). The location and size determination of Fh nanoparticle are displayed in Fig. S4 and Table S2.† Previous studies on iron oxide, namely Mt, in lignocellulose scaffolds found mineralization to occur primarily at the lumen–cell wall interface without major mineral deposition within the cell wall.20
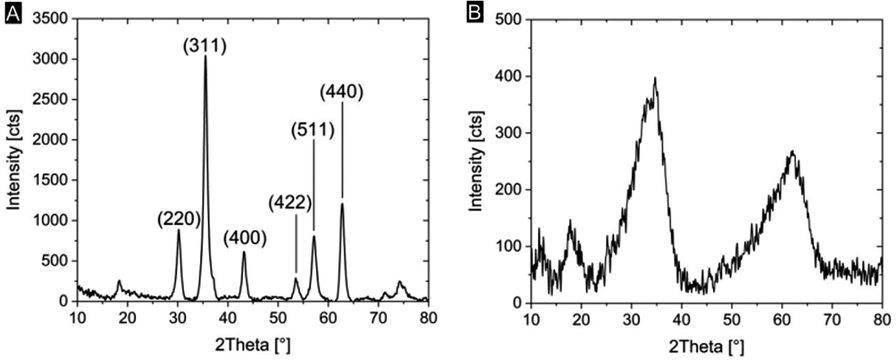 | ||
| Fig. 4 Powder X-ray diffraction from respective mineral phases precipitated in reaction solution. (A) Magnetite powder XRD with labelled Bragg reflections. (B) Two broad XRD peaks indicate the formation of 2-line ferrihydrite. | ||
This result could be due to the higher tendency of particle aggregation at high ionic strengths52,53 or the smaller particle size of Fh compared with Mt, allowing for a larger amount of particle growth within the cell wall micropores. The average pore size of the wood cell wall ranges in the low nanometer regime and changes dynamically depending on the relative humidity.54–57 As previously mentioned, the particle filtering capabilities of a native plant xylem were found to be approximately 20 nm,18 while the average size of synthetic Mt and Fh particles have been found to be 11 nm (ref. 58) and 3 nm,40 respectively. The smaller Fh particle size may account for higher mobility within the cell ultrastructure and limit interparticle aggregation, while the small size of the sub-micropores may prevent Mt particles from forming inside the cell wall. Our data suggests that the confinement of poorly crystalline Fh within the wood cell wall matrix stabilizes the metastable iron oxide phase and preludes particle–particle aggregation to a large extent, as ferrihydrite usually transforms to more stable phases over a couple of days.31
Arsenate adsorption
In the present paper, we quantified the degree to which iron-mineralized biocomposites are capable of remediating As from drinking water. To accomplish this, we conducted batch and gravity-driven flow-through adsorption testing using As solutions of known concentration. The arsenic levels were varied within the range of those found in contaminated environments that exceeded the EPA limit for drinking waters (10–300 ppb or 10–300 μg L−1) and would therefore be relevant for our decontamination technology. Testing consisted of soaking the composites in 1 mg L−1 or 100 μg L−1 As solutions for varying amounts of time or subjecting cylindrical filters to a continuous gravity-driven flow. The As concentration in the eluent solutions was determined using microwave plasma atomic emission spectroscopy (MP-AES) and inductively coupled plasma mass spectrometry (ICP-MS) after the composites were introduced.The two As adsorption methods simulate standing and running waters and give evidence that the lignocellulose/Fe(OH)3 composite materials have high capacities for As adsorption under any scenario, with Fh composites outperforming Mt composites. In batch incubation adsorption tests with a starting concentration of 1 mg L−1 (=1 ppm), Fh and Mt had absorbed ∼56% and 48% of the initial As concentration after 24 h exposure, respectively (Fig. 5A). The extended incubation time ensured complete water saturation of the modified wood cell walls and diffusion of As into the cell wall nanopores, the mass of filter used and amount of adsorbed AsO4 recorded in these results was subsequently used for the calculation of the adsorption capacity. The adsorption capacity for our Fh mineralized composites was found to be 46.52 μg g−1, which is similar to other reported iron oxide-based materials.5 Previous research reported on iron oxide/oxyhydroxides absorbent materials with significantly higher binding capacities,19 however, the less straightforward separation of these materials after use and the higher cost of activated carbon may offset the benefit of increased As binding. While these results demonstrate the composites' As binding capacity, this method still requires a substantial amount of time for complete As removal.
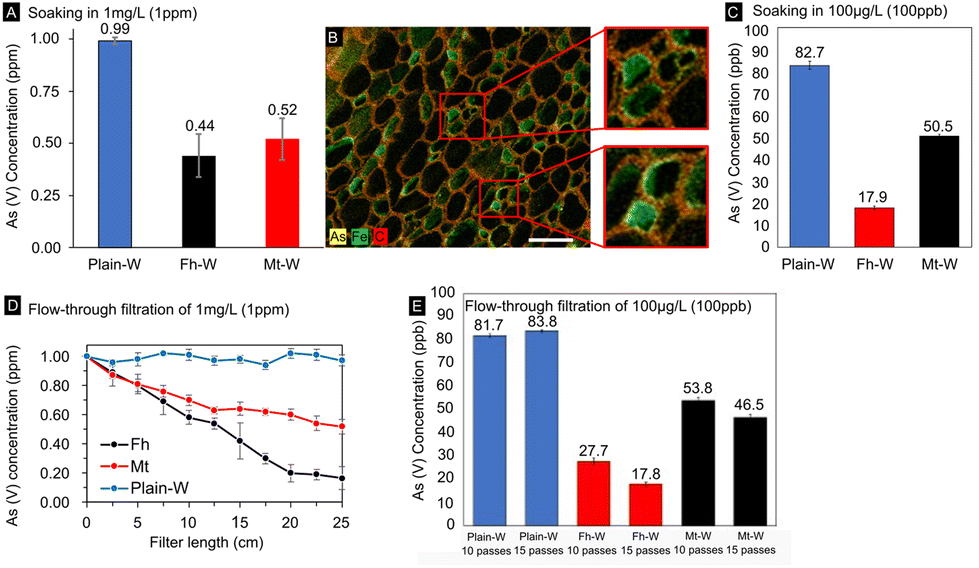 | ||
| Fig. 5 Adsorptive capacity of iron oxide-wood filters tested in (A–C) batch incubation tests and (D and E) gravity-driven flow-through tests. (A) MP-AES data were obtained from batch incubation tests in 1 mg L−1 (1 ppm) As(V) solutions for 24 hours. (B) SEM-EDS mapping of Fh-modified wood material after exposure to 1 mg L−1 (1 ppm) As(V) in batch incubation test. (C) ICP-MS data quantified As(V) concentrations after immersion of filters in 100 μg L−1 (100 ppb) As(V) solutions for 24 hours. (D) As shown by MP-AES, As(V) concentration decreased as a function of filter length as 1 mg L−1 (1 ppm) As(V) solutions of were passed through the samples. (E) ICP-MS data showed a substantial decrease in As(V) levels after gravity-driven flow-through test with 100 μg L−1 (100 ppb) As(V) solutions. | ||
To confirm the immobilization of As within the wood filters, we mapped the elemental distribution after exposure to 1 mg L−1 As(V) in the batch incubation experiments. Our SEM/EDS results clearly demonstrate a colocalization of As and Fe (Fig. 5B), which is consistent with the adsorption of As on poorly crystalline ferrihydrite as well as the surface precipitation of ferric arsenate mineral, FeAsO4·2.4H2O.59 Using EDS microanalysis, we detected up to 2 wt% As immobilized in the scaffolds, with the majority deposited along the middle lamella of the wood fibers and with minor amounts precipitated within the cell wall. The colocalization of As–Fe within the cell wall further indicates that the higher adsorption partially stems from the increased surface area of Fh available for binding interactions.
The second approach was based on cylindrical filters subjected to a gravity-driven flow (Fig. 1D). With a ∼82% and ∼50% reduction of the initial As(V) concentration using Fh- and Mt-modified filters (Fig. 5D), respectively, the flow-through approach was more effective in reducing the As concentration than the batch incubation approach. As a result, the decontamination procedure was completed in a considerably smaller time frame (Fig. 5D and E). Remarkably, Fh performs substantially better than Mt under flow-through conditions, whereby the high degree of As adsorption supports the viability of the material as a rapid low-cost filtration system. The differences in adsorption capacity between the two mineral phases are likely due to a larger exposed surface area and reactivity of Fh, facilitating binding interactions with dissolved arsenate. In addition, Fh was found to be more widely distributed within cell walls and radial parenchyma cells, which are also involved in liquid transport through the hierarchical wood structure. The differences between the two filtration methods highlight the significance of the mineral distribution within the three-dimensional scaffold. We hypothesize that the additional Fh surface area within the sub-microporous cell wall increased binding interactions and therefore As uptake. Another benefit of the flow-through approach is that the entire water sample gets exposed to the iron oxide modified wood filter. The removal rate can likely be further improved by utilizing a vacuum pump to increase the flow rate through the filters in comparison to the tested gravity-driven approach. However, the relationship between increased flow rate and removal efficiency would likely need to be further optimized to compensate for the decreased contact time between As solute and mineral phases. The effects of other cations detected by SEM-EDS microanalysis on the adsorption of As were not investigated. Relatively high levels of K were measured within the scaffold due to the use of KOH in synthesis. Similarly, adsorption tests were only performed at near-neutral pH and may require further efficacy testing across wider pH ranges.
After confirming As removal in soaking and flow-through tests, both types of experiments were repeated with a more environmentally relevant starting concentration for arsenic contamination of 100 μg L−1 (100 ppb), a tenth of the previous concentration. At these levels, As is well below the threshold for detection with our MP-AES instrument without further sample preparation or introduction modules for the instrument. Therefore, low-concentration As samples were analyzed by ICP-MS. Even at lower As starting concentration, significant As removal was observed. After 24 hours of exposure for the batch incubation trials, Fh- and Mt-modified wood filters had removed 82.1% and 49.5% of the As in solution (Fig. 5A). Similar results were seen in the flow-through trials, at 25 cm filter length 72.3% of the As had been removed by the Fh composites and 43.6% by the Mt composites. At 37.5 cm filter length, 82.2% and 53.5% removal was achieved for Fh and Mt respectively (Fig. 5D). Both flow-through tests with Fh-based composites achieved a reduction to <20 ppb, which is slightly above the WHO limit for drinking water, while Mt based composites only achieved a reduction to <50 ppb. ICP-MS measurements were also used to assess Fe levels to rule out leaching of the Fe-containing mineral phases during filtration (Fig. S6†). In the flow-through trials, we detected higher Fe levels with Fh-containing filters in comparison to the Mt-functionalized filters, while Mt leaches more Fe in soaking experiments. In all analyzed samples, Fe concentrations never exceeded the EPA limit of 0.3 mg L−1.
Taken together, the As removal efficiency of the hybrid iron oxide/wood filters resembles other technologies based on more costly or less abundant materials while providing significant confinement of As within the bulk structure.24 The proposed gravity-driven flow-through technology is easy to use in resource-limited settings without vacuum pumps. An added benefit is that pristine xylem filters can remove bacteria and other micro-sized particles from drinking water.18 One practical advantage consists in the ease of separation once the filter material is saturated with contaminants. We did not attempt recycling or regenerating the As-loaded materials after multiple filtration cycles. With regards to hazardous waste disposal, the U. S. Environmental Protection Agency recommends discarding As-contaminated solid materials and residuals in landfills or settling ponds.60 Our present research as well as other studies have shown that iron oxide particles immobilized inside the wood cell wall structure do not release harmful levels of Fe ions into the aqueous environment.20 This means that the re-release of arsenic from the composite material would be prolonged compared to iron-containing nanomaterials and would instead be tied to the rate of wood degradation. Under normal operating conditions, the wood cell wall is resilient to breakdown beyond the lifetime of the filter material, but the rate of wood decay strongly depends on environmental factors like tree species, temperature and precipitation.61,62 With 46.52 μg g−1, the As(V) adsorption capacity of our xylem-based filter materials is comparable or higher than many literature studies,5,63 but lower than more sophisticated scaffold-based technologies requiring a larger number of chemicals or processing steps.19,24 To combine the high adsorption capacity of iron oxide modified activated carbon19 with the cellular hierarchy of biological materials, one could consider preparing functionalized filter materials from carbonized wood. Freely floating nanoparticles, such as zero-valent iron (ZVI),5 ferrihydrite,63 or Fe3O45 nanoparticles, present an alternative technology for absorbing contaminants from drinking water. Although these nanomaterials have substantially higher As adsorption capacities, namely 12 mg g−1,5 285 μg g−1,63 and 46.06 mg g−1,5 respectively, they entail several practical disadvantages for local drinking water purification compared to our scaffold-based delivery mechanism. Apart from the need to remove As-loaded nanoparticles from potable water by ultracentrifugation, membrane filtration or the application of magnetic fields, ultrafine particles may agglomerate in solution,5 exert harmful effects on the biota64,65 or allow absorbed arsenic to leach into groundwater.66 Generally speaking, iron oxides are some of the most common naturally occurring minerals in soils, marine67 and freshwater sediments68 and thus do not pose any harm to the aquatic ecosystem – in contrast, elevated iron oxide concentrations can provide ecosystem benefits by mitigating toxic sulfide and limiting eutrophication of water bodies.69 However, the presence of other ions, such as phosphates, in the complex matrix of ground water could impact the removal efficiency of the filter towards As through competitive surface binding, as demonstrated for other iron-based filter materials.70 As the removal of excess phosphorous can also be desirable, the number of filtration cycles or length of filter could likely be raised to result in the removal of both compounds in waters with high P levels.
Conclusions
The contamination of drinking water with hazardous arsenic is a common problem in both developing and industrialized countries. In this study, we developed a straightforward and cost-effective decontamination technology to address dangerously high levels of arsenic that can be used as batch incubation filter or gravity-driven flow-through filter. To this end, a lightweight and widely available lignocellulosic material (balsa wood) was chemically modified by precipitating nanocrystalline iron oxides inside the cell walls or at the cell wall interfaces. By varying the reaction chemistry, we achieved a cell-wall specific deposition and stabilization of poorly crystalline ferrihydrite, whereas magnetite primarily formed at the lumen/cell wall interface. X-ray microCT, SEM-EDS mappings and confocal Raman mappings provided the distribution of the mineral phases from the macro- to the sub-micrometer level. Remarkably, the mineral distribution within the cell wall architecture, as well as the innate surface reactivity of the mineral phase had an impact on the As adsorption capacity, with Fh clearly outperforming Mt. Based on our results, these low-cost hybrid materials bear significant potential as easy-to-use filtering material for water-borne pollutants in resource-limited settings. Apart from arsenate(V), our technology may address a variety of current and emerging environmental contaminants that are known to adsorb on iron oxide mineral phases, including phosphate,39,71 polycyclic aromatic hydrocarbons (PAH),72 perfluoroalkyl and polyfluoroalkyl substances (PFAS),73 or heavy metals, such as lead and cadmium.41Author contributions
SS, SF and BD: data curation, investigation, formal analysis, validation. SS and VM: writing – original draft. VM: conceptualization, project administration, supervision. VM, SF: funding acquisition. SS, SF, VM: writing – review & editing. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was primarily funded by institutional start-up funds to VM. In addition, VM thanks the National Science Foundation (NSF2137663, Division of Materials Research) for partial financial support. The acquisition of a Transmission Electron Microscope was supported by a Department of Defense instrumentation/equipment grant (W911NF2110147) to VM. SF is indebted to FAU's Office of Undergraduate Research and Inquiry (OURI) for a Summer Undergraduate Research Fellowship and an Undergraduate Research Grant. We thank the Owls Imaging Lab at FAU High School (FAUHS) for providing access to research equipment. Research technician Jamie Knaub is acknowledged for technical support during X-ray microtomography scans. We acknowledge Dr Alexander Franco Hernandez, Research Assistant Professor at the Advanced Materials Engineering Research Institute (AMERI) of Florida International University for collecting X-ray powder diffraction data and for providing access to FE-SEM. ICP-MS measurements were performed in the OHSU Elemental Analysis Core at Oregon Health and Science University with partial support from NIH (S10OD02849). We acknowledge Dr Martina Ralle and Sophia Miller for ICP-MS data collection.References
- J. D. Ayotte, L. Medalie, S. L. Qi, L. C. Backer and B. T. Nolan, Environ. Sci. Technol., 2017, 51, 12443–12454 CrossRef CAS PubMed.
- R. R. Reddy, G. D. Rodriguez, T. M. Webster, M. J. Abedin, M. R. Karim, L. Raskin and K. F. Hayes, Water Res., 2020, 170, 115325 CrossRef CAS PubMed.
- A. H. Smith, E. O. Lingas and M. Rahman, Bull. W. H. O., 2000, 78, 1093–1103 CAS.
- Q. Wei, B. Leblon and A. La Rocque, Can. J. For. Res., 2011, 41, 2120–2140 CrossRef.
- L. Hao, M. Liu, N. Wang and G. Li, RSC Adv., 2018, 8, 39545–39560 RSC.
- M. F. Hughes, Toxicol. Lett., 2002, 133, 1–16 CrossRef CAS PubMed.
- R. Y. Ning, Desalination, 2002, 143, 237–241 CrossRef CAS.
- S.-A. Schmidt, E. Gukelberger, M. Hermann, F. Fiedler, B. Großmann, J. Hoinkis, A. Ghosh, D. Chatterjee and J. Bundschuh, J. Hazard. Mater., 2016, 318, 671–678 CrossRef CAS PubMed.
- A. Abejón, A. Garea and A. Irabien, Sep. Purif. Technol., 2015, 144, 46–53 CrossRef.
- T. Gurunathan, S. Mohanty and S. K. Nayak, Composites, Part A, 2015, 77, 1–25 CrossRef CAS.
- M. Borrega, P. Ahvenainen, R. Serimaa and L. Gibson, Wood Sci. Technol., 2015, 49, 403–420 CrossRef CAS.
- P. Fratzl and R. Weinkamer, Prog. Mater. Sci., 2007, 52, 1263–1334 CrossRef CAS.
- M. Schubert, G. Panzarasa and I. Burgert, Chem. Rev., 2023, 123, 1889–1924 CrossRef CAS PubMed.
- Y. Ding, Z. Pang, K. Lan, Y. Yao, G. Panzarasa, L. Xu, M. Lo Ricco, D. R. Rammer, J. Y. Zhu, M. Hu, X. Pan, T. Li, I. Burgert and L. Hu, Chem. Rev., 2023, 123, 1843–1888 CrossRef CAS PubMed.
- S. Vitas, T. Keplinger, N. Reichholf, R. Figi and E. Cabane, J. Hazard. Mater., 2018, 355, 119–127 CrossRef CAS PubMed.
- Z. Cheng, H. Guan, J. Meng and X. Wang, ACS Omega, 2020, 5, 14096–14103 CrossRef CAS PubMed.
- S. He, C. Chen, G. Chen, F. Chen, J. Dai, J. Song, F. Jiang, C. Jia, H. Xie, Y. Yao, E. Hitz, G. Chen, R. Mi, M. Jiao, S. Das and L. Hu, Chem. Mater., 2020, 32, 1887–1895 CrossRef CAS.
- M. S. H. Boutilier, J. Lee, V. Chambers, V. Venkatesh and R. Karnik, PLoS One, 2014, 9, e89934 CrossRef PubMed.
- Z. Liu, F.-S. Zhang and R. Sasai, Chem. Eng. J., 2010, 160, 57–62 CrossRef CAS.
- V. Merk, M. Chanana, N. Gierlinger, A. M. Hirt and I. Burgert, ACS Appl. Mater. Interfaces, 2014, 6, 9760–9767 CrossRef CAS PubMed.
- R. M. Cornell and U. Schwertmann, The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses, John Wiley & Sons, 2003 Search PubMed.
- J.-C. Lee, E. J. Kim, H.-W. Kim and K. Baek, Geoderma, 2016, 270, 76–82 CrossRef CAS.
- H. J. Shipley, S. Yean, A. T. Kan and M. B. Tomson, Environ. Toxicol. Chem., 2009, 28, 509–515 CrossRef CAS PubMed.
- A. A. Kumar, A. Som, P. Longo, C. Sudhakar, R. G. Bhuin, S. Sen Gupta, Anshup, M. U. Sankar, A. Chaudhary, R. Kumar and T. Pradeep, Adv. Mater., 2017, 29(7), 1604260 CrossRef PubMed.
- V. Merk, M. Chanana, T. Keplinger, S. Gaan and I. Burgert, Green Chem., 2015, 17, 1423–1428 RSC.
- T. Keplinger, E. Cabane, M. Chanana, P. Hass, V. Merk, N. Gierlinger and I. Burgert, Acta Biomater., 2015, 11, 256–263 CrossRef CAS PubMed.
- M. Frey, D. Widner, J. S. Segmehl, K. Casdorff, T. Keplinger and I. Burgert, ACS Appl. Mater. Interfaces, 2018, 10, 5030–5037 CrossRef CAS PubMed.
- O. N. Shebanova and P. Lazor, J. Raman Spectrosc., 2003, 34, 845–852 CrossRef CAS.
- M. Hanesch, Geophys. J. Int., 2009, 177, 941–948 CrossRef CAS.
- U. Schwertmann and R. M. Cornell, Iron Oxides in the Laboratory: Preparation and Characterization, Wiley, Weinheim, Chichester, 2nd edn, 2000 Search PubMed.
- U. Schwertmann, J. Friedl and H. Stanjek, J. Colloid Interface Sci., 1999, 209, 215–223 CrossRef CAS PubMed.
- U. C. Schwertmann, Iron Oxides, 2003, pp. 95–110 Search PubMed.
- L. Dyer, P. D. Fawell, O. M. G. Newman and W. R. Richmond, J. Colloid Interface Sci., 2010, 348, 65–70 CrossRef CAS PubMed.
- A. Manceau and W. P. Gates, Clays Clay Miner., 1997, 45, 448–460 CrossRef CAS.
- A. Manceau, Clay Miner., 2010, 45, 225–228 CrossRef CAS.
- A. Manceau and V. A. Drits, Clay Miner., 1993, 28, 165–184 CrossRef CAS.
- M. Villacís-García, M. Ugalde-Arzate, K. Vaca-Escobar, M. Villalobos, R. Zanella and N. Martínez-Villegas, Bol. Soc. Geol. Mex., 2015, 67, 433–446 CrossRef.
- N. Y. Acelas, C. Hadad, A. Restrepo, C. Ibarguen and E. Flórez, Inorg. Chem., 2017, 56, 5455–5464 CrossRef CAS PubMed.
- H. Wang, J. Zhu, Q.-L. Fu, J.-W. Xiong, C. Hong, H.-Q. Hu and A. Violante, Pedosphere, 2015, 25, 405–414 CrossRef CAS.
- M. Villalobos and J. Antelo, Rev. Int. Contam. Ambiental, 2011, 27, 139–151 CAS.
- K. Rout, M. Mohapatra and S. Anand, Dalton Trans., 2012, 41, 3302–3312 RSC.
- M. T. Tyree, Nature, 2003, 423, 923 CrossRef CAS PubMed.
- J. S. Sperry, Int. J. Plant Sci., 2003, 164, S115–S127 CrossRef.
- A. Scholz, M. Klepsch, Z. Karimi and S. Jansen, Front. Plant Sci., 2013, 4(56), 1–11 Search PubMed.
- P. Hass, F. K. Wittel, S. A. McDonald, F. Marone, M. Stampanoni, H. J. Herrmann and P. Niemz, Holzforschung, 2010, 64, 639–644 CAS.
- E.-S. Jang and C.-W. Kang, J. Wood Sci., 2022, 68, 31 CrossRef CAS.
- M. T. Tyree and M. H. Zimmermann, Xylem Structure and the Ascent of Sap, Springer, Berlin, Heidelberg, 2002, pp. 27–48 Search PubMed.
- S. Tanaka, M. Seki, T. Miki, I. Shigematsu and K. Kanayama, J. Wood Sci., 2015, 61, 543–551 CrossRef CAS.
- H. K. Burr and A. J. Stamm, J. Phys. Colloid Chem., 1947, 51, 240–261 CrossRef CAS PubMed.
- G. N. Christensen, Aust. J. Appl. Sci., 1951, 2, 440–453 CAS.
- J. Ma, Z. Ji, X. Zhou, Z. Zhang and F. Xu, Microsc. Microanal., 2013, 19, 243–253 CrossRef CAS PubMed.
- B. A. Legg, M. Zhu, L. R. Comolli, B. Gilbert and J. F. Banfield, Environ. Sci. Technol., 2014, 48, 13703–13710 CrossRef CAS PubMed.
- J. Liu, S. M. Louie, C. Pham, C. Dai, D. Liang and Y. Hu, Environ. Res., 2019, 172, 552–560 CrossRef CAS PubMed.
- J. Fahlén and L. Salmén, Biomacromolecules, 2005, 6, 433–438 CrossRef PubMed.
- L. A. Donaldson, H. W. Kroese, S. J. Hill and R. A. Franich, J. Microsc., 2015, 259, 228–236 CrossRef CAS PubMed.
- M. Nopens, U. Sazama, S. König, S. Kaschuro, A. Krause and M. Fröba, Sci. Rep., 2020, 10, 9543 CrossRef CAS PubMed.
- X. Li and Z. Zhao, Wood Sci. Technol., 2020, 54, 1241–1251 CrossRef CAS.
- M. C. Mascolo, Y. Pei and T. A. Ring, Materials, 2013, 6, 5549–5567 CrossRef CAS PubMed.
- Y. Jia, L. Xu, Z. Fang and G. P. Demopoulos, Environ. Sci. Technol., 2006, 40, 3248–3253 CrossRef CAS PubMed.
- U. S. E. P. Agency, The Arsenic Rule: Water Treatment Plant Residuals, https://www.epa.gov/sites/default/files/2015-09/documents/train6-residuals.pdf Search PubMed.
- O. Hararuk, W. A. Kurz and M. Didion, For. Ecosyst., 2020, 7, 36 CrossRef.
- K. A. Pietsch, D. Eichenberg, K. Nadrowski, J. Bauhus, F. Buscot, W. Purahong, B. Wipfler, T. Wubet, M. Yu and C. Wirth, Oikos, 2019, 128, 701–715 CrossRef.
- O. S. Thirunavukkarasu, T. Viraraghavan and K. S. Subramanian, Water Qual. Res. J., 2001, 36, 55–70 CrossRef CAS.
- R. Mukherjee, R. Kumar, A. Sinha, Y. Lama and A. K. Saha, Crit. Rev. Environ. Sci. Technol., 2016, 46, 443–466 CrossRef CAS.
- P. N. Owens, R. J. Batalla, A. J. Collins, B. Gomez, D. M. Hicks, A. J. Horowitz, G. M. Kondolf, M. Marden, M. J. Page and D. H. Peacock, River Res. Appl., 2005, 21, 693–717 CrossRef.
- S. Shankar, U. Shanker and Shikha, Sci. World J., 2014, 2014, 304524 Search PubMed.
- R. M. Chambers, J. W. Fourqurean, S. A. Macko and R. Hoppenot, Limnol. Oceanogr., 2001, 46, 1278–1286 CrossRef CAS.
- D. C. Cooper, A. L. Neal, R. K. Kukkadapu, D. Brewe, A. Coby and F. W. Picardal, Geochim. Cosmochim. Acta, 2005, 69, 1739–1754 CrossRef CAS.
- C. P. Slomp, S. J. Van der Gaast and W. Van Raaphorst, Mar. Chem., 1996, 52, 55–73 CrossRef CAS.
- J. Lalley, C. Han, X. Li, D. D. Dionysiou and M. N. Nadagouda, Chem. Eng. J., 2016, 284, 1386–1396 CrossRef CAS.
- M. Mallet, K. Barthélémy, C. Ruby, A. Renard and S. Naille, J. Colloid Interface Sci., 2013, 407, 95–101 CrossRef CAS PubMed.
- T. B. Boving and K. Neary, J. Contam. Hydrol., 2007, 91, 43–57 CrossRef CAS PubMed.
- H. Campos-Pereira, D. B. Kleja, C. Sjöstedt, L. Ahrens, W. Klysubun and J. P. Gustafsson, Environ. Sci. Technol., 2020, 54, 15722–15730 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00326d |
| This journal is © The Royal Society of Chemistry 2024 |