
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFlow-driven enhancement of neodymium and dysprosium separation from aqueous solutions†
Qingpu
Wang
 a and
Chinmayee V.
Subban
*ab
a and
Chinmayee V.
Subban
*ab
aEnergy and Environment Directorate, Pacific Northwest National Laboratory, Seattle, Washington 98109, USA
bDepartment of Materials Science and Engineering, University of Washington, Seattle, Washington 98195, USA. E-mail: chinmayee.subban@pnnl.gov
First published on 12th February 2024
Abstract
Selective extraction of rare earth elements (REEs) from waste NdFeB magnets and natural mineral sources has been challenging due to the similar properties of neodymium (Nd) and dysprosium (Dy). Current separation methods mainly include solvent extraction and organic ligand-based selective precipitation, which are chemical- and energy-intensive in addition to the long separation times required to reach thermodynamic equilibrium. Here, we demonstrate a laminar co-flow method that relies on flow-induced non-equilibrium conditions to selectively precipitate Dy3+ from aqueous solutions containing mixed Nd3+ and Dy3+ at various ratios. The concentration of reactant sodium dibutyl phosphate for selective precipitation was identified based on the differences in the spatiotemporal dynamics of the Nd3+ and Dy3+ precipitates. Under optimized conditions, our method showed increased Dy purity in the precipitate product at significantly shorter reaction times, compared to commonly used convective bulk mixing. We found a nearly Dy-pure (99.9%) precipitate from starting mixtures of Nd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Dy in 50:50 and 30:70 ratios. Our single-step method is efficient and environmentally friendly and does not require harmful organic solvents or difficult to synthesize complex ligands.
:Dy in 50:50 and 30:70 ratios. Our single-step method is efficient and environmentally friendly and does not require harmful organic solvents or difficult to synthesize complex ligands.
Sustainability spotlightThe widespread deployment of renewable energy technologies depends on reliable and sustainable supply of critical materials. For example, REE-containing high-performance permanent magnets are essential components of electric vehicles and wind turbines. Beyond primary ores, recycling waste magnets to selectively recover individual REEs could offer a secondary source that is reliable and can reduce the supply chain risks. However, established REE separation methods rely heavily on novelty organic ligands and solvents that increase process costs and introduce waste disposal concerns. An all-aqueous process that uses commodity chemicals to selectively precipitate the REE would eliminate these issues. In this work, we report such a method, where just by changing the flow and mixing conditions of an aqueous Dy and Nd salt solution, we achieve higher rates of selective Dy-precipitation and higher product purities. Our results highlight the benefits of considering beyond conventional stirred mixing strategies for advancing REE separation and recycling. |
1. Introduction
A growing coalition of countries and corporations are pledging to achieve net-zero greenhouse gas emissions to address the global climate crisis.1 As a result, there is an exponentially increasing demand for clean energy technologies and the materials that they rely on. Rare earth elements (REEs), especially Nd and Dy, are deemed as highly critical to clean energy by the U.S. Department of Energy.2 For example, neodymium-iron-boron (NdFeB) permanent magnets are key components in generators of wind turbines and in motors of electric vehicles. However, the availability of these materials is effectively restricted to few mine sites in China that supply a majority of the global REE market.3,4 To mitigate such supply risks, ideally developing domestic mine sites with sustainable extraction processes is necessary, but that would cost a significant amount of time given the permitting and regulatory approvals needed. A more promising near-term avenue is exploring secondary REE sources through recycling end-of-life REE-containing magnets and components.The recovery of REEs from waste NdFeB magnets is mainly through mature hydrometallurgical procedures.5 Typically, the powdered magnet scrap is digested in an acidic solution and treated with precipitating reactants, yielding an iron (hydr)oxide solid and a mixed REE-rich leachate after filtration.6 However, the subsequent separation of individual REEs has been especially difficult due to their highly similar physical and chemical properties. Conventional REE separation methods have been mainly focused on solvent extraction that relies on ion exchange, ion complexation, or ion solvation.7–11 Generally, these processes are organic solvent- and energy-intensive as well as time-consuming. Bogart et al.12,13 reported a one-step solubility-based REE separation method using size-sensitive apertures formed by metal–ligand complexes and showed higher separation factors in the presence of external magnetic fields.14 Conceptually similar work has been reported using various organic ligands.15–19 Most of the organic-ligand-based methods are able to separate multiple light and heavy REE pairs, but often involve difficult to scale-up ligand synthesis or require a strictly water-free environment, posing engineering challenges in industrial and practical settings. Besides purely organic solvent systems, REE separations have been performed at the interface of aqueous-organic phases.20,21 There have also been a limited number of studies in all-aqueous environments including a protein-based process,22 a selective oxalate solubilization method,23 and selective precipitation or crystallization with dibutyl phosphate,24 borate,25 and tetrazolate.26
All the above-mentioned REE separation methods rely on the differences in equilibrium constants. Reaching the thermodynamic equilibrium is often time-consuming for selective precipitation even when accelerated by elevating the reaction temperature. Studies have demonstrated higher REE separation factors when far-from-equilibrium or non-equilibrium conditions are used in organic solvents.27,28 To the best of our knowledge, there have been no studies on REE separation in all-aqueous systems under non-equilibrium conditions. Non-equilibrium reaction conditions can be induced with the use of stimuli (e.g., temperature, pressure) or fields (e.g., flow, electric). A simple strategy for non-equilibrium selective crystallization is altering flow conditions and using laminar co-flow of reactants to create significantly different ion concentration profiles at the liquid/liquid interface compared with bulk stirred mixing (i.e., equilibrium conditions). This flow-induced non-equilibrium method has been previously used to investigate materials synthesis,29–32 crystal growth,33–35 and origins of life at submarine hydrothermal vents.36–38
In our recent work, we reported the use of a laminar co-flow method (LCM) for selective precipitation of Mg2+ from natural seawater.39 Such selectivity achieved through flow-induced non-equilibrium does not need chemical- or energy-intensive membranes, solvents, or adsorbents, making it easy to scale-up and implement. Here, we apply LCM for REE separation in aqueous media at room temperature. Our goal is to develop an environmentally friendly approach that can enable efficient recovery of REEs from high-concentration secondary sources such as spent NdFeB permanent magnets (20–35 wt% REEs). Although the majority of them is Nd, the magnets generally contain smaller amounts of other REEs (praseodymium Pr, terbium Tb, and gadolinium Gd, and Dy) to either increase the intrinsic room-temperature magnetic coercivity or improve the high-temperature performance.40,41 Given that most NdFeB magnets commonly use Dy to prevent demagnetization, here we focus our investigation on LCM separation of Nd and Dy. To achieve selective precipitation. We chose dibutyl phosphate (DBP) among the other phosphorus ligands (e.g., tributyl phosphate,27 di-(2-ethylhexyl)phosphoric acid42) because of its high water-solubility.24 By directly co-flowing dilute solutions of DBP and NdCl3/DyCl3 using LCM, we observed precipitate formation at the interface of the reactant solutions. However, the precipitates of Nd(DBP)3 and Dy(DBP)3 were nearly indistinguishable from each other and from samples obtained using a conventional, stirred bulk mixing method (BMM) by powder X-ray diffraction (Fig. 1a, b and S1†). As determined by the Scherrer equation, the crystallite domain size of Nd(DBP)3 in LCM was found to be three times larger than that in BMM, whereas the sizes of Dy(DBP)3 in LCM and BMM were similar (Table S1†). Furthermore, their scanning electron micrographs showed hollow tubular structures with different average tube sizes with Nd(DBP)3 forming larger tubes than Dy(DBP)3 (Fig. 1c and d). This subtle morphological variance suggests the difference in Nd3+ and Dy3+ precipitation rates, which can be leveraged for separation.
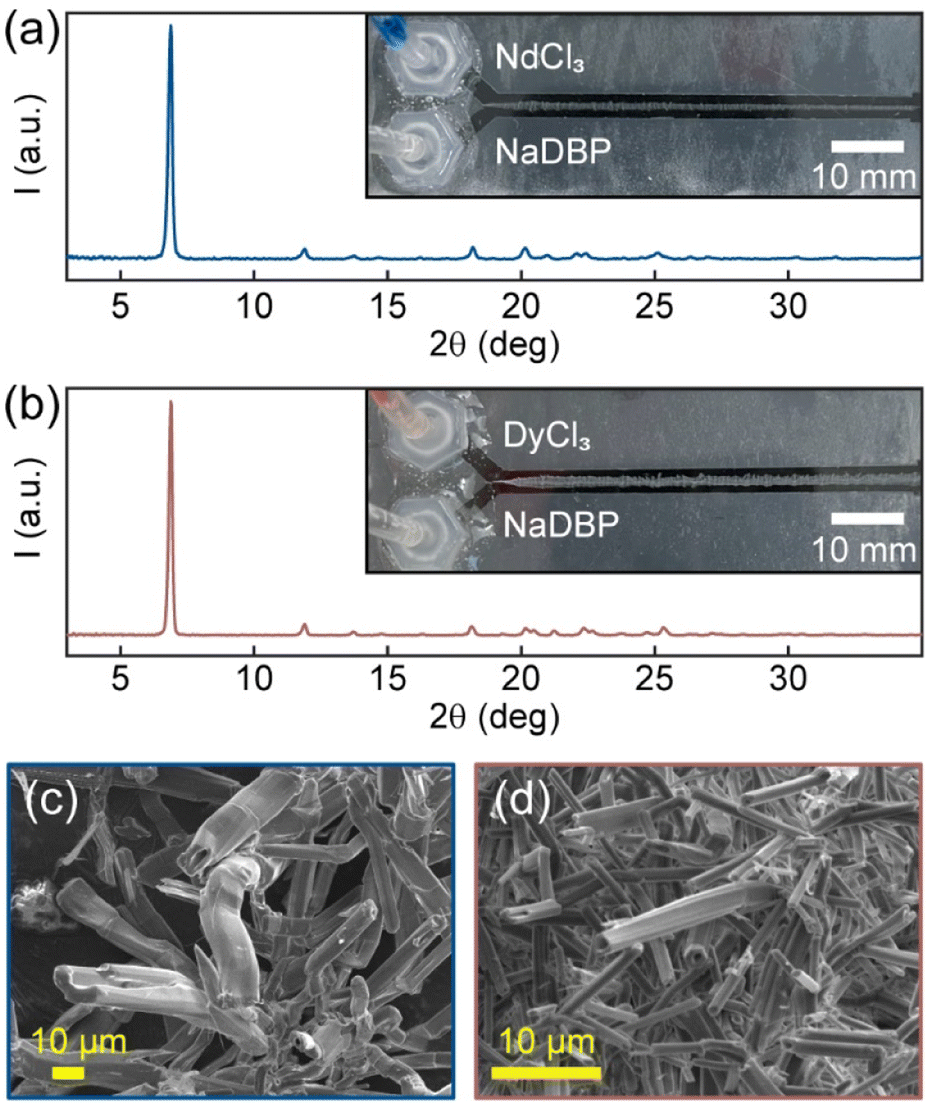 | ||
| Fig. 1 Simple precipitation experiments in two-inlet microfluidic devices. Powder XRD patterns (a and b) and SEM images (c and d) of the pure Nd(DBP)3 (a and c) and Dy(DBP)3 (b and d) precipitates obtained using LCM. Insets are photographs of the resulting precipitates in microfluidic channels after 45 min of co-flow. | ||
In this paper, we designed a three-inlet microfluidic channel to first visualize the two precipitation processes simultaneously, aiming to understand the differences in their spatiotemporal dynamics with different NaDBP concentrations. Then, we used the information to identify concentration conditions for selective precipitation of Dy(DBP)3 from a mixed solution of NdCl3 and DyCl3 with various Nd:Dy ratios. To identify how non-equilibrium conditions induced by LCM result in different products vs. equilibrium stirred mixing, we conducted parallel bulk mixing experiments. Our results highlight the benefits of LCM both for reaction rates and purity of the resulting precipitate.
2. Experimental
2.1. Chemicals
Dibutyl phosphate (HDBP, (CH3(CH2)3)2HPO4, 97%, Thermo Scientific Chemicals), sodium hydroxide (NaOH, Fisher Scientific), neodymium chloride hydrate (NdCl3·H2O, 99.9% REO, Thermo Scientific Chemicals), dysprosium chloride(DyCl3, anhydrous, 99.9% Dy, Fisher Chemical), and nitric acid (HNO3, Trace Metal grade, Fisher Chemical) were used as received. All solutions were prepared with deionized water (resistivity, 18.2 MΩ cm) filtered by a water purification system (Milli-Q IQ 7000).In a 100 mL glass bottle (PYREX), 25 mL of 4.0 M NaOH solution was added to 20.5 mL of HDBP and stirred for 1 h. The equimolar mixing of the two reactants yielded NaDBP (about 1 M) which is more soluble than HDBP.24
2.2. Non-equilibrium experiments (LCM in microfluidic devices)
To investigate flow-induced non-equilibrium Nd/Dy separation, we designed and constructed microfluidic devices based on previously reported methods.32,39 The device (Fig. 2a) consisted of a cut parafilm membrane sandwiched between two square polycarbonate plates measuring 75 × 75 mm2 (thickness, 1.6 mm). A three-pronged Y-shaped pattern was designed to align with the three inlets and one outlet on the polycarbonate plates. This customized pattern was cut on the parafilm using a computer-controlled cutting machine (Silhouette Portrait 3). The assembled device was heated on a hot plate to slightly melt the parafilm that, after cooling down, glued the plates together without requiring external fastening during use. The cut area on the parafilm membrane created a thin channel with a width of 5 mm and a length of 188 mm. The resulting microfluidic device with one layer of parafilm (thickness: 0.13 mm) held a volume about 0.12 mL (Fig. 2b). With four layers of parafilm between the plates, the volume of the microfluidic channel was increased to 0.49 mL. Note that the simple two-inlet microfluidic devices43 in Fig. 1a and b were prepared using microscope glass slides (25 × 75 mm2) as the bottom plate with a channel volume of 0.02 mL.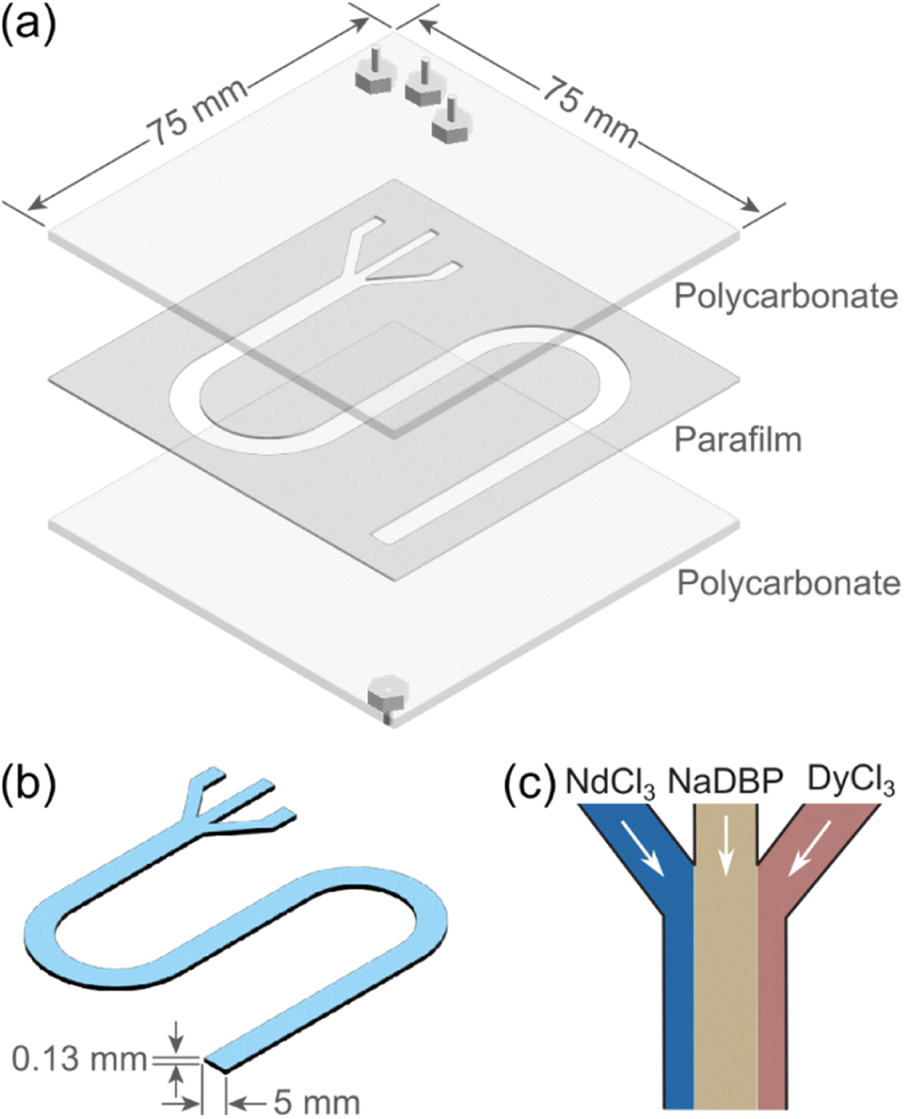 | ||
| Fig. 2 Three-inlet microfluidic device for separation. (a) Deconstructed view. (b) Reaction chamber with a single layer of parafilm. (c) Solution configuration used to investigate precipitation dynamics at the two different reactive interfaces. | ||
Three syringes containing reactant solutions were connected to the inlets of the microfluidic device through plastic tubing (Tygon, S3 B-44-4X, inner diameter, 1.6 mm). The solutions were simultaneously injected using programmable syringe pumps (New Era, NE-300) into the channel that was prefilled with DI water to avoid potential, undesired effects at air/liquid interfaces. A constant flow rate of 1 mL h−1 per syringe was used for all experiments. The injection time varied from 45 min to 30 h.
The configuration of the reactant solution delivery depended on the purpose of the experiments. For recording the precipitation dynamics, the middle inlet was connected to a NaDBP solution of various concentrations (1.2 to 120 mM), while the side inlets were linked to 4 mM NdCl3 and 4 mM DyCl3, respectively (Fig. 2c). For the selective precipitation experiments, the middle inlet was connected to a mixed solution of 4 mM NdCl3 and various concentrations of DyCl3, while each of the side inlet was connected to a 1.2 mM NaDBP solution.
2.3. Equilibrium experiments (bulk mixing in vials)
To simulate conventional equilibrium stirred mixing, referred to henceforth as the bulk mixing method (BMM), 5 mL of NaDBP solutions were added to 5 mL NdCl3 and/or DyCl3 solutions in a 20 mL glass vial. The concentration conditions and the reaction time were identical to those of their LCM counterpart samples. After reaction, the mixture was centrifuged at 10000 rpm for 3 min. The precipitate was dried under ambient conditions.
2.4. Image analysis
The progress of the precipitate formation in the microfluidic device was recorded using an optical microscope (Thermo Fisher Scientific, EVOS FL Auto). The microscopic image data were analyzed using in-house scripts in MATLAB (MathWorks, R2022b).2.5. Characterization
After the co-flow experiment was finished, the precipitate product was readily available for collection by disassembly of the microfluidic device. The precipitates were dried overnight on the polycarbonate plates under ambient conditions. The samples were manually ground using a mortar and pestle for the powder X-ray diffraction (XRD) measurements using a Bruker D8 Discover Microfocus diffractometer at an average scan rate of 0.37 deg s−1.For scanning electron microscopy (SEM, FEI Sirion XL30) the precipitates themselves are electrically conductive, but imaging the samples on the original non-conductive glass substrate prevents potential alterations to their morphologies during transfer, and hence the as-formed precipitates on the glass substrate were sputter-coated with Au/Pd at a 60/40 ratio before imaging.
The concentrations of Nd and Dy in the dried samples were determined using inductively coupled plasma mass spectrometry (ICP-MS, PerkinElmer NexION 2000). For the ICP-MS measurements, dried precipitates were digested in 35% HNO3 overnight and diluted to concentrations lower than 25 ppb. The Nd calibration curve was obtained using dilutions from a single-component standard (HPS, 100035-2), while the Dy calibration curve was obtained using dilutions from a multi-component standard (HPS, ICP-MS-68A). The calibration window was 0.05 to 40 ppb for both Nd and Dy (Fig. S2†).
3. Results and discussion
To identify relevant conditions for separation of Nd and Dy, we first investigate and characterize the differences in the precipitation dynamics of Nd(DBP)3 and Dy(DBP)3. Using the experimental setup shown in Fig. 2c, we co-injected three reactant solutions into a three-inlet microfluidic channel at a constant flow rate. The middle solution contained NaDBP of various concentrations (1.2, 4, 12, 36, and 120 mM), while the two sides were 4 mM NdCl3 (left) and 4 mM DyCl3 (right). The configuration simultaneously generated two reactive interfaces where the following two chemical reactions were expected to occur respectively,| Nd3+ + 3DBP− ⇌ Nd(DBP)3 |
| Dy3+ + 3DBP− ⇌ Dy(DBP)3 |
During the experiment, we observed these precipitation reactions by optical microscopy imaging. The viewing window was about 1 cm away from the mixing point of the three solutions (Fig. S3†). Image sequences in Fig. 3a show three frames at 5, 15, and 45 min for each concentration condition.
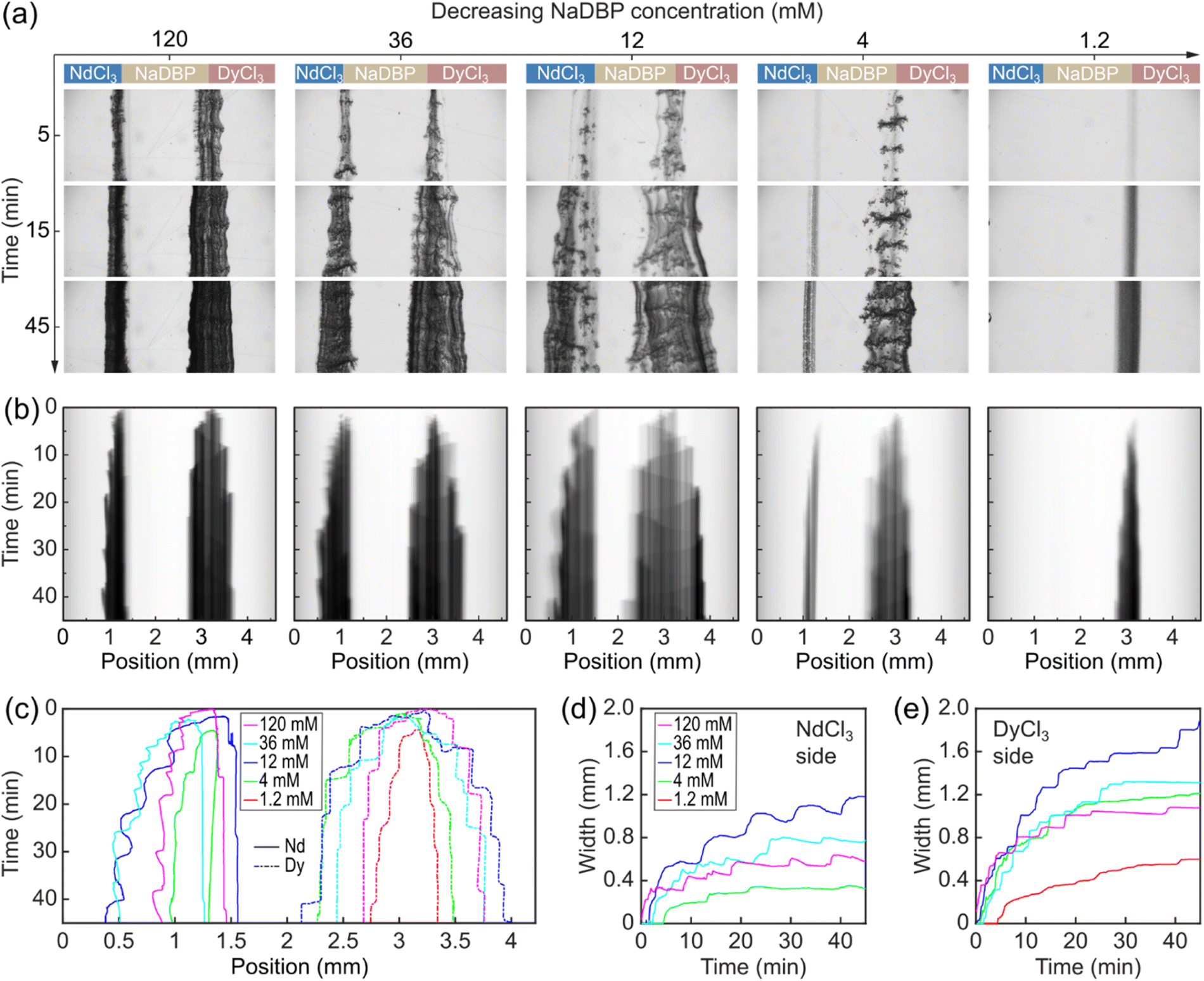 | ||
| Fig. 3 Differences in precipitation dynamics of Nd(DBP)3 and Dy(DBP)3. (a) Temporal image sequence of precipitation progress in the microfluidic channel for different NaDBP concentrations. Field of view: 4.6 mm × 1.8 mm. (b) Time–space plots of the growing precipitates. The five columns in (a and b) from left to right represent [NaDBP] = 120, 36, 12, 4, 1.2 mM, respectively. (c) Stacked contours obtained from the time–space plots. (d and e) Precipitate width as a function of time for Nd(DBP)3 (d) and Dy(DBP)3 (e). | ||
Under the stoichiometric condition ([NaDBP] = 12 mM, the 3rd column), two blocks of precipitates formed at the two interfaces as soon as the solutions met. The calculated Reynolds number (Re) in the channel is 0.3, indicating laminar flow. The lack of turbulence allowed precipitates to accumulate at the interfaces, and the precipitate growth occurred perpendicular to the flow direction (downward in Fig. 3a). We observed inhomogeneity in the porosity and the optical density (image darkness) of precipitates both along and perpendicular to the flow direction. The different porosities in the field of view (4.6 mm × 1.8 mm) could be attributed to initial erratic nucleation sites at the reactive interfaces that largely affect the precipitate particle growth and thus the overall appearance.
An increase of NaDBP concentration from 12 mM to 36 mM caused the precipitates to appear denser (a darker color) and narrower (the 2nd column in Fig. 3a). The former observation is expected because an increased number of nucleation sites occurred with a higher reactant concentration. The latter is counterintuitive and probably due to the high compactness of precipitates that effectively blocked the large-scale flow of solutions. Further increasing the NaDBP concentration showed the same trends (the 1st column in Fig. 3a), which suggests the shrink in precipitate width as the NaDBP concentration increases was not accidental. On the other hand, a decrease in NaDBP concentration from 12 mM to 4 mM resulted in a decrease in both compactness and thickness of the precipitates (the 4th column in Fig. 3a). Further decreasing the NaDBP concentration to 1.2 mM resulted in a much smaller amount and narrower Dy(DBP)3 precipitate, and surprisingly, the absence of the Nd(DBP)3 precipitate at the NdCl3/NaDBP interface. Note that all concentration conditions investigated here are in the supersaturation regime for both Nd(DBP)3 and Dy(DBP)3 under equilibrium (see calculated saturation indices in Table S2 in the ESI†).
Furthermore, we quantitatively characterized the differences in the spatiotemporal dynamics of the Nd(DBP)3 and Dy(DBP)3 precipitates. The image datasets for each NaDBP concentration were collected at 5 s intervals for 45 min (see the timelapse video in Movie S1 in ESI†). We averaged each individual image along the flow direction given the varying precipitate widths, generating a 1-pixel-wide color profile spanning 2048 pixels which corresponded to 4.6 mm. Then, these color profiles were stacked chronologically into the time–space plots showing continuous changes in both interiors and edges of the precipitate walls (Fig. 3b). We observed sinusoidal wave-like features in the interior of all Dy(DBP)3 precipitates that were absent in the Nd(DBP)3 (e.g., the 3rd column of Fig. 3b). These waves likely correspond to the propagating growth front oscillation inside the precipitate due to periodic changes in the pressure and the subchannel width.43 In addition, the Dy(DBP)3 precipitate grew in both directions and centred around the initial solution interface, whereas Nd(DBP)3 thickened unidirectionally towards the NdCl3 solution. Similar bidirectional precipitate growth has been previously observed in an inorganic–organic hybrid precipitate system,43 while unidirectional precipitate growth is akin to those reported in metal hydroxides.32,44–46 These thickening dynamics were further analysed as shown in Fig. 3c which plots the measured edge positions of the two precipitates as a function of NaDBP concentrations. The data show plateau-like features for both edges of Dy(DBP)3 that were approximately symmetric around the initial NaDBP/DyCl3 interface. During the periods when the precipitate edges were not evolving, slight dissolution of Dy(DBP)3 was observed on the DyCl3 edge but not on the NaDBP edge. In contrast, the edges on the NdCl3 side of Nd(DBP)3 precipitates showed large-scale step-wise progression, while edges on the NaDBP side also advanced in a similar fashion (about 110 μm in 45 min). This relatively small increase in precipitate thickness suggests that the growth mechanism of Nd(DBP)3 is different from the strictly unidirectional growth of metal hydroxide precipitates whose edges on the NaOH side typically remained completely stationary for several hours. Given that both the Nd(DBP)3 and Dy(DBP)3 precipitates thickened bidirectionally in simple two-inlet channels (insets of Fig. 1a and b), we believe that the decrease in the subchannel width due to the rapid bidirectional thickening of adjacent Dy(DBP)3 precipitates caused the seemingly unidirectional growth of Nd(DBP)3 in the three-inlet channels. Moreover, the differences in the border movements are represented as the width of the Nd(DBP)3 and Dy(DBP)3 precipitates (Fig. 3d and e). The temporal evolution of the precipitate widths indicates a more significant dissolution in the Nd(DBP)3 precipitate than Dy(DBP)3 under the same NaDBP concentration conditions. We found that the widest precipitates were obtained with 12 mM NaDBP (the stoichiometric ratio), and that the width of Dy(DBP)3 precipitates was consistently larger than that of Nd(DBP)3.
In the following, we investigate selective precipitation using the laminar co-flowing setup with a constant NaDBP concentration of 1.2 mM where DyCl3 exclusively precipitated. We introduced a mixture of 4 mM NdCl3 and 4 mM DyCl3 in the middle of the three inlets, and a solution of 1.2 mM NaDBP on each side (Fig. 4a). To increase the reaction volume, we used a device with ∼4× the channel height (using four layers of parafilm, Fig. S4†). This setup generated two identical reactive interfaces in the channel and significantly increased the contact areas of the reactant solutions (an increase of 28 fold from the initial design in Fig. 1a and b) and thus the precipitate output. After 16 h of co-flow, the resulting precipitates formed two nearly identical and symmetric-looking precipitate layers (Fig. 4b). Representative micrographs from the middle of each column clearly show a decreasing trend in the amount and compactness of the precipitate downstream (Fig. 4d–f). This trend was mainly caused by the decrease in the Nd/DyCl3 concentration and the interfacial concentration gradient along the flow direction. The changes in the precipitate appearance along the length of the flow channel suggest possible differences in the elemental composition (Nd:Dy) of the precipitate as a function of position. Thus, dried precipitates were collected from each column (defined in Fig. S5†) and stored separately in small glass vials at the end of the experiment (Fig. 4c) for analytical characterization.
 | ||
| Fig. 4 Selective precipitation of Dy(DBP)3. (a) Solution configuration for two identical reactive interfaces. (b and c) Photographs of the resulting precipitates in the microfluidic device (b) and transferred to small vials (c). Outer diameter of the vial: 12 mm. (d–f) Micrographs collected at different columns along the channel. The red, green, and blue color borders for the images correspond to the location indicated by the same color-coded boxes in (b). Field of view: 4.4 mm × 0.8 mm. In this experiment, the solutions of 1.2 mM NaDBP (left and right), 4 mM NdCl3 and 4 mM DyCl3 (middle) were co-injected for 16 h. | ||
To evaluate the performance of selective precipitation, we measured the Dy purity of precipitate samples collected from LCM and BMM. The Dy purity was calculated as
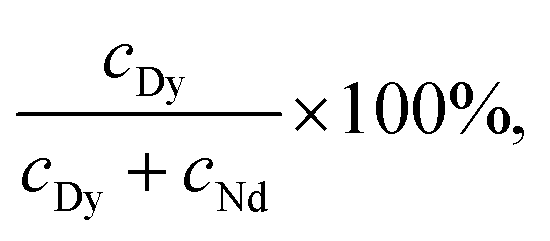
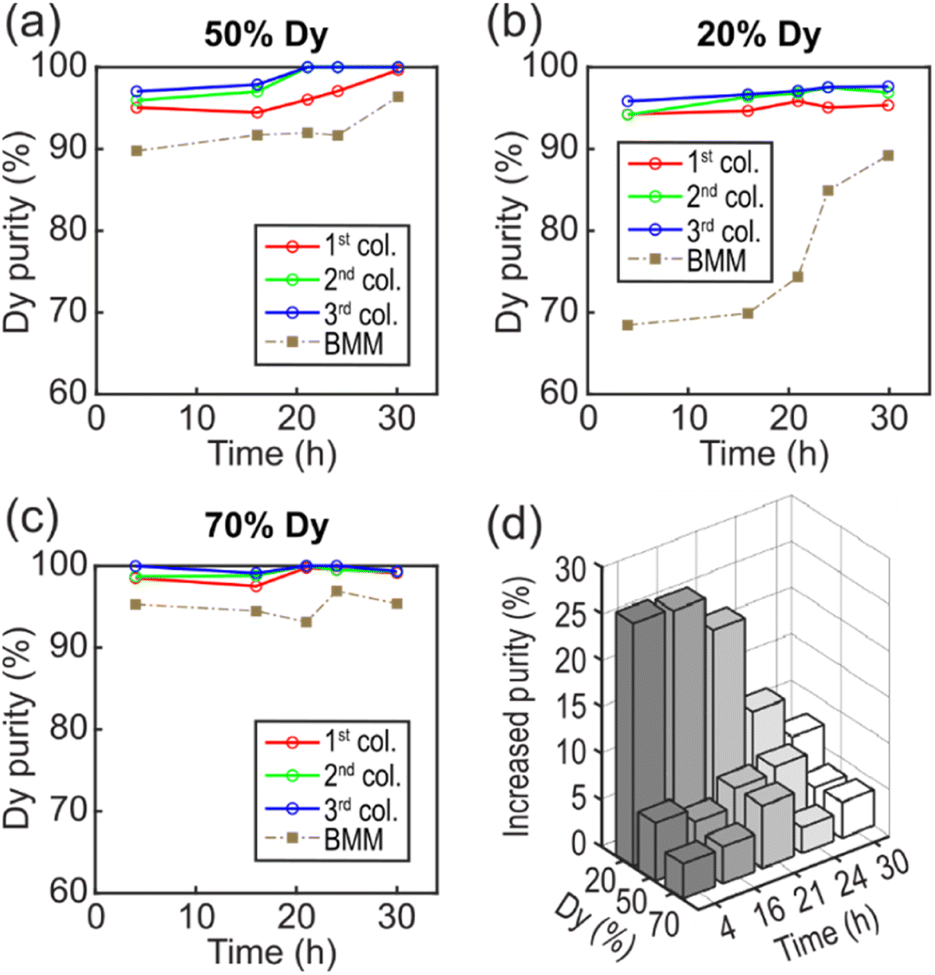 | ||
| Fig. 5 Comparison of separation performance. (a–c) Dy purity as a function of reaction time for three different initial Dy ratio at 50% (a), 20% (b), and 70% (c). The red, green, and blue hollow circles represent data points for LCM experiments at the 1st, 2nd, and 3rd columns, respectively. The gray solid squares are BMM experiments. (d) Increased purity by LCM as a function of reaction time and initial Dy ratio. | ||
As the ratio of Nd:Dy is source dependent and variable,48 we performed selective precipitation using solutions of different Nd:Dy ratios representative of compositions found across primary ores and permanent magnets. The Nd:Dy ratio was varied by keeping the NdCl3 constant at 4 mM while changing the DyCl3 concentration. For a high Nd:Dy ratio (20% Dy), we prepared a solution of 4 mM NdCl3 and 1 mM DyCl3 simulating the REE leachate of commonly used NdFeB magnets.40 Our results show that LCM yielded a high Dy purity of 95.8% in 4 h and 97.6% in 30 h (Fig. 5b). In comparison, BMM only yielded a purity of 68.5% in 4 h. For a low Nd:Dy ratio (70% Dy), we used a solution consisting of 4 mM NdCl3 and 9.3 mM DyCl3 simulating Dy-rich natural minerals.49 The lowest Dy purity was 98.5% in 4 h, and longer reaction times yielded purities ranging from 97.5% to 100% for LCM, while the highest purity of 96.9% found in BMM required a reaction time of 24 h (Fig. 5c). The trend of increasing Dy purity with increasing column number was also found for these two solution compositions. This result indicates that the decrease in the concentration (gradient) along the flow direction could benefit the selective precipitation of Dy. Note that the ICP-MS measured values of 24.2%, 46.7%, and 75.0% Dy in the feed solutions slightly differed from the intended 20%, 50%, and 70% Dy feed compositions (Fig. 5), but the impact of this small but consistent offset is expected to be negligible in terms of observed trends in Dy precipitation rates and purity.
To compare the overall performance of LCM and BMM, we plotted the increase in Dy purity as functions of reaction time and the initial Dy ratio (Fig. 5d). The increased purity was defined as the difference between the average purity of three columns for LCM and the purity for BMM. The most significant improvement was found with an initial composition of 20% Dy and a reaction time of 4 h. In addition, we obtained 0.76 mg of dried samples in LCM and 0.12 mg in BMM with a 4 h reaction time using a 20% Dy solution, suggesting that LCM also presents a more efficient way for product recovery without requiring centrifugation. The higher yield in LCM was partially due to a smaller contact area, i.e., the walls of the channel, where precipitates formed compactly with little to no loss during sample collection. Overall, LCM showed noticeable purity improvements over BMM for all solution compositions (20% to 70% Dy) and all reaction times (4 h to 30 h). We attribute the selective precipitation of Dy to the periodic precipitation–dissolution processes that occur at the reactive interfaces.
4. Conclusions
In summary, we demonstrated the use of the LCM approach for the selective precipitation of Dy3+ from mixed aqueous solutions of Nd/DyCl3. By visualizing the spatiotemporal dynamics, we obtained the differences of Nd(DBP)3 and Dy(DBP)3 precipitate formation for different NaDBP concentrations under laminar co-flow. Under flow-driven non-equilibrium conditions, Nd(DBP)3 precipitates might not form even under supersaturation when Dy(DBP)3 precipitates formed. With the optimized NaDBP concentration condition, LCM showed enhanced selectivity for Dy (quantified as precipitate purity) and significantly shorter reaction times to generate precipitates for a broad range of Nd:Dy ratio feed solutions, compared to BMM. This improvement was the most pronounced for low Dy-containing feedstock where a precipitate product with 95.8% Dy was obtained from an initial solution with 20% Dy.
Our results show the benefits of using flow-induced non-equilibrium conditions to achieve enhanced separation of REEs over the stirred-mixing equilibrium conditions commonly used in industry. The higher separation efficiencies are achieved purely by altering the mixing conditions and not with the use of any membranes or sorbents, differentiating our method from existing ones. The aqueous chemistry and use of commodity reactants for precipitation make our process environmentally friendly, while the enhanced selectivity and ability to achieve higher precipitation rates across a spectrum of Nd/Dy compositions indicates the approach to be broadly applicable for diverse feedstocks. This work is the first report on REE separations using flow-driven non-equilibrium and introduces the optimization rationale to extend the method to diverse chemistries. However, further research is necessary to truly evaluate the scalability and versatility of the approach for extraction and recovery of REEs from real world feedstocks, which introduce higher compositional complexities. Additionally, developing a device design with reliable precipitate recovery and a cost–benefit analysis of laminar vs. stirred mixing, which are the focus of our future efforts, will be critical to determining the relevance of the approach to industrial-scale separations.
Author contributions
Qingpu Wang: conceptualization, investigation, formal analysis, visualization, writing – original draft. Chinmayee V. Subban: conceptualization, funding acquisition, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Laboratory Directed Research and Development Program (LDRD) at Pacific Northwest National Laboratory (PNNL), with partial support under the Non-Equilibrium Transport Driven Separations Initiative (NETS). PNNL is a multiprogram national laboratory operated for DOE by Battelle under contract DE-AC05-76RLO1830. Part of this work was conducted at the Molecular Analysis Facility, a National Nanotechnology Coordinated Infrastructure (NNCI) site at the University of Washington operated with partial support from the National Science Foundation via awards NNCI-2025489 and NNCI-1542101. The authors acknowledge Dr Elias Nakouzi for valuable discussions and Dr Samantha L. Young for assistance with Scherrer analysis.Notes and references
- N. Höhne, M. J. Gidden, M. den Elzen, F. Hans, C. Fyson, A. Geiges, M. L. Jeffery, S. Gonzales-Zuñiga, S. Mooldijk, W. Hare and J. Rogelj, Nat. Clim. Change, 2021, 11, 820–822 CrossRef.
- U. S. Department of Energy, Critical Materials Assessment, 2023 Search PubMed.
- A. R. Chakhmouradian and F. Wall, Elements, 2012, 8, 333–340 CrossRef CAS.
- U.S. Geological Survey, Mineral Commodity Summaries 2023, Report 2023, Reston, VA, 2023 Search PubMed.
- L. Talens Peiró and G. Villalba Méndez, JOM, 2013, 65, 1327–1340 CrossRef.
- S. Van Loy, M. A. R. Önal, K. Binnemans and T. Van Gerven, Hydrometallurgy, 2020, 191, 105154 CrossRef CAS.
- W. D. Jamrack, Rare Metal Extraction by Chemical Engineering Techniques: International Series of Monographs on Chemical Engineering, Elsevier, 2014 Search PubMed.
- F. Xie, T. A. Zhang, D. Dreisinger and F. Doyle, Miner. Eng., 2014, 56, 10–28 CrossRef CAS.
- H.-S. Yoon, C.-J. Kim, K.-W. Chung, S.-D. Kim, J.-Y. Lee and J. R. Kumar, Hydrometallurgy, 2016, 165, 27–43 CrossRef CAS.
- B. Dewulf, N. K. Batchu and K. Binnemans, ACS Sustainable Chem. Eng., 2020, 8, 19032–19039 CrossRef CAS PubMed.
- S. Riaño and K. Binnemans, Green Chem., 2015, 17, 2931–2942 RSC.
- J. A. Bogart, C. A. Lippincott, P. J. Carroll and E. J. Schelter, Angew. Chem., Int. Ed., 2015, 54, 8222–8225 CrossRef CAS PubMed.
- J. A. Bogart, B. E. Cole, M. A. Boreen, C. A. Lippincott, B. C. Manor, P. J. Carroll and E. J. Schelter, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14887–14892 CrossRef CAS PubMed.
- R. F. Higgins, T. Cheisson, B. E. Cole, B. C. Manor, P. J. Carroll and E. J. Schelter, Angew. Chem., Int. Ed., 2020, 59, 1851–1856 CrossRef CAS.
- J. J. M. Nelson, T. Cheisson, H. J. Rugh, M. R. Gau, P. J. Carroll and E. J. Schelter, Commun. Chem., 2020, 3, 7 CrossRef CAS.
- T. Cheisson, B. E. Cole, B. C. Manor, P. J. Carroll and E. J. Schelter, ACS Sustainable Chem. Eng., 2019, 7, 4993–5001 CrossRef CAS.
- B. E. Cole, I. B. Falcones, T. Cheisson, B. C. Manor, P. J. Carroll and E. J. Schelter, Chem. Commun., 2018, 54, 10276–10279 RSC.
- H. Ya Gao, W. Li Peng, P. Pan Meng, X. Feng Feng, J. Qiang Li, H. Qiong Wu, C. Sheng Yan, Y. Yang Xiong and F. Luo, Chem. Commun., 2017, 53, 5737–5739 RSC.
- A. Masuya-Suzuki, K. Hosobori, R. Sawamura, Y. Abe, R. Karashimada and N. Iki, Chem. Commun., 2022, 58, 2283–2286 RSC.
- J. G. O'Connell-Danes, B. T. Ngwenya, C. A. Morrison and J. B. Love, Nat. Commun., 2022, 13, 4497 CrossRef PubMed.
- K. R. Johnson, D. M. Driscoll, J. T. Damron, A. S. Ivanov and S. Jansone-Popova, JACS Au, 2023, 3, 584–591 CrossRef CAS PubMed.
- Z. Dong, J. A. Mattocks, G. J. P. Deblonde, D. Hu, Y. Jiao, J. A. Cotruvo Jr and D. M. Park, ACS Cent. Sci., 2021, 7, 1798–1808 CrossRef CAS PubMed.
- D. Prodius, M. Klocke, V. Smetana, T. Alammar, M. Perez Garcia, T. L. Windus, I. C. Nlebedim and A.-V. Mudring, Chem. Commun., 2020, 56, 11386–11389 RSC.
- Y. Tasaki-Handa, Y. Abe, K. Ooi, H. Narita, M. Tanaka and A. Wakisaka, J. Phys. Chem. B, 2016, 120, 12730–12735 CrossRef CAS PubMed.
- X. Yin, Y. Wang, X. Bai, Y. Wang, L. Chen, C. Xiao, J. Diwu, S. Du, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, Nat. Commun., 2017, 8, 14438 CrossRef CAS.
- Z. Bai, B. Scheibe, J. M. Sperling and T. E. Albrecht-Schönzart, Inorg. Chem., 2022, 61, 19193–19202 CrossRef CAS PubMed.
- A. A. Kopyrin, A. A. Fomichev and M. A. Afonin, Radiochemistry, 2007, 49, 277–280 CrossRef CAS.
- N. Sui, S. Miao, K. Cui, F. Meng and K. Huang, J. Rare Earths, 2024, 42(1), 200–209 CrossRef CAS.
- N. Calvo Galve, A. Abrishamkar, A. Sorrenti, L. Di Rienzo, M. Satta, M. D'Abramo, E. Coronado, A. J. de Mello, G. Mínguez Espallargas and J. Puigmartí-Luis, Angew. Chem., Int. Ed., 2021, 60, 15920–15927 CrossRef CAS PubMed.
- Y. Xia, S. Sevim, J. P. Vale, J. Seibel, D. Rodríguez-San-Miguel, D. Kim, S. Pané, T. S. Mayor, S. De Feyter and J. Puigmartí-Luis, Nat. Commun., 2022, 13, 7006 CrossRef CAS PubMed.
- S. Sevim, A. Sorrenti, C. Franco, S. Furukawa, S. Pané, A. J. deMello and J. Puigmartí-Luis, Chem. Soc. Rev., 2018, 47, 3788–3803 RSC.
- B. C. Batista and O. Steinbock, J. Phys. Chem. C, 2015, 119, 27045–27052 CrossRef CAS.
- M. Emmanuel, D. Horváth and Á. Tóth, CrystEngComm, 2020, 22, 4887–4893 RSC.
- G. Laffite, C. Leroy, C. Bonhomme, L. Bonhomme-Coury, E. Letavernier, M. Daudon, V. Frochot, J. P. Haymann, S. Rouzière, I. T. Lucas, D. Bazin, F. Babonneau and A. Abou-Hassan, Lab Chip, 2016, 16, 1157–1160 RSC.
- Y. Zeng, J. Cao, Z. Wang, J. Guo and J. Lu, Cryst. Growth Des., 2018, 18, 1710–1721 CrossRef CAS.
- R. Hudson, R. de Graaf, M. Strandoo Rodin, A. Ohno, N. Lane, S. E. McGlynn, Y. M. A. Yamada, R. Nakamura, L. M. Barge, D. Braun and V. Sojo, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 22873–22879 CrossRef CAS PubMed.
- Q. Wang and O. Steinbock, ChemCatChem, 2020, 12, 63–74 CrossRef CAS.
- Q. Wang, L. M. Barge and O. Steinbock, Chem.–Eur. J., 2019, 25, 4732–4739 CrossRef CAS PubMed.
- Q. Wang, E. Nakouzi, E. A. Ryan and C. V. Subban, Energy Environ. Sci. Lett., 2022, 9, 645–649 CAS.
- X. Du and T. E. Graedel, J. Ind. Ecol., 2011, 15, 836–843 CrossRef CAS.
- A. Kumari and S. K. Sahu, Sep. Purif. Technol., 2023, 317, 123527 CrossRef CAS.
- Y. Tasaki-Handa, Y. Abe, K. Ooi, H. Narita, M. Tanaka and A. Wakisaka, Sep. Purif. Technol., 2016, 157, 162–168 CrossRef CAS.
- Q. Wang and O. Steinbock, Chem.–Eur. J., 2019, 25, 10427–10432 CrossRef CAS PubMed.
- Y. Ding, B. Batista, O. Steinbock, J. H. E. Cartwright and S. S. S. Cardoso, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 9182–9186 CrossRef CAS PubMed.
- J. A. Nogueira, B. C. Batista, M. A. Cooper and O. Steinbock, Angew. Chem., Int. Ed., 2023, 62, e202306885 CrossRef CAS PubMed.
- Q. Wang, M. R. Bentley and O. Steinbock, J. Phys. Chem. C, 2017, 121, 14120–14127 CrossRef CAS.
- X. Liu and R. H. Byrne, Geochim. Cosmochim. Acta, 1997, 61, 1625–1633 CrossRef CAS.
- N. Oono, M. Sagawa, R. Kasada, H. Matsui and A. Kimura, J. Magn. Magn. Mater., 2011, 323, 297–300 CrossRef CAS.
- A. Jordens, Y. P. Cheng and K. E. Waters, Miner. Eng., 2013, 41, 97–114 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00403a |
| This journal is © The Royal Society of Chemistry 2024 |