
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStability study of a superbase-derived ionic liquid [mTBNH][OAc] with enhanced cellulose dissolution ability: thermal and natural degradation†
Ivan
Melikhov
a,
Irina
Sulaeva
 ab,
Stefano
Barbini
a,
Markus
Bacher
a,
Dev
Sriranganadane
a,
Ilkka
Kilpeläinen
c,
Thomas
Rosenau
a and
Antje
Potthast
*a
ab,
Stefano
Barbini
a,
Markus
Bacher
a,
Dev
Sriranganadane
a,
Ilkka
Kilpeläinen
c,
Thomas
Rosenau
a and
Antje
Potthast
*a
aUniversity of Natural Resources and Life Sciences, Vienna, (BOKU), Department of Chemistry, Institute for Chemistry of Renewable Resources, Konrad-Lorenz Strasse 24, 3430 Tulln an der Donau, Austria. E-mail: antje.potthast@boku.ac.at
bUniversity of Natural Resources and Life Sciences, Vienna, (BOKU), Core Facility Analysis of Lignocellulosics (Alice), Konrad-Lorenz Strasse 24, 3430 Tulln an der Donau, Austria
cUniversity of Helsinki, Department of Chemistry, Helsinki Institute of Sustainability Science (HELSUS), Yliopistonkatu 3, 00014, Helsingin Yliopisto, Finland
First published on 16th May 2024
Abstract
Ionic liquids (ILs) have the potential to revolutionize the production of cellulose fibers by offering more sustainable and eco-friendly processes compared to conventional spinning techniques, which might involve the use of hazardous chemicals and demand significant energy inputs. At the same time, it is crucial to acknowledge that the lack of a comprehensive understanding of the ILs' stability and the nature of possible byproducts remains the central concern associated with their application. In this study, an investigation into the stability of the novel superbase-derived IL, [mTBNH][OAc], under different conditions was carried out. Specifically, the IL was subjected to thermal ageing at 95 °C to investigate intrinsic thermal instabilities in the presence of water and wood polysaccharides. Moreover, natural ageing at room temperature in the absence of light was also examined. Changes in the composition of the IL during ageing processes were monitored and byproducts were identified by means of Nuclear Magnetic Resonance (NMR) spectroscopy and Supercritical Fluid Chromatography coupled with high-resolution Quadrupole Time-of-Flight Mass Spectrometry (SFC-QToF-MS). Under thermal ageing, hydrolysis products, with the six-membered ring being opened and a urea structure being formed, dominated. Upon long-term ageing, either of the two heterocyclic rings was oxygenated, under the introduction of hydroxyl and keto groups, with subsequent ring-opening and elimination reactions.
Sustainability spotlightIonic liquids (ILs) are seen as promising green solvents for cellulose and, therefore, hold a potential for the development of new sustainable cellulose fiber production technologies. To optimize these technologies, the stability of ILs has to be assessed. The aim of the present work was to develop an analytical technique for ionic liquid stability analysis and for detection of its degradation products present in minor quantities. The method developed was successfully applied to access the stability of a new IL [mTBNH][OAc], which was recently proved to be suitable for spinning of high-quality fibers from sustainable feedstock. Our work aligns with the following UN sustainable development goal: responsible consumption and production (SDG 12). |
Introduction
In the context of a growing demand for sustainable and environmentally friendly materials, cellulose fibers are seen as a promising alternative to traditional synthetic fibers due to their renewable nature and biodegradability.1 As we all know, current approaches to cellulose fibers can have limitations in terms of efficiency, scalability, environmental impact, and costs, which challenge further expansion of their production.2 For example, for cotton growing practices, which substantially vary depending on the geographical location, the water consumption for the production of 1 kilogram of fibers, in some cases, can spike up to 22 tons. This aggravates the already critical water scarcity issue.3,4 Furthermore, the utilization of fertilizers in cotton cultivation contributes to the contamination of both surface and groundwater.3 As for regenerated fibers, i.e., cellulose which undergoes a dissolution/spinning sequence, a major concern relates to the use of hazardous chemicals, such as carbon disulfide in rayon fiber production, and the release of pollutants into air and water.5 In contrast to rayon manufacture, the lyocell process is considered to be more environmentally friendly: cellulose is directly dissolved in N-methylmorpholine-N-oxide (NMMO) without chemical derivatization, followed by extrusion through a spinneret, stretching within an air gap, and ultimate regeneration within a spin bath composed only of water. However, the inherent instability of the solvent NMMO6 requires the incorporation of stabilizers, which can cause discoloration of the spinning dope and eventually of the fibers,7 necessitating additional washing and bleaching procedures.Given the current need to address environmental issues and to cover the growing “cellulose gap”,8 which signifies an increasing demand for man-made cellulosic fibers, the development of new technologies for cellulose fiber production has become a critical concern. In this context, ionic liquids (ILs) hold substantial promise as a prominent “green” solvent for cellulose due to their low melting point, low volatility and flammability, tuneable properties, and high dissolution power.
Even though the discovery of ILs' ability to directly dissolve cellulose was reported by Charles Graenacher as far back as 90 years ago,9 active research targeting the processing of cellulose using ILs has only gained significant attention in the past two decades. The exponential growth of publications concerning the use of ILs for cellulose dissolution was triggered by Swatloski et al. who demonstrated that 1-butyl-3-methylimidazolium chloride can dissolve cellulose up to a concentration of 25 wt% under microwave heating without any activation or pretreatment.10 Nowadays, the scope of utilizing ILs as media for the valorisation of cellulose continually expands each year. On a research scale, this spans processes such as hydrolysis into reducing sugars11 for further fermentation or biotechnological conversions, hydrogenation to sugar alcohols,12 and conversion to important chemical building blocks, such as 5-hydroxymethylfurfural (HMF),13 levulinic acid,14 or lactic acid.15 Future will show whether all these potential applications will find their way into large-scale production.
Despite the wide range of potential combinations of cations and anions, only a limited number of ILs have been identified as effective solvents for cellulose fiber production. The so-called first-generation ILs, based on 1,3-dialkylimidazolium, have demonstrated excellent cellulose dissolution capabilities. However, they have also exhibited undesired reactivity towards cellulose, particularly at the C2 position of the imidazolium moiety, where it can react with the reducing end of the cellulose polymer chain forming a carbon–carbon bond.16,17 Also the anions are not necessarily behaving in an inert way. Acetate, for instance, causes cellulose acetylation as a side reaction.18,19 The newly formed cellulosic byproducts can impact the physical and rheological properties of the cellulose/IL mixture. This can extend to potential damage or fibrillation of the regenerated fibers, and at the same time impede the recovery of the IL. Also, the impact of these impurities on the applicability of the products, especially in the physiological and medical field, is so far an open issue.
In more recent studies, protic superbase-derived ILs (SILs) have been proved to be the media of choice in the production of high-performance textile fibers, ultimately leading to the development of novel fiber manufacture processes, such as Ioncell-F.20 Owing to their superior solubilization properties, protic SILs hold great potential for a wide range of applications in materials science, including cellulose dissolution, as well as various upstream cellulose forming applications (fibers, film, cellulosic bodies).21–23 As for sustainable fiber production, SILs contribute to an increased process efficiency by enabling the utilization of a broader spectrum of starting materials, including waste fibers and newspapers,24–26 which distinguishes them positively from the conventional NMMO-based lyocell fiber production. Moreover, the use of hemicellulose-rich pulp, which is commonly avoided in rayon and lyocell processes due to unwanted hemicellulose reactivity with solvent systems,27,28 has been demonstrated to yield fibers with good properties when using SILs.29,30 These findings indicate that SILs bear the potential to improve the entire fiber production process by addressing the resource efficiency issue.
Among the variety of SILs, bicyclic amidine- and guanidine-based structures, such as 1,5-diazabicyclo[4.3.0]non-5-ene acetate [DBNH][OAc] and 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-enium acetate [mTBDH][OAc] used in the Ioncell-F process, emerged as excellent cellulose solvents. They allow for high cellulose concentration in the dope and relatively low operating temperatures during dissolution and spinning; this results in milder conditions, which mitigate cellulose degradation, enhance fiber yield, and improve strength properties.20 However, the recycling issue of SILs has yet to be solved.
The search for novel organic ILs that can be efficiently synthesized from readily available and inexpensive starting materials remains ongoing. Recently, another guanidine-based SIL, [mTBNH][OAc], comprising a mixture of two isomers (1 and 2 in Fig. 1), has been identified as a promising candidate from both the synthesis and the cost perspectives. This IL is currently under active investigation for its potential as a solvent in pulp processing31 and cellulose fiber manufacturing,29,32,33 development of recycling methods,34–36 and its use as reaction medium for efficient cellulose modification.37 One of the primary challenges in the development towards large-scale application is the imperative need for a comprehensive characterisation of its stability, side reactions, and possible formation of degradation products. The enrichment of the solvent with degradation products may reduce its solvation efficacy and cause recycling problems, resulting in the increase of overall production costs.
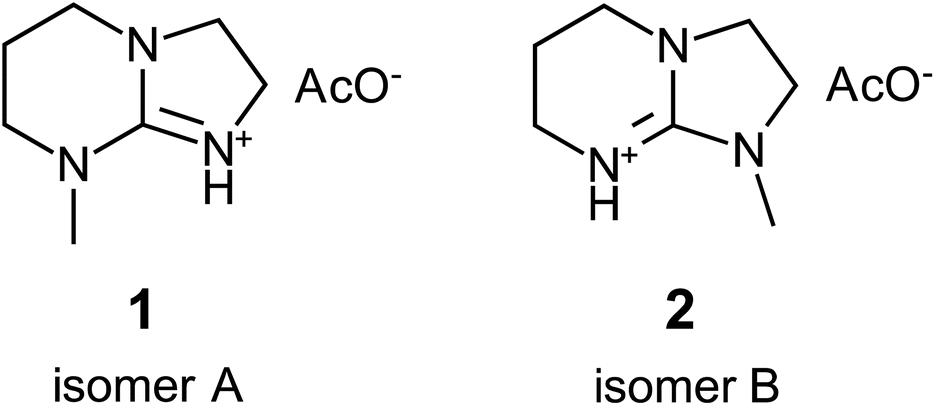 | ||
| Fig. 1 The two isomeric forms of the ionic liquid [mTBNH][OAc], distinguished by the N-methylation site: 5-methyl-1,5,7-triaza-bicyclo[4.3.0]non-6-enium acetate (1, isomer A) and 7-methyl-1,5,7-triaza-bicyclo[4.3.0]non-6-enium acetate (2, isomer B). | ||
The stability of commonly used ILs, such as imidazolium-based, quaternary-ammonium, and quaternary phosphonium, has been extensively addressed in the literature. These studies included stability tests performed under different conditions depending on ILs' application, including thermal, electrochemical and radiolytic environments.38–40 At the same time, the stability of SILs has not been extensively explored. Thus, the objective of the present research was to gain understanding of the degradation mechanisms and the stability profile of [mTBNH][OAc], contributing to the overall knowledge about the stability of SILs, particularly in the context of their application for cellulose fiber production. This was done by a combination of analytical methods: Supercritical Fluid Chromatography coupled with high-resolution Quadrupole Time-of-Flight Mass Spectrometry (SFC-QToF-MS) and Nuclear Magnetic Resonance (NMR) spectroscopy for byproduct separation and identification, respectively. These methods were used to characterize the individual degradation products and to identify the main degradation pathways and kinetics for [mTBNH][OAc].
Materials and methods
Thermal ageing experiment
The SIL [mTBNH][OAc] was synthesized as a mixture of two isomers (see Fig. 1) according to previous work.35 For the thermal ageing experiment, five IL samples were prepared: neat IL, IL with 10 wt% water (UHPLC/MS grade, Fisher Scientific GmbH, Vienna, Austria), IL with 40 wt% water, IL + 2 wt% xylan (Beechwood xylan, Lenzing AG, Austria), and IL + 1 wt% cellulose (bleached beech sulfite dissolving pulp, Lenzing AG, Austria). Each sample was placed in a GC vial, sealed, and aged in an oven (Memmert 100–800, Schwabach, Germany) at 95 °C for a period of 20 days.SFC-QTof-MS analysis
All MS experiments were conducted in positive mode using a high-resolution hybrid mass spectrometer (XEVO G2-XS QToF, Waters Corporation, Milford, MA, USA) in combination with a Supercritical Fluid Chromatography (SFC) system (ACQUITY UPC2, Waters Corporation, Milford, MA, USA). The separation and investigation of the degradation products of the naturally aged IL were performed with a 2-picolylamine column (Torus 2-PIC, pore size 130 Å, particle size 1.7 μm, 3 mm × 100 mm, Waters Corporation, Milford, MA, USA) equipped with a precolumn (Torus 2-PIC VanGuard, pore size 130 Å, particle size 1.7 μm, 2.1 mm × 5 mm, Waters Corporation, Milford, MA, USA) under the following conditions: column temperature: 45 °C, autosampler temperature: 20 °C, injection volume: 1.0 μL, flow rate: 1.2 mL min−1, automated back-pressure regulator (ABPR) pressure: 2000 psi (137.9 bar).The mobile phase consisted of supercritical carbon dioxide (>99.995%, Linde AG, Austria) as solvent A; solvent B was a methanol/acetonitrile mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v, LC-MS grade, Fisher Scientific (Austria) GmbH, Vienna) containing 25 mM ammonium formate (pH = 5.7, >99%, LC-MS grade, VWR). The gradient of solvent B was set as follows: 0 min, 1%; 1 min, 1%; 10 min, 30%; 10.50 min, 40%; 11.50 min, 40%; 12 min, 1%, 15 min, 1% (curve 6). After each injection, the injector needle was purged with methanol (600 μL) and 2-propanol (200 μL). The column effluent was mixed with a makeup solvent system (methanol/2-propanol/water mixture, 7:2:1, v/v, LC-MS grade, Fisher Scientific GmbH, Vienna, Austria), delivered by the isocratic solvent manager. A decreasing flow rate of the makeup solvent system was set based on the increasing percentages of the modifier in the chromatography run: 0 min, 0.3 mL min−1; 10 min, 0.2 mL min−1; 11.5 min, 0.1 mL min−1; 12 min, 0.3 mL min−1; 15 min, 0.3 mL min−1.
:1, v/v, LC-MS grade, Fisher Scientific (Austria) GmbH, Vienna) containing 25 mM ammonium formate (pH = 5.7, >99%, LC-MS grade, VWR). The gradient of solvent B was set as follows: 0 min, 1%; 1 min, 1%; 10 min, 30%; 10.50 min, 40%; 11.50 min, 40%; 12 min, 1%, 15 min, 1% (curve 6). After each injection, the injector needle was purged with methanol (600 μL) and 2-propanol (200 μL). The column effluent was mixed with a makeup solvent system (methanol/2-propanol/water mixture, 7:2:1, v/v, LC-MS grade, Fisher Scientific GmbH, Vienna, Austria), delivered by the isocratic solvent manager. A decreasing flow rate of the makeup solvent system was set based on the increasing percentages of the modifier in the chromatography run: 0 min, 0.3 mL min−1; 10 min, 0.2 mL min−1; 11.5 min, 0.1 mL min−1; 12 min, 0.3 mL min−1; 15 min, 0.3 mL min−1.
The mass range used was 50 to 2000 m/z with the following tuning parameters: capillary voltages: 3.0 V; sampling cone: 40 V; source offset: 80 V; source temperature: 150 °C; desolvation temperature: 500 °C; cone gas (N2) flow: 48 L h−1; and desolvation gas flow (N2): 1020 L h−1. The lock mass for automated mass correction was based on the monoisotopic mass of leucine–enkephaline (Waters Corporation, Milford, MA, USA): positive [M + H]+ = 556.2771. A scan time of 1 s, interval of 20 s, mass window of ±0.5 Da, infusion flow rate of 3.0 μL min−1, and capillary voltage of 1.80 kV were set as LockSpray.
The MS/MS experiments were conducted using a fast DDA (data-dependent acquisition) approach. The switching threshold for the MS survey was set to 15000, meaning that MS/MS acquisition begins when the intensity of an individual ion rises above this threshold. The MS survey scanning condition was set to 0.25 s in the centroid format. The maximum number of ions selected for MS/MS from a single MS survey scan was limited to 7, and the scan rate for MS/MS was set to 0.2 s, also in the centroid format. The “MS/MS stop” feature was enabled when the total ion current (TIC) accumulated to 100000 or when 0.25 s had elapsed, whichever occurred first. Argon (5.0, Linde AG, Austria) was utilized as the collision gas for MS/MS experiments, with a ramping collision energy ranging from 25 to 55 eV.
The fit configuration % (fit conf.%) indicates the confidence in the fit to the isotopic pattern: the higher the value, the better is the confidence in the elemental composition. Also, MS/MS experiments were carried out during the analysis to obtain the fragmentation patterns.
Flash chromatography purification
900 mg of the naturally aged sample of the SIL [mTBNH][OAc] was used for separation and purification on silica gel to elucidate the molecular structures of degradation products by means of NMR. The dry loading procedure was chosen due to the low solubility of the IL sample in the eluant THF/heptane (8:2, v/v). The gradient used for column chromatography separation started from THF/heptane (8:2, v/v) to pure THF in 37 fractions (740 mL). Then, MeOH was introduced as a more polar solvent in a 10% ratio, and a gradient from THF/MeOH (9:1, v/v) to pure MeOH with a 10% increase of MeOH every 100 mL was applied until 93 fractions were collected into 20 mL test tubes.
Fractions 17–20 contained a substance providing a spot visible at 254 nm on TLC, with an m/z of 152.0833 (C7H10N3O+) eluting at 3.09 min in SFC-QToF-MS and were pooled and dried under reduced pressure, yielding 1.5 mg of 7-methyl-4,5-dihydro-1,3a,7-triaza-1-indenium-6(7H)-one (Table 1, peak i). Fractions 27–34 contained a substance that gave a TLC spot visible at 366 nm with an m/z of 150.0678 (C7H8N3O+), eluting at 3.40 min in SFC-ESI-QToF-MS, and were pooled and dried under reduced pressure resulting in 1 mg of 7-methyl-1,3a,7-triaza-1-indenium-4(7H)-one (Table 1, peak x).
| Peak | Retention time (min) | Observed mass (m/z) | Error (ppm) | Elemental composition | Fit conf. (%) |
|---|---|---|---|---|---|
| Impurities present in the fresh sample | |||||
| a | 5.44 | 138.1035 | 2.9 | C7H12N3+ | 100 |
| b | 5.91 | 154.0984 | 2.6 | C7H12N3O+ | 100 |
| c | 7.01 | 126.1033 | 1.6 | C6H12N3+ | 100 |
| d | 7.58 | 154.1349 | 3.2 | C8H16N3+ | 100 |
| e | 7.77 | 154.1348 | 2.6 | C8H16N3+ | 100 |
| f | 7.84 | 154.1347 | 1.9 | C8H16N3+ | 99.97 |
| g | 7.95 | 170.1298 | 2.9 | C8H16N3O+ | 99.99 |
| h | 8.83 | 154.1346 | 1.3 | C8H16N3+ | 100 |
|
|||||
| Degradation by-products from natural ageing | |||||
| i | 3.09 | 152.0827 | 2.0 | C7H10N3O+ | 100 |
| j | 3.94 | 154.0984 | 2.6 | C7H12N3O+ | 100 |
| k | 4.26 | 168.1137 | 0 | C8H14N3O+ | 100 |
| l | 4.76 | 154.0983 | 1.9 | C7H12N3O+ | 100 |
| m | 5.33 | 154.0983 | 1.9 | C7H12N3O+ | 100 |
| n | 5.55 | 154.0983 | 1.9 | C7H12N3O+ | 100 |
| o | 6.47 | 212.1397 | −0.9 | C10H18N3O2+ | 98.86 |
| p | 7.89 | 170.0925 | −2.9 | C7H12N3O2+ | 99.96 |
| q | 9.20 | 156.1139 | 1.3 | C7H14N3O+ | 99.93 |
| r | 9.38 | 182.1296 | 1.6 | C9H16N3O+ | 99.84 |
| s | 9.45 | 156.1142 | 3.2 | C7H14N3O+ | 99.75 |
| t | 11.16 | 172.1086 | 0 | C7H14N3O2+ | 92.35 |
|
|||||
| Degradation by-product detected after isolation with flash chromatography | |||||
| x | 3.40 | 150.0669 | 1.3 | C7H8N3O+ | 100 |
|
|||||
| Two isomers of mTBNH starting SIL | |||||
| 1 | 8.54 | 140.1223 | 25 | C7H14N3+ | — |
| 2 | 8.39 | 140.1223 | 25 | C7H14N3+ | — |
Fractions 85–93 were pooled and dried under reduced pressure resulting in a fraction of 50.4 mg, which was dissolved in 7 mL of heptane/i-PrOH/MeOH/water 7.1/57/21.4/14.3 (v/v). This mixture (2.5 mL) was subjected to a second column purification on silica gel using MeOH with 2.5% triethylamine (TEA) as the mobile phase. The product 3-[2-(methylamino)-2-imidazolidinylium-1-yl]propionate (Table 1, peak t) had an m/z of 172.1086, eluting at 11.16 min in SFC-QToF-MS.
Identification of the degradation products by nuclear magnetic resonance (NMR) spectroscopy
A Bruker Avance II 400 instrument (1H resonance at 400.13 MHz, and 13C resonance at 100.62 MHz equipped with a 5 mm N2-cooled broadband cryoprobe head (Prodigy) with z-gradient) was used for the NMR analysis. 1H NMR data were collected with 32k data points, multiplied with a Gaussian window function prior to Fourier transformation. The acquisition time was 2.3 s with a 1 s relaxation delay. 13C spectrum with WALTZ16 1H decoupling was acquired using 64k data points. Signal-to-noise enhancement was achieved by multiplication of the FID with an exponential window function (lb = 1 Hz). All two-dimensional experiments were performed with 1k × 256 data points, while the number of transients (2–16 scans) and the sweep widths were optimized individually. The HSQC experiment was performed in edited mode using adiabatic pulsing for inversion of 13C and GARP-sequence for broadband 13C-decoupling, optimized for 1J(CH) = 145 Hz. To obtain higher resolution in the 13C-dimension, two separate HMBC spectra were acquired with band-selective pulses optimized either for the carboxyl region (δC 150–180 ppm) or the aliphatic region (δC 15–55 ppm). For the NOESY spectrum a mixing time of 0.5 s was used. The data were processed and analyzed using Bruker TopSpin versions 3.6 or higher. The spectra were recorded in CD3OD for the isolated natural degradation products and in DMSO-d6 for the thermally aged samples. Chemical shifts were expressed in δ ppm values referenced to residual solvent (non-deuterated) signals (CD3OD: 3.31 ppm for 1H, 49.0 ppm for 13C, and DMSO-d6: 2.49 ppm for 1H, 39.6 ppm for 13C). Coupling constants are given in Hz.Thermogravimetric analysis (TGA)
Thermal analysis of [mTBNH][OAc] and [mTBDH][OAc] was performed by using a thermogravimetric analyzer TG 209 F1 Iris (Netzsch, Germany). Samples of 15–20 mg were placed in crucibles and heated from 30 °C to 350 °C at a rate of 10 °C min−1 and nitrogen flow of 40 mL min−1. The data were processed using NETZSCH Proteus Thermal Analysis software, v. 8.0.2.Water content analysis
Water content was determined by volumetric Karl Fischer (KF) titration at 25 ± 1 °C with a Karl Fischer titrator DL18 (Mettler-Toledo, Greisensee, Switzerland) applying the one-component technique with CombiTitrant 5 (VWR International, Vienna, Austria). Dry methanol (VWR International, Vienna, Austria) was used as the solvent. The sample solution was injected directly into the Karl Fischer electrolyte cell using a syringe. The amount of the injected sample solution was calculated based on the weight difference of the syringe before and after the sample injection. The standard deviation for KF titration was calculated from three replicate measurements.Results and discussion
The degradation of the SIL [mTBNH][OAc], as of any other IL, is a complex phenomenon that is influenced by various factors. An in-depth understanding of this process is crucial for assessing the economic viability of an IL-based fiber production. The specific fiber processing conditions, such as elevated temperatures, the presence of water, and the use of process chemicals, can accelerate the degradation of ILs and lead to the formation of undesirable degradation products. During fiber spinning, heating is often employed to enable complete dissolution of the cellulosic pulp.6,26 It can trigger thermal degradation of both the IL and the pulp's polysaccharides (cellulose and minor hemicellulose components). Furthermore, the presence of water and pulp constituents can influence the underlying chemical degradation pathways and the extent of IL degradation. Apart from this, the long-term stability of an IL under ambient conditions needs to be thoroughly investigated before scaling up the process: it determines the IL's shelf life and contributes to the overall economic viability of the process. The influence of these factors on the SIL's stability has been explicitly addressed in the following context.Thermal degradation
Amidine (DBN) and guanidine (mTBD) bases exhibit a tendency to undergo hydrolysis already under mild conditions, resulting in the formation of aminolactams and aminoureas, respectively.41 In a recent study, it was demonstrated that this hydrolytic process for the SILs [mTBDH][OAc] and [DBNH][OAc] is accompanied by subsequent acetylation reactions at a relatively moderate temperature (85–105 °C),42 involving the hydrolyzed byproducts and acetic acid derived from the acetate anions. In our study, the objective was to investigate the extent of hydrolytic degradation of the novel SIL [mTBNH][OAc] at different water concentrations. This analysis permits a comparison of its stability against the ILs currently employed in the IONCELL technology, namely [mTBDH][OAc] and [DBNH][OAc]. Apart from the presence of water, also the impact of xylan and cellulose on the thermal stability of [mTBNH][OAc] was addressed.Despite the visual disparities in chromophore formation (different color depths) observed among samples aged under different conditions, the byproducts resulting from hydrolytic degradation of [mTBNH][OAc] were the same across all thermally aged samples (Fig. 2). This consistency was confirmed by NMR and MS measurements. The identified hydrolytic degradation pathways for both isomers are shown in Scheme 1 (detailed peak assignment can be found in the ESI, Fig. S1–S11, Tables S1 and S2†).
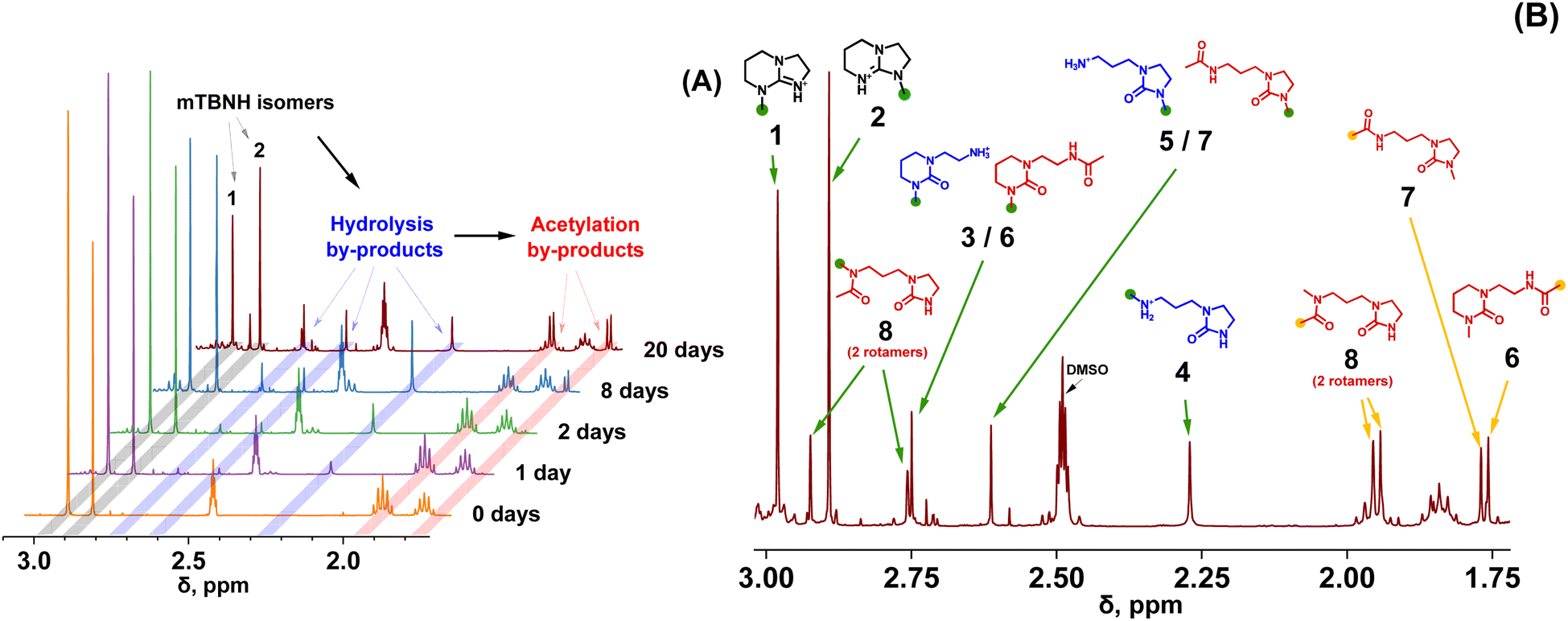 | ||
| Fig. 2 Thermal (hydrolytic) degradation of [mTBNH][OAc] with 10 wt% water at 95 °C monitored at different times by 1H NMR (A). 1H NMR spectra of the IL sample aged 20 days at 95 °C in the presence of 10 wt% water, with assigned degradation products (B). | ||
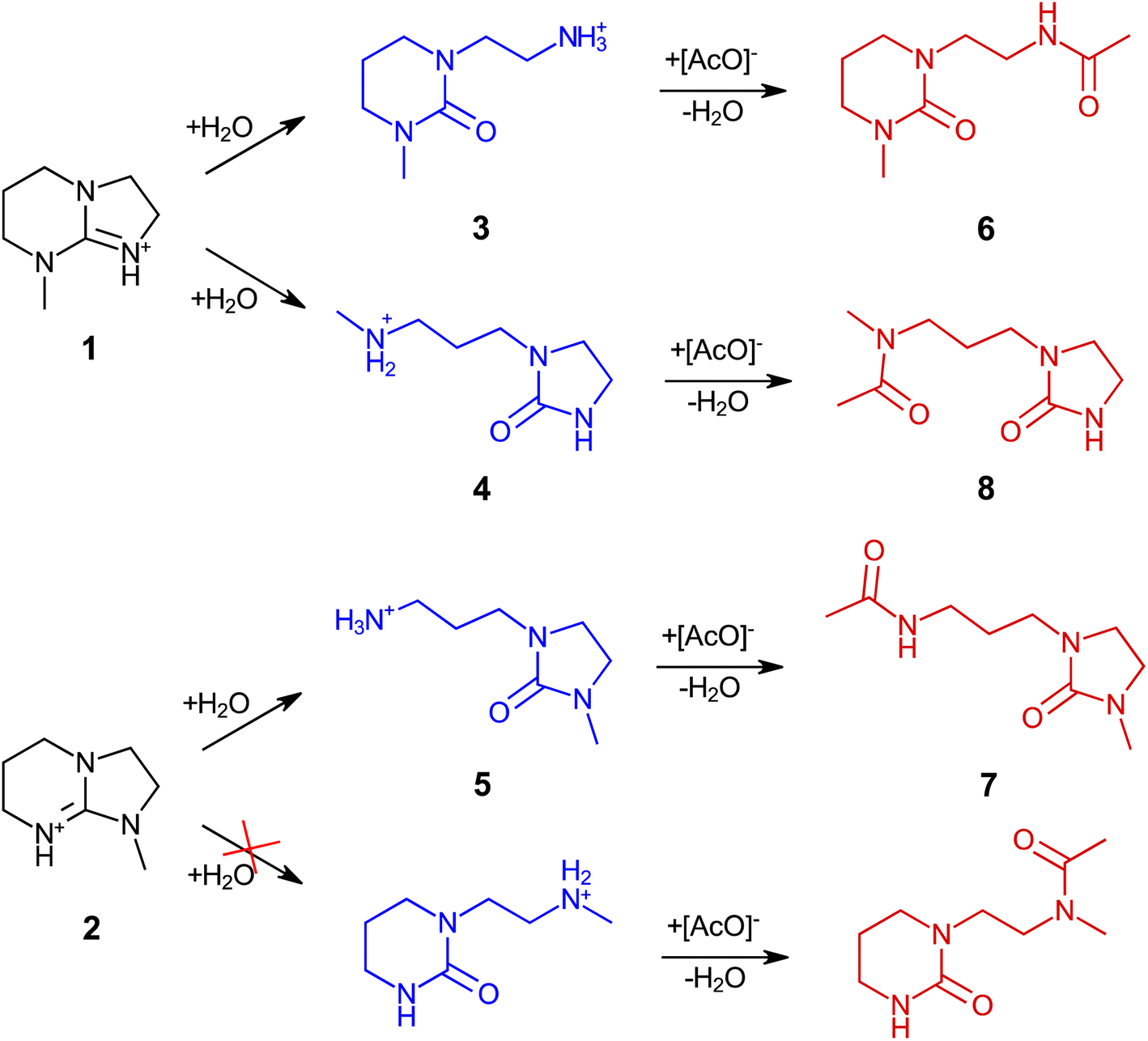 | ||
| Scheme 1 Degradation pathways of the [mTBNH][OAc] isomers during thermal ageing, comprising initial hydrolysis and subsequent acetylation. The degradation pathway leading to the opening of the five-membered ring of isomer B (2) was not observed. | ||
The N-methyl groups in isomers A and B of [mTBNH][OAc] produce distinct, intense singlets in the 1H NMR spectra at 2.98 ppm and 2.89 ppm, respectively, as shown in Fig. 2. Hydrolysis at 95 °C induces ring opening of the guanidine, resulting in the formation of five- or six-membered cyclic urea compounds that contain an additional either primary or secondary amine substituent. This process is reflected in a change of the chemical shift of the N–CH3 singlets. For isomer A, two hydrolysis byproducts are observed at 2.75 ppm (3) and 2.27 ppm (4), whereas for isomer B a single byproduct appears at 2.61 ppm (5). The N-methylated five-membered ring apparently had higher hydrolytic stability than its N-methylated six-membered counterpart.
Traces of acetylation byproducts originating from the primary hydrolysis products (amines 3 and 5) became apparent already after one day of thermal ageing. These byproducts show two characteristic acetyl-CH3 singlets at 1.75 ppm and 1.77 ppm (for 6 and 7, respectively). The N–CH3 peaks of 6 and 7 overlap with those of 3 and 5.
Secondary amines such as N-methylamines are usually more basic than their structurally similar primary parent amines, which translates into a relative dominance of the protonated forms and less free amines being present for N-acylation. Thus, acetylation of secondary amine 4 occurred at a slower rate compared to primary amines, and formation of acetylation byproduct 8 only became evident after 8 days of thermal ageing. Hindered rotation of the acetamide C–N bond in 8 resulted in two atropisomers (rotational isomers). Their occurrence was confirmed by the observation of cross-peaks in NOESY NMR spectra (Fig. S11†) with the two corresponding N–CH3 signals (2.92 ppm and 2.76 ppm). The peaks of the two atropisomeric acetyl-CH3 groups in 8 (1.95 ppm and 1.97 ppm) overlap with a multiplet from aliphatic methylene protons (C–CH2–C) in the six-membered-ring of the guanidine structure of isomer A (1), but nevertheless allow clear distinction, as seen for the 20 day-aged sample in Fig. 2.
Similar to SIL [mTBDH][OAc],42 each isomer of [mTBNH][OAc] may follow one of two pathways leading to the opening of the cyclic guanidine structure (Scheme 1). As mentioned, hydrolytic opening of the N-methylated five-membered ring was not observed. Consequently, isomer B degraded to a much smaller extent than isomer A.
The degree of decomposition was approximated by the N–CH3 peak integrals of the known/identified structures, including peaks for the two mTBNH isomers, the hydrolysis byproducts (3–5), and the acetylation byproducts (6–8). A few additional minor peaks were detected within the N–CH3 spectral range. Their concentration was rather small and the structural elucidation of the corresponding trace products did not succeed. Nevertheless, as their contribution was not considered, the actual degradation values might be slightly higher than those based only on the identified N–CH3 peaks. The results of [mTBNH][OAc] stability investigations are shown in Fig. 3 and 4.
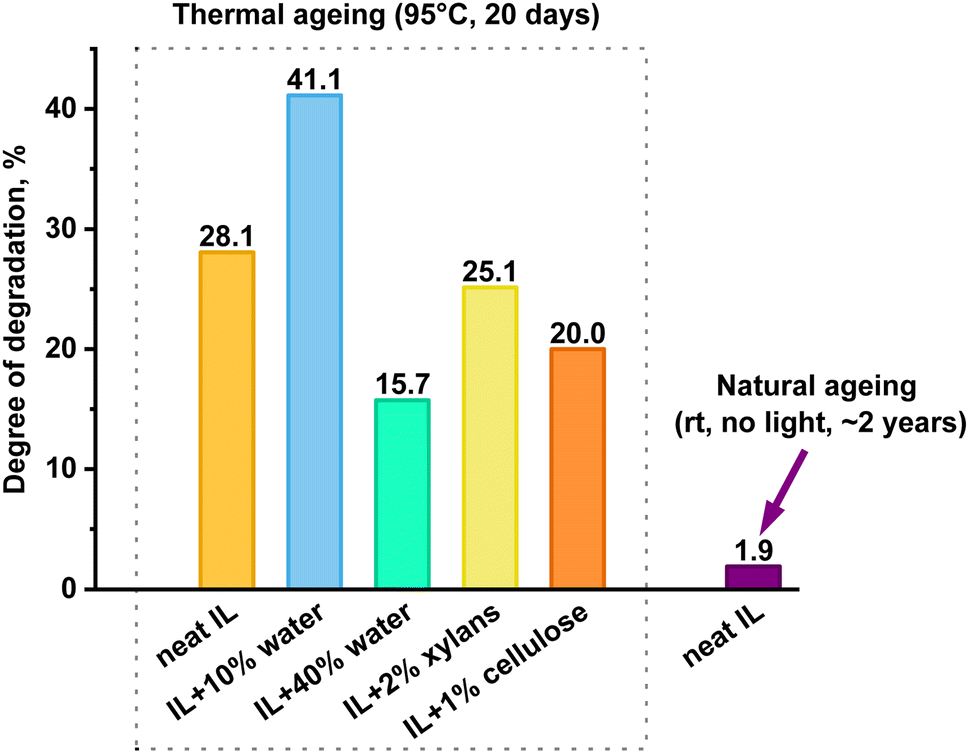 | ||
| Fig. 3 Extent of decomposition of [mTBNH][OAc] in the presence of different additives, calculated from 1H NMR peak integrals. | ||
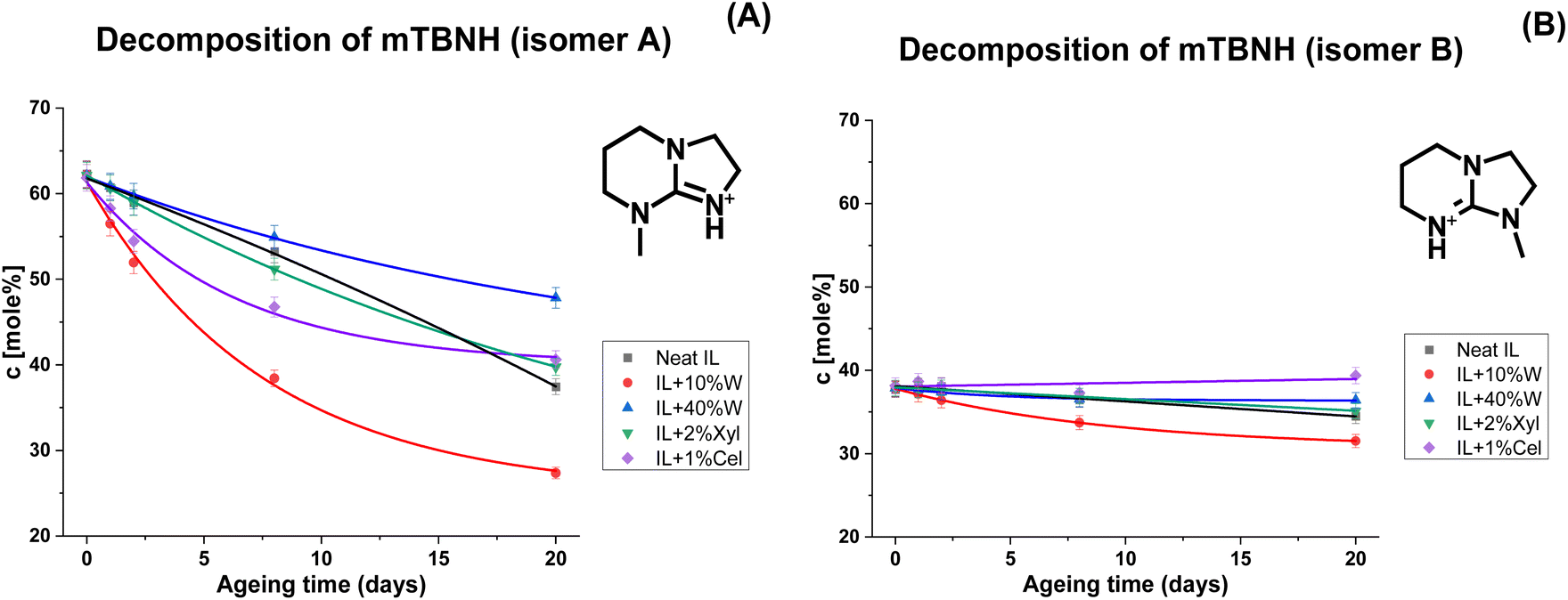 | ||
| Fig. 4 Kinetics of thermal degradation of [mTBNH][OAc] isomers A (A) and B (B) in the presence of different additives, based on 1H NMR measurements. | ||
The presence of wood polysaccharides is a key factor in terms of inducing yellowing, which is discussed in an upcoming account, while they seem to have a slightly stabilizing effect on [mTBNH][OAc] upon hydrolysis (Fig. 3). Water emerged as the most influential factor governing hydrolytic SIL stability. Even minor amounts of water were found to promote the hydrolytic degradation of the IL. It should be noted, however, that SIL hydrolysis is initiated by nucleophilic attack at the bridging carbon atom (C-6), with hydroxy anions being much stronger nucleophiles than the water molecules present. In the organic medium with small amounts of water present, the OH− concentration is the result of a complex interplay of factors. This explains the independence of the hydrolysis rate on the actual water concentration, which was significantly larger in the presence of only 10% water than with 40% water present. Traces of water present in the neat IL sample (0.69 ± 0.04 wt%) are sufficient to promote its hydrolytic degradation at elevated temperatures.
Similar stability studies on [mTBDH][OAc] revealed that the highest degradation rate of ILs was detected in the presence of 10 wt% water,42 which agrees with our present findings. A computational study for mTBD42 utilizing Aspen Plus showed that the combined activity coefficient of water and [mTBDH][OAc] reaches its peak at approximately 10 wt% water.
The position of the N-methyl group in the guanidine structure significantly influences the stability of the corresponding isomers. This is evident from the kinetic curves in Fig. 4 and the NMR spectra observed for the IL sample containing 10 wt% of water (Fig. 2). The A/B isomer ratio, which started at 1.65:1 for the fresh sample, changed to 0.87:1 for the sample aged for 20 days, indicating a much higher degradation rate for isomer A. Exploring alternative approaches for synthesizing [mTBNH][OAc], in a way that substantially favors the formation of isomer B over isomer A, holds significant potential for improving overall thermal stability and, thus, suitability for a lyocell-like spinning process.
During the initial phase of thermal ageing, the presence of cellulose leads to a faster degradation of the IL compared to the neat sample (Fig. 4A), while xylan in the system does not noticeably alter the kinetics of the [mTBNH][OAc] degradation. However, after 20 days of ageing, the extents of degradation for both samples with wood polysaccharides are found to be lower than that of the neat IL (Fig. 3).
The intrinsic thermal instabilities of guanidine and amidine-based SILs result in compositional changes during the process of thermal, evaporative water removal from the washing liquid of the coagulation bath, significantly affecting the solubilization properties of the recycled solvents. [DBNH][OAc] was shown to lose its ability to dissolve cellulose once the concentration of hydrolysis products reached 5 wt%.43 In contrast, [mTBDH][OAc] is capable of dissolving 13 wt% cellulose and ensures a stable fiber spinning process even in the presence of 20 wt% of hydrolysis products.
A comparison of the degradation between [mTBNH][OAc] and the IONCELL SILs ([mTBDH][OAc] and [DBNH][OAc]), as shown in Fig. 5, indicates the superior thermal stability of the former IL under the specified conditions (95 °C, 20 days of ageing, 10 wt% water). However, the limiting extent of degradation that [mTBNH][OAc] can tolerate – while retaining the ability to dissolve a specific quantity of cellulose and to produce high-quality spun fibers – remains a question for further exploration. Notably, under mentioned conditions the commonly used IL [Emim][OAc] didn't exhibit any sign of degradation (Fig. S13†).
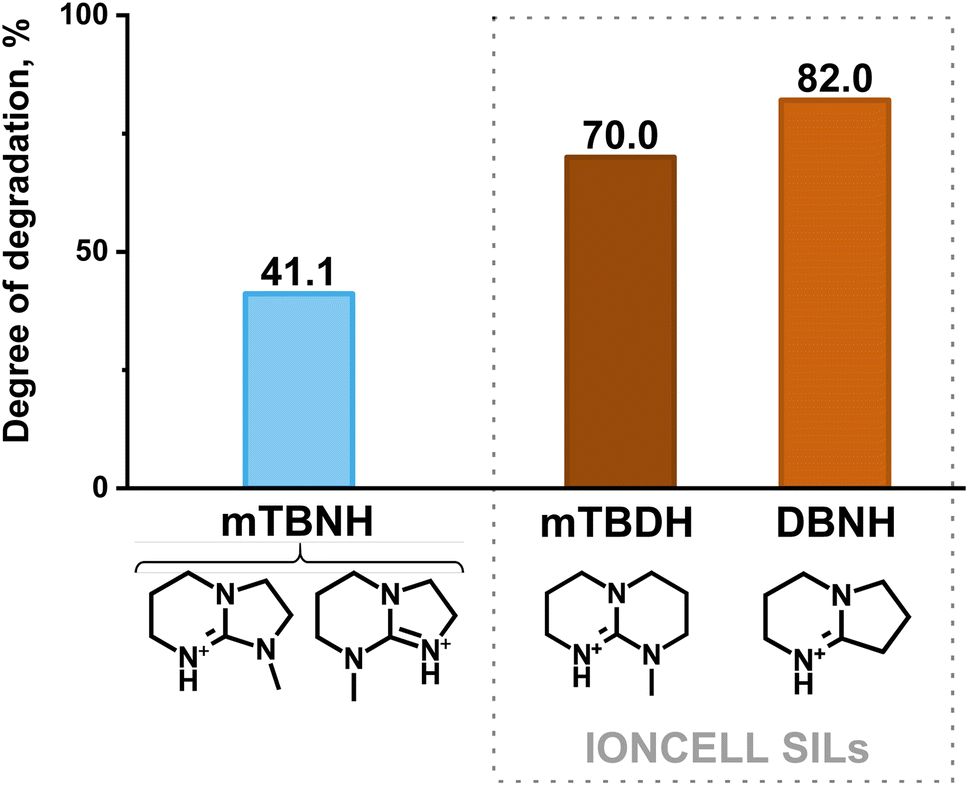 | ||
| Fig. 5 Comparison of the extent of degradation (in % of the starting IL) of [mTBNH][OAc] upon thermal ageing (95 °C, 20 days) in the presence of 10 wt% water (measured in the present work) with IONCELL SILs [mTBDH][OAc] and [DBNH][OAc] (measured by Schlapp-Hackl et al.42). The degree of decomposition of IONCELL SIL [mTBDH][OAc] at 20 days (480 h) was calculated by approximating two experimental results (for 438.23 h – 68.66%, and for 507.92 h – 71.02%), while for [DBNH][OAc] the degradation degree at 480 h was measured.42 | ||
The short-term thermal stability of the investigated ILs was also assessed by TGA; the data are provided in Fig S14 and S15.† The onset temperature (Tonset) for [mTBNH][OAc] was 235 °C. The onset temperatures for IONCELL SILs [mTBDH][OAc] and [DBNH][OAc] were 242 °C (measured in this study) and 182 °C (reported by Kuzmina et al.44), respectively. It is worth noting that values for Tonset obtained from TGA exaggerate the actual long-term thermal stability of SILs, as the actual formation of degradation products starts at significantly lower temperatures.44,45 Moreover, from cellulose fiber production perspective it is more relevant to investigate the stability of IL samples at temperatures close to operating conditions, which is, in the case of [mTBNH][OAc], below 100 °C.
Natural degradation
The degradation extent of the naturally aged sample was determined using 1H NMR spectra based on changes in the peak area ratio between the combined isomer signals (at 2.98 ppm and 2.89 ppm) and the acetate anion (at 1.66 ppm). According to the analysis, [mTBNH][OAc] showed great stability under ambient conditions: the degradation degree remained below 2% after two years of shelf-storage (Fig. 3).However, despite the relatively small extent of natural degradation, it remains crucial to address this process in more detail, as the presence of trace byproducts can cause variability in solvent composition and thus fluctuations in the quality and properties of the resulting spun fibers.
Standard techniques, such as NMR, were not suitable for the detection of minor amounts of impurities in the ppm and ppb range. To analyse degradation products in this concentration range, an SFC-QToF-MS approach was developed instead (Fig. 6). Through the injection of 2 ng of IL, the method allowed for qualitative detection of trace constituents and the accurate determination of their elemental composition with high confidence (>92% certainty) and low divergence from the theoretical mass (<3.2 ppm), as given in Table 1. This provided information on specific chemical changes during the degradation process and on any carry-overs from SIL synthesis.
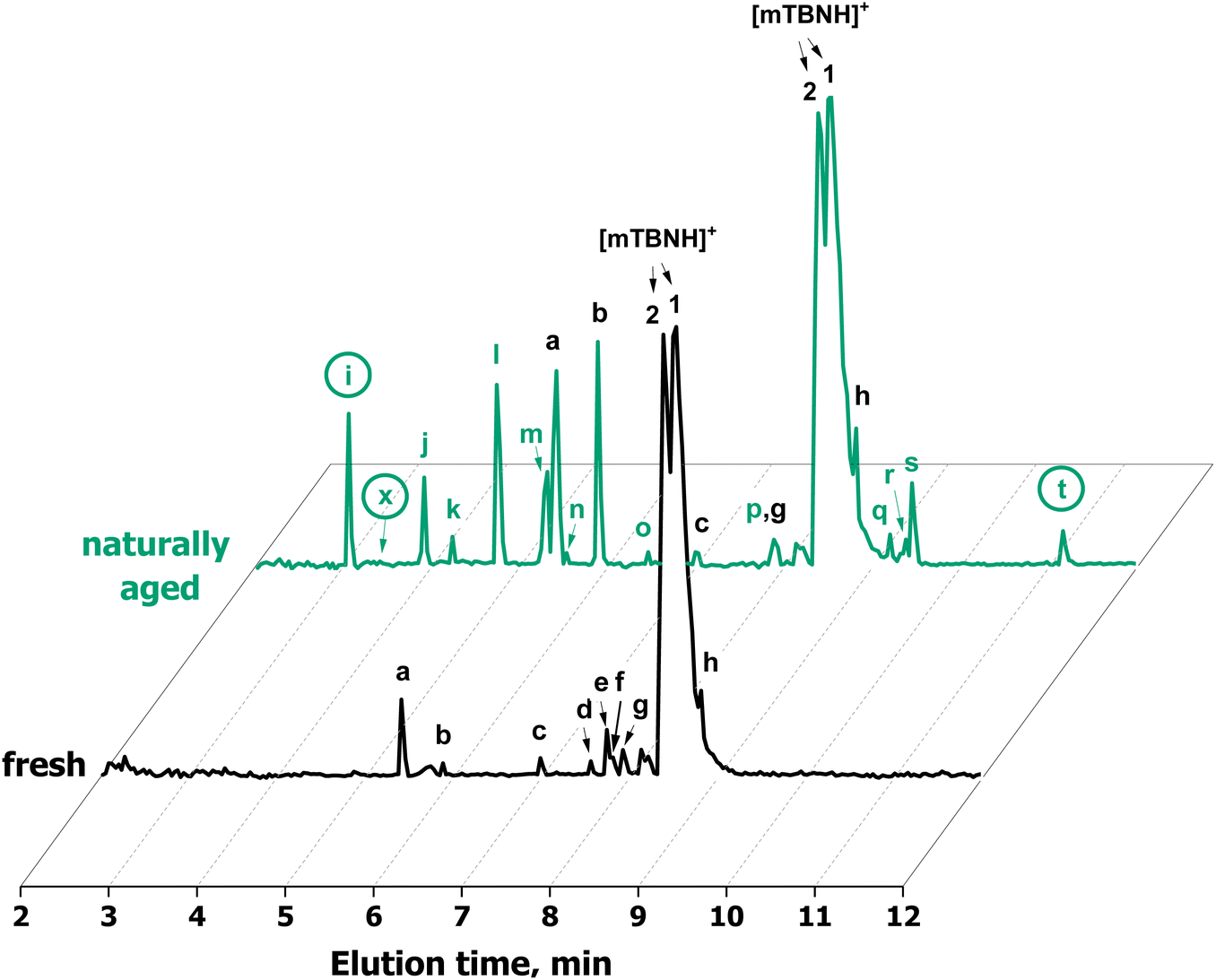 | ||
| Fig. 6 SFC-QToF-MS chromatograms of fresh and aged (RT, absence of light, over 2 years) [mTBNH][OAc] samples. Sample concentration is 400 ppm, injection volume is 5 μL. The circled peaks correspond to structures confirmed with NMR spectroscopy. Peak x corresponds to the compound which was detected only after an isolation with flash column chromatography. | ||
Through SFC-QToF-MS analysis, impurities originating from the synthesis stage (homologs of mTBNH, peaks c–f and h) were detected. Additionally, the high similarity of MS/MS spectra between peak e and g suggests that the former is an oxidation product derived from one of these homologous side products.
Two peaks present in the fresh sample showed significant changes in the intensity during natural ageing: peak a with the elemental composition C7H12N3+ and peak b with the elemental composition C7H12N3O+. This suggests that these two compounds originate from the natural ageing process rather than from the IL synthesis stage.
The scan of the naturally aged IL sample with SFC-QToF-MS showed the formation of new peaks compared to the fresh sample. Based on the elemental composition of these byproducts, which contain 1 or 2 additional oxygen atoms while having the same number or fewer hydrogen atoms, all these compounds constitute oxidation products of the original SIL.
Although QToF-MS is a powerful analytical technique that provides information about the elemental composition of the detected compounds, it has inherent limitations in comprehensive structure elucidation. Therefore, the additional use of complementary techniques, such as NMR, remains essential for establishing the chemical structure. However, NMR analysis requires much larger sample amounts. At the same time, isolation and concentration of degradation products of the IL using conventional approaches pose challenges due to their extremely low concentrations. To address this issue, a small-scale separation protocol was optimised in terms of the best-performing mobile and stationary phases. The method implies the use of micropipette columns followed by the detection of products using SFC-QToF-MS. This approach allowed for better handling of low-concentration samples and facilitated the identification and analysis of the IL degradation products.
Through this approach, the structures of the compounds corresponding to peak i (9, 7-methyl-4,5-dihydro-1,3a,7-triaza-1-indenium-6(7H)-one) and peak t (10; 3-[2-(methylamino)-2-imidazolidinylium-1-yl]propionate) were elucidated (Fig. 6, Table 1, Scheme 2 and Fig. S12†). Both compounds are derivatives of isomer A of mTBNH. The degradation pathways leading to their formation were proposed based on the high-resolution QToF-MS (elemental composition) and NMR results (Scheme 2). According to the proposed mechanism, the compound corresponding to peak t was formed by hydrolytic cleavage of the amide bond of the oxidised byproduct with elemental composition C7H12N3O+, resulting in the opening of the six-membered ring of the bicyclic guanidine structure. The other degradation product (peak i) with the confirmed structure was derived from the oxidised product with elemental composition C7H12N3O2+ (peak p) upon water elimination and concomitant aromatization of the five-membered ring to form an imidazole moiety.
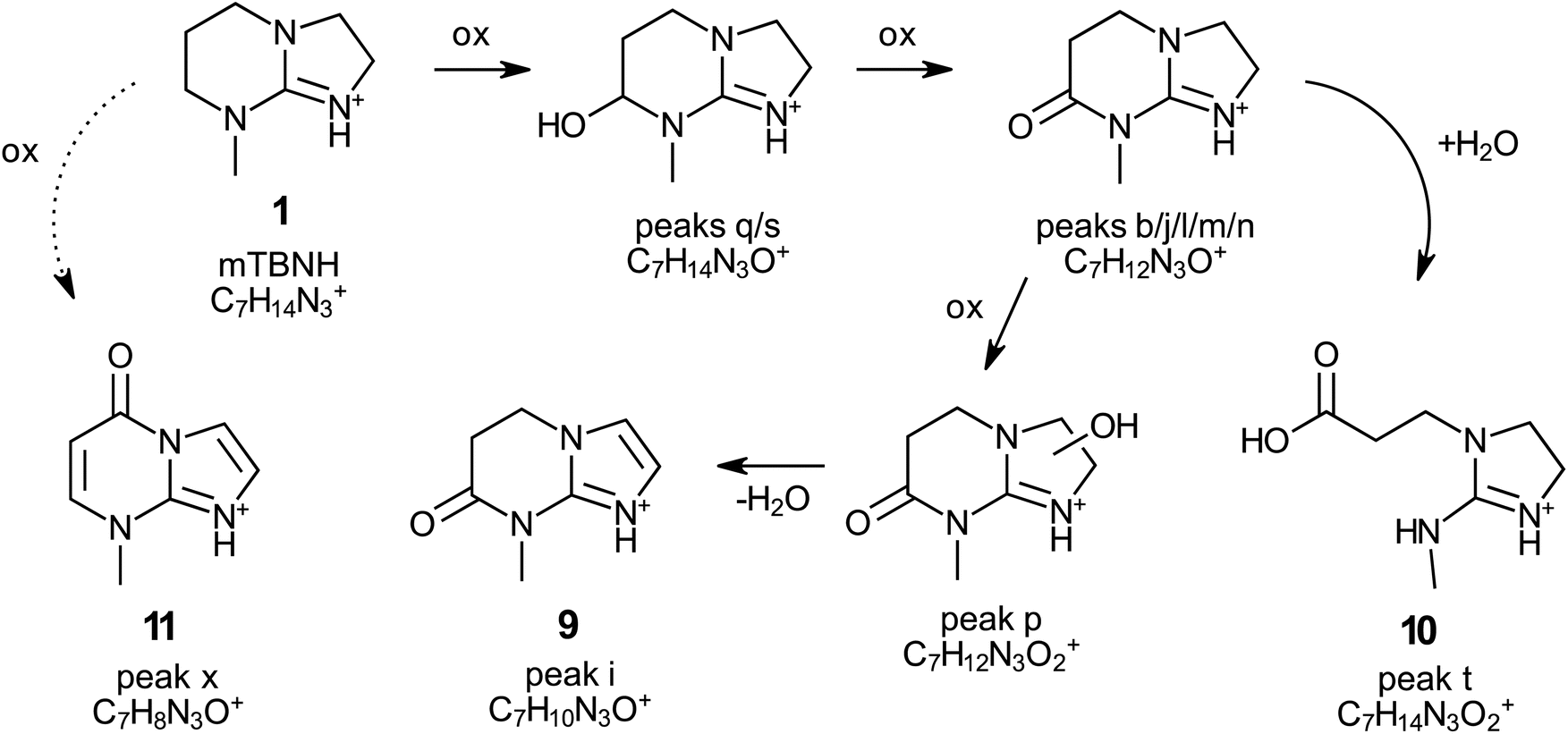 | ||
| Scheme 2 Proposed formation pathway of the products of degradation of [mTBNH][OAc] during natural ageing (rt, no light, over 2 years). Structures of compounds 9–11 were confirmed with NMR (Fig. S12†). All compounds exist as mesomeric structures (not shown in the scheme). | ||
The structure of another compound was revealed by means of NMR analysis after its isolation by flash chromatography (11, peak x, 7-methyl-1,3a,7-triaza-1-indenium-4(7H)-one). The bicyclic structure containing two additional olefinic carbon–carbon bonds and a carbonyl group compared to the mTBNH isomer A indicates that quite severe oxidation occurred. Introduction of the amide oxygen means a formal change of the carbon atom's oxidation number from −2 to ±0, and from −2 to −1 in the case of conversion of an aliphatic methylene carbon to an olefinic one. The olefinic structures are formed by introduction of a hydroxy group to either of the two carbons, followed by water elimination under the formation of the double bond. This elimination process is favoured by conjugation (to the amide carbonyl in the six-membered ring) or aromatization (in the imidazole ring). The absence of a corresponding peak at 3.40 minutes in the chromatogram of the aged sample (Fig. 6) suggests that this specific byproduct exists either in extremely low concentration, or possesses poor ionizability, or perhaps a combination of both factors. However, by utilizing extracted ion chromatography (XIC) a distinct peak of this compound was observed in the naturally aged sample.
While the SFC-QToF-MS method itself is highly sensitive, it should be noted that the number of detected peaks can vary depending on search parameters, particularly when their intensities approach the noise threshold. Considering the complexity of the analysed sample that might contain additional degradation products in trace amounts, we focused our analysis on the most intense peaks. It is worth noting that a more detailed examination of peaks at the noise level could potentially offer more byproduct structures. Nevertheless, given that the overall concentration of all degradation products within the naturally aged sample did not exceed 2% (Fig. 3), this approach allowed us to prioritize the most significant compounds for further investigation. It is safe to state that the major process causing byproduct formation upon natural ageing of the studied SIL is oxidation, most likely in an autoxidation by atmospheric oxygen. The introduction of hydroxy and keto groups at aliphatic methylene positions, as observed, is typical for such a process, and also the confirmation of such autoxidative processes upon ageing of 1,3-dialkylimidazolium ILs46 supports this mechanistic proposal.
Conclusions
In the present work, we investigated the stability of the novel SIL, [mTBNH][OAc], under various conditions. Through NMR measurements, we monitored chemical changes in the IL during thermal treatment at 95 °C for a duration of 20 days, both in the presence of water and wood polysaccharides. Furthermore, we examined the natural ageing of the SIL over a two-year period, employing both NMR and the SFC-QToF-MS methods for analysis.It was shown that the stability of mTBNH is influenced by the position of the N-methyl group in the guanidine bicyclic structure, with isomer B (with the N of the five-membered ring methylated) being more stable. While the presence of wood polysaccharides did not significantly affect the rate or pathway of thermal degradation, the impact of water was substantial. In the presence of 10 wt% water, a maximum degradation extent of 41% was observed by the end of the thermal ageing experiment. Notably, mTBNH demonstrated enhanced thermal stability as compared to common structurally similar SILs, e.g., mTBDH. However, the precise limits of degradation that this IL can withstand while retaining its cellulose-dissolving capacity and ensuring the sustainable production of spun fibers remain to be determined in future application studies. It was demonstrated that the primary hydrolysis (ring opening) products were acetylated in a follow-up step. The chemical structures of the hydrolysis and acetylation product were established and confirmed.
More sensitive analytical methods, capable of detecting very minor quantities of analytes, were required for the study of natural ageing processes of the SIL, due to the small amount of degradation products formed (less than 2% over two years). We successfully utilized SFC-QToF-MS to detect degradation products, determine their elemental composition, and optimize flash chromatography separation conditions for further NMR-based structural elucidation. Our findings suggest that autoxidation plays a significant role in the degradation of [mTBNH][OAc] under storage conditions at room temperature and in the absence of light. These processes cause introduction of carbonyl and hydroxy groups, the latter also being eliminated in subsequent steps to produce olefinic double bonds. These mechanistic insights contribute to our understanding of the stability and degradation mechanisms of this novel class of ionic liquids.
Optimizing the separation conditions and performing flash chromatography purification are necessary for the structural elucidation of a compound by NMR. However, these steps can be extremely time-consuming and laborious. For assessing the stability and purity of the SIL, a rapid scan using SFC-QToF-MS may be sufficient. Moreover, the high sensitivity of the developed method can be employed to investigate crucial parameters of spinning systems, including the detection of residual IL amounts on spun fibers, a critical aspect for optimizing the washing step in the production process.
Author contributions
Ivan Melikhov: investigation, data curation, formal analysis, writing – original draft, writing – review & editing; Irina Sulaeva: data curation, formal analysis, writing – review & editing; Stefano Barbini: investigation, formal analysis, writing – original draft, writing – review & editing; Markus Bacher: data curation, formal analysis; Dev Sriranganadane: formal analysis, investigation; Thomas Rosenau: formal analysis, writing – review & editing; Ilkka Kilpeläinen: resources, writing – review & editing; Antje Potthast: conceptualization, data curation, funding acquisition, project administration, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was developed within the scope of the GRETE project funded by the Bio-Based Industries Joint Undertaking under the European Union’s Horizon 2020 research and innovation program under grant agreement No. 837527 – GRETE – H2020-BBI-JTI-2018. The equipment was kindly provided by the BOKU Core Facility ALICE. The BOKU doctoral school ABC&M and the Austrian Biorefinery Center Tulln (ABCT-II) are gratefully acknowledged for their support. Dr Jan Janesh is gratefully acknowledged for his support with TGA analysis and DI Nadine Kohlhuber for the assistance with water content analysis.References
- D. Eichinger, Lenzinger Ber., 2012, 90, 1–7 Search PubMed.
- S. Rana, S. Pichandi, S. Parveen and R. Fangueiro, in Roadmap to Sustainable Textiles and Clothing: Eco-Friendly Raw Materials, Technologies, and Processing Methods, ed. S. S. Muthu, Springer Singapore, Singapore, 2014, pp. 239–276 Search PubMed.
- E. M. Kalliala and P. Nousiainen, Autex Res. J., 1999, 1, 8–20 CrossRef.
- A. K. Chapagain, A. Y. Hoekstra, H. H. G. Savenije and R. Gautam, Ecol. Econ., 2006, 60, 186–203 CrossRef.
- D. P. Kelly and N. A. Smith, in Advances in Microbial Ecology, ed. K. C. Marshall, Springer US, Boston, MA, 1990, pp. 345–385 Search PubMed.
- T. Rosenau, A. Potthast, H. Sixta and P. Kosma, Prog. Polym. Sci., 2001, 26, 1763–1837 CrossRef CAS.
- T. Rosenau, A. Potthast, I. Adorjan, A. Hofinger, H. Sixta, H. Firgo and P. Kosma, Cellulose, 2002, 9, 283–291 CrossRef CAS.
- F. Haemmerle, Lenzinger Ber., 2011, 89, 12–21 CAS.
- C. Graenacher, US Pat., 1924238, 1933 Search PubMed.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- R. M. Wahlström and A. Suurnäkki, Green Chem., 2015, 17, 694–714 RSC.
- A. Fukuoka and P. L. Dhepe, Angew Chem. Int. Ed. Engl., 2006, 45, 5161–5163 CrossRef CAS PubMed.
- C. Li, Z. Zhang and Z. K. Zhao, Tetrahedron Lett., 2009, 50, 5403–5405 CrossRef CAS.
- H. Ren, Y. Zhou and L. Liu, Bioresour. Technol., 2013, 129, 616–619 CrossRef CAS PubMed.
- F.-F. Wang, J. Liu, H. Li, C.-L. Liu, R.-Z. Yang and W.-S. Dong, Green Chem., 2015, 17, 2455–2463 RSC.
- G. Ebner, S. Schiehser, A. Potthast and T. Rosenau, Tetrahedron Lett., 2008, 49, 7322–7324 CrossRef CAS.
- M. T. Clough, K. Geyer, P. A. Hunt, S. Son, U. Vagt and T. Welton, Green Chem., 2015, 17, 231–243 RSC.
- T. Zweckmair, H. Hettegger, H. Abushammala, M. Bacher, A. Potthast, M.-P. Laborie and T. Rosenau, Cellulose, 2015, 22, 3583–3596 CrossRef CAS.
- H. Abushammala, H. Hettegger, M. Bacher, P. Korntner, A. Potthast, T. Rosenau and M.-P. Laborie, Cellulose, 2017, 24, 2767–2774 CrossRef CAS.
- H. Sixta, A. Michud, L. Hauru, S. Asaadi, Y. Ma, A. W. T. King, I. Kilpeläinen and M. Hummel, Nord. Pulp Pap. Res. J., 2015, 30, 43–57 CrossRef CAS.
- X. Li, H. Li, Z. Ling, D. Xu, T. You, Y.-Y. Wu and F. Xu, Macromolecules, 2020, 53, 3284–3295 CrossRef CAS.
- X. Niu, S. Huan, H. Li, H. Pan and O. J. Rojas, J. Hazard. Mater., 2021, 402, 124073 CrossRef CAS PubMed.
- T. Kakko, A. W. T. King and I. Kilpeläinen, Cellulose, 2017, 24, 5341–5354 CrossRef CAS.
- S. Asaadi, M. Hummel, S. Hellsten, T. Härkäsalmi, Y. Ma, A. Michud and H. Sixta, ChemSusChem, 2016, 9, 3250–3258 CrossRef CAS PubMed.
- Y. Ma, M. Hummel, I. Kontro and H. Sixta, Green Chem., 2018, 20, 160–169 RSC.
- A. Michud, M. Tanttu, S. Asaadi, Y. Ma, E. Netti, P. Kääriainen, A. Persson, A. Berntsson, M. Hummel and H. Sixta, Text. Res. J., 2016, 86, 543–552 CrossRef CAS.
- I. S. F. Mendes, A. Prates and D. V. Evtuguin, Carbohydr. Polym., 2021, 273, 118466 CrossRef CAS PubMed.
- X. Jiang, Y. Bai, X. Chen and W. Liu, J. Bioresour. Bioprod., 2020, 5, 16–25 CrossRef.
- E. Spönla, S. Hannula, T. Kamppuri, U. Holopainen-Mantila, I. Sulaeva, A. Potthast, A. Harlin, S. Grönqvist and J. Rahikainen, Cellulose, 2023, 30, 11407–11423 CrossRef.
- M. Sturm, J. K. J. Helminen, A. Honkasalo, A. Roselli, N. Von Weymarn and I. Kilpeläinen, J. Appl. Polym. Sci., 2023, 140, e53901 CrossRef CAS.
- P. Ahokas, A. Khakalo, A. Jaiswal, T. Koso, A. W. T. King, E. Spönla, A. Harlin and H. Orelma, ACS Sustain. Chem. Eng., 2023, 11, 15919–15930 CrossRef CAS.
- T. Kamppuri, J. Rahikainen, S. Hannula, E. Spönla, A. Potthast, I. Sulaeva, S. Grönqvist and A. Harlin, presented in part at NWBC 2022: The 10th Nordic Wood Biorefinery Conference, Helsinki, Finland, October, 2022. Search PubMed.
- S. Hannula, Master’s thesis (tech), University of Oulu, 2021.
- M. A. R. Martins, F. H. B. Sosa, I. Kilpeläinen and J. A. P. Coutinho, Fluid Phase Equilib., 2022, 556, 113414 CrossRef CAS.
- F. H. B. Sosa, P. J. Carvalho and J. A. P. Coutinho, Ind. Eng. Chem. Res., 2022, 61, 14626–14636 CrossRef CAS PubMed.
- F. H. B. Sosa, I. Kilpeläinen, J. Rocha and J. A. P. Coutinho, Fluid Phase Equilib., 2023, 573, 113857 CrossRef CAS.
- A. R. Todorov, A. W. T. King and I. Kilpeläinen, RSC Adv., 2023, 13, 5983–5992 RSC.
- Z. Xue, L. Qin, J. Jiang, T. Mu and G. Gao, Phys. Chem. Chem. Phys., 2018, 20, 8382–8402 RSC.
- B. Wang, L. Qin, T. Mu, Z. Xue and G. Gao, Chem. Rev., 2017, 117, 7113–7131 CrossRef CAS PubMed.
- C. Maton, N. De Vos and C. V. Stevens, Chem. Soc. Rev., 2013, 42, 5963 RSC.
- A. M. Hyde, R. Calabria, R. Arvary, X. Wang and A. Klapars, Org. Process Res. Dev., 2019, 23, 1860–1871 CrossRef CAS.
- I. Schlapp-Hackl, J. Witos, K. Ojha, P. Uusi-Kyyny, V. Alopaeus and H. Sixta, Ind. Eng. Chem. Res., 2022, 61, 259–268 CrossRef CAS.
- S. Elsayed, B. Viard, C. Guizani, S. Hellsten, J. Witos and H. Sixta, Ind. Eng. Chem. Res., 2020, 59, 20211–20220 CrossRef CAS.
- O. Kuzmina, J. Bhardwaj, S. R. Vincent, N. D. Wanasekara, L. M. Kalossaka, J. Griffith, A. Potthast, S. Rahatekar, S. J. Eichhorn and T. Welton, Green Chem., 2017, 19, 5949–5957 RSC.
- Y. Huang, Z. Chen, J. M. Crosthwaite, S. N. V. K. Aki and J. F. Brennecke, J. Chem. Thermodyn., 2021, 161, 106560 CrossRef CAS.
- H. Hettegger, J. Zhang, M. Koide, U. Rinner, A. Potthast, Y. Gotoh and T. Rosenau, Fibers, 2022, 10, 50 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00087k |
| This journal is © The Royal Society of Chemistry 2024 |