
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Membrane-immobilized transaminases for the synthesis of enantiopure amines†
Hippolyte
Meersseman Arango
a,
Xuan Dieu Linh
Nguyen
a,
Patricia
Luis
b,
Tom
Leyssens
 a,
David
Roura Padrosa
c,
Francesca
Paradisi
c and
Damien P.
Debecker
*a
a,
David
Roura Padrosa
c,
Francesca
Paradisi
c and
Damien P.
Debecker
*a
aInstitute of Condensed Matter and Nanosciences (IMCN), Université catholique de Louvain (UCLouvain), Place Louis Pasteur, 1, 1348 Louvain-La-Neuve, Belgium. E-mail: damien.debecker@uclouvain.be
bMaterials & Process Engineering (iMMC-IMAP), Université catholique de Louvain (UCLouvain), Place Sainte Barbe 2, 1348 Louvain-la-Neuve, Belgium
cDepartment of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, Bern, Switzerland
First published on 19th September 2024
Abstract
For the manufacture of enantiopure amines, greener synthesis processes are needed. Transaminases (TAs) are able to produce chiral amines with excellent enantioselectivity and in mild conditions, and can be immobilized to target stability, recoverability, and reusability. In the perspective of process intensification, we propose to study TA immobilization onto polymeric membranes. Two main immobilization strategies were investigated, requiring prior membrane surface functionalization. On the one hand, a polyacrylonitrile (PAN) membrane surface was partially hydrolyzed and coated with polyethyleneimine (PEI) to electrostatically trap TAs. On the second hand, a polypropylene (PP) membrane was coated with polydopamine (PDA), which was subsequently modified with glycerol diglycidyl ether (GDE) in order to covalently graft TAs. The successful membrane functionalization was confirmed by surface characterization techniques (infrared spectroscopy, X-ray photoelectron spectroscopy, contact angle measurements, and scanning electron microscopy). Enzyme leaching was observed from the functionalized PAN membrane, highlighting the need to post-treat the reversibly immobilized TAs to improve their anchoring. The covalent coupling of TAs with PEI using glutaraldehyde (GA) was found highly effective to avoid leaching and to increase the enzyme loading, without affecting the specific activity of the biocatalyst. Similarly, the covalent grafting of TA onto functionalized PP membranes yielded very efficient biocatalysts (retaining 85% specific activity with respect to soluble TA) displaying perfect recyclability throughout successive cycles. Immobilizing either the S-selective HeWT or the R-selective TsRTA resulted in robust heterogeneous biocatalysts with antagonist enantioselectivities. Thus, chiral amine synthesis can be performed effectively with biocatalytic membranes, which paves the way to intensified continuous flow synthesis processes.
Sustainability spotlightChiral amines are essential building blocks for the manufacture of commercial drugs. Their current synthesis via multi-step batch processes catalysed by organometallic homogeneous catalysts is associated with a high e-factor. Alternatively, transaminases are able to enantioselectively produce chiral amines in mild conditions. Yet, biocatalytic routes face unfavourable thermodynamics, poor stability, and high cost. Current efforts aim at designing robust heterogenized biocatalysts amenable to continuous processes. Here, we report effective methods to immobilize transaminases onto macroporous polymeric membranes. The biocatalytic membranes show high activity and are fully reusable. They can even operate without additional co-factor. These new objects are well-suited for upcoming intensified hybrid flow processes, concatenated with separation. Research on greener synthesis organic synthesis aligns with UN SDG #3 and #12. |
1 Introduction
Chemical processes dedicated to the production of active pharmaceutical ingredients (APIs) are known to generate significant amount of waste per unit mass of product (high e-factor).1 The specific case of the synthesis of chiral amines, which are essential building blocks for the pharmaceutical industry,2–4 is a prominent illustration of this issue. Industrially, the synthesis of chiral amines is operated via multi-step batch processes which usually feature low overall yield, produce large amounts of waste, and are energy-intensive. They are typically catalysed by organometallic homogeneous catalysts based on toxic and depleted heavy metals (Ru, Rh, Pd) which usually operate at relatively high temperature, are not 100% enantioselective, and are difficult to recover.5,6 In this context, it is of particular interest to develop more sustainable chiral amine synthesis methods.7,8Biocatalytic routes have gained considerable attention in the last decades as potentially effective and sustainable alternatives. Remarkably, amine transaminases (TAs) catalyse the direct synthesis of chiral amines from pro-chiral ketones, using cheap and readily available amino donors (e.g. amino-acids) through transamination, with excellent enantioselectivity and in mild conditions. TAs are catching the eye as tremendous achievements have been made recently, both at the fundamental and applied levels.9–17 Industrial applications of biocatalytic transamination, however, remain scarce for TAs since they are usually employed as free enzymes in solution, which display limited stability. Batch processes utilizing such free enzymes do not allow easy catalyst separation, recovery, and reuse.18–20 Thus, immobilization strategies are often proposed.21–24 Additionally, thermodynamic limitations and substrate/product inhibitions tend to limit the applicability of transaminases in asymmetric synthesis of enantiopure amines.25
To overcome these limitations, scientists aim at enhancing the TA robustness and at developing equilibrium shifting strategies. The first point can be achieved through enzyme immobilization, as the resulting heterogeneous biocatalysts are often more versatile and amenable to more productive flow processes. The second point usually relies on using a large amino donor excess or on consuming/removing the (co)product during reaction.26,27 Besides the widely reported multi-enzymatic cascade reactions28 or non-catalytic consecutive reactions29 that can be used to push the equilibrium of the transamination reaction towards the production of the target amine, one alternative possibility is the physical separation of one of the transamination products towards another phase in the system. For example, in situ (co)product removal (ISPR) strategies were recently employed in batch with free transaminases to drive the reaction towards the formation of valuable chiral molecules.30,31 In these examples, the acetophenone co-product was removed from the aqueous phase reaction medium by liquid–liquid extraction (using an organic co-solvent), or the targeted chiral amine was selectively crystallized by salt formation.
When aiming to perform such reactions in continuous flow, possibly coupled with product separation, membrane technologies can be of particular interest.32–34 Membrane contactors are known to offer operational flexibility, large and tunable interfacial area, modular linear scale-up which allows easy concatenation with other operations, compactness, and low energy consumption. Therefore, researchers have implemented membrane contactors at the outlet of the transamination flow reactor to separate their outputs.35–37 In these processes, membranes are solely employed as separation unit for downstream processing and the transaminases are immobilized separately (onto classical supports) and packed into distinct fixed-bed reactors.
Taking this to the next level, it would be of particular interest to immobilize enzymes directly onto active membrane supports, and hence to develop bifunctional membranes allowing to simultaneously host the immobilized enzymes and perform the product separation to intensify the transamination process. The immobilization of enzymes onto polymeric membranes has already been reported with lipases, carbonic anhydrase, and glucose oxidases.38–43 Recently, Howdle et al. developed an electrospun polycarvone acrylate di-epoxide/polyvinylidene fluoride (PCADE/PVDF) membrane and exploited it for the immobilization of the TA from Halomonas elongata (HeWT).44 This epoxy-functionalized membrane allowed 61.9% immobilization yield and 43.6% of specific activity recovery (no TA leaching), paving the way for potential application in combined reaction-separation processes.
In the perspective of designing effective hybrid chemical processes (i.e. combining reaction on immobilized enzymes and in situ separation through a membrane), it is essential to first master the step of enzyme immobilization on conventional polymeric membranes that are routinely employed industrially. Such supports differ from usual enzyme carriers such as porous silica, or resins beads (i.e. typically 100 μm particles, with average pore size of 20–60 nm (ref. 45)), in the sense that polymeric membranes tend to display lower specific surface area available for immobilization,46,47 resulting in potentially lower enzyme loadings.48 Also, their surface is usually not directly amenable to enzyme grafting, so that chemical functionalization is needed. Thus, it is of prime importance to develop robust enzyme immobilization strategies on these membranes, with the aim to optimize enzyme loading, preserve specific activity of immobilized enzymes, and avoid leaching.
In this context, we turned our attention to the immobilization of two transaminases (the S-selective TA from Halomonas elongata (HeWT)49 and the R-selective TA from Thermomyces stellatus (TsRTA)50) onto commercially available polymeric microporous membranes. Polyacrylonitrile membranes (PAN) and polypropylene (PP) were selected as commercially available and industrially relevant membranes showing good mechanical resistance and featuring respectively hydrophilic and hydrophobic surface chemistry. We leverage electrostatic interactions and covalent grafting strategies to avoid leaching. The membrane carriers are characterized at different stages of the preparation. After TA immobilization, using a model kinetic resolution, we show that these functional materials exhibit high catalytic performance (specific activity), minor leaching and excellent reusability. This paves the way to a future use in flow mode hybrid processes, possibly concatenated with purification strategies.
2 Experimental
2.1 Materials
4′-Bromoacetophenone (BAP; ≥98%), R-4-bromo-α-methylbenzylamine (R-BMBA; 99%), S-4-bromo-α-methylbenzylamine (S-BMBA; 99%), hydrochloric acid (37 wt%, aqueous solution), pyridoxal 5′-phosphat]e hydrate (PLP; ≥98%), 4-bromo-α-methylbenzylamine (rac-BMBA; 98%), dimethylsulfoxide (DMSO; ≥99.9%), glycerol diglycidyl ether (GDE; technical grade), carbonate-bicarbonate buffer capsules, Bradford reagent were purchased from Sigma-Aldrich. Sodium hydroxide (NaOH; ≥99%), N-2-hydroxyethylpiperazine-N′-2-ethane sulphonic acid (HEPES; ≥ 99.5%), HEPES sodium salt (≥99%), pyruvic acid sodium salt (≥99.9%), 2-(N-morpholino)-ethane sulphonic acid sodium salt (MES sodium salt; ≥99%), glutaraldehyde (GA; 25 wt% aqueous solution) were purchased from Carl Roth. Branched polyethyleneimine (PEI; 50 wt%, aqueous solution, M.N. 60,000) was purchased from Acros Organics. Dichloromethane (HPLC grade) and ethanol (absolute) were purchased from VWR Chemicals. 1-Phenyl-2-propanol (>98%), tris(hydroxymethyl)aminomethane (tris; >99%), 3-hydroxytyramine hydrochloride (dopamine hydrochloride; 98%), tert-butyl methyl ether (MTBE; >99%), 3,3-diphenylpropionic acid (3-DPPA; 97%) were purchased from Tokio Chemical Industry. Commercial polyacrylonitrile membranes (PAN) were purchased from Snyder filtration company (USA). Commercial polypropylene membranes (PP) were purchased from 3 M (USA). Transaminases HeWT (from Halomonas elongata) and TsRTA (from Thermomyces stellatus) were expressed and lyophilized as previously described by Paradisi et al.,49,50 and then used as cell-free extracts. Distilled water was applied for all synthesis and treatment processes.2.2 Transaminase immobilization onto polyacrylonitrile membrane (PAN)
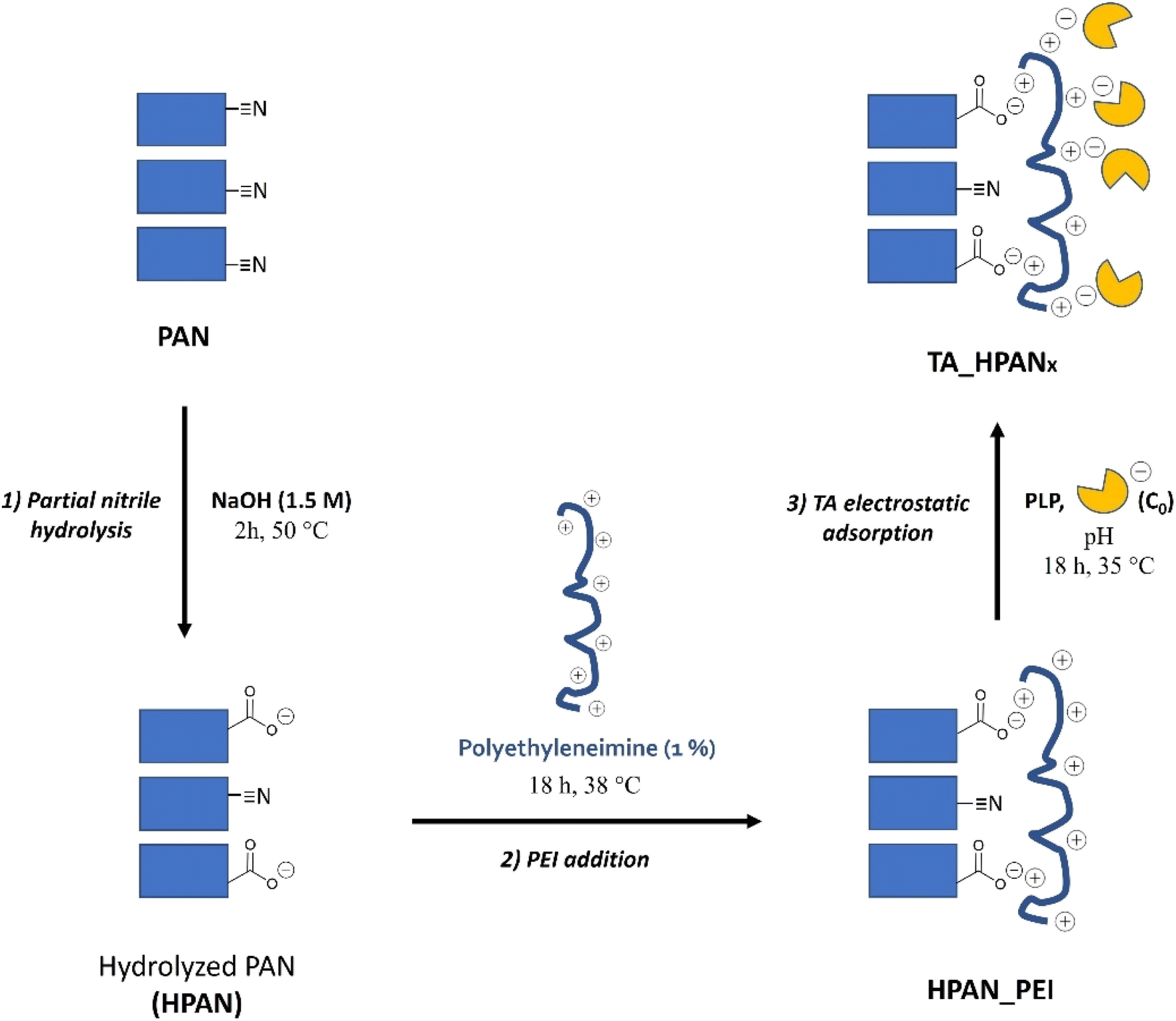 | ||
| Fig. 1 Schematic representation of PAN functionalization and TA immobilization procedures, leading to TA_HPAN_xa or TA_HPAN_xb. In the case of TA_HPAN_2a, TA_HPAN_2b, TA_HPAN_3a and TA_HPAN3b, an additional post-treatment was applied (with SA or GA, respectively) before the rinsing step. | ||
This TA immobilization was performed on each PAN membrane support employed in this study (i.e. PAN, HPAN, HPAN_PEI), in order to evaluate the impact of the different steps of functionalization on the catalytic performance of the resulting immobilized TAs. Depending on the immobilization pH, TAs immobilized on pristine PAN were labelled as TA_PANa (if pH was 8) or TA_PANb (if pH was 5.5). Similarly, TAs immobilized on HPAN were labeled as TA_HPANxa or TA_HPANxb, where x =/stand for TAs immobilized on HPAN, x = 1 for HPAN_PEI (without post-treatment), x = 2 for HPAN_PEI (with SA post-treatment) and x = 3 HPAN_PEI (with GA post-treatment), respectively.
2.3 Transaminase immobilization onto polypropylene membrane (PP)
Second, the obtained PP_PDA membrane was modified with a bisepoxide coupling agent (glycerol diglycidyl ether; GDE) to confer an appropriate linker arm for the subsequent covalent grafting of the enzyme56 (Fig. 2, step 1). The PP_PDA was immersed in 50 mL of a 100 mg per mL GDE solution (in ethanol) and stirred for 18 hours (unless stated otherwise) at room temperature. The resulting PP_PDA_GDE membrane was then rinsed with 50 mL of ethanol for 1 hour first, then with 100 mL distilled water for 1 hour (repeated three times).
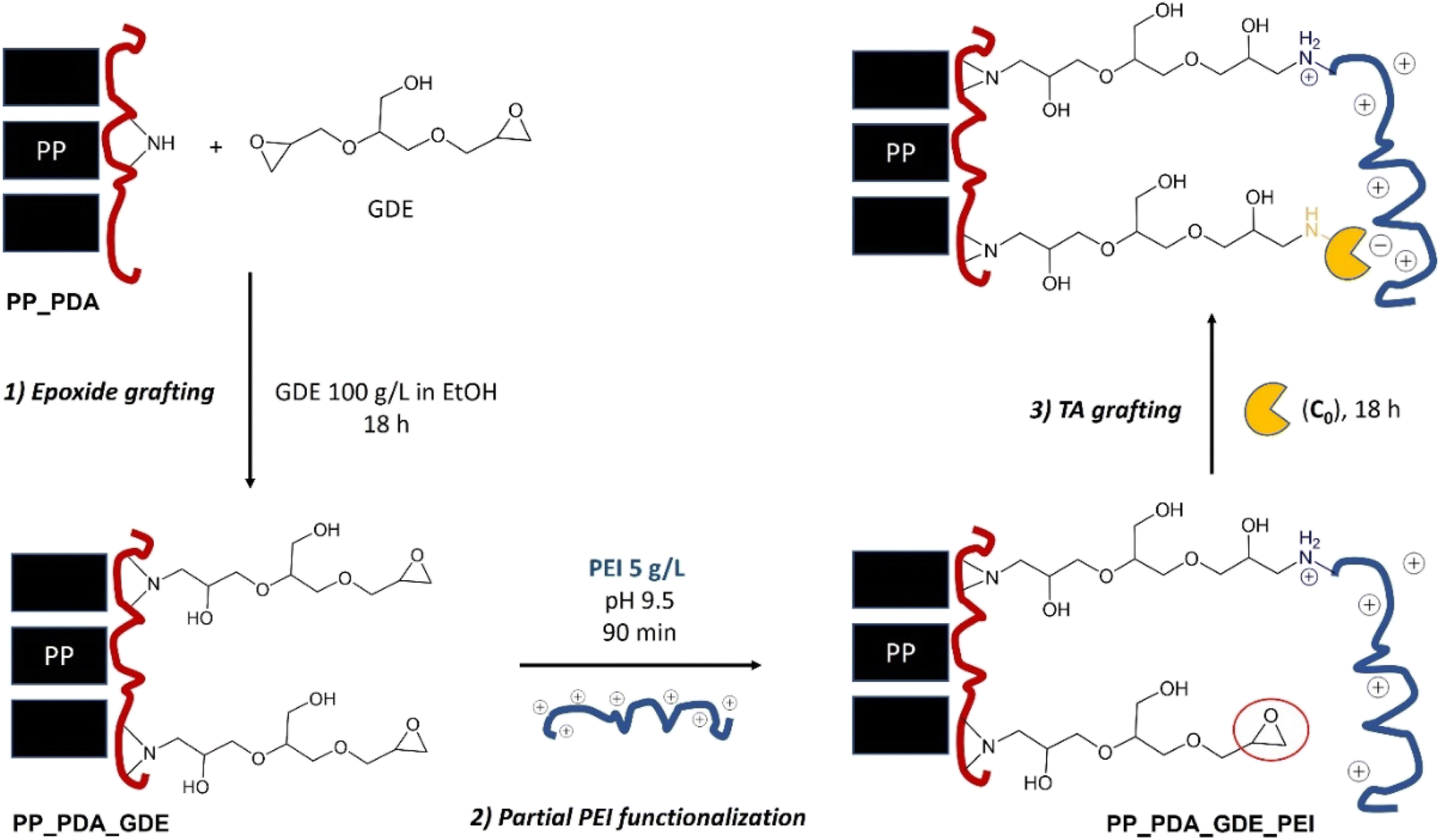 | ||
| Fig. 2 Schematic representation of PP_PDA functionalization and TA immobilization procedures on PP-based membranes, leading to TA_PPy catalysts. In some cases, the protocole was modified by adding an additional step (i.e. PLP immobilization) after the third step. | ||
Third, in order to drive the covalent grafting of the transaminase on the epoxy linker arm, the PP_PDA_GDE membrane was partially functionalized with polyethyleneimine (prior to enzyme immobilization; Fig. 2, step 2). Thus, the PP_PDA_GDE was immersed into 50 mL of a 5 mg per mL PEI solution in carbonate/bicarbonate 0.1 M buffer at pH 9.5 and stirred for 90 minutes (unless stated otherwise) at room temperature.
In a variation of this protocol, we attempted to prepare self-sufficient biocatalysts. Inspired López-Gallego et al.,57 we immobilized the enzyme onto PP_PDA_GDE_PEI (with either 0.1 mM or 1 mM PLP, sodium pyruvate 10 mM in HEPES 0.1 M buffer pH 8) and then rinsed the resulting membrane three time (5 mL sodium pyruvate 10 mM in HEPES 0.1 M buffer pH 8), and directly incubated it with PLP (1 mM in HEPES 10 mM pH 8 for 90 minutes at room temperature under gentle stirring). Additional rinsing was applied again (four times 5 mL sodium pyruvate 10 mM in HEPES 0.1 M buffer pH 8, 30 minutes). The obtained membranes were denoted TA_PP3_SSz, where z is the concentration of PLP (in mM) used during the TA immobilization step. The amount of PLP effectively loaded onto the membrane was evaluated by UV absorption (see ESI†).
2.4 Characterization of the membrane carriers
Both pristine and functionalized PAN and PP membranes were characterized by infrared spectroscopy, X-ray photoelectron spectroscopy, contact angle measurements, and scanning electron microscopy. Experimental details on these characterization techniques are provided as ESI.†2.5 Characterization of enzyme-loaded membranes
Enzyme loadings on the membranes were evaluated by mass balance. Typically, a functionalized membrane was incubated in a 5 mL (V0) TA solution of known concentration, C0 [mg mL−1]. After immobilization, the membrane was removed from the reactor and the TA concentration in the remaining solution (C1) was measured by Bradford titration (see ESI†). The membrane was then washed with 5 mL (V0) of rinsing solution, and the enzyme concentration in the resulting solution was measured (C2). The same procedure was applied to the next rinsing solutions, leading to measure C3 and C4. The immobilized enzyme loading (L) was determined by eqn (1). The immobilization yield (%) is defined as the ratio between immobilized TA (L) and the total TA mass introduced during the immobilization (5 × C0).| L = V0 × (C0 − C1 − C2 − C3 − C4) [mg] | (1) |
2.6 Biocatalytic testing
The kinetic resolution of BMBA was used as a model reaction to assess the catalytic activity of free and membrane-immobilized transaminases. Racemic BMBA was reacted with pyruvate, to produce BAP and either L-or D-alanine leaving unreacted R- or S-BMBA when using an S- or a R-selective TA, respectively (Fig. 3). When catalytic tests were run with immobilized transaminases, one disk of 5 cm2 of membrane support was employed.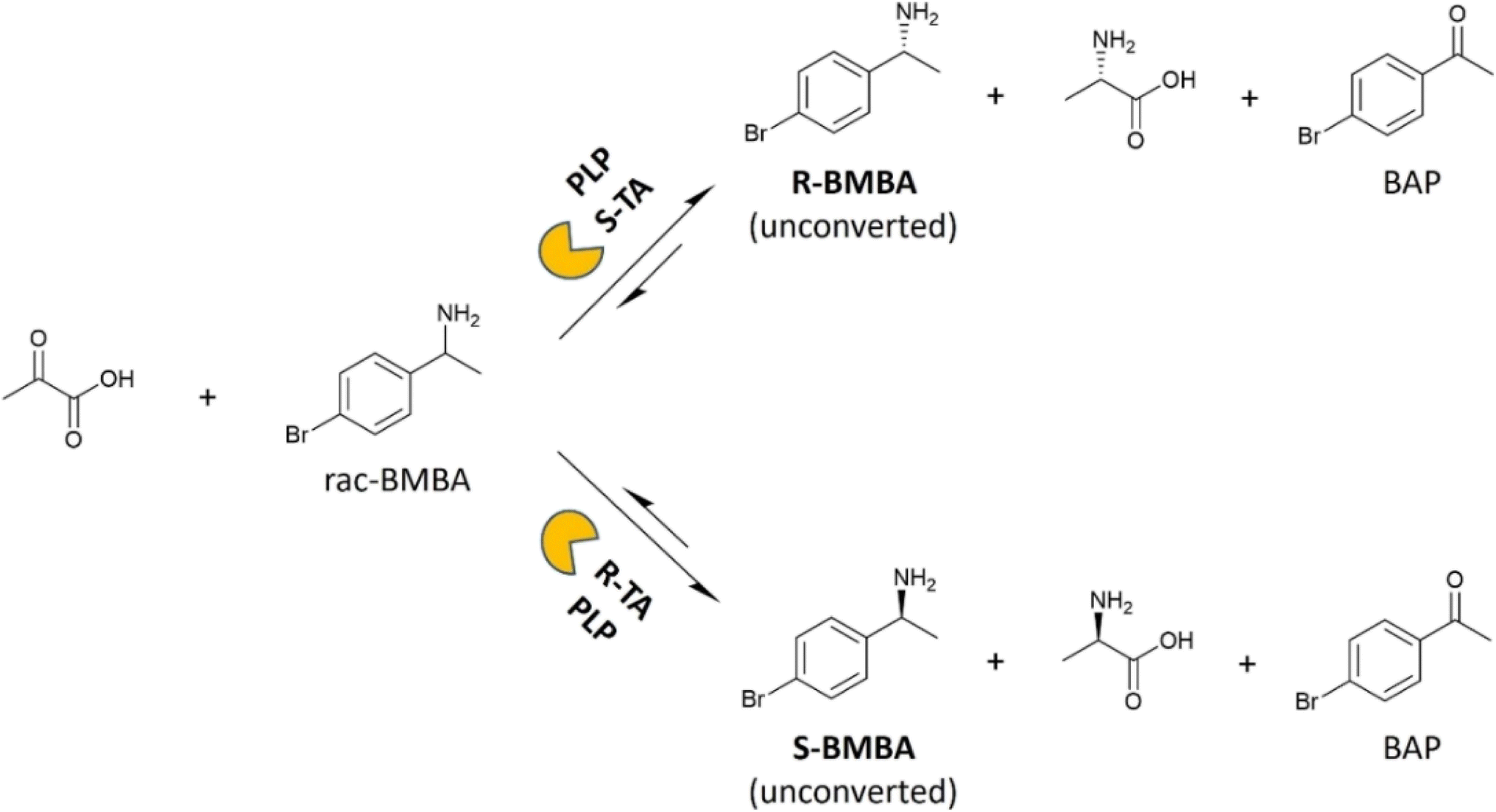 | ||
| Fig. 3 Model transamination (kinetic resolution of racemic BMBA with pyruvic acid) implemented to study the membrane-immobilized TAs specific activity. Precisely, this represents a kinetic resolution performed by a S-selective TA (up) and a R-selective TA (down). Typical reaction conditions were 37 °C, HEPES buffer 0.1 M pH 8, PLP 1 mM (or in the absence of PLP), sodium pyruvate 10 mM, racemic BMBA 10 mM, 1-phenyl-2-propanol 6 mM (as internal standard), DMSO 3% (v/v), 5 mL total volume in a batch reactor (5 mL round bottom glass flask) under moderate magnetic stirring. | ||
The progress of the reaction was followed by analyzing 100 μL samples taken from the reaction medium. 10 μL of sodium hydroxide (2 M) was added and the mixture was vortexed for 5 seconds. 400 μL of dichloromethane was then added to the aqueous phase, the sample was vortexed for 15 seconds and left to rest for 5 minutes to allow extraction of BAP, BMBA and 1-phenyl-2-propanol into the organic phase. This extraction step was repeated twice and the organic phases were pooled and analyzed by gas chromatography (see ESI†).
The yield is defined as the proportion of rac-BMBA converted into BAP (%). The maximum theoretical yield for the kinetic resolution is thus 50%. The specific activity is defined as the number of μmol of 4′-bromacetophenone formed per minute per mg of immobilized enzymes and evaluated by eqn (2), where L is the immobilized enzyme loading (determined by mass balance via the Bradford method (mg)) and, t is the reaction time (min). Specific activity was always determined in the kinetic regime (initial activity), after 15 minutes of reaction. The residual specific activity (%) is defined as the ratio between the specific activity of the immobilized TA and the specific activity of free TA (at identical enzyme concentration, in the same reaction conditions).
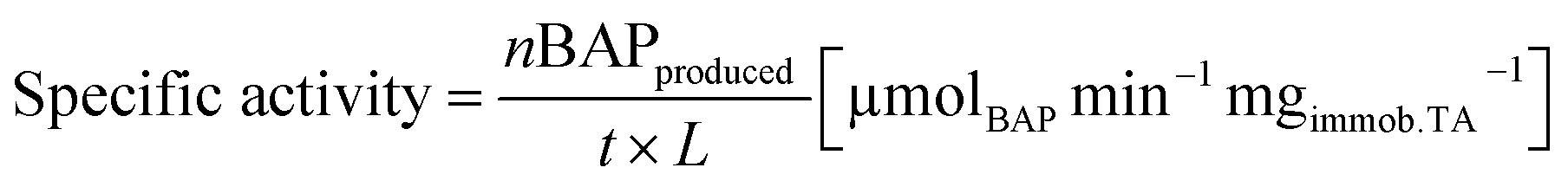 | (2) |
After 24 hours of reaction, the solid membrane was removed and the concentration of leached enzyme was evaluated via the Bradford method (ESI†). The leached TA fraction (%) is defined as the ratio between the mass of leached TA after one catalytic test and the initial immobilized TA loading (L). 50 mM 3,3-diphenylpropionic acid (3-DPPA) was added to the reaction medium in order to crystallize with the remaining BMBA (in the form of a BMBA:DPPA salt).30 After 20 hours of crystallization, crystals were filtered, washed twice with 5 mL distilled water, then once with 5 mL tert-butyl methyl ether (MTBE) to remove residual BAP, and then dried at room temperature overnight. Semi-quantification of BMBA enantiomers was then determined using Chiral High-Performance Liquid Chromatography (Chiral-HPLC), by dissolving the obtained crystals in the mobile phase (95% isohexane/5% 2-propanol/0.1% diethylamine) (see ESI†).
3 Results and discussion
3.1 Surface characterization of the pristine and modified membrane supports
The two membrane types (PAN and PP) have been modified and functionalized in order to bring the needed anchoring points for enzyme immobilization. Here, we describe these chemical modifications and provide detailed characterization of the membranes that are used in the next section to immobilize TA.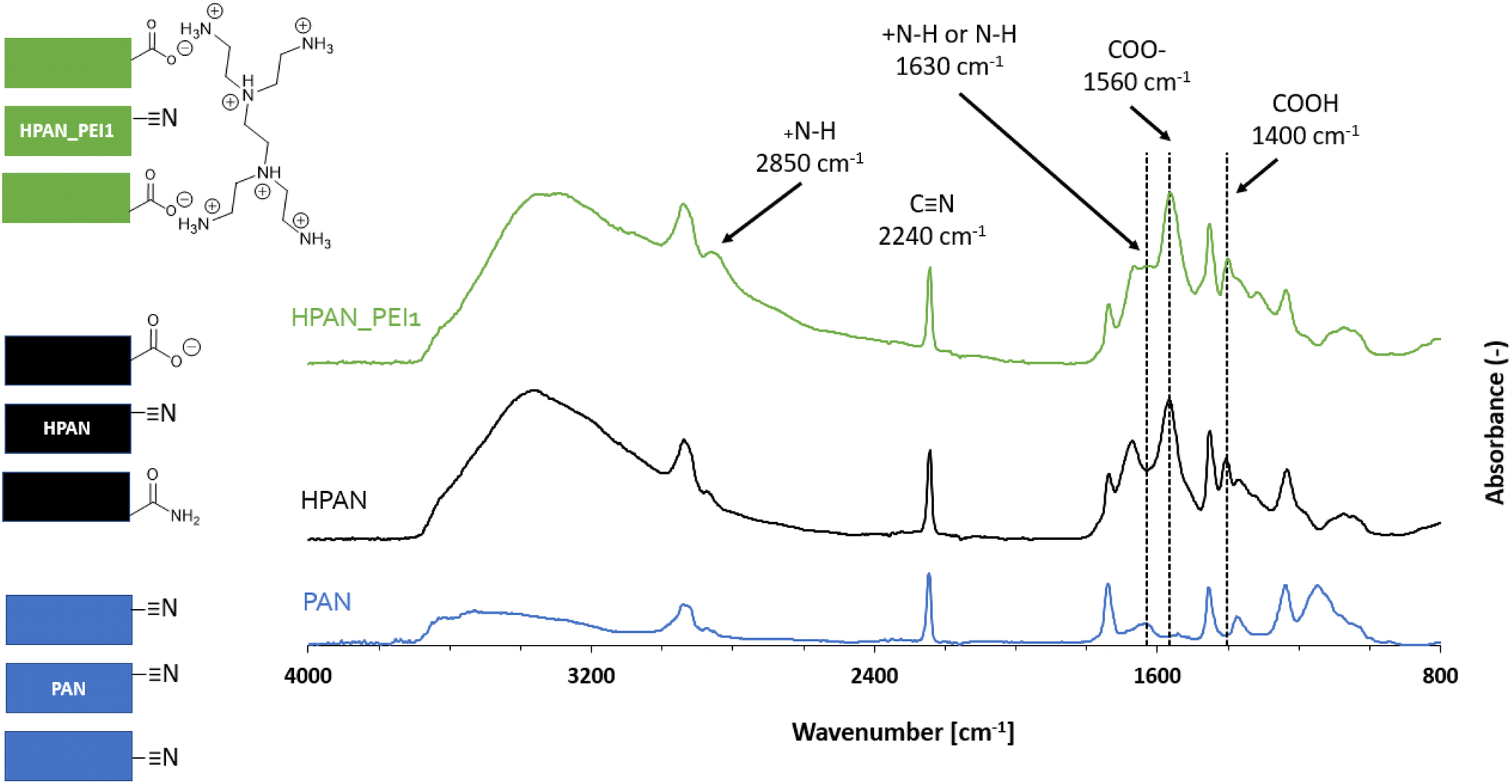 | ||
| Fig. 4 ATR-FTIR spectra (and schematic representation) of the pristine (blue), hydrolyzed (black) and PEI-functionalized PAN (in green) membrane. | ||
Based on previous reports,51,59 we applied a mild hydrolysis treatment (120 minutes with NaOH 1.5 M at 50 °C) in order to favor the formation of COO− surface groups while preserving the HPAN membrane mechanical properties. Expectedly, new IR peaks highlighted the presence of carboxylic acid/carboxylate moieties at 1560, 1400 and around 3300 cm−1,51,59 along with amides groups (characteristic peak at 1670 cm−1) coming from the partial surface hydrolysis of nitriles.
The subsequent addition of polyethyleneimine (HPAN_PEI1) was confirmed by the appearance of two small peaks attributed to amines and amine salts (at 1630 and 2850 cm−1).60 Additional surface-sensitive in situ infrared experiments (DRIFTS; see Fig. S2†) were performed on HPAN_PEI1 at 120 °C (to get rid of the broad O–H stretching band from 2800 to 3600 cm−1 due to surface hydration). It revealed characteristic peaks of amine (3420 and 2850 cm−1) as well as alkane (2925 and 1450 cm−1) and nitrile (2240 cm−1) moieties, which confirmed the results obtained from ATR-FTIR.
Characterization by XPS (Fig. S3†) showed that the pristine PAN surface was partly oxidized (Table 1, entry 1), which confirmed the qualitative ATR-FTIR observations. Consequently, the N/C ratio obtained at the PAN surface is lower (0.25) than the theoretical one (0.33). As expected, the basic hydrolysis of PAN (Fig. S4†) resulted in an increase of the O/C ratio and in a decrease of the N/C due to the conversion of nitrile moieties into amides and carboxylates moieties (Table 1, entry 2). Addition of PEI by electrostatic adsorption at the HPAN surface (Fig. S5†) logically led back to an increase of the surface N content.
| Membrane carrier | Mole fraction (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Naa | Caa | Cla | Fa | O | N | C | N/C | O/C | |
| a These elements were detected in significant amounts (in some samples), but their presence is exclusively due to contaminations (either present on the original commercial membranes, or generated during the experiments). b Below detection limit. c The value of the obtained ratio was <0.01. | |||||||||
| PAN | 0.5 | bdl | bdl | 1.9 | 8.9 | 17.8 | 70.9 | 0.25 | 0.13 |
| HPAN | 1.3 | bdl | bdl | 2.1 | 13.8 | 14.9 | 67.9 | 0.22 | 0.20 |
| HPAN_PEI | bdl | bdl | bdl | 2.6 | 16.4 | 16.4 | 64.6 | 0.25 | 0.25 |
| PP | bdl | bdl | bdl | bdl | 1.8 | bdl | 98.2 | —c | 0.02 |
| PP_PDA | bdl | 1.8 | bdl | bdl | 24.1 | 8.0 | 66.1 | 0.12 | 0.36 |
| PP_PDA_GDE | bdl | bdl | 2.8 | 1.1 | 28.0 | 6.0 | 62.1 | 0.10 | 0.45 |
| PP_PDA_GDE_PEI | bdl | bdl | 1.9 | 0.6 | 20.7 | 12.2 | 64.6 | 0.19 | 0.32 |
In the next steps, the PP_PDA membrane was functionalized with GDE and then with PEI. The signature peak of the epoxy groups (i.e. symmetric ring stretching, expected at 1250 cm−1) was not clearly observed on the PP_PDA_GDE spectrum, which might suggest an opening of the epoxy rings prior to the grafting of GDE, resulting in the presence of diol groups. Consistently, the small shoulder observed at 1090 cm−1 may correspond to the C–C–O symmetric stretch of secondary alcohols present in the diols. However, upon functionalisation the membrane was turned hydrophilic (see water contact angle (WCA) analyses, Fig. S7†) which creates large bands in the 3400 and 1630 cm−1 regions, hampering the observation of signature bands for amines, epoxides, or diols.
In XPS, pristine PP membranes (Fig. S8†) showed nearly exclusively aliphatic C–(C,H) signal (Fig. S9†), no nitrogen, and only traces of oxygen. Upon addition of PDA (Fig. S10†) signals for oxygen and nitrogen logically appeared. Notably, the N/C ratio of the PP_PDA reaches a similar value to that of the theoretical value of the polydopamine polymer (N/CPDA = 0.125), suggesting the formation of a PDA coating of at least 10 nm thickness at the PP surface.66 As expected, the grafting of GDE (Fig. S11†) on the amine residues present at the PP_PDA surface increased the oxygen surface concentration at the expense of nitrogen (Table 1, line 6), and the addition of PEI on PP_PDA_GDE (Fig. S12†) resulted in a marked increase in the nitrogen content (and N/C ratio) (Table 1, line 7).
Scanning electron microscopy (SEM) allowed to verify that the morphology of the PP membrane was preserved after functionalization: the surface of pristine PP and PP_PDA_GDE_PEI showed similar porosity (Fig. 5a and b), which confirmed that the membrane remains porous after functionalization. No change was pictured on cross-sections images either (Fig. 5c and d), which indicates that the membrane porosity was intact.
 | ||
| Fig. 5 SEM images showing the surface from a top view (left) and the cross-section (right) and of the pristine PP and PP_PDA_GDE_PEI membranes. | ||
3.2 Transamination with membrane-immobilized TAs
TAs were immobilized on the membranes described above and tested in the kinetic resolution of BMBA. Fig. 6 shows the activity (in terms of BAP yield) displayed by the different heterogeneous biocatalysts obtained by the immobilization of TsRTA on the different membrane supports. When using the pristine PP or PAN membranes as supports for the enzyme, the activity was virtually nil. However, upon functionalization – and depending on the parameters of functionalization and immobilization (vide infra) – significant biocatalytic activity was observed. In general, PP-immobilized TAs exhibited superior performance as compared to the PAN-immobilized TAs. Similar activity trends were obtained when employing HeWT as immobilized transaminase on these supports (Fig. S13†). As matter of comparison, activity profiles and specific activities obtained with soluble TAs (HeWT and TsRTA) are shown in Fig. S14.†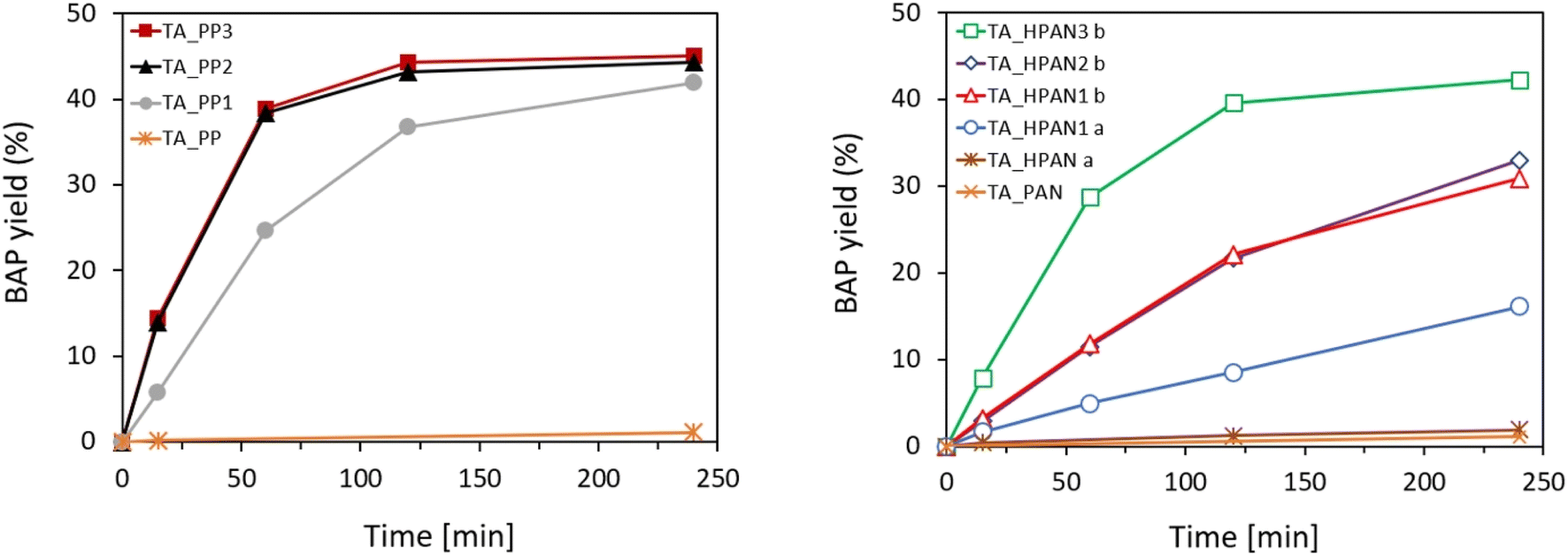 | ||
| Fig. 6 Activity profiles displayed by the different PP-immobilized (left) and PAN-immobilized (right) TA biocatalysts in the kinetic resolution of BMBA. TA immobilization was always applied using a nominal enzyme concentration of C0 = 0.25 mg mL−1, and TsRTA was used as TA. Reaction conditions: 10 mM rac-BMBA, 10 mM pyruvate, 1 mM PLP in 0.1 M HEPES pH 8 buffer, 37 °C (5 mL reaction volume). | ||
To interpret the raw yields obtained with the different enzyme-loaded membranes, complementary indicators must be considered. Table 2 gathers the immobilization yield, specific activity recovery, and leaching fraction displayed by the obtained membrane-immobilized TAs, for both immobilization strategies. Regarding the HPAN_PEI immobilized biocatalysts, it can be observed that the immobilization yield is boosted when the TA immobilization was performed at pH 5.5 (HeWT_HPAN1b (entry 2) and TsRTA_HPAN1b (entry 6)) rather than 8 (HeWT_HPAN1a (entry 1) and TsRTA_HPAN1a (entry 5)). This can be explained by the fact the PEI is more positively charged at low pH and favors the electrostatic adsorption of a larger amount of TA at the membrane surface. Accordingly, the observed activity is higher. Importantly, the specific activity (activity normalized by the amount of immobilized TA on the membrane) was the same, which indicates that, on average, the intrinsic activity of each additional immobilized transaminases was maintained. However, leaching after catalytic test was important, highlighting the need of post-treatment strategies to improve the anchoring of the immobilized TAs at the membrane surface.
| Entry | Immobilized TA | Membrane carrier | Immob. pH | Post-treatment | TA immob. Yielda (%) | Sp. activity recoveryb,d (%) | Leaching fractionc (%) |
|---|---|---|---|---|---|---|---|
a
 .
b Sp. activity recovery (%) = (Sp. activityimmTA/Sp. activityfree TA) × 100. At the considered enzyme concentrations, the specific activities of free TsRTA and free HeWT were of 0.74 μmol min−1 mg−1 and 0.76 μmol min−1 mg−1, respectively.
c TA leaching fraction (%) = (TA leaching after test/TA loading) × 100.
d Some experiments have been made in triplicate (n = 3) and always showed relatively small standard deviations. .
b Sp. activity recovery (%) = (Sp. activityimmTA/Sp. activityfree TA) × 100. At the considered enzyme concentrations, the specific activities of free TsRTA and free HeWT were of 0.74 μmol min−1 mg−1 and 0.76 μmol min−1 mg−1, respectively.
c TA leaching fraction (%) = (TA leaching after test/TA loading) × 100.
d Some experiments have been made in triplicate (n = 3) and always showed relatively small standard deviations.
|
|||||||
| 1 | HeWT_HPAN1a | HPAN_PEI | 8 | — | 24 | 12 ± 1.4 | 6 |
| 2 | HeWT_HPAN1b | HPAN_PEI | 5.5 | — | 58 | 12 ± 2.3 | 14 |
| 3 | HeWT_HPAN2b | HPAN_PEI | 5.5 | SA | 56 | 12 | 7 |
| 4 | HeWT_HPAN3b | HPAN_PEI | 5.5 | GA | 75 | 22 ± 1.6 | 1 |
| 5 | TsRTA_HPAN1a | HPAN_PEI | 8 | — | 23 | 19 ± 1.7 | 6 |
| 6 | TsRTA_HPAN1b | HPAN_PEI | 5.5 | — | 46 | 19 ± 2.7 | 14 |
| 7 | TsRTA_HPAN2b | HPAN_PEI | 5.5 | SA | 44 | 18 | 7 |
| 8 | TsRTA_HPAN3b | HPAN_PEI | 5.5 | GA | 64 | 36 ± 2.1 | 2 |
| 9 | HeWT_PP1 | PP_PDA | 8 | — | 60 | 19 ± 2.6 | 19 |
| 10 | HeWT_PP2 | PP_PDA_GDE | 8 | — | 84 | 27 ± 2.3 | 2 |
| 11 | HeWT_PP3 | PP_PDA_GDE_PEI | 8 | — | 62 | 45 ± 2.1 | 2 |
| 12 | TsRTA_PP1 | PP_PDA | 8 | — | 40 | 39 ± 2.9 | 15 |
| 13 | TsRTA_PP2 | PP_PDA_GDE | 8 | — | 73 | 59 ± 3.8 | 3 |
| 14 | TsRTA_PP3 | PP_PDA_GDE_PEI | 8 | — | 54 | 85 ± 3.3 | 2 |
Inspired by Shi et al.,40 we attempted to entrap the immobilized TA into a polymeric matrix formed by sodium alginate (SA, see Fig. 7; bottom). This biopolymer is able to electrostatically interact with the PEI layer, bringing additional negative charges that can in principle help stabilizing the enzyme. This post-treatment was found to preserve the enzyme loading and the specific activity, and concomitantly to reduce enzyme leaching (Table 2, compare HeWT_HPAN2b (entry 3) and TsRTA_HPAN2b (entry 7) to HeWT_HPAN1b (entry 2) and TsRTA_HPAN1b (entry 6), respectively).
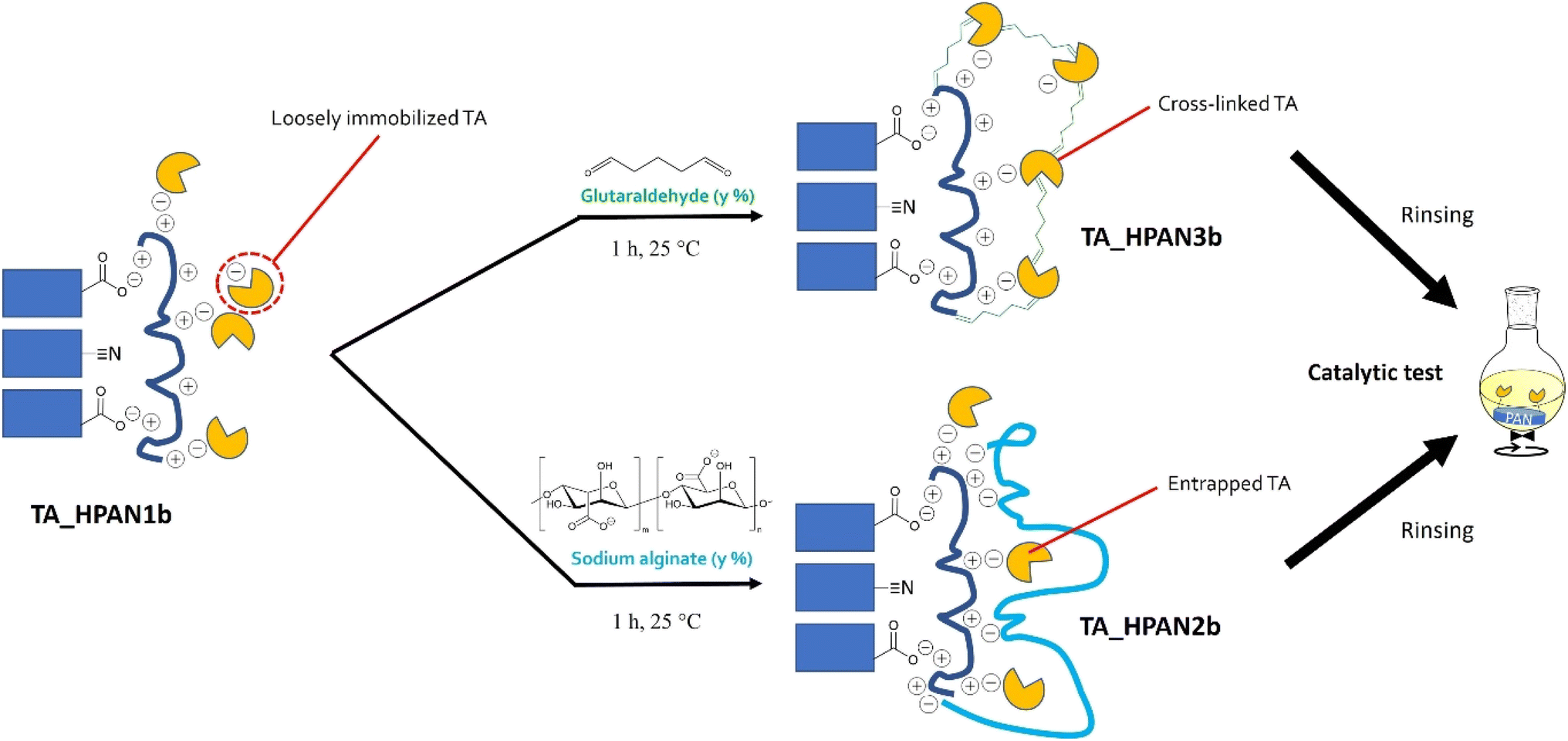 | ||
| Fig. 7 Schematic representation of post-treatment strategies applied on TA_HPAN1 resulting in TA_HPAN2 and TA_HPAN3 biocatalysts. | ||
Alternatively, inspired by Shi et al.40 and Paradisi et al.,67 we attempted to covalently bind the enzymes to the PEI layer using glutaraldehyde (GA, see Fig. 7, top) as a coupling agent. Such post-treatment strategy was found to (i) boost the enzyme loading (by securing the fixation of otherwise loosely attached TAs to the membrane surface), (ii) enhance the specific activity, and (iii) drastically curb the extent of enzyme leaching (Table 2, compare HeWT_HPAN3b (entry 4) and TsRTA_HPAN3b (entry 8) to HeWT_HPAN1b (entry 2) and TsRTA_HPAN1b (entry 6), respectively). The surge in specific activity after treating with GA seems surprising, for cross-linking is known to rigidify the enzymes structure, and it is often argued to be the cause of partial deactivation (e.g. in cross-linked enzyme aggregates).67 Yet the measurements were repeated (immobilization, activity assays, and Bradford tests to determine the loading) and the improvement was verified to be statistically significant (see the standard deviations in Table 2). Similar beneficial effect of such GA cross-linking of PEI-immobilized enzymes have been previously documented, with positive effects on specific activity (with lipases68,69), or on stability and reusability (with TAs70). In fact, here, TA enzymes are not only cross-linked together but also bound to PEI via GA. We surmise that bonding occurs preferentially with PEI (rather than cross-linking). Hence, one hypothesis is that the higher specific activity obtained for TAs_HPAN3b is linked to a more favorable (more hydrated, less constrained) chemical microenvironment conferred by the PEI layer to the immobilized TAs.71
Simple adsorption of TA on PP_PDA membranes led to important leaching (entry 9 and 12). However, using the covalent immobilization approach with GDE, TA leaching was significantly reduced (Table 2, compare HeWT_PP2 (entry 10) and TsRTA_PP2 (entry 13) with HeWT_PP1 (entry 9) and TsRTA_PP1 (entry 12), respectively). This highlights the beneficial role of the epoxy functions, able to immobilize the TA via covalent coupling.72 Interestingly, TA_PP2 also showed greater immobilization yield and specific activity with respect to TA_PP1 biocatalyst. Such enhanced specific activity obtained with GDE-immobilized TAs has already been observed in literature, and it was attributed to the hydrophilic and appropriate length of the epoxy linker-arm.73 Further functionalization with PEI resulted in a lower immobilization yield, but a higher specific activity (Table 2, compare HeWT_PP3 (entry 11) and TsRTA_PP3 (entry 14) with HeWT_PP2 (entry 10) and TsRTA_PP2 (entry 13) respectively). The enhanced specific activity recovery displayed by TA_PDA_GDE_PEI can be tentatively attributed to a more favorable (hydrated) chemical microenvironment conferred by the PEI layer to the immobilized TAs.71
In both immobilization methods, TsRTA displayed lower enzyme immobilization yields, but higher specific activity, as compared to HeWT (a more visual comparison is shown in Fig. S15 and S16†).
The immobilization and catalytic performance obtained with TsRTA_HPAN3b and TsRTA_PP3 as shown in Table 2 are the highest obtained in this study. In fact, various experimental parameters of the functionalization and immobilization steps have been studied systematically and optimized (see ESI, Fig. S17–S19†) to lead to the results reported in Table 2. Overall, the catalytic performance of these membrane-immobilized TAs compares well with other immobilized TAs described in literature. Indeed, typical transaminase immobilization via covalent grafting on metal-derivatized epoxy resins yields only 30–50% recovered specific activity.72,74 Additionally, our best-performing membrane-immobilzed biocatalysts also achieve similar or better immobilization efficiencies compared to a series of TAs immobilized on a variety of different supports (Table S2†), including polycarvone acrylate di-epoxide (PCADE)-functionalized membranes, 2D-zeolites and functionalized lignin.
3.3 Robustness, reusability and enantioselectivity of the biocatalytic membranes
In order to assess if the activity displayed by the developed immobilized TAs can only be attributed to heterogeneous catalysis, hot-filtration-tests were performed on both optimized TsRTA_HPAN3b and TsRTA_PP3 biocatalysts (Fig. 8). The TA-loaded membrane was removed from the reaction media after a short reaction time (15 minutes), and the activity was monitored and compared to a classical catalytic test (i.e. in which the membrane was not removed). Some residual activity could be detected after the removal of TsRTA_HPAN3b (Fig. 8a), which highlights the fact that a small fraction of leached TsRTA contributed to the observed activity. Such leaching fraction was either estimated to 5.1% (in the form of immobilized TsRTA) or 1.8% (in the form of soluble TsRTA), based on the slope between 15- and 60 minutes activity points. This can be explained by a slow hydrolysis of the imine bonds between GA and TsRTA or PEI. On the other hand, the hot-filtration test exhibited a completely flat profile after the removal of TsRTA_PP3 biocatalyst (Fig. 8b), demonstrating that, in this case, only heterogeneous catalysis is involved in the observed activity.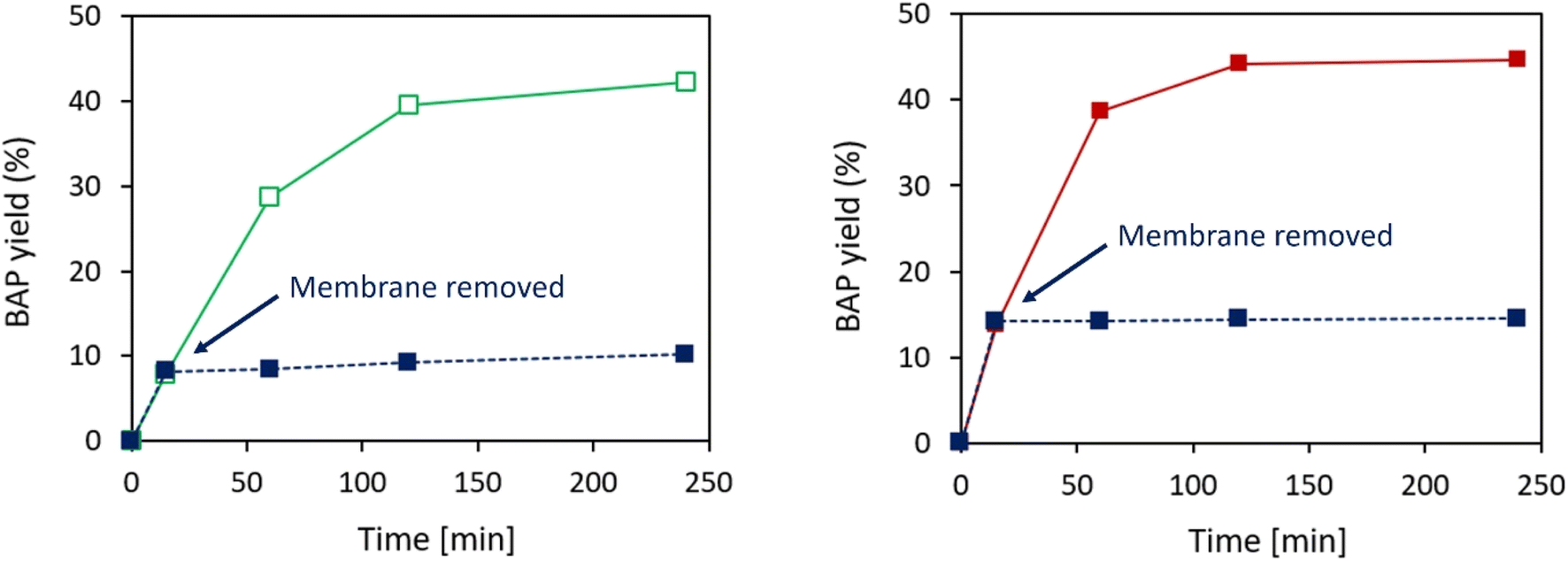 | ||
| Fig. 8 Hot-filtration tests performed on TA_HPAN3b (left) and TA_PP3 (right), performed at equal immobilization concentration (C0 = 0.25 mg per mL TA, using TsRTA as TA). Full curves represent the standard catalytic tests while dotted curves represent the hot-filtration tests (i.e. in which the membrane-immobilized TAs were removed after 15 minutes). Reaction conditions: 10 mM rac-BMBA, 10 mM pyruvate, 1 mM PLP in 0.1 M HEPES pH 8 buffer, 37 °C (5 mL reaction volume). | ||
The two selected catalysts were also tested in 4 successive catalytic cycles to assess their recyclability. At the end of each cycle, the membrane-immobilized TAs were washed twice with 5 mL of buffer solution (i.e. HEPES 0.1 M pH 8 containing PLP 1 mM, pyruvate 10 mM), and then immersed into a fresh reaction medium. The obtained reaction profiles (Fig. 9) unambiguously show that the membrane discs were recyclable. In all cases, the same final conversion (close to thermodynamic equilibrium) could be reached. More importantly, specific activity (approached by initial activity) was not affected throughout the successive catalytic cycles. This result paves the way toward a possible use of membrane-immobilized enzymes in continuous flow processes. Such robustness and recyclability was also confirmed with the S-selective HeWT enzyme (Fig. S20†), as no significant decrease of specific activity could be observed throughout the cycles.
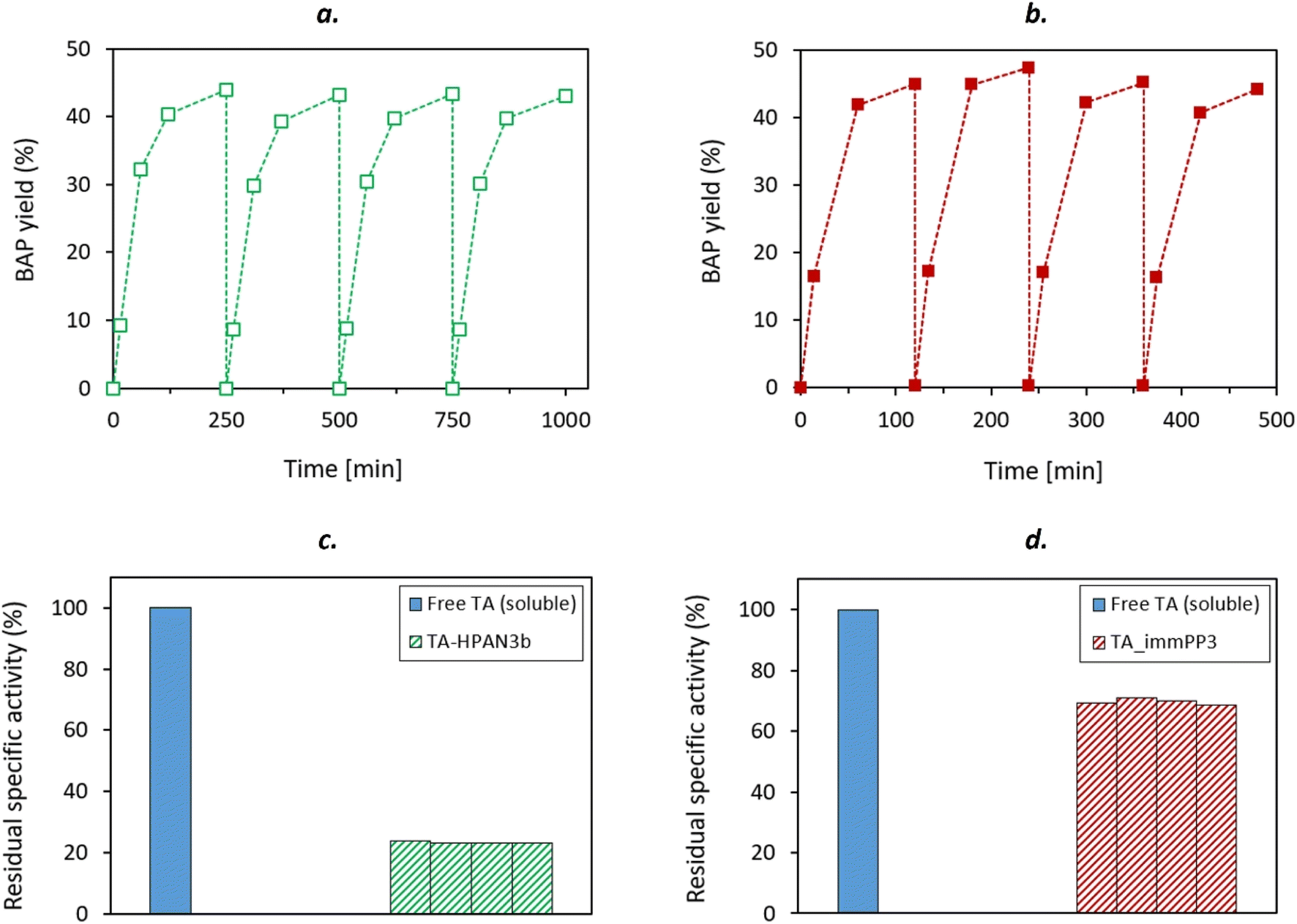 | ||
| Fig. 9 Activity profiles (a) and (b) and residual specific activities (c) and (d) obtained from the recyclability tests performed with TA_HPAN3b (left) and TA_PP3 (right) at equal immobilization concentration (C0 = 0.5 mg per mL TA, using TsRTA as TA). Each bar shown in (c) and (d) represent the residual specific activity (with respect to free TA), measured at each catalytic cycle (computed after 15 minutes, i.e. the first point of the activity profiles shown in (a) and (b)). Reaction conditions: 10 mM rac-BMBA, 10 mM pyruvate, 1 mM PLP in 0.1 M HEPES pH 8 buffer, 37 °C (5 mL reaction volume). | ||
Another interesting aspect to investigate was the ability of the biocatalytic membrane to work in the absence of externally added co-factor (PLP). Such ability has already been reported on PEI-coated supports, onto which both PLP and TA could be co-immobilized.57,75,76 This aspect would be particularly important in the perspective of a continuous flow membrane reactor, since it would allow to get rid of the costly PLP feed during the operation. Hence, TsRTA_PP3 (for which only the TA immobilization step is done in the presence of PLP) was tested in successive catalytic cycles without adding PLP to the reaction media (Fig. 10). In such case, the residual activity dropped after each reaction cycle (i.e. down from 78% to 23% after five cycles). This suggests a significant PLP leaching leading to immobilized TA deactivation. In order to overcome this problem, we slightly adapted the immobilization process. Inspired from López-Gallego et al.57 we implemented a two-step immobilization. Briefly, after performing classical enzyme immobilization (as always, in the presence of PLP, i.e. 0.1 mM or 1 mM), a subsequent step of PLP immobilization was performed at lower ionic force in order to favor the co-factor grafting at the PEI-coated membrane surface. The resulting membrane was tested in 8 successive catalytic cycles without PLP addition, and exhibited much higher stability as compared to TA_PP3. In particular, TA_PP3_SS0.1 did not show any activity drop. That remarkable stability displayed by TA_PP3_ SS0.1 might be explained by the higher PLP loading achieved for this catalyst (0.97 μmol as compared to TA_PP3_ SS1, 0.85 μmol) (Table S3†). These results suggest that upon this two-step immobilization strategy (and employing 0.1 mM PLP for TA immobilization step), PLP is suitably provided to the enzyme (i.e. available for the transamination catalytic act) in satisfying amounts. It is noteworthy that performing TA immobilization with 0.1 mM (instead of 1 mM) of PLP also enabled to boost the TA loading and increase the overall activity, but it lowered the specific activity of the catalyst (Table S3,† line 3).
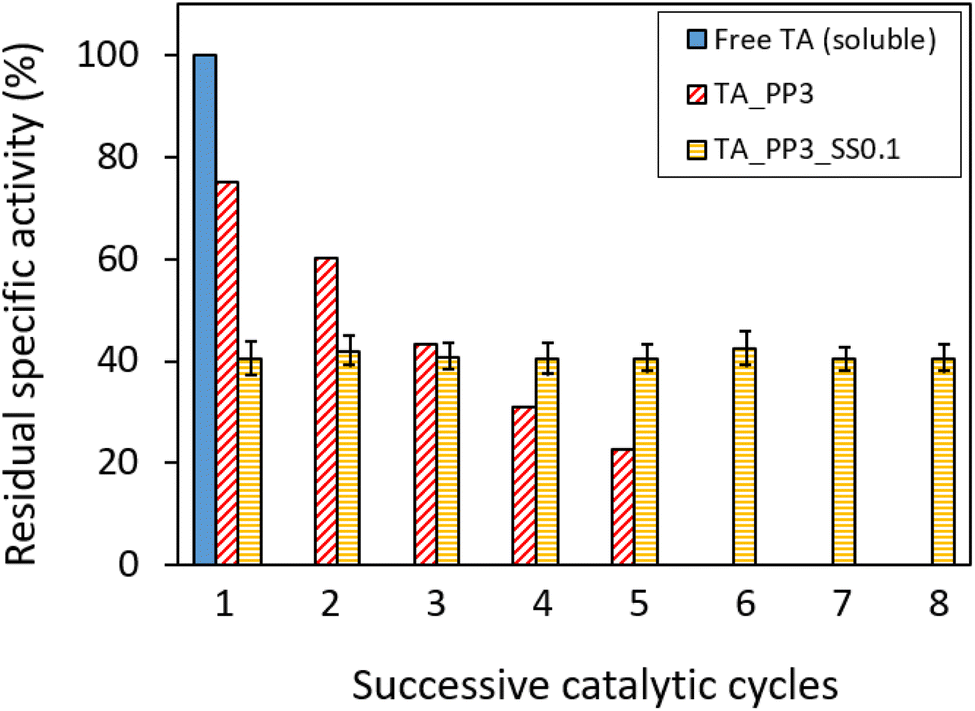 | ||
| Fig. 10 Recyclability tests performed on TA_PP3 (red dashed bars) and TA_PP3_SS0.1 (yellow dashed bars) without PLP addition in the reaction media. For all three catalysts, TsRTA immobilization concentration was 0.5 mg mL−1. Each catalytic cycle was separated by three buffer washings. Each bar represents the residual specific activity (with respect to free TsRTA), measured from the initial activity at each catalytic cycle (computed after 15 minutes). The error bars show the standard deviation measured on 3 different catalytic tests (n = 3). Reaction conditions: 10 mM rac-BMBA, 10 mM pyruvate, 0 mM PLP (for TA_PP3 and TA_PP3_SS0.1) or 1 mM PLP (for free TA) in 0.1 M HEPES pH 8 buffer, 37 °C (5 mL reaction volume). | ||
Finally, chiral HPLC analyses allowed us to confirm that the immobilization process did not affect the biocatalysts enantioselectivity (Table 3). To this aim, the produced BMBA enantiomers obtained when employing the two best-performing immobilization strategies (namely TA_HPAN3b and TA_PP3) were analyzed and quantified. For all four membrane-immobilized TAs studied, only one BMBA enantiomer was detected, suggesting that the obtained biocatalytic membranes are enantioselective. Since the investigated reaction is a kinetic resolution (i.e. starting from a racemic mixture), the unconverted BMBA enantiomer (e.g. R-BMBA for HeWT, S-BMBA for TsRTA) was always detected by chiral HPLC (Fig. S21–23†).
| TA_HPAN3b | TA_PP3 | |
|---|---|---|
| HeWT | >99% | >99% |
| TsRTA | >99% | >99% |
The “greenness” of the biocatalytic approach presented here can in theory be compared to common chemo-catalytic processes (see ESI, Fig. S24–S26†) used for chiral amine synthesis, for example by comparing E-factors.8 Rough estimations (see ESI, Table S4†) show that the reaction itself (considering the reagents, solvent, and catalysts) is characterized by similar values of E-factors. Yet, it is noteworthy that the biocatalytic strategy produces enantiopure products, which is not the case of the other methods. Further purifications (e.g. preferential crystallizations, catalyst removal, chiral chromatography) will be required in the chemo-catalytic processes, which will markedly increase the overall E-factor of such chemo-catalytic processes. Purification (not accounted for in these calculations due to lack of information) is known to be a major driver for the overall (environmental) cost of the process of chiral amine synthesis.9,77 We therefore anticipate a significant advantage of the biocatalytic process when the whole process is considered.
4 Summary and conclusion
We report two efficient immobilization routes to obtain robust membrane-immobilized transaminases. These immobilization strategies consist of the TA electrostatic adsorption and covalent grafting, on polyethyleneimine (PEI)-coated polyacrylonitrile (PAN) and functionalized polypropylene (PP) membranes, respectively.As important enzyme leaching was observed on the electrostatically immobilized TAs, post-treatment strategies of the electrostatically immobilized TAs were applied to improve TA anchoring at the functionalized PAN membrane surface. Among the developed strategies, the covalent binding of TAs and of the PEI layer using glutaraldehyde (GA) gave the most satisfying results (high specific activity, minor leaching). On the other hand, the TA covalent grafting on functionalized PP membranes yielded even more efficient biocatalysts (higher specific activity) displaying enhanced robustness (no leaching) and full recyclability. Importantly, these two strategies allowed to efficiently immobilize two different TAs (the S-selective HeWT, and the R-selective TsRTA), resulting in stereo-divergent biocatalytic membranes. Additionally, co-immobilization of TA and PLP was also achieved on functionalized PP membranes by adapting the immobilization protocol, which resulted in highly reusable membrane-immobilized biocatalysts capable of catalyzing transaminations in the absence of externally added PLP. Such self-sufficient ability should be attractive from an industrial point of view, since it should help increasing the cost-efficiency and reducing the E-factor of the transamination process. Thus, all in all, both studied routes to immobilize TAs on membranes led to functional biocatalytic materials exhibiting perfect enantioselectivity, high catalytic performance, minor leaching and excellent reusability. This paves the way to a future use in flow mode hybrid processes.
The transfer and implementation of such biocatalytic membranes in continuous flow (as a flat-sheet membrane reactor) has now to be carried out. Ultimately, more challenging transamination reactions (i.e. asymmetric synthesis) should be tackled with this immobilized TA, by taking benefit of the ability of the membrane carrier to act as a separation unit for (co)product removal.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The chiral HPLC analyses were performed by Ir. Laurent Collard. SEM analyses were performed by Ir. Sara Chergaoui. This work was funded by Actions de Recherche Concerté (ARC) under the project « PURE » (20/25–108) and by the Fonds de la Recherche Scientifique (FNRS) under the project CDR J.0044.20.References
- L. J. Diorazio, P. Richardson, H. F. Sneddon, A. Moores, C. Briddell and I. Martinez, ACS Sustain. Chem. Eng., 2021, 9, 16862–16864 CrossRef CAS.
- M. D. Patil, G. Grogan, A. Bommarius and H. Yun, Catalysts, 2018, 8, 254 CrossRef.
- D. Ghislieri and N. J. Turner, Top. Catal., 2014, 57, 284–300 CrossRef CAS.
- H. M. Arango, L. van den Biggelaar, P. Soumillion, P. Luis, T. Leyssens, F. Paradisi and D. P. Debecker, React. Chem. Eng., 2023, 8, 1505–1544 RSC.
- T. C. Nugent and M. El-Shazly, Adv. Synth. Catal., 2011, 353, 804 CrossRef CAS.
- P. Kelefiotis-Stratidakis, T. Tyrikos-Ergas and I. V. Pavlidis, Org. Biomol. Chem., 2019, 17, 1634–1642 RSC.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998, vol. 2 Search PubMed.
- D. P. Debecker, K. Kuok, M. Hii, A. Moores, L. M. Rossi, B. Sels, D. T. Allen and B. Subramaniam, ACS Sustain. Chem. Eng., 2021, 9, 4936–4940 CrossRef CAS.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS.
- I. Slabu, J. L. Galman, R. C. Lloyd and N. J. Turner, ACS Catal., 2017, 7, 8263–8284 CrossRef CAS.
- F. Guo and P. Berglund, Green Chem., 2017, 19, 333–360 RSC.
- E. E. Ferrandi and D. Monti, World J. Microbiol. Biotechnol., 2017, 34, 13 CrossRef PubMed.
- D. J. Wallace, I. Mangion and P. Coleman, in Comprehensive Accounts of Pharmaceutical Research and Development: from Discovery to Late-Stage Process Development Volume 1, American Chemical Society, 2016, vol. 1239, pp. 1–36 Search PubMed.
- M. Girardin, S. G. Ouellet, D. Gauvreau, J. C. Moore, G. Hughes, P. N. Devine, P. D. O'Shea and L.-C. Campeau, Org. Process Res. Dev., 2013, 17, 61–68 CrossRef CAS.
- S. J. Novick, N. Dellas, R. Garcia, C. Ching, A. Bautista, D. Homan, O. Alvizo, D. Entwistle, F. Kleinbeck, T. Schlama and T. Ruch, ACS Catal., 2021, 11, 3762–3770 CrossRef CAS.
- C. K. Chung, P. G. Bulger, B. Kosjek, K. M. Belyk, N. Rivera, M. E. Scott, G. R. Humphrey, J. Limanto, D. C. Bachert and K. M. Emerson, Org. Process Res. Dev., 2014, 18, 215–227 CrossRef CAS.
- M. Burns, C. A. Martinez, B. Vanderplas, R. Wisdom, S. Yu and R. A. Singer, Org. Process Res. Dev., 2017, 21, 871–877 CrossRef CAS.
- L. Tamborini, P. Fernandes, F. Paradisi and F. Molinari, Trends Biotechnol., 2018, 36, 73–88 CrossRef CAS PubMed.
- A. I. Benítez-Mateos, M. L. Contente, D. R. Padrosa and F. Paradisi, React. Chem. Eng., 2021, 6, 599–611 RSC.
- M. Romero-Fernández and F. Paradisi, Curr. Opin. Chem. Biol., 2020, 55, 1–8 CrossRef.
- L. van den Biggelaar, P. Soumillion and D. P. Debecker, RSC Adv., 2019, 9, 18538–18546 RSC.
- L. Van den Biggelaar, P. Soumillion and D. P. Debecker, Catalysts, 2017, 7, 54 CrossRef.
- M. P. Thompson, I. Peñafiel, S. C. Cosgrove and N. J. Turner, Org. Process Res. Dev., 2019, 23, 9–18 CrossRef CAS.
- W. Khanam and N. C. Dubey, Mater. Today Chem., 2022, 24, 100922 CrossRef CAS.
- A. Gomm and E. O'Reilly, Curr. Opin. Chem. Biol., 2018, 43, 106–112 CrossRef CAS PubMed.
- R. E. Meadows, K. R. Mulholland, M. Schürmann, M. Golden, H. Kierkels, E. Meulenbroeks, D. Mink, O. May, C. Squire, H. Straatman and A. S. Wells, Org. Process Res. Dev., 2013, 17, 1117–1122 CrossRef CAS.
- M. D. Truppo, J. D. Rozzell, J. C. Moore and N. J. Turner, Org. Biomol. Chem., 2008, 7, 395–398 RSC.
- K. E. Cassimjee, C. Branneby, V. Abedi, A. Wells and P. Berglund, Chem. Commun., 2010, 46, 5569–5571 RSC.
- A. P. Green, N. J. Turner and E. O'Reilly, Angew. Chem., Int. Ed., 2014, 53, 10714–10717 CrossRef CAS PubMed.
- D. Hülsewede, M. Tänzler, P. Süss, A. Mildner, U. Menyes and J. von Langermann, Eur. J. Org Chem., 2018, 2018, 2130–2133 CrossRef.
- L. Frodsham, M. Golden, S. Hard, M. N. Kenworthy, D. J. Klauber, K. Leslie, C. Macleod, R. E. Meadows, K. R. Mulholland, J. Reilly, C. Squire, S. Tomasi, D. Watt and A. S. Wells, Org. Process Res. Dev., 2013, 17, 1123–1130 CrossRef CAS.
- P. Luis, Fundamental Modeling of Membrane Systems, Elsevier, 1st edn., 2018 Search PubMed.
- R. Gérardy, D. P. Debecker, J. Estager, P. Luis and J.-C. M. Monbaliu, Chem. Rev., 2020, 120, 7219–7347 CrossRef.
- W. Li, J. Estager, J.-C. M. Monbaliu, D. P. Debecker and P. Luis, J. Chem. Technol. Biotechnol., 2020, 95, 2311–2334 CrossRef CAS.
- J. S. Shin, B. G. Kim, A. Liese and C. Wandrey, Biotechnol. Bioeng., 2001, 73, 179–187 CrossRef CAS.
- J.-S. Shin and B.-G. Kim, Biotechnol. Bioeng., 1997, 55, 348–358 CrossRef CAS.
- G. Rehn, P. Adlercreutz and C. Grey, J. Biotechnol., 2014, 179, 50–55 CrossRef CAS PubMed.
- A. H. Rather, R. S. Khan, T. U. Wani, M. A. Beigh and F. A. Sheikh, Biotechnol. Bioeng., 2022, 119, 9–33 CrossRef CAS PubMed.
- J. Manuel, M. Kim, R. Dharela, G. S. Chauhan, D. Fapyane, S.-J. Lee, I. S. Chang, S.-H. Kang, S.-W. Kim and J.-H. Ahn, J. Biomed. Nanotechnol., 2015, 11, 143–149 CrossRef CAS PubMed.
- Y. Li, H. Wang, J. Lu, A. Chu, L. Zhang, Z. Ding, S. Xu, Z. Gu and G. Shi, Bioresour. Technol., 2019, 274, 9–17 CrossRef CAS.
- G. M. Rios, M. P. Belleville, D. Paolucci and J. Sanchez, J. Membr. Sci., 2004, 242, 189–196 CrossRef CAS.
- J. Luo, S. Song, H. Zhang, H. Zhang, J. Zhang and Y. Wan, Eng. Life Sci., 2020, 20, 441–450 CrossRef CAS.
- C. Molina-Fernández, A. Péters, D. P. Debecker and P. Luis, Biochem. Eng. J., 2022, 187, 108639 CrossRef.
- U. Montanari, D. Cocchi, T. M. Brugo, A. Pollicino, V. Taresco, M. Romero Fernandez, J. C. Moore, D. Sagnelli, F. Paradisi, A. Zucchelli, S. M. Howdle and C. Gualandi, Polymers, 2021, 13, 1804 CrossRef CAS PubMed.
- Resindion srl, https://www.resindion.com/relizyme.php?content=technical-information, accessed 3 January 2024.
- S. Chergaoui, D. P. Debecker, T. Leyssens and P. Luis, Cryst. Growth Des., 2023, 23, 6418–6430 CrossRef CAS.
- A. Tejeda-Mansir, R. M. Montesinos and R. Guzmán, J. Biochem. Biophys. Methods, 2001, 49, 1–28 CrossRef CAS.
- E. Lalli, J. S. Silva, C. Boi and G. C. Sarti, Membranes, 2019, 10, 1 CrossRef PubMed.
- L. Cerioli, M. Planchestainer, J. Cassidy, D. Tessaro and F. Paradisi, J. Mol. Catal. B: Enzym., 2015, 120, 141–150 CrossRef CAS.
- C. M. Heckmann, L. J. Gourlay, B. Dominguez and F. Paradisi, Front. Bioeng. Biotechnol., 2020, 8, 707 CrossRef PubMed.
- L. Pérez-Álvarez, L. Ruiz-Rubio, I. Moreno and J. L. Vilas-Vilela, Polymers, 2019, 11, E1843 CrossRef.
- H. Zhang, J. Luo, S. Li, Y. Wei and Y. Wan, Langmuir, 2018, 34, 2585–2594 CrossRef CAS.
- Z. Niu, Y. Zhao, W. Sun, S. Shi and Y. Gong, Appl. Surf. Sci., 2016, 386, 41–50 CrossRef CAS.
- J. Yang, M. A. C. Stuart and M. Kamperman, Chem. Soc. Rev., 2014, 43, 8271–8298 RSC.
- S. Gámez, E. de la Torre and E. M. Gaigneaux, Chem. Eng. J., 2022, 427, 131820 CrossRef.
- S. P. de Souza, I. I. Junior, G. M. A. Silva, L. S. M. Miranda, M. F. Santiago, F. L.-Y. Lam, A. Dawood, U. T. Bornscheuer and R. O. M. A. de Souza, RSC Adv., 2016, 6, 6665–6671 RSC.
- A. I. Benítez-Mateos, M. L. Contente, S. Velasco-Lozano, F. Paradisi and F. López-Gallego, ACS Sustain. Chem. Eng., 2018, 6, 13151–13159 CrossRef.
- IR Spectrum Table, https://www.sigmaaldrich.com/BE/en/technical-documents/technical-article/analytical-chemistry/photometry-and-reflectometry/ir-spectrum-table, accessed 8 August 2022.
- G. Zhang, H. Meng and S. Ji, Desalination, 2009, 242, 313–324 CrossRef CAS.
- B. Smith, Infrared Spectral Interpretation: A Systematic Approach, CRC Press, 1998 Search PubMed.
- J. Cai, T. Chen, L. Cui, Q. Jia, M. Liu, R. Zheng, G. Yan, D. Wei and J. Liu, Int. J. Hydrogen Energy, 2020, 45, 6592–6603 CrossRef CAS.
- H. Lee, N. F. Scherer and P. B. Messersmith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12999–13003 CrossRef CAS.
- J. Jiang, L. Zhu, L. Zhu, B. Zhu and Y. Xu, Langmuir, 2011, 27, 14180–14187 CrossRef CAS PubMed.
- Z.-Y. Xi, Y.-Y. Xu, L.-P. Zhu, Y. Wang and B.-K. Zhu, J. Membr. Sci., 2009, 327, 244–253 CrossRef CAS.
- B. Smith, Spectroscopy, 2017, 32, 19–23 CAS.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- D. Roura Padrosa, Z. Nisar and F. Paradisi, Catalysts, 2021, 11, 520 CrossRef.
- S. Arana-Peña, N. S. Rios, C. Mendez-Sanchez, Y. Lokha, D. Carballares, L. R. B. Gonçalves and R. Fernandez-Lafuente, Int. J. Biol. Macromol., 2020, 145, 856–864 CrossRef PubMed.
- J. M. Palomo, R. L. Segura, G. Fernandez-Lorente, R. Fernandez-Lafuente and J. M. Guisán, Enzyme Microb. Technol., 2007, 40, 704–707 CrossRef CAS.
- S. Velasco-Lozano, E. Jackson, M. Ripoll, F. López-Gallego and L. Betancor, Int. J. Biol. Macromol., 2020, 164, 4318–4328 CrossRef CAS PubMed.
- A. Vander Straeten, D. Lefèvre, S. Demoustier-Champagne and C. Dupont-Gillain, Adv. Colloid Interface Sci., 2020, 280, 102161 CrossRef CAS PubMed.
- M. Planchestainer, M. L. Contente, J. Cassidy, F. Molinari, L. Tamborini and F. Paradisi, Green Chem., 2017, 19, 372–375 RSC.
- E. Abaházi, P. Sátorhelyi, B. Erdélyi, B. G. Vértessy, H. Land, C. Paizs, P. Berglund and L. Poppe, Biochem. Eng. J., 2018, 132, 270–278 CrossRef.
- C. M. Heckmann, B. Dominguez and F. Paradisi, ACS Sustain. Chem. Eng., 2021, 9, 4122–4129 CrossRef CAS.
- S. Velasco-Lozano, A. I. Benítez-Mateos and F. López-Gallego, Angew. Chem., Int. Ed., 2017, 56, 771–775 CrossRef CAS PubMed.
- B. Reus, M. Damian and F. G. Mutti, J. Flow Chem., 2024, 14, 219–238 CrossRef CAS.
- J. Yang, A. Buekenhoudt, M. V. Dael, P. Luis, Y. Satyawali, R. Malina and S. Lizin, Org. Process Res. Dev., 2022, 26, 2052–2066 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00293h |
| This journal is © The Royal Society of Chemistry 2024 |