
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective hydrodeoxygenation of oxygenated aromatic molecules using a molecular palladium catalyst covalently bound to a solid SiO2 support†
Jake G.
Tillou
a,
Joseph J.
Kuchta
III
a,
Nathan
Thornburg
 b,
Santosh K.
Balijepalli
c and
Aaron K.
Vannucci
*a
b,
Santosh K.
Balijepalli
c and
Aaron K.
Vannucci
*a
aDepartment of Chemistry and Biochemistry, University of South Carolina, Columbia, SC 29208, USA. E-mail: vannucci@mailbox.sc.edu
bDepartment of Chemical Engineering, University of South Carolina, Columbia, SC 29208, USA
cOffice of Vice President for Research, University of South Carolina, Columbia, SC 29208, USA
First published on 22nd July 2024
Abstract
The selective hydrodeoxygenation of lignin derived aromatics represents an important step towards the valorization of biomass. With this goal in mind, we synthesized a hybrid molecular/heterogeneous catalyst comprised of a (2,6-bis(1-methylbenzimidazolyl)pyridine-4′-aminopropyltrisiloxane)palladium(II) molecular catalyst covalently bound to a solid silica support through the siloxane functional group. A series of model complexes containing C–O bonds typically found in lignin biomass were explored and varying degrees of C–O bond hydrogenation were achieved. The stable covalent binding of the catalyst to the support was attributed to the observed long catalyst lifetimes which led to over 6000 catalytic turnovers without catalyst deactivation. Spectroscopic characterization of the catalyst pre- and post-catalytic reactions shows the catalyst maintains molecular integrity under the reaction conditions examined. The catalyst also exhibited complete selectivity for hydrodeoxygenation over ring hydrogenation of oxygenated aromatic molecules.
Sustainability spotlightCurrently, lignin is an untapped resource that is typically discarded as an unfavorable by-product of valorization of cellulose and hemicellulose. Lignin is a highly aromatic branched polymer and could be utilized for the production of fuels and commodity chemicals. Recent advances in lignin-depolymerization technology have allowed for the high yield of oxygenated aromatic monomers and dimers. These oxygenated functionalities result in decreased energy density and stability. The selective hydrodeoxygenation of oxygenated aromatic molecules while avoiding ring hydrogenation would represent a step forward in the sustainable conversion of biomass. This work utilizes hybrid molecular catalysts to target product selectivity. Our work emphasizes the importance of following UN sustainable development goals: affordable and clean energy (SDG 7) and responsible consumption and production (SDG 12). |
Introduction
Biomass is the largest source of renewable liquid fuels and commodity chemicals. Lignocellulosic biomass is composed of cellulose, hemicellulose, and lignin (Fig. 1).1 Cellulose and hemicellulose are carbohydrate polymers whereas lignin is a highly branched, oxygenated, aromatic polymer.1 Currently, cellulose and hemicellulose are utilized for bioethanol and paper production amongst many other products while lignin conversion is relegated to being burned on-site for energy for cellulose conversion.2 As such, lignin has been treated as a lower value by-product of lignocellulose valorization.3 The density of aromatic subunits in lignin, however, make it a potential source for sustainable commodity chemicals and liquid fuels.3 In fact, the effective conversion of lignin biomass to commodity chemicals has been identified as an essential step towards the further commercial utilization of biomass, which would represent a move towards a more sustainable chemical industry.4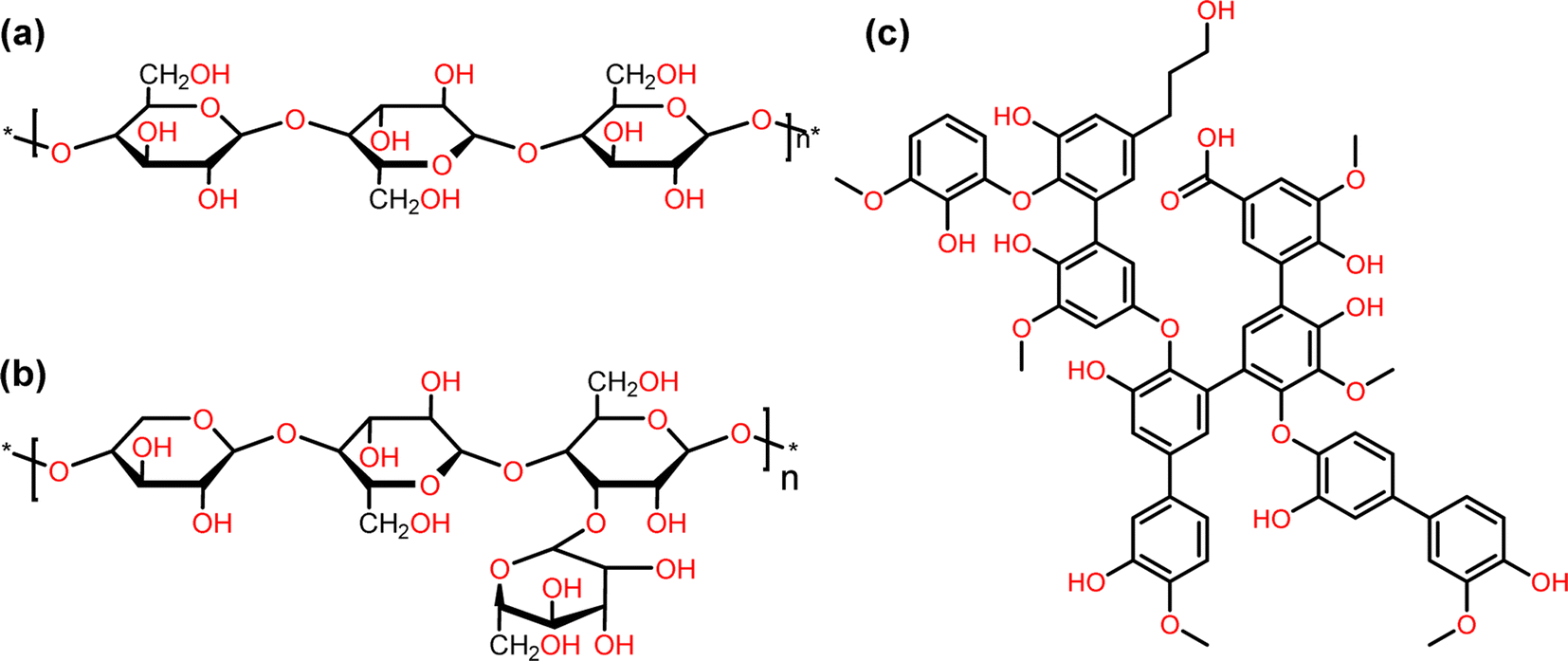 | ||
| Fig. 1 Representation of the structure of cellulose (a), hemicellulose (b), and lignin (c). | ||
The first step in lignin conversion is the depolymerization of lignin biomass. Recent advances have shown that effective depolymerization of lignin can be achieved when degraded and/or condensed lignin structures are avoided.3 High yields (>50%) of monomeric and dimeric aromatic structures can now readily be achieved during the lignin depolymerization process.5–8 Many, if not all, of these low molecular weight aromatic structures, however, have a high oxygen content, as shown on the left side of Fig. 2. These oxygenated aromatic units have a lower energy density compared to deoxygenated counter parts and also have a low stability due to being peroxide formers.9,10 Deoxygenation of these aromatic units is typically achieved through complete hydrogenation leading to lower value, non-aromatic products as shown in the hydrogenation step of Fig. 2.11–16 Synthesis of higher value aromatic chemicals from lignin biomass therefore typically requires a second dehydrogenation step. In addition, reports have shown that catalysts that promote complete hydrogenation of oxygenated aromatics often times suffer from catalyst deactivation through coking.17–19 Therefore, direct hydrodeoxygenation without ring hydrogenation (Fig. 2) would result in higher value commodity chemicals, higher-octane fuels, and would require fewer steps toward valorization.7
 | ||
| Fig. 2 Representation of the deoxygenation of lignin model substrates by either complete hydrogenation followed by dehydrogenation, or direct hydrodeoxygenation. | ||
The selective deoxygenation of lignin-derived molecules, also known as hydrodeoxygenation (HDO), aims to maintain the aromatic structure of the molecules during the deoxygenation process. Research in the field of deoxygenation of biomass is commonly conducted using heterogeneous metallic nanoparticle catalysts.20–22 High conversion of lignin derived compounds can be achieved with heterogeneous catalysts, but ring hydrogenated products are also common.5,21 High yields for HDO products have been recently achieved using heterogeneous nanoparticle catalysts by either modifying the catalyst surface or using flow reactors to minimize the time on stream for the oxygenated reactants.23–26 Furthermore, recent trends in the field show that minimizing H2 pressures leads to a higher selectivity towards HDO over ring hydrogenation during deoxygenation reactions performed by heterogeneous catalysts.5 These results all point towards HDO selectivity being increased by minimizing the interaction between the aromatic rings of lignin-derived monomers and the extended surfaces of nanoparticle hydrogenation catalysts.
With the hypothesis that extended nanoparticle surfaces lead to undesired ring hydrogenation reactions, researchers have begun to explore molecular catalysts for selective HDO of lignin-derived biomass. Molecular catalysts offer a “single site” for reactivity, and thus can avoid reactivity that occurs when the aromatic rings of the biomass subunits interact with extended catalyst surfaces. Thus, molecular catalysts that exhibit selective reactivity towards C–O bonds, over interaction with aromatic rings, should lead to selective hydrodeoxygenation. Selectivity for deoxygenated aromatic products over ring hydrogenated products, in fact, has been achieved with molecular catalysts.27–29 Homogeneous ruthenium or iridium complexes have been reported to selectively convert vanillyl alcohol to methyl guaiacol without ring hydrogenation and the reactivity of the complexes could be tuned through ligand variations.27,30 A homogenous polypyridyl complex was also capable of selectively deoxygenating aromatic alcohols, aldehydes, and ketones under mild conditions.28 While these catalysts can display high selectivity for the desired products, they are difficult to adopt into industrial applications due to difficult post reaction separations compared to their heterogeneous counterparts. By immobilizing molecular catalysts onto solid, heterogenous supports, as exemplified by Fig. 3, the issue of post reaction separations can be overcome.29 These molecular-heterogeneous systems, also known as hybrid catalysts, may combine the advantages of molecular catalyst selectivity with heterogeneous catalysts ease of separation.29,31 These advantages point toward hybrid catalysts for biomass conversion, however, molecular catalysts are often not as stable as heterogeneous catalysts and the conditions required for catalytic HDO can lead to molecular catalyst decomposition and/or leaching from the support surface.32
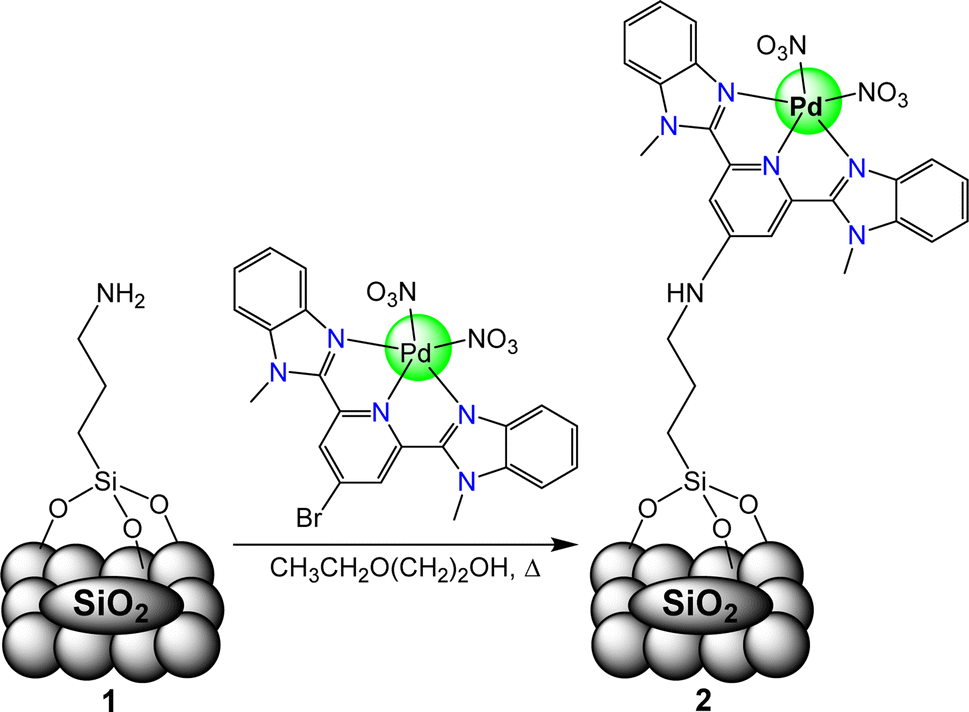 | ||
| Fig. 3 Graphical representation of the synthesis of the catalyst structure on the silica support. (1) 3-(Aminopropyl)triethoxysilane covalently bound to A300 support. (2) The hybrid catalyst chloro(2,6-bis(1-methylbenzimidazolyl)pyridine-4′-aminopropyl trisiloxane) palladium(II) nitrate/A300. | ||
For hybrid catalysts, there are two aspects of stability that must be considered. The first aspect being the binding of the molecular catalyst to the support. Stable binding prevents the molecular catalyst structure from detaching from the support.33 Acidic moieties have widely been used to bind organometallic compounds to metal oxide supports.34 These acidic moieties are susceptible to desorption from the support surface in polar organic solvents or aqueous conditions.35 The second aspect of hybrid catalyst stability is that of the structure of the molecular catalyst itself. Reports have shown that molecular catalysts can decompose into metallic nanoparticles at elevated temperatures, and that these catalyst decomposition products can remain catalytically active.32,36 Decomposition routes can also include leaching of the metal from the ligand into the reaction solution.37 Characterization of the hybrid catalysts before and after the catalytic reactions along with examining the reaction solution under catalytic conditions are important tests for determining catalyst stability.37
To address these stability issues of hybrid catalysts while also aiming to achieve high reactivity, we have designed a palladium hybrid catalyst with a 2,6-bis(1-methylbenzimidazolyl)pyridine (Mebimpy) ligand bound to SiO2 through silyl groups. Silyl binding to metal oxide supports occurs through covalent bonds to the support and has been shown to exhibit increased stability compared to acidic linking groups.38 The Mebimpy ligand was chosen due to previously exhibiting both increased stability and catalytic activity when directly compared to related terpyridine-containing catalysts.39 Here we show (2,6-bis(1-methylbenzimidazolyl)pyridine-4′-aminopropyltrisiloxane)palladium(II) nitrate [Pd(Mebimpy-APTS)](NO3)2 catalyst immobilized on a silica support through silyl binding exhibits increased stability and reactivity compared to previously reported molecular and hybrid HDO catalysts.28,32
Results and discussion
The synthesis of the catalyst, shown in Fig. 3, was performed stepwise and adapted from previously reported synthesis.40–43 The aerosil 300 silica surface, represented by SiO2 in Fig. 3, was functionalized with 3-(aminopropyl)triethoxysilane by stirring overnight in methanol at room temperature. Silane functionalized A300 (1) was isolated via vacuum filtration and washed with methanol to yield a white solid. The palladium molecular catalyst was synthesized by refluxing 2,6-bis(1′-methylbenzimidazolyl)-4-bromopyridine with palladium nitrate in ethanol. The molecular catalyst was covalently bound to the silane via a coupling reaction in 2-ethoxyethanol at reflux for 6 hours. Molecular catalyst attachment to the silane-functionalized silica (2) was characterized by XPS, solid-state NMR (Fig. S1†) and IR spectroscopy (Fig. S2†). Solid-state 13C NMR data shows evidence for Mebimpy presence on the A300 silica surface with peaks corresponding to aromatic carbons above 120 ppm and peaks corresponding to the aminopropyl linker between 70 and 0 ppm.38 The IR spectrum shows evidence of C–H stretching frequencies below 1550 cm−1. The XPS spectrum of the catalyst as synthesized is presented in Fig. 4a. The Pd3d5/2 binding energy was determined to be 338.2 eV which is consistent with a Pd(II) species bound to non-oxide anionic ligands,44 and the binding energy agrees with Pd bound to NO3 anions.45 This binding energy is also comparable to previously reported molecular palladium complexes bound to solid supports.46 Taken together, the spectroscopic characterization of the hybrid catalyst supports structure 2 illustrated in Fig. 3. The hybrid catalyst was also examined by a powder XRD (Fig. S3†) and ICP-MS. X-ray diffraction patterns of [Pd(Mebimpy-APTS)(NO3)2]/A300 showed no metallic palladium nanoparticles present within the limit of detection of the instrument.47 The amount of palladium present in a sample of [Pd(Mebimpy-APTS)Cl]Cl/A300 was determined via ICP-MS as 2.0 wt%. This is equivalent to 6.3 × 10−7 mol of catalyst per m2 of support, which is consistent with previously reported hybrid catalysts loadings.29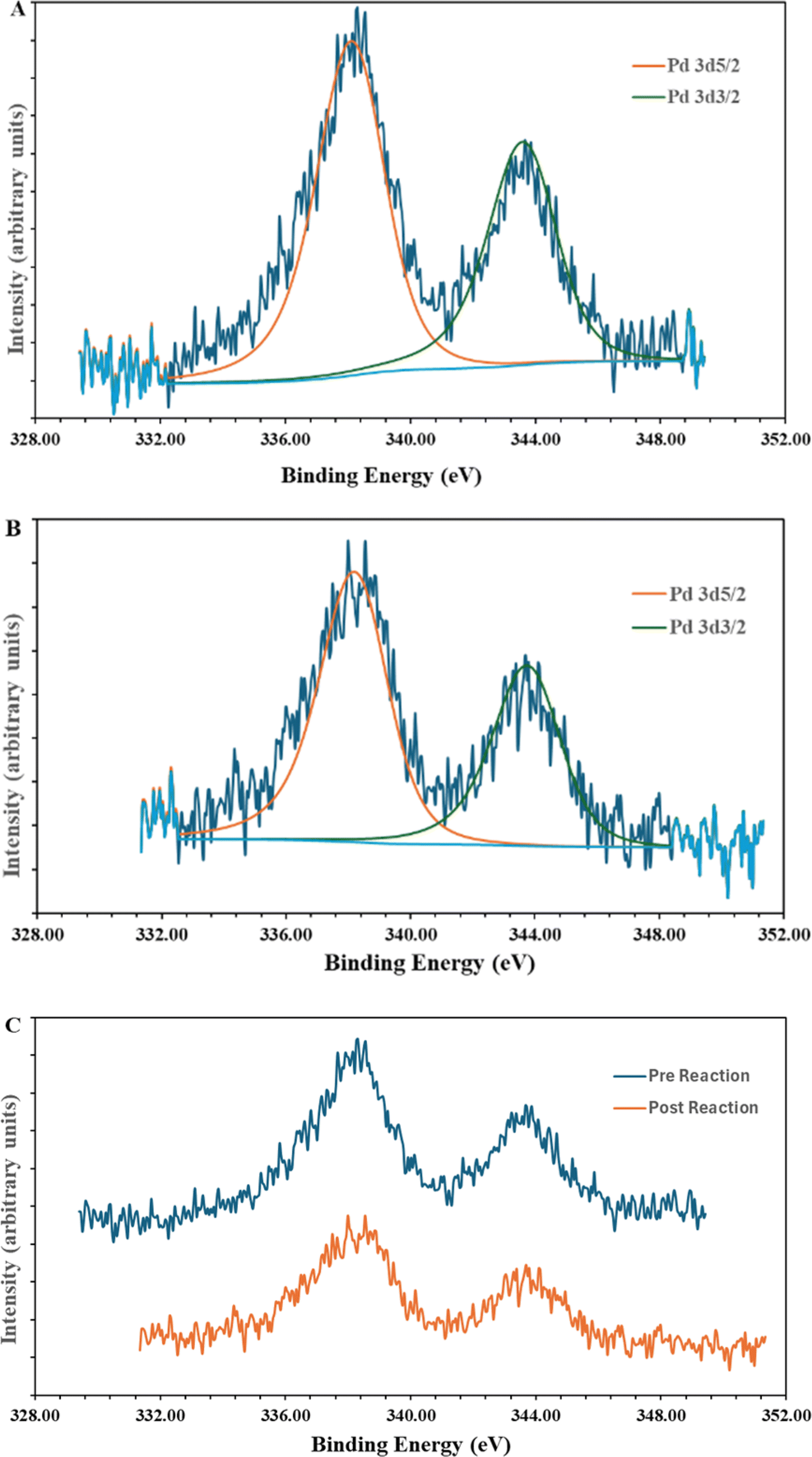 | ||
| Fig. 4 XPS spectra of hybrid catalyst 2. (A) Catalyst as synthesized; (B) catalyst post catalytic reaction; (C) overlay XPS spectrum of as synthesized (pre) catalyst, and catalyst post catalytic reaction. | ||
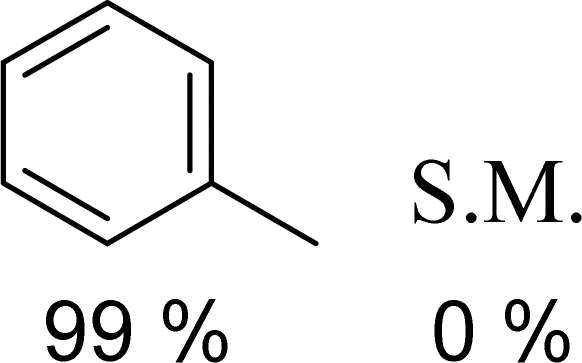
To initially explore the reactivity and stability of the catalyst, we examined the catalytic HDO of benzyl alcohol, which allows for direct comparison to previously reported studies.29,32,48 Initial reactions were performed in a stainless steel reactor pressurized with 20 bar of H2 and heated to 100 °C as had been previously reported.29 Reactions were conducted in n-dodecane solvent with a 0.1 M concentration of benzyl alcohol. The solvent was chosen due to its relatively high boiling point and non-coordinating nature.29 As shown in Table 1, reactions under these conditions for both 4 hours (entry 1) and 1 hour (entry 2) resulted in complete conversion of the benzyl alcohol substrate within the detection limits of our GC-MS. The only observed product was toluene, indicating complete selectivity towards HDO over ring hydrogenation. This level of conversion equates to 1250 turnovers of the catalyst. No evidence for ring hydrogenation or decarbonylation was observed via liquid injection GC-MS analysis of the post reaction mixture. The amount of toluene produced in the reaction was determined through calibration curves (see ESI†) and resulted in a reaction carbon balance of >99%, indicating direct conversion of benzyl alcohol to toluene.
|
|
|||
|---|---|---|---|
| Entry | H2 pressure | Reaction duration (h) | % yield toluene |
| a Conditions: 0.1 M benzyl alcohol in 25 mL of n-dodecane with 0.08 mol% (100 mg of solid catalyst at 2.0 wt% Pd) catalyst with respect to benzyl alcohol in a stainless steel reactor at 100 °C unless otherwise noted. All yields were repeated in triplicate and are ±3%. Products determined by GC-MS analysis. b Reaction conducted in a two-neck round bottom flask. c Reaction performed at 25 °C. d No catalyst added to the reaction mixture. | |||
| 1 | 20 bar | 4 | 99% |
| 2 | 20 bar | 1 | 99% |
| 3 | 10 bar | 1 | 99% |
| 4 | 1 bar | 1 | 99% |
| 5b | 1 bar | 1 | 99% |
| 6b | 1 bar | 0.5 | 95% |
| 7b | 1 bar | 0.25 | 40% |
| 8b,c | 1 bar | 1 | 0% |
| 9b | 0 bar | 1 | 0% |
| 10b,d | 1 bar | 1 | 1% |
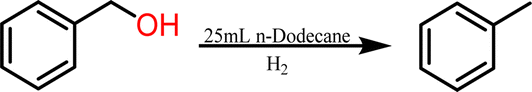
To examine the conditions needed to observe catalytic HDO, first the hydrogen pressure of the reaction was varied as shown in entries 2–4 in Table 1. The reactivity and selectivity of the catalysts was maintained when decreasing the hydrogen pressure from 20 bar to 1 bar under otherwise identical conditions. This is an advantageous result as performing reactions under lower hydrogen pressures conserves H2 gas and previous reports have shown that excess H2 pressures can lead to increased ring hydrogenated products for the conversion of lignin derived biomass.5 Additionally, reactions performed near one atmosphere pressure allows for performing reactions in standard laboratory glassware which may allow for easier reaction monitoring and sampling. Entry 5 in Table 1 shows the results of an HDO reaction performed in a glass round-bottom flask under one atm of H2 gas. Once again complete conversion of benzyl alcohol to toluene was achieved with near unity carbon balance. Next, the reaction duration was varied (entries 6 and 7). A reaction performed for 30 minutes resulted in 95% yield of toluene. GC-MS analysis of the post reaction mixture showed roughly 5% of the starting benzyl alcohol remained without observation of any other major product. Further reducing the reaction time to 15 minutes once again led to only toluene production and unreacted benzyl alcohol. While complete HDO selectivity for the conversion of benzyl alcohol to toluene has been achieved, it has not been achieved at these short timescales nor under these low H2 pressures.29 Control reactions show that elevated temperature (entry 8), hydrogen atmosphere (entry 9), and catalyst (entry 10) are necessary for the observed HDO reactivity.
With general conditions in hand where the catalyst exhibited stability, a kinetic study was performed to compare to previous results.29 A reaction with benzyl alcohol was prepared and performed under identical conditions to entry 5 of Table 1. Small aliquots (∼0.5 mL) of the reaction mixture were taken every 5 minutes for GC-MS analysis. After 30 minutes, solvent and substrate were replenished to mimic starting conditions for the reaction. This was repeated for five cycles and the samples were analyzed via GC-MS. The benzyl alcohol (BA) consumption across all five cycles is plotted in Fig. 5 and in each case the only measurable product observed was toluene. Individual plots of the consumption of benzyl alcohol can be found in Fig. S8–S12.† The data show a linear, first order dependance with respect to benzyl alcohol. This is consistent with a previously reported hybrid molecular HDO catalyst.29 This consistent, first-order reactivity over five cycles indicates hybrid catalyst stability during the reaction cycles. Small variances in kinetics between aliquot additions can be attributed to a buildup of the toluene product slowing reactivity through equilibrium effects and variances in substrate concentrations between runs.
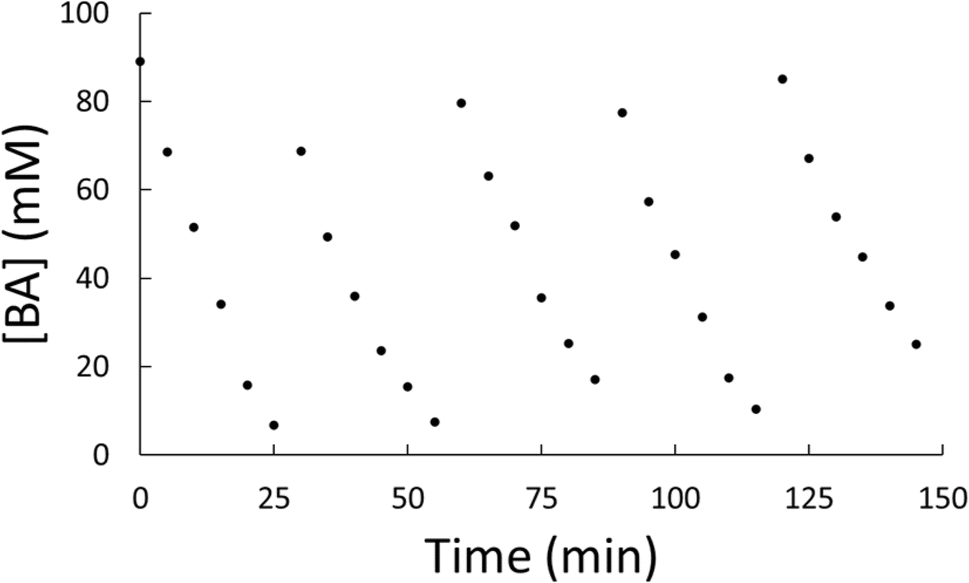 | ||
| Fig. 5 Plot of the measured concentration of benzyl alcohol, [BA], versus time for five reaction cycles of the same catalyst. | ||
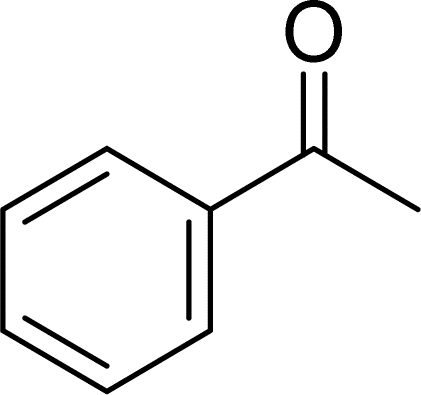
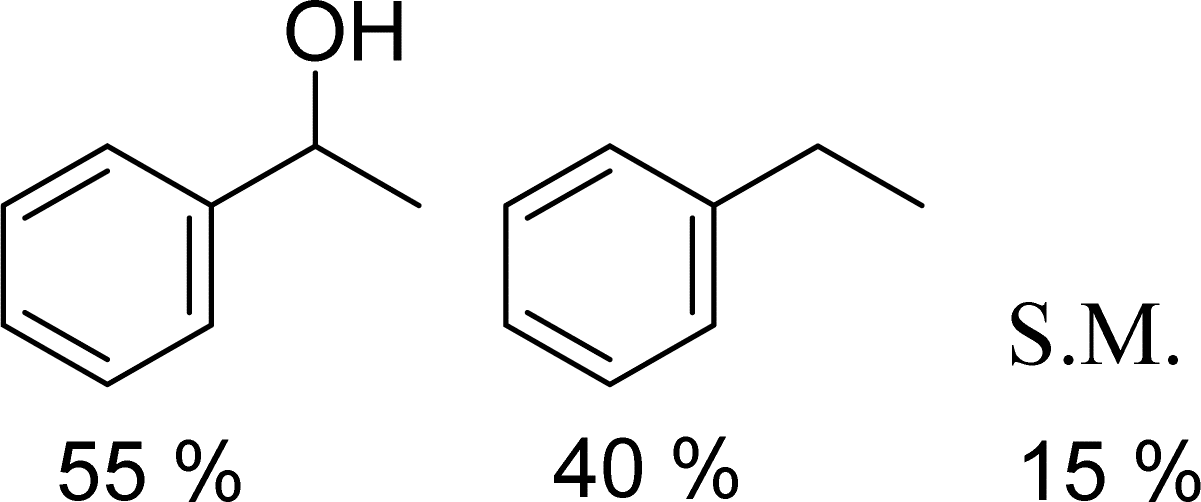
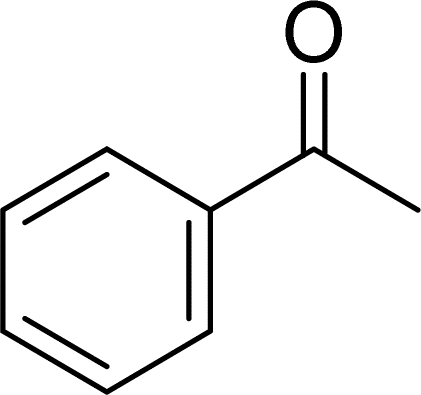
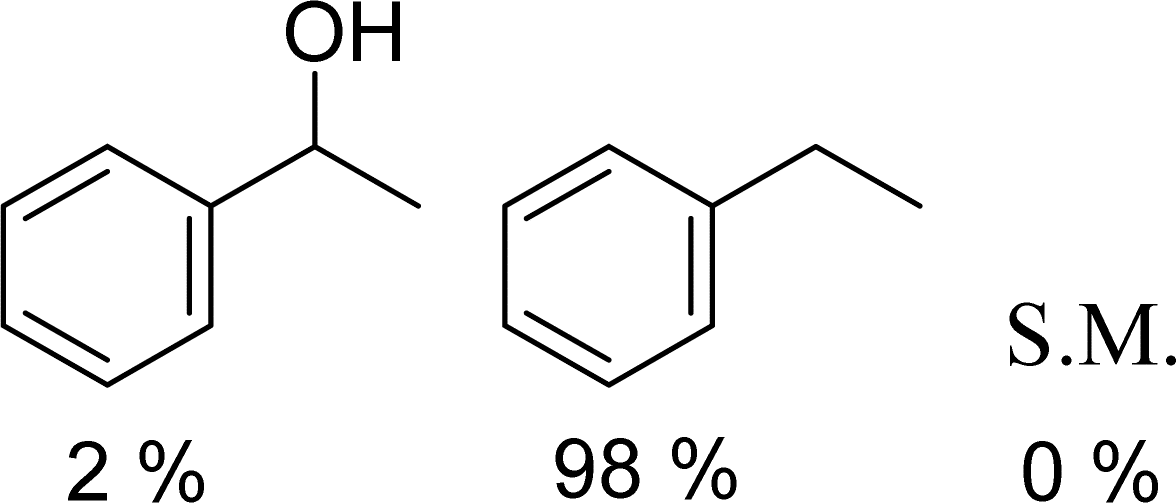
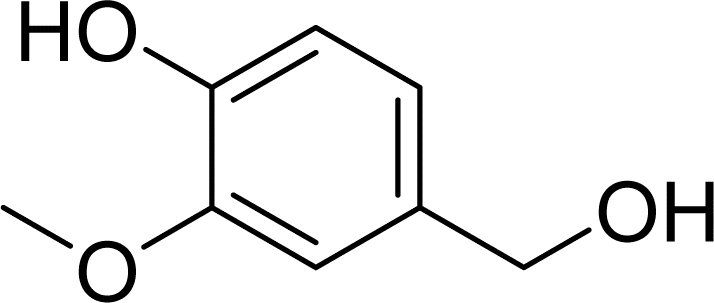
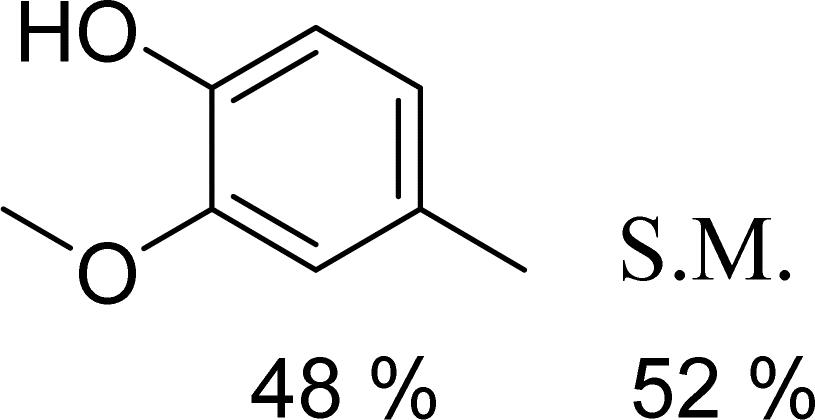
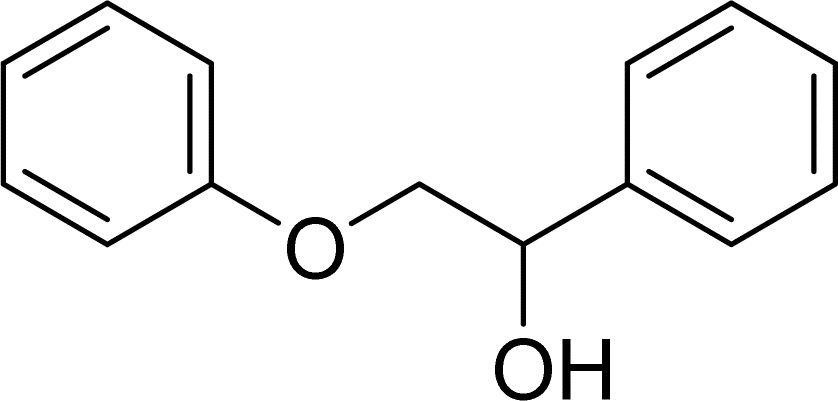
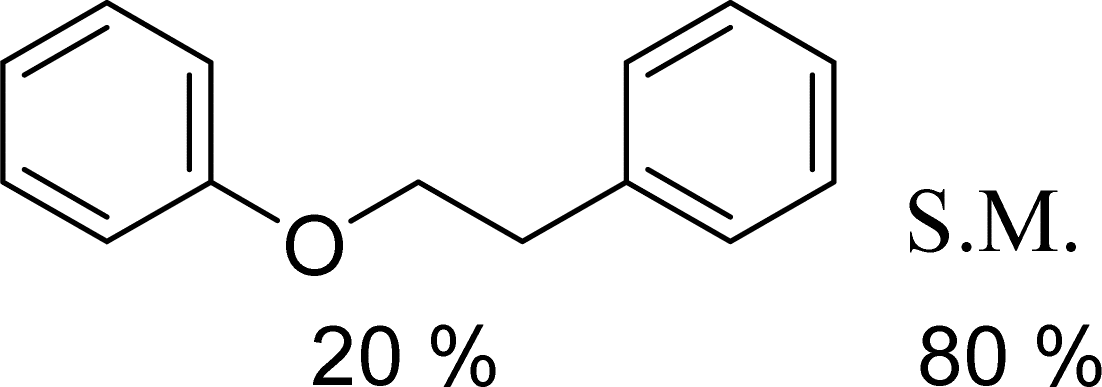
With general conditions in hand to achieve HDO on benzyl alcohol, we examined additional oxygenated aromatics for HDO reactivity. Acetophenone, 2-phenoxy-1-phenylethanol, and vanillyl alcohol were chosen as alternative aromatic oxygenates (Table 2). A two-hour reaction with acetophenone as the substrate resulted in high conversion and the formation of both ethylbenzene and phenylethane-1-ol. This product mixture is most likely due to the stepwise hydrogenation of the carbon–oxygen double bond which first converts the ketone to an alcohol, followed by the deoxygenation of the alcohol to form ethylbenzene as has been previously reported.28 This stepwise hypothesis is supported by performing the HDO of acetophenone for 6 hours. The longer reaction time led to complete consumption of the starting ketone and a decrease in the alcohol product with a concurrent increase in the deoxygenated ethylbenzene product. Reactions with vanillyl alcohol resulted in the formation of creosol via the deoxygenation of the benzylic C–O bond in vanillyl alcohol. While this reaction is directly analogous to the deoxygenation of benzyl alcohol, under identical conditions, less HDO reactivity was observed for vanillyl alcohol compared to benzyl alcohol. This reactivity trend may give insight into a possible reaction mechanism, as vanillyl alcohol is more electron rich than benzyl alcohol. The relative lack of reactivity between the catalyst and vanillyl alcohol may imply a metal hydride active species as metal hydrides are less prone to reactivity with electron rich substrates.49 Reactions with 2-phenoxy-1-phenylethanol resulted in the formation of phenethoxybenzene. This reaction was performed at 200 °C for 3.5 hours and no ring hydrogenation products were observed. This selectivity highlights the advantages of hybrid catalysts for HDO as ring hydrogenation is avoided as long as the catalyst maintains molecular integrity.
| Substrate | Temperature (°C) | Reaction length (h) | Products% yield |
|---|---|---|---|
| a Conditions: 0.1 M substrate in 25 mL of n-dodecane with 0.08 mol% catalyst with respect to substrate at 1 bar H2 pressure unless otherwise noted. S.M. is starting material remaining. All yields were repeated in triplicate and are ±5%. | |||
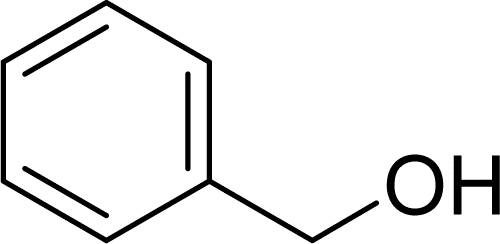
|
100 | 1 |
|
|
100 | 2 |
|
|
100 | 6 |
|
|
100 | 1 |
|
|
200 | 3.5 |
|
To explore the stability of the catalyst post reaction samples were analyzed via XPS, solid-state 13C NMR, and IR spectroscopy, and ICP-MS and powder XRD and compared to pre-reaction characterization. The post reaction XPS spectrum is shown in Fig. 4B. The Pd3d5/2 binding energy is consistent with a Pd(II) species bound to non-oxo anionic ligands. Furthermore, the overlay of the pre- and post-reaction XPS spectra is shown in Fig. 4C. There is no considerable shift or change in the XPS spectra after the catalytic reaction, indicating the Pd has maintained a consistent oxidation state and coordination sphere. This highlights the importance of ligand design, such as utilizing a tridentate ligand, to ensure the Pd maintains molecular integrity. To further support molecular integrity during the reaction, a filtration of a reaction was performed while the reaction was heated, and the reaction solution was analyzed with ICP-MS for leached Pd in solution. The analysis showed that 0.5 ppm Pd was present in the solution, which equates to roughly 0.6% of the Pd present in the hybrid catalyst on the SiO2 support. While this small amount of Pd may be active, it likely cannot account for all of the observed reactivity as no induction period is observed in the reaction and rapid hydrodeoxygenation occurs under mild conditions as shown in Table 1. In addition, the ICP-MS does not determine the nature of the Pd and the detected palladium could be molecular catalyst in solution. Post-reaction solid-state 13C NMR (Fig. S4†) and IR spectroscopy (Fig. S5†) after reaction display peaks corresponding to the molecular catalyst analogous to the pre-reaction spectra. This highlights the advantage and importance of the covalently bound silanol binding groups. Commonly employed acidic binding groups are only bound to the oxide supports via a hydrogen bonding network50 and this leads to hybrid catalysts that as susceptible to detaching from the surface of the support.51 In the powder XRD data, no peaks were observed corresponding to metallic palladium post reaction within the limit of detection of the instrument. This consistency of data between the pre- and post-reaction catalyst characterization suggests that the molecular catalyst is stable under the chosen HDO conditions and that the covalent bonding to the SiO2 support helps maintain the hybrid catalyst structure. This stability is an improvement compared to a previously reported molecular hybrid catalyst utilizing a terpyridine ligand bound to SiO2 support through an acidic binding group.29
Conclusions
A palladium hybrid catalyst bearing a bis-benzimidazole ligand covalently bound to a silica support was designed, characterized, and tested for catalytic hydrodeoxygenation of aromatic molecules related to lignin biomass. The ligand structure and covalent binding to the support led to a hybrid catalyst that exhibited molecular stability up to 200 °C. NMR, IR, and XPS spectroscopies along with ICP-MS supported the catalyst stability. Catalytic cycling and consistent product formation and selectivity for thousands of catalytic turnovers show the potential for hybrid catalysts in the field of biomass conversion. The selective deoxygenation of aromatic molecules without ring hydrogenation represents a direct and more sustainable route for the conversion of lignin biomass. An emphasis on increased reactivity towards difficult to activate bonds, such as phenolic C–O bonds, is needed for the next generation of selective HDO hybrid catalysts. The design principles examined within this study – single-site catalyst reactivity, binding stability of the molecular catalyst to the support and ligand design to promote reactivity – will aid in future catalyst design in the study of biomass conversion and beyond.Experimental
Materials
Methanol (99.9%, Fisher), ethanol (99.9%, Fisher), 2-ethoxyethanol (99.9%, Honeywell) and n-dodecane, (anhydrous, 99%, Thermo) were purchased and used without any further purification as solvents for catalyst synthesis and catalytic testing. Chelidamic acid monohydrate (>95%, TCI), N-methyl-1,2-phenylenediamine (97%, Thermo), polyphosphoric acid (84%, Alfa Aesar), sodium carbonate (Fisher), phosphorus(V) oxybromide (98%, BTC), and sodium hydroxide (Fisher) were used to synthesize the chosen ligand. Aerosil 300 (A300, Evonik) was used as the metal oxide support. (3-Aminopropyl)trimethoxysilane, (APTES, 97%, Thermo) was used to immobilize the ligand to the support. Palladium(II) nitrate (Sigma) and hydrochloric acid (37%, Fisher) were used to synthesize the catalyst. Benzyl alcohol (98%, Fisher), vanillyl alcohol (>98%, TCI), m-cresol (99%, Alfa Aesar), and 2-phenoxy-1-phenylethanol (97%, Ambeed) were used as substrates in catalytic testing.Instrumentation
Gas Chromatography-Mass Spectrometry (GC-MS) analysis was performed using a Shimadzu QP-2010S with a 30 m Rxi-5ms Restek separation column with a 0.25 mm inner diameter. The oven temperature program was started at 40 °C for 0.5 min, followed by a 10 °C min−1 increase in temperature up to 140 °C. The temperature was held for 2 min., followed by a 25 °C min−1 increase in temperature up to 300 °C, at which the temperature was held for 5 min. Solid-state NMR analysis was taken on a Bruker Avance III-HD (500 MHz). Inductively coupled plasma-mass spectrometry (ICP-MS, Finnigan ELEMENT XR double focusing magnetic field) was used with rhenium as the internal standard. A quartz torch and injector (Thermo Fisher Scientific) and 0.2 mL min−1 Micromist U-series nebulizer (GE, Australia) were used for sample injection. Infrared (IR) spectroscopy was obtained using a PerkinElmer Spectrum 100 FT-IT Spectrometer. All XRD data was collected using a Rigaku Miroflex Benchtop XRD with a solid state, high sensitivity D/tex detector. CuKα radiation was utilized at a sampling range of 20–80° 2θ at 3° per minute.The surface electronic environments of the catalysts were investigated with the AXIS Ultra DLD XPS (Kratos Analytical) instrument. The XPS system is equipped with a non-monochromatic Al Kα source (1486.8 eV) operated at 120 W X-ray power, a hemispherical analyzer, a charge neutralizer, a catalysis cell, and a load lock chamber for rapid introduction of samples without breaking vacuum. The X-rays were incident at an angle of 45°, with respect to the surface normal. Analysis was performed at a pressure of ∼1 × 109 torr and high-resolution core level spectra were acquired in the constant analyzer energy mode using a pass energy of 40 eV and a 0.05 eV step size. The XPS experiments were performed by using a low energy electron beam, directed at the sample, for charge neutralization. The curve fitting procedure was carried out using the Casa XPS software, and the peak approximation was carried out by a combination of Gaussian–Lorentzian functions, with subtraction of Shirley-type background. The binding energy (BE) scale of the scans are corrected in reference to the C ls peak at 285 eV (adventitious carbon from contamination) and given with an accuracy of ±0.2 eV.
Synthesis
2,6-Bis(1′-methylbenzimidazolyl)-4-hydroxypyridine (Mebimpy-OH) was synthesized from a reported literature procedure.42 To an oven dried round-bottom flask, n-methyl-1,2-phenylenediamine (1.92 mL, 16.38 mmol), chelidamic acid (1.50 g, 8.19 mmol), and polyphosphoric acid (20 mL) were added. The flask was heated to 220 °C and stirred for 12 hours. The hot mixture was poured into ice cold deionized water to yield a blue precipitate. The precipitate was filtered and resuspended in deionized water at 70 °C. Sodium carbonate was added to the solution until pH became 7. The solution was then filtered, washed with deionized water, and allowed to dry to yield a light grey powder.2,6-Bis(1′-methylbenzimidazolyl)-4-bromopyridine (Mebimpy-Br) was synthesized from a reported literature procedure.43 Mebimpy-OH (250 mg, 0.70 mmol) was dissolved in molten phosphorus(V) oxybromide (2.5 g, 8.7 mmol) at 140 °C under nitrogen. The reaction was stirred for 16 h and allowed to cool to room temperature. 40 mL of Milli-Q water was added to the reaction mixture. The mixture was stirred at 80 °C for 1 h. A 1 M NaOH solution was added dropwise until the solution was alkaline. The solution was filtered to yield a gray solid.
(2,6-Bis(1′-methylbenzimidazolyl)-4-bromopyridine)palladium(NO3)2 [Pd(Mebimpy-Br)(NO3)2] was synthesized from an modified literature procedure.41 Mebimpy-Br (50 mg, 0.121 mmol) and Pd(NO3)2 (28 mg, 0.122 mmol) were combined in a round bottom flask and were refluxed in ethanol (150 mL) at 90 °C for 3 h. The reaction was allowed to cool to room temperature and the solution was filtered to remove unreacted Pd(NO3)2. The solid was rinsed with a small amount of deionized water to remove the product from the filter paper. The product was obtained as a green solid via rotary evaporation of the filtrate. The green solid was used without further purification for further catalyst synthesis. 3-(Aminopropyl) triethoxysilane/A300 (APTES/A300) was prepared from an altered literature procedure.40 3-(Aminopropyl)triethoxysilane (300 μL, 1.3 mmol) and A300 (3.0 g) were stirred at room temperature in methanol (300 mL) for 16 h. The functionalized silica was filtered and washed in triplicate with methanol. The APTES/A300 was allowed to dry overnight and crushed into a fine powder with a mortar and pestle.
(2,6-Bis(1-methylbenzimidazolyl)pyridine-4′-aminopropyltrisiloxane)palladium(II) nitrate/A300 ([Pd(Mebimpy_APTS)(NO3)2]/A300) was synthesized from a modified literature procedure.40 To a 250 mL round bottom flask, APTES/A300 (1 g), [Pd(Mebimpy-Br)(NO3)2] (50 mg, 0.121 mmol), and 2-ethoxyethanol (50 mL) were added. The mixture was refluxed at 140 °C for 6 h and allowed to cool to room temperature. The resulting solid was filtered via vacuum filtration and rinsed with fresh ethanol in triplicate. The solid was allowed to dry under vacuum and crushed into a fine powder using a mortar and pestle. The amount of palladium present in a sample of [Pd(Mebimpy-APTS)Cl]Cl/A300 (3) was determined via ICP-MS as 2.0 wt%
Catalytic testing - general reaction procedures
High pressure reactions (≥1 atm) were conducted in a 450 mL Parr Pressure Vessel. Typical experiments utilized benzyl alcohol (260 μL, 0.10 M) as the substrate with chloro(2,6-bis(1-methylbenzimidazolyl)pyridine-4′-aminopropyltrisoloxane)palladium(II) nitrate/A300 (100 mg/0.08 mol% Pd catalyst with respect to the benzyl alcohol substrate) in 25 mL of n-dodecane. High pressure reactions were properly sealed in the reaction vessel and purged with nitrogen three times followed by three purging cycles of hydrogen at the chosen reaction pressure (1–20 bar H2). The reaction vessel was then filled with the desired pressure of H2. A heating jacket was used to reach the desired reaction temperatures. Reactions were stirred at 300 rpm for the duration of the reactions. The catalyst was separated from the reaction mixture via vacuum filtration, rinsed with hexanes, and allowed to dry for post reaction analysis.Reactions at roughly atmospheric pressure were conducted in a two neck round bottom flask. Typical experiments utilized benzyl alcohol (260 μL, 0.10 M) as the substrate with chloro(2,6-bis(1-methylbenzimidazolyl)pyridine-4′-aminopropyltrisoloxane)palladium(II) chloride/A300 (0.08 mol%) in 25 mL of n-dodecane. One neck was sealed with a rubber septum. The other neck was connected to a reflux condenser to minimize evaporation of desired products. The sealed system was purged with nitrogen for 10 min, then the system was purged with hydrogen for 10 min. The reaction mixture was heated to the desired temperature using either an oil bath (≤150 °C) or a sand bath (>150 °C). Catalyst was separated from the reaction via vacuum filtration for post reaction analysis.
Calibration curves for product concentration determination were made as assays containing the starting substrate at 0.1 M concentrations and product concentrations varied from 0 M up to 0.1 M concentrations in dodecane solvent.
Catalytic testing – recyclability/low conversion and sampling
All recyclability testing was conducted in a two neck round bottom flask. The flask was prepared as stated above without benzyl alcohol. Once the flask, catalyst and solvent reached the desired reaction temperature, benzyl alcohol (260 μL, 0.10 M) was injected. Every 5 min., 0.5 mL of the reaction solution was sampled using a syringe. Every 30 min. (6 samples), 3 mL of 0.10 M benzyl alcohol in n-dodecane was injected into the reaction and H2 was bubbled for 5 seconds to replace the solvent lost from sampling and replenish consumed substrate. This process was repeated for 3–5 cycles.Filtration of the reaction solution while under heating was performed after performing the reaction at 100 °C for 6 hours. The reaction solution was passed through a medium glass frit Buchner funnel at 100 °C and the filtrate was analyzed using ICP-MS.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Acknowledgement is made to the ACS GCI Pharmaceutical Roundtable Research Ignition Grant for support of this research. The XPS facility receives financial support from the University of South Carolina Office of the Vice President for Research.References
- M. P. Pandey and C. S. Kim, Chem. Eng. Technol., 2011, 34, 29–41 CrossRef CAS.
- S. De, B. Saha and R. Luque, Bioresour. Technol., 2015, 178, 108–118 CrossRef CAS.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- A. Bohre, S. Dutta, B. Saha, M. M. Abu-Omar and A. C. S. Sus, Chem. Eng., 2015, 3, 1263–1277 CAS.
- J. G. Tillou, C. J. Ezeorah, J. J. Kuchta, S. C. D. Dissanayake Mudiyanselage, J. D. Sitter and A. K. Vannucci, RSC Sustainability, 2023, 1, 1608–1633 RSC.
- Y. Jing, L. Dong, Y. Guo, X. Liu and Y. Wang, ChemSusChem, 2020, 13, 4181–4198 CrossRef CAS PubMed.
- J. Zhang, J. Sun and Y. Wang, Green Chem., 2020, 22, 1072–1098 RSC.
- R. Shu, R. Li, B. Lin, C. Wang, Z. Cheng and Y. Chen, Biomass Bioenergy, 2020, 132, 105432 CrossRef CAS.
- T. Renders, G. Van den Bossche, T. Vangeel, K. Van Aelst and B. Sels, Curr. Opin. Biotechnol., 2019, 56, 193–201 CrossRef CAS.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- S. Mukundan, R. K. Chowdari and J. Beltramini, Adv. Sustainable Syst., 2021, 5, 2000243 CrossRef CAS.
- L. Dong, Y. Xin, X. Liu, Y. Guo, C.-W. Pao, J.-L. Chen and Y. Wang, Green Chem., 2019, 21, 3081–3090 RSC.
- H. Ohta, K. Tobayashi, A. Kuroo, M. Nakatsuka, H. Kobayashi, A. Fukuoka, G. Hamasaka, Y. Uozumi, H. Murayama, M. Tokunaga and M. Hayashi, Chem.–Eur. J., 2019, 25, 14762–14766 CrossRef CAS PubMed.
- S. Echeandia, P. L. Arias, V. L. Barrio, B. Pawelec and J. L. G. Fierro, Appl. Catal., B, 2010, 101, 1–12 CrossRef CAS.
- X. Wang and R. Rinaldi, Angew. Chem., Int. Ed., 2013, 52, 11499–11503 CrossRef CAS PubMed.
- B. Feng, H. Kobayashi, H. Ohta and A. Fukuoka, J. Mol. Catal. A: Chem., 2014, 388–389, 41–46 CrossRef CAS.
- X. Zhang, W. Tang, Q. Zhang, Y. Li, L. Chen, Y. Xu, C. Wang and L. Ma, Fuel, 2018, 215, 825–834 CrossRef CAS.
- G. Zhou, P. A. Jensen, D. M. Le, N. O. Knudsen, A. D. Jensen and A. C. S. Sus, Chem. Eng., 2016, 4, 5432–5440 CAS.
- N. Yan, Y. Yuan, R. Dykeman, Y. Kou and P. J. Dyson, Angew. Chem., Int. Ed., 2010, 49, 5549–5553 CrossRef CAS.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS.
- X. Wu, Q. Ge and X. Zhu, Catal. Today, 2021, 365, 143–161 CrossRef CAS.
- Q. Lai, C. Zhang and J. H. Holles, Appl. Catal., A, 2016, 528, 1–13 CrossRef CAS.
- C. Liu, C. Zhou, Y. Wang, X. Liu, L. Zhu, H. Ma, Z. Zhou and F. Qi, Proc. Combust. Inst., 2021, 38, 4345–4353 CrossRef CAS.
- G. Kumar, E. Nikolla, S. Linic, J. W. Medlin and M. J. Janik, ACS Catal., 2018, 8, 3202–3208 CrossRef CAS.
- A. H. Jenkins and J. W. Medlin, Acc. Chem. Res., 2021, 54, 4080–4090 CrossRef CAS PubMed.
- P. Hao, D. K. Schwartz and J. W. Medlin, ACS Catal., 2018, 8, 11165–11173 CrossRef CAS.
- W. Yao, S. Das, N. A. DeLucia, F. Qu, C. M. Boudreaux, A. K. Vannucci and E. T. Papish, Organometallics, 2020, 39, 662–669 CrossRef CAS.
- N. A. DeLucia, N. Das, S. Overa, A. Paul and A. K. Vannucci, Catal. Today, 2018, 302, 146–150 CrossRef CAS.
- N. A. DeLucia, A. Jystad, K. V. Laan, J. M. M. Tengco, M. Caricato and A. K. Vannucci, ACS Catal., 2019, 9, 9060–9071 CrossRef CAS.
- W. Yao, C. A. Buell, A. Kuppravalli, A. K. Vannucci and E. T. Papish, Organometallics, 2023, 42, 2806–2812 CrossRef CAS.
- P. J. Ayare, S. A. Gregory, R. J. Key, A. E. Short, J. G. Tillou, J. D. Sitter, T. Yom, D. W. Goodlett, D.-C. Lee, F. M. Alamgir, M. D. Losego and A. K. Vannucci, Green Chem., 2021, 23, 9523–9533 RSC.
- J. G. Tillou and A. K. Vannucci, J. Organomet. Chem., 2021, 944, 121848 CrossRef CAS.
- R. J. Key and A. K. Vannucci, Organometallics, 2018, 37, 1468–1472 CrossRef CAS.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- K. Hanson, M. K. Brennaman, H. Luo, C. R. K. Glasson, J. J. Concepcion, W. Song and T. J. Meyer, ACS Appl. Mater. Interfaces, 2012, 4, 1462–1469 CrossRef CAS.
- J. Huang, J. D. Blakemore, D. Fazi, O. Kokhan, N. D. Schley, R. H. Crabtree, G. W. Brudvig and D. M. Tiede, Phys. Chem. Chem. Phys., 2014, 16, 1814–1819 RSC.
- N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS.
- X. Jia, H. S. Nedzbala, S. R. Bottum, J. F. Cahoon, J. J. Concepcion, C. L. Donley, A. Gang, Q. Han, N. Hazari, M. C. Kessinger, M. R. Lockett, J. M. Mayer, B. Q. Mercado, G. J. Meyer, A. J. Pearce, C. L. Rooney, R. N. Sampaio, B. Shang and H. Wang, Inorg. Chem., 2023, 62, 2359–2375 CrossRef CAS PubMed.
- Z. Chen, J. J. Concepcion, J. F. Hull, P. G. Hoertz and T. J. Meyer, Dalton Trans., 2010, 39, 6950–6952 RSC.
- Z. Singh, P. R. Donnarumma and M. B. Majewski, Inorg. Chem., 2020, 59, 12994–12999 CrossRef CAS PubMed.
- Shivakumaraiah and N. M. N. Gowda, J. Chem. Res., 2005, 2005, 505–507 CrossRef.
- P. B. J. S. Onge, T.-C. Chen, A. Langlois, A. Younus, I. Jo Hai, B.-H. Lin, Y.-C. Chiu and S. Rondeau-Gagné, J. Mater. Chem. C, 2020, 8, 8213–8223 RSC.
- K. Cheng and B. Tieke, RSC Adv., 2014, 4, 25079–25088 RSC.
- M. Brun, A. Berthet and J. C. Bertolini, J. Electron Spectrosc. Relat. Phenom., 1999, 104, 55–60 CrossRef CAS.
- T. L. Barr, J. Vac. Sci. Technol., A, 1991, 9, 1793–1805 CrossRef CAS.
- L. Fernández-García, M. Blanco, C. Blanco, P. Álvarez, M. Granda, R. Santamaría and R. Menéndez, J. Mol. Catal. A: Chem., 2016, 416, 140–146 CrossRef.
- K. O'Connell and J. R. Regalbuto, Catal. Lett., 2015, 145, 777–783 CrossRef.
- C.-H. Lien and J. W. Medlin, J. Phys. Chem. C, 2014, 118, 23783–23789 CrossRef CAS.
- E. S. Wiedner, M. B. Chambers, C. L. Pitman, R. M. Bullock, A. J. M. Miller and A. M. Appel, Chem. Rev., 2016, 116, 8655–8692 CrossRef CAS PubMed.
- K. L. Materna, R. H. Crabtree and G. W. Brudvig, Chem. Soc. Rev., 2017, 46, 6099–6110 RSC.
- A. K. Vannucci, L. Alibabaei, M. D. Losego, J. J. Concepcion, B. Kalanyan, G. N. Parsons and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20918–20922 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00333k |
| This journal is © The Royal Society of Chemistry 2024 |