
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lignocellulosic biomass valorisation: a review of feedstocks, processes and potential value chains and their implications for the decision-making process†
Britt
Segers
 a,
Philippe
Nimmegeers
abc,
Marc
Spiller
def,
Giorgio
Tofani
g,
Edita
Jasiukaitytė-Grojzdek
g,
Elina
Dace
hi,
Timo
Kikas
j,
Jorge M.
Marchetti
k,
Milena
Rajić
l,
Güray
Yildiz
m and
Pieter
Billen
*ae
a,
Philippe
Nimmegeers
abc,
Marc
Spiller
def,
Giorgio
Tofani
g,
Edita
Jasiukaitytė-Grojzdek
g,
Elina
Dace
hi,
Timo
Kikas
j,
Jorge M.
Marchetti
k,
Milena
Rajić
l,
Güray
Yildiz
m and
Pieter
Billen
*ae
aIntelligence in Processes, Advanced Catalysts and Solvents (iPRACS), Faculty of Applied Engineering, University of Antwerp, Groenenborgerlaan 171, 2020 Antwerp, Belgium
bEnvironmental Economics (EnvEcon), Department of Engineering Management, Faculty of Business and Economics, University of Antwerp, Prinsstraat 13, 2000 Antwerp, Belgium
cFlanders Make@UAntwerp, 2000 Antwerp, Belgium
dBiobased Sustainability Engineering (SUSTAIN), Department of Bioscience Engineering, University of Antwerp, 2020 Antwerpen, Belgium
eCentre for Advanced Process Technology for Urban Resource Recovery (CAPTURE), Frieda Saeysstraat 1, 9052 Gent, Belgium
fVITO WaterClimateHub, Wetenschapspark 1, 8400 Oostende, Belgium
gDepartment of Catalysis and Chemical Reaction Engineering, National Institute of Chemistry, Hajdrihova 19, 1000 Ljubljana, Slovenia
hBaltic Studies Centre, Kokneses Prospekts 26-2, Riga, LV-1014, Latvia
iInstitute of Microbiology and Biotechnology, University of Latvia, Jelgavas iela 1, Riga, LV-1004, Latvia
jBiosystems Engineering, Institute of Forestry and Engineering, Estonian University of Life Sciences, Kreutzwaldi 56, Tartu, 51014, Estonia
kFaculty of Science and Technology, Norwegian University of Life Sciences, Drøbakveien 31, 1432 Ås, Norway
lDepartment of Management in Mechanical Engineering, Faculty of Mechanical Engineering, University of Niš, Aleksandra Medvedeva 14, 18000 Niš, Serbia
mDepartment of Materials Engineering and Production, Faculty of Mechanical Engineering, Bialystok University of Technology, Wiejska 45C, 15-351 Bialystok, Poland
First published on 10th October 2024
Abstract
Several studies have reported the importance of transforming the current fossil-based economy into a bio-economy. Lignocellulosic biomass, as the most abundant renewable feedstock, has high potential. However, in practice, its use is limited to energy generation. This study aims to provide an overview of the potential lignocellulosic valorisation pathways and identify the next steps that should be taken to move towards a bio-economy. The study reviews the lignocellulosic biomass feedstocks and their compositional differences, depending on the type, valorisation processes, and value chains that can be created by selecting the respective valorisation processes. The study shows the abundance of pathways that can be created when attempting to link lignocellulosic biomass with a high diversity of compositions to the many potential end-products that can be created. Due to this abundance, selecting the optimal biomass-end-product combinations for the development of a sustainable bio-economy is challenging. Current state-of-the-art process-based assessment methods (TEA/LCA) have limited genericity, as they are only valid for specific processes at a specific time and place. As a result, using these types of assessments to try to find optimal biomass-end-product combinations would require too much time, data, and expertise. This creates the need to shift away from process-based assessments to state-based assessments, where the path of least thermodynamic resistance is sought.
Sustainability spotlightTransitioning from the current fossil-based economy into a bio-based economy shows high potential to fill in the gap created by the decreasing fossil resource supply and increasing demand for their derived end products. Lignocellulosic biomass, the most abundant renewable feedstock, is investigated to fill this role by valorising their main constituents (cellulose, hemicellulose, and lignin) in a variety of end products (fuels, chemicals, and materials), creating an abundance of potential biomass-end product combinations. This review proposes a novel state-based assessment to facilitate the decision-making process to identify these optimal combinations and implement the bio-economy in a sustainable way. This work aligns with the following UN sustainable development goals: affordable and clean energy (SDG7), responsible consumption and production (SDG12), and climate action (SDG13). |
Introduction
Up to 2022, around 80% of all energy is provided by fossil fuels,1 which remains the dominant feedstock for fuels and value-added chemicals.2 As a result, despite ongoing efforts to develop renewable energy technologies, the current global economy is still dependent on fossil-based feedstocks.Owing to the very long regeneration time of fossil feedstocks and their much shorter consumption time, the supply of fossil feedstocks is limited. Due to this time difference, the CO2 produced when fossil resources are used is considered a net emission.3,4 Furthermore, these CO2 emissions are considered to be the largest contributor to climate change by trapping heat in the atmosphere.1,5
Net-zero ambitions set by governments have been developed to reduce greenhouse gas (GHG) emissions by 2050,6 with Europe aiming for climate neutrality6 and the U.S. for net-zero emissions,7 while China aims for carbon neutrality by 2060,8 resulting in an expected decline of fossil refining capacity, particularly in Europe, of 4–14% (2030) and 19–58% (2040), with a global decrease of 9–23% by 2040.9 In addition to the declining supply of fossil resources, demand for fossil-derived end-products is expected to increase, leading to rising prices for end-products.9 To face future demand and rising prices, alternative resources are essential and will become more competitive as backstop technologies.10 Biomass has shown potential to fulfil this role as an alternative resource.
The ever-increasing CO2 emissions have led to the need for a renewable feedstock with a regeneration time that is comparable to the fuel consumption time and biochemical production with the potential to replace the fossil counterpart, creating the so-called bio-economy concept.3,4,11 Biomass is a renewable carbon source and can be considered a good alternative to its fossil counterpart. As plants absorb CO2 from the atmosphere through photosynthesis and convert it into carbohydrates, ideally no additional carbon is emitted. In reality, the valorisation processes to obtain end-products from biomass still require energy that currently comes from CO2-emitting resources.12
The EU has highlighted the importance of the transition to a bio-economy by developing a strategy to strengthen the bio-based sector, establishing new local biorefineries, and understanding the environmental limits of the bio-economy, which are being further developed by local authorities.13,14
Biomass can generally be considered as the total amount of material derived from living organisms in a given space at a given time. These living organisms are also known as organic matter, and are important sources of carbon.4,15 Multiple sources of biomass exist, which can be divided into six categories: energy crops, agricultural products and waste, wood and forestry, municipal waste, aquatic biomass, and industrial waste and co-products.15–22Fig. 1 provides an overview of the primary sources of biomass. First, the category of energy crops includes non-food crops on surplus or degraded land with high energy potential.16–19,21 Second, the agricultural products and residues category includes primary crops (including food crops) and green/horticultural residues.15–18,20–22 Third, wood and forestry biomass are biomass that can be harvested specifically or as a residue.15–22 Fourth, municipal waste is a category that includes organic waste, such as food and materials, both liquid and solid.15–22 Next, the aquatic biomass category is a combination of algae, sludge, and wastewater.16,19,20 Finally, industrial waste and co-products include a variety of waste, e.g., animal waste, manure, slaughterhouse by-products, gases, post-fermentation microbial biomass, etc.15,17,18,20–23
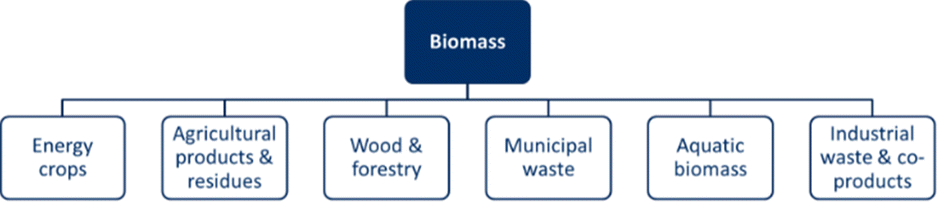 | ||
| Fig. 1 Overview of the different biomass streams. | ||
At the same time, these six categories include several types of biomass based on its composition. One specific type is lignocellulosic biomass, whose possible sources include woody plant species, energy crops, agricultural residues, grasses, and industrial waste from certain factories (e.g., pulp industry).21,24–27
Lignocellulosic biomass can be converted into various end-products using pretreatment and conversion methods. The conventional biomass conversion involves technologies analogous to those utilised in petrochemistry.28 A biorefinery using biomass as a feedstock is thus conceptually analogous to a petroleum refinery.24 Specifically, a lignocellulosic biorefinery uses lignocellulosic feedstocks.29 This type of biorefinery has high potential because the feedstocks are available in large quantities, and are not limited to one region of the world.30 However, there are differences within biorefineries in terms of the feedstock they use. In the past, lignocellulosic biorefineries focused on the valorisation of polycarbohydrates (cellulose and hemicelluloses), considering lignin as a component to be eliminated.31 More recently, the need for a full valorisation of biomass with a zero-waste approach requires a greater focus on the separation of lignin in a so-called “lignin-first biorefinery”.32
Lignocellulosic biorefineries have the potential to become an alternative to fossil-based refineries.30 These types of refineries are considered to be one of the most powerful approaches to producing fuels, platform chemicals, and materials.31 Furthermore, the use of lignocellulosic biomass has enabled the creation of second-generation biorefineries that use non-food biomass as a feedstock.33–35 In 2019, more than 40 lignocellulosic biorefineries were already in operation across Europe.36
There are certain advantages to using lignocellulosic biomass to produce the end-products that are generally produced by their fossil counterpart. In addition to reducing the use of non-renewable resources, bio-based feedstocks can be either biodegradable or reusable, rather than being used for energy consumption after a single use. After multiple cycles of use, energy recovery is still possible.18
Biofuels can be compared to their petrochemical counterparts. In general, biofuels use the same process as fossil fuels, where energy is released when the fuels are burned. However, there is a major difference when considering the formation of biofuels and fossil fuels. Biofuels release the energy stored in the biomass during its short lifetime. Fossil fuels, on the contrary, take much longer to regenerate.15 Biofuels can be produced from lignocellulosic biomass, which is considered the most abundant renewable feedstock and is cheaper than crude oil.25,26,28,33 The use of cellulose originating from lignocellulose enables the production of second-generation biofuels. Compared to first-generation biofuels, which use food-competing resources such as corn and soy,21,29,37,38 these second-generation biofuels do not compete with food.24,29 It should be noted that not every type of fuel can be produced from lignocellulosic biomass. Due to its low oil content, biodiesel is not preferred.39
Despite its many advantages, there are still some trade-offs in using lignocellulosic biomass instead of fossil resources. First, depending on the origin of the feedstock (pristine biomass or lignocellulosic waste), other impurities (e.g., metals and plastic derivatives) may be present and the structural complexity of the biomass itself makes the conversion process more difficult.40 This conversion process is further complicated by the intrinsic heterogeneity of lignocellulosic biomass and its components, particularly lignin.32 Second, the overuse of biomass could lead to a loss of biodiversity.41 Third, market price fluctuations make it difficult to estimate biomass prices.36 Next, fuels derived from biomass are in an oxidised state and require a deoxygenation step to become a drop-in fuel.42,43 Meanwhile, for chemical production, it is difficult to achieve high yields in an energy-efficient manner.44 Finally, many biorefinery processes are still in the early stages of development, and are therefore not as mature as fossil refineries, which are smaller in scale and in higher cost. In general, there are still many barriers that make it difficult for lignocellulosic biorefineries to become the main technology for our daily needs.
Lignocellulosic biomass is a large group within biomass, with great diversity in the composition of both the biomass itself and its potential end-products. The composition of lignocellulosic biomass is determined by the type, whereas the end-products can have a broad or narrow range of composition, depending on the application. Ideally, biomass-end-product combinations are created, as shown in Fig. 2, where biomass is linked to an end-product via a conversion pathway. By creating such biomass-end-product combinations, an optimal pathway to a specific end-product can be determined for each lignocellulosic biomass feedstock. Conversely, the optimal process and lignocellulosic biomass can be determined for a specific desired end-product. Due to the large number of combinations in terms of the biomass type, composition, potential end-products, and conversion pathways, this is not a straightforward task.
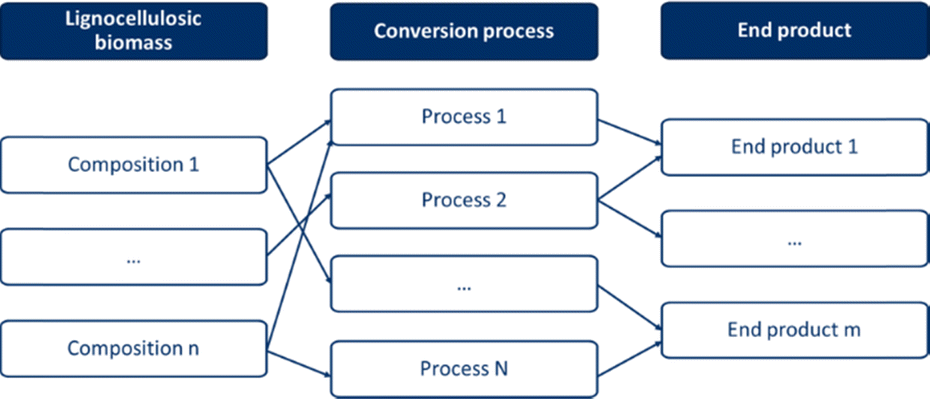 | ||
| Fig. 2 Lignocellulosic biomass valorisation pathways. | ||
This work focuses on the main biopolymers present in lignocellulosic biomass (cellulose, hemicelluloses, and lignin). The valorisation of other components such as ash and extractives is less developed. This research aims to address the complexities of implementing a lignocellulose-based economy as an alternative to the current fossil-based economy. The current literature mainly focuses on specific case studies, investigating a specific biomass in a specific process, and on specific end-product(s), and lacks a global overview and potential to select optimal biomass-end-product combinations. Furthermore, the application of current state-of-the-art quantitative assessments lacks in applicability to the development of such biomass-end-product combinations. Many developments are currently taking place in the field of quantitative assessments with the introduction of ex-ante Life Cycle Assessments (LCA),45 learning effects in Techno-Economic Assessments (TEA),46 Geospatial Environmental Techno-Economic Assessments (ETEA),47 Superstructure Optimisation,48etc. However, these types of assessments still focus on specific processes, creating a need for novel assessment methods that can easily compare potential biomass-end-product combinations. This study proposes the development of new quantitative state-based assessment methods based on properties available at each Technology Readiness Level (TRL) as a shortcut to the current state-of-the-art assessments. Statistical Entropy Analysis (SEA)49 is a more recent assessment method whose applicability has been investigated in various fields, ranging from resource efficiency,50 quantification of recyclability,51 material flow analysis,52 and linking assessment to circular economy principles.53 In addition, a variety of feedstocks have been studied, ranging from metals to e-waste and plastics. Although SEA has not yet been applied to the valorisation of lignocellulosic biomass, the potential it has shown in different sectors and for a variety of feedstocks is promising for this novel application.
Lignocellulosic biomass
What is lignocellulosic biomass?
Lignocellulosic biomass consists mainly of three polymers: cellulose, hemicelluloses, and lignin. Cellulose is a linear (C6H10O5)n polymer formed by linking monomeric D-glucose and dimeric cellobiose through β-1,4-glycosidic bonds. While cellulose is generally crystalline, a combination of both amorphous and crystalline regions can be observed in plants or biomass. The degree of polymerisation depends on the type of lignocellulosic biomass. In contrast to cellulose, hemicelluloses are a combination of various monomers, consisting of five different C5 (xylose and arabinose) and C6 sugars (galactose, glucose, and mannose). The exact composition of hemicelluloses varies with the type of lignocellulosic biomass. Furthermore, hemicelluloses are amorphous due to its heterogeneous and complex structure. One of the main functions of hemicelluloses is to create links between cellulose and lignin. Lignin is a three-dimensional structure of three phenyl propanoic monomers (coniferyl, sinapyl, and p-coumaryl alcohols), and is therefore an aromatic heteropolymer. Once part of the polymeric lignin structures, the three monolignols are referred to as guaiacyl, syringyl, and p-hydroxy-phenyl subunits, respectively. Similar to hemicelluloses, the exact composition of lignin also varies with the type of lignocellulosic biomass. The main function of lignin is to provide structural support and resistance to environmental influences, and it is also considered to be the glue within lignocellulosic biomass.19,24,29,36,39,54–58 The chemical structures of all three constituents are shown in Fig. 3.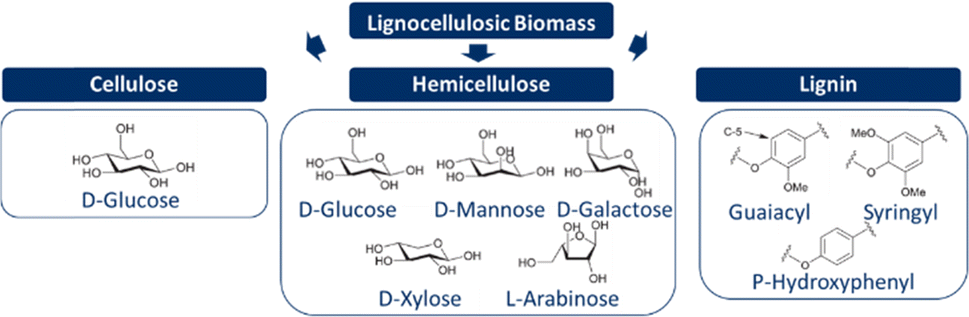 | ||
| Fig. 3 Constituents of the lignocellulosic biomass and their chemical structure. | ||
Types of/and categorisation of lignocellulosic biomass based on composition
As mentioned, the exact composition of lignocellulosic biomass depends on the feedstock source, which is generally divided into five categories, four of which are virgin biomass: energy crops (a type of herbaceous and woody biomass specifically selected for energy generation), grasses/herbaceous crops, softwoods, and hardwoods. The fifth category can be defined as secondary streams generated by factories and populations.24,29,30,40,54,55,57–63Fig. 4 provides an overview of the five categories of lignocellulosic biomass, showing the compositional differences between these categories due to the multiple polymers from which hemicelluloses and lignin are composed. In terms of hemicelluloses, softwoods are dominated by mannose sugars,24,30,55,57,61,62 whereas xylose sugars are the main hemicellulose sugars in grasses57,61 and hardwoods.24,30,55,57,61,62 With regards to lignin, softwoods contain predominately guaiacyl units.30,40,54,55,57,61–65 For hardwoods, syringyl units are also significantly present.30,40,54,55,57,61–65 Finally, for grasses/herbaceous crops, p-hydroxy-phenyl groups are present as well.30,40,54,57,61,63–65 | ||
| Fig. 4 Lignocellulosic biomass categories and their general composition shown here schematically demonstrate that the composition of the lignocellulosic biomass is dependent on the category specifically for hemicellulose and lignin. | ||
To valorise lignocellulosic biomass, it is essential to know the exact composition of the biomass in terms of its constituents, especially when chemicals are targeted. On average, 35–51% (d.b.) of biomass is cellulose, 20–33% are hemicelluloses, and 13–30% is lignin.19,20,28,33,37–39,55–57,61,66 Furthermore, these fractions differ between the types and subtypes of lignocellulosic biomass. The composition of the three polymers in different lignocellulosic biomass types is shown in Table 1, while the composition of the lignocellulosic subtypes (specific feedstocks within a given type) is provided in Tables S1–S4 in the ESI.†
| Cellulose | Hemicelluloses | Lignin | Reference | ||||
|---|---|---|---|---|---|---|---|
| Lower limit | Upper limit | Lower limit | Upper limit | Lower limit | Upper limit | ||
| Energy crops | 21% | 54% | 5% | 30% | 5% | 10% | 24 |
| Grasses | 25% | 40% | 23% | 50% | 10% | 30% | 24, 26, 30, 37, 40, 54, 63 and 65–67 |
| Softwoods | 40% | 50% | 19% | 35% | 21% | 35% | 24, 26, 30, 37, 40, 54, 55, 63 and 65 |
| Hardwoods | 38% | 55% | 17% | 40% | 16% | 30% | 24, 26, 30, 37, 40, 54, 55, 63 and 65 and 67 |
| Secondary streams | 25% | 29% | 18% | 20% | 17% | 21% | 29, 39, 57, 66 and 67 |
For each of the lignocellulosic biomass types, cellulose is present in the highest concentration. However, energy crops and softwoods show a significantly higher value than food/feed residues. The highest hemicellulose fraction is found in grasses, while the highest lignin fraction is found in softwoods.
Energy crops can all be placed in a different category in terms of composition; miscanthus (Miscanthus giganteus), switchgrass (Panicum virgatum), giant reed (Arundo donax), and pennisetum (Pennisetum) are examples of grasses, while eucalyptus (Eucalyptus globulus), willow, and poplar are examples of hardwoods.59,60,68
In addition to the distribution of the main constituents (cellulose, hemicelluloses, and lignin), the distribution of monolignols is an important parameter for valorisation. For example, guaiacyl-rich biomass contains more stable C–C bonds than syringyl-rich biomass, which means that the overall stability of the lignocellulosic biomass is different, affecting the valorisation process.40 The monolignol distribution is strongly related to the type of lignocellulosic biomass, as mentioned in Fig. 4. Table 2 shows the composition range of each monolignol. Energy crops and secondary streams are not included. No data were available for these feedstocks, as they are a mixture of grass/herbaceous, softwood, and hardwood materials, causing relevant fluctuations of the content. The lower and upper limits of the ranges for each type are determined in the same way as for the distribution of cellulose, hemicelluloses, and lignin. As shown in Fig. 4, softwoods contain mainly guaiacyl units, hardwoods contain guaiacyl and syringyl units, and grasses contain the three subunits. The biomass also consists of other materials, such as extractives and inorganics. The content of these materials can be less than 5% of the total biomass in the case of woody biomass, and around 10–15% in the case of herbaceous crops.69,70 However, this review will focus on the valorisation of biopolymers, the most valuable and studied part of lignocellulosic biomass.
How can the lignocellulosic biomass be valorised?
 | ||
| Fig. 5 Lignocellulosic biomass valorisation products in one of the possible classifications showing the different end products, although many materials such as polymers are synthesised from chemicals, indicating the complexity of the lignocellulosic biomass valorisation process. | ||
Lignin needs to be removed due to its complex structure, which hinders the ability to break down lignocellulose. Lignin is bonded by strong ether linkages and carbon–carbon bonds.56 This strength depends on the amount of β-O-4 caused by the distribution of monolignols, which creates a difference between softwoods, hardwoods, and grasses.54 For example, hardwoods are syringyl-rich, resulting in fewer stable C–C bonds compared to softwoods that are guaiacyl-rich.40
Lignin can be modified or partially separated, dissolved, and removed.28,30,37,38 Not all methods allow lignin to be recovered. However, ensuring this recovery would potentially provide multiple revenue streams, thus improving the economic viability.33,61 A selective fractionation technology is required to achieve the separation of lignin and (hemi)cellulose.32
In addition to lignin removal, pretreatment technologies serve other purposes, each with the primary goal of increasing the accessibility for degradation.26,36,39,66 An ideal pretreatment method would have the following characteristics: reduce cellulose crystallinity,19,21,26,38,39 reduce cellulose degree of polymerisation,21 increase biomass surface area,19,38 address hemicelluloses content,19,21,26,38,57 degrade lignin if not removed,19,21,26,57 address particle size,21,36 increase porosity,21,39 produce few inhibitors and by-products,33,57 change chemical composition,39etc. Furthermore, the pretreatment should be suitable for treating a variety of lignocellulosic feedstocks.33 The methods can be divided into four categories: physical, chemical, physicochemical, and biological pretreatments.73–76 Each of these categories contains a variety of options, as shown in Fig. 6. The outcome of the pretreatment depends on the composition of the lignocellulosic biomass, and therefore, on the type of lignocellulosic feedstock.36 Due to the wide variety of pretreatment methods, it is possible to select the most appropriate method or combination of methods for the chosen conversion process or end-product.25 The method is selected based on the type of lignocellulosic biomass, economic feasibility, and environmental impact.33 In general, a pretreatment method consumes power and heat.33,39 In addition, not all technologies are environmentally friendly or sustainable due to the need for high temperatures or the use of chemicals.29
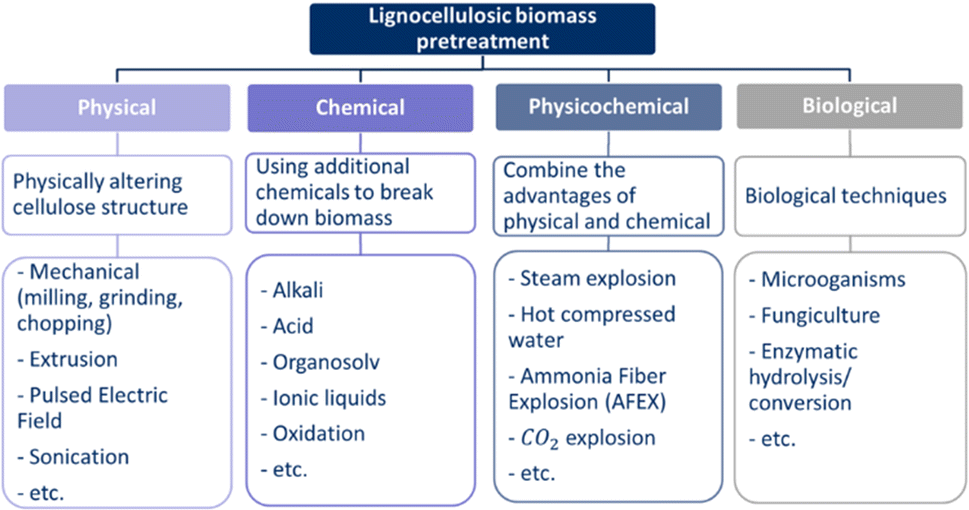 | ||
| Fig. 6 Pretreatment methods prior to lignocellulosic biomass valorisation divided into the four different categories shown with a short description and examples. | ||
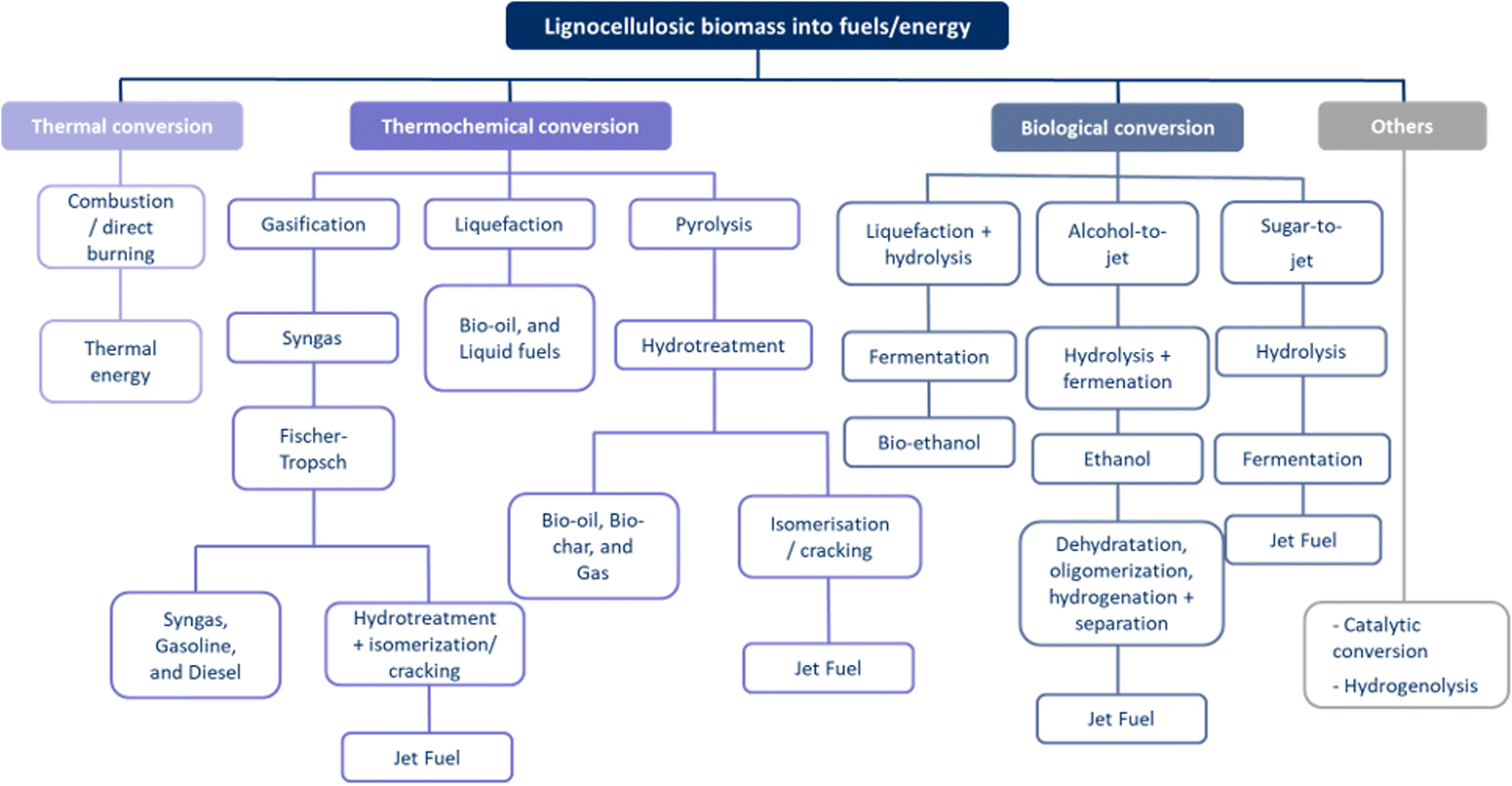 | ||
| Fig. 7 Pathways for converting lignocellulosic biomass into fuels/energy, indicating the different types of conversion methods that can potentially be applied with the different intermediate steps, and their end products, showing the high variety of options creating the complex valorisation process. | ||
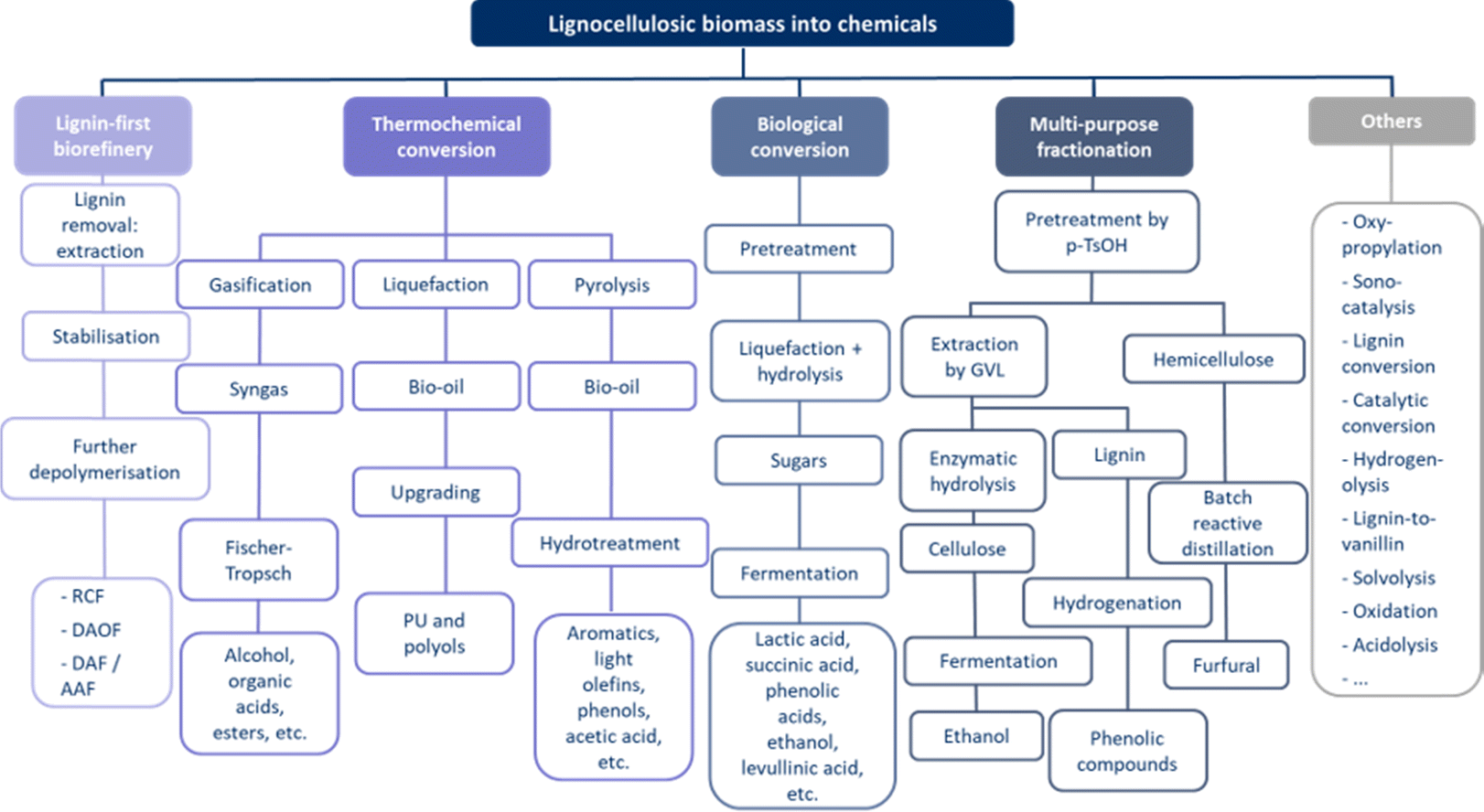 | ||
| Fig. 8 Pathways for converting lignocellulosic biomass into chemicals, indicating the different types of conversion methods that can potentially be applied with the different intermediate steps, and their end products, showing the high variety of options creating the complex valorisation process. | ||
Pyrolysis can produce fuels (fuel intermediates) from biomass in liquid (bio-oil), solid (bio-char), and gaseous (non-condensable gasses) forms.19,25,29,30,33,36,38,41,57,58,63,65,66 Both virgin biomass and secondary streams, e.g., lignin extracted by organosolv pretreatment, can be considered.58,63 Controlling the operating conditions influences the composition and yield of the products obtained. Some important parameters are temperature, pressure, residence time of vapours and biomass feed, and reactor configuration.66 Various types of pyrolysis can be distinguished in Table 3. In most cases, bio-oil,3,30,33,36,38,41,58,63,65,66 bio-char,29,30,33,38,58,63,66 and non-condensable flue gasses33,38,58,63 are produced. However, it is difficult to control the distribution of these fractions, mainly due to the complexity of the feedstock.63 An upgrading step is required to produce drop-in fuels. Further processing options include hydrotreatment, a combination of hydrotreatment, hydrocracking and/or hydroisomerisation, methanol synthesis, catalytic pyrolysis (zeolite upgrading), or aqueous-phase processing.30,33,41,58,66 These reactions can produce upgraded bio-oil,3,19,29,30,33,36,38,41,57,58,63,65 bio-char,30,33,38,41,63,66 drop-in fuels,19,25,30,33,38,41,58 methane,30 bio-jet fuel,41,66 heavy fuel,33 and hydrogen.38,57,58
| Temperature | Residence time | Other parameters | Yield | Reference | |
|---|---|---|---|---|---|
| Pyrolysis | 300–1000 °C | 58 wt% bio-oil | 29, 33, 58, 63 and 65 | ||
| 42 wt% bio-char | |||||
| Flash | 800–1000 °C | <1 s | Particle size 3 mm | 75 wt% bio-oil | 33, 38 and 66 |
| Highest heating rate | 12 wt% bio-char | ||||
| Chemicals and gasses | |||||
| Fast | 400–600 °C | 0.5–5 s | 3000–1000 °C min−1 | 70–80% bio-oil | 3, 30, 33, 38, 58 and 66 |
| 10–20% gas, 15–25% bio-char | |||||
| Intermediate | Low | 5–30 min | Low heating rate | Bio-char, bio-oil and gas | 33 and 38 |
| Slow/carbonisation | Low | Days | Low heating rate | Bio-char | 33 and 38 |
| Vacuum | ±300 °C | Negative P | Bio-char | 66 | |
| Lignin depolymerisation | Up to 800 °C | 66 | |||
| Pretreated | 150–400 °C | >6–10% phenols, bio-oil | 29 and 63 | ||
| Ultra-rapid | <0.5 s | Chemicals, gas | 38 | ||
| Hydrous vs. hydropyrolysis | 416–500 °C | 45 min vs. <2 min | Gas vs. bio-oil | 38 and 58 | |
| Catalytic fast | 416–500 °C | Catalyst: Metal oxides/zeolite | High quality bio-oil rich in aromatics and phenolics | 30, 58 and 66 |
Hydrotreatment, hydrocracking, and/or hydroisomerisation of the bio-oil produces upgraded (bio-jet) fuels that approach the drop-in standards. Such upgrading steps require catalysts, namely CoMo/γ-Al2O3 and NiMo/γ-Al2O3 for hydrotreatment, and CoMoS/Al2O3 or HZSM-5 for the hydrocracking.66 Another route to bio-jet fuel is through the deoxygenation of bio-oil.66 Methanol synthesis produces bio-jet fuel by reacting the bio-oil and gas with hydrogen.41 Heavy or transport fuel can be produced by hydrotreatment, which takes place at 127–500 °C using high-pressure hydrogen. Supported transition metal or sulphide catalysts are required as catalysts.33,58 Fast pyrolysis combined with hydrotreatment is a simple, cost-effective and efficient process for the production of liquid hydrocarbon fuels.58 Hydrodeoxygenation (HDO) upgrades the bio-oil by stabilizing, deoxygenating, and increasing the energy density.30,58 Aqueous-phase processing separates bio-oil, followed by an aqueous-phase reforming or aqueous-phase dehydration/hydrogenation process of the aqueous fraction to hydrogen or hydrocarbons.58 By using fast pyrolysis, the need for an upgrading step can be avoided, and liquid fuel can be directly produced.33 Catalytic fast pyrolysis uses zeolite-based heterogeneous catalysts to directly upgrade the primary pyrolysis vapours to a fuel intermediate. Cracking is performed at 350–500 °C and atmospheric pressure.30,58 Bio-char can be separated by a cyclone and used as a fuel.33 The desired fractions can be separated by distillation.66
In addition to fuels and/or fuel intermediates, chemicals are also possible end-products of pyrolysis,25,29,30,33,38,57,63 requiring a pretreatment for the valorisation of the secondary streams. For example, organosolv and acid–alkali treatments that remove lignin are possible.29,30,63 Similar to the pyrolysis for fuel production, the yield varies, depending on the operating conditions. Table 4 lists the operating conditions depending on the type of pyrolysis. Bio-oil is formed containing small carbonyl compounds (acetic acid, acetaldehyde, hydroxyaldehydes, etc.), sugar-derived compounds (furfural, levoglucosan, anhydrosugars, etc.), and lignin-derived compounds.30 In some cases, such as the pyrolysis of lignin, a catalyst is required, such as zeolites, HZSM-5, or mordenite, which influences the composition of the resulting bio-oil.30,63 Zeolite catalysis, either in situ or ex situ, promotes the formation of aromatics and light olefins. HZSM-5 as a catalyst gives a high monomer yield with an increased selectivity for monomeric aromatics.42,78 In the case of pyrolysis of lignin, after pretreatment, there was a difference between organosolv lignin, and a combination of organosolv and kraft lignin. Without a catalyst, monomer yields were 17–18% and 3–7%, respectively.30 After pyrolysis, bio-oil,30,33 phenolics,30 and monomer fractions30 may undergo an upgrading step.25,30,33 This may involve hydrotreatment, hydrodeoxygenation (HDO), or zeolite cracking.30 These upgrading reactions can produce chemicals,25,33,38,57 cycloalkanes,30 phenolic compounds29,30,63 from tar,38 acetic acid,29 aromatic compounds,29,30 light olefins,30,57 and bio-char.30
| Temperature | Residence time | Other parameters | Yield | Reference | |
|---|---|---|---|---|---|
| Pretreated | 200–400 °C | >6–10% phenol | 29 and 63 | ||
| Fast (in general, catalytic and/or pretreated) | 400–600 °C | 1–2 s | 300–1000 °C min−1 | 75% liquid: wide range monomer | 30 and 63 |
| Flash | <1 s | Bio-oil, chemicals, gas | 38 | ||
| Ultra-rapid | <0.5 s | Chemical, gas | 38 |
In zeolite cracking, the upgrading step is part of the pyrolysis step, which is referred to as catalytic fast pyrolysis.30,79 Hydrodeoxygenation (HDO) of phenolic monomers leads to the generation of cycloalkanes, aromatics, and phenols. The formation of these products was determined by the operating pressure and temperature. High temperatures (300–500 °C) and low hydrogen pressures favour aromatics, as the reaction takes place mainly in the gas phase. Lower temperatures (100–400 °C) and high hydrogen pressures favour cycloalkanes in the liquid phase. Phenol formation can occur in both the gas and liquid phases. Phenolic compounds can in turn be converted to cycloalkanes with high yields using a catalytic system.30
Gasification with a focus on fuel formation3,19,25,29,30,33,57,58,65,66 aims to produce a maximum yield of gaseous compounds (i.e., producer gas or syngas) and a minimum amount of char and condensed hydrocarbons (i.e., tar).33Table 5 provides an overview of the process conditions used for the gasification. The produced gas is a mixture of gases, such as CO, H2, CO2, CH4, etc.19,29,30,33,58,66 A liquid fraction (tar) and a solid (char) fraction are also obtained.66 Solid particles are removed from the gas stream by using a cyclone, while other impurities are removed by water scrubbing or solid sorption.33 Syngas,19,29,65 diesel and gasoline,3,30,33,57,66 fuel,19,25,30,33,66 bio-jet fuel (SAF: Sustainable Aviation Fuel),66 hydrocarbons,33,58,66 hydrogen33,57,66 are possible end-products of these processes.
| Temperature | Residence time | Other parameters | Yield | Reference | |
|---|---|---|---|---|---|
| Gasification | 600–1000 °C | 3–4 s | 1–40 bar | Syngas | 3 and 33 |
| Fixed bed | 100–800 °C | Downdraft, updraft, or crossflow | Gas for heating and power | 33 | |
| Fluidised bed | 1000 °C | Circulating, dual, or bubbling | Syngas, liquid fuels, heat and power, electricity, co-firing | 33 | |
| Entrained bed | 1400–1500 °C | Liquid fuel | 33 |
Syngas can be further upgraded by Fischer–Tropsch, or methanol/dimethyl ether synthesis.3,25,30,33,58,65,66 In a Fischer–Tropsch synthesis,3,30,33,58,66 CO reacts with H2 to produce mainly linear and branched hydrocarbons according to reaction (1). The length of the carbon chain (n) depends on the operating conditions,66 which are given in Table 6. The required catalyst can vary, e.g., metal catalysts (Fe, Co-based, Ni, Cu, Fe, Co, Rh, and Ru).33,66 Other catalysts that have been studied are Fe2O3 nanoparticles inside TiO2 nanotubes, Co/ZrO2-SiO2 bimodal catalyst, Co-SiO2, and α-alumina impregnated with Co and Re.66 Additional steps are required to produce bio-jet fuel, namely a hydrotreating and isomerisation/cracking.66 Biomass-based jet fuel is not expected to differ from fossil-based jet fuel because the properties of the feedstock are destroyed in the gasification step.66 Biomass gasification without a catalyst is not economically feasible due to the presence of methane and tar in the syngas. However, catalytic biomass gasification improves efficiency by controlling the composition of the product.33
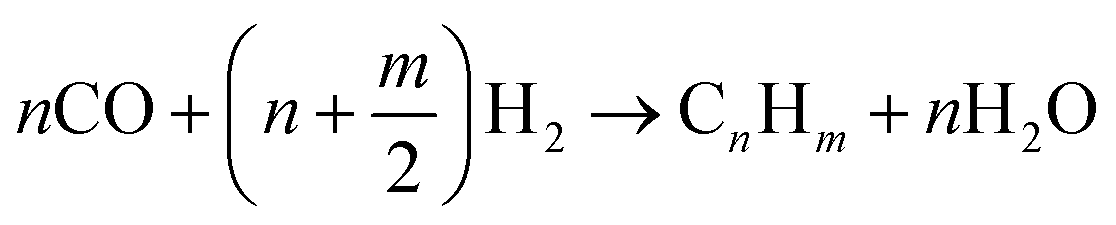 | (1) |
Fischer–Tropsch reaction of syngas.66
Gasification of biomass into syngas can be used to produce chemicals as well.25,30,33,57 To produce the desired chemicals, an upgrading step using Fischer–Tropsch synthesis is required.25,30,33 Depending on the operating conditions and the type of catalyst, long- or short-chain molecules are obtained, as shown in Table 6.33 In this case, however, chemicals25,30,33 such as, alcohols,33,57 hydrocarbons,33 olefins,57 organic acids, and esters,33 are formed.
Liquefaction30,57,58 (or hydrothermal liquefaction (HTL)3,41) is a fragmentation technology that can be applied to both wet and dry lignocellulosic biomass.41 A temperature range of 250–450 °C, a pressure of 5–20 bar, a catalyst, and a solvent are required to perform liquefaction. These temperatures are lower than those required for pyrolysis or gasification. Higher temperatures may limit the gas formation, while lower temperatures will not decompose the cellulose.30 The reaction can be carried out in water or other solvents, such as methanol, ethanol, isopropanol, or a defined mixture of these. Water favours C1- and C2-substitutes, while alcohols favour C3 side chains. To increase the yield of bio-oil, basic catalysts are mainly preferred, namely Na2CO3, K2CO3, KOH and Ca(OH)2.30 Bio-oil,3,30,41,57 bio-char,30,41 and gases30,41 are obtained from the liquefaction process. These can be further upgraded in order to make them compatible with fossil feedstocks.30 Final products of upgrading can include bio-oil,3,30,41,57,58 liquid fuels,30,41,58 hydrogen,57 and methane.57 Compared to pyrolysis, liquefaction has a lower TRL.41 In addition to fuels, liquefaction can lead to the production of chemicals.28 The operating conditions for the liquefaction of lignocellulosic biomass into chemicals are given in Table 7. Both acid (sulphuric acid) and basic catalysts can be used. However, the latter is less corrosive to the metal equipment. Polyethylene glycol and glycerol are required as solvents. A high solvent-to-biomass ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 5:1 is required. There are two competing reactions that occur during the liquefaction process: recondensation reactions and liquefaction reactions. As a result, there is a risk of reduced efficiency. This parameter can be improved by prior separation of lignin from cellulose and the use of catalysts. Liquefaction of cellulose is slow due to the complicated reactions that take place. Cellulose is decomposed by solvolytic reactions into glucose or other cellulose derivatives. The reaction with the liquefaction solvents forms glycoside derivatives, which can further react to form levulinic acid and/or levulinates. The efficiency of the liquefaction reaction is determined by the composition, structure, and morphology of the lignocellulosic biomass. Softwoods react faster than hardwoods, although recondensation reactions are also faster. This results in the formation of polyols, which can be used to produce PUs (polyurethanes).28
:1 to 5:1 is required. There are two competing reactions that occur during the liquefaction process: recondensation reactions and liquefaction reactions. As a result, there is a risk of reduced efficiency. This parameter can be improved by prior separation of lignin from cellulose and the use of catalysts. Liquefaction of cellulose is slow due to the complicated reactions that take place. Cellulose is decomposed by solvolytic reactions into glucose or other cellulose derivatives. The reaction with the liquefaction solvents forms glycoside derivatives, which can further react to form levulinic acid and/or levulinates. The efficiency of the liquefaction reaction is determined by the composition, structure, and morphology of the lignocellulosic biomass. Softwoods react faster than hardwoods, although recondensation reactions are also faster. This results in the formation of polyols, which can be used to produce PUs (polyurethanes).28
Saccharification has the potential to produce fuels.19,21,26,31,33,36,38,39,57,61,62,68 In most cases, pretreatment is used prior to the reaction.19,21,26,33,36,38,39,57,61,62 However, in some cases (CBP, consolidated bioprocessing), the reaction can be performed without pretreatment.61 The lignocellulosic biomass can be deconstructed using dilute or concentrated acids, such as sulphuric acid.19,36,61 However, there is no single best pretreatment for all types of lignocellulosic biomass.19 Enzymatic hydrolysis of cellulose produces the sugars, glucose and oligosaccharides.19,21,26,31,33,36,38,39,57,61,62,68 Enzymes such as cellulase act as catalysts.19,21,26,31,33,36,38,39,57,61,62,68 Three types of enzymes are used: endoglucanase (EF) or 1,4-β-D-glucan-4-glucanohydrolase, exoglucanase (including 1,4-β-D-glucan-4-glucanohydrolase and 1,4-β-D-glucan cellobiohydrolase), and β-glucosidase (BGL) or β-glucoside glucohydrolase.21,36 Cellulose degradation involves multiple reaction steps. First, the β-glycoside linkages of the inner regions of cellulose are hydrolysed into oligosaccharides. Second, new chain ends are formed. This is followed by the hydrolysis of glucose or cellobiose, the hydrolysis of the microcrystalline structure, and finally the cleavage of soluble cellodextrins and cellobiose into glucose.21 If both hemicelluloses and cellulose go through enzymatic hydrolysis, glucose and pentose sugars are obtained.36,57,61,62
The sugars produced can be refined by fermentation (reaction (2)).19,21,26,31,33,36,38,39,57,61,62,68 Microorganisms assist the fermentation process.19,26,33,36,57,61,62,68 Microorganisms can be genetically modified for fermentation (e.g., Clostridium thermocellum, Caldicellulosiruptor sp., Monilia sp., Paecilomyces sp., and Neurospora crassa),33 or fungal species (e.g., Fusarium, Rhizopus, Monilia, Neurospora and Paecilomyces).57 After fermentation, a distillation takes place in order to separate the ethanol.57 In addition to bio-ethanol,19,21,26,31,33,36,38,39,57,61,62,68 other liquid fuels19,26,36,38 can be produced, such as biodiesel,57 bio-butanol,39,57,62,68 methane,57 and alkanes and alkenes.36
| C6H12O6 → 2C2H5OH + 2CO2 | (2) |
Fermentation of glucose to ethanol.80
Biological/biochemical conversions can also be used to produce chemicals.19,28,29,33,36,38,39,57,61,62 Pretreatment of the biomass is necessary for the production of chemicals.19,24,28,29,36,38,39,57,61 The polysaccharides, mainly cellulose, are depolymerised to glucose and oligosaccharides.19,28 This is followed by liquefaction and hydrolysis, catalysed by enzymes such as cellulase.19,29,36,38,39,57,61,62 The glucose and sugars19,28,29,33,36,38,57,61,62 are further converted by fermentation.19,24,33,36,38,39,57,61,62 Fermentation also requires a catalyst as well, which in this case are enzymes that can be produced from specifically engineered microorganisms.19,28,38,61,62 Besides the valorisation of polysaccharides, ligninolytic enzymes consisting of laccase, and lignin, manganese and versatile peroxides enable the valorisation of lignin towards high value chemicals (vanillin). These enzymes enable the degradation and delignification process.33,38,81,82 Various chemicals,19,28,33,36,57,61,62 organic acids,24 carboxylic acids,36 levulinic acid and/or levulinates,24 lactic acid19,36,38,39,62 (green solvents and PLA via lactic acid36), succinic acid,19,36,38 propionic acid, butyric acid, hexanoic acid,36 phenolic acids (vanillic, ferulic, p-coumaric),29,81 butanol, propanediol,39 and bio-ethanol19,33,61 are obtained. Instead of fermentation, other (non-biological) processes can be used to produce chemical derivatives, such as furfural and xylitol.28,39,61
For both fuel and chemical production, there are three schemes for saccharification and fermentation. First, separate saccharification and fermentation (SSF) involves enzymatic hydrolysis, fermentation of C6 sugars, and fermentation of C5 sugars, which occur in separate reaction vessels.21,33,38,57 Second, simultaneous hydrolysis and fermentation (SHF) combines enzymatic hydrolysis and fermentation of C6 sugars in a single vessel, while the fermentation of C5 sugars takes place in separate vessels.21,33,38,57 Finally, simultaneous saccharification and co-fermentation (SSCF) combines enzymatic hydrolysis, fermentation of C6 sugars and fermentation of C5 sugars in a single vessel.21,38,57 However, each of these schemes produces the enzyme (cellulase) in a separate system.21 When cellulase production, hydrolysis, and fermentation are combined, it is called consolidated bioprocessing (CBP) or direct microbial conversion (DMC).21,33,38
Other biological/biochemical conversions focus specifically on the production of advanced fuels83 such as bio-jet fuels through the alcohol-to-jet and sugar-to-jet processes.66,84,85
Alcohol-to-jet starts with a pretreatment that removes lignin from the polysaccharides.66,84 The first part of this process is the hydrolysis and fermentation of the pretreated biomass.66,84 Enzymatic hydrolysis at 40–50 °C and pH 4–5 releases the sugars (glucose, xylose, arabinose, galactose, and mannose).66 Possible enzymes are endoglucanases, exoglucanase or cellobiohydrolase, β-glucosidase, glucuronidase, acetylesterase, xylanase, and β-glucosidase.66 The sugars are fermented to ethanol by microorganisms (bacteria, yeast, or fungi).66,84 Fermentation is challenging because the ethanol yield is determined by the microorganisms selected (Saccharomyces cerevisiae, Zymomonas mobilis), modified strains (Escherichia coli, S. cerevisiae, and Z. mobilis), Candida shehatae, or newly developed microorganisms.84
The second part consists of dehydration, oligomerisation, and hydrogenation of ethanol, followed by the separation of the desired fractions, which is a non-biochemical upgrading step.66,84 In the dehydration step, ethanol is first converted to ethylene and water using an acid catalyst (reaction (3)). High temperatures (180–300 °C) in combination with zeolites, alumina, and silica–alumina catalysts promote the degradation of ethanol.84 Second, various ethylene molecules are converted into long-chain linear olefins by an oligomerisation through reaction (4). For bio-jet fuel, a C8–C16 carbon-chain is desirable. However, the length is influenced by the catalyst used. Ni_ALSBA-15 is a mesoporous catalyst that produces C4–C10 olefins at 150 °C and 3.0 MPa. Covalent organic frameworks supporting Ni catalysts produce C4–C8 olefins at 50 °C and 15 bar. MIL-100(Cr), a chromium catalyst in an organometallic framework, forms C6, C8, and C10 olefins. Chromium-based complexes with bis(benzimidazole-methyl) amine ligands can produce up to 10% olefins that are larger than C8. C10–C55 olefins can be produced by ionic liquid catalysis.84 Next, alkanes (in the same range as the alkenes) are produced from the saturated hydrocarbon molecules by hydrogenation.84
 | (3) |
Dehydration of ethanol to ethylene.84
 | (4) |
Oligomerisation of ethylene to long-chain linear olefins.84
Last, the desired fractions are separated.84 Each section (ethanol production, dehydration, and ethylene oligomerisation) has its own separation equipment. For ethanol production, the by-products are separated by distillation until the azeotrope (96 wt%) is obtained. Higher purity can be achieved by extractive distillation or pervaporation. The dehydration step requires three separation steps: quenching, scrubbing, and drying. After oligomerisation, a heat-based separation such as distillation purifies the hydrocarbons.84 The bio-jet fuel66,84 is separated from the by-products (naphtha and green diesel).84
Sugar-to-jet,66,84 similar to alcohol-to-jet, starts with a pretreatment that removes lignin.66 The biomass is hydrolysed and sugars are extracted.66 The sugars are then directly converted without the formation of ethanol.66,84 Fermentation may be followed by hydrotreating and/or hydrocracking/hydroisomerisation.84 A variety of products (pentadecane, farnesene, fatty esters, and fatty acids) can be produced, depending on the microorganisms.84 In addition to fermentation, catalytic upgrading to bio-jet fuels with hydrogenation, reforming, condensation or dehydration and oligomerisation is possible.84 Each of these conversion pathways has the common goal of producing bio-jet fuel.66,84,85
For example, the mevalonate pathway (reaction (5)) yields farnesene (C15H24), which can be hydrogenated to farnesane. Bio-jet fuel is obtained by hydrocracking and hydroisomerisation.84
| C6H12O6 + 6H2O → 6CO2+H2 | (5) |
| C6H12O6 + 4.8H2O → 0.4C15H24 + 6H2O | (6) |
Mevalonate pathway to farnesene.84
Depending on the conversion pathway, a separation technology is required.84 For example, conversion to farnesene requires de-emulsification, liquid–liquid centrifugation, and flash, which may be followed by distillation for hydrogenation of farnesene, hydrocracking, and hydroisomerisation.84
The general process of a lignin-first biorefinery starts with the removal of lignin by solvolysis or acid-catalysed reactions, similar to an organosolv pretreatment.86 The main difference between organosolv and the lignin-first approach is the fact that condensation reactions are limited. Under the conditions in which lignin is extracted by most other pretreatments, recalcitrant polymers are produced, resulting in a low-quality lignin. The pretreated lignocellulosic biomass is stabilised, and condensation reactions of the reactive species are prevented. The stabilised intermediate is a mixture of depolymerised lignin and carbohydrate pulp. If necessary, a further stabilisation step is added. After the fractionation step, downstream processing includes lignin separation by liquid–liquid extraction, membrane filtration, distillation, and column chromatography. Potential end-products include monophenolic-derivatives, HMF, levulinic acid, furfural, bio-ethanol, glucose, valuable chemicals, and energy-density fuels. However, some challenges can arise from the lack of standards for the feedstock, analysis, and process evaluation, making it difficult to compare results and methodologies. Yield calculations are complicated by the heterogeneous nature of the lignocellulosic biomass.31,32,54
The first and best known type of lignin-first biorefinery is Reductive Catalytic Fractionation (RCF), which involves integrated biomass fractionation and lignin depolymerisation.32,40,54,63,72,87–93 In the first step, lignin is removed by solvolytic extraction. As in all lignin-first biorefineries, what is generally considered to be the pretreatment, in this case the organosolv-like process, is integrated into the valorisation process. An organic solvent, such as short-chain alcohols (C1–C4) and cyclic ethers (dioxane), is combined with water to extract the lignin in an organosolv-like process. Simultaneously, the lignin is depolymerised to aromatic monomers, in contrast to the condensed lignin produced by most pretreatment processes.31,32,40,54,63,72,87–89,92 The reductive catalytic fractionation itself proceeds along two pathways, depending on the temperature, pH, and solvent polarity. Relatively severe conditions (250 °C) favour solvolytic depolymerisation, in which the role of the catalyst is limited to stabilisation. Less severe conditions (190 °C) have slower solvolytics and the catalyst will perform solvolytics, moving towards catalytic hydrogenolysis.40 In general, RCF takes place at 180–250 °C for 2–6 h in the presence of a catalyst.40,54,87,88,90 Typical catalysts are heterogeneous redox-active catalysts, such as carbon-supported catalysts (Pd/C, Ru/C, Ni/C, Cu-PMO, ZnPd/C, beta zeolite, ZnMoO4/MCM-41), and precious metals.31,40,54,63,72,87–91 To further reduce the reaction conditions, alternative reducing agents can be used in addition to high-pressure H2, such as indirect H-sources from the solvent or directly from lignocellulose.32,40,54,63,87 In such hydrogen gas-free reactions, the use of RANEY®-Ni catalysts has been investigated, showing promising results in improving the hydrogen transfer that takes place.94–98 Over the years, mechanochemistry utilising mechanical forces (e.g., ball milling to activate chemical bonds) has attracted much research interest for the depolymerisation of lignin.99 This technique has the advantage of being solvent-free, and it can be used on lignocellulose or its biopolymers (polysaccharides and lignin) for their fractionation and depolymerisation.100 This first step produces a cellulose-rich polysaccharide pulp with reduced C5–C6 sugars, and depolymerised monophenols and oligomers derived from lignin. In the second step, the lignin-derived intermediates are stabilised to prevent the formation of condensed lignin.31,32,40,54,63,72,87,89,91,92 Various phenolic monomers, dimers, and oligomers are formed as lignin oil,32,40,54,63,87–92 which are potential building blocks for value-added aromatic chemicals, fuels, polymers, and pharmaceuticals.54,89 Cellulose and hemicellulose derivatives are also produced with a variety of C5-polyols and furans as carbohydrate pulp.40,54,91,92
The main focus of the RCF process is the depolymerisation of lignin without the production of condensed lignin, resulting in high yields and selectivity.31,54,90 However, in addition to lignin-derived components, a solid carbohydrate pulp (C5–C6 sugars) is also obtained, which can be further valorised by enzymatic hydrolysis and fermentation to bio-ethanol.54,93 Prior to the valorisation of both fractions, separation steps are required to separate the lignin oil from the pulp and possibly other impurities.40,89 As RCF is still an emerging technology, challenges such as feedstock, location, catalyst, solvent, and reactor selection remain.32,54,89,92
The second type of lignin-first biorefinery is called Dithionite-Assisted Organosolv Fractionation (DAOF).72,87 As the name suggests, sodium dithionite (Na2S2O4) is added to the reaction as an alternative to precious metal catalysts and hydrogen gas in RCF. The reaction takes place at 150–250 °C (10 °C min−1) for 0–6 h in a n-butanol/water solvent, and N2-atmosphere (1–30 bar). Important parameters include temperature, reaction time, n-butanol/water ratio, N2-pressure, dithionite loading, and solvent/biomass ratio. The organosolv process, which is generally regarded as pretreatment, is integrated into the valorisation process for DAOF, as it is for all lignin-first biorefineries. A solid (carbohydrate pulp) and liquid fraction is formed, which must be separated by centrifugation. Further separation of the liquid fraction into an organic (lignin oil) and aqueous (non-condensed carbohydrate) phase is possible. Both the solid and organic liquid fractions can be purified into potential end-products, while the aqueous liquid fraction is considered wastewater. The carbohydrate pulp (solid) can be upgraded to paper, sugar, ethanol, or other solvents, while the lignin oil contains a variety of phenolic monomers, dimers and oligomers that can used as building blocks in other chemical processes.72,87
In addition to the two lignin-first biorefineries mentioned above, there are other possibilities with diol-assisted fractionation and aldehyde-assisted fractionation. Alternative solvents, catalysts, and reagents are used, resulting in a different composition of the lignin oil produced.32,87
An example of multi-purpose fractionation is where the hemicelluloses are first separated via p-TsOH (p-toluene sulphonic acid). The liberated hemicelluloses are then subjected to a batch reactive distillation to produce furfural. Hemicelluloses are separated by solid–liquid separation with p-TsOH at mild temperatures between 90–130 °C for 60 min (while hydrolysing to xylose). Batch Reactive Distillation (BDR) is performed at 150 °C for 40 minutes, and the furfural is immediately removed by dehydration to improve its yield.71
Second, lignin is removed by an extraction with GVL (γ-valerolactone), after which phenolic compounds are produced by hydrogenation. A binary mixture of GVL-H2O is used to extract lignin at 120 °C for 60 min without loss of functionalities for both lignin and cellulose. The two fractions are separated by filtration. Lignin is depolymerised by hydrogenation at 230 °C for 15 h at a pressure of 8–1.5 MPa to obtain bio-oil containing phenols, guaiacols, and syringols.71
Last, the cellulose stream is hydrolysed and fermented to ethanol. The cellulose-rich solid is directly degraded to glucose by enzymatic hydrolysis/saccharification at 30 °C for 72 hours, followed by fermentation at 30 °C for 48 hours to ethanol. This is very similar to the hydrolysis and fermentation mentioned above in the section on biological/biochemical conversion.71
| Reaction | Similar to | End product | Reference | |
|---|---|---|---|---|
| Catalytic conversion | Depolymerisation, hydrotreating, oxidation, liquid-phase reforming | Reactions using catalyst | Fuels and high-value chemicals | 25 |
| Oxypropylation | Propylene oxide grafted onto macromolecular structure | Polyols | 28 | |
| Sonocatalysis | Combining catalyst with sonification | Combined with other processes | Depending on application | 25 |
| Hydrogenolysis | Cleavage ether bonds: solvolytic delignification and depolymerisation | Lignin-first | Phenols, bio-oil, cresols, aromatic hydrocarbons | 29 and 65 |
| Lignin-to-vanillin | Oxidation, depolymerisation, hydrolysis | Catalytic lignin conversion | Phenolic aldehyde: vanillin | 65 |
| Non-hydrogen reductive depolymerisation | Reductive depolymerisation: hydrogenolysis, dehydration, hydrogenation | Catalytic conversion, lignin-first | Phenolic monomers | 65 |
| Solvolysis | (Non)-catalytic depolymerisation solvent | Reaction using solvent | Phenols, value-added chemicals | 63 and 65 |
| Oxidation | Oxidation depolymerisation using oxidants | Phenols, benzylic aldehydes, acids | 29 and 65 | |
| Acidolysis | Fractionation/depolymerisation of oxidized lignin | Depolymerisation with acid | Aromatic monomers | 63 and 65 |
Current state-based assessment methods for the valorisation of lignocellulosic biomass
Lignocellulosic biomass is very diverse in composition and can be valorised through a plethora of conversion methods, as outlined in the above, to produce a variety of end-products, which complicates valorisation in the current economy.101 An example of the complexity of lignin is given by Tišma et al., where wheat straw can be converted into vanillin, other monophenolic compounds, bio-oil, etc.29 Conversely, vanillin can be produced from biomass other than wheat straw, such as sugar beet pulp, rice straw, and corn stover.29 Current studies have mainly focused on a specific biomass, a specific process, or a specific end-product. In order to create optimal biomass-end-product combinations as shown in Fig. 2, the focus needs to shift to the selection of an end-product depending on the available biomass source; or vice versa, the selection of a resource for a desired end product. Different types of decisions need to be made regarding the valorisation of lignocellulosic biomass, depending on the activities of the stakeholders. For example, an agricultural company may have residual biomass to offer to industry, but needs to decide what end-product to make from it in order to understand who to approach. Conversely, companies currently producing fossil-based end products may need to find new feedstocks to replace fossil resources, but again need to decide which alternative bioresource can replace the fossil counterpart. The abundance of valorisation options and the high diversity in composition make the selection of the optimal biomass-end-product combinations for different situations a challenge, as it depends on many different factors. In order to decide on the optimal biomass-end-product combination, the environmental impact and economic viability should be assessed quantitatively. The current state-of-the-art quantitative assessment methods, which are used to make decisions for a specific process and thus focus on specific assessments, are Life Cycle Assessment (LCA) and Techno-Economic Assessment (TEA), or combinations of these in an Environmental Techno-Economic Assessment (ETEA).102 LCA quantifies the environmental impacts over the entire life cycle of a product, process, or service. Four key steps involve the goal and scope definition, life cycle inventory (LCI), life cycle impact assessment (LCIA), and interpretation. A continuous interaction between these steps creates the iterative process that is an LCA.103,104TEA estimates the economic viability by calculating the costs, revenues and profits from market data, process flow diagrams and mass and energy balances. The costs fall into two categories: CAPEX (capital expenditure) and OPEX (operating expenditure). Revenues are determined by the scale of the end product and any by-products.
Important economic parameters used in these assessments are the net present value (NPV), return on investment, pay-back time, etc.105 For interpretation, a sensitivity analysis is required to identify the key influencing parameters (e.g., raw material costs, utilities, selling prices), and an uncertainty analysis is required to quantify the uncertainty on the environmental and economic indicators resulting from the uncertainty of parameters.103,104
Both TEA and LCA can be combined in the ETEA framework as they share the same technological backbone. The process of an ETEA starts with a market study, followed by a process flow diagram/mass and energy balance, an environmental analysis together with an economic analysis, followed by interpretation, and finally back to the market study, after which the process is repeated.102 LCA and TEA provide the current decision-making process for biomass, as shown in Fig. 9. However, specific boundary and background systems define these methods, along with the specific technologies, geographical regions, and other assumptions. Because of these assumptions, studies carried out by different experts are not directly comparable, leading to a lack of genericity.106 Furthermore, a large amount of data and detailed information is required to carry out these assessments. Information is not available for novel technologies at low TRLs. Scenario, sensitivity, and uncertainty analyses can be used to estimate technology change and future performance. However, again, assumptions have to be made.45,46,107 These assessments provide a wealth of information, but the results are only valid for a specific process, in a specific location at a specific time, and are therefore process-specific. Due to the wide variety of lignocellulosic biomass types, conversion pathways, and end-products, finding the optimal biomass-end-product combinations using process-specific assessments requires too much expertise, time, and data.
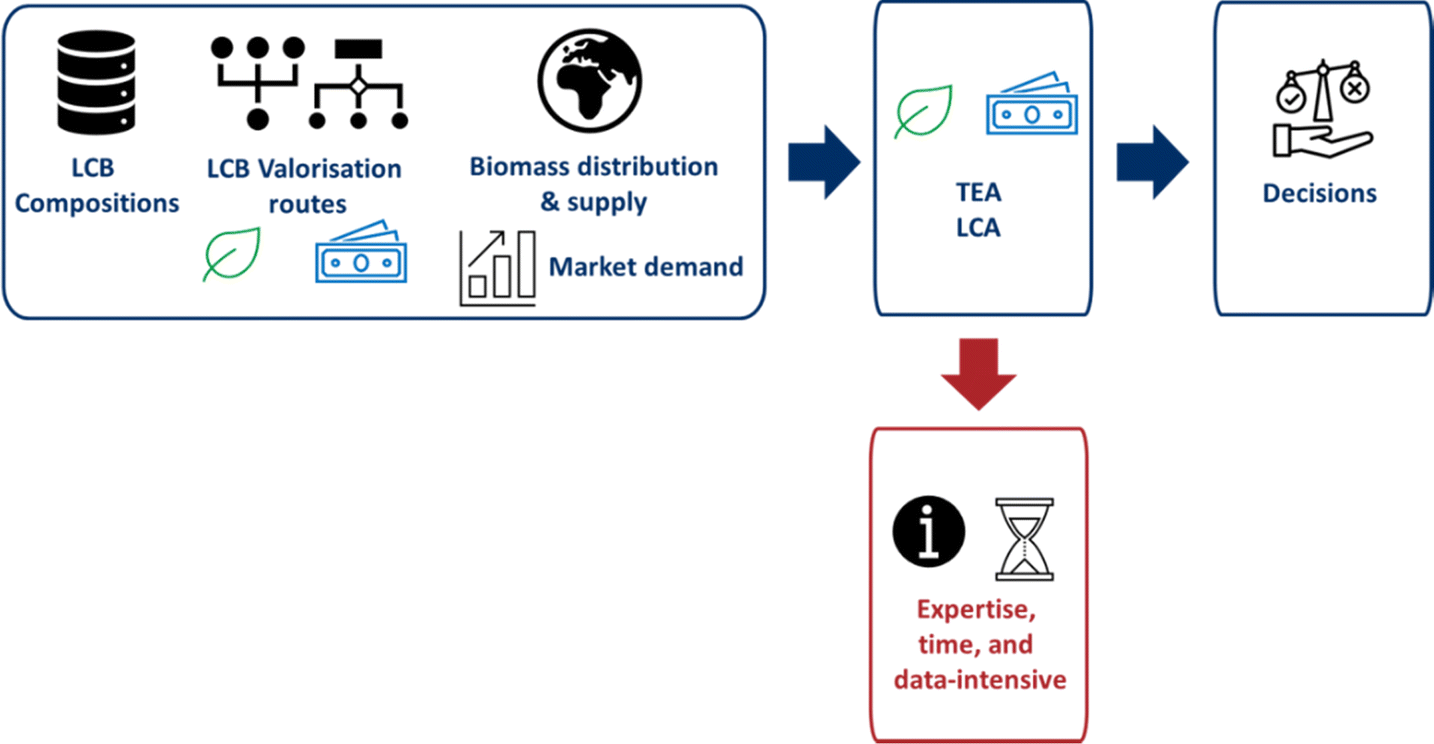 | ||
| Fig. 9 Current decision-making process for lignocellulosic biomass valorisation. | ||
Recent advances have extended the above assessments to include future impacts at low TRL. However, many aspects require further research.108 In terms of LCA, some recommendations are made specifically for biomass applications, such as the addition of land-use and (indirect) land-use change, carbon release at end-of-life, etc.106 Land-use change takes into account the emissions that occur when the original use of the land is changed to one that was previously used for other biomass applications. This gives a one-sided result that only considers the carbon savings compared to the petrochemical counterpart, without considering the additional carbon costs. If this land-use change leads to a shift in the original use of the land, this original use needs to relocate, resulting in an indirect land-use change at another location that must be allocated (e.g., deforestation, cropland intensification).106,109,110 Other possibilities include ex-ante LCA in order to assess environmental impacts for low TRL processes. For industrial scale impacts, estimates can be made based on lab-scale results to provide better insight into the future performance of the process.45 Although ex-ante LCA focuses on emerging technologies, most of these technologies have defined system boundaries, unlike novel low TRL processes, and therefore require further research on their implementation.108
Regarding TEA, learning effects can (or rather should) be taken into account. When investigating novel technologies, it is not considered that these technologies will be optimised over time and become easier to apply, thus reducing costs.46 These learning effects need to be considered when assessing the applicability of novel technologies using learning curves. The review published by Thomassen et al. (2020) shows that there are different types of learning effects, such as learning-by-doing, learning-by-searching, learning-by-using, learning-by-interaction and scale effects, which have different impacts that need to be taken into account. Furthermore, learning curves are mostly based on historical data, which limits their applicability to specific sectors.46 Lignocellulosic biomass valorisation has only a limited amount of historical information to develop these learning curves, so there is a need to further develop these learning effects.
Vasilakou et al. (2023) proposed a framework that implements learning effects for a structural approach to TEA of advanced biofuels. In order to implement these learning curves, it is first necessary to define what they entail, together with the associated definitions. Two different learning curves have been considered: single-component and multi-component learning curves. The single-component learning curve focuses on the cost of production, while the multi-component learning curve divides the cost into smaller parts, each with a different learning rate. By calculating the learning curves, a minimum learning rate required to achieve the desired cost reduction can be calculated.111
When assessing the sustainability of a process, the objective is to maximise the economic viability and minimise the environmental impact. In multi-objective optimisation, the often conflicting outcomes of economic viability and environmental impact are traded off. Pareto aggregation is proposed for this purpose. However, as the name suggests, multiple objectives and indicators are required, which complicates the decision-making process.112
Geospatial ETEA considers the influence of geospatial parameters, such as biomass supply, prices, tax rates, and salaries, depending on the geospatial location. A clear influence on both economic viability and environmental impact can be identified by the difference in minimum selling price (MSP) and greenhouse gas (GHG) emissions.47
In addition to more traditional assessments, such as TEA and LCA, alternative methods exist for assessing the environmental impact and economic viability of a process. An exergo-economic analysis is an example of such an assessment. An exergy analysis assesses the quality of energy, i.e., exergy is a quantitative measure of energy quality. The First Law of Thermodynamics states the conservation of energy, i.e., energy cannot be created or destroyed. However, it can be altered in form, and thus lose quality. This loss of quality is what exergy analysis is all about. Two types of exergy can be calculated: exergy losses and exergy efficiencies. Exergy losses focus on the loss of potential to produce the desired product, while exergy efficiencies focus on how close the process is to the ideal.113 These two types can be linked, as a reduction in exergy losses leads to an increase in exergy efficiency.114 An exergo-economic analysis combines the results of the exergy analysis with an economic assessment. These assessments can determine avoidable and unavoidable exergy losses, and investment cost rates. A ratio of energy and exergy losses to the net present value (NPV) can be determined.115,116
Another trend is the use of superstructure optimisation to make decisions in process design, moving from simulation to optimisation of a studied process or selection of an optimal production route.48 This is a superstructure of different processes (discrete decision variables) and for each of these processes, there are operating conditions. A variety of superstructure optimisation techniques exist, and the appropriate technique must be selected for each process. There is a superstructure of different processes (discrete decision variables) and for each of these processes, there are operating conditions. For a selected feedstock, technology, and product, a superstructure is created containing compound and technological information, after which simulations and data estimation are performed to create an optimisation model, leading to the development of an optimal strategy. Finally, the production of a selected end-product from a selected feedstock is optimised.48,117,118 Examples where this has been applied include the use of CO2-to-fuel strategies,117 and even for lignin valorisation.118
Machine learning has also been explored for predicting environmental impacts. However, this is highly dependent on the data used to train it, and requires a lot of time and data.119 Fozer et al.120 proposed a hybrid predictive high-throughput sustainability screening to make predictions for low TRL processes. This screening used a sequential application of data-driven hybrid prediction, starting with Artificial Neural Networks (ANN), followed by Response Surface Methodology (RSM), and Desirable Optimisation Method (DOM). Again, a large amount of data is required to perform such sustainability screening. Nonetheless, while all of the above are valuable tools for high-level decision-making, it is highly unlikely that these complex methods would be available or affordable for the micro-level decisions of the agricultural company mentioned before. In addition, the considerable amount of data required to make these decisions is either unavailable, widely dispersed or available at different resolutions.
Of the data required, four types are critical to the development of lignocellulosic biomass valorisation pathways using process-specific assessments: geospatial information, biomass composition, market information (supply and demand), and process technologies. Geospatial information includes all data relating to the spatial distribution of the biomass. Biomass composition influences product composition. Biomass supply and demand determine whether sufficient biomass is available, and influence the preferred valorisation pathway. Lastly, current and future process technologies create the potential pathways, but this requires a very good understanding of what the process looks like.121
The above assessments can provide a variety of information that is important for economic and environmental evaluation. Nonetheless, the assessments are process-specific. Providing an overview of valorisation pathways and their potential requires too much time based on process-specific assessments only. A more generic and easily applicable methodology for assessing environmental sustainability and economic viability is needed to address these challenges. This methodology should be based solely on common and readily available information, and therefore be applicable at low TRL.
From process-based assessments to state-based assessments
The limitations identified require a shift from process-based assessments to state-based assessments. These assessments focus on the properties of the initial and final states of a process without looking at the path taken. Specifically for biomass, these assessments allow us to start from a given resource and look at the potential end-products, or to start from a given end-product and look at the potential resources. By using state-based rather than process-based assessments, the time and complexity of the assessments can be drastically reduced, making them available for cheaper micro-level decisions, although some accuracy is sacrificed. Three important parameters need to be considered in the decision-making process available at any TRL: composition, geospatial distribution, and market share. Recent research shows the potential of (statistical) entropy for environmental and economic impact assessments.Entropy is a thermodynamic function defined by Carnot and Clausius, among others. Thermodynamic entropy approaches explore the relationship between heat and energy, which can be translated into entropy.122 On the other hand, Boltzmann and Planck defined entropy independently of energy,123 which was further developed by Shannon as a statistical function.124 Currently, both definitions of entropy are considered separately. The aim of this research is to establish a link between the statistical definitions of entropy and energy.
Entropy, as defined by Shannon, is a statistical function that calculates the average uncertainty of a distributed variable for any distribution.124 Shannon entropy is defined by the following formula: 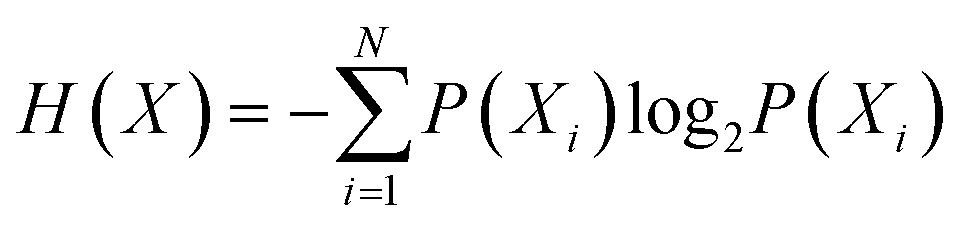 , where X is a randomly distributed variable, Xi is a possible outcome/value of X, and P(Xi) is the probability that X = Xi. Furthermore, the sum of all probabilities is equal to “1”
, where X is a randomly distributed variable, Xi is a possible outcome/value of X, and P(Xi) is the probability that X = Xi. Furthermore, the sum of all probabilities is equal to “1” 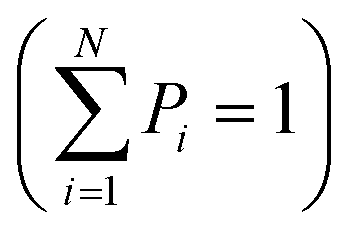 . Two extreme scenarios can occur: a minimum and a maximum entropy. The entropy is minimum when H = 0 with the lowest possible uncertainty. Only one P(Xi) is not “0” and is therefore equal to “1”. A maximum entropy, with the highest possible uncertainty, is reached when all possible probabilities are equal and uniformly distributed. Therefore, P(Xi) = 1/N for a given N, resulting in an entropy of H = logN. The maximum entropy is not a fixed value and depends on N. All possible values between the minimum and the maximum are a degree of uncertainty. Thus, the lower the entropy (closer to H = 0), the lower the uncertainty; and conversely, the higher the entropy (closer to H = logN), the higher the uncertainty.
. Two extreme scenarios can occur: a minimum and a maximum entropy. The entropy is minimum when H = 0 with the lowest possible uncertainty. Only one P(Xi) is not “0” and is therefore equal to “1”. A maximum entropy, with the highest possible uncertainty, is reached when all possible probabilities are equal and uniformly distributed. Therefore, P(Xi) = 1/N for a given N, resulting in an entropy of H = logN. The maximum entropy is not a fixed value and depends on N. All possible values between the minimum and the maximum are a degree of uncertainty. Thus, the lower the entropy (closer to H = 0), the lower the uncertainty; and conversely, the higher the entropy (closer to H = logN), the higher the uncertainty.
Shannon entropy serves as the basis of the Statistical Entropy Analysis (SEA) proposed by Rechberger and Brunner.49 They translated Shannon entropy to material flows to quantify their complexity. In calculating the statistical entropy of material flows, probabilities are replaced by concentrations in the material flow, such as mass fractions. The Shannon entropy is fitted to the following formula: 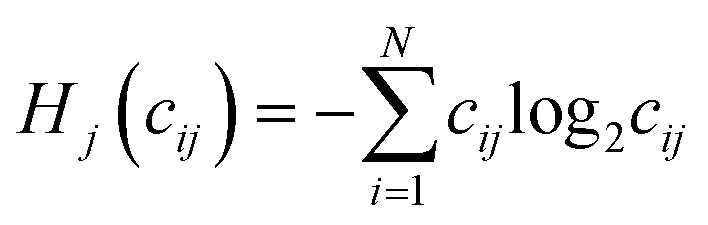 , where i is the index for the different chemical compounds in material flow j in the system for which Hj is calculated, N is the total amount of chemical compounds in the system, and cij is the mass fraction of compound i in material flow j. For the mass fractions, the sum of all mass fractions is “1”
, where i is the index for the different chemical compounds in material flow j in the system for which Hj is calculated, N is the total amount of chemical compounds in the system, and cij is the mass fraction of compound i in material flow j. For the mass fractions, the sum of all mass fractions is “1” 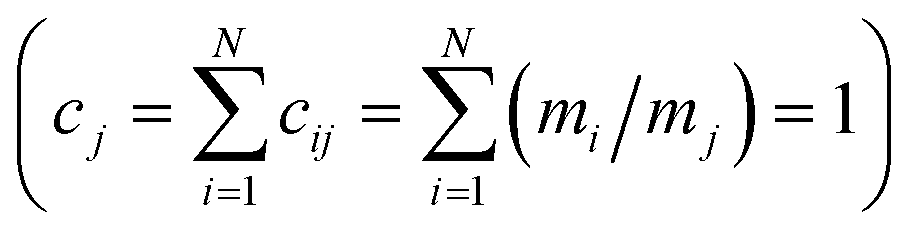 . In the case of SEA, a low entropy corresponds to a highly concentrated mass flow (low uncertainty) and a high entropy corresponds to a highly diluted mass flow (high uncertainty). The maximum entropy is obtained when all substances are uniformly distributed (c1j = c2j = … = cNj), and can be calculated as Hmax = log2N, where N is the number of different compounds. This method has already been applied to the resource efficiency of inorganic materials and metals,50 to the quantification of the recyclability of e-waste,51 and to the analysis of material flows for express plastic packaging.52
. In the case of SEA, a low entropy corresponds to a highly concentrated mass flow (low uncertainty) and a high entropy corresponds to a highly diluted mass flow (high uncertainty). The maximum entropy is obtained when all substances are uniformly distributed (c1j = c2j = … = cNj), and can be calculated as Hmax = log2N, where N is the number of different compounds. This method has already been applied to the resource efficiency of inorganic materials and metals,50 to the quantification of the recyclability of e-waste,51 and to the analysis of material flows for express plastic packaging.52
SEA can be further extended to assess multi-component systems, called Multilevel SEA (MSEA), where statistical entropy is calculated at the component and product levels.125 This method has been applied to the study of exentropy, using both statistical entropy and exergy analysis to determine recyclability, where different recycling processes are compared in terms of mass and energy conservation.126,127 The integration of energy calculations and statistical entropy calculations is currently being investigated to explore potential applications in the assessment of recyclability indicators.128,129 In addition, statistical entropy-based definitions have been evaluated to quantify the separation complexity of mixed-plastic waste streams.130 Furthermore, the link between statistical entropy and LCA is being investigated to develop a new methodology for assessing circularity in recycling, taking into account both the concentration of materials and their environmental impact.131 This study highlights the potential of linking the results of process-based assessments with statistical entropy. Other studies have focused on linking statistical entropy to circularity and the (carbon) circular economy from other perspectives by combining entropy with yield characteristics.53 Statistical entropy has not yet been applied to biomass valorisation, but its broad applicability, for example, in metallurgy and plastic packaging shows great potential for extending the scope of statistical entropy to biomass.
Furthermore, statistical entropy could be applied to compare the energy required to (i) valorise a given biomass into a variety of end products, and (ii) produce a selected end product from a variety of (bio)resources. Using statistical entropy as a proxy for energy, the path with minimum thermodynamic resistance can be determined, taking into account the three predefined parameters for the decision-making process at any TRL, namely the compositional profile, the geospatial availability of the biomass, and the available market. As this is a novel method, statistical entropy has not yet been applied in this context. To do this, the input data required for the state-based assessment must be unified and translated into entropies.
The first set of input data is biomass composition. The compositional profile of the biomass source and the end-product must be translated into entropies called compositional entropy. Statistical entropy determines the complexity of the input stream, the biomass itself, and the output stream, the end-product, through mass balances. In addition to calculating entropy over an entire process, it is possible to look at specific process steps to indicate which are the critical steps in terms of complexity change and energy requirements.
The second set of input data is the geospatial biomass distribution. This again needs to be translated into entropies called geospatial entropy. The geospatial distribution of biomass location reflects concentration or probability. Recent studies have focused on different definitions of geospatial entropy, from which the most appropriate definitions for biomass valorisation need to be identified.132
The third, and last parameter, can be integrated in the geospatial entropy, as this represents the biomass availability.
The two levels of entropy mentioned above represent different pieces of information that need to be related. These levels represent (i) where to locate a biorefinery or source of biomass, and (ii) what biomass to source. In order to develop a state-based assessment, the entropy levels need to be integrated into an energy ranking where the energy-entropy link is established. This includes how the entropy levels relate to each other and how they compare to energy. This will also establish the energy-economy link and the energy-climate impact link to ensure validity against TEA and LCA results. The aim is not to replace LCA and TEA, but to fill the gaps where these methods are not applicable.
Conclusions
This review has highlighted the different lignocellulosic biomass valorisation pathways currently in use or under investigation. These include differences in the composition of lignocellulosic biomass types, potential end-products, pretreatments, and conversion methods. The evidence from this study suggests that each of these valorisation pathways has its own implications and potential. However, no optimal pathways are suggested for different situations linking biomass sources to end-products. The high dependency on many factors complicates this process. Looking at the current state-of-the-art process-based assessment methods to determine whether processes are economically viable and environmentally sustainable, there are still limitations, mainly in terms of data availability, and time, data and expertise intensity. A state-based assessment, resulting in an energy ranking based on statistical entropy, shows potential in addressing the current limitations. This study focuses on shifting away from the current state-of-the-art process-based assessment and creating a novel, easily applicable state-based assessment framework that supports faster and less data-intensive decision-making for (bio)resource valorisation. In doing so, decisions in two directions can be better supported; considering a given biomass feedstock that can potentially produce a variety of end products, and vice versa, producing a selected end product from a variety of (bio)resources. This limits the need for complex and detailed data, and drastically reduces the time needed to perform these assessments, potentially leading to accelerated development of the current bio-economy. However, the aim is not to replace TEA and LCA, but to fill the gaps where these methods are not applicable. Future work is required to determine the extent to which statistical entropy can serve as a proxy for the energy requirements of these processes, and potentially fill the current gaps to create biomass-end-product combinations.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review. The data supporting this article have been included as part of the ESI.†Author contributions
Britt Segers: writing – original draft, writing – review & editing, conceptualization, methodology, data curation, formal analysis, validation, visualization. Philippe Nimmegeers: writing – original draft, writing – review & editing, conceptualization, methodology, data curation, validation, visualization, supervision. Marc Spiller: writing – review & editing, methodology, resources, supervision. Giorgio Tofani: writing – original draft, writing – review & editing. Edita Jasiukaitytė-Grojzdek: writing – original draft, writing – review & editing. Elina Dace: writing – original draft, writing – review & editing. Timo Kikas: writing – original draft, writing – review & editing. Jorge M. Marchetti: writing – original draft, writing – review & editing. Milena Rajić: writing – original draft, writing – review & editing. Güray Yildiz: writing – original draft, writing – review & editing. Pieter Billen: writing – original draft, writing – review & editing, conceptualization, methodology, resources, supervision. All have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was carried out within the framework of the ADV_BIO project financed by the FOD Economie – Energietransitiefonds/SPF Économie – Fonds de Transition Energétique, call 2019–2020 subsidies. Philippe Nimmegeers holds a FWO senior postdoctoral fellowship (grant number: 1215523N) granted by FWO Vlaanderen/Research Foundation Flanders. Support was also received from the Special Research Fund of the University of Antwerp (BOF) for the MAZE BOF-SEP project (grant number: 47134). The collaboration between international authors was facilitated by COST Action 20133 FULLRECO4US.Notes and references
- M. Denchak, Fossil Fuels: the Dirty Facts, https://www.nrdc.org/stories/fossil-fuels-dirty-facts, accessed January, 2023 Search PubMed.
- O. Yishai, S. N. Lindner, J. G. de la Cruz, H. Tenenboim and A. Bar-Even, Curr. Opin. Chem. Biol., 2016, 35, 1–9 CrossRef CAS PubMed.
- R. Höfer, in Renewable Resources for Surface Coatings, Inks and Adhesives, Royal Society of Chemistry, 2022, vol. 72 Search PubMed.
- D. J. Roddy, Interface Focus, 2013, 3, 20120038 CrossRef PubMed.
- H. Ritchie, M. Roser and P. Rosado, Energy, https://ourworldindata.org/energy, accessed January, 2023 Search PubMed.
- Going Climate-Neutral by 2050: A Strategic Long-Term Vision for a Prosperous, Modern, Competitive and Climate-Neutral EU Economy, https://op.europa.eu/en/publication-detail/-/publication/92f6d5bc-76bc-11e9-9f05-01aa75ed71a1, accessed March, 2024 Search PubMed.
- The Long-Term Strategy of the United States: Pathways to Net-Zero Greenhouse Gas Emissions by 2050, https://www.whitehouse.gov/wp-content/uploads/2021/10/us-long-term-strategy.pdf, accessed June, 2024 Search PubMed.
- China's Achievements, New Goals and New Measures for Nationally Determined Contributions, https://unfccc.int/sites/default/files/NDC/2022-06/China%E2%80%99s%20Achievements%2C%20New%20Goals%20and%20New%20Measures%20for%20Nationally%20Determined%20Contributions.pdf, accessed June, 2024 Search PubMed.
- Refining in the Energy Transition through 2040, https://www.mckinsey.com/industries/oil-and-gas/our-insights/refining-in-the-energy-transition-through-2040, accessed March, 2024 Search PubMed.
- C. F. Sepulveda, Int. Rev. Econ. Educ., 2020, 35, 100194 CrossRef.
- Bioeconomy, https://research-and-innovation.ec.europa.eu/research-area/environment/bioeconomy_en, accessed January, 2023 Search PubMed.
- J. J. Bozell and M. K. Patel, ACS, New York, 2006 Search PubMed.
- Report from the Commission to the European Parliament, the Council, the European Economic and Social Committee and the Committee of the Regions, https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=COM%3A2022%3A283%3AFIN, accessed September, 2023 Search PubMed.
- Bioeconomy in Flanders: the Vision and Strategyof the Government of Flanders for a Sustainable and Competitive Bioeconomy in 2030, https://publicaties.vlaanderen.be/view-file/13586, accessed January, 2023 Search PubMed.
- The Main Types of Biomass Energy, https://theearthproject.com/biomass-energy-types/, accessed December, 2022 Search PubMed.
- Biomass Resources, https://www.energy.gov/eere/bioenergy/biomass-resources, accessed December, 2022 Search PubMed.
- Categories of Biomass Material, https://www.geo.fu-berlin.de/en/v/iwm-network/learning_content/watershed-resources/ressource_biomass/categories_material/index.html, accessed December, 2022 Search PubMed.
- M. Hoogwijk, A. Faaij, R. van den Broek, G. Berndes, D. Gielen and W. Turkenburg, Biomass Bioenergy, 2003, 25, 119–133 CrossRef.
- E. W. Qian, in Research Approaches to Sustainable Biomass Systems, ed. S. Tojo and T. Hirasawa, Academic Press, Boston, 2014, pp. 181–204, DOI:10.1016/B978-0-12-404609-2.00007-6.
- U. Javourez, M. O'Donohue and L. Hamelin, Biotechnol. Adv., 2021, 53, 107857 CrossRef CAS PubMed.
- K. Merklein, S. S. Fong and Y. Deng, in Biotechnology for Biofuel Production and Optimization, ed. C. A. Eckert and C. T. Trinh, Elsevier, Amsterdam, 2016, pp. 291–324, DOI:10.1016/B978-0-444-63475-7.00011-X.
- Biomass at a Glance, https://www.need.org/wp-content/uploads/2019/10/BiomassAtAGlance_11x17.pdf, accessed December, 2022 Search PubMed.
- A. Stikane, E. Dace and E. Stalidzans, New Biotechnol., 2022, 70, 109–115 CrossRef CAS PubMed.
- A. Yousuf, D. Pirozzi and F. Sannino, in Lignocellulosic Biomass to Liquid Biofuels, ed. A. Yousuf, D. Pirozzi and F. Sannino, Academic Press, 2020, pp. 1–15, DOI:10.1016/B978-0-12-815936-1.00001-0.
- E. Kuna, R. Behling, S. Valange, G. Chatel and J. C. Colmenares, Chemistry and Chemical Technologies in Waste Valorization, 2017, pp. 1–20 Search PubMed.
- P. Das, Itä-Suomen Yliopisto, 2019 Search PubMed.
- A. Haile, G. G. Gelebo, T. Tesfaye, W. Mengie, M. A. Mebrate, A. Abuhay and D. Y. Limeneh, Bioresour. Bioprocess., 2021, 8, 1–22 CrossRef PubMed.
- X. Ge, C. Chang, L. Zhang, S. Cui, X. Luo, S. Hu, Y. Qin and Y. Li, in Advances in Bioenergy, ed. Y. Li and X. Ge, Elsevier, 2018, vol. 3, pp. 161–213 Search PubMed.
- M. Tišma, M. Bucić-Kojić and M. Planinić, Chem. Biochem. Eng. Q., 2021, 35, 139–156 CrossRef.
- W. Schutyser, T. Renders, G. Van den Bossche, S. Van den Bosch, S. F. Koelewijn, T. Ennaert and B. F. Sels, Nanotechnol. Catal., 2017, 537–584 CAS.
- Z. Sun, J. Cheng, D. Wang, T. Q. Yuan, G. Song and K. Barta, ChemSusChem, 2020, 13, 5199–5212 CrossRef CAS PubMed.
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. S. Luterbacher, J. Ralph, R. Rinaldi, Y. Román-Leshkov, J. S. Samec, B. F. Sels and F. Wang, Energy Environ. Sci., 2021, 14, 262–292 RSC.
- I. u. Haq, K. Qaisar, A. Nawaz, F. Akram, H. Mukhtar, Y. Xu, M. W. Mumtaz, U. Rashid, W. A. W. A. K. Ghani and T. S. Y. Choong, Catalysts, 2021, 11, 309 CrossRef.
- J. Langeveld, J. Dixon and J. Jaworski, Crop Sci., 2010, 50, S142–S151 CrossRef.
- A. Blasi, A. Verardi, C. G. Lopresto, S. Siciliano and P. Sangiorgio, Recycling, 2023, 8, 61 CrossRef.
- M. S. Singhvi and D. V. Gokhale, Appl. Microbiol. Biotechnol., 2019, 103, 9305–9320 CrossRef CAS PubMed.
- D. Tocco, C. Carucci, M. Monduzzi, A. Salis and E. Sanjust, ACS Sustain. Chem. Eng., 2021, 9, 2412–2432 CrossRef CAS.
- J. N. Putro, F. E. Soetaredjo, S.-Y. Lin, Y.-H. Ju and S. Ismadji, RSC Adv., 2016, 6, 46834–46852 RSC.
- M. Tayyab, A. Noman, W. Islam, S. Waheed, Y. Arafat, F. Ali, M. Zaynab, S. Lin, H. Zhang and W. Lin, Appl. Ecol. Environ. Res, 2018, 16, 225–249 CrossRef.
- T. Renders, G. Van den Bossche, T. Vangeel, K. Van Aelst and B. Sels, Curr. Opin. Biotechnol., 2019, 56, 193–201 CrossRef CAS PubMed.
- S. Shapiro-Bengtsen, L. Hamelin, L. Bregnbæk, L. Zou and M. Münster, Energy Environ. Sci., 2022, 15, 1950–1966 RSC.
- G. Yildiz and W. Prins, Energy Fuels, 2022, 37, 805–832 CrossRef.
- S. van Dyk, J. Su, J. D. Mcmillan and J. Saddler, Biofuels, Bioprod. Biorefin., 2019, 13, 760–775 CrossRef CAS.
- Z. Sun, B. Fridrich, A. De Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- M. Buyle, B. Maes, S. Van Passel, K. Boonen, A. Vercalsteren and A. Audenaert, J. Cleaner Prod., 2021, 313, 127921 CrossRef.
- G. Thomassen, S. Van Passel and J. Dewulf, Renewable Sustainable Energy Rev., 2020, 130, 109937 CrossRef.
- K. Vasilakou, P. Nimmegeers, P. Billen and S. Van Passel, Renewable Sustainable Energy Rev., 2023, 187, 113743 CrossRef CAS.
- L. Mencarelli, Q. Chen, A. Pagot and I. E. Grossmann, Comput. Chem. Eng., 2020, 136, 106808 CrossRef CAS.
- H. Rechberger and P. H. Brunner, Environ. Sci. Technol., 2002, 36, 809–816 CrossRef CAS PubMed.
- H. Rechberger and T. E. Graedel, Ecol. Econ., 2002, 42, 59–72 CrossRef.
- X. Zeng and J. Li, J. Cleaner Prod., 2016, 131, 156–162 CrossRef.
- Y. Tang, J. Mao, G. Yu, J. Li and J. Wang, Environ. Sci. Pollut. Res., 2024, 31, 28939–28949 CrossRef PubMed.
- F. Compart and M. Gräbner, Circ. Econ. Sustainability, 2024, 4, 2169–2197, DOI:10.1007/s43615-023-00339-1.
- E. Paone, T. Tabanelli and F. Mauriello, Curr. Opin. Green Sustainable Chem., 2020, 24, 1–6 CrossRef.
- R. M. Rowell, Handbook of Wood Chemistry and Wood Composites, CRC Press, 2005 Search PubMed.
- E. Hughes, The Lignin Challenge, https://edu.rsc.org/feature/the-lignin-challenge/2000124, accessed December, 2022 Search PubMed.
- Z. Anwar, M. Gulfraz and M. Irshad, J. Radiat. Res. Appl. Sci., 2014, 7, 163–173 CAS.
- H. Wang, J. Male and Y. Wang, ACS Catal., 2013, 3, 1047–1070 CrossRef CAS.
- G. D. Saratale, R. G. Saratale, J. R. Banu and J.-S. Chang, in Biohydrogen, ed. A. Pandey, S. V. Mohan, J.-S. Chang, P. C. Hallenbeck and C. Larroche, Elsevier, 2nd edn, 2019, pp. 247–277, DOI:10.1016/B978-0-444-64203-5.00010-1.
- Lignocellulosic Crops for Production of Advanced Biofuels, accessed December, 2022 Search PubMed.
- A. Brandt, J. Gräsvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550–583 RSC.
- C. K. Nitsos, T. Choli-Papadopoulou, K. A. Matis and K. S. Triantafyllidis, ACS Sustain. Chem. Eng., 2016, 4, 4529–4544 CrossRef CAS.
- X. Liu, F. P. Bouxin, J. Fan, V. L. Budarin, C. Hu and J. H. Clark, ChemSusChem, 2020, 13, 4296–4317 CrossRef CAS PubMed.
- M. N. Belgacem and A. Gandini, Monomers, Polymers and Composites from Renewable Resources, Elsevier, 2011 Search PubMed.
- Q. Mei, X. Shen, H. Liu and B. Han, Chin. Chem. Lett., 2019, 30, 15–24 CrossRef CAS.
- C. Gutiérrez-Antonio, A. G. Romero-Izquierdo, F. I. Gómez-Castro and S. Hernández, in Production Processes of Renewable Aviation Fuel, ed. C. Gutiérrez-Antonio, A. G. Romero-Izquierdo, F. I. Gómez-Castro and S. Hernández, Elsevier, 2021, pp. 129–169, DOI:10.1016/B978-0-12-819719-6.00005-5.
- W. Geng, R. Narron, X. Jiang, J. J. Pawlak, H.-m. Chang, S. Park, H. Jameel and R. A. Venditti, Cellulose, 2019, 26, 3219–3230 CrossRef CAS.
- L. Kou, Y. Song, X. Zhang and T. Tan, Bioresour. Technol., 2017, 241, 424–429 CrossRef CAS PubMed.
- S. V. Vassilev, D. Baxter, L. K. Andersen and C. G. Vassileva, Fuel, 2013, 105, 40–76 CrossRef CAS.
- G. Tofani, I. Cornet and S. Tavernier, Biomass Convers. Biorefin., 2022, 1–16 Search PubMed.
- H. Ji, C. Dong, G. Yang and Z. Pang, ACS Sustain. Chem. Eng., 2018, 6, 15306–15315 CrossRef CAS.
- F. Brienza, K. Van Aelst, F. Devred, D. Magnin, M. Tschulkow, P. Nimmegeers, S. Van Passel, B. F. Sels, P. Gerin and D. P. Debecker, Chem. Eng. J., 2022, 450, 138179 CrossRef CAS.
- J. Baruah, B. K. Nath, R. Sharma, S. Kumar, R. C. Deka, D. C. Baruah and E. Kalita, Front. Energy Res., 2018, 6, 141 CrossRef.
- J. K. Singh, P. Vyas, A. Dubey, C. P. Upadhyaya, R. Kothari, V. V. Tyagi and A. Kumar, Front. Biosci., 2018, 10, 350–371 CrossRef PubMed.
- A. K. Kumar and S. Sharma, Bioresour. Bioprocess., 2017, 4, 1–19 CrossRef PubMed.
- M. Garedew, F. Lin, B. Song, T. M. DeWinter, J. E. Jackson, C. M. Saffron, C. H. Lam and P. T. Anastas, ChemSusChem, 2020, 13, 4214–4237 CrossRef CAS PubMed.
- S. Kusch and M. Morar, ProEnvironment ProMediu, 2009, vol. 2 Search PubMed.
- G. Yildiz, F. Ronsse, R. Van Duren and W. Prins, Renewable Sustainable Energy Rev., 2016, 57, 1596–1610 CrossRef CAS.
- G. Yildiz, F. Ronsse and W. Prins, in Fast pyrolysis of biomass: advances in science and technology, ed. K. Wang and R. C. Brown, Royal Society of Chemistry, 2017, vol. 50, pp. 200–230, 10.1039/9781788010245-00200.
- C. R. Soccol, V. Faraco, S. G. Karp, L. P. S. Vandenberghe, V. Thomaz-Soccol, A. L. Woiciechowski and A. Pandey, in Biofuels: Alternative Feedstocks and Conversion Processes for the Production of Liquid and Gaseous Biofuels, ed. A. Pandey, C. Larroche, C.-G. Dussap, E. Gnansounou, S. K. Khanal and S. Ricke, Academic Press, 2nd edn, 2019, pp. 331–354, DOI:10.1016/B978-0-12-816856-1.00014-2.
- S. K. Shin, Y. J. Ko, J. E. Hyeon and S. O. Han, Bioresour. Technol., 2019, 289, 121728 CrossRef CAS PubMed.
- M. Singhvi and B. S. Kim, Biotechnol. Appl. Biochem., 2021, 68, 459–468 CrossRef CAS PubMed.
- S. Shanmugam, A. Hari, A. Pugazhendhi and T. Kikas, Energies, 2023, 16, 4998 CrossRef CAS.
- C. Gutiérrez-Antonio, A. G. Romero-Izquierdo, F. I. Gómez-Castro and S. Hernández, in Production Processes of Renewable Aviation Fuel, ed. C. Gutiérrez-Antonio, A. G. Romero-Izquierdo, F. I. Gómez-Castro and S. Hernández, Elsevier, 2021, pp. 93–127, DOI:10.1016/B978-0-12-819719-6.00004-3.
- C. Gutiérrez-Antonio, A. G. Romero-Izquierdo, F. I. Gómez-Castro and S. Hernández, in Production Processes of Renewable Aviation Fuel, ed. C. Gutiérrez-Antonio, A. G. Romero-Izquierdo, F. I. Gómez-Castro and S. Hernández, Elsevier, 2021, pp. 55–91, DOI:10.1016/B978-0-12-819719-6.00003-1.
- P. P. Thoresen, L. Matsakas, U. Rova and P. Christakopoulos, Bioresour. Technol., 2020, 306, 123189 CrossRef CAS PubMed.
- F. Brienza, Extending the scope of reductive lignin depolymerization toward new feedstocks and innovative non-metal approaches, UCL-Université Catholique de Louvain, 2022 Search PubMed.
- D. V. Sahayaraj, A. Lusi, A. J. Kohler, H. Bateni, H. Radhakrishnan, A. Saraeian, B. H. Shanks, X. Bai and J.-P. Tessonnier, Energy Environ. Sci., 2023, 16, 97–112 RSC.
- A. W. Bartling, M. L. Stone, R. J. Hanes, A. Bhatt, Y. Zhang, M. J. Biddy, R. Davis, J. S. Kruger, N. E. Thornburg and J. S. Luterbacher, Energy Environ. Sci., 2021, 14, 4147–4168 RSC.
- E. M. Anderson, R. Katahira, M. Reed, M. G. Resch, E. M. Karp, G. T. Beckham and Y. Román-Leshkov, ACS Sustain. Chem. Eng., 2016, 4, 6940–6950 CrossRef CAS.
- Y. Liao, S.-F. Koelewijn, G. Van den Bossche, J. Van Aelst, S. Van den Bosch, T. Renders, K. Navare, T. Nicolaï, K. Van Aelst and M. Maesen, Science, 2020, 367, 1385–1390 CrossRef CAS PubMed.
- M. Tschulkow, T. Compernolle, S. Van den Bosch, J. Van Aelst, I. Storms, M. Van Dael, G. Van den Bossche, B. Sels and S. Van Passel, J. Cleaner Prod., 2020, 266, 122022 CrossRef CAS.
- T. Nicolaï, W. Arts, S. Calderon-Ardila, R. Smets, M. Van Der Borght, J. Thevelein and B. F. Sels, ACS Sustain. Chem. Eng., 2024, 12, 8353–8365 CrossRef.
- F. Brienza, K. Van Aelst, F. Devred, D. Magnin, B. F. Sels, P. Gerin, I. Cybulska and D. P. Debecker, ChemSusChem, 2023, 16, e202300103 CrossRef CAS PubMed.
- Z. Cao, M. Dierks, M. T. Clough, I. B. Daltro de Castro and R. Rinaldi, Joule, 2018, 2, 1118–1133 CrossRef CAS PubMed.
- R. Rinaldi, R. T. Woodward, P. Ferrini and H. J. Rivera, J. Braz. Chem. Soc., 2019, 30, 479–491 CAS.
- I. Graça, R. T. Woodward, M. Kennema and R. Rinaldi, ACS Sustain. Chem. Eng., 2018, 6, 13408–13419 CrossRef.
- M. Kennema, I. B. D. de Castro, F. Meemken and R. Rinaldi, ACS Catal., 2017, 7, 2437–2445 CrossRef CAS.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- M. Kessler and R. Rinaldi, Front. Chem., 2022, 9, 816553 CrossRef PubMed.
- F. Brienza, D. Cannella, D. Montesdeoca, I. Cybulska and D. P. Debecker, RSC Sustainability, 2024, 2, 37–90 RSC.
- G. Thomassen, M. Van Dael, S. Van Passel and F. You, Green Chem., 2019, 21, 4868–4886 RSC.
- K. Chomkhamsri, M. Wolf and R. Pant, in Towards Life Cycle Sustainability Management, ed. M. Finkbeiner, Springer, 2011, pp. 107–117, DOI:10.1007/978-94-007-1899-9_11.
- S. Hellweg and L. Milà i Canals, Science, 2014, 344, 1109–1113 CrossRef CAS PubMed.
- R. Sinnott and G. Towler, in Chemical Engineering Design, Butterworth-Heinemann, 2020, pp. 275–369 Search PubMed.
- G. Bishop, D. Styles and P. N. L. Lens, Resour., Conserv. Recycl., 2021, 168, 105451 CrossRef CAS.
- D. Laner, O. Cencic, N. Svensson and J. Krook, Environ. Sci. Technol., 2016, 50, 6882–6891 CrossRef CAS PubMed.
- M. Buyle, A. Audenaert, P. Billen, K. Boonen and S. Van Passel, Sustainability, 2019, 11, 5456 CrossRef.
- V. Piemonte and F. Gironi, Environ. Sci. Technol., 2011, 30, 685–691 CAS.
- J. H. Schmidt, B. P. Weidema and M. Brandão, J. Cleaner Prod., 2015, 99, 230–238 CrossRef.
- K. Vasilakou, P. Nimmegeers, G. Thomassen, P. Billen and S. Van Passel, Appl. Energy, 2023, 344, 121263 CrossRef.
- K. Vasilakou, P. Billen, S. Van Passel and P. Nimmegeers, Energy Convers. Manage., 2024, 303, 118184 CrossRef CAS.
- I. Dincer and M. A. Rosen, in Exergy, ed. I. Dincer and M. A. Rosen, Elsevier, 2nd edn, 2013, pp. 21–30, DOI:10.1016/B978-0-08-097089-9.00002-4.
- M. Mehrpooya, M. Khalili and M. M. M. Sharifzadeh, Renewable Sustainable Energy Rev., 2018, 91, 869–887 CrossRef CAS.
- D. L. Tinoco-Caicedo, E. Feijoó-Villa, J. Calle-Murillo, A. Lozano-Medina and A. M. Blanco-Marigorta, in Computer Aided Chemical Engineering, ed. M. Türkay and R. Gani, Elsevier, 2021, vol. 50, pp. 353–358 Search PubMed.
- M. Bayat and M. Ozalp, in Exergetic, Energetic and Environmental Dimensions, ed. I. Dincer, C. O. Colpan and O. Kizilkan, Academic Press, 2018, pp. 383–402, DOI:10.1016/B978-0-12-813734-5.00022-6.
- T. N. Do, C. You, H. Chung and J. Kim, Comput. Chem. Eng., 2023, 170, 108136 CrossRef CAS.
- A. J. Robinson, A. Giuliano, O. Y. Abdelaziz, C. P. Hulteberg, A. Koutinas, K. S. Triantafyllidis, D. Barletta and I. De Bari, Bioresour. Technol., 2022, 364, 128004 CrossRef CAS PubMed.
- J. Kleinekorte, J. Kleppich, L. Fleitmann, V. Beckert, L. Blodau and A. Bardow, ACS Sustain. Chem. Eng., 2023, 11, 9303–9319 CrossRef CAS.
- D. Fozer, P. Nimmegeers, A. J. Toth, P. S. Varbanov, J. J. Klemeš, P. Mizsey, M. Z. Hauschild and M. Owsianiak, Environ. Sci. Technol., 2023, 57, 13449–13462 CrossRef CAS PubMed.
- K. Lokesh, L. Ladu and L. Summerton, Sustainability, 2018, 10, 1695 CrossRef.
- I. Müller, A History of Thermodynamics: the Doctrine of Energy and Entropy, Springer Science & Business Media, 2007 Search PubMed.
- S. Goldstein and J. L. Lebowitz, Phys. D, 2004, 193, 53–66 CrossRef.
- C. E. Shannon, Bell Syst. Tech. J., 1948, 27, 379–423 CrossRef.
- A. Parchomenko, D. Nelen, J. Gillabel, K. C. Vrancken and H. Rechberger, Resour., Conserv. Recycl., 2020, 161, 104925 CrossRef.
- M. Vierunketo, A. Klemettinen, M. A. Reuter, A. Santasalo-Aarnio and R. Serna-Guerrero, iScience, 2023, 26, 108237 CrossRef CAS PubMed.
- M. Vierunketo, A. Klemettinen, M. A. Reuter, A. Santasalo-Aarnio and R. Serna-Guerrero, J. Cleaner Prod., 2024, 471, 143435 CrossRef CAS.
- P. Nimmegeers, A. Parchomenko, P. De Meulenaere, D. R. D'hooge, P. H. Van Steenberge, H. Rechberger and P. Billen, Sustainability, 2021, 13, 3553 CrossRef CAS.
- C. Moyaert, Y. Fishel, L. Van Nueten, O. Cencic, H. Rechberger, P. Billen and P. Nimmegeers, ACS Sustain. Chem. Eng., 2022, 10, 11719–11725 CrossRef CAS.
- P. Nimmegeers and P. Billen, ACS Sustain. Chem. Eng., 2021, 9, 9813–9822 CrossRef CAS.
- G. Tas, A. Klemettinen and R. Serna-Guerrero, ChemSusChem, 2024, e202400376 CrossRef CAS PubMed.
- C. Moyaert, P. Nimmegeers, B. Mellouk, D. Voordeckers, P. De Meulenaere and P. Billen, Environ. Sci.: Adv., 2024, 3, 314–331 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00342j |
| This journal is © The Royal Society of Chemistry 2024 |