
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtomic layer deposition of Ru nanoclusters on Ba–LaCeOx: a highly efficient catalyst for ammonia synthesis under mild conditions†
Kaiying
Wang‡
 a,
Baitang
Jin‡
b,
Xiaoqing
He
cd and
Xinhua
Liang
*ab
a,
Baitang
Jin‡
b,
Xiaoqing
He
cd and
Xinhua
Liang
*ab
aDepartment of Energy, Environmental & Chemical Engineering, Washington University in St. Louis, St. Louis, Missouri 63130, USA. E-mail: Xinhua.Liang@wustl.edu
bLinda and Bipin Doshi Department of Chemical and Biochemical Engineering, Missouri University of Science and Technology, Rolla, Missouri 65409, USA
cElectron Microscopy Core Facility, University of Missouri, Columbia, Missouri 65211, USA
dDepartment of Mechanical and Aerospace Engineering, University of Missouri, Columbia, Missouri 65211, USA
First published on 7th October 2024
Abstract
Ammonia synthesis has significant implications for global energy and environmental issues, driving the need for highly active catalysts that operate under mild conditions. This study reports the successful deposition of uniform ∼1.0 nm metallic ruthenium (Ru) nanoclusters onto Ba–LaCeOx particles via atomic layer deposition (ALD). The catalytic performance of the ALD-prepared Ru nanoclusters was assessed for ammonia synthesis and compared with two catalysts produced by conventional incipient wetness impregnation. For the ALD-prepared Ru nanoclusters, a pre-reaction H2-reduction step induced partial encapsulation of suboxide species on Ru sites due to strong metal–support interactions, limiting Ru nanocluster sintering and maintaining a reduced Ru size of 1.7 nm. The electron donation from the reduced support to Ru sites imparted an electron-rich character, which facilitated the weakening of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond and promoted the rate-determining step of ammonia synthesis. The ALD-Ru catalysts exhibited competitive ammonia synthesis activity under milder conditions, compared to the impregnated catalysts, with a lower requirement for initial reaction temperature. These results highlight the potential of ALD-synthesized Ru nanoclusters as highly efficient catalysts for low-temperature ammonia production, offering a promising avenue for advancing ammonia synthesis technologies.
N bond and promoted the rate-determining step of ammonia synthesis. The ALD-Ru catalysts exhibited competitive ammonia synthesis activity under milder conditions, compared to the impregnated catalysts, with a lower requirement for initial reaction temperature. These results highlight the potential of ALD-synthesized Ru nanoclusters as highly efficient catalysts for low-temperature ammonia production, offering a promising avenue for advancing ammonia synthesis technologies.
Sustainability spotlightAddressing the global need for sustainable energy sources and industrial processes, our research focuses on ammonia synthesis using ruthenium catalysts. This work advances sustainable chemistry by enabling ammonia production under milder conditions, significantly reducing energy consumption and carbon emissions compared to conventional methods. The use of ruthenium catalysts allows for ammonia synthesis at lower temperatures and pressures, enhancing process efficiency and reducing environmental impact. Additionally, the minimal amount of ruthenium required in catalyst promotes resource conservation. This research aligns with UN Sustainable Development Goals 7 (Affordable and Clean Energy), 9 (Industry, Innovation, and Infrastructure), and 13 (Climate Action) by contributing to cleaner industrial processes, innovative catalytic technologies, and climate change mitigation through reduced carbon emissions in ammonia production. |
1. Introduction
Ammonia, a cornerstone of the global fertilizer industry, sustains approximately 70% of the world's food production.1,2 Its role extends beyond agriculture, emerging as a viable energy carrier due to its superior energy density (15.6 MJ L−1 for liquid NH3, 1.7 times that of liquid H2), carbon neutrality, and logistical advantages in transportation and storage. This positions ammonia as a key player in energy storage solutions, leveraging existing infrastructure for its distribution and handling.3 The synthesis of ammonia, a process that combines nitrogen and hydrogen, is inherently exothermic and benefits thermodynamically from lower temperatures and higher pressures.1 Traditionally, the Haber–Bosch process, reliant on iron-based catalysts, has been the standard method for ammonia production.4 However, this process, operating at temperatures above 450 °C and pressures exceeding 20 MPa, is a significant energy consumer and carbon dioxide emitter, accounting for 2% of global energy usage and 3% of CO2 emissions.5In response to these challenges, ruthenium-based catalysts have been identified as a promising alternative, offering enhanced activity under less severe conditions: below 400 °C and under 10 MPa. This shift could dramatically reduce both energy requirements and carbon emissions.6,7 The development of highly active ruthenium catalysts for ammonia synthesis at these milder conditions has been a focus of recent research. The effectiveness of these catalysts is further improved by the choice of support materials, which can modify the electronic properties of ruthenium, thereby enhancing the overall reaction. Mixed oxide composites have been applied as the support for Ru-based catalysts, such as CeO2–La2O3,8 CeO2–MgO,9 BaO–CeO2,10 and La2O3–Pr2O3.11 Recent studies have reported that Ru/LaCeOx and Ru/BaO–LaCeOx exhibited superior activity in NH3 synthesis under mild conditions. It has been suggested that further reduction of the support could enhance electron donation from the support,12–15 enhancing the electron-rich property of Ru and accelerating N2 activation on Ru sites. During the high-temperature H2 reduction, the reducible support can be partially reduced by H2 spillover from the transition metal sites.16–18
A key to optimizing ruthenium-based catalysts lies in the precise engineering of the active sites, particularly the B5-type sites found in ruthenium nanoparticles. These sites, characterized by a cluster of five ruthenium atoms, exhibit a high affinity for nitrogen, facilitating the breaking of the strong NN triple bond via electron donations to the π-orbitals of N2 under mild conditions. The optimal performance is observed with hemispherical ruthenium nanoparticles approximately 2 nm in diameter, where the concentration of B5-type sites is maximized.19 The method of catalyst preparation plays a crucial role in achieving this configuration. Atomic layer deposition (ALD), a technique that allows for the controlled deposition of metal nanoparticles, emerges as a superior method for preparing ruthenium catalysts.20,21 By enabling precise control over particle size and the metal–support interface, ALD offers a pathway to highly efficient ammonia synthesis under mild conditions. To the best of our knowledge, this is the first report on utilizing ALD-synthesized Ru catalysts for ammonia synthesis.
Another strategy to enhance ammonia synthesis activity is to perform electronic modification by incorporating a basic promoter into Ru catalysts. The electronic modification approach has significantly improved ammonia synthesis activity.22–24 The mechanism involves the transfer of electrons from the basic components to the Ru metal. Subsequently, the transfer of electrons from Ru to the antibonding π-orbitals of N2 weakens the NN bond, facilitating NN cleavage and promoting ammonia formation. Infrared spectroscopy studies have confirmed the weakening of the NN bond by doping with strong basic oxides, with Cs2O reported as the most effective promoter. Consequently, most highly active Ru catalysts contain Cs2O as a promoter.25,26 However, CsOH, which may form in the presence of H2O impurities in the reactants, has a low melting point (272 °C) and can migrate on the catalyst particle surface or vaporize under reaction conditions, eventually leading to catalyst degradation.23 On the other hand, BaO has also been reported as an effective promoter, and barium–ruthenium supported on activated carbon (Ba–Ru/AC) catalysts have been employed in commercial industrial processes. Horiuchi et al. also reported that ruthenium supported on barium titanate (Ru/BaTiO3) and barium–ruthenium supported on magnesium oxide (Ba–Ru/MgO) exhibited comparable high catalytic activity to cesium–ruthenium supported on magnesium oxide (Cs–Ru/MgO) for ammonia synthesis.27 These findings suggest that barium is a more favorable promoter compared to cesium for enhancing the ammonia synthesis activity of ruthenium-based catalysts, as it offers superior thermal stability and resistance to deactivation under reaction conditions.
In this work, we employed BaO–LaCeOx as the catalyst support and utilized the ALD technique to deposit size-controllable Ru nanoclusters on this support for ammonia synthesis under mild conditions. Compared to conventional Ru catalysts prepared by incipient wetness (IW) impregnation using Ru3(CO)12 or RuCl3 precursors, the ALD-prepared Ru/BaO–LaCeOx catalyst demonstrated competitive activity for ammonia synthesis.
2. Experimental
2.1. Catalyst preparation
The BaO–LaCeOx support was synthesized by a coprecipitation method for LaCeOx, followed by an impregnation method for Ba addition. An aqueous solution of La(NO3)3·6H2O and Ce(NO3)3·6H2O was pumped into a concentrated ammonia aqueous solution (28 wt%) to precipitate lanthanum and cerium hydroxides. After filtration and washing with deionized water, the precipitate was added into an aqueous solution of Ba(OH)2, which was stirred for 1 hour. The resulted slurry was evaporated at 80 °C to obtain a powder precursor. This powder was then calcined in air flow at 500 °C, followed by calcination in static air at 700 °C. The final BaO–LaCeOx support, denoted as BLCO, had a molar composition of Ba![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :La:Ce = 0.1:0.45:0.45. Ru nanoclusters were deposited on the BaO–LaCeOx support by ALD employing a custom-built fluidized bed reactor, as detailed in our previous reports.28–30 Bis(cyclopentadienyl)ruthenium(II) (RuCp2, Strem, 99%) and O2 were used as the metal precursor and oxidant, respectively. Prior to ALD, the BaO–LaCeOx particles were loaded into the ALD reactor and heated overnight at 150 °C under vacuum to remove physiosorbed moisture. The ALD process was conducted at 400 °C, with N2 (Airgas, 99.9%) serving as the carrier gas and purge gas for RuCp2 delivery and reactor purging. Each ALD cycle consisted of the following sequence: RuCp2 precursor dose, N2 purge, reactor evacuation, O2 dose, N2 purge, and reactor evacuation. A total of 30 RuCp2 ALD cycles were performed on the BaO–LaCeOx support to deposit the Ru nanoclusters, and the catalyst was denoted as Ru/BLCO_ALD. The loading of Ru for the Ru/BLCO_ALD was 0.5 wt% from inductively coupled plasma-optical emission spectroscopy (ICP-OES).
:La:Ce = 0.1:0.45:0.45. Ru nanoclusters were deposited on the BaO–LaCeOx support by ALD employing a custom-built fluidized bed reactor, as detailed in our previous reports.28–30 Bis(cyclopentadienyl)ruthenium(II) (RuCp2, Strem, 99%) and O2 were used as the metal precursor and oxidant, respectively. Prior to ALD, the BaO–LaCeOx particles were loaded into the ALD reactor and heated overnight at 150 °C under vacuum to remove physiosorbed moisture. The ALD process was conducted at 400 °C, with N2 (Airgas, 99.9%) serving as the carrier gas and purge gas for RuCp2 delivery and reactor purging. Each ALD cycle consisted of the following sequence: RuCp2 precursor dose, N2 purge, reactor evacuation, O2 dose, N2 purge, and reactor evacuation. A total of 30 RuCp2 ALD cycles were performed on the BaO–LaCeOx support to deposit the Ru nanoclusters, and the catalyst was denoted as Ru/BLCO_ALD. The loading of Ru for the Ru/BLCO_ALD was 0.5 wt% from inductively coupled plasma-optical emission spectroscopy (ICP-OES).
For comparative purposes, ruthenium nanoclusters were also deposited onto the BaO–LaCeOx support using conventional IW impregnation technique with two different ruthenium precursors with ∼0.5 wt% Ru loading. In the first method, ruthenium tris(carbonyl) cluster (Ru3(CO)12) dissolved in tetrahydrofuran (THF) was employed as the precursor. The BaO–LaCeOx support was impregnated with the Ru3(CO)12/THF solution under continuous stirring for 12 hours. Subsequently, the solvent was removed via rotary evaporation. The impregnated sample was dried overnight in an oven at 80 °C and then calcined at 500 °C for 5 hours in a muffle furnace. This catalyst was denoted as Ru/BLCO_Ru3(CO)12. In the second method, ruthenium trichloride hydrate (RuCl3·xH2O) dissolved in deionized water was utilized as the precursor solution for impregnation of the BaO–LaCeOx support under stirring for 1 hour. The impregnated sample was dried overnight at 80 °C in an oven, followed by calcination at 500 °C for 5 hours in a muffle furnace. This catalyst was labeled as Ru/BLCO_RuCl3.
2.2. Ammonia synthesis reaction
A homemade stainless steel fixed-bed reactor (O.D. = 12.7 mm and I.D. = 9.5 mm) was employed for ammonia synthesis testing. In a typical experiment, ∼0.32 g of catalyst was physically mixed with 3.2 g of quartz sand and loaded into the reactor, supported on a quartz wool plug (∼50 mg). A K-type thermocouple (Omega Engineering) was positioned in the middle of the reactor to measure the catalyst temperature. High-precision mass flow controllers (Brooks Instruments) regulated the reactant gas flow rates. All inlet gases were passed through a rigorous moisture and oxygen filter system (CRS ZPure LS O2/H2O filter) before entering the reactor, effectively eliminating the presence of water vapor and oxygen. The reactor effluent was analyzed by an online gas chromatograph (SRI 8610C) equipped with a 6-foot Hayesep D column for NH3, CO2, and H2O analysis, and a 6-foot molecular sieve 13X column for N2, H2, CO, and CH4 analysis, using a thermal conductivity detector (TCD). Prior to reaction, the catalysts underwent in situ reduction at 500 °C for 3 hours under a 20 mL min−1 flow of H2 (UHP grade, Airgas). After reduction, the feed was switched to a reactant mixture of N2/H2 (25/75 vol%, UHP grade, Airgas) and the reactor was pressurized to the desired conditions. A back-pressure regulator was used to control the total system pressure, which was monitored by a pressure gauge. The activity was evaluated using the turnover frequency (TOF), which was normalized by both the Ru loading amount and the dispersion of Ru deposited on the supports. The TOF was calculated using the following equation:31where rNH3 represents the molar production rate of NH3 (mol s−1) at the reactor outlet, MRu is the atomic mass of Ru (g mol−1), mcat denotes the weight of the catalyst packed into the reactor (g), D represents the dispersion of Ru deposits. The dispersion (D) was estimated using the equation D = 12.9/d,17 where d is the average Ru particle size (nm) obtained from transmission electron microscopy (TEM) analysis of the reduced samples.
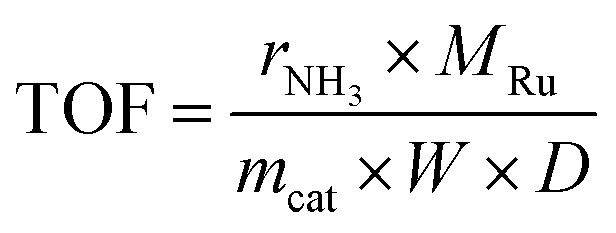
2.3. Catalyst characterizations
The morphology of the fresh and spent catalysts was examined by TEM using an FEI Tecnai F20 instrument. X-ray photoelectron spectroscopy (XPS) was conducted on a Kratos Axis 165 spectrometer to probe the chemical states of the catalysts. Crystalline phases were identified by X-ray diffraction (XRD) using a Philips X'Pert Multi-purpose Diffractometer. The ruthenium loading was quantified by ICP-OES using a PerkinElmer 2000D instrument. Prior to analysis, the samples were digested in aqua regia solution using a 16-position Titan MPS microwave digestion system. Hydrogen temperature-programmed reduction (H2-TPR) experiments were performed using a Micromeritics AutoChem II 2920 instrument to investigate the reducibility of the catalysts. The samples were pretreated at 300 °C under Ar flow, followed by cooling to room temperature. TPR analysis was then carried out by ramping the temperature from room temperature to 1000 °C with a rate of 10 °C min−1 under a 10 vol% H2/Ar gas mixture. Hydrogen temperature-programmed desorption (H2-TPD) analysis was conducted using a Micromeritics AutoChem II 2920 instrument to examine the desorption of chemisorbed H2 from the catalyst surface. The procedure involved exposing the sample to H2 at 500 °C for 3 hours, followed by a 1 hour hold in H2 at 50 °C. The catalyst was then purged in 30 mL min−1 of Ar for 1 hour before being heated to 600 °C for the final desorption step.3. Results and discussion
The morphology and structural characteristics of fresh Ru catalysts synthesized via different preparation methods were investigated utilizing TEM, as depicted in Fig. 1. In Fig. 1a, the Ru particle size of the fresh Ru/BLCO_ALD catalyst was approximately 1.0 nm. Regarding the fresh Ru/BLCO_Ru3(CO)12 catalyst (Fig. 1b), no discernible particle structure was observed, suggesting that the RuO2 sites were predominantly present in an amorphous layer structure. Yao et al. also reported that an amorphous RuO2 structure was obtained when utilizing Ru3(CO)12 as the metal precursor.32 This observation was attributed to the intermolecular forces inherent to Ru3(CO)12 and its strong interaction with the support material. As for the fresh Ru/BLCO_RuCl3 catalyst (Fig. 1c), the size of the Ru nanoclusters was approximately 1.4 nm. Therefore, the morphological and structural characteristics of the fresh Ru catalysts exhibited distinct variations contingent upon the different synthesis methods employed, underscoring the impact of preparation techniques on the resulting catalyst properties.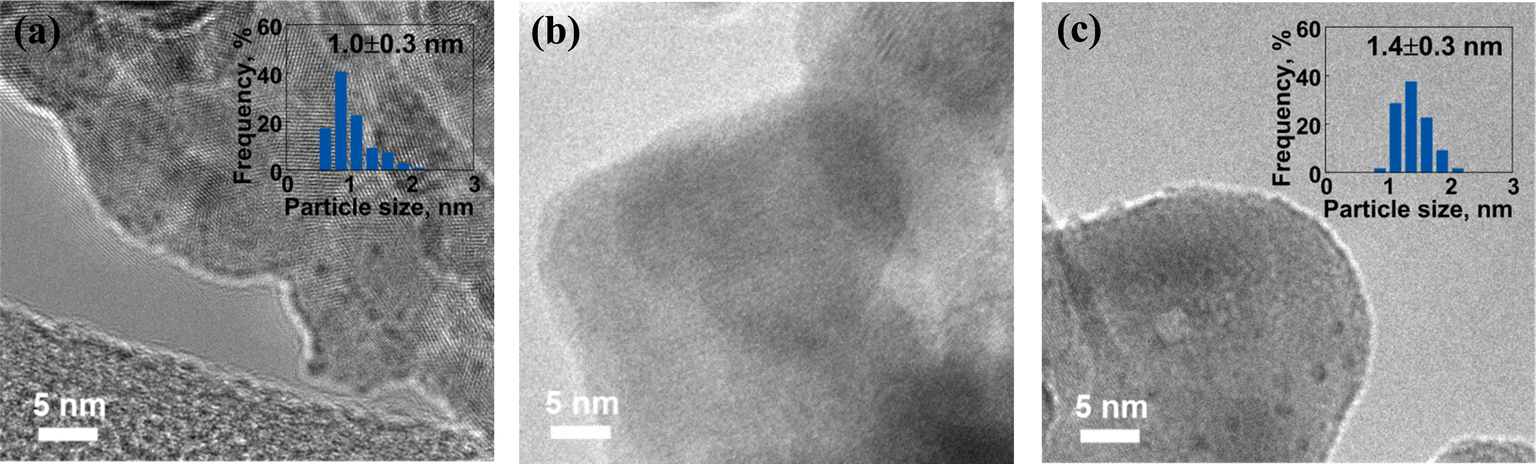 | ||
| Fig. 1 TEM images of (a) fresh Ru/BLCO_ALD, (b) fresh Ru/BLCO_Ru3(CO)12, and (c) fresh Ru/BLCO_RuCl3. | ||
High-resolution XPS spectra were acquired to investigate the chemical states of C, Ru, O, Ce, Ba, and La in three fresh catalysts prepared by different synthesis methods, as shown in Fig. 2 and S1.† The spectra were deconvoluted and calibrated by fixing the adventitious carbon (Cadv) at 284.5 eV. Notably, Ru 3p spectra were also recorded to deconvolute the overlapping C 1s and Ru 3d. For C 1s and Ru 3d spectra in Fig. 2a, the C 1s spectra were deconvoluted into adventitious carbon at 284.5 eV, C–O at 286.4 eV, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 288.5 eV,33,34 while Ru 3d spectra were deconvoluted into Ru at 280.2 eV, RuO2 at 282.0 eV for Ru 3d5/2, and Ru at 283.9 eV and RuO2 at 285.2 eV for Ru 3d3/2. Notably, the Ru nanoclusters prepared by the ALD method consisted of both Ru and RuO2, whereas the Ru nanoclusters in the IW-prepared catalysts with Ru3(CO)12 or RuCl3 as Ru precursor only consisted of RuO2. For the Ru 3p spectra in Fig. 2b, the Ru 3p peaks were deconvoluted into Ru at 461.6 eV, RuO2 at 463.7 eV for Ru 3p3/2, and Ru at 483.7 eV and RuO2 at 485.6 eV for Ru 3p3/2, respectively. Based on the deconvolution results for Ru 3p and Ru 3d, a consistent conclusion was drawn that the Ru nanoclusters prepared by ALD consisted of metallic Ru and oxidized RuO2, which was different from the catalysts prepared by the traditional IW method. The distinct chemical states of Ru sites originated from the unique ALD growth, and the deposition of metallic Ru by ALD has been reported in numerous studies.35–38 For the ammonia synthesis reaction, the unique chemical states of the ALD-prepared Ru sites were found to exhibit better interactions with the support in reducing atmosphere, which will be discussed later.
O at 288.5 eV,33,34 while Ru 3d spectra were deconvoluted into Ru at 280.2 eV, RuO2 at 282.0 eV for Ru 3d5/2, and Ru at 283.9 eV and RuO2 at 285.2 eV for Ru 3d3/2. Notably, the Ru nanoclusters prepared by the ALD method consisted of both Ru and RuO2, whereas the Ru nanoclusters in the IW-prepared catalysts with Ru3(CO)12 or RuCl3 as Ru precursor only consisted of RuO2. For the Ru 3p spectra in Fig. 2b, the Ru 3p peaks were deconvoluted into Ru at 461.6 eV, RuO2 at 463.7 eV for Ru 3p3/2, and Ru at 483.7 eV and RuO2 at 485.6 eV for Ru 3p3/2, respectively. Based on the deconvolution results for Ru 3p and Ru 3d, a consistent conclusion was drawn that the Ru nanoclusters prepared by ALD consisted of metallic Ru and oxidized RuO2, which was different from the catalysts prepared by the traditional IW method. The distinct chemical states of Ru sites originated from the unique ALD growth, and the deposition of metallic Ru by ALD has been reported in numerous studies.35–38 For the ammonia synthesis reaction, the unique chemical states of the ALD-prepared Ru sites were found to exhibit better interactions with the support in reducing atmosphere, which will be discussed later.
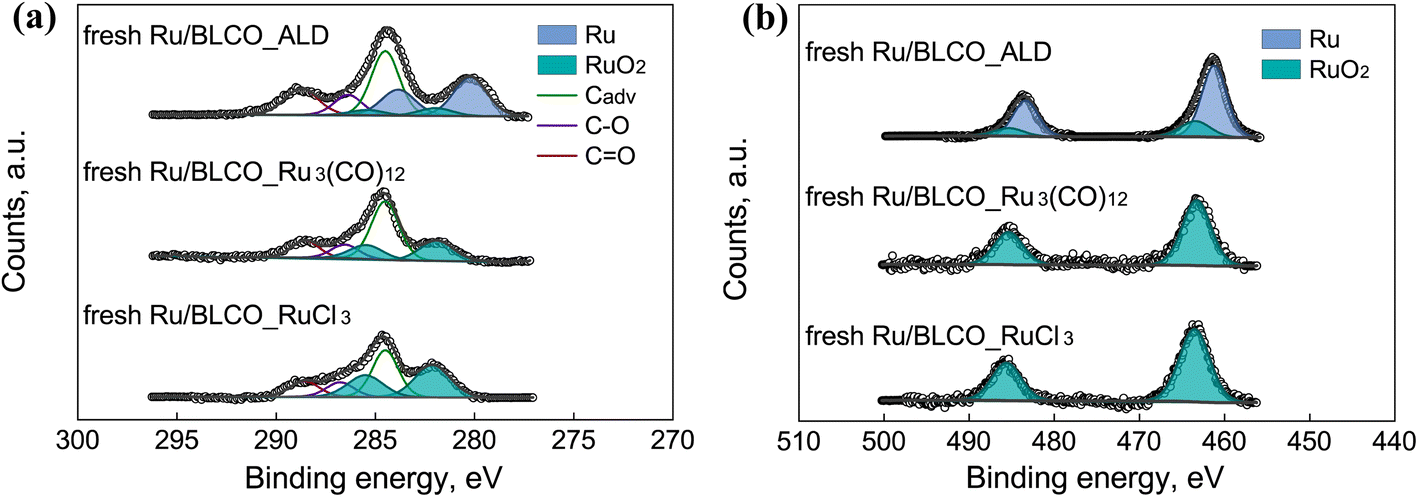 | ||
| Fig. 2 XPS spectra of (a) C 1s + Ru 3d and (b) Ru 3p of Ru/BLCO_ALD, fresh Ru/BLCO_Ru3(CO)12, and fresh Ru/BLCO_RuCl3. | ||
H2-TPR was performed to investigate the interaction between Ru sites and the BaO–LaCeOx support for catalysts prepared via different preparation methods. As shown in Fig. 3, the BaO–LaCeOx support revealed two distinct reduction peaks. The first peak, centered at approximately 420 °C, can be attributed to the reduction of surface Ce(IV) species to Ce(III). This lower reduction temperature, compared to pure CeO2, which typically exhibits its first reduction peak above 490 °C,39–41 indicates enhanced reducibility of the surface cerium ions. This enhancement can be ascribed to the incorporation of Ba and La into the CeO2 lattice, which likely introduces oxygen vacancies and modifies the local electronic structure of Ce ions.42,43 The second reduction peak, observed at around 800 °C, corresponds to the reduction of bulk Ce(IV) to Ce(III). Interestingly, this peak position remained relatively unchanged, compared to pure CeO2, suggesting that the addition of Ba and La primarily affects the surface properties of the material while leaving the bulk reduction characteristics largely unaltered. During the ammonia synthesis process, where the reduction and reaction temperatures exceeded that required for surface Ce(IV) reduction, the redox behavior of surface Ce cations played a crucial role in influencing the catalyst properties and performance through strong metal–support interactions (SMSI).44,45 For the supported Ru catalysts, the TPR profiles revealed distinct differences based on the preparation methods. A new reduction peak appeared at ∼120 °C for the Ru/BLCO_Ru3(CO)12 and Ru/BLCO_RuCl3 catalysts, which was assigned to the reduction of RuO2 species.46–48 The sharp peak at ∼120 °C corresponded to free RuO2, while the broader peak at >150 °C indicated RuO2 interacting with the BaO–LaCeOx support. In contrast, no RuO2 reduction peak was observed for Ru/BLCO_ALD, consistent with the metallic state of Ru in this catalyst. Moreover, the surface Ce(IV) reduction peaks differed among the catalysts. The peak shifted to lower temperatures of ∼300 °C for Ru/BLCO_ALD and ∼280 °C for Ru/BLCO_Ru3(CO)12, indicating enhanced Ce(IV) reducibility due to the Ru-support interactions. However, for Ru/BLCO_RuCl3, the surface Ce(IV) reduction remained at ∼420 °C, suggesting insignificant interaction between Ru and the support. With the assistance of Ru, the CeO2 was reduced to CeOx suboxide and thus the mutual interaction between Ru and the reducible oxide support will reconstruct the metal–support interface structure with intimate bonding during the reduction. The improved Ce(IV) reducibility for ALD and Ru3(CO)12-derived catalysts was attributed to hydrogen spillover from Ru sites during reduction, facilitated by smaller Ru particle sizes and SMSI. Consequently, Ce(IV) was reduced to Ce(III), forming a partially reduced CeOx suboxide that migrated towards Ru nanoclusters and partially encapsulated the Ru nanoclusters due to SMSI. This encapsulated structure enabled electron transfer from the oxygen-deficient CeOx to Ru sites, enriching the electron density of Ru active sites. Therefore, the different preparation methods resulted in varying degrees of metal–support interactions and Ru particle sizes, which significantly influenced the catalytic properties through modifying the electronic structure of Ru active sites via SMSI effects.
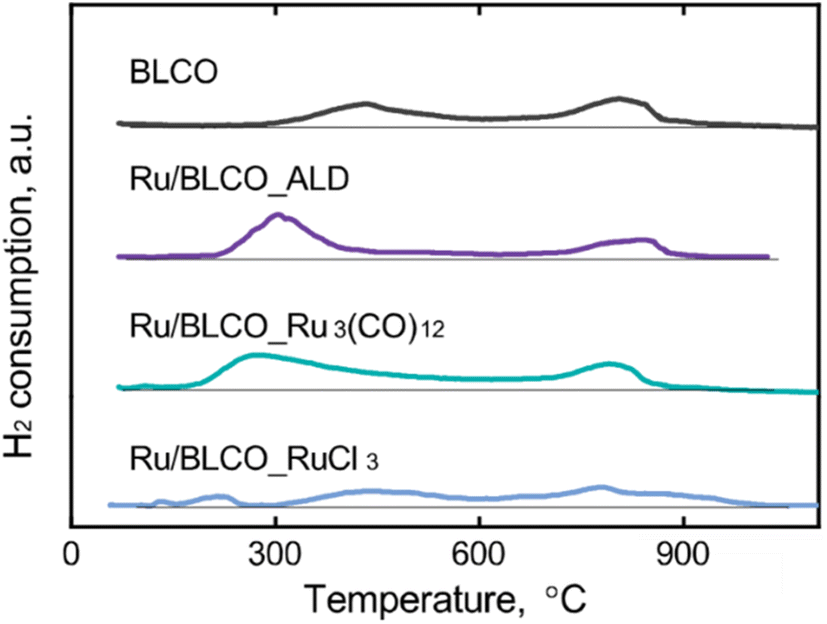 | ||
| Fig. 3 H2-TPR profiles BLCO support, Ru/BLCO_ALD, Ru/BLCO_Ru3(CO)12, and Ru/BLCO_RuCl3. | ||
To investigate the hydrogen adsorption/desorption characteristics of Ru catalysts and the BLCO support, H2-TPD experiments were conducted.49,50Fig. 4 illustrates the TPD profiles of the samples. The BLCO support exhibited the highest hydrogen desorption temperature among all samples tested. Upon introduction of 0.5 wt% Ru, a notable decrease in desorption temperature was observed. For instance, the Ru/BLCO_ALD catalyst demonstrated a hydrogen desorption temperature of approximately 90 °C, significantly lower than the temperature of 280 °C observed for the bare BLCO support. This indicates that the presence of Ru substantially facilitates hydrogen desorption. Among the Ru-containing catalysts, Ru/BLCO_ALD displayed the lowest desorption temperature, followed by Ru/BLCO_RuCl3 and Ru/BLCO_Ru3(CO)12. This trend suggests that Ru/BLCO_ALD may exhibit superior performance due to its facilitated hydrogen desorption characteristics. Quantitative analysis of hydrogen adsorption capacities revealed that the BLCO support adsorbed 1.9 cm3 g−1 of hydrogen, while the Ru-containing catalysts showed increased capacities. Ru/BLCO_RuCl3, Ru/BLCO_Ru3(CO)12, and Ru/BLCO_ALD exhibited adsorption capacities of 2.4, 4.0, and 5.7 cm3 g−1, respectively. The incorporation of Ru significantly enhanced the hydrogen adsorption capacity of the catalysts. This observation aligns with previous research indicating that more reduced ceria can adsorb larger quantities of hydrogen.9,51 XPS analysis in the following section corroborated these findings. The Ce(III) content was determined to be approximately 27.6% for Ru/BLCO_ALD, compared to 22.0% for the reduced Ru/BLCO_Ru3(CO)12 catalyst and 19.1% for the reduced Ru/BLCO_RuCl3 catalyst. This higher Ce(III) content in Ru/BLCO_ALD is consistent with its enhanced hydrogen adsorption capacity. Therefore, the superior performance of the Ru/BLCO_ALD catalyst can be attributed to its facilitated hydrogen desorption, as evidenced by the lower desorption temperature, and increased hydrogen adsorption capacity, likely due to a higher degree of ceria reduction. These findings underscore the importance of catalyst preparation methods in optimizing hydrogen interaction and, consequently, ammonia synthesis activity.
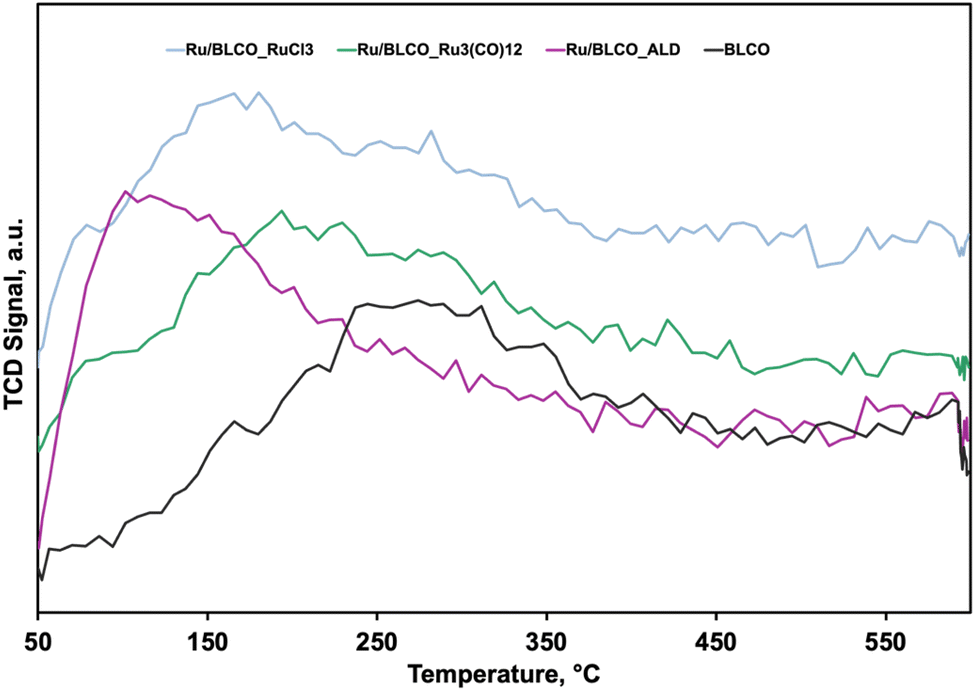 | ||
| Fig. 4 H2-TPD profiles BLCO support, Ru/BLCO_ALD, Ru/BLCO_Ru3(CO)12, and Ru/BLCO_RuCl3. | ||
The morphology and structure of the reduced catalysts were examined using TEM, as shown in Fig. 5 and S2.† After H2 reduction, the size of Ru sites increased due to the sintering during the high-temperature reduction process. For the ALD-prepared Ru/BLCO_ALD catalyst, the Ru size of the reduced catalyst was at 1.7 nm, compared to 1.0 nm of the as-prepared catalyst. The Ru size of the reduced Ru/BLCO_Ru3(CO)12 catalyst was 2.5 nm, and the Ru size of the reduced Ru/BLCO_RuCl3 catalyst was about 3.2 nm, which were larger than the Ru size of the reduced catalyst prepared by the ALD method. The smaller size for ALD-prepared Ru size indicated that sintering was less significant, which could be ascribed to the SMSI effect. As discussed for the results of XPS and H2-TPR, the ALD-prepared Ru sites were mainly in the metallic state, whereas the Ru nanoclusters in the other two IW-prepared catalysts were in an oxidized RuO2 state. The metallic state Ru nanoclusters enhanced SMSI formation during the high-temperature reduction process, which limited the sintering of Ru sites. In addition, the Ru/BLCO_Ru3(CO)12 catalyst seemed to have a stronger SMSI effect than the Ru/BLCO_RuCl3 catalyst, consistent with the H2-TPR results. Regarding the ammonia synthesis reaction, the size of Ru nanoclusters is a crucial factor, particularly the Ru hemispherical nanoparticles with suitable size at ∼2 nm, which are considered optimal for the activation sites such as B5 sites.19 Besides, some interacting structures were noticed between the metallic Ru nanoparticles and the oxide support, as shown in Fig. S2,† which are different from the fresh catalyst morphology. These structures may result from the reduction process via SMSI. In this work, the ALD-prepared Ru/BLCO_ALD catalyst, with a suitable Ru size, was expected to exhibit excellent catalytic performance.
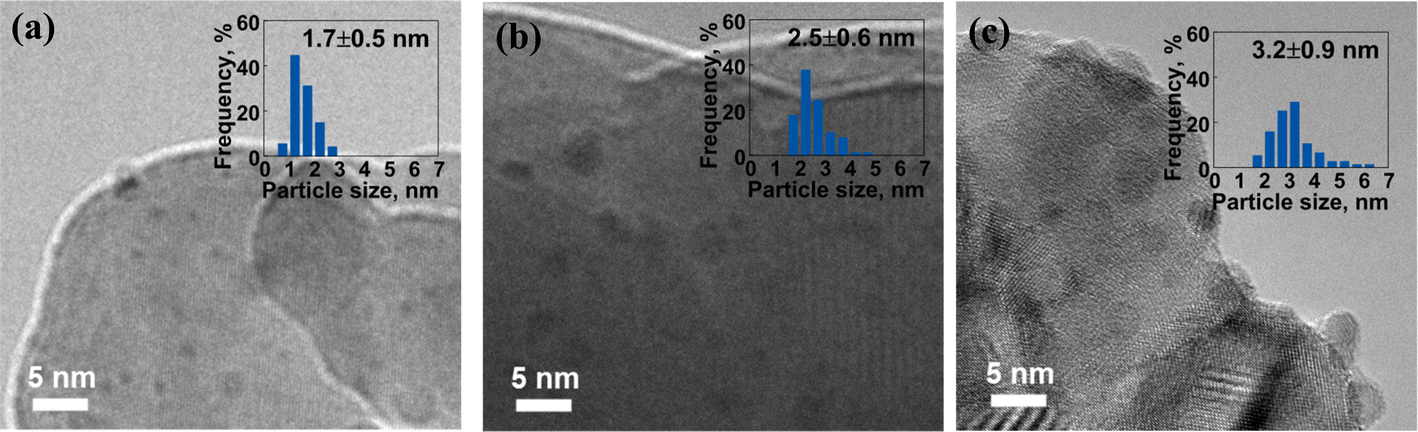 | ||
| Fig. 5 TEM images of (a) reduced Ru/BLCO_ALD, (b) reduced Ru/BLCO_Ru3(CO)12, and (c) reduced Ru/BLCO_RuCl3 catalyst. | ||
High-resolution XPS spectra were conducted to determine the chemical states of C, Ru, O, Ce, Ba, and La after the reduction process for the three catalysts prepared by different methods, as shown in Fig. 6 and S3.† The Ba 3d and La 3d XPS spectra presented in Fig. S3† do not exhibit significant differences among the various catalysts, which can be attributed to several factors. Firstly, Ba and La species are generally less susceptible to reduction by H2 compared to Ce, potentially resulting in minimal changes in their oxidation states during the reduction process. Secondly, the ex situ XPS analysis may limit our ability to detect subtle changes in the chemical states of these elements under reaction conditions. The observed Ba 3d binding energy of approximately 780.2 eV for both fresh and reduced catalysts is consistent with the presence of BaO, as reported in previous studies.52 However, the exact role and chemical state of Ba in Ru-based ammonia synthesis catalysts remain subjects of ongoing debate in the scientific community. For instance, Truszkiewicz et al. proposed that barium undergoes partial reduction during pretreatment, resulting in an active promoter phase composed of a mixture of Ba0 and BaO. This hypothesis suggests a more complex interplay between the metallic and oxide forms of barium in the catalytic process.53 Aika et al. suggested that barium might exist not only as BaO but also as barium hydroxide. The relative proportions of these two compounds can fluctuate depending on temperature and the partial pressure of water in the inlet stream. Intriguingly, they observed higher ammonia synthesis activities under conditions favoring BaO as the predominant barium-containing phase.54 But the authors did not exclude the possibility that barium also modified the ruthenium surface, which indicated that barium may be a simultaneous electron and structural promotion.55 In our specific case, the possibility of Ba(OH)2 formation can be effectively ruled out due to the use of an O2/H2O filter in the inlet gas stream. While our ex situ XPS analysis did not reveal significant changes in the Ba 3d binding energy after reduction, it's crucial to note that this does not preclude the possibility of barium undergoing partial reduction under H2 pretreatment conditions. Interestingly, our surface composition analysis, as presented in Table S1,† reveals a notable change in the Ba/Ru surface ratio after H2 reduction. For the fresh Ru/BLCO_ALD catalyst, we observed a Ba/Ru ratio of 1.66. This ratio increased to 2.24 after reduction, indicating a significant surface enrichment of barium species. This observation aligns with the concept of barium acting as a structural promoter in our catalyst system. Given the complexity of this system and the limitations of ex situ characterization techniques, it is evident that the precise mechanism of barium promotion is intimately linked to the state of the promoter under working conditions. To elucidate this mechanism fully, advanced in situ and operando characterization techniques are required.
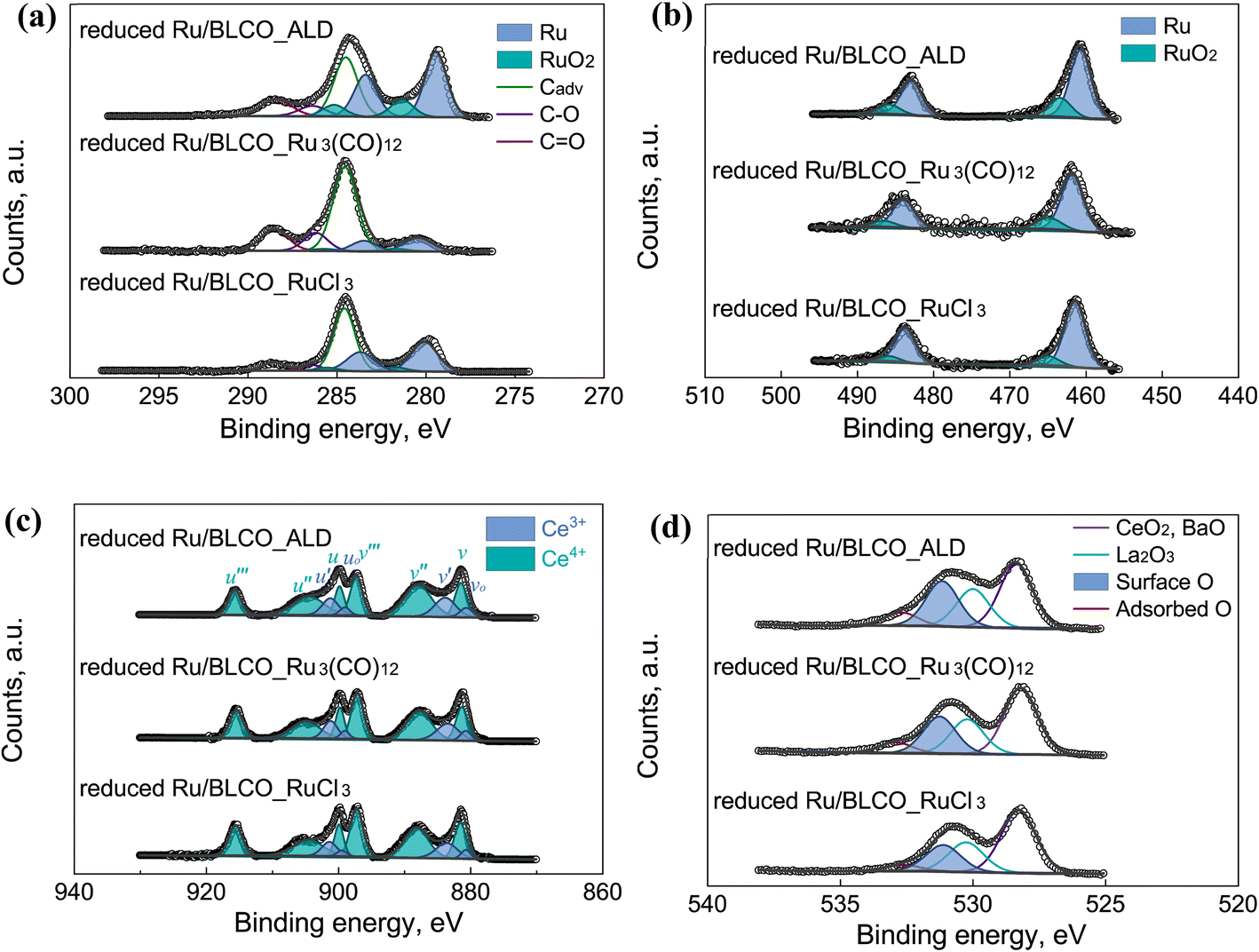 | ||
| Fig. 6 XPS spectra of (a) C 1s + Ru 3d, (b) Ru 3p, (c) Ce 3d, and (d) O 1s of the reduced Ru/BLCO_ALD, reduced Ru/BLCO_Ru3(CO)12, and reduced Ru/BLCO_RuCl3 catalysts. | ||
Fig. 6a depicts the spectra of C 1s and Ru 3d for the reduced catalysts, and Fig. 6b depicts the spectra of Ru 3p for the reduced catalysts. In Fig. 6a, the C 1s spectra were deconvoluted into adventitious carbon at 284.5 eV, C–O at 286.4 eV, and CO at 288.5 eV. The adventitious carbon was calibrated at 284.5 eV. Regarding Ru, all the Ru 3p and Ru 3d profiles consisted of both metallic Ru and a small amount of RuO2. Based on the XPS results of the fresh catalysts and reduced catalysts, RuO2 in the catalysts was reduced to the metallic Ru during the H2-reduction process. From the Ru 3d5/2 region, the peak of the metallic Ru in the reduced Ru/BLCO_ALD was observed at 279.4 eV, which was lower than 280.1 eV for the fresh Ru/BLCO_ALD catalyst, 280.2 eV for the reduced Ru/BLCO_Ru3(CO)12 catalyst, and 280.1 eV for the reduced Ru/BLCO_RuCl3 catalyst. Similar phenomena were observed for the Ru 3p3/2 region; the metallic Ru peak for Ru/BLCO_ALD was at 460.9 eV, whereas the metallic Ru peak was at 461.4 eV for fresh Ru/BLCO_ALD, 461.5 eV for reduced Ru/BLCO_Ru3(CO)12, and 461.4 eV for reduced Ru/BLCO_RuCl3. The difference confirmed the special shift of Ru chemical states for the Ru/BLCO_ALD catalyst after the H2-reduction process. The lower binding energy of Ru was ascribed to the electron donation from the support, as reported as the electronic metal–support interaction. Comparing different preparation methods, the metallic Ru sites of the ALD-prepared Ru/BLCO_ALD catalyst might contribute to the construction of electronic metal–support interaction during the reduction process. The electron-rich state of Ru has been reported to enhance the dissociative adsorption of N2 on Ru sites.56 The relative concentration of Ru and RuO2 is listed in Table S2.† Ru% for the fresh Ru/BLCO_ALD catalyst was ∼72.3%, whereas the refresh Ru/BLCO_Ru3(CO)12 and Ru/BLCO_RuCl3 catalysts were composed entirely of RuO2. Ru% for the reduced Ru/BLCO_ALD catalyst was ∼77%, which was lower than ∼82% for the reduced Ru/BLCO_Ru3(CO)12 catalyst and ∼87% for the reduced Ru/BLCO_RuCl3 catalyst. The lower reduction degree of Ru for the reduced Ru/BLCO_ALD catalyst resulted from the partial encapsulation of suboxide due to SMSI during the H2-reduction process.57,58
Based on the Ce 3d and O 1s spectra, the Ce valence and the oxygen species were studied to demonstrate the mutual effect of ALD-deposited Ru on the support. For the Ce 3d spectra in Fig. 6c, the Ce(IV) peaks consisted of Ce 3d5/2 peaks (v at 881.3 eV, v′′ at 887.8 eV, and v′′′ at 897.2 eV) and Ce 3d3/2 peaks (u at 899.8 eV, u′′ at 904.7 eV, and u′′′ at 915.6 eV), while Ce(III) consisted of Ce 3d5/2 peaks (vo at 880.8 eV and v′ at 883.5 eV) and Ce 3d3/2 peaks (uo at 899.1 eV and u′ at 901.4 eV).45,59 During the H2-reduction process, Ce(IV) was partially reduced to Ce(III), forming the CeOx suboxide. Especially, the H2 spillover from Ru sites could promote the reduction of Ce(IV) and lead to the encapsulation of CeOx on Ru nanoclusters, due the SMSI effect. Under this circumstance, the CeOx interacted mutually with Ru sites, which could influence the catalytic performance. In this work, the reduced Ru/BLCO_ALD catalyst had a higher concentration of Ce(III) than the reduced Ru/BLCO_Ru3(CO)12 and reduced Ru/BLCO_RuCl3 catalysts. The Ce(III) content (calculating from vo% + v′% + uo% + u′%) was found to be ∼27.6%, as compared to ∼22.0% for the reduced Ru/BLCO_Ru3(CO)12 catalyst and ∼19.1% for the reduced Ru/BLCO_RuCl3 catalyst. Besides, the independent u′′′ peak was taken as the diagnostic peak for Ce(IV) content, and the u′′′% was 8.5% for the reduced Ru/BLCO_ALD catalyst, which was lower than 10.2% for the reduced Ru/BLCO_Ru3(CO)12 catalyst and 11.1% for the reduced Ru/BLCO_RuCl3 catalyst. The lower Ce(IV) and higher Ce(III) contents of Ru/BLCO_ALD indicate that the special Ru nanoclusters prepared by ALD provided a stronger H2 spillover effect, enhancing the reduction of Ce(IV) to Ce(III). As discussed in the previous section, the Ru nanoclusters prepared by ALD were in the metallic state, and the bonding between Ru and BaO–LaCeOx support was formed during the ALD process. These factors were conducive to enhancing the H2 spillover effect and achieving a deeper reduction of the oxidized Ce species. This stronger interaction between the metallic Ru and the reduced BaO–LaCeOx support further enhanced the overall catalytic performance. The atomic composition results are listed in Table S1.† Based on the XPS surface result, higher Ce/Ru trend can be observed after H2-reduction, which indicates the structure change of Ce species and possible partial decoration of CeOx on the Ru nanoparticles.
For the O 1s spectra in Fig. 6d, the oxygen species could be fitted into lattice O of La2O3 and BaO, lattice O of CeO2, surface O, and adsorbed O. The lattice O of CeO2 and BaO was located at ∼528.6 eV,60–62 and the lattice oxygen of La2O3 was located at ∼530.3 eV.63 The surface oxygen was located at ∼531.2 eV, which consisted of surface low-coordination O, hydroxyl O, and oxygen vacancy sites;64,65 especially, the oxygen sites in Ce(III)–O were also reported as oxygen vacancy position.62,66 The adsorbed O was located at ∼532.7 eV, mainly originating from adsorbed oxygen-containing molecules (e.g., H2O and CO2).64 Notably, the surface O species for the reduced Ru/BLCO_ALD catalyst reached 28.1%, which is significantly higher than 23.2% for the reduced Ru/BLCO_Ru3(CO)12 catalyst and 21.3% for the reduced Ru/BLCO_RuCl3 catalyst. The increased surface oxygen species was ascribed to the formation of surface oxygen vacancies of the suboxide that was induced by the H2-reduction process. Studies have demonstrated that the formation of suboxide and oxygen vacancy could enable the electron transfer to the interfacial Ru metallic sties by charge compensation. During the ammonia synthesis reaction, the excess electron from the suboxide and oxygen vacancies contributed to the weakening of NN bond on the Ru nanoclusters, which is the rate determining step of the ammonia synthesis reaction. Therefore, the Ru sites with a negative charge provide higher activity for the dissociation adsorption of N2 and could achieve higher ammonia synthesis activity.
Fig. 7 depicts the catalytic performance of ammonia synthesis under different conditions (gas hourly space velocity (GHSV) of 3000 and 6000 mL gcat−1 h−1, 1.0 and 2.8 MPa) using catalysts prepared by three different methods. At 6000 mL gcat−1 h−1, the ammonia synthesis rate increased with the increasing temperature for all the catalysts. However, the ammonia synthesis rate of the Ru/BLCO_ALD catalyst first increased but then decreased at 3000 mL gcat−1 h−1, as the reaction rate reached the equilibrium conversion. For instance, the highest ammonia synthesis rate of the Ru/BLCO_ALD catalyst was obtained at 425 °C under 2.8 MPa and 3000 mL gcat−1 h−1, with the rate decreasing when the temperature further increased to 450 °C. The ammonia synthesis rates of the Ru/BLCO_Ru3(CO)12 and Ru/BLCO_RuCl3 catalysts increased as the temperature increased within the tested range due to their low activity, which was far from equilibrium. Besides, the ammonia synthesis rates were higher at 6000 mL gcat−1 h−1 than at 3000 mL gcat−1 h−1. For example, at 400 °C and 2.8 MPa, the ammonia synthesis rate for Ru/BLCO_ALD was 9.2 mmolNH3 gcat−1 h−1 at 6000 mL gcat−1 h−1, which was higher than 7.8 mmolNH3 gcat−1 h−1 at 3000 mL gcat−1 h−1. This indicates that the overall catalyst performance at 3000 mL gcat−1 h−1 might be limited by the low GHSV. Comparing the catalytic performance of different catalysts, the catalytic activity followed the following order: Ru/BLCO_ALD > Ru/BLCO_Ru3(CO)12 > Ru/BLCO_RuCl3 under different conditions, indicating that the Ru nanoclusters prepared by ALD exhibited better activity than the catalysts prepared by the traditional IW method. For instance, at 2.8 MPa and 3000 mL gcat−1 h−1, the ammonia synthesis rate for Ru/BLCO_ALD was 7.8 mmolNH3 gcat−1 h−1, which was higher than 5.5 mmolNH3 gcat−1 h−1 for Ru/BLCO_Ru3(CO)12 and 3.8 mmolNH3 gcat−1 h−1 for Ru/BLCO_RuCl3. For the catalysts prepared by the traditional IW method, Ru/BLCO_Ru3(CO)12 exhibited higher performance than Ru/BLCO_RuCl3, which was consistent with previous report that a Ru-based catalyst prepared using Cl-free Ru precursor was more active than that prepared using Cl-containing Ru precursor, as some of the Cl− could remain on the Ru surfaces.25,67
 | ||
| Fig. 7 Catalytic activities of ammonia synthesis using Ru/BLCO_ALD, Ru/BLCO_Ru3(CO)12, and Ru/BLCO_RuCl3 under different conditions: (a) 2.8 MPa and 3000 mL gcat−1 h−1, (b) 2.8 MPa and 6000 mL gcat−1 h−1, (c) 1.0 MPa and 3000 mL gcat−1 h−1, and (d) 1.0 MPa and 6000 mL gcat−1 h−1. Note: the dash line shows the equilibrium ammonia synthesis rate. | ||
TOF is a crucial metric that provides insights into the intrinsic activity of catalysts, allowing for a more accurate comparison between different catalytic systems. In this study, we evaluated the TOF values for our prepared catalysts at 6000 mL gcat−1 h−1. The obtained TOF values at 350 °C for Ru/BLCO_ALD, Ru/BLCO_Ru3(CO)12, and Ru/BLCO_RuCl3 catalysts are 5.5 × 10−3 s−1, 3.1 × 10−3 s−1, and 1.3 × 10−3 s−1, respectively. These results clearly demonstrate the competitive intrinsic activity of the Ru/BLCO_ALD catalyst, which exhibited a TOF value approximately 1.8 times of Ru/BLCO_Ru3(CO)12 and 4.2 times of Ru/BLCO_RuCl3 under the same reaction conditions. Further investigation revealed a positive correlation between temperature and TOF values for all catalysts, with Ru/BLCO_ALD showing the most pronounced effect. For the Ru/BLCO_ALD catalyst, the TOF increased from 5.5 × 10−3 s−1 at 350 °C to 9.1 × 10−3 s−1 at 375 °C, representing a 65.5% increase. When the temperature was further raised to 400 °C, the TOF reached 1.3 × 10−2 s−1, marking a substantial 136.4% increase from the initial value at 350 °C. Arrhenius plots were constructed for the ammonia synthesis reactions catalyzed by Ru/BLCO_ALD, Ru/BLCO_Ru3(CO)12, and Ru/BLCO_RuCl3 (Fig. S4†). The apparent activation energies (Ea) were calculated to be 60, 68, and 71 kJ mol−1 for Ru/BLCO_ALD, Ru/BLCO_Ru3(CO)12, and Ru/BLCO_RuCl3 catalysts, respectively. Notably, the Ea value of Ru/BLCO_ALD (60 kJ mol−1) was significantly lower than that reported for Cs+/Ru/MgO_500red (100 kJ mol−1).68 Moreover, it was comparable to the values reported in the literature for other high-performance catalysts, such as 10 wt% Ru/Ca(NH2)2 (59 kJ mol−1)69 and Ru/La0.5Ce0.5O1.75_650red (64 kJ mol−1).68 The lower apparent activation energy exhibited by Ru/BLCO_ALD suggests a reduced energy barrier for the rate-determining step in the ammonia synthesis reaction. This reduction in Ea is likely another key factor contributing to the high ammonia synthesis rate observed for the Ru/BLCO_ALD catalyst. As described in the previous section, the Ru/BLCO_ALD had a smaller Ru size and electron-rich property arising from the electronic metal–support interaction, compared to the other two catalysts prepared by the IW method. As a result, the Ru nanoclusters by ALD activated the N2 molecules more effectively and exhibited better catalytic performance in ammonia synthesis.70
To comprehensively evaluate the performance of our Ru/BLCO_ALD catalyst, we conducted a thorough comparison with previously reported catalysts from the literature, as presented in Table 1. To ensure a fair and standardized comparison, we normalized the ammonia production rates to per gram of ruthenium, accounting for the varying ruthenium contents employed across different studies. The performance of our Ru/BLCO_ALD catalyst demonstrated competitive activity, compared to many conventional ruthenium nanoparticle catalysts reported in the literature, including Ru/C,79 Ru/0.3Cs–MgO,81 and Ba–Ru/γC–Al2O3,75 under comparable reaction conditions. This enhanced performance can be attributed to the unique properties conferred by the ALD technique, such as uniform ruthenium dispersion, SMSI, and optimized electronic properties of the ruthenium species. However, it is important to note that some recently reported ruthenium single-atom catalysts (SACs), such as Ru/HZ SAC79 and Ru/CeO2 SAC,76 exhibited higher catalytic activity than our Ru/BLCO_ALD catalyst. The superior performance of these single-atom catalysts could be primarily attributed to the complete exposure of ruthenium atoms, effectively achieving a theoretical dispersion of 100%. This maximizes the utilization efficiency of the precious metal and provides an abundance of active sites for the reaction. It is crucial to note that the ammonia synthesis mechanism differs significantly between nanoparticle catalysts and SACs. Ruthenium nanoparticles typically follow a direct dissociation mechanism, with B5-type surface sites playing a pivotal role in N2 activation. In contrast, SACs operate via an associative mechanism, which can be more energetically favorable under certain conditions.79 The remarkable performance of SACs in ammonia synthesis is highly encouraging and opens new avenues for catalyst design using ALD. This technique's precision and versatility make it an ideal method for preparing single atom catalysts.82–85 Building upon our current findings, our future research will focus on optimizing ALD parameters to synthesize single-atom ruthenium catalysts. This approach aims to combine the benefits of our current system with the advantages of single-atom catalysis. By precisely controlling the ALD process, we anticipate creating a new generation of catalysts that maximize ruthenium atom efficiency and catalytic performance. These next-generation catalysts hold promise for achieving exceptional activity under mild reaction conditions while simultaneously reducing ruthenium usage, thereby addressing both performance and economic considerations in industrial ammonia production.
| Samples | T (°C) | P (MPa) | Rate (mmolNH3 gRu−1 h−1) | References |
|---|---|---|---|---|
| Ru/BLCO_ALD | 350 | 1 | 430 | This work |
| Ru/BLCO_ALD | 400 | 1 | 830 | This work |
| Ru/BLCO_Ru3(CO)12 | 400 | 1 | 658 | This work |
| Ru/BLCO_RuCl3 | 400 | 1 | 578 | This work |
| Ru/Ti0.18–Ce | 400 | 1 | 767.6 | 71 |
| Ru/CeO2-w | 400 | 1 | 754 | 72 |
| Ru/LaCe–C | 400 | 1 | 406 | 73 |
| Ru–CeO2-r | 400 | 1 | 510 | 74 |
| Ba–Ru/gC–Al2O3 | 400 | 1 | 332 | 75 |
| Ru/CeO2 SAC | 400 | 1 | 1058.8 | 76 |
| Ru/Pr2O3 | 400 | 0.9 | 304 | 23 |
| Ba–Ru SAs/S-1 | 400 | 0.1 | 514.6296 | 77 |
| Ru/C12A7:e− | 400 | 0.1 | 715 | 78 |
| Ru/HZ SAC | 300 | 1 | 1260 | 79 |
| Ru/C | 400 | 1 | 300 | 79 |
| Ru/Ba–Ca(NH2)2 | 360 | 0.9 | 604 | 22 |
| Ru–Ba/Al2O3 | 400 | 1 | 144.34 | 80 |
| Ru/Ca(NH2)2 | 340 | 0.1 | 96 | 69 |
| Ru/0.2Cs–MgO | 400 | 1 | 717 | 81 |
Fig. 8 illustrates the impact of pressure on the catalytic performance of ammonia synthesis employing different catalysts synthesized via three distinct methods at 350 °C and a GHSV of 6000 mL gcat−1 h−1. For the Ru/BLCO_RuCl3 catalyst, the ammonia synthesis rate exhibited a slight increase from 0.95 to 1.54 mmolNH3 gcat−1 h−1 as the pressure rose from 1.0 to 4.2 MPa. The Ru/BLCO_ Ru3(CO)12 catalyst demonstrated a more substantial increase, from 1.68 to 3.72 mmolNH3 gcat−1 h−1, over the same pressure range. The Ru/BLCO_ALD catalyst exhibited the most significant enhancement, with the ammonia synthesis rate increasing from 2.15 to 6.6 mmolNH3 gcat−1 h−1 as the pressure increased from 1.0 to 4.2 MPa. Hydrogen poisoning at elevated hydrogen partial pressures may be a limiting factor for the Ru/BLCO_RuCl3 catalysts in ammonia synthesis.86,87 In such scenarios, excess hydrogen adatoms can occupy the active sites on the Ru catalyst, inhibiting the efficient dissociative adsorption of N2 on the Ru surface, thereby suppressing the reactivity of the Ru/BLCO_RuCl3 catalyst for ammonia synthesis. Conversely, for the Ru/BLCO_ALD and Ru/BLCO_Ru3(CO)12 catalysts, the hydrogen spillover effect observed for Ru, as confirmed by H2-TPR experiments, can mitigate the adsorption of hydrogen on the Ru surface, reducing hydrogen poisoning of the Ru species.88 Furthermore, it has been reported that the stored hydrogen atoms are reversible and can participate in ammonia synthesis.78,89 Therefore, the judicious selection of the synthesis method and precursor for Ru catalysts can alleviate their susceptibility to hydrogen poisoning, particularly at elevated partial hydrogen pressures, thereby enhancing their catalytic activity for ammonia synthesis.
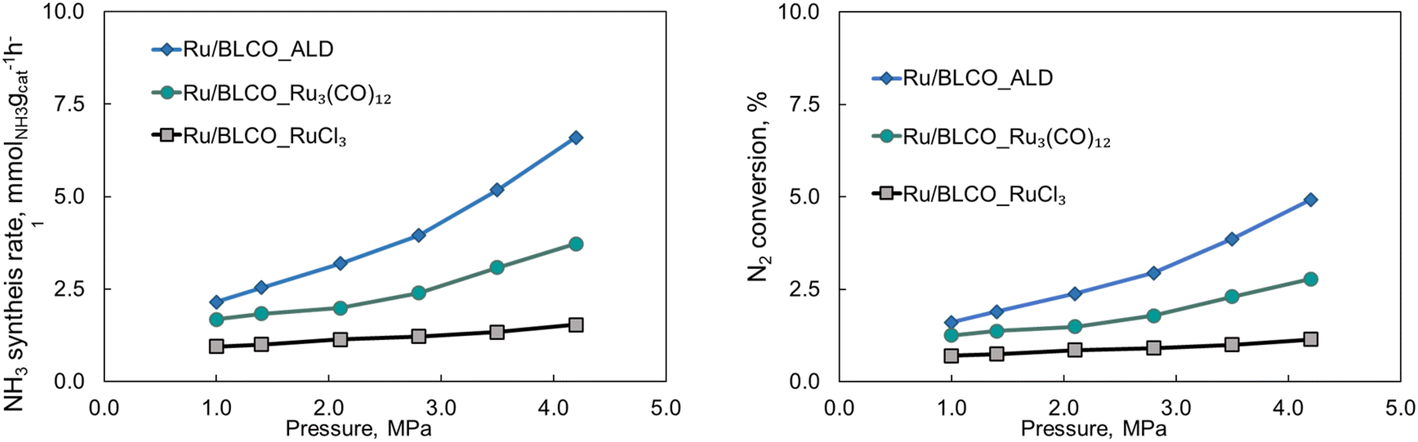 | ||
| Fig. 8 Effects of pressure on the catalytic activities for ammonia synthesis using Ru/BLCO_ALD, Ru/BLCO_Ru3(CO)12, and Ru/BLCO_RuCl3 at 350 °C and 6000 mL gcat−1 h−1. | ||
The stability of catalysts is a crucial factor for their practical applications in industrial processes. To evaluate the stability under harsh reaction conditions, the best-performing Ru/BLCO_ALD catalyst was subjected to an extended test for ammonia synthesis at 350 °C, 4.2 MPa, and a high GHSV of 6000 mL g−1 h−1. As shown in Fig. 9, during the 85 hours test, the Ru/BLCO_ALD catalyst exhibited remarkable stability, maintaining a consistent nitrogen conversion of approximately 5%. Besides, we also conducted another 20 h stability test using the spent catalyst of the 85 h test, and the performance was also stable for the spent catalysts (Fig. S5†). The high stability of the Ru/BLCO_ALD catalyst can be attributed to the SMSI effects, as confirmed by H2-TPR and XPS analyses. The SMSI phenomenon involves the migration of partially reduced metal oxide species from the support onto the metal nanoparticles, forming a thin oxide overlayer that can enhance the catalyst's stability and activity.90Scheme 1 visually represents the SMSI process in ammonia synthesis. The stability of the Ru/BLCO_ALD catalyst under such demanding conditions is noteworthy, as ammonia synthesis typically requires high temperatures and pressures, which can lead to catalyst deactivation over time due to various mechanisms, such as sintering, coking, or poisoning. The stable performance observed in this study suggests that the Ru/BLCO_ALD catalyst is robust and can maintain its catalytic activity for an extended period, even at high temperatures, pressures, and gas flow rates. It is worth noting that the stability test was conducted for 105 hours, which is a significant duration for evaluating catalyst performance under industrial-like conditions. However, further long-term stability studies over hundreds or thousands of hours is necessary to fully assess the catalyst's lifetime and potential for commercial applications.
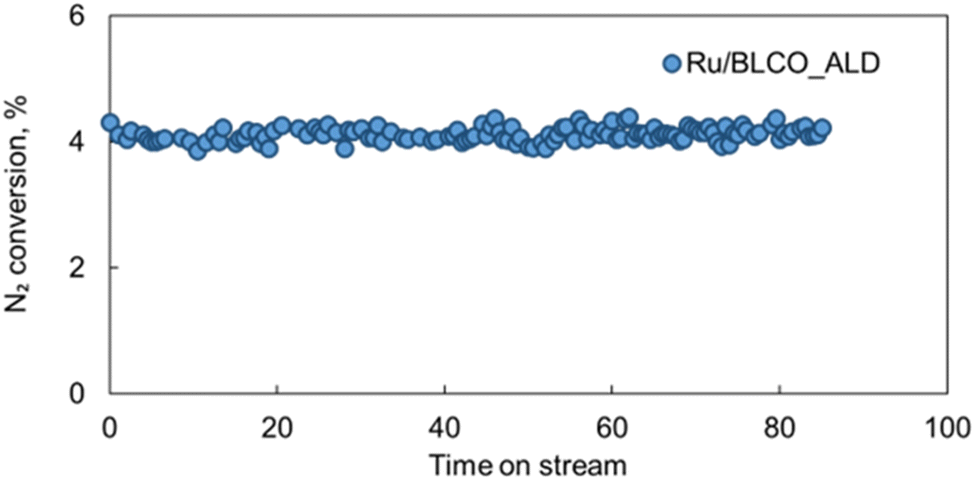 | ||
| Fig. 9 Stability test of Ru/BLCO_ALD for ammonia synthesis under conditions of 350 °C, 4.2 MPa, and 6000 mL gcat−1 h−1 for a total of 85 hours. | ||
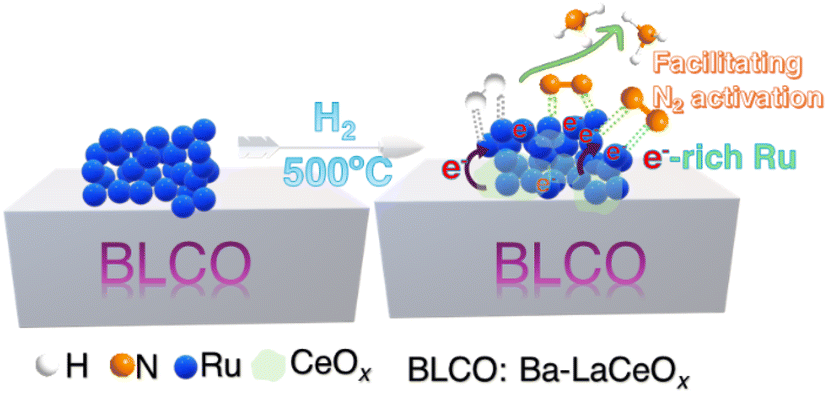 | ||
| Scheme 1 The SMSI enhanced ammonia synthesis process. | ||
4. Conclusion
In this work, Ru/Ba–LaCeOx (Ru/BLCO) catalysts for ammonia synthesis were synthesized via three distinct methods: ALD utilizing RuCp2 as the precursor, and IW employing Ru3(CO)12 and RuCl3 as ruthenium precursors. The ALD technique yielded highly dispersed metallic Ru nanoclusters with a uniform size of 1.0 nm, whereas the IW method resulted in larger Ru nanoclusters predominantly in the RuO2 phase. For the ALD-prepared Ru/BLCO catalyst, SMSI effects were induced during the hydrogen reduction process, leading to partial encapsulation of the Ru nanoclusters by a suboxide layer. This encapsulation mitigated detrimental sintering of the Ru nanoclusters and facilitated electron donation from the reduced support to the Ru sites, enabling the formation of an electronic metal–support interaction between Ru and the reduced support. The electronic metal–support interaction increased the negative charge density on the Ru sites. The excess electrons on the Ru sites weakened the NN bond, thereby enhancing the rate-determining step for ammonia synthesis. Consequently, the ALD-prepared Ru/BLCO catalysts exhibited competitive catalytic activity for ammonia synthesis under milder conditions and lower initial temperature requirements compared to the IW-prepared catalysts, demonstrating promising potential for practical applications in ammonia synthesis catalysts.
Data availability
The data will be made available on request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the U.S. National Science Foundation (Award Number 2306177) and Linda and Bipin Doshi endowment of Missouri University of Science and Technology.References
- V. S. Marakatti and E. M. Gaigneaux, ChemCatChem, 2020, 12, 5838–5857 CrossRef CAS.
- A. Mani, T. Budd and E. Maine, RSC Sustainability, 2024, 2, 903–927 RSC.
- A. Valera-Medina, H. Xiao, M. Owen-Jones, W. I. David and P. Bowen, Prog. Energy Combust. Sci., 2018, 69, 63–102 CrossRef.
- N. Salmon and R. Bañares-Alcántara, RSC Sustainability, 2023, 1, 923–937 RSC.
- S. A. Matlin, G. Mehta, S. E. Cornell, A. Krief and H. Hopf, RSC Sustainability, 2023, 1, 1704–1721 RSC.
- N. Saadatjou, A. Jafari and S. Sahebdelfar, Chem. Eng. Commun., 2015, 202, 420–448 CrossRef CAS.
- Q. Wang, J. Guo and P. Chen, J. Energy Chem., 2019, 36, 25–36 CrossRef.
- X. Luo, R. Wang, J. Ni, J. Lin, B. Lin, X. Xu and K. Wei, Catal. Lett., 2009, 133, 382–387 CrossRef CAS.
- X. Wang, J. Ni, B. Lin, R. Wang, J. Lin and K. Wei, Catal. Commun., 2010, 12, 251–254 CrossRef CAS.
- X.-L. Yang, W.-Q. Zhang, C.-G. Xia, X.-M. Xiong, X.-Y. Mu and B. Hu, Catal. Commun., 2010, 11, 867–870 CrossRef CAS.
- Y. Ogura, K. Tsujimaru, K. Sato, S.-I. Miyahara, T. Toriyama, T. Yamamoto, S. Matsumura and K. Nagaoka, ACS Sustain. Chem. Eng., 2018, 6, 17258–17266 CrossRef CAS.
- Y. Ogura, K. Sato, S.-I. Miyahara, Y. Kawano, T. Toriyama, T. Yamamoto, S. Matsumura, S. Hosokawa and K. Nagaoka, Chem. Sci., 2018, 9, 2230–2237 RSC.
- Y. Ogura, T. Asai, K. Sato, S.-I. Miyahara, T. Toriyama, T. Yamamoto, S. Matsumura and K. Nagaoka, Energy Technol., 2020, 8, 2000264 CrossRef CAS.
- K. Sato, S.-i. Miyahara, Y. Ogura, K. Tsujimaru, Y. Wada, T. Toriyama, T. Yamamoto, S. Matsumura and K. Nagaoka, ACS Sustain. Chem. Eng., 2020, 8, 2726–2734 CrossRef CAS.
- W. Han, Z. Li and H. Liu, J. Rare Earths, 2019, 37, 492–499 CrossRef CAS.
- X. Fan, K. Wang, X. He, S. Li, M. Yu and X. Liang, Carbon Resour. Convers., 2024, 7, 100184 CrossRef CAS.
- Y. Guo, S. Mei, K. Yuan, D.-J. Wang, H.-C. Liu, C.-H. Yan and Y.-W. Zhang, ACS Catal., 2018, 8, 6203–6215 CrossRef CAS.
- C. Li, M. Li, Y. Zheng, B. Fang, J. Lin, J. Ni, B. Lin and L. Jiang, Appl. Catal., B, 2023, 320, 100184 Search PubMed.
- C. J. H. Jacobsen, S. Dahl, P. L. Hansen, E. Törnqvist, L. Jensen, H. Topsøe, D. V. Prip, P. B. Møenshaug and I. Chorkendorff, J. Mol. Catal. A: Chem., 2000, 163, 19–26 CrossRef CAS.
- B. Zhang and Y. Qin, ACS Catal., 2018, 8, 10064–10081 CrossRef CAS.
- L. Cao and J. Lu, Catal. Sci. Technol., 2020, 10, 2695–2710 RSC.
- M. Kitano, Y. Inoue, M. Sasase, K. Kishida, Y. Kobayashi, K. Nishiyama, T. Tada, S. Kawamura, T. Yokoyama, M. Hara and H. Hosono, Angew. Chem., 2018, 57, 2648–2652 CrossRef CAS.
- K. Sato, K. Imamura, Y. Kawano, S. I. Miyahara, T. Yamamoto, S. Matsumura and K. Nagaoka, Chem. Sci., 2017, 8, 674–679 RSC.
- W. Li, S. Wang and J. Li, Chem.–Asian J., 2019, 14, 2815–2821 CrossRef CAS.
- S.-Y. Chen, C.-L. Chang, M. Nishi, W.-C. Hsiao, Y. I. A. Reyes, H. Tateno, H.-H. Chou, C.-M. Yang, H.-Y. T. Chen, T. Mochizuki, H. Takagi and T. Nanba, Appl. Catal., B, 2022, 310, 121269 CrossRef CAS.
- W. Rarogpilecka, E. Miskiewicz, S. Jodzis, J. Petryk, D. Lomot, Z. Kaszkur, Z. Karpinski and Z. Kowalczyk, J. Catal., 2006, 239, 313–325 CrossRef CAS.
- Y. Horiuchi, G. Kamei, M. Saito and M. Matsuoka, Chem. Lett., 2013, 42, 1282–1284 CrossRef CAS.
- Z. Shang, S. Li, L. Li, G. Liu and X. Liang, Appl. Catal., B, 2017, 201, 302–309 CrossRef CAS.
- B. Jin, K. Wang, H. Yu, X. He and X. Liang, Chem. Eng. J., 2023, 459, 141611 CrossRef CAS.
- K. Wang, X. He, J. C. Wang and X. Liang, Nanotechnology, 2022, 33, 215602 CrossRef CAS.
- H. Kim, A. Jan, D. H. Kwon, H. I. Ji, K. J. Yoon, J. H. Lee, Y. Jun, J. W. Son and S. Yang, Small, 2023, 19, e2205424 CrossRef.
- D. Liu, T. Ding, L. Wang, H. Zhang, L. Xu, B. Pang, X. Liu, H. Wang, J. Wang, K. Wu and T. Yao, Nat. Commun., 2023, 14, 1720 CrossRef CAS.
- B. Jin, Z. Shang, S. Li, Y.-B. Jiang, X. Gu and X. Liang, Catal. Sci. Technol., 2020, 10, 3212–3222 RSC.
- B. Jin, S. Li and X. Liang, Fuel, 2021, 284, 119082 CrossRef CAS.
- S.-J. Park, W.-H. Kim, W. Maeng and H. Kim, Microelectron. Eng., 2008, 85, 39–44 CrossRef CAS.
- X. Jiang, T. M. Gür, F. B. Prinz and S. F. Bent, Chem. Mater., 2010, 22, 3024–3032 CrossRef CAS.
- E. Färm, S. Lindroos, M. Ritala and M. Leskelä, Chem. Mater., 2012, 24, 275–278 CrossRef.
- J. Hämäläinen, M. Ritala and M. Leskelä, Chem. Mater., 2014, 26, 786–801 CrossRef.
- Z. Ni, X. Djitcheu, X. Gao, J. Wang, H. Liu and Q. Zhang, Sci. Rep., 2022, 12, 5344 CrossRef CAS.
- A. Jan, J. Shin, J. Ahn, S. Yang, K. J. Yoon, J. W. Son, H. Kim, J. H. Lee and H. I. Ji, RSC Adv., 2019, 9, 27002–27012 RSC.
- Z. Ma, S. Zhao, X. Pei, X. Xiong and B. Hu, Catal. Sci. Technol., 2017, 7, 191–199 RSC.
- K. Singh, R. Kumar and A. Chowdhury, Mater. Today: Proc., 2018, 5, 22993–22997 CAS.
- B. M. Reddy, L. Katta and G. Thrimurthulu, Chem. Mater., 2009, 22, 467–475 CrossRef.
- B. Fang, F. Liu, C. Zhang, C. Li, J. Ni, X. Wang, J. Lin, B. Lin and L. Jiang, ACS Sustain. Chem. Eng., 2021, 9, 8962–8969 CrossRef CAS.
- B. Lin, B. Fang, Y. Wu, C. Li, J. Ni, X. Wang, J. Lin, C.-t. Au and L. Jiang, ACS Catal., 2021, 11, 1331–1339 CrossRef CAS.
- J. Zhou, Z. Gao, G. Xiang, T. Zhai, Z. Liu, W. Zhao, X. Liang and L. Wang, Nat. Commun., 2022, 13, 327 CrossRef CAS.
- K. Shun, K. Mori, S. Masuda, N. Hashimoto, Y. Hinuma, H. Kobayashi and H. Yamashita, Chem. Sci., 2022, 13, 8137–8147 RSC.
- D. Fu, X. Wu, B. Cui, Y. Guo, H. Wang, J. Han, Q. Ge and X. Zhu, ChemCatChem, 2021, 13, 4814–4823 CrossRef CAS.
- Y. Baik, M. Kwen, K. Lee, S. Chi, S. Lee, K. Cho, H. Kim and M. Choi, J. Am. Chem. Soc., 2023, 145, 11364–11374 CrossRef CAS.
- B. Lin, Y. Liu, L. Heng, X. Wang, J. Ni, J. Lin and L. Jiang, Ind. Eng. Chem. Res., 2018, 57, 9127–9135 CrossRef CAS.
- L. Zhang, J. Lin, J. Ni, R. Wang and K. Wei, Catal. Commun., 2011, 15, 23–26 CrossRef CAS.
- M. Hattori, T. Mori, T. Arai, Y. Inoue, M. Sasase, T. Tada, M. Kitano, T. Yokoyama, M. Hara and H. Hosono, ACS Catal., 2018, 8, 10977–10984 CrossRef CAS.
- E. Truszkiewicz, W. Raróg-Pilecka, K. Schmidt-Szałowski, S. Jodzis, E. Wilczkowska, D. Łomot, Z. Kaszkur, Z. Karpiński and Z. Kowalczyk, J. Catal., 2009, 265, 181–190 CrossRef CAS.
- H. S. Zeng, K. Inazu and K.-I. Aika, J. Catal., 2002, 211, 33–41 CrossRef CAS.
- A. Cao, V. J. Bukas, V. Shadravan, Z. Wang, H. Li, J. Kibsgaard, I. Chorkendorff and J. K. Norskov, Nat. Commun., 2022, 13, 2382 CrossRef CAS.
- N. Kuganathan, H. Hosono, A. L. Shluger and P. V. Sushko, J. Am. Chem. Soc., 2014, 136, 2216–2219 CrossRef CAS.
- X. Wang, X. Yang, G. Pei, J. Yang, J. Liu, F. Zhao, F. Jin, W. Jiang, H. Ben and L. Zhang, Carbon Energy, 2023, 6, e391 CrossRef.
- B. M. Stühmeier, R. J. Schuster, L. Hartmann, S. Selve, H. A. El-Sayed and H. A. Gasteiger, J. Electrochem. Soc., 2022, 169, 034519 CrossRef.
- B. Jin, S. Li and X. Liang, Ind. Eng. Chem. Res., 2021, 10377–10386 Search PubMed.
- O. Karslıoğlu, L. Trotochaud, I. Zegkinoglou and H. Bluhm, J. Electron Spectrosc. Relat. Phenom., 2018, 225, 55–61 CrossRef.
- G. Zhou, H. Liu, K. Cui, A. Jia, G. Hu, Z. Jiao, Y. Liu and X. Zhang, Appl. Surf. Sci., 2016, 383, 248–252 CrossRef CAS.
- F. Meng, Z. Fan, C. Zhang, Y. Hu, T. Guan and A. Li, J. Mater. Sci., 2017, 33, 444–451 CAS.
- P. Fleming, R. A. Farrell, J. D. Holmes and M. A. Morris, J. Am. Ceram. Soc., 2010, 93, 1187–1194 CrossRef CAS.
- S. Chen, L. Zeng, H. Tian, X. Li and J. Gong, ACS Catal., 2017, 7, 3548–3559 CrossRef CAS.
- K. Wang, X. He and X. Liang, Int. J. Hydrogen Energy, 2024, 66, 195–207 CrossRef CAS.
- K. Wang, S. Li, M. Yu and X. Liang, Energies, 2024, 17, 839 CrossRef CAS.
- A. Miyazaki, I. Balint, K.-I. Aika and Y. Nakano, J. Catal., 2001, 204, 364–371 CrossRef CAS.
- Y. Ogura, K. Sato, S. I. Miyahara, Y. Kawano, T. Toriyama, T. Yamamoto, S. Matsumura, S. Hosokawa and K. Nagaoka, Chem. Sci., 2018, 9, 2230–2237 RSC.
- Y. Inoue, M. Kitano, K. Kishida, H. Abe, Y. Niwa, M. Sasase, Y. Fujita, H. Ishikawa, T. Yokoyama, M. Hara and H. Hosono, ACS Catal., 2016, 6, 7577–7584 CrossRef CAS.
- C. Li, Y. Shi, Z. Zhang, J. Ni, X. Wang, J. Lin, B. Lin and L. Jiang, J. Energy Chem., 2021, 60, 403–409 CrossRef CAS.
- C. Li, Z. Zhang, Y. Zheng, B. Fang, J. Ni, J. Lin, B. Lin, X. Wang and L. Jiang, Chem. Eng. Sci., 2022, 251, 117434 CrossRef CAS.
- C. Li, M. Li, Y. Zheng, B. Fang, J. Lin, J. Ni, B. Lin and L. Jiang, Appl. Catal., B, 2023, 320, 121982 CrossRef CAS.
- C. Li, Y. Zheng, M. Li, B. Fang, J. Lin, J. Ni, X. Wang, B. Lin and L. Jiang, Int. J. Hydrogen Energy, 2022, 47, 23240–23248 CrossRef CAS.
- B. Lin, Y. Liu, L. Heng, X. Wang, J. Ni, J. Lin and L. Jiang, Ind. Eng. Chem. Res., 2018, 57, 9127–9135 CrossRef CAS.
- B. Lin, L. Heng, H. Yin, B. Fang, J. Ni, X. Wang, J. Lin and L. Jiang, Ind. Eng. Chem. Res., 2019, 58, 10285–10295 CrossRef CAS.
- B. Lin, Y. Wu, B. Fang, C. Li, J. Ni, X. Wang, J. Lin and L. Jiang, Chin. J. Catal., 2021, 42, 1712–1723 CrossRef CAS.
- J.-Z. Qiu, J. Hu, J. Lan, L.-F. Wang, G. Fu, R. Xiao, B. Ge and J. Jiang, Chem. Mater., 2019, 31, 9413–9421 CrossRef CAS.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S. W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef CAS.
- X. Wang, L. Li, Z. Fang, Y. Zhang, J. Ni, B. Lin, L. Zheng, C.-t. Au and L. Jiang, ACS Catal., 2020, 10, 9504–9514 CrossRef CAS.
- B. Lin, L. Heng, B. Fang, H. Yin, J. Ni, X. Wang, J. Lin and L. Jiang, ACS Catal., 2019, 9, 1635–1644 CrossRef CAS.
- L. Liu, X. Zhang, X. Ju, J. Feng, J. Wang and P. Chen, Dalton Trans., 2021, 50, 12074–12078 RSC.
- X. Wang, B. Jin, Y. Jin, T. Wu, L. Ma and X. Liang, ACS Appl. Nano Mater., 2020, 3, 2867–2874 CrossRef CAS.
- J. Fonseca and J. Lu, ACS Catal., 2021, 11, 7018–7059 CrossRef CAS.
- K. Wang, X. Wang and X. Liang, ChemCatChem, 2020, 13, 28–58 CrossRef.
- L. Zhang, M. N. Banis and X. Sun, Natl. Sci. Rev., 2018, 5, 628–630 CrossRef CAS.
- B. Lin, Y. Wu, B. Fang, C. Li, J. Ni, X. Wang, J. Lin and L. Jiang, Chin. J. Catal., 2021, 42, 1712–1723 CrossRef CAS.
- S. Wu, Y.-K. Peng, T.-Y. Chen, J. Mo, A. Large, I. McPherson, H.-L. Chou, I. Wilkinson, F. Venturini, D. Grinter, P. Ferrer Escorihuela, G. Held and S. C. E. Tsang, ACS Catal., 2020, 10, 5614–5622 CrossRef CAS.
- J. Huang, M. Yuan, X. Li, Y. Wang, M. Li, J. Li and Z. You, J. Catal., 2020, 389, 556–565 CrossRef CAS.
- Y. Kobayashi, Y. Tang, T. Kageyama, H. Yamashita, N. Masuda, S. Hosokawa and H. Kageyama, J. Am. Chem. Soc., 2017, 139, 18240–18246 CrossRef CAS.
- C. Li, F. Liu, Y. Shi, Y. Zheng, B. Fang, J. Lin, J. Ni, X. Wang, B. Lin and L. Jiang, ACS Sustain. Chem. Eng., 2021, 9, 4885–4893 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00350k |
| ‡ Equal contribution to this work. |
| This journal is © The Royal Society of Chemistry 2024 |