
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComparison of traditional and mechanochemical production processes for nine active pharmaceutical ingredients (APIs)
Emília P. T. Leitão *
*
Hovione Farmaciência, S.A., Sete Casas, Loures 2674-506, Portugal. E-mail: eleitao@hovione.com
First published on 22nd October 2024
Abstract
The pharmaceutical industry plays a crucial role in enhancing life expectancy and the quality of life. However, drug production generates waste, contributing to environmental impacts such as Greenhouse Gas (GHG) emissions and imposing a significant economic burden. In response to these challenges, researchers are actively exploring alternative sustainable processes and technologies that are efficient, selective, robust, and cost-effective. This review compares conventional and mechanosynthesis methods for nine active pharmaceutical ingredients (APIs) described in the literature, which contain some of the most common reactions found in APIs. The objective of the study is to determine the methodology that best adheres to the core principles of green chemistry. The analysis indicates that mechanosynthesis more closely adheres to these core principles, including waste prevention, safer chemical use, energy efficiency, and better green metrics (AE, CE, RME, PMI, E-factor, and cE-factor). While not all mechanosynthesis reactions adhere to all 12 principles, they generally conform to more principles than traditional solution-based reactions.
Sustainability spotlightThe increasing concern about climate change prompted the United Nations (UN) to create the 17 Sustainable Development Goals (SDGs). Meeting Goal 3 (good health and well-being) relies on the pharmaceutical industry's production of APIs for medicine production. However, API manufacturing results in significant waste, causing companies adverse environmental and economic impacts. This study demonstrates the benefits of mechanochemistry, an eco-friendly technology, compared to traditional solution-based methods using green metrics. It shows that the use of mechanochemistry in API production is in line with Goal 9 (as it is a solvent-free methodology), Goal 12 (due to lower energy requirements for processes leading to decreased dependence on fossil fuels), and Goal 13 (by reducing wastes which result in less carbon emissions). |
Introduction
The global population was 7.6 billion in 2017 and is projected to increase to 8.6 billion by 2030, 9.8 billion by 2050, and 11.2 billion by 2100.1 In the past, people's life expectancy was less than 30 years, but with the development of healthcare systems, access to medical care has improved significantly. The pharmaceutical industry has played a vital role in this development by discovering, developing, manufacturing, and marketing effective disease treatments, preventive vaccines, and medicines that improve the quality of life for people with chronic diseases.2,3 Due to the growing and aging population, the pharmaceutical industry needs to create a broader range of products to meet the increasing demand and improve healthcare systems.4 Drug manufacturing is known for producing a higher amount of waste and by-products than other sectors in the chemical industry. This can be seen from its E-factor, shown in Table 1.5This difference can be attributed to the multiple steps involved in the process, the use of stoichiometric reagents, and large amounts of solvents.5 Pfizer reported in 1994 that the pharmaceutical industry produces 25–100 kg of waste for every kilogram of API (Active Pharmaceutical Ingredients) it manufactures. This practice remained standard in 2005.6 Constable and co-workers reported that the solvents alone make up roughly 80–90% of the mass in pharmaceutical and fine chemical operations.7 Alternative reaction media or ‘green solvents’ have been suggested for organic synthesis, but their potential environmental and health risks remain unclear.8 Additionally, they are expensive and not economical to use. Despite being environmentally friendly, their disposal and/or recycling have an associated carbon footprint.
Over the years, various initiatives have emerged to design cost-effective and eco-friendly processes. In 1998, Paul Anastas and John Warner introduced the 12 principles of Green Chemistry as guidance for designing chemical products and processes. The principles address toxicity (reducing the hazard), feedstocks (use of renewable resources), designing safer products (non-toxic products by design), biodegradability (enhancing breakdown at the end of life), energy (reducing the energy needs), accidents (eliminating accidents), efficiency (shorter processes synthesis).9 These principles apply to organic chemistry, biochemistry, inorganic, analytical, physical, and chemical engineering.10 To reinforce the use of these principles, the ACS Green Chemical Institute (GCI) joined forces with pharmaceutical companies to establish a Pharmaceutical Roundtable to promote green chemistry and engineering in the industry, focusing on creating a sustainable environment.11 As a result of this need, mechanochemistry emerged and was recognized by IUPAC as one of the ten technologies that can change our planet.12
The IUPAC defines a mechanochemical reaction as: “a chemical reaction induced by the direct absorption of mechanical energy” (shearing, stretching, and grinding are typical methods for mechano-chemical generating of reactive sites).13 According to Laszlo Takacs, mechanochemical reactions are not a recent discovery. He claims that the first mechanochemical reaction, along with the first description of a process for obtaining a pure metal from a compound, can be traced back to a book called “De Lapidibus” (On Stones) by Theophrastus of Eresus (371–286 BC), a student and successor of Aristotle at the Lyceum in Athens. Theophrastus wrote, “native cinnabar was rubbed with vinegar in a copper mortar with a copper pestle yielding the liquid metal”.14 Recently, the synthesis of cinnabar has been reproduced.15 Several centuries after, in 1820, Faraday induced the mechanochemical reduction of AgCl with Zn, Sn, Fe and Cu using trituration in a mortar.16 In 1835–1932, Wilhelm Ostwald first classified mechanochemistry as a branch of chemistry along with thermochemistry, electrochemistry, and photochemistry.17 Nowadays, mechanochemistry is applied to other areas, such as catalysis,18 fertilizers,19 inorganic synthesis,20 polymers,21 waste management,22,23 pharmaceutical co-crystals,24 cosmetics,25 nanocomposites,26 etc.
The field of mechanochemistry has gained significant attention over the years, as evidenced by the increasing number of publications and patents that contain either “mechanochemistry” or “mechanochemical” in their description, as shown in Fig. 1.
 | ||
| Fig. 1 Yearly publications in mechanochemistry (papers, abstracts, patents, and communications). Data extracted from the SciFinder database (accessed on 2024.01.12). | ||
Mechanochemical processes can occur without solvents or in the presence of small amounts of solvents (liquid-assisted grinding, LAG) or involving gaseous reactants. Mechanochemical processes present distinct advantages compared to traditional solution-based reactions. They offer exceptional environmental sustainability and cost-effectiveness by eliminating or reducing the need for solvents. Moreover, they facilitate secure reactions without the use of hazardous solvents, consequently minimizing waste and greenhouse emissions. Mechanochemical processes exhibit higher yields, have shorter reaction times, and provide better control over product selectivity. Furthermore, they offer access to new product libraries not achievable through other methods.27–30 Mechanochemical synthesis can be performed using ball milling (BM) equipment.31 Ball milling is typically a batch-processing method with relatively low production rates.32 However, this method has some challenges, such as the high-temperature localized spots caused by friction, known as the “hot-spot” theory. These spots may not be compatible with grinding reactions. Even slight temperature changes can significantly impact reaction rates and decomposition. However, this theory has not been refuted.28 Furthermore, product contamination with metal due to abrasion or leaching of the milling media during the process has been reported as another challenge,28,33,34 because the amount of metals allowed in an API is regulated by the European Medicines Agency (EMA).35 However, this challenge can be overcome using zirconium oxide36 or Teflon reactors instead of stainless steel.37 Another challenge is that the ball mill may experience shutdown times, and decanting the product can be difficult, depending on the physical nature of the material (e.g., caked products versus free-flowing powders).32 However, it has been reported that some of these challenges can be addressed using twin-screw extrusion (TSE), a technique to transition to continuous processes, enabling scalable mechanochemical synthesis.
This review compares the synthesis of nine active pharmaceutical ingredients described in the literature with some of the most common reactions found in APIs, using solution-based and mechanosynthesis procedures, to determine which process better aligns with the core principles of green chemistry, including waste prevention, safer chemical use, improved energy efficiency, and greater atom efficiency. To assess the efficiency and environmental impact of each process, the green metrics such as atom economy (AE), carbon efficiency (CE), reaction mass efficiency (RME), environmental factor (E-factor), complete E-factor (cE-factor, which includes water in the calculations),38,39 and process mass intensity (PMI) were used.38,40 The equations to calculate the referred metrics are depicted in Table 2.38–40
| Metrics | Green metrics equation | Optimum value |
|---|---|---|
| Atom economy |  |
100% |
| Carbon efficiency | 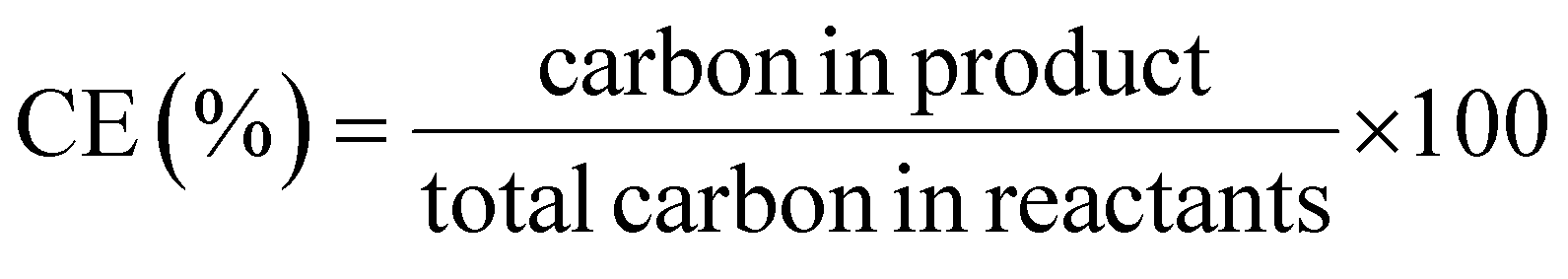 |
100% |
| Reaction mass efficiency | 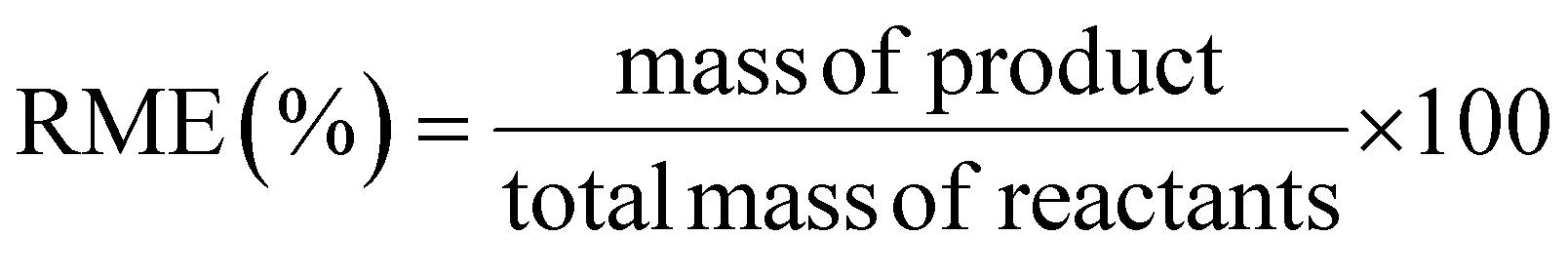 |
100% |
| Process mass intensity | 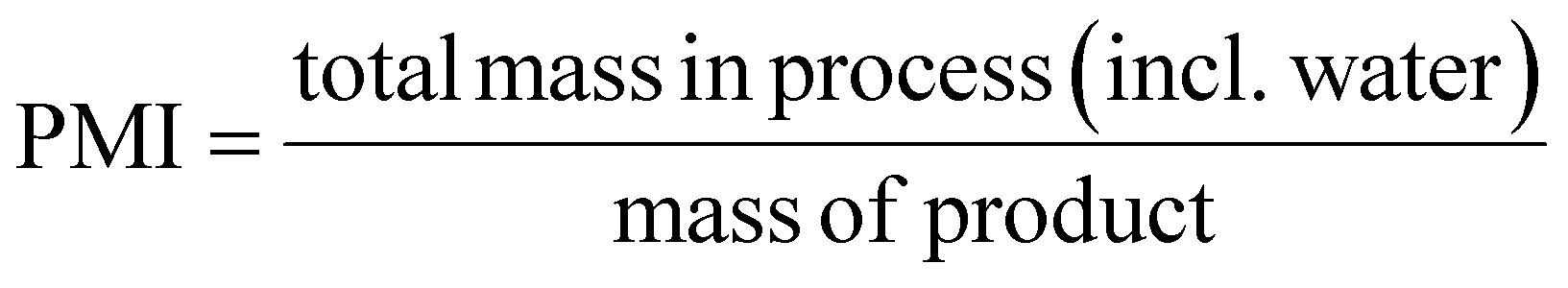 |
1 |
| PMI = E factor + 1 | ||
| E-factor | 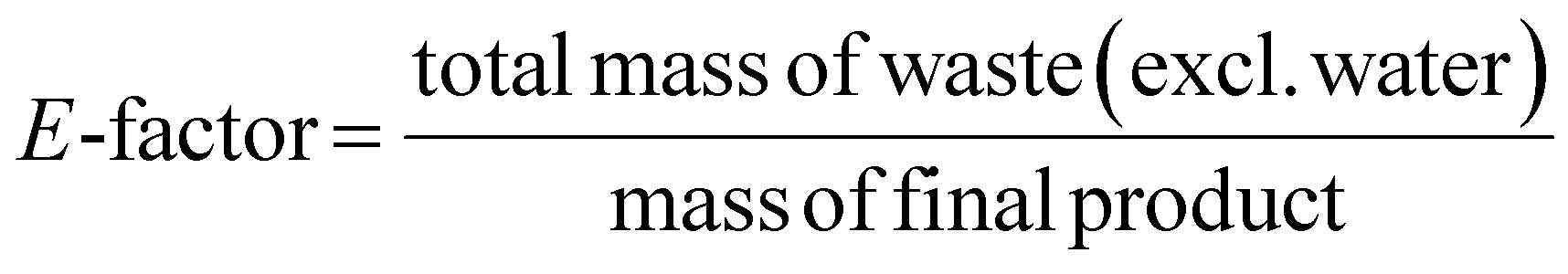 |
0 |
| Complete E-factor | 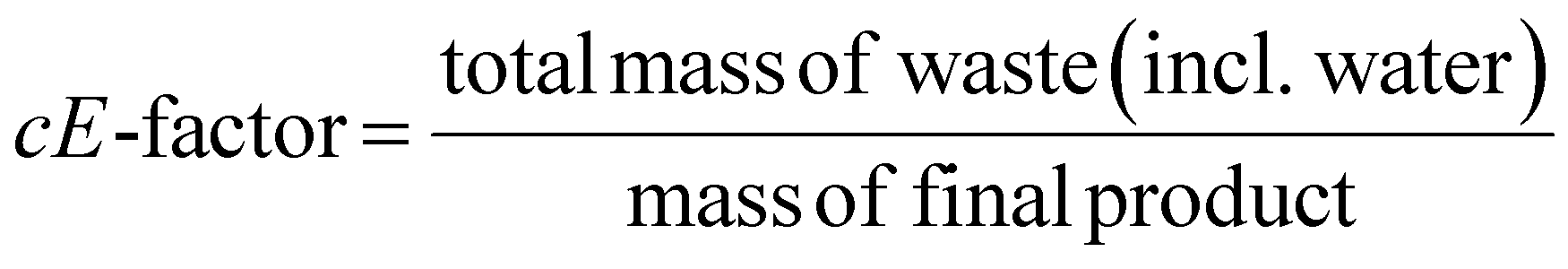 |
0 |
Teriflunomide
Teriflunomide (1) is a drug that treats relapsing forms of multiple sclerosis (RMS; RRMS). It is available commercially under the tradename Aubagio® by Sanofi and has been approved for use in various regions, including Europe, North America, Latin America, and Australia.41,42 In 1996, Bartlett and Kämmerer from Hoechst Aktiengesellschaft claimed the preparation of 1. The process consisted of reacting 5-methyl isoxazole-4-carbonyl chloride 2 with 4-(trifluoromethyl) aniline hydrochloride 3 in acetonitrile to obtain leflunomide 4. Leflunomide 4 was then hydrolyzed with an aqueous sodium hydroxide solution in methanol. The product obtained was isolated by filtration and dried in the air, producing a pure product with 85% overall yield, Scheme 1.43 | ||
| Scheme 1 Teriflunomide 1 route synthesis described by Bartlett and Kämmerer.43 | ||
Several manufacturing methods have been developed over the years using different materials.44 In 2012, Métro and co-workers reported the mechanochemical synthesis of teriflunomide 1 in a Retsch PM100 Planetary Mill using a stainless-steel grinding bowl with fifty stainless steel balls (5 mm diameter). The process involved two steps: first, carboxylic acid 5 was activated with CDI (carbonyldiimidazole) at 500 rpm for 20 minutes, followed by a reaction with amine hydrochloride 3, which was ground for 5 hours at 500 rpm. During this step, there was a 1 minute break every 10 minutes, along with an inversion of the rotation direction after each break (Scheme 2).45
 | ||
| Scheme 2 Mechanosynthesis of teriflunomide described by Métro and co-workers.45 | ||
The method enabled the purification and recovery of the desired product without using solvents, except water, to suspend and isolate the product after pH adjustment. The rate of aniline acylation increased compared to reactions in solution. Teriflunomide was obtained with 81% yield starting from 4-(trifluoromethyl)aniline (or 73% yield considering the 5-methylisoxazole-4-carboxylic acid as the starting raw material).45 This process avoided using thionyl chloride, a hazardous and toxic reagent, to prepare compound 2.46
In 2023, Lavayssiere and Lamaty described the mechanosynthesis of teriflunomide in an Xplore Pharma Melt parallel co-rotating twin-screw extruder. The method involved mixing carboxylic acid 5, COMU [(1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate; 1.0 equiv.], 4-(trifluoromethyl) aniline (1 equiv.), DIPEA (N,N-diisopropylethylamine or Hünig's base, 1.1 equiv.), and acetonitrile (0.6 equiv.) at 30 °C with a screw rotation speed of 200 rpm for 2 hours. The process resulted in an 85% conversion by HPLC (High-Performance Liquid Chromatography) and an 80% yield after precipitation in water, isolation, and drying.47 The reaction time, overall molar yield, and green metrics for each process are summarized in Table 3.
| Process | Reaction time (h) | Molar yield (%) | E-factor (kg kg−1) | cE-factor (kg kg−1) | AE (%) | RME (%) | CE (%) | PMI (kg kg−1) |
|---|---|---|---|---|---|---|---|---|
| a Aging time, after the pH adjustment. | ||||||||
| Bartlett process | 1st Rx – 0.3 | 85 | 18.8 | 22.9 | 78.7 | 49.2 | 62.3 | 23.9 |
| Solution | 2nd Rx – 0.5 | |||||||
| Métro process | 1st Rx – 0.3 | 73 | 0.5 | 15.0 | 83.2 | 64.8 | 77.7 | 16.0 |
| Planetary ball mill | 2nd Rx – 5 | |||||||
| Lavayssiere process | 1st Rx – 2 | 80 | 5.2 | 5.2 | 93.7 | 75.0 | 80.0 | 6.2 |
| Twin-screw extruder | 2nd Rx – 24a | |||||||
The Bartlett and Kämmerer process has been shown to offer shorter reaction times and higher yields than processes initiated from carboxylic acid (mechanochemical processes).43 The use of mechanochemistry by Métro and colleagues allowed them to use only water in their process.45 They combined the two reactions into a single operation without isolating the intermediate, thus eliminating the need for organic solvents. This methodology also enabled Lavayssiere and Lamaty to significantly reduce the amount of acetonitrile used in their process.47 The reduction of acetonitrile is important because this solvent is classified as a class 2 solvent according to ICH Guidelines. Its use in pharmaceutical products must be restricted to protect consumers from potential adverse effects.48 Furthermore, acetonitrile is vulnerable to supply shortages as a byproduct of acrylonitrile production. In addition to its supply volatility, acetonitrile poses challenges in waste management and receives poor life cycle management ratings.49 Besides that, reducing solvents resulted in lower E-factors and PMI, thereby reducing waste disposal and energy consumption. Additionally, mechanochemical processes resulted in higher carbon incorporation from the reactants into the final product (CE) and the reactants themselves into the final product, as demonstrated by the higher AE. Furthermore, the RME outperforms the base solution process by accounting for yield and excess reagent usage. In the Bartlett and Kämmerer process, the calculation did not account for the water used to wash the wet cake in the initial step.43 Similarly, in Métro and co-workers' process,45 the amounts of acetonitrile used to dissolve the wet cake and the water used to precipitate the product were not considered as they were not specified.
Ftivazide
Ftivazide (6, isonicotinic acid vanillylidenehydrazide) is a derivative of isoniazid, used to treat tuberculosis in the Soviet Union in 1953 and is still in clinical practice in the Russian Federation.50 Ftivazide can be synthesized using the Sudha and co-workers process. The process involves condensation of vanillin 7 (4-hydroxy-3-methoxybenzaldehyde) with isonicotinic acid hydrazide 8 (1 equiv.) catalyzed by glacial acetic acid (0.05 equiv.) at reflux, resulting in the desired compound 6 in 90% yield, Scheme 3.51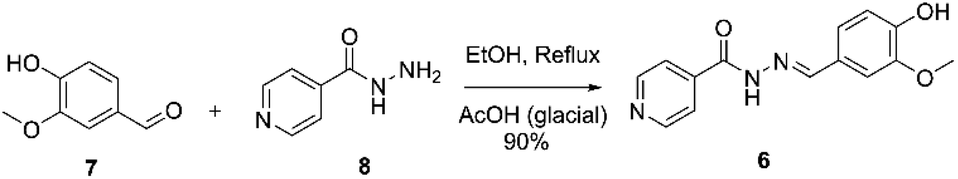 | ||
| Scheme 3 Synthetic procedure of ftivazide described by Sudha and co-workers.51 | ||
More recently 2017, Soubhye and co-workers reported the synthesis of ftivazide by refluxing isonicotinic acid hydrazide 8 and aldehyde 7 for only 3 hours in ethanol with 100% yield.52
In 2014, Oliveira and co-workers described the mechanosynthesis of ftivazide 6. They milled 8 (1 equiv.), aldehyde 7 (1 equiv.), and the catalyst p-toluenesulfonic acid (0.5 equiv.) in a vibratory ball mill (Pulverisette 0). The ball mill was equipped with a single stainless-steel ball, 50 mm in diameter and 500 g in weight, in a semi-spherical vessel 9.5 cm in diameter. The plate vibrated at a frequency of 50 Hz and an amplitude of 2.0 mm for two hours. According to the authors, the TLC (Thin Layer Chromatography) analysis indicated the consumption of the reagents and the appearance of hydrazone 6 as a result. The product obtained was washed with NaHCO3 solution and dried, leading to a 99% yield with no byproducts detected.53 The reaction time, molar yield, and green metrics for each process are summarized in Table 4.
| Process | Reaction time (h) | Yield (%) | E-Factor (kg kg−1) | cE-factor (kg kg−1) | AE (%) | RME (%) | CE (%) | PMI (kg kg−1) |
|---|---|---|---|---|---|---|---|---|
| a The amount of ethanol used to wash the wet cake was not specified and, therefore, was not considered in the calculation.b The amount of aqueous NaHCO3 used to remove the catalyst was not specified and therefore was not considered in the calculation. | ||||||||
| Sudha process | 3 | 90 | 9.1a | 9.1a | 93.8 | 84.8 | 90.4 | 10.1a |
| Solution | ||||||||
| Soubhye solution process | 3 | 100 | 1.5 | 1.5 | 93.8 | 93.8 | 100.0 | 2.5 |
| Solution | ||||||||
| Oliveira process | 2 | 99 | 0.4b | 0.4b | 93.8 | 93.3 | 99.6 | 1.4b |
| Vibratory ball mill | ||||||||
The mechanochemical process exhibits a shorter reaction time, with the equipment supplying the necessary energy for the reaction, while the reactions involving Soubhye and Sudha are conducted at reflux temperature. The mechanochemical process is notably more environmentally sustainable than the Sudha process, as indicated by its superior green metrics, lower E-factors and PMI, and higher RME. However, the green metrics are on par with Soubhye's optimized solution process, potentially indicating an overvaluation of Oliveira's process due to the unspecified amount of aqueous NaHCO3 employed for catalyst removal. It is noteworthy that the Soubhye process utilizes ethanol, whereas the Oliveira process uses water, presenting a significant difference in waste treatment.
Axitinib
Axitinib 9 is a drug developed by Pfizer and approved in 2012 under the trade name Inlyta®. It is used in the United States to treat patients with advanced renal cell carcinoma (RCC) who have already undergone prior treatment with sunitinib or a cytokine but have not shown improvement.54 Axitinib may be prepared according to the process described by Chekal and co-workers using the second-generation synthesis (2014) developed to shorten the process and address the Pd control strategy (Scheme 4).55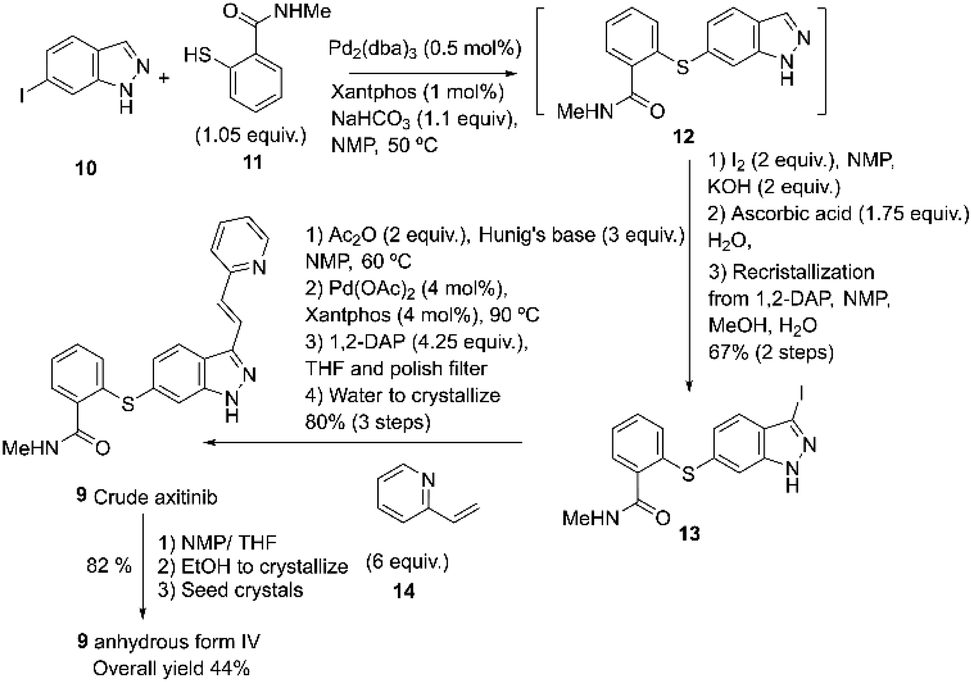 | ||
| Scheme 4 Second-generation synthesis of axitinib described by Chekal and co-workers.55 | ||
This approach uses Xantphos as a supporting ligand for Pd to accomplish the Migita coupling between 10 and 11, resulting in intermediate 12. Compound 13 is formed in situ by adding the iodine and base, followed by a Heck reaction between 13 and 14 at 90 °C. After the workup, crude axitinib 9 is recrystallized from NMP/THF/ETOH. The axitinib 9 is isolated as a white crystalline solid with 99.9% purity and an overall yield of 44%.55
In 2015, Zhai and co-workers developed an alternative process using CuI/1,10-phenanthroline/K2CO3 as a catalyst in the Heck-type and C–S coupling reactions. This alternative process aimed to reduce the high cost of the palladium catalyst. Axtinib was obtained free from impurities and meeting regulatory requirements. However, the overall yield of this process was lower (39%) than that of the second-generation process, confirming that palladium is a more efficient catalyst than copper.56
In 2018, Yu and co-workers reported the mechanosynthesis of axitinib starting with commercially available 6-bromo-1H-indazole 15 (Scheme 5).57
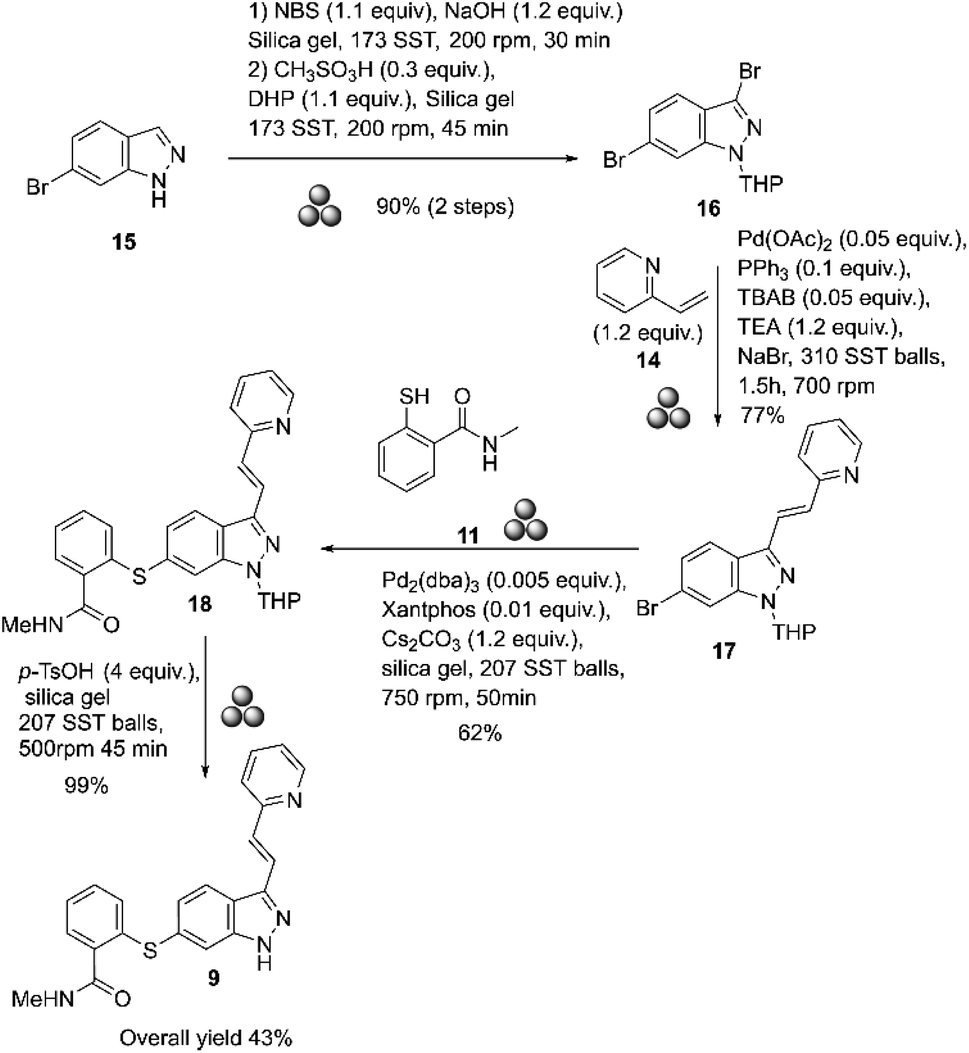 | ||
| Scheme 5 Mechanosynthesis of axitinib described by Yu and co-workers.57 | ||
The reactions were conducted in a high-energy ball mill (Fritsch GmbH Planet Mill Pulverisette 7) using 80 mL stainless-steel grinding vessels. The bromination at the 3-position was done by mixing substrate 15 with NBS, NaOH, and silica gel (4.0 g) at 200 rpm for 30 minutes. The resulting product was purified by column chromatography. This product was then reacted with 3,4-dihydropyran, CH3SO3H, and silica gel (4.0 g) at 200 rpm for 45 minutes to give 3,6-dibromo-1-(tetrahydro-2H-pyran-2-yl)-1H-indazole 16 in 90% yield, after purification by column chromatography. The Heck coupling was performed selectively by mixing the substrate 16 with 14, Pd(OAc)2, PPh3, TEA (triethylamine), TBAB (tetrabutylammonium bromide), and NaBr at 700 rpm for 1.5 hours. At the end of the reaction, the product obtained was purified by column chromatography on silica gel (4.0 g), which afforded 77% of compound 17. The Migita coupling reaction was selectively performed by mixing 17 with Pd2(dba)3, Xantphos, CsCO3 and silica gel (4.0 g) at 750 rpm for 50 minutes. After purification by column chromatography, a 62% yield of THP-axitinib 18 was obtained. Finally, the deprotection of axitinib was carried out by mixing substrate 18 with p-TsOH and silica gel (4.0 g) at 500 rpm for 45 min. The product obtained was purified by column chromatography, which afforded axitinib at 99%.
The residual Pd content in the axitinib was determined to be no more than 2 ppm by ICP (Inductively Coupled Plasma) analysis. According to the authors, this mechanochemical protocol provided a solvent-free, highly efficient, and tractable alternative for synthetic procedures57 compared to the synthetic methods described by Chekal (Scheme 4).55 In Yu and co-workers' process, all the intermediates and final products were purified using column chromatography, a time-consuming process that should be avoided whenever possible. Therefore, the environmental impact comparison was only assessed for the reaction step. Table 5 summarizes the molar yield and environmental metrics for both the Yu and Chekal processes.
| Process | Yield (%) | AE (%) | RME (%) | CE (%) |
|---|---|---|---|---|
| a The complete E-factor, cE-factor, and PMI were not calculated due to insufficient information. | ||||
| Chekal process | 44 | 50.2 | 12.4 | 19.7 |
| Solution | ||||
| Yu process | 43 | 52.8 | 21.8 | 30.2 |
| Planetary ball mill | ||||
Based on these results, the Yu and co-workers' process is more environmentally friendly than the Chekal at the reaction stage, due to its higher AE, RME, and CE.57 In addition, in this case, the mechanochemical process avoided using NMP (N-methyl pyrrolidone), a solvent classified as a class 2 that must be limited in pharmaceutical products due to potential adverse effects.58 Additionally, it's important to avoid NMP as it can lead to serious health effects, such as miscarriages, reduced fertility, and damage to the liver, kidneys, immune system, and nervous system.59 It is also considered to be reprotoxic and is labeled with the health hazard statement ‘H360D: may damage the unborn child’.60 For this reason, pharmaceutical sectors were recommended to find more environmentally friendly substitutes.61
Nitrofurantoin and dantrolene
Nitrofurantoin (19, Furadantin®; Berkfuran®; Chemiofuran®; Cyanti®; Cysti®; Fua-Med®; Furachel®; Furala®; etc.)62 (Fig. 2) is a wide-spectrum antibiotic accessible since 1953 and is utilized widely to treat urinary tract infections as it usually stays active against drug-resistant uropathogen. The use of nitrofurantoin 19 has increased exponentially since new guidelines have repositioned it as first-line therapy for uncomplicated lower urinary tract infection (UTI).63,64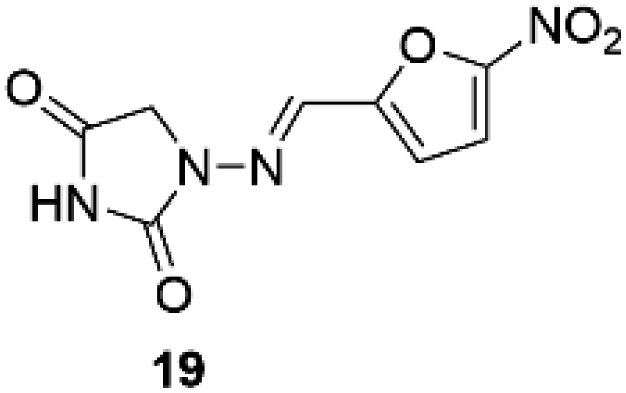 | ||
| Fig. 2 Chemical structure of nitrofurantoin 19. | ||
Dantrolene (20, Dantrium®) (Fig. 3), from Norwich Pharmacal, initially discovered as an efficient and specific skeletal muscle myorelaxant, is nowadays the only clinically available agent for treating malignant hyperthermia (MH) and a substrate for breast cancer-resistant protein.63
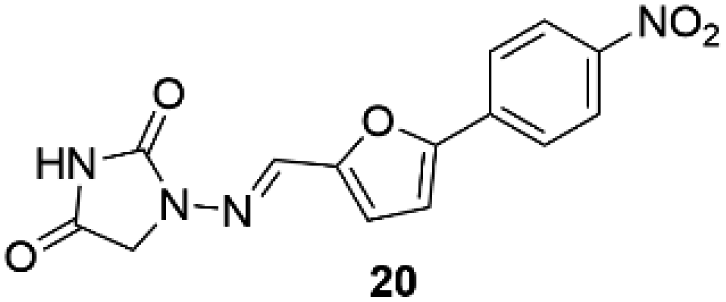 | ||
| Fig. 3 Chemical structure of dantrolene 20. | ||
Nitrofurantoin 19 may be prepared as described by Hayes. The process consisted of reacting 1-aminohydatoyn sulfate 21 with 5-nitro-2-furaldehyde diacetate 22, Scheme 6.65
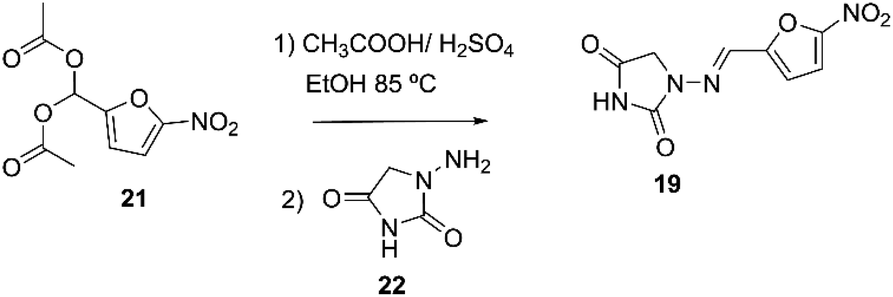 | ||
| Scheme 6 Synthetic procedure of nitrofurantoin described by Hayes.65 | ||
Over the years, other synthetic pathways have emerged, such as the process described by Li and co-workers from Shandong Fangxing Science and Technology Development Co Ltd in 2007, starting from 5-nitrofurfural diethyl acetal with HCl/water.66
Dantrolene 20, can be prepared as described by Fabry and co-workers. The process involves reacting 1-aminohydantoin (1.1 equiv.) in 0.67 M HCl with 5-(4-nitrophenyl)furan-2-carbaldehyde (1 equiv.) in DMF (N-dimethylformamide; 5 mL) at 0 °C. The mixture was stirred for one hour at room temperature. Then, 10 mL of water was added, and the resulting precipitate was filtered and washed with 50 mL of water. The collected solid was dried under reduced pressure to yield dantrolene with 96% yield (Scheme 7).67
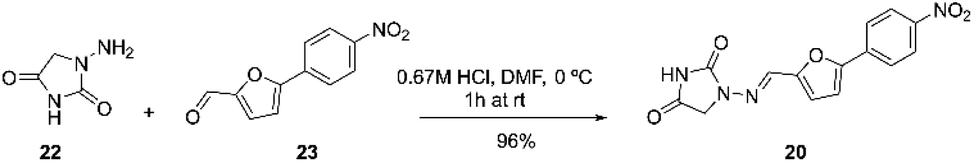 | ||
| Scheme 7 Synthetic procedure of dantrolene described by Fabry and co-workers.67 | ||
In 2018, Colacino and co-workers reported the mechanosynthesis of nitrofurantoin using a ball mill, with the best results obtained in a SPEX Mill 8000. Nitrofurantoin 19 was prepared in 15 minutes with a 95% yield. The product was directly recovered as a powder from the jar without any post-synthetic treatment, except for removing the water produced during the reaction in vacuo at room temperature over P2O5. Dantrolene 20 was also prepared using a planetary and an SPEX mill, with almost identical yields obtained (90%) in both cases, independent of the mill type (planetary or SPEX) and the process parameters used, Scheme 8.64
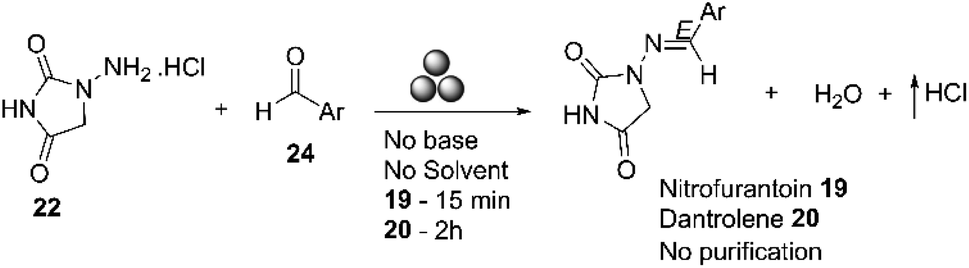 | ||
| Scheme 8 Preparation of nitrofurantoin 19 and dantrolene 20 by mechanochemistry described by Colacino and co-workers.64 | ||
Nitrofurantoin 19 and dantrolene 20 were prepared using a zero-waste procedure. This method did not require additional reagents to activate the reactants, additives, catalysts, or solvents.64 In their study, Colacino and co-workers compared the environmental impact of using mechanochemistry versus solution-based methods for nitrofurantoin and dantrolene.64 They found that the mechanochemical processes were more environmentally friendly than solution-based processes.
In 2021, Crawford and co-workers showed that dantrolene and nitrofurantoin could be successfully produced through mechanosynthesis in TSE. Nitrofurantoin was synthesized starting from 1-aminohydantoin hydrochloride and 5-nitro-2-furaldehyde (1 equiv.) using Liquid-Assisted Grinding (LAG) with CH3CN (0.5× v/w) at room temperature and a screw speed of 30 rpm. After 1 hour, the screw speed was increased to 55 rpm because no product was coming out from the extruder. The E-isomer was obtained exclusively with a Space-Time Yield (STY) of 6.8 × 103 kg m−3 day−1, and no Z-isomer was detected. They also demonstrated through a control experiment that the solid reagents do not react when placed in contact for 90 minutes, confirming the need for the mechanical effects of the extruder for the reaction to occur. The hydrochloric acid produced in the reaction was removed using gas vents along the length of the barrel. The gas was released through an airtight connection and transferred to a basic aqueous solution for neutralization.68 According to the authors, this HCl byproduct has the potential for valorization.
Dantrolene was synthesized starting from 1-aminohydantoin hydrochloride and 5-(4-nitrophenyl)furfural (1 equiv.) with a torque of 1.0 Nm and a specific screw configuration. ATR-IR spectroscopy (Attenuated Total Reflectance Infrared) indicated high conversions. However, some starting material was still observed.68 According to the authors, the process requires further mechanistic study to allow the reaction to reach completion, which may require a mild base.
As shown in Table 6, mechanochemical processes are more cost-effective and environmentally friendly than solution-based processes due to their ability to produce significantly less waste, as evidenced by their lower E-factor and PMI. Furthermore, the mechanochemical process employed in the production of dantrolene offers the advantage of circumventing the use of DMF (class 2 solvent by ICH Guidelines). Due to potential adverse effects, DMF should be limited in pharmaceutical products.58
| API | Process | t (h) | Yield (%) | E-factor (kg kg−1) | AE (%) | PMI (kg kg−1) |
|---|---|---|---|---|---|---|
| a Residence time. | ||||||
| 19 | Li process | 8 | 95 | 16 | 81 | 17 |
| Solution | ||||||
| Colacino process | 0.25 | 95 | 0.3 | 81 | 1.3 | |
| Planetary ball mill or SPEX mill | ||||||
| 19 | Crawford process | 0.7a | 100 | 0.5 | 81 | 1.5 |
| Twin screw extruder | ||||||
| 20 | Fabry process | 1 | 95 | 239 | 85 | 240 |
| Solution | ||||||
| Colacino process | 2 | 90 | 0.3 | 85 | 1.3 | |
| Planetary ball mill or SPEX mill | ||||||
| 20 | Crawford process | 0.7a | 87 | 0.3 | 85 | 1.3 |
| Twin screw extruder | ||||||
It is also considered reprotoxic and “may damage the unborn child”.60 Due to potential health risks, the European Commission has introduced a regulation banning the use of DMF as of December 12, 2023. Consequently, pharmaceutical sectors have initiated the quest for more environmentally friendly substitutes.61
Rufinamide
Rufinamide 25, marketed as Banzel® or Inovelon®, was approved to treat Lennox–Gastaut syndrome (LGS), a severe form of childhood epilepsy. Meier developed rufinamide at Ciba-Geigy Corporation in the United States, later known as Novartis.68–71 Rufinamide may also be prepared, as reported by Kankan, by reacting 2,6-difluorobenzyl bromide 26 with sodium azide in water at 70 °C for 30 hours to obtain 2,6-difluorobenzyl azide 27. Compound 27 then reacts with methyl propiolate 28 to produce methyl 1-(2,6-difluorobenzyl)-1H-1,2,3-triazole-4-carboxylate 29, which is hydrolyzed with aqueous ammonia to produce rufinamide 25, Scheme 9.72 | ||
| Scheme 9 Novartis medicinal chemistry route of rufinamide.72 | ||
Padmaja and Chanda recently summarized several synthesis that have emerged over time to improve the process.73 In 2022, Bhattacherjee and co-workers reported a regiospecific, environmentally benign mechanochemical grinding for Huisgen's 1,3-dipolar cycloaddition of azide 27 and alkyne 33 to give triazole 34 using Cu-beads. This led to an easy workup technique without generating unwanted waste and good yield, Scheme 10.74
 | ||
| Scheme 10 Preparation of rufinamide by mechanochemistry described by Bhattacherjee and co-workers.74 | ||
In 2022, Gómez-Carpintero and co-workers reported the mechanosynthesis of rufinamide as a one-pot sequence of three steps under ball milling and solvent-free conditions.75 A mixture of 2-(bromomethyl)-1,3-difluorobenzene and sodium azide was subjected to ball milling at 25 Hz using a single stainless-steel ball (15 mm diameter) for 60 min. Methyl propiolate and powdered copper were added to the crude mixture and submitted to ball milling for 15 min at 25 Hz. Finally, calcium nitride, indium trichloride, and methanol were added, and the resulting mixture was milled at 30 Hz for an additional 90 min. The product was purified by washing it with distilled water and then dried via vacuum filtration. The resulting solid was finally crystallized in ethanol/H2O to give rufinamide as a white solid with a 47% overall yield. Although low-molecular-weight azides are known to be shock-sensitive, no explosions or increased exothermicity were observed because the reactions were conducted in a sealed steel container, Scheme 11.75
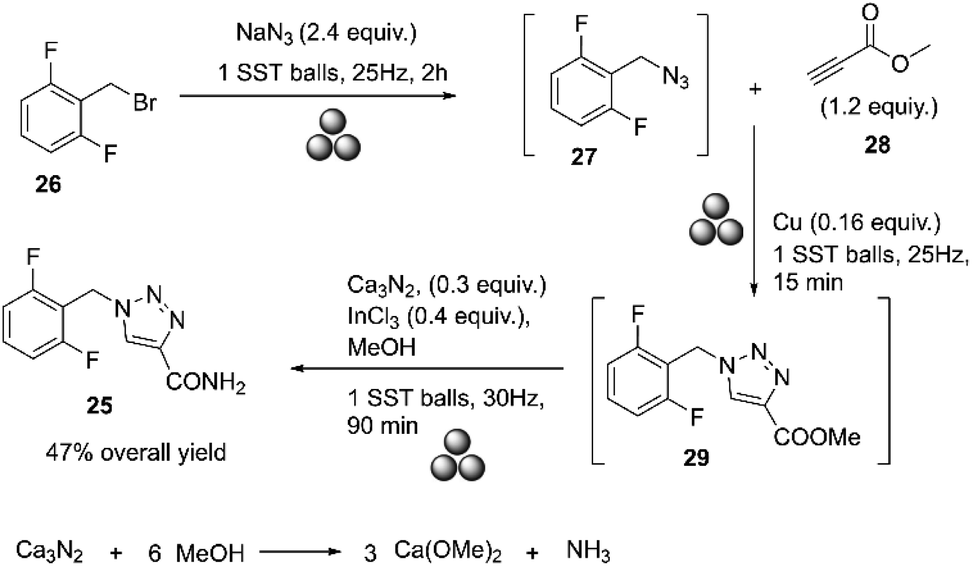 | ||
| Scheme 11 Mechanochemical one-pot synthesis of rufinamide described by Gómez-Carpintero.75 | ||
According to the authors, this synthesis offers several advantages: operational simplicity, good overall yield, solvent-free reaction conditions, avoiding hazardous solvents, and expensive catalysts.75 The insufficient information provided for the workup steps prevented the calculation of the cE-factor, E-factor, and PMI. The comparison of environmental impact indicates that combining mechanochemical and solution-based processes (Bhattacherjee and co-workers process) has a better yield and environmental performance than each process individually. This combination leads to a higher integration of reactants and carbon in the final product and high reactant efficiency, as demonstrated by the higher AE, CE, and RME values in Table 7. Furthermore, mechanochemistry eliminates the need for dichloromethane (DCM),58 a solvent associated with substantial hazards such as neurotoxic, cardiotoxic, and carcinogenic effects, rendering it injurious to tissues. Its hazardous nature has led government agencies and safety organizations worldwide to heavily regulate its usage.76 Consequently, pharmaceutical industries are currently seeking alternatives to this solvent for their processes.
| Process | Yield (%) | AE (%) | RME (%) | CE (%) |
|---|---|---|---|---|
| a The complete E-factor, cE-factor, and PMI were not calculated due to insufficient information. | ||||
| Kankan process | 52 | 63.8 | 32.9 | 47.8 |
| Solution | ||||
| Bhattacherjee process | 76 | 44.6 | 20.2 | 79.6 |
| Solution + planetary ball mill | ||||
| Gómez-Carpintero process | 47 | 34.2 | 15.2 | 27.5 |
| MM 400 mixer mill | ||||
Moclobemide
Moclobemide 35, developed by Hoffmann-La Roche, was first launched in Sweden in 1989 as Aurorix®,77 in Canada as Manerix®78 and later in over 50 countries worldwide for treating depression. However, it was not introduced in the USA until 2002. According to Hoffmann-La Roche, the decision not to enter the US market was based on competition with widely used selective serotonin reuptake inhibitors (SSRIs).79Moclobemide 35 can be prepared, as reported by Braddock and co-workers in 2018, by direct amidation of the 4-chlorobenzoic acid 36 with 4-(2-aminoethyl)morpholine 37 in the presence of tetramethoxysilane 38 in toluene at reflux temperature with 99% yield, Scheme 12.80
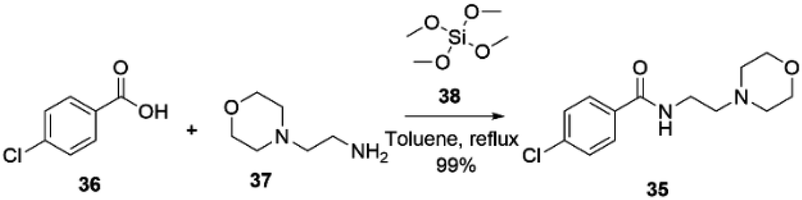 | ||
| Scheme 12 Synthesis of moclobemide described by Braddock and co-workers.80 | ||
In 2022, Lavayssiere and Lamaty reported the synthesis of moclobemide in an Xplore Pharma Melt parallel co-rotating twin screw extruder. A mixture of 4-chlorobenzoic acid 36 (1 equiv.), 4-aminoethyl morpholine (1 equiv.), COMU (1.0 equiv.), DIPEA (1.1 equiv.) and acetonitrile (3.3× v/w) at 30 °C. The screw rotation speed was set at 200 rpm and the manual fold gate turned towards the recirculation pipe. After 10 min of recirculation, the manual fold gate was turned to extrusion mode and a purple gel was recovered with 99% conversion. The workup procedure involved dissolving the product, evaporating the acetonitrile, adjusting the pH, extracting with ethyl acetate, and drying the organic phase over MgSO4. After repeated crystallization from the mother liquors, the desired product was obtained with a 95% yield.47
In 2023, Stolar and co-workers reported a solvent-free thermo-mechanochemical method for directly coupling 4-chlorobenzoic acid 36 and amine 37, which avoids activators and additives, giving water as the only by-product of the reaction. This process involves milling 36 with 37 for one hour at 190 °C in a vibratory ball mill using 12 mL volume stainless steel jars with two 7 mm balls of the same material. Then cool to room temperature before an additional hour of milling at 190 °C. This results in a complete conversion to moclobemide 35 isolated in pure form directly from the milling jar (>99% yield). The thermo-mechanochemical amidation process has a 93.7% atom economy and an E-factor of 0.07, making it environmentally efficient,81 compared to the Braddock process, which was 93.7% and 85.0 respectively. The values of E-factors and PMI are already higher for the Braddock process and do not include the amounts of NaCl used to saturate the aqueous phase and the anhydrous sodium sulfate used to dry the organic phase (Table 8).
| Process | Reaction time (h) | Yield (%) | E-factor (kg kg−1) | cE-factor (kg kg−1) | AE (%) | RME (%) | CE (%) | PMI (kg kg−1) |
|---|---|---|---|---|---|---|---|---|
| a Without considering the amount of NaCl needed to saturate the aqueous phase and the anhydrous sodium sulfate required to dry the organic phase.b The amount of materials used in the workup where not specified. | ||||||||
| Braddocka process | 1 | 99 | 85.0 | 140.0 | 93.7 | 92.9 | 99.1 | 141.0 |
| Solution | ||||||||
| Lavayssiereb | 0.5 | 95 | 4.2 | 4.2 | 93.7 | 88.3 | 94.2 | 5.2 |
| Twin screw extruder | ||||||||
| Stolar process | 1 + 1 | 100 | 0.07 | 0.03 | 93.7 | 93.7 | 99.9 | 1.07 |
| Vibratory ball mill | ||||||||
The process metrics of Lavayssiere and co-workers appear to be better than those obtained by the Braddock process. However, it's important to note that the solvents and reagents used during the workup step were not specified. As a result, the E-factor, cE-factor, and PMI may be higher than presented. The RME value indicates that the yield and quantity of reactants need optimization due to their lower value.
Iopamidol
Iopamidol 38 is a non-ionic X-ray contrast agent that was patented in 1975 by SAVAC A.G. and is still currently used worldwide under various trade names such as Iopamiro® (Bracco), Isovue® (Squib), Niopam® (Merck, UK), and Solutrast® (Byk Gulden), Iopamiron® (Schering).82 The synthesis of Iopamidol can be done using a process described by Felder and co-workers.83 The process involves acylating 5-amino-2,4,6-triiodo-isophthalic acid 39 with thionyl chloride at reflux for 6 hours. The resulting mixture is concentrated to dryness, yielding 5-amino-2,4,6-triiodo-isophthalyl chloride 40 in 98% yield. Compound 40 then reacts with L-2-acetoxypropionyl chloride 41 in DMAc (dimethylacetamide) at room temperature, and the residue obtained is crystallized in ice/water and further purified in warm chloroform, resulting in a 98% yield of the desired compound 42. The intermediate obtained 42 reacts with 1,3-dihydroxyisopropylamine 43 in DMAc. After workup, the residue is precipitated in DCM (dichloromethane) and then purified in warm DCM, yielding compound 44 with a 92% yield. The acetoxy group from compound 44 is hydrolyzed in aqueous NaOH at 40 °C. Sequential contact with cation and anion exchange resins caused the removal of salts from the saponification mixture, and the deionized solution was evaporated to dryness. The residue obtained 38 was further purified by recrystallization from ethanol (57% overall yield), Scheme 13.83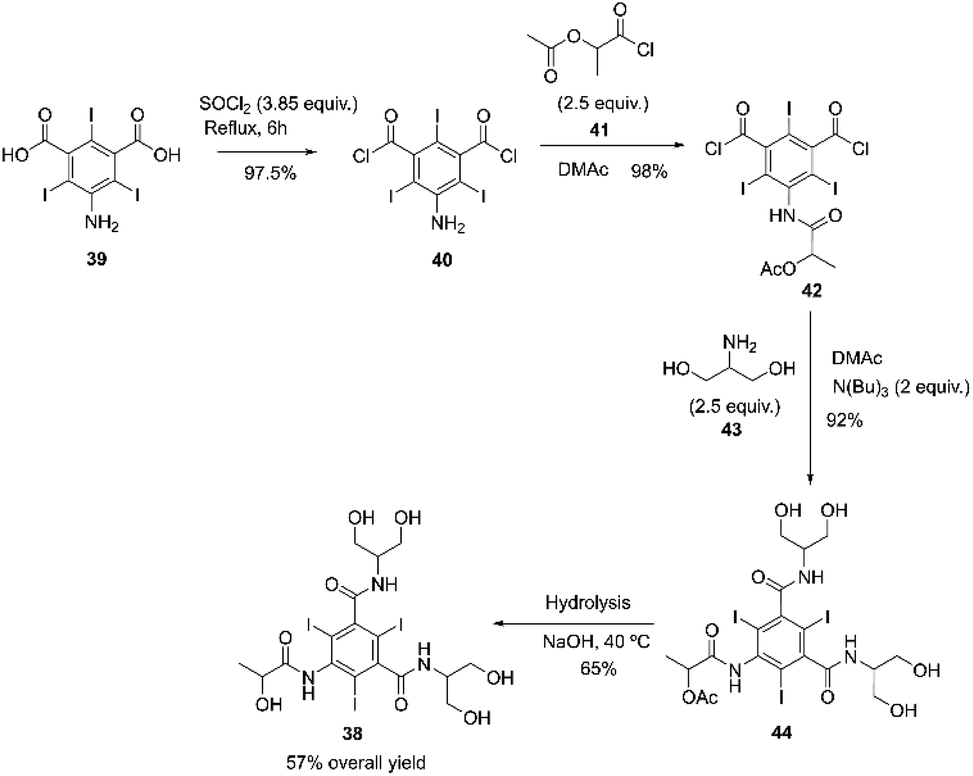 | ||
| Scheme 13 Synthesis of iopamidol described by Felder and co-workers.83 | ||
The above process was used until the early 1980s. However, it was modified due to environmental concerns regarding thionyl chloride's toxicity and hazardous nature, stability issues, and process cost. The new process involved esterifying 5-amino-1,3-benzenedicarboxylic acid 45 with n-butanol or methanol and p-toluenesulfonic acid (PTSA). The resulting compound 46 was then subjected to amidation with serinol 43 in the presence of NaOMe forming compound 47. Compound 47 was iodinated by direct oxidation with iodine monochloride, yielding compound 48.84,85 The hydroxyl groups of compound 48 were selectively acylated with acetic anhydride using DMAP (4-(dimethylamino)pyridine) as a catalyst, forming compound 49. Subsequently, the aniline group of compound 49 was acylated with compound 41, followed by hydrolysis with NaOH of all the acetoxy groups, resulting in the synthesis of iopamidol 38 with an overall yield of 63%, Scheme 14.84,86
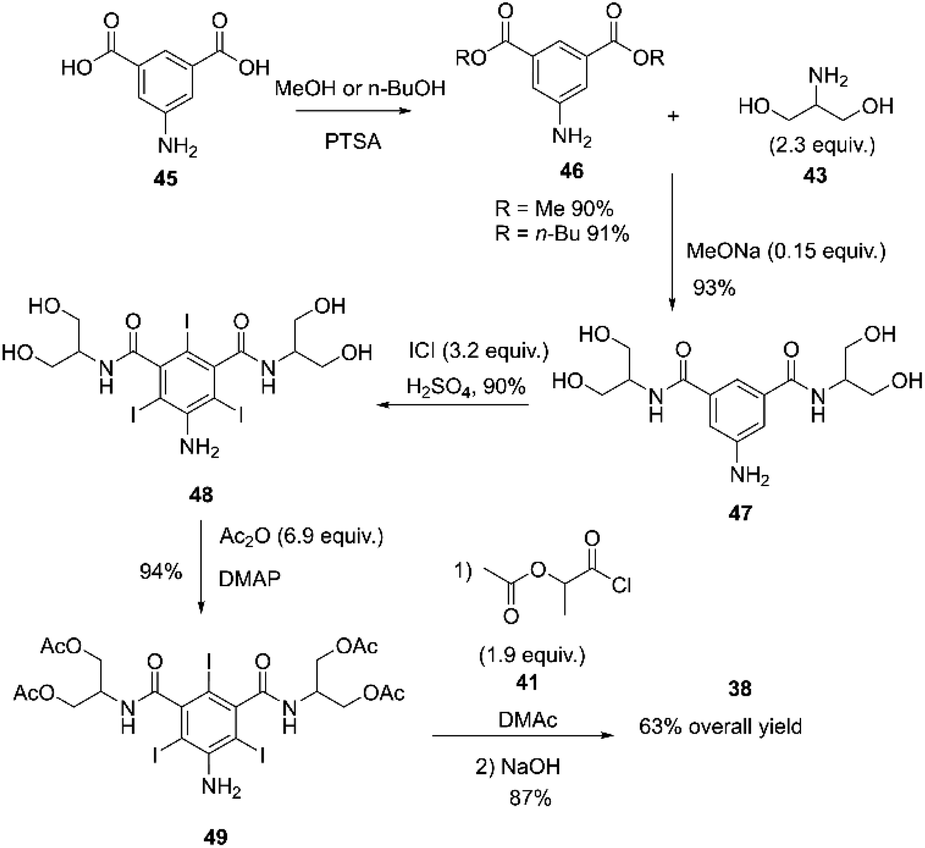 | ||
| Scheme 14 Synthesis of iopamidol described by Anelli and co-workers.84–86 | ||
Barge and co-workers conducted a pilot-scale synthesis of iopamidol in a 5 L ball mill prototype designed by Mr G. Omiccioli. The ball mill was equipped with a water-cooled double-jacket and filled with 300 g of stainless-steel balls, each with a diameter of 15 mm (350 balls). The amidation reaction between the acyl chloride 42 and the amine 43 was carried out in the presence of a small amount of triethanolamine (TEOA) at 30 Hz for 30 minutes. Adding a small amount of base improved the reaction yield to 85% and reduced the excess of serinol from 5 to 2.5 equivalents, making the process easier. This yield was obtained after hydrolyzing intermediate 44 with NaOH at 400 rpm for 10 minutes, and then the workup was performed (Scheme 15).87
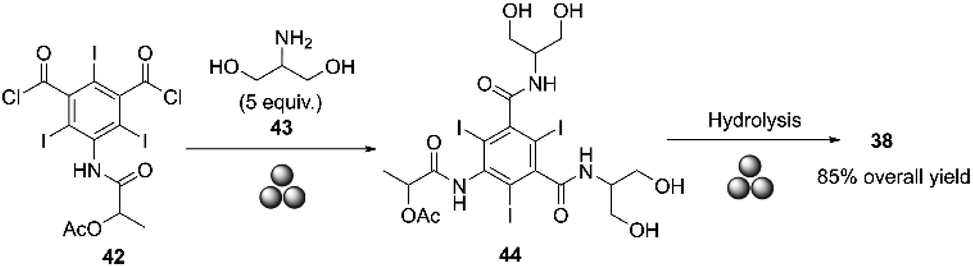 | ||
| Scheme 15 Synthesis of iopamidol by mechanochemistry described by Barge and co-workers.87 | ||
The overall molar yield and green metrics for each process are summarized in Table 9.
| Process | Yield (%) | AE (%) | RME (%) | CE (%) |
|---|---|---|---|---|
| a The complete E-factor, cE-factor, and PMI were not calculated due to insufficient information. | ||||
| Felder process | 57 | 66.4 | 27.2 | 35.6 |
| Solution | ||||
| Anelli process | 63 | 37.6 | 16.9 | 12.4 |
| Solution | ||||
| Barge process | 85 | 87.1 | 56.7 | 51.7 |
| High-speed planetary ball mill | ||||
The mechanochemical process has the advantage of avoiding using DMAc, which is a class 2 solvent.58 Besides that, DMAc as DMF is considered reprotoxic and “may damage the unborn child”.60 Unfortunately, some of the green metrics could not be calculated due to insufficient information. However, based on the possible calculations, mechanosynthesis has a high yield and is more environmentally friendly than solution-based processes. This is due to combining two reactions into a single operation, without isolating the intermediate, eliminating the need to add organic solvents. This reduces waste disposal and energy consumption. Moreover, the mechanochemical process resulted in a higher incorporation of reactants into the final product and carbon incorporation from the reactants into the final product, as demonstrated by the higher values of EA and CE, respectively. Additionally, the RME outperforms the base solution process regarding yield and excess reagent usage. It is important to highlight that the process began with an advanced starting material, which is advantageous as it reduces waste and decreases the process cycle time.
Paracetamol
Paracetamol (50, acetaminophen, N-acetyl-p-aminophenol) is one of the most widely used over-the-counter analgesic antipyretic drugs.88 Due to supply chain issues and localized production, several countries are experiencing shortages of paracetamol. Production has shifted from developed nations due to high processing costs and strict environmental regulations. New, environmentally friendly production methods are needed to reshape drug supplies effectively.89In 2018, Portada and co-workers reported the first mechanical synthesis of paracetamol by milling p-nitrophenol 51 with ammonium formate and Pd/C in a Retsch mill at a frequency of 30 Hz, using a 10 mL Teflon jar and two 10 mm diameter Teflon balls. The p-aminophenol was then acetylated by milling equal amounts of p-aminophenol 52 and acetic anhydride at 30 Hz for 30 minutes, with silica as the milling auxiliary, affording paracetamol in high yield. The Teflon jar was chosen over a stainless steel one to prevent sample contamination with iron, Scheme 16.37
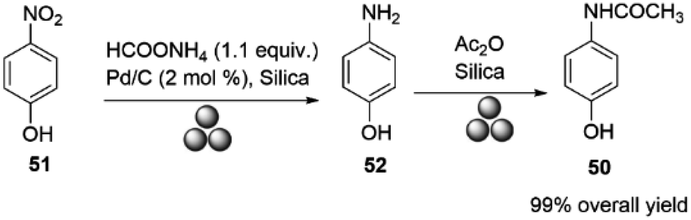 | ||
| Scheme 16 Synthesis of paracetamol described by Portada and co-workers.37 | ||
In 2024, Park and colleagues conducted a study on the mechanochemical hydrogenation of 4-nitrophenol. They compared the catalytic transfer hydrogenation (CTH) of 4-nitrophenol using formic acid and the catalytic hydrogenation using hydrogen gas flow. They used a Retsch MM 200 ball mill for CTH and 400 shaker mills for gas flow hydrogenation. The results showed that the hydrogenation using formic acid was not selective. Hydrogenation under hydrogen gas flow was carried out with acetic anhydride at 20 Hz for 30 minutes with 5 wt% Pd/C catalyst in a 15 mL custom steel milling vessel equipped with gas inlet and outlet ports. Under the optimized conditions (with a small amount of isopropanol and at a hydrogen flow rate of 60 SCCM), the selectivity increased, preventing the undesirable side reactions of 4-aminophenol observed in the formic acid process. Furthermore, they compared ball milling results with traditional stirred tank hydrogenation in a Parr reactor. In the ball mill, paracetamol was obtained with 99% purity and 96% yield, while in the stirred tank pressurized steel autoclave, the yield was 95%.89 The overall molar yield and green metrics for each process are summarized in Table 10.
When comparing the environmental impact of these processes, despite the lower yield obtained in the ball mill Park process, the mechanosynthesis is more eco-friendly than the solution-based process due to the significant reduction of solvents and the use of hydrogen instead of ammonium formate/Pd/C and iron powder as a catalyst. This results in a higher AE and lower cE-factor. The disadvantage of the hydrogenation in the ball mill is that it requires three times more hydrogen than the pressurized stirred tank reactor, resulting in a lower reaction mass efficiency (RME) and a higher PMI. However, this can be overcome by recycling the hydrogen. The green metrics for the stirred tank process can also be improved by performing the reaction in a more concentrated media. It is important to note that the PMI values for Park processes only describe the paracetamol yield directly from the reaction vessel, without considering any separation or purification steps.
Conclusions
Despite being a theoretical assessment, the examples demonstrate that mechanosynthesis creates less waste than solution-based counterparts. This is because solution-based processes typically require larger quantities of solvents for reaction, isolation, and equipment cleaning. In the examples given, around 58–95% of the material's mass is made up of solvents. In contrast, mechanochemical reactions can be carried out in a single step, allowing successive chemical reactions without isolating intermediates. This is advantageous because it reduces the number of steps, saves time and resources, and increases chemical yield by eliminating the need for intermediates lengthy separations, isolations, and purifications.The examples also demonstrate that mechanochemistry enables specific reactions, such as amidation (the most common functional group in nature and in APIs), to take place under safer conditions without using hazardous reagents like thionyl chloride or harsh conditions (e.g., reflux). This method involves activating the carboxylic acid with CDI, followed by its reaction with the amine, as demonstrated in the teriflunomide example. This approach also enables direct amidation reactions between acyl chloride and amine, as shown in the iopamidol example.
Based on the current literature, energy savings are not frequently reported. In general, the impact and shearing forces of the balls create the energy required for reactions, resulting in faster and more energy-efficient processes compared to traditional methods that rely on inputting energy to raise the reaction temperature. Axitinib, nitrofurantoin, dantrolene, and moclobemide (TSE process) serve as examples.
Regarding safety, two examples were presented where shock-sensitive or explosive compounds (azides) and flammable gas (hydrogen) were used without any safety issues. However, it may be challenging to ensure safe use on larger scales. The explosivity of materials should be evaluated because the mechanical energy generated can trigger an explosion. Additionally, the thermal stability of the materials and mixtures should be evaluated, along with the exothermicity of the reactions. This last point should be addressed using different methodologies because, unlike a reaction in a conventional reactor, it is not expected to occur in an adiabatic environment.
In summary, considering all the information herein presented, mechanochemical processes provide substantial cost savings for manufacturers and, consequently, for consumers. Although not all the 12 principles of green chemistry were met when evaluating a process, the examples demonstrated that mechanochemistry fulfilled more principles than its solution-based counterpart. However, implementing mechanochemistry on an industrial scale using ball milling remains challenging. Twin-screw extrusion (TSE) is being advocated as a scalable alternative to address the challenges ball mills pose in manufacturing. The implementation of this technique allows for a smooth transition from batch to continuous processing. However, drawing broader conclusions about this method requires more extensive data. One common challenge for both the BM and the TSE is equipment cleaning, which requires meeting very low acceptable levels of impurities and must not be ignored.
Researchers from the pharmaceutical industry and academia are encouraged to collaborate in developing this technology to benefit the environment and economy.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.References
- Nations United, World Population Prospects: The 2017 Revision. Dep. Econ. Soc. Aff., 2017 Search PubMed.
- A. Sauvy, Cah. Sociol. Demogr. Med., 1987, 27, 191–219 CAS.
- M. S. Lipsky and L. K. Sharp, J. Am. Board Fam. Pract., 2001, 14, 362–367 CAS.
- J. L. Schneider, A. Wilson and J. M. Rosenbeck, Benchmark Int. J., 2010, 17, 421–434 CrossRef.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- A. Larson, K. O’Brien and A. Anderson, Pfizer Pharmaceuticals: Green Chemistry Innovation and Business Strategy, Darden Case No. UVA-ENT-0088, 2017 Search PubMed.
- D. J. C. Constable, C. Jimenez-Gonzalez and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133–137 CrossRef CAS.
- N. Winterton, Clean Technol. Environ. Policy, 2021, 23, 2499–2522 CrossRef PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry : Theory and Practice, Oxford University Press, 1998 Search PubMed.
- A. E. Marteel-Parrish and M. A. Abraham, Green Chem. Eng. A Pathw. To Sustain., 2013, 9780470413, pp. 1–361 Search PubMed.
- ACS Green Chemistry Institute, Converg. PMI Calc., 2014 Search PubMed.
- F. Gomollón-Bel, Chem. Int., 2019, 40, 12–17 Search PubMed.
- A. D. McNaught and A. Wilkinson, in The IUPAC Compendium of Chemical Terminology, International Union of Pure and Applied Chemistry (IUPAC), Research Triangle Park, NC, 2014, p. 903 Search PubMed.
- L. Takacs, J. Miner. Met. Mater. Soc., 2000, 52, 12–13 CrossRef CAS.
- M. Marchini, G. Montanari, L. Casali, M. Martelli, L. Raggetti, M. Baláž, P. Baláž and L. Maini, RSC Mechanochem., 2024, 1, 123–129 RSC.
- L. Takacs, J. Therm. Anal. Calorim., 2007, 90, 81–84 CrossRef CAS.
- L. Takacs, Acta Phys. Pol., A, 2012, 121, 711–714 CrossRef CAS.
- S. Hwang, S. Grätz and L. Borchardt, Chem. Commun., 2022, 58, 1661–1671 RSC.
- E. AlShamaileh, A. E. Al-Rawajfeh and M. Alrbaihat, Open Agric. J., 2018, 12, 11–19 CrossRef CAS.
- V. Declerck, E. Colacino, X. Bantreil, J. Martinez and F. Lamaty, Chem. Commun., 2012, 48, 11778–11782 RSC.
- A. Krusenbaum, S. Grätz, G. T. Tigineh, L. Borchardt and J. G. Kim, Chem. Soc. Rev., 2022, 51, 2873–2905 RSC.
- X. Guo, D. Xiang, G. Duan and P. Mou, Waste Manag., 2010, 30, 4–10 CrossRef PubMed.
- Q. Tan and J. Li, Environ. Sci. Technol., 2015, 49, 5849–5861 CrossRef CAS PubMed.
- A. V. Trask and W. Jones, Crystal Engineering of Organic Cocrystals by the Solid-State Grinding Approach, 2005, pp. 41–70 Search PubMed.
- H. J. Kang, Y. H. Choi, I. W. Joo and J. E. Lee, Bull. Korean Chem. Soc., 2021, 42, 737–739 CrossRef CAS.
- T. F. Grigorieva, A. P. Barinova and N. Z. Lyakhov, J. Nanoparticle Res., 2003, 5, 439–453 CrossRef CAS.
- J. L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed.
- S. Pagola, Crystals, 2023, 13, 124–157 CrossRef CAS.
- F. Cuccu, L. De Luca, F. Delogu, E. Colacino, N. Solin, R. Mocci and A. Porcheddu, ChemSusChem, 2022, 15, e202200362–e202200403 CrossRef CAS PubMed.
- C. Bolm and J. G. Hernández, Angew. Chem., Int. Ed., 2019, 58, 3285–3299 CrossRef CAS PubMed.
- J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC.
- D. E. Crawford and J. Casaban, Adv. Mater., 2016, 28, 5747–5754 CrossRef CAS PubMed.
- J. Jablan, E. Marguí, L. Posavec, D. Klarić, D. Cinčić, N. Galić and M. Jug, J. Pharm. Biomed. Anal., 2024, 238, 115855–115864 CrossRef CAS PubMed.
- A. Bodach, A. Portet, F. Winkelmann, B. Herrmann, F. Gallou, E. Ponnusamy, D. Virieux, E. Colacino and M. Felderhoff, ChemSusChem, 2024, 17, e202301220 CrossRef CAS PubMed.
- International Conference on Harmonisation, ICH Qual. Guidel., 2000, pp. 1–48 Search PubMed.
- R. Tedjini, R. Viveiros, T. Casimiro and V. D. B. Bonifácio, RSC Mechanochem., 2024, 2, 176–180 RSC.
- T. Portada, D. Margetić and V. Štrukil, Molecules, 2018, 23, 3163–3181 CrossRef PubMed.
- R. A. Sheldon, ACS Sustain. Chem. Eng., 2018, 6, 32–48 CrossRef CAS.
- D. J. C. Constable, A. D. Curzons and V. L. Cunningham, Green Chem., 2002, 4, 521–527 RSC.
- C. Jimenez-Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manley, Org. Process Res. Dev., 2011, 15, 912–917 CrossRef CAS.
- J. Paik, Pediatr. Drugs, 2021, 23, 609–613 CrossRef PubMed.
- B. Mehta and Y. Gohil, Pharm. Innov., 2017, 6, 440–449 CAS.
- R. R. Bartlett and F.-J. Kämmerer, US Pat., 5494911, 1996 Search PubMed.
- E. P. T. Leitão and L. M. S. Sobral, J. Biomed. Res. Environ. Sci., 2024, 5, 159–213 CrossRef.
- T.-X. Métro, J. Bonnamour, T. Reidon, J. Sarpoulet, J. Martinez and F. Lamaty, Chem. Commun., 2012, 48, 11781–11783 RSC.
- D. D. Wirth, S. Vikas and P. Jagadish, in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, Chichester, UK, 2017, pp. 1–6 Search PubMed.
- M. Lavayssiere and F. Lamaty, Chem. Commun., 2023, 59, 3439–3442 RSC.
- U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, & Center for Biologics Evaluation and Research, Q3C — Tables and List Guidance for Industry (ICH Revision 4), 2018, https://www.fda.gov/media/133650/download, accessed on 24 August 2024 Search PubMed.
- I. F. McConvey, D. Woods, M. Lewis, Q. Gan and P. Nancarrow, Org. Process Res. Dev., 2012, 16, 612–624 CrossRef CAS.
- A.-F. Villamizar-Mogotocoro, L. Y. Vargas-Méndez and V. V. Kouznetsov, Eur. J. Pharm. Sci., 2020, 151, 105374–105407 CrossRef CAS PubMed.
- N. Sudha, R. Surendran and S. Jeyaram, Indian J. Phys., 2024, 98, 1453–1462 CrossRef CAS.
- J. Soubhye, M. Gelbcke, P. Van Antwerpen, F. Dufrasne, M. Y. Boufadi, J. Nève, P. G. Furtmüller, C. Obinger, K. Zouaoui Boudjeltia and F. Meyer, ACS Med. Chem. Lett., 2017, 8, 206–210 CrossRef CAS PubMed.
- P. F. M. Oliveira, M. Baron, A. Chamayou, C. André-Barrès, B. Guidetti and M. Baltas, RSC Adv., 2014, 4, 56736–56742 RSC.
- K. Tzogani, V. Skibeli, I. Westgaard, M. Dalhus, H. Thoresen, K. B. Slot, P. Damkier, K. Hofland, J. Borregaard, J. Ersbøll, T. Salmonson, R. Pieters, R. Sylvester, G. Mickisch, J. Bergh and F. Pignatti, Oncologist, 2015, 20, 196–201 CrossRef CAS PubMed.
- B. P. Chekal, S. M. Guinness, B. M. Lillie, R. W. McLaughlin, C. W. Palmer, R. J. Post, J. E. Sieser, R. A. Singer, G. W. Sluggett, R. Vaidyanathan and G. J. Withbroe, Org. Process Res. Dev., 2014, 18, 266–274 CrossRef CAS.
- L. H. Zhai, L. H. Guo, Y. H. Luo, Y. Ling and B. W. Sun, Org. Process Res. Dev., 2015, 19, 849–857 CrossRef CAS.
- J. Yu, Z. Hong, X. Yang, Y. Jiang, Z. Jiang and W. Su, Beilstein J. Org. Chem., 2018, 14, 786–795 CrossRef CAS PubMed.
- Pharmacopée Européenne, Eur. Pharmacopoeia 8.0, 2008, pp. 639–646 Search PubMed.
- U.S. Environmental Protection Agency, EPA Proposes Requirements to Protect Workers and Consumers from Exposure to Toxic Solvent N-Methylpyrrolidone, U.S. Environmental Protection Agency, URL: https://www.epa.gov/newsreleases/epa-proposes-requirements-protect-workers-and-consumers-exposure-toxic-solvent-n (accessed 14-10-2024) Search PubMed.
- J. Sherwood, F. Albericio and B. G. de la Torre, ChemSusChem, 2024, 17, 1–6 CrossRef PubMed.
- European Chemicals Agency (ECHA), https://echa.europa.eu/view-article/-/journal_content/title/9109026-58 accessed 24 August 2024 Search PubMed.
- S. Budavari, M. J. O'Neal, A. Smith and P. E. Heckelman, in, The Merck Index, M. & Co, Rahway, New Jersey U.S.A., 11th edn, 1989, p. 946 Search PubMed.
- M. Mahdizade Ari, S. Dashtbin, F. Ghasemi, S. Shahroodian, P. Kiani, E. Bafandeh, T. Darbandi, R. Ghanavati and A. Darbandi, Front. Cell. Infect. Microbiol., 2023, 13, 1–13 Search PubMed.
- E. Colacino, A. Porcheddu, I. Halasz, C. Charnay, F. Delogu, R. Guerra and J. Fullenwarth, Green Chem., 2018, 20, 2973–2977 RSC.
- K. J. Hayes, US Pat. 2610181, 1952 Search PubMed.
- X. Li, F. Zhang, L. Xiubing and Z. Feng, CN Pat., 101450941A, 2007 Search PubMed.
- D. C. Fabry, Y. A. Ho, R. Zapf, W. Tremel, M. Panthöfer, M. Rueping and T. H. Rehm, Green Chem., 2017, 19, 1911–1918 RSC.
- D. E. Crawford, A. Porcheddu, A. S. McCalmont, F. Delogu, S. L. James and E. Colacino, ACS Sustain. Chem. Eng., 2020, 8, 12230–12238 CrossRef CAS.
- C. E. Stafstrom, Neuropsychiatric Dis. Treat., 2009, 5, 547–551 CrossRef CAS PubMed.
- M. Bialer and H. S. White, Nat. Rev. Drug Discovery, 2010, 9, 68–82 CrossRef CAS PubMed.
- R. Meier, US Pat., US4789680, 1986 Search PubMed.
- R. N. Kankan, D. R. Rao and D. R. Birari, US Pat., 8183269, 2012 Search PubMed.
- R. D. Padmaja and K. Chanda, Org. Process Res. Dev., 2018, 22, 457–466 CrossRef CAS.
- D. Bhattacherjee, I. S. Kovalev, D. S. Kopchuk, M. Rahman, S. Santra, G. V Zyryanov, P. Das, R. Purohit, V. L. Rusinov and O. N. Chupakhin, Molecules, 2022, 27, 7784–7795 CrossRef CAS PubMed.
- J. Gómez-Carpintero, C. Cabrero, J. D. Sánchez, J. F. González and J. C. Menéndez, Green Chem. Lett. Rev., 2022, 15, 638–645 CrossRef.
- R. Yogesh, N. Srivastava and B. M. Mulik, Macromol. Symp., 2023, 407 DOI:10.1002/masy.202100502.
- A. J. Scheen, Rev. Med. Liege, 1994, 49, 291–292 CAS.
- E. Weinberg and I. Mallov, Communications, 2016, 6–7 Search PubMed.
- U. Bonnet, CNS Drug Rev., 2002, 8, 283–308 CrossRef CAS PubMed.
- D. C. Braddock, P. D. Lickiss, B. C. Rowley, D. Pugh, T. Purnomo, G. Santhakumar and S. J. Fussell, Org. Lett., 2018, 20, 950–953 CrossRef CAS PubMed.
- T. Stolar, J. Alić, G. Talajić, N. Cindro, M. Rubčić, K. Molčanov, K. Užarević and J. G. Hernández, Chem. Commun., 2023, 59, 13490–13493 RSC.
- S. Budavari, M. J. O'Neal, A. Smith and P. E. Heckelman, in, The Merck Index, Merck & Co, Rahway, New Jersey U.S.A., 11th edn, 1989, p. 731 Search PubMed.
- E. Felder and D. E. Pitre, US Pat. 4001323, 1975 Search PubMed.
- P. L. Anelli, M. Brocchetta, L. Lattuada and F. Uggeri, Pure Appl. Chem., 2012, 84, 485–491 CAS.
- P. L. Anelli, M. Brocchetta, G. Lux and E. Cappelletti, WO Pat., 02/044125A1, 2002 Search PubMed.
- P. L. Anelli, M. Brocchetta, G. Lux and E. Cappelletti, WO Pat., 02/44132A1, 2002 Search PubMed.
- A. Barge, F. Baricco, G. Cravotto, R. Fretta and L. Lattuada, ACS Sustain. Chem. Eng., 2020, 8, 12825–12830 CrossRef CAS.
- S. S. Ayoub, Temperature, 2021, 8, 351–371 CrossRef PubMed.
- J. Park, J. S. Maier, C. Evans, M. Hatzell, S. France, C. Sievers and A. S. Bommarius, Green Chem., 2024, 26, 4079–4091 RSC.
| This journal is © The Royal Society of Chemistry 2024 |