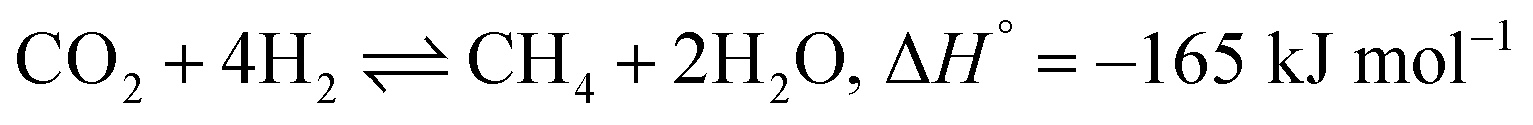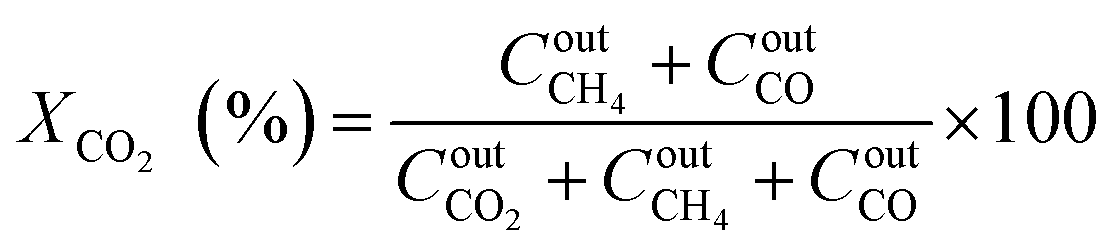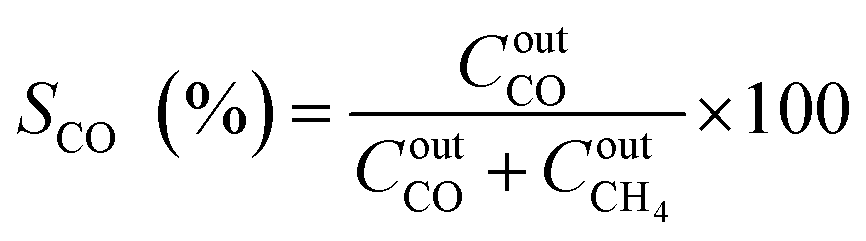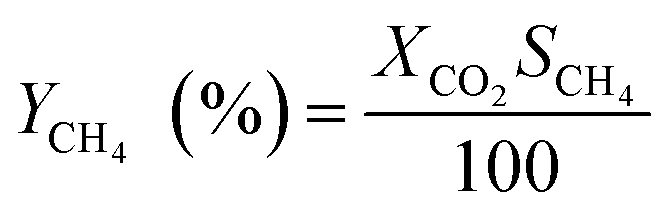Exsolved LaNiRuO3 perovskite-based catalysts for CO2 methanation reaction†
Received
23rd July 2024
, Accepted 5th October 2024
First published on 13th November 2024
Abstract
Hydrogenation of CO2 for methane formation is one of the thermodynamically favorable processes for reducing atmospheric CO2 emissions. The present work demonstrates the synthesis and evaluation of LaNiRuO3 perovskite-derived catalysts for CO2 methanation in both supported and unsupported (bulk) forms. Specifically, two catalysts were prepared: (i) a Ru-substituted LaNiO3 perovskite, LaNi0.9Ru0.1O3, with 10 at% Ru and (ii) a supported version (30% LaNi0.9Ru0.1O3/Al2O3). The catalysts were synthesized through controlled reduction conditions, and they were thoroughly characterized, before and after the exsolution process, using XRD, TEM, XPS, BET, H2-TPR, and H2-TPD techniques. The characterization results indicated that the exsolved LaNi0.9Ru0.1O3/Al2O3 catalyst formed small Ni particles (∼6 nm), resulting in better dispersion (18%) while maintaining a high surface area (141 m2 g−1) and porosity. This catalyst demonstrated a 10% higher CO2 conversion (77%) at a temperature lower by 50 °C (i.e. 400 °C) than the exsolved bulk LaNi0.9Ru0.1O3 perovskite. Both catalysts exhibited over 90% selectivity for CH4 in the 250–450 °C range. The enhanced catalytic performance of the exsolved LaNi0.9Ru0.1O3/Al2O3 catalyst was attributed to the small Ni particle size, better dispersion, and the alumina support's high surface area and basic properties, facilitating the adsorption and dissociation of H2 and CO2. Further long-term stability tests at 400 °C and 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL g−1 h−1 (WHSV) over 54 h revealed that the exsolved LaNi0.9Ru0.1O3/Al2O3 catalyst maintained a 70% CO2 conversion, with the CH4 yield and selectivity above 60% and 95%, respectively. Thus, supporting the perovskite catalyst on Al2O3 demonstrated a pronounced effect on the CO2 conversion rate and CH4 selectivity at lower temperatures along with ensuring the stability of catalyst over extended periods.
000 mL g−1 h−1 (WHSV) over 54 h revealed that the exsolved LaNi0.9Ru0.1O3/Al2O3 catalyst maintained a 70% CO2 conversion, with the CH4 yield and selectivity above 60% and 95%, respectively. Thus, supporting the perovskite catalyst on Al2O3 demonstrated a pronounced effect on the CO2 conversion rate and CH4 selectivity at lower temperatures along with ensuring the stability of catalyst over extended periods.
Sustainability spotlight
To drive toward a zero-carbon society, we need to accelerate the deployment of technologies that can tackle CO2 emissions. This work aspires to deliver an affordable and easy-to-prepare catalytic system that can be used for tunable and efficient CO2 conversion to natural gas without promoting the production of parasitic reactions/products. Perovskites are herein proposed as they have a high scalability potential for translating the research into real-life application. This work aligns well with SDGs 7 (affordable and clean energy), 11 (sustainable cities and communities), and 13 (climate action).
|
1. Introduction
The extensive reliance on fossil fuels has risen significantly as a response to the escalating global energy demands, contributing to the increased release of greenhouse gases (GHGs).1 Carbon dioxide (CO2) has been the major contributor to global warming and reached a global average of 419.3 ppm level in 2023 according to the NOAA's Global Monitoring Lab annual report. Therefore, there have been concentrated research efforts aimed at the capture, utilization, and storage of CO2 (CCUS) to mitigate the consequences.2,3 The hydrogenation of CO2 to methane is an important CO2 utilization process and has certain advantages over other CO2 utilization techniques, such as the reverse water gas shift reaction (RWGS), due to its thermodynamic feasibility.4–8 Sabatier was the first to report the CO2 methanation reaction, which follows eqn (1):9| | 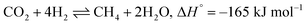 | (1) |
The exothermic nature of CO2 methanation favors the reaction occurring at lower temperatures compared to the RWGS. The equilibrium of the reaction can be achieved at lower temperatures on the catalyst surface by avoiding the kinetic constraints and improving CO2 activation and H2 dissociation.10,11
Many efforts have been invested to develop noble- and non-noble metal-based robust catalysts for the CO2 methanation reaction during the past decade.12 Ni-based supported catalysts are more affordable than and have shown comparable performances to noble-metal based catalysts for this reaction.13,14 The scarcity and expensive nature of noble metals limit their applicability at large-scale industries. Several support materials, such as Al2O3, SiO2, CeO2, and TiO2, for Ni-based catalysts have been studied for the CO2 methanation reaction.12–14 Meanwhile, the metal loading in the catalyst is usually kept low to attain well-dispersed active catalytic sites and prevent agglomeration. The conventional impregnation methods for catalyst preparation are generally inefficient, especially for stabilizing the metal nanoparticles (e.g., Ni) on the support, as the metal particles tends to agglomerate at high temperatures, compromising the catalyst performance. In this context, exsolution has attracted significant attention in the past decade as a promising approach for synthesizing supported catalysts.15,16 In the exsolution process of, in particular, mixed metal oxides, usually the guest metals (that will later serve as active sites) are substituted within the host oxide lattice and will evolve to the surface in the form of metallic nanoparticles under reduction conditions at high temperatures. In this unique process, there is a crystallographic alignment between the exsolved metals and the host oxide phase as the exsolved nanoparticles were initially part of the support crystal lattice. In turn, the exsolved metal nanoparticles are partially socketed to the host oxide phase. Hence, the exsolution process allows for a better control of the metal–support interactions and enhanced structure stability compared to the traditional impregnation synthesis.17–19 It has been noted that perovskite, typically with an ABO3 stoichiometry, is usually selected in the literature as the host oxide lattice for the exsolution process. ABO3-type perovskite structures (A-lanthanide, B-transition metal), as promising oxide materials, have been intensively used in high-temperature catalytic reactions (e.g., redox chemical activation, CO2 and CH4 reforming) due to their thermal stability, reactivity, and economical aspects.20–23 The perovskite structure can be used as a matrix to tailor the design of the metal–support interactions through a careful choice of the composition of A- and B-sites.24–26 Studies have indicated that A-site deficiencies in conjunction with oxygen vacancies destabilize the perovskite structure in such a way that B-site atoms can be exsolved so to maintain the original stoichiometry. The exsolution of perovskite structures can lead to the transition metal being strongly socketed into the oxide support and the exsolved metals will then possess exceptional resistance to agglomeration and thus exhibit better and more stable catalytic activity. On the other hand, partial substitution of the A-site (with alkali and rare-earth metals) or B-site (with transition metals) in the perovskite structure is a proven method for improving the catalytic activity in the derived catalyst systems.27–29 However, unsupported perovskites exhibit a low-surface area with poor dispersion of active metals, which could affect the catalytic activity due to the diffusion limitation of the reaction intermediates. In order to address this limitation observed with perovskites, various studies have been conducted involving dispersing the perovskites on support materials.30–37 Most the studies in the literature have focused on utilizing SiO2 and CeO2 as support materials for LaNiO3 perovskites.30–32 For instance, S. Li et al. selected the LaNi1−xMoxO3 perovskite-type structure and loaded on SiO2 to develop a catalyst for CO2 methanation. Under reduction conditions, MoOx, La2O3, and Ni nanoparticles were formed, which were highly dispersed on the SiO2 surface, resulting in the formation of a Ni/MoOx–La2O3/SiO2 catalyst. As MoOx is active for the reverse water gas shift reaction (RWGS) and Ni can effectively catalyze CO methanation, the resulting Ni/MoOx–La2O3/SiO2 catalyst showed good performance for CO2 methanation.30 Onrubia-Calvo et al.31,32 and T. Zhang37 studied the viability of a LaNiO3 precursor dispersed on a CeO2 support for the CO2 methanation reaction, and observed that the exsolution of Ni nanoparticles occurred during the controlled reduction of LaNiO3/CeO2, which led to the formation of a Ni–La2O3/CeO2 catalyst. This catalyst obtained after the reduction of the LaNiO3/CeO2 catalyst exhibited highly dispersed Ni nanoparticles (∼5 nm), a higher concentration of weak to medium basic sites, and higher accessibility of Ni nanoparticles, which could facilitate CO2 adsorption as well as H2 activation and improve the overall CO2 methanation efficiency. Ultimately, the supported LaNiO3 catalyst on a high-surface-area support exhibited notably better and stable catalytic activity toward the CO2 methanation reaction compared to catalysts produced from bare perovskites and conventional catalysts (Ni supported on SiO2 and CeO2). As Ni-based catalysts exhibit good catalytic activity toward CO2 methanation and as modification of the support by adding La2O3 enhances the metal–support interaction and basicity of the catalyst, most prior studies have selected the LaNiO3 perovskite precursor to disperse on a high-surface-area support for catalytic applications. Nonetheless, to the best of our knowledge, the catalytic activity of partially B-site-substituted LaNiO3 perovskite supported on Al2O3 catalysts for the CO2 methanation reaction has not been studied so far.
In this context, the aim of this work was to utilize the well-known LaNiO3 as a precursor to develop B-site Ru-substituted LaNiO3 perovskite (LaNi1−xRu0xO3), which was then used as a stand-alone catalyst or dispersed on an Al2O3 support. It was anticipated that the addition of Ru to Ni will increase the metal dispersion and enhance the catalytic activity at lower temperature, as per the open literature.38,39 In the present study, catalysts derived from B-site Ru-substituted LaNiO3 perovskite, both bulk and supported on Al2O3, were synthesized and tested for the CO2 methanation reaction. The structural, morphological, and physicochemical properties of the produced catalysts were investigated using XRD, SEM, TEM, XPS, BET, H2-TPR, and H2-TPD techniques. HAADF-STEM and H2-TPD analyses proved that the supported catalysts exhibited better Ni dispersion with a small particle size. Further, the CO2 methanation activity of both catalysts was tested at 25000 mL g−1 h−1 weight hourly space velocity (WHSV) in the temperature range of 200–500 °C along with stability studies for 54 h at 400 °C under the same WHSV conditions. It was found that the supported perovskite catalyst exhibited better catalytic activity compared to that of the unsupported (bulk) one.
2. Experimental section
2.1. Catalyst preparation
2.1.1. LaNiRuO3 perovskite supported on Al2O3.
LaNi0.9Ru0.1O3/Al2O3 was prepared according to the incipient wetness impregnation method.38 Typically, citric acid (complexing agent) and ethylene glycol were added to a 100 mL metal solution with a molar ratio of citric acid (CA):total metal ions (La, Ni, Ru):ethylene glycol (EG) of 1.2:1:0.48 under stirring. The metal nitrates with the molar ratio of La:(Ni + Ru) of 1:1 and Ni:Ru molar ratio of 0.9:0.1 were dissolved in deionized water. The solution was then heated to 80 °C and maintained under vigorous stirring for 15 min. Afterwards, the solution was cooled down to room temperature. Next 3 g of commercial medium porous γ-alumina (PURALOX SBa Series, Sasol) was added as a support to the above mixture with stirring overnight. Then the resulting sample was dried at 80 °C for 6 h and 120 °C for 12 h in the air. In a furnace, the dried sample was calcined in static air at 350 °C for 2 h and 700 °C for 5 h at a 2 °C min−1 rate and then ground to fine powder.
2.1.2 LaNiRuO3 bulk perovskite.
LaNi0.9Ru0.1O3 perovskite was prepared according to the protocol described above without alumina to understand the effect of alumina on the catalyst perovskite phase dispersion and activity.
For convenience, from now onwards, the catalysts will be denoted as LaNiRuO3 and LaNiRuO3/Al2O3 for the LaNi0.9Ru0.1O3 and LaNi0.9Ru0.1O3/Al2O3 compositions, respectively. To exsolve the active metals onto the surface of the bulk perovskite and supported perovskite, both samples were reduced under 10% H2/Ar (30 cm3 min−1) at 800 °C for 1 h to produce the exsolution derivatives of LaNiRuO3 and LaNiRuO3/Al2O3.
2.2. Materials characterization
The crystallinity of the prepared perovskite catalysts was measured using X-ray diffraction (Bruker D2 Phaser) with a scanning range of 10–90° and a rate of 0.05°/0.5 s. The crystallite size was estimated using the Scherrer equation D = 0.94λ/βcosθ, where λ is the Cu Kα X-ray wavelength, β is the full width at half maximum, and θ is the diffraction angle. Morphological and elemental analyses of the catalysts were performed by field-emission scanning electron microscopy (JEOL JSM-7610F) coupled with energy dispersive X-ray spectroscopy (FESEM-EDS). Prior to scanning, the catalysts were mounted onto aluminum stubs with a carbon tape. Then the catalysts were sputter-coated with a thin layer of Au/Pd to prevent the charging effect. High-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) and element mapping were performed utilizing an electron microscope (Titan Themis Z) equipped with a spherical aberration corrector for the probe, and a high throughput (solid angle ∼ 2.4 sr) energy dispersive spectroscopy (EDS) detector. The elemental maps were generated with the post-processing of the STEM-EDS spectrum imaging datasets. Gatan software was utilized to determine the Ni particle size from the HRTEM images. Raman spectroscopy (Horiba Jobin Yvon instrument, red laser (λ = 633 nm) and 50× objective lens) was used to complement the XRD results and provide a deeper insight into the complex lattice substructure. N2 adsorption–desorption measurements were carried out on a 3Flex, Micromeritics at 77 K. The catalysts were degassed under vacuum at 150 °C overnight to remove the weakly adsorbed gas species. The Brunauer–Emmett–Teller (BET) equation was used to calculate the specific surface area (m2 g−1) in the relative pressure range from 0.05 to 0.3. The pore size distributions were derived from the BJH model (desorption section). To determine the reducibility of the different species, H2 temperature-programmed reduction (H2-TPR) experiments were carried out with a Micromeritics AutoChem II 2920 instrument. Here, 90 mg of sample was pretreated with 20% O2/He at 300 °C for 40 min to remove any moisture and clean the surface. Then the sample's reducibility was recorded from room temperature to 800 °C for 10 min under 10% H2/Ar (30 cm3 min−1) with a heating rate of 10 °C min−1 to exsolve Ni–Ru to the perovskite's surface. Using the same instrument, H2 temperature-programmed desorption (H2-TPD) experiments were conducted and surface dispersion of the Ni/Ru metals through H2 chemisorption was estimated using the formula D (%) = 2Vad × 62.93/Mtot as per the literature protocol.40 We considered the atomic weight of the metal as 62.93 mg mmol−1 due to the presence of 90% Ni and 10% Ru in the total catalyst. In the above formula, Vad is the H2 uptake in mmol per gram of catalyst, and Mtot is the total weight percentage of Ni and Ru. About 80 mg of sample was packed into quartz wool in a quartz U-tube and exsolved at 800 °C under a 10% H2/Ar flow (30 cm3 min−1) for 60 min. The sample was purged with Ar (30 cm3 min−1) for 10 min at 800 °C and then continued to cool down to room temperature. Then, the flow was switched to 10% H2/Ar with a total flow of 30 cm3 min−1 to adsorb hydrogen for 30 min. Subsequently, using the same flow rate, the sample was purged with Ar for 40 min to remove weakly adsorbed gases. The H2 desorption signal was recorded as the temperature was raised to 800 °C, then calibrated, and the hydrogen uptake was estimated in mmol g−1. For ICP analysis of the samples, a Thermo Scientific, iCAP™ 7600 ICP-OES system was used to determine the metal content. The instrument was equipped with a Teledyne ASX-560 Autosampler (CETAC Technologies), concentric glass nebulizer, glass cyclonic spray chamber, semi-demountable EMT torch, and 2 mm bore quartz center tube (Duo). The instrumental parameters were: RF power 1.15 kW, nebulizer gas flow 0.5 L min−1, auxiliary gas flow 0.5 L min−1, plasma gas flow 12 L min−1, and sample peristaltic pump 50 rpm. The axial mode of viewing was used in the measurements. Prior to ICP analysis, the samples were digested in a HCl–HNO3 mixture (6:1, v/v) using a microwave oven. Commercial stock solutions of Étalons multi-elements, 100 mg per L Ru in HCl 15%, 10 mg per L La, and 10 mg per L Ni in HNO3 2% were used for calibration of the ICP-OES.
2.3. CO2 methanation catalytic studies
The catalytic performance was carried out in a 0.9 cm fixed-bed tubular microreactor using a H2 to CO2 molar ratio of 4 balanced with He at a total flow rate of 100 mL min−1. Prior to the CO2 methanation reaction, 0.24 g of catalyst was reduced in situ in 10% H2/He (30 cm3 min−1) at 800 °C for 120 min at 1 bar. Utilizing a weight hourly space velocity (WHSV) of 25000 mL g−1 h−1 at 200–500 °C with 50 °C increments, the catalytic activity was recorded every 30 and 60 min per temperature increment, and their average was considered in the calculation using eqn (2)–(6). The composition of the effluent mixed gases was analyzed by a gas chromatography system (Thermo Fisher Scientific) equipped with both a thermal conductivity detector (TCD) and a flame ionization detector (FID). The CO2 conversion, CO and CH4 selectivities, and CO and CH4 yields were investigated. Catalytic stability was evaluated for 54 h time on-stream (TOS) at 400 °C with the same H2 to CO2 ratio and WHSV of 25000 mL g−1 h−1. The catalytic performance was evaluated every hour and determined according to the following equations (eqn (2)–(6)):| | 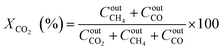 | (2) |
| | 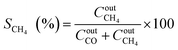 | (3) |
| | 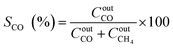 | (4) |
| | 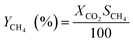 | (5) |
| | 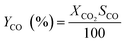 | (6) |
where Couti represents the concentration (e.g., mole fraction) of component i in the reactor's outlet gas flow.
3. Results and discussion
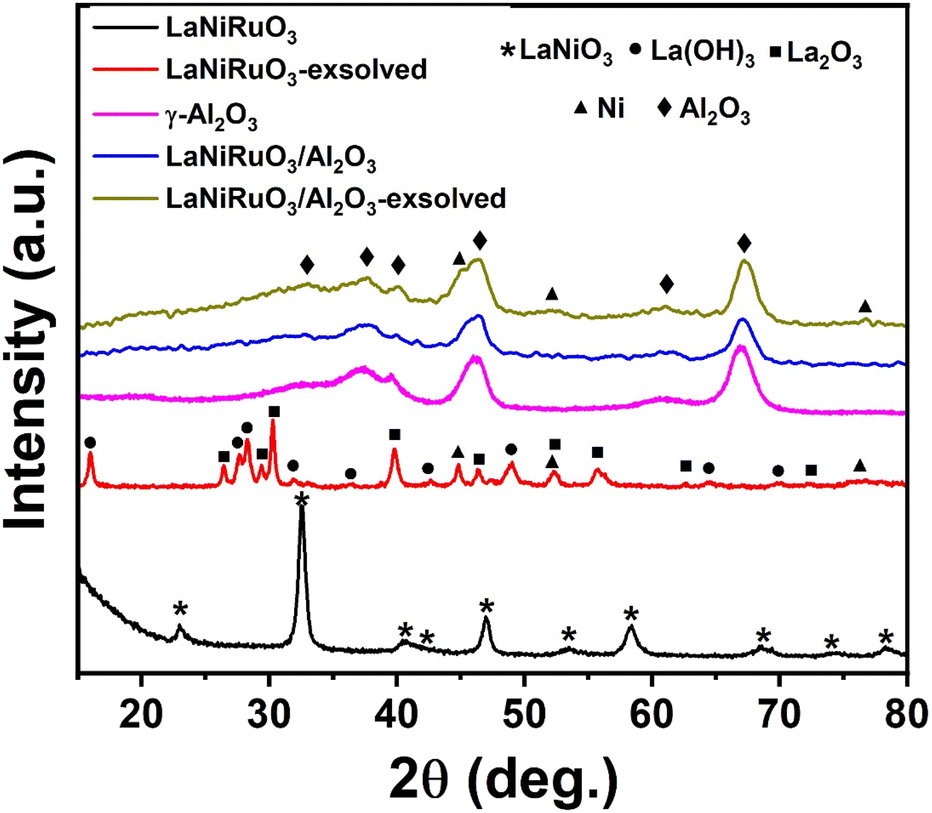
3.1. Structural analyses
The structural characterizations of the as-synthesized bulk perovskite (calcined at 700 °C for 5 h) and exsolved LaNiRuO3 (following reduction under 10% H2/Ar at 800 °C for 60 min) as well as the LaNiRuO3/Al2O3 perovskite derivatives were carried out using XRD; the results are portrayed in Fig. 1. The XRD diffractograms of the as-synthesized bulk LaNiRuO3 sample showed intense peaks at 22.9°, 32.5°, 46.9°, and 58.4°, corresponding to the characteristic peaks of the rhombohedral LaNiO3 perovskite phase (parental perovskite).41 In the case of the calcined/as-synthesized LaNiRuO3/Al2O3, the XRD pattern exhibited an intermediate diffractogram compared to that of the bulk perovskite and the support, with a strong presence of the Al2O3 support phase peaks. This suggests the existence of both phases (perovskite and alumina) in the supported perovskite structure.
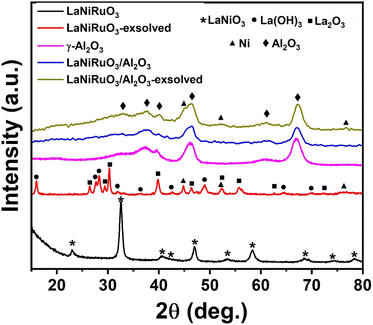 |
| | Fig. 1 XRD diffractograms of the as-synthesized, and exsolved perovskite-derived LaNiRuO3 and LaNiRuO3/Al2O3 catalysts. | |
The reflections of the exsolved LaNiRuO3 showed sharp intense peaks corresponding to La(OH)3 (JCPDS 13-1481),42,43 along with other peaks belonging to La2O3 and Ni phases.38,44,45 However, in the exsolved LaNiRuO3/Al2O3 catalyst, it was observed that the peaks belonging to lanthanum (La(OH)3 and La2O3) may have been masked when the perovskite structure was grown over γ-alumina due to the presence of wide peaks of the alumina phase.46 The formation of the La(OH)3 phase (eqn (7)) in the exsolved perovskite-derivative catalysts could be due to the in situ reaction between La2O3 and H2O molecules formed during the reduction process. A similar kind of behavior was observed in the LaNiO3 perovskites (partially substituting the B-site by Fe and Mn) after reduction.29
| | 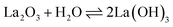 | (7) |
It is worth mentioning that exsolved Ni peaks (which existed in the FCC structure) were inevitably visible after exsolution from the supported perovskite. On the other hand, a slight shift toward the lower-angle side in the Ni peaks was observed in both catalysts post exsolution. This was attributed to the incorporation of larger-sized Ru atoms exsolved from the B-site of the perovskite structure into the Ni lattice, causing a decrease in d-spacing and potentially leading to the formation of a Ni–Ru bimetallic alloy.47 The Ni crystallite size (assuming the Ni–Ru alloy was a Ni single metal for the crystallite size and dispersion calculations) was estimated using the Scherrer equation D = 0.94λ/βcosθ. It was found to be 17.3 nm for the exsolved LaNiRuO3 catalyst but proved challenging to measure for the exsolved LaNiRuO3/Al2O3 catalyst due to the overlap with alumina peaks (though it could be estimated from the HRTEM and H2-TPD analyses). Here, the Tamman temperatures of Ni and Ru had to be mentioned, i.e., at 590 °C and 1089 °C, while the Hüttig temperatures were 245 °C and 544 °C, respectively. Both parameters are very important as they describe the mobility of atoms in the bulk and surface, respectively. So, in this particular case, it was expected that some surface layers of the Ni–Ru bimetallic alloy would have been formed under the exsolution reducing conditions (10% H2/Ar at 800 °C for 60 min).
Further, the morphology of the exsolved catalysts was investigated using scanning electron microscopy (SEM) and high-resolution transmission electron microscopy (HRTEM). The SEM micrographs for both catalysts (Fig. 2a and 3a) showed agglomerates with no particular morphology pattern. The STEM-HAADF and element mapping images of the exsolution-derived catalysts (reduction under 10% H2/Ar at 800 °C for 60 min) revealed large agglomerates in the bulk LaNiRuO3 and relatively smaller particles in the LaNiRuO3/Al2O3-supported catalyst, Fig. 2 and 3, respectively. Furthermore, the distribution of Ni particle sizes was estimated based on the size of at least 20 particles, as identified in Fig. 2j and 3k.
 |
| | Fig. 2 SEM micrograph (a) and STEM-HAADF images along with the corresponding EDS maps (b–j) obtained for the exsolved (10% H2/Ar at 800 °C for 60 min) bulk LaNiRuO3 catalyst. | |
 |
| | Fig. 3 SEM micrograph (a) and STEM-HAADF images and corresponding EDS maps (b–k) obtained for the exsolved (10% H2/Ar at 800 °C for 60 min) supported LaNiRuO3/Al2O3 catalyst. | |
Overall, a consistent trend emerged, with La (green color), Ni (blue color), Ru (yellow color), O (red color), and Al (cyan color) elements exhibiting a homogeneous distribution across all the analyzed areas. Moreover, the Ni nanoparticles were clearly uniformly distributed on the surfaces of La and/or Al surface, regardless of the support presence or not. However, the exsolved bulk LaNiRuO3 perovskite (Fig. 2f, h and j) exhibited a more heterogeneous and larger Ni particle size of approximately 45 nm compared to the alumina-supported one of ∼7 nm (Fig. 3f, g, i and k). It is worth noting that the crystallite size of Ni was found to be 17.3 nm for the exsolved LaNiRuO3 catalyst based on the XRD studies. The difference between the two values stems from the fundamental principles of the two techniques and the features probed by each one. In particular, XRD measures the primary crystallite size while TEM measures the particle size, which consists of one or more crystallites, hence the observed difference. The crystallite size measured using XRD was consistent with the results obtained from H2-TPD chemisorption analysis (discussed later, Table 3). It was evidenced that the use of alumina (surface area of 188 m2 g−1) as a support favored the formation of a smaller Ni nanoparticle size of 7 nm. However, due to the extremely low Ru loading (0.717–2.39 wt%), its high atomic dispersion, and/or its embedding into the Ni lattice (as confirmed by XRD analysis, Fig. 1), it was not possible to detect their individual size, which was suspected to be below the detection limit (up to 1 nm) of the TEM instrument. In order to determine the metal content of the samples, both catalysts (LaNiRuO3 and LaNiRuO3/Al2O3) were analyzed by ICP-OES. The analysis results of ICP-OES are provided with nominal values for comparison in the ESI, Table S1.†
The Raman spectra of the LaNiRuO3 and LaNiRuO3/Al2O3 catalysts (calcined and exsolved) obtained using a red laser with an excitation wavelength of 633 nm are shown in Fig. S1a and b,† respectively. This technique provides a deeper insight into the complex lattice substructure of the catalysts and compliments the structural findings from the XRD. Looking at the Raman spectra from 100 to 3000 cm−1 (Fig. S1a†), all the catalysts, except the LaNiRuO3/Al2O3 catalyst, showed an intense peak at 822 cm−1, corresponding to the redox behavior (Ni3+ to Ni4+) due to the addition of Ru to the perovskite structure.48 The peaks at 100–150, 200–400, 450–700 cm−1 corresponded to the vibrations of A-site cation (labeled in blue in Fig. S1b†), B–O vibrations (labeled in yellow in Fig. S1b†), and BO6 octahedra vibrations (labeled in green in Fig. S1b†) in the ABO3 perovskite structures respectively.49 Schober et al. studied the complex physical properties of perovskite LaNiO3 films on the LaAlO3 substrate via Raman spectroscopy.50 The Raman spectra revealed the A1g and Eg peaks of LaNiO3 on LaAlO3. Similarly, Wyss et al. used Raman spectroscopy (λ = 660 nm and 785 nm) to investigate perovskite-type LaNiO3 thin film on a (100) pc-oriented LaAlO3 partially covered by a PAuM.51 The Raman spectra obtained from the areas without porous gold (Au) membranes were consistent with the peaks obtained from LaNiRuO3 and LaNiRuO3/Al2O3 catalysts, as shown in Fig. S1.† The Raman spectra of the LaNiRuO3/Al2O3 catalysts showed peaks corresponding to the LaAlO3 phase, which were not observed with the LaNiRuO3 catalyst. This might be due to the substitution of Ni (B-sites in the ABO3 structure) with Al, coming from γ-Al2O3, during the synthesis. It is noteworthy to mention that in the case of the LaNiRuO3/Al2O3 catalyst (calcined), the presence of γ-Al2O3 masked the peaks of other phases; however, the peaks of the other phases could be noticed with careful analysis of the Raman spectrum. In the case of the exsolved catalysts (100–700 cm−1), La2O3 and La(OH)3 phases were both observed in the Raman spectra (Fig. S1b†), which were labeled with a circle (●) and triangle (▲), respectively. This coincides with the XRD findings of the exsolved perovskite-derived catalysts. The formation of La(OH)3 phase was inferred to be formed from an in situ reaction between La2O3 and H2O molecules formed during the reduction process. The peaks at 126.11, 185.05, 336.19, and 442.52 cm−1 were associated with La2O3 vibrational modes.52 Namely, cubic and hexagonal varieties,53 La–O vibration,54 and Eg ν1 mode,55 respectively. Probably, La2O3 was formed when the Ni was exsolved by treating LaNiO3 perovskite at high temperatures and in a reducing environment into La2NiO4 and La2O3.56 Additionally, the vibrational mode of La(OH)3 with the space group P63/m was observed in the exsolved samples.57 The bands around 282, 340, and 451 cm−1 represented the active vibrational modes of A1g, E2g, and E1g, respectively.58 The first two aforementioned bands correspond to the anion translation behavior, while the latter corresponds to the liberation of –OH dipole.58 It is noteworthy to mention, that the blue shift exhibited by the exsolved samples could be correlated to the exsolved Ni, which changed the chemical bond length of the molecules by inducing defects and oxygen vacancies in the perovskite structure.
3.2. Surface species analysis
The surface of the exsolved catalysts was examined using X-ray photoelectron spectroscopy (XPS) to reveal the nature of the chemical bonding of the different elements as well as their chemical environment. Fig. 4 illustrates the XPS survey spectra and elemental spectra corresponding to La 3d, C 1s, Ru 3d, and O 1s. The binding energies and surface atomic percentages were determined from peak fitting using Avantage software as shown in Table 1.
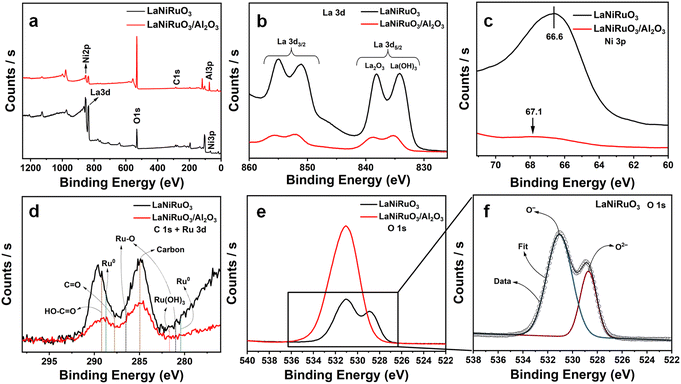 |
| | Fig. 4 (a) Survey spectra, (b) high-resolution XPS spectra of La 3d, (c) Ni 3p, (d) C 1s + Ru 3d, (e) O 1s high-resolution XPS spectra for both exsolved samples for unsupported LaNiRuO3 and supported LaNiRuO3/Al2O3, along with the (f) O 1s deconvoluted spectrum for LaNiRuO3. | |
Table 1 XPS peak positions of the identified components in the high-resolution elemental spectra for the exsolved bulk LaNiRuO3 and supported LaNiRuO3/Al2O3a
| Sample |
Core level |
Peak BE (eV) |
Atomic (%) |
Ni + Ru/La |
|
Ru: it was not possible to trace this due to the low amount of loading.
|
| LaNiRuO3 |
La 3d5 |
834.4 |
70.6 |
0.034 |
| C 1s |
285.0 |
3.7 |
| Ni 3p |
66.5 |
2.4 |
| O 1s |
530.9 |
23.3 |
| LaNiRuO3/Al2O3 |
La 3d5 |
835.7 |
15.9 |
0.006 |
| C 1s |
285.0 |
2.1 |
| Ni 3p |
66.7 |
0.1 |
| O 1s |
531.1 |
58.9 |
| Al 2p |
74.2 |
21.8 |
The survey spectra in Fig. 4a showed La, Ni, Al, and O elemental peaks in the respective catalysts. The Ni and La peaks were dominant in the case of the LaNiO3 catalyst, while the same peaks were overshadowed by Al due to the existence of 70% Al2O3 support in the LaNiRuO3/Al2O3 catalyst. In Fig. 4b, two typical La 3d doublet peaks, i.e., La 3d3/2 and La 3d5/2,59,60 could be observed, corresponding to the presence of La3+ ions in the oxide environment of La2O3 at 838.2 eV and La(OH)3 at 834.2 eV for both perovskites. However, there was a higher quantity observed in the LaNiRuO3 surface, as indicated by the intensity of the spectrum and the atomic percentage (at%) values (Table 1). Fig. 4c shows that the Ni 3p peak position was shifted to a higher binding energy in the LaNiRuO3/Al2O3 catalyst compared to that of the LaNiRuO3 catalyst. This proves that the exsolved Ni/Ru atoms had a strong interaction and better dispersion (Table 3) with the support in the Al2O3-supported catalyst compared to pristine perovskite catalyst during the reduction process. This kind of interaction is highly desirable for improving the catalytic performance. Fig. 4d shows the Ru core level spectra in both perovskites. It is known that the Ru 3d overlaps with the C 1s spectra, therefore requiring a meticulous deconvolution to untangle the contributions from Ru and C. According to the literature, metallic ruthenium and its oxide forms, Ru3+ and Ru2+, were identified.61–63 The low/minute loading of Ru (2.4 wt% in LaNiRuO3 and 0.72 wt% in LaNiRuO3/Al2O3), coupled with the tangled Ru/C photoelectron spectra and the inherent limitations of the TEM and XPS techniques, posed challenges in accurately quantifying Ru in both the catalysts. The high-resolution C 1s XPS spectra were resolved into two distinct components identified as C–C (285.0 eV) and –COOH (289.2 eV), respectively.61 The O 1s spectrum revealed a peak split in the LaNiRuO3 perovskite, as shown in the O 1s deconvolution in Fig. 4f. This split corresponded to two different oxygen environments: more surface-adsorbed oxygen (O−) at a higher binding energy (BE = 531.1 eV) and lattice oxygen (O2−) at 529.0 eV.60 In contrast, the alumina-supported perovskite (LaNiRuO3/Al2O3, Fig. 4e) was predominantly characterized by surface-adsorbed oxygen (ex. OH, CO32−), suggesting an enhanced contribution of the surface –OH groups from alumina surface-adsorbed oxygen species in the spectra.
3.3. Physisorption and chemisorption studies
3.3.1 N2 physisorption.
The analysis of the main textural properties (Table 2) of the as-synthesized perovskites was conducted using isothermal N2-adsorption at 77 K. According to the IUPAC classification, both perovskite-based catalysts exhibited characteristic features of type IV isotherms (Fig. 5), associated with a hysteresis loop due to capillary condensation in the mesopores.64,65Table 2 summarizes the textural properties of the as-synthesized perovskites. The specific surface areas of LaNiRuO3 and LaNiRuO3/Al2O3 were 4 and 141 m2 g−1, respectively, with the former displaying a smaller pore volume and larger pore size compared to the latter, as clearly shown in Fig. 5b. The high SBET and Vp of the alumina-supported sample contributed to the exsolution of smaller Ni nanoparticles, as indicated in the HRTEM analysis (Fig. 3).65
Table 2 Specific surface areas (SBET), pore volumes (Vp), and pore sizes for the as-synthesized LaNiRuO3 and LaNiRuO3/Al2O3 perovskite catalysts
| Sample |
S
BET (m2 g−1) |
V
p (cm3 g−1) |
Pore sizea (nm) |
|
Desorption average pore diameter (4 V A−1 by BET).
|
| LaNiRuO3 |
4 |
0.051 |
48.3 |
| LaNiRuO3/Al2O3 |
141 |
0.37 |
10.6 |
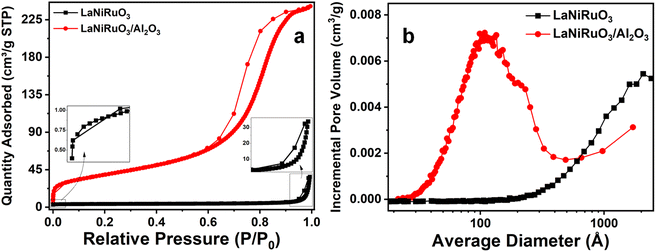 |
| | Fig. 5 (a) N2 adsorption–desorption isotherms and (b) pore-size distribution curves of the LaNiRuO3 and LaNiRuO3/Al2O3 perovskites. | |
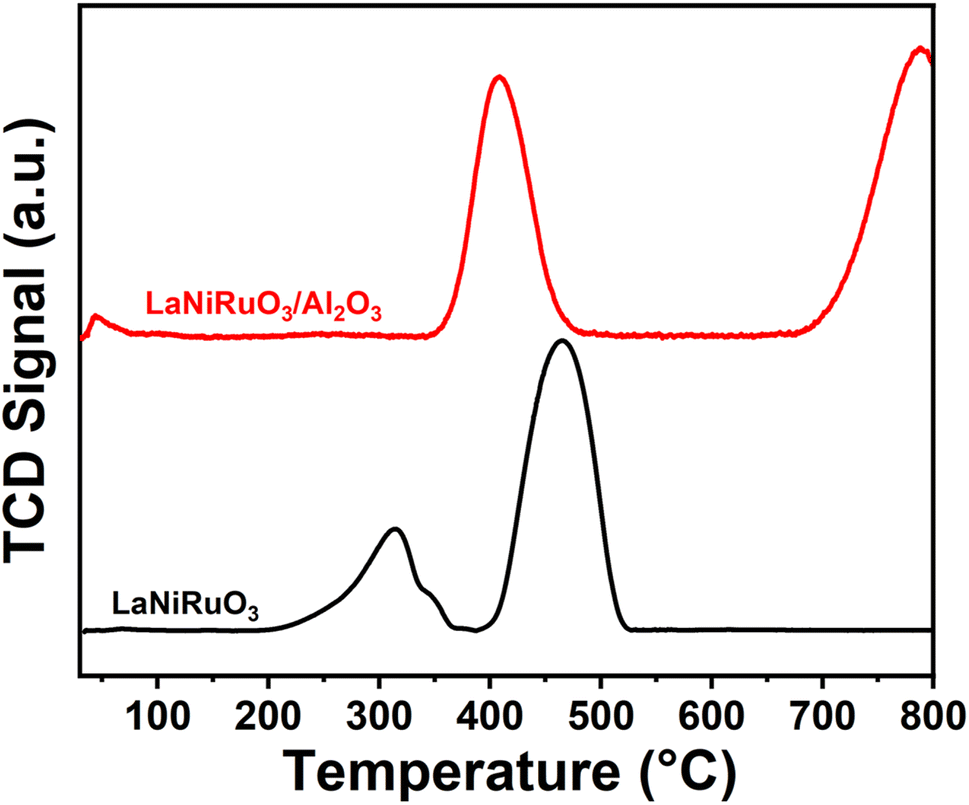
3.3.2 Redox behavior.
The positions of the peaks in temperature-programmed reduction under hydrogen (H2-TPR) have been reported previously to be influenced by oxygen defects, the crystallite size, bonding strength, transition metal state/location, and doping level of the perovskites.66,67 The reducibility of the fresh catalysts was obtained by performing H2-TPR experiments from room temperature to 800 °C (Fig. 6). A small reduction peak observed below 100 °C in both the perovskites studied indicated the reduction of Ru present in the structure. The TPR profile of bulk LaNiRuO3 perovskite displayed two main peaks centered at around 320 °C and 465 °C, which were ascribed to the reduction of LaNiRuO3 and the intermediate phase La2NiO4 (formed during the reduction process) according to eqn (8)–(10).68 The reduction temperatures were lower compared to the bulk LaNiO3 structure.68 This was due to the fact that the presence of Ru in the B-site causes a weakening of the Ni–O bonds in the perovskite structure and hence the structure can be reduced at lower temperatures.| | | 4LaNiO3 + 2H2 → La4Ni3O10 + Ni + 2H2O | (8) |
| | | La4Ni3O10 + 3H2 → La2NiO4 + 2Ni + La2O3 + 2H2O | (9) |
| | | La2NiO4 + 2H2 → Ni + La2O3 + H2O | (10) |
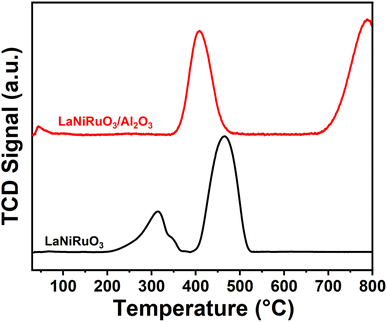 |
| | Fig. 6 H2-TPR profiles of the LaNiRuO3 and LaNiRuO3/Al2O3 perovskites. | |
On the other hand, the LaNiRuO3/Al2O3 perovskite displayed two peaks centered at ∼400 °C and a second peak above 750 °C, respectively. The former was due to the reduction of Ru3+ to Ru2+ and/or Ni3+ to Ni2+ in the LaNiO3 (Ni3+) phase (as detected by XRD, Fig. 1). According to the literature, NiO and RuO2 reduction takes place around 350 °C and 200 °C, respectively. Deviations from this reduction temperatures demonstrate the M–O bond in the perovskite structure. The second peak in the case of the H2-TPR of the LaNiRuO3/Al2O3 catalyst corresponded to the reduction of the highly stable NiAl2O4 phase formed during calcination, since neither Al2O3 nor La–Al2O3 supports are reducible at this temperature.65 These results suggest that there was a strong interaction observed between Ni and Al2O3 phase during the reduction process and hence the observed better dispersion (18%, Table 3) with the small Ni particle size (7 nm, Table 3 and Fig. 3k).
Table 3 H2-TPD adsorption quantities, Ni dispersion, and Ni particle size
| Catalyst |
H2 consumption (mmol g−1) |
Dispersion (%) |
Ni size (nm) |
|
Ni particle size detected using HRTEM.
Ni crystallite size determined using Scherrer's formula.
|
| LaNiRuO3 |
0.1238 |
6 |
16 (45a, 17.3b) |
| LaNiRuO3/Al2O3 |
0.1097 |
18 |
6 (7a) |
3.3.3 H2 chemisorption studies.
Complementary to the Ni crystallite size and the Ni particle size obtained through the XRD studies (using Scherrer's equation) and HRTEM, respectively, the nanoparticles size was estimated by measuring the hydrogen uptake during H2 chemisorption (H2-TPD) measurements (Fig. 7). The three peaks (α, β, and γ) in the 25–800 °C range were considered for analysis, where the α peak corresponded to the weakly adsorbed hydrogen species, and β and γ peaks to the chemisorbed H atoms with different coordination environments.69 Overall H2 uptakes of 0.124 mmol g−1 and 0.110 mmol g−1 were observed for LaNiRuO3 and LaNiRuO3/Al2O3 catalysts, respectively. The dispersion of Ni in the LaNiRuO3 (6.1%) perovskite was three times less than it was for LaNiRuO3/Al2O3 (18.0%), as shown in Table 3. Regarding the Ni particle size, it is worth mentioning that the Ni particle size derived from H2-TPD (6 nm) was in good agreement with the one derived from the HRTEM and EDS analyses (7 nm, Fig. 3k), which depicts the same improved dispersion of Ni and the smaller NPs in the exsolved LaNiRuO3/Al2O3 perovskite catalyst. However, the Ni size (16 nm) derived from the H2-TPD chemisorption results over the exsolved bulk LaNiRuO3 catalyst was not aligned with the HRTEM and EDS analyses, which showed a much larger Ni particle (45 nm); XRD studies for the latter gave a primary crystallite size of 17.3 nm. This has to do with the intrinsic differences in the technique's principles.
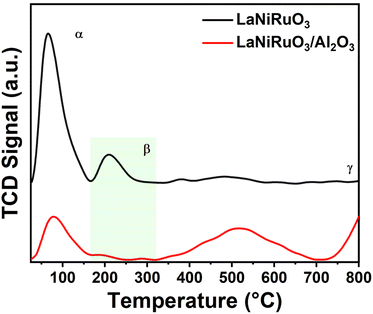 |
| | Fig. 7 H2-TPD profiles of the reduced LaNiRuO3 and LaNiRuO3/Al2O3 perovskites. | |
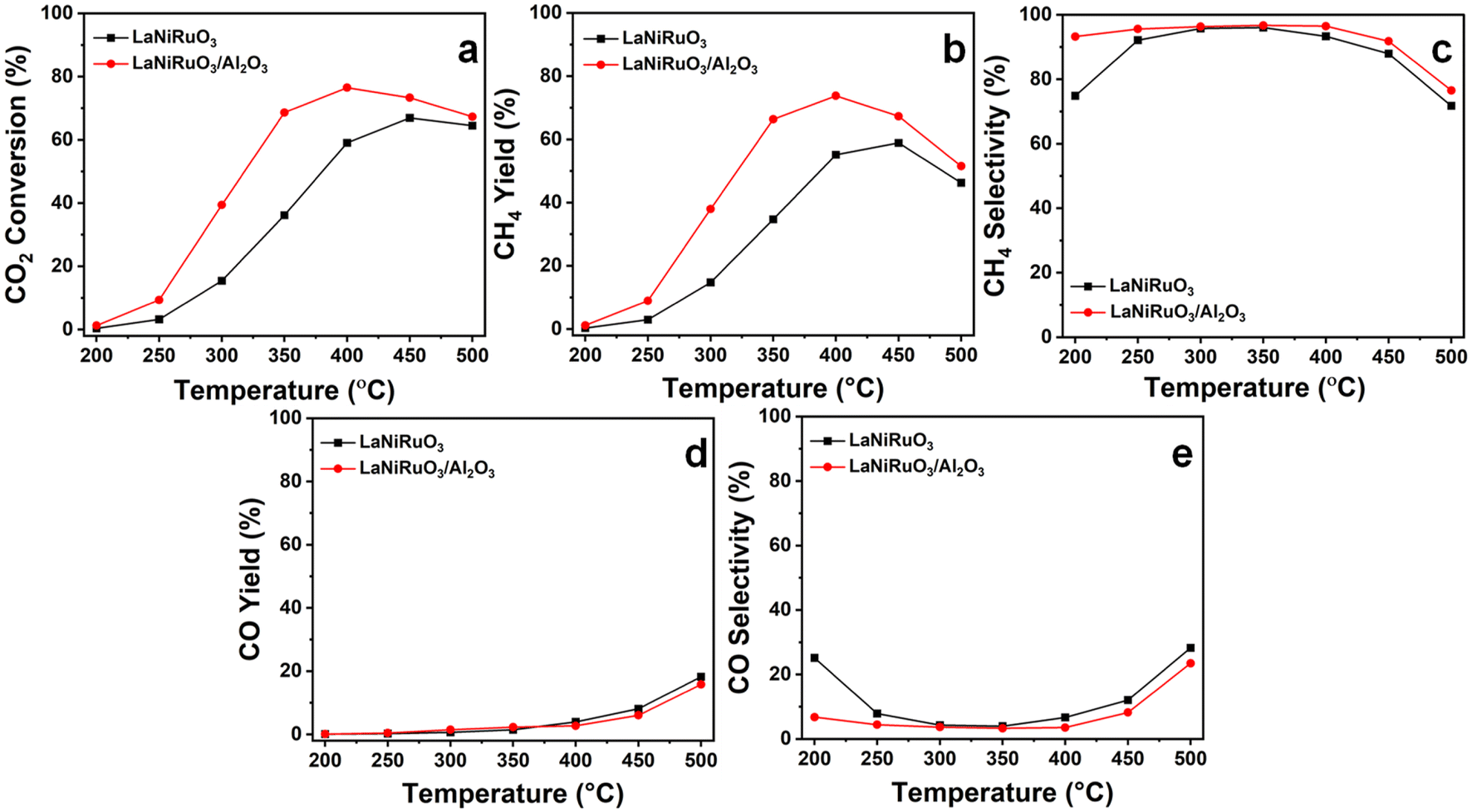
3.4. CO2 methanation activity and stability tests
Fig. 8 demonstrates the catalytic performance of the exsolved LaNiRuO3 and LaNiRuO3/Al2O3 catalysts at temperatures ranging from 200 °C to 500 °C under the following reaction conditions: WHSV = 25000 mL g−1 h−1, H2:CO2 = 4, and P = 1 bar. As illustrated in Fig. 8a, the CO2 conversion increased steadily with the temperature from 200 °C to 450 °C for the LaNiRuO3 catalyst with an optimum of 67% at 450 °C; a same trend was observed for the LaNiRuO3/Al2O3 catalyst in the 200 °C to 400 °C temperature range with an optimum conversion of 77% at 400 °C. The methane yield (Fig. 8b) was found to increase with supporting the LaNiRuO3 on alumina from 59% to 74% with a three-fold less CO yield and CO selectivity (Fig. 8d and e). Both the catalysts demonstrated a methane selectivity of ≥92% (Fig. 8c) between 250 °C and 400 °C, and 200 °C to 450 °C for LaNiRuO3 and LaNiRuO3/Al2O3, respectively.
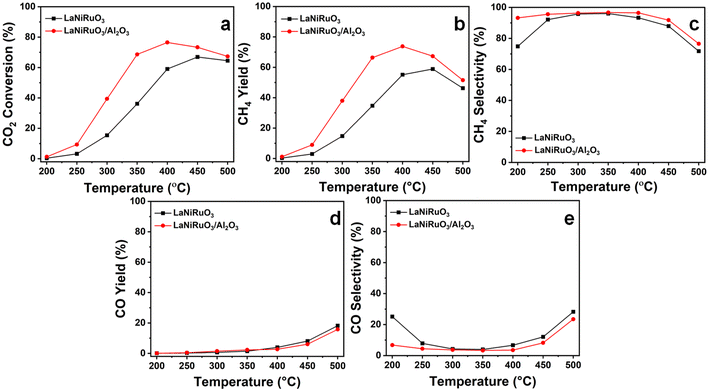 |
| | Fig. 8 Catalytic performance of the exsolved LaNiRuO3 and LaNiRuO3/Al2O3 catalysts at various temperatures: (a) CO2 conversion, (b) CH4 yield, (c) CH4 selectivity, (d) CO yield, and (e) CO selectivity. Reaction conditions: WHSV = 25000 mL g−1 h−1; H2:CO2 molar ratio = 4; P = 1 bar. | |
The structural sensitivity for the CO2 methanation activity has been an important aspect since the invention of the reaction. Some research has shown structural insensitivity with different Ni loadings in NiAl2O3 catalysts, while some reports have clearly shown the effects of the Ni particle size, metal–support interactions, and the basic nature of the support in the catalysts.70,71 For example, Vogt el al. showed the effect of the Ni particle size on the CO2 methanation activity of Ni/SiO2 catalysts in the range of 1–7 nm.72 Similarly, Blanco et al. studied the effect of the particle size on the CO2 reduction reaction by increasing the doping content of Ni on perovskite-based catalysts and achieved the highest catalytic activity with the samples containing Ni crystallites under 10 nm.73 In the present study, the superior performance of LaNiRuO3/Al2O3 could be attributed to the small size and better dispersion of Ni particles, as proven by HRTEM (7 nm) and H2-TPD (6 nm), and the abundance of active surface sites. Also, the amphoteric nature of Al2O3 might have provided more basic sites, which favors CO2 adsorption and H2 dissociation on active catalytic sites. In addition, further increasing the reaction temperature above 400 °C was observed to decrease the methane selectivity, probably due to the promotion of the endothermic RWGS reaction (eqn (11)) and the formation of CO, as described in eqn (11).65
| | 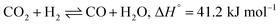 | (11) |
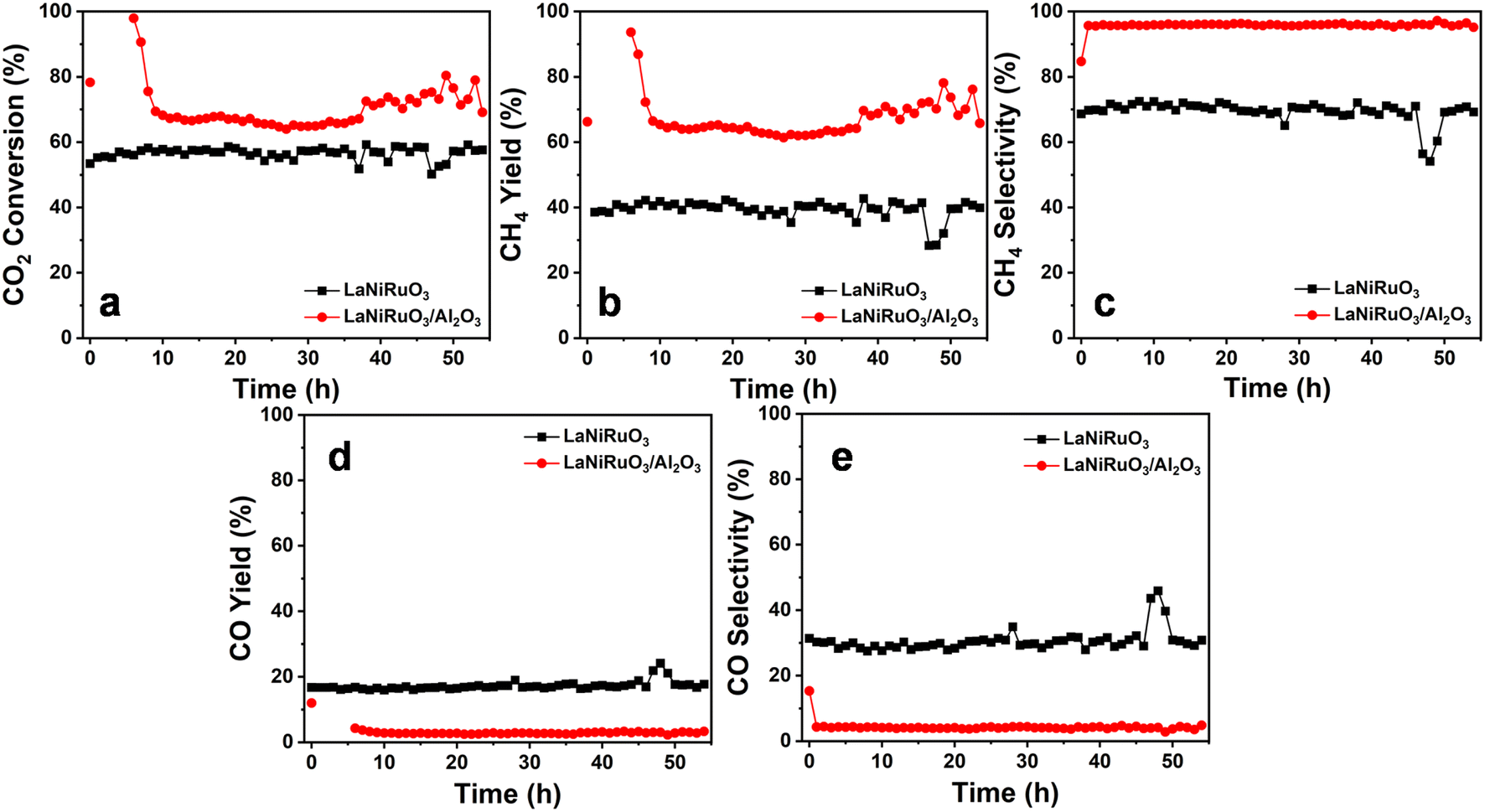
The study of catalyst stability under CO2 methanation is essential for industry applications, whereby the catalyst resistance toward sintering and/or oxidation of the metallic sites and possible deactivation is evaluated. Therefore, a 54 h stability test under a stream reaction was carried out at 400 °C (the temperature was chosen based on the catalytic activity profile) by monitoring the CO2 conversion, as well as both the CO and CH4 selectivity and yield (activity descriptor). The catalytic stability results are shown in Fig. 9. The CO2 conversions illustrated in Fig. 9a, for the LaNiRuO3 and LaNiRuO3/Al2O3 catalysts were found to be 69% and 78% in the first 10 min, respectively. On average, the catalytic performance demonstrated the good stability in the conversion of CO2 and CH4 selectivity with few anomalies for 40 h on stream. It could also be observed that the alumina support facilitated better CO2 conversions with a high CH4 yield and low CO yield, as indicated in Table 4. To study the sintering of the metal phase and the coke formation following the TOS experiments, the spent catalysts were characterized using XRD and Raman spectroscopy. From the XRD patterns of the spent catalysts (Fig. S2†), it could be seen that the intensity of the diffraction peaks related to the Ni phase did not significantly increase. The Ni crystallite size in the case of the LaNiRuO3 catalyst (calculated using Scherrer's equation) was found to be around 19 nm, which was slightly larger than that of the fresh catalyst (17.3 nm). However, the particle size of Ni in the case of the LaNiRuO3/Al2O3 catalyst could not be accurately calculated due to the overlapping of the Al2O3 diffraction peaks with the Ni phase. It is important to note that the minimal increase in Ni particle size after the TOS experiments indicated good stability against sintering under the studied conditions. The spent catalysts were further studied by Raman spectroscopy. The intensity ratio of the D and G bands (ID/IG) is typically used as a descriptor to provide information on the nature of carbon formed on the catalyst surface. The D and G bands at ∼1331 and 1583 cm−1 corresponded to the amorphous carbon or the disordered graphite in the multi-walled carbon nanotubes, and the sp2 hybridization, which was related to the ordered carbon atoms in hexagonal sheets.74 However, the prominent D and G bands could not be observed for the spent catalysts, indicating the minimal coke formation. Due to the complexity of the structure and overlapping of the bands, as shown in Fig. S2c,† it was difficult to accurately compare the nature of the carbon formed on the surface of the different catalysts. Overall, concerning the confinement of the Ni exsolved particles, in the case of the supported system, it can be stated that the strong metal–support interaction prevented the Ni sintering, which in turn was less prone to metal agglomeration and coke formation, signifying the high activity of the LaNiRuO3/Al2O3 catalyst for the long run of 54 h of the TOS. The results of this work confirm that the better dispersion, small crystallite size of the active Ni-site achieved, and the basic nature of the support promoted the excellent catalytic activity and stability in the exsolved LaNiRuO3/Al2O3 catalyst toward the CO2 methanation reaction.
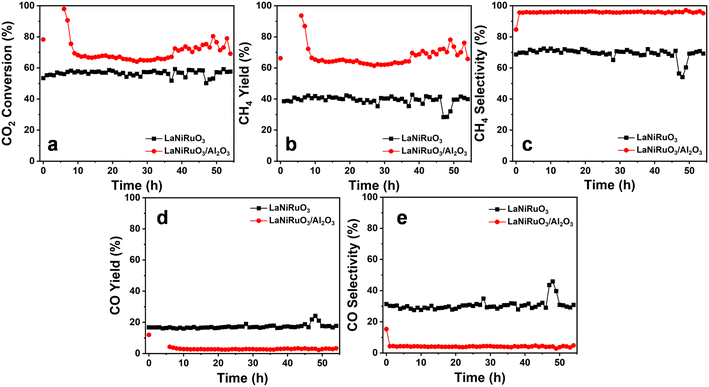 |
| | Fig. 9 Long-term stability tests at 400 °C for the exsolved LaNiRuO3 and LaNiRuO3/Al2O3 catalysts, (a) CO2 conversion, (b) CH4 yield, (c) CH4 selectivity, (d) CO yield, (e) CO selectivity. Reaction conditions: WHSV = 25000 mL g−1 h−1; H2:CO2 molar ratio = 4; P = 1 bar. | |
Table 4 Average catalytic performance at 54 h time on-stream for the methanation reaction at 400 °C
| Catalyst |
Conversion CO2 (%) |
Selectivity CH4 (%) |
Selectivity CO (%) |
CH4 yield (%) |
CO yield (%) |
| LaNiRuO3 |
57 |
69 |
31 |
39 |
17 |
| LaNiRuO3/Al2O3 |
70 |
96 |
4 |
67 |
3 |
4. Conclusions
In this study, we focused on the synthesis of catalysts derived from B-site Ru-substituted LaNiO3 perovskites supported on alumina and their applicability for the CO2 methanation reaction. In particular, catalysts comprising 10% Ni-site substituted by Ru in the LaNiO3 perovskite (LaNi0.9Ru0.1O3) and the same supported on γ-alumina (LaNi0.9Ru0.1O3/Al2O3) were prepared by a wetness impregnation method using citric acid and ethylene glycol. The structural, morphological, textural, surface, and chemisorption properties were studied for the as-synthesized and exsolved perovskites. XRD analysis revealed the successful formation of the perovskite phase in LaNi0.9Ru0.1O3 and a complete overlapping of the Al2O3 phase in the case of the as-synthesized supported perovskite. The exsolved catalysts exhibited Ni–Ru bimetallic alloy formation after reduction. The HADAAF-STEM and H2-TPD analyses confirmed that the Ni nanoparticles exsolved from the supported catalyst showed 3 times better dispersion (18%) with almost a half particle size (7 nm) compared to that of the pristine perovskite-derived catalyst. The Al2O3 support promoted the formation of smaller-sized Ni nanoparticles with better dispersion. The H2-TPR profiles suggested the reduction of the perovskite phase in the case of LaNi0.9Ru0.1O3 and promoted strong perovskite–support interaction by forming the NiAl2O4 phase during the reduction process in the supported perovskite. Further, the exsolved LaNi0.9Ru0.1O3/Al2O3 catalyst exhibited better catalytic activity, with a CO2 conversion rate of 77% and 96% CH4 yield at 400 °C; while the exsolved pristine perovskite showed a 67% CO2 conversion rate with a 92% CH4 yield at 450 °C. The supported catalyst not only showed better CO2 conversion and CH4 yields at lower temperatures, it also exhibited a 3-fold less CO yield and selectivity than that of the pristine catalyst. The superior catalytic performance of the supported catalyst could be attributed to the better dispersion and small-sized nanoparticles formed during the reduction process, which can avail more catalytic active sites. Also, the amphoteric nature of the Al2O3 support might have provided more basic sites and helped achieve better CO2 adsorption and H2 dissociation. Furthermore, both catalysts exhibited highly stable catalytic activity for a period of 54 h time on stream performed at 400 °C. Overall, the LaNi0.9Ru0.1O3/Al2O3 perovskite-derived catalyst is a promising candidate exhibiting high and stable CO2 methanation activity for extended periods under a wide range of catalytic conditions.
Data availability
The data supporting this manuscript are available from the corresponding author upon request.
Conflicts of interest
Authors declare no conflict of interest.
Acknowledgements
The authors acknowledge funding of this project by Khalifa University of Science and Technology through the grants RC2-2018-024. This research is also supported by ASPIRE, the technology program management pillar of Abu Dhabi's Advanced Technology Research Council (ATRC), via the ASPIRE Virtual Research Institute Award No. VRI20-07.
References
- S. Wang, G. Li and C. Fang, Renewable Sustainable Energy Rev., 2018, 81, 2144–2159 CrossRef.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC.
- A. Porta, R. Matarrese, C. G. Visconti, L. Castoldi and L. Lietti, Ind. Eng. Chem. Res., 2021, 60, 6706–6718 CrossRef CAS.
- G. I. Siakavelas, N. D. Charisiou, S. AlKhoori, A. A. AlKhoori, V. Sebastian, S. J. Hinder, M. A. Baker, I. V. Yentekakis, K. Polychronopoulou and M. A. Goula, Appl. Catal., B, 2021, 282, 119562 CrossRef CAS.
- G. I. Siakavelas, N. D. Charisiou, A. AlKhoori, S. AlKhoori, V. Sebastian, S. J. Hinder, M. A. Baker, I. V. Yentekakis, K. Polychronopoulou and M. A. Goula, J. CO2 Util., 2021, 51, 101618 CrossRef CAS.
- A. I. Tsiotsias, N. D. Charisiou, A. AlKhoori, S. Gaber, V. Stolojan, V. Sebastian, B. van der Linden, A. Bansode, S. J. Hinder, M. A. Baker, K. Polychronopoulou and M. A. Goula, J. Energy Chem., 2022, 71, 547–561 CrossRef CAS.
- E. P. Komarala, A. A. Alkhoori, X. Zhang, H. M. Cheng and K. Polychronopoulou, J. Energy Chem., 2023, 86, 246–262 CrossRef CAS.
- A. A. Alkhoori, O. Elmutasim, A. A. Dabbawala, M. A. Vasiliades, K. C. Petallidou, A.-H. Emwas, D. H. Anjum, N. Singh, M. A. Baker, N. D. Charisiou, M. A. Goula, A. M. Efstathiou and K. Polychronopoulou, ACS Appl. Energy Mater., 2023, 6, 8550–8571 CrossRef CAS.
- P. J. Lunde, Ind. Eng. Chem. Process Des. Dev., 1974, 13, 226–233 CrossRef CAS.
- Y. Guo, S. Mei, K. Yuan, D. J. Wang, H. C. Liu, C. H. Yan and Y. W. Zhang, ACS Catal., 2018, 8, 6203–6215 CrossRef CAS.
- J. Lin, C. Ma, Q. Wang, Y. Xu, G. Ma, J. Wang, H. Wang, C. Dong, C. Zhang and M. Ding, Appl. Catal., B, 2019, 243, 262–272 CrossRef CAS.
- A. I. Tsiotsias, N. D. Charisiou, I. V. Yentekakis and M. A. Goula, Nanomaterials, 2021, 11, 28 CrossRef CAS PubMed.
- C. S. Chen, C. S. Budi, H. C. Wu, D. Saikia and H. M. Kao, ACS Catal., 2017, 7, 8367–8381 CrossRef CAS.
- T. A. Le, M. S. Kim, S. H. Lee, T. W. Kim and E. D. Park, Catal. Today, 2017, 293–294, 89–96 CrossRef CAS.
- K. Kousi, C. Tang, I. S. Metcalfe, D. Neagu, K. Kousi, S. Metcalfe, C. Tang and D. Neagu, Small, 2021, 17, 2006479 CrossRef CAS PubMed.
- M. Buharon, S. Singh, E. P. Komarala and B. A. Rosen, CrystEngComm, 2018, 20, 6372–6376 RSC.
- J. Zhang, M. R. Gao and J. L. Luo, Chem. Mater., 2020, 32, 5424–5441 CrossRef CAS.
- D. Zubenko, S. Singh and B. A. Rosen, Appl. Catal., B, 2017, 209, 711–719 CrossRef CAS.
- M. Barreau, D. Salusso, J. Zhang, M. Haevecker, D. Teschner, A. Efimenko, F. Bournel, J.-J. Gallet, E. Borfecchia, K. Sobczak, C. Petit and S. Zafeiratos, J. Mater. Chem. A, 2024, 12, 7605–7621 RSC.
- H. S. Lim, G. Kim, Y. Kim, M. Lee, D. Kang, H. Lee and J. W. Lee, Chem. Eng. J., 2021, 412, 127557 CrossRef CAS.
- T. Wang, X. Qian, D. Yue, X. Yan, H. Yamashita and Y. Zhao, Chem. Eng. J., 2020, 398, 125638 CrossRef CAS.
- S. Singh, D. Zubenko and B. A. Rosen, ACS Catal., 2016, 6, 4199–4205 CrossRef CAS.
- M. Lee, H. S. Lim, Y. Kim and J. W. Lee, Energy Convers. Manage., 2020, 207, 112507 CrossRef CAS.
- Y. Nishihata, J. Mizuki, T. Akao, H. Tanaka, M. Uenishi, M. Kimura, T. Okamoto and N. Hamada, Nature, 2002, 418(6894), 164–167 CrossRef CAS PubMed.
- S. Singh, D. Zubenko and B. A. Rosen, ACS Catal., 2016, 6, 4199–4205 CrossRef CAS.
- M. Safdar, N. Shezad, F. Akhtar and H. Arellano-García, RSC Adv., 2024, 14, 20240–20253 RSC.
- C. A. Franchini, W. Aranzaez, A. M. Duarte de Farias, G. Pecchi and M. A. Fraga, Appl. Catal., B, 2014, 147, 193–202 CrossRef CAS.
- T. Maneerung, K. Hidajat and S. Kawi, Int. J. Hydrogen Energy, 2017, 42, 9840–9857 CrossRef CAS.
- E. P. Komarala, I. Komissarov and B. A. Rosen, Catalysts, 2020, 10, 27 CrossRef CAS.
- S. Li, S. Guo, D. Gong, N. Kang, K.-G. Fang and Y. Liu, Int. J. Hydrogen Energy, 2019, 44, 1597–1609 CrossRef CAS.
- J. A. Onrubia-Calvo, B. Pereda-Ayo, J. A. González-Marcos, A. Bueno-López and J. R. González-Velasco, Catal. Sci. Technol., 2021, 11, 6065–6079 RSC.
- J. A. Onrubia-Calvo, A. Bermejo-López, S. Pérez-Vázquez, B. Pereda-Ayo, J. A. González-Marcos and J. R. González-Velasco, Fuel, 2022, 320, 123842 CrossRef CAS.
- L. Zhang, L. Zhang, Y. Sun and B. Jiang, J. Colloid Interface Sci., 2021, 596, 455–467 CrossRef CAS PubMed.
- B. J. Hare, D. Maiti, Y. A. Daza, V. R. Bhethanabotla and J. N. Kuhn, ACS Catal., 2018, 8, 3021–3029 CrossRef CAS.
- T. Li, Z.-Y. Zhang, D.-C. Luo, B.-Y. Xu, R.-J. Zhang, J.-L. Yao, D. Li and T. Xie, Nano Res., 2024, 17, 7945–7956 CrossRef CAS.
- Y. Zhu, Y. Liu, Q. Ai, G. Gao, L. Yuan, Q. Fang, X. Tian, X. Zhang, E. Egap, P. M. Ajayan and J. Lou, ACS Mater. Lett., 2022, 4, 464–471 CrossRef CAS.
- T. Zhang, J. Tian, Y. Zhou, J. Zeng, X. Sun and Z. Gong, Int. J. Hydrogen Energy, 2024, 71, 1081–1089 CrossRef CAS.
- S. Li, D. Gong, H. Tang, Z. Ma, Z. T. Liu and Y. Liu, Chem. Eng. J., 2018, 334, 2167–2178 CrossRef CAS.
- S. Hwang, J. Lee, U. G. Hong, J. H. Baik, D. J. Koh, H. Lim and I. K. Song, J. Ind. Eng. Chem., 2013, 19, 698–703 CrossRef CAS.
- A. I. Tsiotsias, N. D. Charisiou, A. Alkhoori, S. Gaber, V. Sebastian, S. J. Hinder, M. A. Baker, K. Polychronopoulou and M. A. Goula, J. CO2 Util., 2022, 61, 102046 CrossRef CAS.
- S. Li, D. Gong, H. Tang, Z. Ma, Z. T. Liu and Y. Liu, Chem. Eng. J., 2018, 334, 2167–2178 CrossRef CAS.
- A. Singh, V. Palakollu, A. Pandey, S. Kanvah and S. Sharma, RSC Adv., 2016, 6, 103455–103462 RSC.
- A. Bin Yousaf, M. Imran, M. Farooq and P. Kasak, Sci. Rep., 2018, 8(1), 1–9 Search PubMed.
- P. Li, C. Tian, W. Yang, W. Zhao and Z. Lü, Front. Mater. Sci., 2019, 13(3), 277–287 CrossRef.
- Q. Yang, G. Liu and Y. Liu, Ind. Eng. Chem. Res., 2018, 57, 1–17 CrossRef CAS.
- R. Prins, J. Catal., 2020, 392, 336–346 CrossRef CAS.
- G. Chen, S. Desinan, R. Rosei, F. Rosei and D. Ma, Chem.–Eur. J., 2012, 18, 7925–7930 CrossRef CAS PubMed.
- H. Cheraparambil, M. Vega-Paredes, Y. Wang, H. Tüysüz, C. Scheu and C. Weidenthaler, J. Mater. Chem. A, 2024, 12, 5194–5203 RSC.
- J. Shi, X. Liu, F. Zhu, W. Tian, Y. Xia, T. Li, R. Rao, T. Zhang and L. Liu, J. Materiomics, 2022, 8, 719–729 CrossRef.
- A. Schober, J. Fowlie, M. Guennou, M. C. Weber, H. Zhao, J. Íñiguez, M. Gibert, J.-M. Triscone and J. Kreisel, APL Mater., 2020, 8, 061102 CrossRef CAS.
- R. M. Wyss, G. Kewes, P. Marabotti, S. M. Koepfli, K.-P. Schlichting, M. Parzefall, E. Bonvin, M. F. Sarott, M. Trassin, M. Oezkent, C.-H. Lu, K.-P. Gradwohl, T. Perrault, L. Habibova, G. Marcelli, M. Giraldo, J. Vermant, L. Novotny, M. Frimmer, M. C. Weber and S. Heeg, Nat. Commun., 2024, 15, 5236 CrossRef CAS PubMed.
- C. Bilel, R. Jbeli, I. Ben Jemaa, A. Boukhachem, F. Saadallah, M. Amlouk and H. Ezzaouïa, J. Mater. Sci.: Mater. Electron., 2020, 31, 7398–7410 CrossRef CAS.
- A. Orera, G. Larraz and M. L. Sanjuán, J. Eur. Ceram. Soc., 2013, 33, 2103–2110 CrossRef CAS.
- J. Cui and G. A. Hope, J. Spectrosc., 2015, 2015, 1–8 CrossRef CAS.
- J. H. Denning and S. D. Ross, J. Phys. C: Solid State Phys., 1972, 5, 1123–1133 CrossRef CAS.
- P. Cao, P. Tang, M. F. Bekheet, H. Du, L. Yang, L. Haug, A. Gili, B. Bischoff, A. Gurlo, M. Kunz, R. E. Dunin-Borkowski, S. Penner and M. Heggen, J. Phys. Chem. C, 2022, 126, 786–796 CrossRef CAS PubMed.
- S. R. Sanivarapu, J. B. Lawrence and G. Sreedhar, ACS Omega, 2018, 3, 6267–6278 CrossRef CAS PubMed.
- Q. G. Zeng, Z. J. Ding, Z. M. Zhang and Y. Q. Sheng, J. Phys. Chem. C, 2010, 114, 4895–4900 CrossRef CAS.
- C. Ye, R. Wang, H. Wang and F. Jiang, BMC Chem., 2020, 14, 1–13 CrossRef PubMed.
- Y. Jeon, D. H. Park, J. Il Park, S. H. Yoon, I. Mochida, J. H. Choy and Y. G. Shul, Sci. Rep., 2013, 3(1), 1–8 Search PubMed.
- J. Gomez-Bolivar, I. P. Mikheenko, R. L. Orozco, S. Sharma, D. Banerjee, M. Walker, R. A. Hand, M. L. Merroun and L. E. Macaskie, Front. Microbiol., 2019, 10, 438962 Search PubMed.
- E. A. Paoli, F. Masini, R. Frydendal, D. Deiana, C. Schlaup, M. Malizia, T. W. Hansen, S. Horch, I. E. L. Stephens and I. Chorkendorff, Chem. Sci., 2014, 6, 190–196 RSC.
- L. O. Paulista, J. Albero, R. J. E. Martins, R. A. R. Boaventura, V. J. P. Vilar, T. F. C. V. Silva and H. García, Catalysts, 2021, 11, 461 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- J. A. Onrubia-Calvo, A. Bermejo-López, S. Pérez-Vázquez, B. Pereda-Ayo, J. A. González-Marcos and J. R. González-Velasco, Fuel, 2022, 320, 123842 CrossRef CAS.
- K. Rida, A. Benabbas, F. Bouremmad, M. A. Peña, E. Sastre and A. Martínez-Arias, Appl. Catal., A, 2007, 327, 173–179 CrossRef CAS.
- N. Mota, M. C. Alvarez-Galván, R. M. Navarro, S. M. Al-Zahrani, A. Goguet, H. Daly, W. Zhang, A. Trunschke, R. Schlögl and J. L. G. Fierro, Appl. Catal., B, 2012, 113–114, 271–280 CrossRef CAS.
- J. A. Onrubia-Calvo, A. Bermejo-López, S. Pérez-Vázquez, B. Pereda-Ayo, J. A. González-Marcos and J. R. González-Velasco, Fuel, 2022, 320, 123842 CrossRef CAS.
- M. Cai, J. Wen, W. Chu, X. Cheng and Z. Li, J. Nat. Gas Chem., 2011, 20, 318–324 CrossRef CAS.
- D. Beierlein, D. Häussermann, M. Pfeifer, T. Schwarz, K. Stöwe, Y. Traa and E. Klemm, Appl. Catal., B, 2019, 247, 200–219 CrossRef CAS.
- M. Romero-Sáez, A. B. Dongil, N. Benito, R. Espinoza-González, N. Escalona and F. Gracia, Appl. Catal., B, 2018, 237, 817–825 CrossRef.
- C. Vogt, E. Groeneveld, G. Kamsma, M. Nachtegaal, L. Lu, C. J. Kiely, P. H. Berben, F. Meirer and B. M. Weckhuysen, Nat. Catal., 2018, 1(2), 127–134 CrossRef CAS.
- A. Blanco, J. Caroca, R. Tamayo, M. Flores, M. Romero-Sáez, R. Espinoza-González and F. Gracia, Fuel, 2022, 320, 123954 CrossRef CAS.
- A. Shahnazi and S. Firoozi, J. CO2 Util., 2021, 45, 101455 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Aseel G. S.
Hussien
ab,
Dalaver H.
Anjum
ab,
Aseel G. S.
Hussien
ab,
Dalaver H.
Anjum