
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe engineering of CO2 hydrogenation catalysts for higher alcohol synthesis
Angie F. J.
Tan
a,
Muhammad Dody
Isnaini
ab,
Muenduen
Phisalaphong
b and
Alex C. K.
Yip
 *a
*a
aMacDiarmid Institute for Advanced Materials and Nanotechnology, Department of Chemical and Process Engineering, University of Canterbury, Christchurch, New Zealand. E-mail: alex.yip@canterbury.ac.nz; Tel: +64-3-3694086
bBio-Circular-Green-economy Technology & Engineering Center, BCGeTEC, Department of Chemical Engineering, Faculty of Engineering, Chulalongkorn University, Bangkok, 10330, Thailand
First published on 6th November 2024
Abstract
Anthropogenic CO2 emissions have drawn significant attention in recent years. Using CO2 as feedstock for chemical processes has become a key solution in overall closed carbon cycles for a vision of a circular carbon economy. CO2 hydrogenation to higher alcohols has emerged as one of the most promising CO2 conversion pathways for mitigating CO2 emissions and producing value-added chemicals. The present review critically discusses the most recent cutting-edge catalyst development in higher alcohol synthesis (HAS), focusing on the influence of different metals, promoters, and supports according to the contributions of different active species in modern catalyst configurations. Particularly, the critical roles of oxygen vacancies and the reaction mechanisms shed light on the rational design of the next-generation CO2 hydrogenation catalysts.
 Angie F. J. Tan | Angie Tan, completed her BSc(Hons) in Industrial Chemistry at the University of Malaysia Sabah, Malaysia, in 2021. She is a PhD candidate and a researcher in the Department of Chemical and Process Engineering at the University of Canterbury, New Zealand. Her research focuses on tackling one of the most pressing global challenges: reducing carbon dioxide emissions by transforming them into valuable, sustainable products. |
 Muhammad Dody Isnaini | Muhammad Dody Isnaini, born in 1996, earned his B.Eng. (2018) in Chemical Engineering at Lambung Mangkurat University, Indonesia, where his research focused on the adsorption of dyes and heavy metals. He is a PhD candidate under the guidance of Prof. Muenduen Phisalaphong and Prof. Bunjerd Jongsomjit in the Department of Chemical Engineering, Chulalongkorn University, Thailand. His doctoral work expands into CO2 capture and catalytic CO2 conversion, aiming to promote green technologies and the development of sustainable materials for a more environmentally responsible future. |
 Muenduen Phisalaphong | Professor Muenduen Phisalaphong has worked in the Department of Chemical Engineering, Faculty of Engineering, Chulalongkorn University since 1989. She received her PhD (Chemical Engineering) from Colorado State University, USA in 1999. She has been working in Biochemical Engineering for more than 30 years. Currently, her research focuses on natural polymer processing and surface modification. Her research interests include technologies/processes for developing biomass-derived composites for biomedical applications, green catalysis, agricultural and environmental applications, adsorption, packaging, and electronics. At present, she has published ∼90 research articles. |
 Alex C. K. Yip | Professor Alex Yip completed his BE(Hon) in Chemical Engineering at UNSW, Australia 2003. He earned his PhD in 2009 from the Hong Kong University of Science and Technology, specializing in heterogeneous catalysis. He now leads the Energy and Environmental Catalysis Group at the University of Canterbury, New Zealand. His research focuses on catalyst design, specifically targeting the conversion of biomass, syngas, and CO2 into valuable chemicals and sustainable fuels. He has published over 110 articles and has served on international journal editorial boards, in addition to his role as an assessor for the MBIE Endeavour Fund (NZ) and ACS Research Fund (USA). |
Sustainability spotlightUtilizing CO2 as feedstock integrated with renewable H2 is one of the most promising pathways for a closed carbon loop cycle for the circular carbon economy. This review provides a critical assessment on the most emerging CO2 hydrogenation catalyst designs, discussing the importance of appropriate combination of metal active sites, supports, and promotors for tailoring metal interfaces, metal support interactions, and acid–base sites, etc. The review also delves into the reaction pathways that balance between dissociative and nondissociative CO activation, and promote CO insertion and C–C coupling for higher alcohol formation. We believe the readers will find the insights in this review paper enlightening and useful for tailoring advanced catalysts with precise design. |
Introduction
Excessive carbon dioxide (CO2) emissions, one of the primary greenhouse gases, have caused serious environmental concerns, which result in global warming and climate change. The atmospheric CO2 concentration has continued to rise over the past decades, exceeding 410 ppm.1 Establishing a practical framework for mitigating CO2 emissions has been periodically discussed at the governmental level. Most nations have signed the Paris Agreement during the UN Climate Change Conference (COP21), which emphasizes limiting global warming to well below 2 degrees Celsius by restricting cumulative CO2 emissions to less than one trillion tons of CO2.2 According to recent climate reports, the global average temperature is projected to rise by over 2 °C by 2050 and more than 4 °C by 2100.3 Considerable attention has been directed towards stabilizing atmospheric CO2 levels, not only by reducing CO2 emissions but also by effectively utilizing CO2. CO2, as the main industrial C1 carbon source, can be utilized in a wide range of applications.Over the past few decades, there has been a significant rise in research studies on CO2 hydrogenation, demonstrating the effectiveness of CO2 hydrogenation reactions in reducing CO2 emissions. CO2 hydrogenation of valuable chemicals has been widely explored using catalytic CO2 conversion technologies such as thermocatalytic conversion, photocatalytic conversion, and electrocatalytic conversion.4 Thermocatalytic conversion, among these technologies, is the most promising due to its high efficiency and great opportunities in large-scale applications.5,6 Thermocatalytic CO2 conversion also promotes a more sustainable approach by utilizing renewable green hydrogen generated via wind power, photovoltaic cells, or excess nuclear power from water electrolysis systems.7–9
CO2 hydrogenation is an excellent strategy that mitigates CO2 release from chemical processes and produces valuable chemicals and fuels, including methane, methanol, ethanol, C2–C4 alcohols, etc., depending on the nature of the specific catalytic active sites and the reaction conditions.10 Particularly, the CO2-to-methanol route has been a promising industrial process due to the significant demand for methanol.11 On the other hand, higher alcohols (C2–C4 alcohols) offer benefits over methanol, including greater energy density,12 lower azeotropic behaviour,13 and less water affinity,12 making them strong alternatives for methanol substitutes in fuels and fuel additives. Also, synthesizing higher alcohols, especially ethanol, at low temperatures is thermodynamically more desirable than methanol due to lower Gibbs free energy and a greater equilibrium constant value.14 Additionally, higher alcohols are used as solvents for resins, fats, waxes, ethers, and gums, as well as useful feedstock and intermediates for various chemicals and pharmaceuticals.15
Despite these benefits, achieving high conversion of CO2 and high selectivity of higher alcohols has remained challenging. An advanced catalyst design is required to promote carbon chain growth and overcome the high kinetic energy barrier associated with CO insertion through C–C coupling into the carbon chain, ultimately facilitating the formation of ethanol and higher alcohols.16,17 To increase CO2 conversion and selectivity for higher alcohols, it is essential to modify the metal active sites, the metal–metal interfaces, the metal-support interactions, and the physical and chemical characteristics of the catalysts to promote key reaction intermediates through balancing, suppressing or synchronizing reactions including reverse water–gas shift (RWGS), Fischer–Tropsch (FT) synthesis, CO2 hydrogenation to alkanes and alkenes, methanol synthesis, and higher alcohol synthesis (HAS).
There have been critical reviews regarding CO2 hydrogenation for HAS.14,18,19 Cui et al.18 centered on the various catalytic systems and reaction mechanisms involved by the thermodynamic and kinetic analyses in the conversion of CO2 into alcohols with discussion focusing on the structure–activity relationship, which was relatively comparable to Latsiou et al.19 but the latter focused solely on HAS. In contrast, Xu et al.14 focused on advancements in catalyst design and performance by categorizing them into different catalyst families with a brief discussion on the promoter and support effect and some information on reaction mechanisms. This review aims to provide a comprehensive and systematic analysis of the critical knowledge and research gap that impacts the rational design of effective catalysts for CO2 hydrogenation to higher alcohols. By summarizing the recent research advancements, we present an integrated and in-depth investigation into the roles of different types of metals, promoters, and supports in catalyst system development in HAS through extensive case-by-case studies. We attempt to address this from the viewpoint of stabilizing active phases and structure–performance relationships to improve the production of higher alcohols, hoping that this review may provide insights on the precise engineering of the interplay between active metal sites, supports and promoters at an atomic scale for optimizing HAS catalytic performance. In addition, this review also underlines the influence of oxygen vacancies and emphasizes the reaction mechanism based on in situ characterizations and theoretical calculations. Finally, the conclusion and future perspective are explored for future investigation in HAS.
Catalyst design and development
Research on CO2 hydrogenation for HAS can be categorized into four main groups: (1) modified Cu-based methanol synthesis catalysts,20,21 (2) modified Co- or Fe-based FT synthesis catalysts,22–27 (3) noble metal-based (Rh, Pd, Ir, Au, and Pt) catalysts,28–32 and (4) Mo-based catalyst,33,34 as listed in Table 1. Cu-based catalysts are well-known candidates in methanol synthesis and RWGS due to their superior performance and cost-effectiveness.54–56 It is now widely recognized that metallic Cu promotes non-dissociative CO activation and its insertion step, thus producing mainly CO and methanol instead of higher alcohols. Modifications of Cu-based catalysts have been developed to enhance the selectivity of higher alcohols.18 Highly dispersed Cu nanoparticles embedded in the Na-beta zeolite (Cu@Na-beta) were synthesized and tested for CO2 hydrogenation reaction in a fixed bed reactor.35 The Cu@Na-beta catalyst showed an excellent ethanol selectivity of 79% at 300 °C and 2.1 MPa and a space-time yield (STY) of 398 mg gcat−1 h−1 at a CO2 conversion of approximately 18%. The authors proposed that the Na-beta can result in the confinement effect of Cu nanoparticles and restrict the formation of unwanted byproducts. Electropositive Cu species were detected in small amounts in the complex Cu@Na-beta catalyst using X-ray photoelectron spectroscopy (XPS) and Cu LMM auger spectra analysis, but none were found in the other prepared catalysts, confirming the cooperative interaction between Cu and Na-beta. The high catalytic performance can be attributed to the synergistic interaction between Cu nanoparticles and the zeolite framework. An et al.21 reported that the combination of cooperative CuI sites on a Zr12 cluster with a metal–organic framework (MOF) exhibited a high ethanol selectivity of >99% with the addition of alkali metal as promoters. CuI species are identified as the active sites for ethanol formation, and the role of intimidate CuI–CuI dual sites stabilized by MOF is crucial for inducing the H2 activation in ethanol synthesis, resulting in the formation of (Cu2+–H−)2 followed by promoting the C–C coupling of methanol and formyl species. Meanwhile, alkali cation (Cs+) creates an electron-rich environment for the Cu center to stabilize the formyl intermediates. This results in the formation of CH3CHO, which can undergo reductive elimination, producing methanol, ethanol, and water and regenerating the CuI–CuI sites.| Catalyst | T (°C) | P (MPa) | CO2/H2 ratio | GHSV (mL gcat−1 h−1) | XCO2 (%) | SCO (%) | SCxHy (%) | SMeOH (%) | SHAa (%) | STYHAa | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Selectivity and space-time yield of a specific product are indicated in parentheses. | |||||||||||
| Cu@Na-beta | 300 | 2.1 | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 :3 |
12000 |
18.0 | 21.0 | — | — | 79.0 (ethanol) | 398 mg gcat−1 h−1 (ethanol) | 35 |
| Na–CuCo-9 | 330 | 4.0 | 1:1 |
5000 | 20.1 | 26.5 | 53.8 | — | 26.8 (ROH) | 80.8 mg gcat−1 h−1 (C3+OH) | 36 |
| 4.6 K–CuMgZnFe | 320 | 5.0 | 1:3 |
6000 | 30.4 | 30.6 | 52.4 | 1.3 | 15.9 | 69.6 mg gcat−1 h−1 | 20 |
| CuFeZn-0.7PDA | 310 | 4.0 | 1:3 |
7200 | 10.6 | 43.2 | 22.0 | — | 34.8 (ROH) | 58.2 mg gcat−1 h−1 | 16 |
| Cs–Cu0.8Fe1.0Zn1.0 | 330 | 5.0 | 1:3 |
4500 | 36.6 | — | — | — | 19.8 | 1.47 mmol gcat−1 h−1 | 37 |
| CuZnAl/K–CuMgZnFe | 320 | 5.0 | 1:3 |
6000 | 42.3 | 13.8 | 67.6 | 1.3 | 17.4 | 2.24 mmol gcat−1 h−1 | 38 |
| Cr (1%)–CuFe | 320 | 4.0 | 1:3 |
6000 | 38.4 | 14.8 | — | — | 29.2 | 104.1 mg gcat−1 h−1 | 39 |
| 2% Na–Co/SiO2 | 310 | 5.0 | 1:3 |
6000 | 53.2 | — | — | — | 12.9 (ROH) | 1.10 mmol gcat−1 h−1 (ethanol) | 40 |
| CoAlOx-600 | 140 | 4.0 | 1:3 |
Tank | — | — | — | — | 92.1 (ethanol) | 0.44 mmol gcat−1 h−1 (ethanol) | 27 |
| Co/La4Ga2O9 | 270 | 3.5 | 1:3 |
3000 | 4.6 | 15.4 | 34.6 | 15.3 | 34.7 (ethanol) | — | 41 |
| 5Co/S-1 | 250 | 2.0 | 1:3 |
4670 | 13.9 | 0 | 58.0 | — | 27.0 (ethanol) | 0.83 mmol gcat−1 h−1 (ethanol) | 42 |
| 2.5K5Co–In2O3 | 380 | 4.0 | 1:3 |
2250 | 36.6 | 80.8 | — | — | — | 3.73 mmol gcat−1 h−1 | 43 |
| 2% Na–Fe@C/5% K–CuZnAl | 320 | 5.0 | 1:3 |
4500 | 39.2 | 9.4 | — | — | 35.0 (ethanol) | — | 25 |
| FeNaS-0.6 | 320 | 3.0 | 1:3 |
8000 | 32.0 | 20.7 | — | — | 12.8 | 78.5 mg gcat−1 h−1 | 23 |
| K-0.82–FeIn/Ce–ZrO2_900 | 300 | 10.0 | 1:3 |
4500 | 29.6 | 13.4 | 53.2 | 4.7 | 28.7 | — | 44 |
| ZnFe2O4/Fe–Zn–Na# | 320 | 5.0 | 1:3 |
12000 |
29.1 | 21.8 | — | — | 18.6 | 137.7 mg gcat−1 h−1 | 45 |
| Ir1–In2O3 | 200 | 6.0 | 1:5 |
Tank | — | — | — | — | 99.7 (ethanol) | 0.99 mmol gcat−1 h−1 (ethanol) | 46 |
| Pd2/CeO2 | 240 | 3.0 | 1:3 |
3000 | 9.2 | — | — | 0.8 | 99.2 (ethanol) | 59.2 mg gcat−1 h−1 (ethanol) | 47 |
| Pd2Cu NPs/P25 | 200 | 3.2 | 1:3 |
Tank | — | — | — | — | 92.0 (ethanol) | 41.50 mmol gcat−1 h−1 (ethanol) | 48 |
| PdFe-6.9 (37 nm) | 300 | 6.0 | 1:3 |
6000 | 33.3 | 27.9 | — | — | 19.1 | 86.5 mg gcat−1 h−1 | 49 |
| 0.3K–1Pd/Fe2O3 | 320 | 4.0 | 1:3 |
6000 | 30.0 | 23.0 | — | — | 13.4 | 52.2 mg gcat−1 h−1 | 26 |
| 0.19Na–Rh@S-1 | 250 | 5.0 | 1:3 |
6000 | 10.0 | — | — | — | 24.0 (ethanol) | 72.00 mmol gRh−1 h−1 (ethanol) | 50 |
| 2Rh0.5Fe0.5Na/CeO2 | 250 | 3.0 | 1:3 |
6000 | 9.8 | 32.1 | 15.9 | 27.3 | 21.5 (ethanol) | 63.5 mmol gRh−1 h−1 (ethanol) | 51 |
| Na–Rh–FeOx/ZSM-5 | 200 | 1.0 | — | 3600 | 7.0 | — | — | — | 97.0 (ethanol) | 1.10 mmol gcat−1 h−1 (ethanol) | 52 |
| K0.2Rh0.2/βMo2C | 150 | 6.0 | 1:3 |
Tank | — | — | — | — | 72.1 (ethanol) | 0.03 mmol gcat−1 h−1 (ethanol) | 53 |
Co-based catalysts have been extensively developed as FT synthesis catalysts due to their strong hydrogenation ability.57 Interestingly, several studies suggested that the co-existence of Co0 and CoOx phases in Co-based catalysts can promote the synthesis of ethanol. For instance, Wang et al.27 demonstrated that the co-existing phases of Co0 and CoOx in CoAlOx catalysts are favored in ethanol synthesis. The authors discovered that the different reduction temperatures significantly affected the catalyst performance. CoAlOx-600 catalyst, which the catalyst reduced at 600 °C, exhibited an outstanding ethanol selectivity of 92.1% and a space-time yield of 0.444 mmol gcat−1 h−1, achieving the best catalytic performance compared to other CoAlOx catalysts, which were reduced at a temperature between 300 °C and 650 °C (Fig. 1). Moreover, methanol, n-propanol and n-butanol were also obtained in this catalyst, further validating the effectiveness of Co-based catalysts in CO2 hydrogenation to higher alcohols. Using the extended X-ray absorption fine structure (EXAFS) spectroscopy, it was observed that the characteristic peak of metallic Co0 showed up after reduction at 600 °C with the co-existence of CoOx species. This suggests that more hydrogen activation can occur due to the presence of metallic Co0, resulting in higher catalytic performance. However, CoAlOx catalysts reduced at temperatures higher than 600 °C, with more metallic sites and a relatively small amount of CoOx species, encountered an increase in methanol yield but a decrease in ethanol yield.
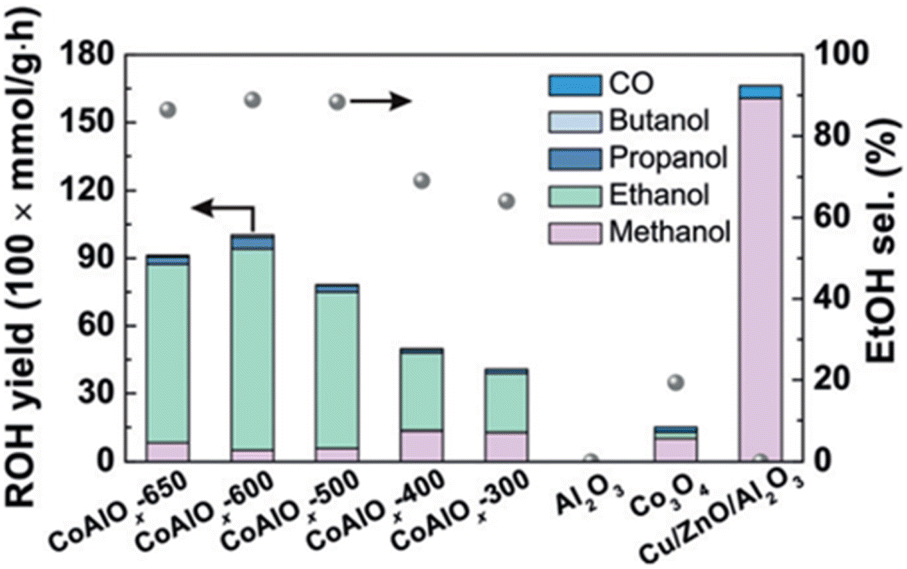 | ||
| Fig. 1 Catalytic performance of various catalysts based on their alcohols yields and ethanol selectivity. Reprinted with permission from ref. 27. Copyright 2018 John Wiley and Sons, Inc. | ||
Similar findings were reported by Ding et al.,42 indicating the significance of co-existing Co0 and CoOx species in facilitating ethanol formation. The authors successfully synthesized silicalite-1 (S-1) with abundant silanols supported on Co species with 5 wt% and 15 wt% of Co loadings, while the sample synthesized using the same procedure but without the addition of NaOH in the precursor gel was denoted as S–S. Of interest is the capability of this novel approach to utilize strong metal-support interaction (SMSI), thus forming Si–O–Co chemical bonds that can stabilize the co-existence Co0 and CoO species. The co-existence Co0 and CoO on Co/S-1 was evidenced using characterization techniques such as X-ray diffraction (XRD), CO adsorption (CO-IR), and quasi in situ XPS analysis. The catalytic performance of the 5 wt% and 15 wt% Co loadings supported on S-1 showed similar results (Fig. 2). This is consistent with the comparable amount of exposed active sites, i.e., 38.4 μmol g−1 and 38.2 μmol g−1 for 15Co/S-1 and 5Co/S-1, respectively, calculated from H2-TPD. Besides, 15Co/S-1 and 5Co/S-1 also showed similar results for ethanol selectivity (26% and 27%) and CO2 conversion (12.1% and 13.9%). A volcano-shaped relationship between CO2 conversion and the reduction temperature was concluded, highlighting the importance of an optimal reduction temperature to balance the metallic Co0 and CoO sites. CO2 conversion increased as the reduction temperature rose from 200 °C to 300 °C but decreased at higher reduction temperatures. Notably, the Co species in 5Co/S-1 was fully reduced into metallic Co0 at 400 °C. The increase in CO2 conversion can be explained by the increase in metallic Co0 sites, which enhance the dissociation activation of hydrogen on the surface and facilitate the breaking of the C–O bond. However, at higher reduction temperature, aggregation of metals may occur, leading to catalyst deactivation.25 For the same reason, although 5Co/S-1 exhibited superior catalytic activity during the initial phase of the reaction, but it lost stability over time due to gradual reduction of CoOx to Co0 under reaction conditions, causing the aggregation of cobalt nanoparticles. In summary, the so-called protection provided by Si–O–Co bond may be able to postpone the reduction of CoOx to Co0, but it is not capable of completely preventing CoOx from fully reducing into Co0, which ultimately affects the catalyst stability.
 | ||
| Fig. 2 CO2 conversion and product selectivity by Co/S-1 and Co/S–S catalysts with different 5 and 15 wt% Co contents. Reprinted with permission from ref. 42. Copyright 2024 Elsevier. | ||
Previous studies have shown that Pd-based catalysts are effective for CO2 hydrogenation to methanol and have recently gained attention in HAS. For instance, Lou et al.47 reported CO2 hydrogenation to ethanol over Pd/CeO2 catalysts. In their study, Pd dimers (Pd2/CeO2) and Pd nanoparticles (nano-Pd/CeO2) were supported on CeO2 with nanorods morphology. Pd2/CeO2 catalyst with the unique geometric structure of Pd dimers exhibited impressive catalytic activity with 9.2% of CO2 conversion, 99.2% of high ethanol selectivity, and STY of 45.6 gethanol gPd−1 h−1 at 240 °C and 3 MPa. The STY of atomically dispersed Pd dimers on the CeO2 catalyst was 90 times higher than that of nano-Pd/CeO2 with STY of 0.5 gethanol gPd−1 h−1 because CO was the primary product formed over nano-Pd/CeO2 catalyst. High-angle dark-field scanning transmission electron microscope (HAADF-STEM) images further revealed the absence of Pd clusters and particles on both fresh and used Pd2/CeO2 catalysts, hypothesizing that Pd species are atomically and uniformly distributed on the CeO2 support. To the best of our knowledge, the formation of agglomerated Pd clusters and particles can result in a decrease in higher alcohol synthesis. They conclude that the catalytic performance is sensitive to different Pd nanoparticle sizes, with single-atom catalyst (SAC) of Pd dimers exhibiting the best catalytic performance. Therefore, this means that tailoring the active sites of a catalyst at an atomic scale can remarkably promote HAS. Meanwhile, the CO adsorption strength, considered the key factor in determining product selectivity, was investigated through in situ diffuse reflectance infrared Fourier transform spectroscopy experiments using CO as a probe molecule (CO-DRIFTS). The result demonstrated that the Pd species in Pd2/CeO2 catalyst were mostly Pd dimers with no neighbouring Pd atoms, which exhibited stronger CO adsorption strength. Claiming that the stronger binding strength of CO over Pd2/CeO2 catalyst can effectively inhibit CO desorption and trigger the C–C coupling reactions between CO and CH3 intermediate, the formation of ethanol is, therefore, enhanced.
A similar concept has been implemented using an Ir-based catalyst to demonstrate that highly dispersed atomic active sites can promote ethanol generation. In particular, bifunctional Ir1–In2O3 SAC was synthesized through a wet-chemical approach, and the CO2 hydrogenation reactions were carried out in an aqueous solution.46 By impregnating Ir onto the In2O3 support, oxygen vacancy (Ov) is induced by partially reducing In2O3 in the H2 atmosphere, as shown in Fig. 3. As a result, the adjacent oxygen vacancy can bind with monoatomic Ir to form Lewis acid–base pair. The partially reduced In2O3 and Lewis acid–base pair act as two active sites, which can enhance the CO2 adsorption and activation into CO* intermediates. In situ DRIFTS experiment results showed further evidence supporting the existence of Irδ+–CO* intermediates, while the in situ Fourier transform infrared spectroscopy (FTIR) confirmed the existence of CH3O*–Ov intermediates. It is concluded that Ir1–In2O3 SAC promotes the C–C coupling by coupling the Irδ+–CO* intermediates with the CH3O*–Ov intermediates to enhance ethanol selectivity. The CO2 hydrogenation experiment achieved a high selectivity to ethanol (>99%) and a turnover frequency (TOF) of up to 481 h−1 in this study. The authors also discovered that the ethanol formation was suppressed from 85.3% to 5.7% when the Ir loading increased from 0.2 wt% to 1.0 wt%, which can be ascribed to the agglomeration of Ir nanoparticles and the over-reduction of In2O3.
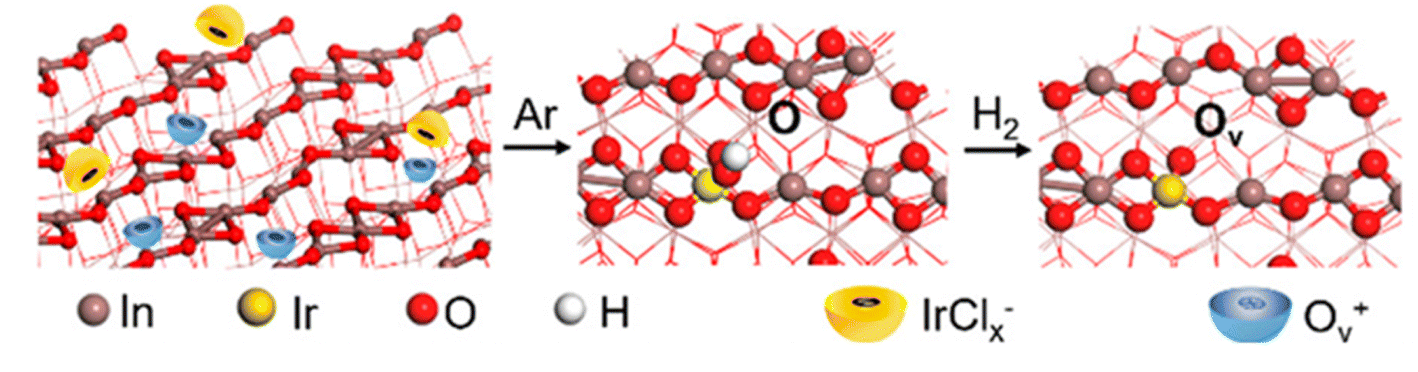 | ||
| Fig. 3 Schematic illustration of synthesis procedure for Ir1–In2O3 SAC. The diagram depicts the generation of Ov by partially reducing In2O3 under H2 atmosphere prior to impregnating of the Ir. These Ov then act as the anchoring site for single Ir atoms and promote the trapping of IrCl62−. Reprinted with permission from ref. 46. Copyright 2020 American Chemical Society. | ||
Besides monometallic catalyst systems, bimetallic catalyst systems have recently emerged as promising candidates for CO2 hydrogenation to higher alcohols. Several studies have shown that introducing a second metal into Cu or noble metal can alter the electronic structure of the catalyst and improve the catalytic performance. Irshad et al.36 proposed that in the synthesized Cu–Co bimetallic catalyst, Cu and Co active sites each serve their distinct roles. Na-promoted bimetallic Cu–Co-y catalysts, combining the capabilities of methanol synthesis and C–C coupling, were prepared with different Cu loadings (y = Cu/(Cu + Co) × 10) via coprecipitation method and investigated for the production of higher alcohols. In simpler terms, the Cu site is accountable for the rapid RWGS to produce CO,58 and, subsequently, experiencing methanol synthesis. In contrast, Co site is responsible for forming CHx intermediates through C–O dissociation.59 Subsequently, CH3CO* is formed through C–C coupling between the CO produced and CHx intermediates, which, after deep hydrogenation, produce acetaldehyde and, ultimately, ethanol or n-butanol.60,61 Based on their conclusion, acetaldehyde is observed as the key intermediate in the synthesis of n-butanol. The “best case” catalyst in their studies was Na-CuCo-9 catalyst, which demonstrated a high C3+OH/ROH fraction of 73.5%, giving a high STYC3+OH of 80.8 mg gcat−1 h−1 while the overall alcohol selectivity was 58% with CO2 conversion of 22.1%. Besides, Na-the CuCo-9 catalyst exhibited superior long-term stability without any detrimental effect on its catalytic performance. However, this study did not explore the role of Na as an alkali promoter. The role of alkali metal as a promoter will be further addressed in a later section. On the other hand, Pd–Cu nanoparticles with different composition ratios and supports for ethanol synthesis were investigated in a slurry batch reactor.48 By adjusting to the ideal Pd/Cu ratio and catalyst support, the Pd2Cu/P25 catalyst displayed a high selectivity to ethanol of 92.5% with a TOF value of 359 h−1. The study, however, did not emphasize the formation of the active phase for HAS; instead, it focused on the presence of charge transfer between Pd and Cu. The formation of electronic interaction caused by charge transfer can improve the reduction of surface oxides to their metallic states, which the authors strongly believed to be the active sites for CO2 activation, thus benefiting the overall CO2 hydrogenation reaction. Pd2Cu nanoparticles deposited on various supports such as SiO2, CeO2, Al2O3, and P25 were also investigated, and the Pd2Cu supported on the P25 catalyst emerged as the best, demonstrating the highest TOF and ethanol selectivity out of the four catalysts, which can be attributed to the presence of oxygen vacancies.
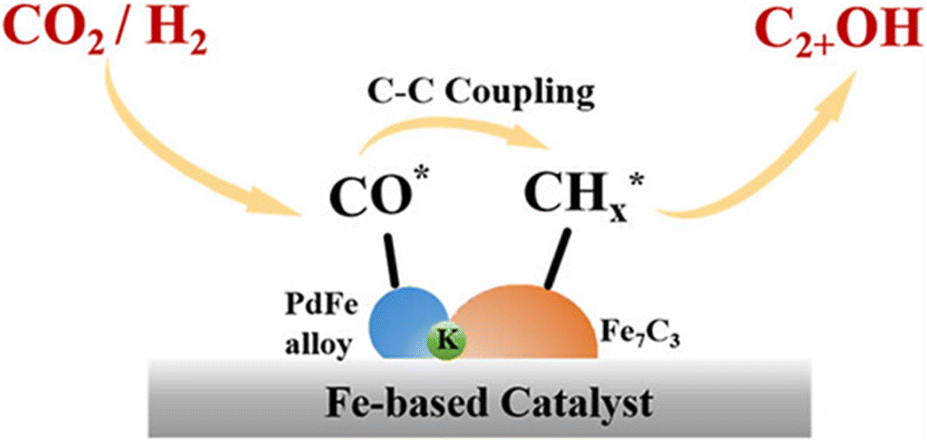
Another exciting study is the synthesis of a bimetallic PdFe catalyst.49 Iron carbides (FeCx) are well known as one of the active phases, which positively affects HAS activity, although it is not very active at low CO partial pressures.62 Combining FeCx phases with metal species can form metal–FeCx interfaces to promote HAS activity. It is presumed that the synergistic effects between PdFe alloy and Fe5C2 exist, forming PdFe alloy–Fe5C2 interfaces that can eventually promote HAS catalytic performance. Under reaction condition of 300 °C, 5 MPa and WHSV = 6000 mL gcat−1 h−1, higher alcohol selectivity of 26.5% and STY of 86.5 mg gcat−1 h−1 were obtained over PdFe catalyst with Pd loading of 6.9 wt%. The authors demonstrated for the first time that transition metal or alloy could induce the formation of deep iron carbidization without the aid of an alkali ion as a promoter. The presence of PdFe alloy and Fe5C2 was verified through in situ XRD experiments, showing that Fe2O3 in spent PdFe-6.9% catalyst was instantly converted to Fe3O4 under hydrogen reduction condition and completely transformed into Fe5C2 under CO2 hydrogenation reaction process within 30 minutes. The XRD analysis also demonstrated that the formation of Fe5C2 phase appeared when the Pd loading ≥4.3 wt% under CO2 hydrogenation reaction condition. At the same time, PdFe alloy was formed when reduced Fe atoms from Fe2O3 migrated to the Pd surface. The formation of PdFe alloy–Fe5C2 interfaces was confirmed through HAADF-STEM images of the spent catalyst. Further evidence of the PdFe alloy–Fe5C2 interface formation was shown when the authors compared the HAS activity with the reference catalyst (physically mixing 6.9 Pd/γ-Al2O3 + Fe2O3 catalyst), which demonstrated no HAS activity. Overall, PdFe alloy mainly generates CO by the RWGS reaction, while Fe5C2 is essential for CO dissociation and carbon chain propagation. The adsorbed alkyl is then reacted with CO at the PdFe alloy–Fe5C2 interfaces, which is subsequently hydrogenated to ethanol. The authors concluded that PdFe alloy–Fe5C2 interfaces are primarily responsible for HAS in their bimetallic system. A study by Yang et al.45 also observed a similar effect, identifying carburized metal-based catalysts (ZnFe2O4/Fe–Zn–Na#) as active phases for CO2 hydrogenation in HAS. Unlike Wang et al.49 research, in which Fe5C2 was reported to be induced after hydrogen reduction and during the CO2 hydrogenation reaction process, Yang et al.45 proposed a tandem catalysis strategy by synthesizing Fe–Zn–Na# catalyst (pre-reduced using H2 for 8 hours) and physically mixing it with ZnFe2O4 catalyst to generate FeCx. However, similar to Wang et al.49 findings, the Fe5C2 phase also appeared after a few hours under reaction conditions as indicated by XRD characterization results. Additionally, CO2 conversion rapidly increased from 0.3% to 31.8% after 2 h time on stream (TOS) at 300 °C. Higher alcohol selectivity showed a similar pattern, sharply rising from 1.1% to 16.4%. Mössbauer spectroscopy analysis and XRD peaks indicated that the iron species in the spent ZnFe2O4/Fe–Zn–Na# catalyst were primarily Fe3O4, ZnFe2O4 and Fe5C2. The authors inferred that Fe3O4 is responsible for CO formation (RWGS reaction), ZnFe2O4 favors the formation of oxygenates, while Fe5C2 enhances C–C coupling reaction and initiates CO insertion to produce higher alcohols.
Wang et al.25 utilized the multifunctional catalyst of 2% Na–Fe@C/5% K–CuZnAl using a tandem catalysis strategy for CO2 hydrogenation to ethanol. The Fe@C catalyst was synthesized by pyrolyzing the Fe-based MOFs under a nitrogen atmosphere to promote uniformly distributed Fe active sites due to the periodic arrangement of organic linkers and metallic centers and to control the physicochemical properties of the MOF-derived catalyst. Na doping on a Fe-based catalyst is considered to favour alkene synthesis. Na doping enhances the adsorption of acidic CO2 due to the formation of a Fe–C bond by electron transfer from Na–Fe@C to CO2 molecules. Thus, the selectivity of ethanol was slightly enhanced with a slight additional amount of HA produced by adding 2% Na to Fe@C (a 2% Na–Fe@C catalyst). The CuZnAl catalyst was combined with the Na–Fe@C catalyst, which was prepared by mixing two catalytic granules, to boost the synthesis of ethanol from CO2 hydrogenation. The ethanol selectivity increased from 14% (over a 2% Na–Fe@C catalyst) to 35% (over a 2% Na–Fe@C/5% K–CuZnAl multifunctional catalyst), as shown in Fig. 4. The Na–Fe@C catalyst promotes the reaction of RWGS (over iron oxide, Fe3O4) and FTS (over iron carbide, mainly Fe5C2), while the addition of the K–CuZnAl catalyst contributes to not only the Fe crystal structure and the catalytic network but also a highly efficient catalyst for the synthesis of methanol to form the intermediate species (formate and CHxO*).
 | ||
| Fig. 4 Product distributions of CO2 hydrogenation (a) 2% Na–Fe@C catalyst and (b) 2% Na–Fe@C/5% K–CuZnAl multifunctional catalyst (b). Reprinted with permission from ref. 25. Copyright 2021 American Chemical Society. | ||
Furthermore, it is possible that the reaction intermediates produced from one catalytic site could diffuse onto the interface of another catalytic site to synthesize the product over a multifunctional catalyst during the tandem process. The authors suggested that the reaction intermediates (formate and CHxO*) could desorb from K–CuZnAl and diffuse onto the interface of the Fe-based catalyst for the possible formation of ethanol through C–C coupling. Moreover, it is necessary to conduct a more in-depth theoretical analysis and in situ characterization for multifunctional catalysts due to the highly complex process. Furthermore, the intimacy mode of the 2% Na–Fe@C/5% K–CuZnAl in the cascade reaction produces the highest selectivity and conversion through the granule-mixing mode with 1 g of quartz sand, which may be attributed to active site coverage in the physical-mixing mode of the 2% Na–Fe@C/5% K–CuZnAl. The granule-mixing mode allows for an increase in the spatial distance between distinct catalytic components. The 2% Na–Fe@C/5% K–CuZnAl multifunctional catalyst achieved an ethanol selectivity of 35% with a CO2 conversion of 39.2%.
Role of promoters or additives
In CO2 hydrogenation, alkali metals, alkaline earth metals, and transition metals can act as promoters or additives to enhance the overall catalytic activity.63 Some research reported that introducing alkali metals can significantly inhibit hydrocarbon formation by suppressing alkylation reaction, providing opportunities for higher alcohol production.44 Some reported that alkali metals could promote structural effects by stabilising active sites or interfaces.40 The promotional effect of alkali metals in HAS was intensively studied by different research groups. For example, Zhang et al.40 synthesized Co/SiO2 catalysts incorporating alkali metals. Under a CO2 hydrogenation reaction condition of 250 °C, 5 MPa, and WHSV = 6000 mL gcat−1 h−1, Co/SiO2 catalyst without any alkali metal promoter and Li–Co/SiO2 produced only hydrocarbon. In contrast, high selectivity in alcohols, olefins, and CO was achieved over Na–Co/SiO2 and K–Co/SiO2 with a decrease in CH4 selectivity compared to Co/SiO2 and Li–Co/SiO2 catalysts. Among the four catalysts, the Na-doped Co/SiO2 catalyst exhibited the best alcohol selectivity and was further investigated for its role in HAS. It was demonstrated that CO2 conversion and hydrocarbon selectivity decreased with higher Na contents increasing from 0 to 5 wt% (Fig. 5). The highest alcohol selectivity (12.5%) was obtained over an optimum Na content of 2 wt% Na–Co/SiO2 catalyst with ethanol STY of 0.47 mmol gcat−1 h−1. The most exciting finding in this study was that CO pulse titration experiments showed that introducing Na content improved Co2C metal dispersion, which aligned with HAADF-STEM and EDX elemental mapping images. Also, a reduction of corresponding particle size was observed. Other observations, such as EXAFS fitting spectra of Co K-edge, further confirmed the improved metal dispersion and reduced particle size as there was a significant decrease in Co–Co coordination numbers in Co2C from 6.5 to 1.9 when Na content increased from 0 to 5 wt%. All these characterisation results suggested that the Na promoter has a structural effect on Co2C, which agrees well with other research, highlighting the exceptional ability of alkali metals to exhibit structural effects that stabilize active metal or interface sites.64 The authors also concluded that an interaction is formed between Na and Co2C. This interaction leads to stable Na–Co2C active sites by generating Na–Co bonds, decreasing particle size, and enhancing the metal dispersion. As a result, RWGS reaction and ethanol STY are improved. Furthermore, an optimized amount of Na is also important in HAS because an excess amount of Na with too strong interaction would weaken the CO adsorption strength and hinder CO insertion, thus leading to high CO selectivity, which is unbeneficial to HAS. | ||
| Fig. 5 Catalytic performance over Co/SiO2 catalysts with different alkali metals and Na contents (a) CO2 conversion and product selectivity with different alkali metals (b) CO2 conversion and product selectivity Na contents (c) alcohol distribution and ethanol STY with different Na contents. Reprinted with permission from ref. 40. Copyright 2021 Elsevier. | ||
A similar trend was observed in the structure–performance relationship between the Na content and ethanol formation by Zhang et al.50 They prepared a series of catalysts with different amounts of Na-promoted Rh nanoparticles embedded in zeolite silicalite-1, denoted as xNa–Rh@S-1, where x was the weight loading of Na. By comparing the catalytic performance with different Na content, 0.19Na–Rh@S-1 with a moderate amount of Na content displayed the best catalytic performance in ethanol generation among all, with CO2 conversion of 10% and ethanol selectivity of 24%. Although the results were generally lower than other published works on HAS, the ethanol STY over 0.19Na–Rh@S-1 was surprisingly high, achieving 72 mmol gcat−1 h−1. C3+ alcohols were also detected, but the selectivity was not ideal, being less than 5%. Consistent with prior research, an excessive amount of Na can lead to severe deposition of Na species on the surface Rh sites, causing high coverage by Na+ on the catalyst surface. Meanwhile, the confinement effect by the rigid framework of zeolite S-1 successfully prevents Rh nanoparticles from agglomeration and restricts the sintering of Na-promoted Rh nanoparticles, supported by TEM results. Hence, Na–Rh@S-1 achieved exceptional long-term stability under reaction conditions. The experimental results (XPS and CO2 chemisorption) also pointed out the role of Na promoter in inducing the Rh+ generation, which can coexist with Rh0 species in the catalyst, thus causing a promotional effect in ethanol synthesis. H2-TPR profiles observed that the reduction peaks with Na addition became broader and split into two peaks with increasing Na contents. Besides, the reduction peaks also experienced a right shift to a higher temperature with increasing Na loadings. Taken together, this implies that incorporating Na contents can inhibit the complete reduction of Rh species. Besides, the experiment data also showed that the presence of Na as a promoter improved the CO2 chemisorption performance, which the presence of strong CO2 adsorption strength and Rh+ species can minimize the H2 adsorption and dissociation ability, thereby suppressing hydrocarbon formation and enhance C–O bond activation and CO insertion for C–C propagation.65,66
Xu et al.20 demonstrated that incorporating a moderate amount of K into CuMgZnFe, abbreviated as K-CMZF catalysts, could effectively boost the production of HAS. It was observed that the selectivity of higher alcohols and STY increased when K loading increased from 0.1 to 4.6 wt% but then decreased at K loading of 17.6 wt%. Optimized K-CMZF catalyst with 4.6 wt% K loading displayed the highest CO2 conversion of 30.4% and higher alcohols STY of 69.6 mg gcat−1 h−1. Based on Anderson–Schulz–Flory (ASF) calculations, the value of chain growth probability (α) increased when K loading increased, highlighting that adding K can enhance the chain growth ability of K-CMZF catalysts. The existence of K promoter not only maintains a great balance between non-dissociative and dissociative CO activation but also controls the hydrogenation capacity, thus ensuring the participation of adequate *CHx and *CO species in C–C coupling reactions while restricting *CHx hydrogenation termination reaction to promote *CHxCO/*CHxCHO hydrogenation to higher alcohols.
Witoon et al.43 systematically investigated the promoter effect on CO2 hydrogenation over K–Co promoted In2O3 catalysts. Adding K and Co into In2O3 significantly boosts the C–C chain growth and CO insertion, thus increasing the higher alcohol selectivity, but had only a minor effect on CO2 conversion. On the other side, both Co- and K-promoted In2O3 catalysts showed a clear difference in product selectivity, wherein the K-promoted In2O3 catalyst was highly selective towards CO and CH3OH synthesis, while the Co-promoted In2O3 favored the production of C2+ hydrocarbons over higher alcohols. The authors postulated that the addition of K reduces the weak and medium H2 adsorption, thereby suppressing the hydrogenation of CH3OH and formation of CH4 from CO. Contrarily, characterization results revealed a mixture of Co0 and CoO in the Co species of Co-promoted In2O3. Co0 sites facilitate the formation of 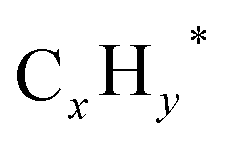 species, which subsequently react with migrating CO* on the CoO surface to form C2+OH products. However, the faster hydrogenation of
species, which subsequently react with migrating CO* on the CoO surface to form C2+OH products. However, the faster hydrogenation of 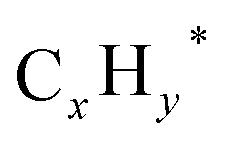 species over CO insertion owing to weak H2 adsorption effectively promotes C2+ hydrocarbon formation rather than C2+ oxygenates formation. To enhance the HAS activity, K–Co promoted In2O3 catalysts with varying weight percent of K and Co were synthesized. As a result, K–Co promoted In2O3 (2.5 wt% K and 5 wt% Co) demonstrated superior HAS catalytic performance with higher alcohols STY of 169.6 g kgcat−1 h−1 and a higher alcohols distribution of 87.4% among the overall alcohols formed. The improved HAS activity can be attributed to the synergistic interaction between K, Co, and In2O3. The introduction of K and Co contributes to generating K–O–Co sites, which reduces weak H2 adsorption and causes a stronger interaction of H2 with the catalyst surface, hence enabling the manipulation of higher alcohol selectivity.
species over CO insertion owing to weak H2 adsorption effectively promotes C2+ hydrocarbon formation rather than C2+ oxygenates formation. To enhance the HAS activity, K–Co promoted In2O3 catalysts with varying weight percent of K and Co were synthesized. As a result, K–Co promoted In2O3 (2.5 wt% K and 5 wt% Co) demonstrated superior HAS catalytic performance with higher alcohols STY of 169.6 g kgcat−1 h−1 and a higher alcohols distribution of 87.4% among the overall alcohols formed. The improved HAS activity can be attributed to the synergistic interaction between K, Co, and In2O3. The introduction of K and Co contributes to generating K–O–Co sites, which reduces weak H2 adsorption and causes a stronger interaction of H2 with the catalyst surface, hence enabling the manipulation of higher alcohol selectivity.
Zhang et al.39 examined the dependence of C2+OH selectivity on the Cr loading level of the Cr–CuFe catalyst, in which the maximum C2+OH selectivity was obtained at Cr = 1%. In detail, Cr-promoted CuFe-based catalysts with K additive were synthesized via the sol–gel method, and the role of Cr as a dopant was further investigated. It was inferred that the interaction between Cu and Fe becomes stronger with the Cr additive. All the CuFe-based catalysts contained the same amount of K as the catalytic performance on Cr (1%)–CuFe catalyst without the K promoter exhibited higher selectivity towards methane and C2–C4 alkanes whereas decrease in olefins and higher alcohols selectivity. This is consistent with previous research, indicating the role of alkali metals in preventing over-hydrogenation and hydrocarbon formation. With the aid of alkali metal, catalyst alkalinity is improved, therefore enhancing CO2 adsorption and activation.20 An appropriate Cr loading amount can enhance H2 adsorption and activation while weakening CO adsorption strength on the CuFe catalyst. This suggests that Cr additives can effectively mitigate CO over-dissociation, inhibiting the formation of excessive iron carbide species (FeCx) from metallic Fe. Supported by Fe (2p) XPS and 57Fe Mössbauer spectroscopy, FeCx characteristics were observed over the Cr (1%)–CuFe catalyst compared to the undoped CuFe catalyst. Besides, the amount of Cu0 (23.2%) was lower, and the amount of Cu+ and Cu2+ (76.8%) was higher in Cr (1%)–CuFe catalyst than that of the undoped CuFe catalyst (30.4% of Cu0, 69.6% of Cu+ and Cu2+), as indicated by Cu LMM Auger electron spectra. These findings confirmed the improvement of interactions between Cu and Fe species, which are conducive to forming Cu–FeCx interfaces, the active sites of the catalyst for HAS.37 Excessive Cr loading has a negative effect on HAS because the Cr species would heavily cover the catalyst surface, and the interaction of CHx with non-dissociative CO over Cu–FeCx interfaces will be inhibited. The reaction mechanism proposed that C–C coupling between the interaction between CHx and non-dissociative CO over Cu–FeCx interfaces produces more acetate and acetaldehyde intermediates, enhancing the synthesis of higher alcohols.
Ji et al.51 explored the effects of different transition metals and alkali metals on Rh/CeO2 catalyst for CO2 hydrogenation in ethanol synthesis. Fe and Na doping as dual promoters demonstrated superior catalytic activity for CO2 hydrogenation with high ethanol selectivity of 29.8% and ethanol STY of 116.7 mmol gcat−1 h−1 at 250 °C and 3 MPa. The optimum amounts of Fe and Na in the RhFeNa/CeO2 catalyst, denoted as 2Rh0.5Fe0.5Na/CeO2, were both 0.5 wt%, achieving the highest catalytic performance and Rh dispersions with Rh loading of 2 wt%. H2-TPR and CO2-TPD profiles showed that adding Fe could enhance the Rh metal dispersion by showing increased H2 consumption, while adding Na was also shown to increase H2 consumption and promote the reduction of Rh species to metallic Rh. Other additives such as Li, Cs, and K suppressed the reduction of Rh species. It was concluded that adding Na and Fe as dual promoters improve the overall Rh metal dispersion and form strong interactions with Rh, thus enhancing the SMSI. FTIR studies of chemisorbed CO and XPS results further revealed that Na and/or Fe additions could enhance the CO adsorption ability, which increased the formation of Rh+ active sites. The mechanism studies observed that there were more intermediates of CO* and HCOO* produced on the Fe and Na-promoted Rh/CeO2 catalysts compared to the unpromoted catalysts, indicating that the doping of Fe and Na promotes dissociation and hydrogenation capacity of CO2, hence resulting in higher ethanol production, as shown in Fig. 6. However, there was a slight decrease in catalytic activity in the early stage of the reaction, which then became stable after 50 hours. The characterization results of the spent catalyst remained the same as those of the fresh catalyst. The only difference detected was that thermogravimetric analysis (TGA) showed a relatively small weight loss of 6.4% at temperatures ranging between 150 °C and 250 °C, which can be attributed to the desorption of water and organic compound of the spent catalyst surface.
 | ||
| Fig. 6 In situ DRIFTS of various catalysts after switching the feed gas from CO2 to CO2 + H2, followed by returning to CO2 at 250 °C and 0.1 MPa (a) 2Rh/CeO2 (b) 2Rh0.5Fe/CeO2 (c) 2Rh0.5Na/CeO2 and (d) 2Rh0.5Fe0.5Na/CeO2. Reprinted with permission from ref. 51. Copyright 2024 Elsevier. | ||
Yao et al.23 also studied the dual promoter effect by incorporating Na and S into monometallic iron catalysts for HAS. The authors proposed that the sulfur existed in the form of sulfate plays a pivotal role in tuning the CO activation through strong synergistic interaction between Na and S, which increases the amount of adsorbed CO to bind with alkyl species on Na-promoted Fe sites, forming higher alcohols. Experimental results in catalytic performance showed that FeNa catalyst exhibited alcohol selectivity of less than 5% while FeNa catalyst promoted with 0.6 wt% of sulfur through precipitation method, denoted as FeNaS-0.6 catalyst, achieved the maximum alcohol selectivity of 16.1% with more than 98% of C2+OH fraction in total alcohols. The authors also synthesized the FeNaS-im catalyst by introducing the same sulfate content as the FeNaS-0.6 catalyst on FeNa via the wet impregnation method to study the promotional effects of sulfate. The higher alcohol selectivity of the FeNaS-im catalyst was slightly lower than that of the FeNaS-0.6 catalyst, which can be ascribed to the disadvantage of the impregnation method, resulting in non-uniform sulfate distribution and weak interactions between Fe species and sulfate species. It is well-documented that dissociative and non-dissociative CO activation are essential for HAS. In this study, the presence of sulfate with strong electron-withdrawing ability offers additional Fe sites for non-dissociative CO adsorption. At the same time, adding Na helps maintain the CO dissociative ability of Fe sites, highlighting the importance of maintaining a kinetic balance between dissociative and non-dissociative CO activation in HAS.
Role of catalyst supports
Catalyst support plays a pivotal role in CO2 hydrogenation by enhancing metal dispersion, stabilizing active sites, regulating catalyst surface interfaces, influencing key intermediates to alter reaction mechanisms, and improving adsorption or activation capabilities.67 In addition, the construction of SMSI has been demonstrated to effectively stabilize active sites under CO2 hydrogenation reaction conditions.68,69 The SMSI effect is widely acknowledged for its significant influence on the stability, activity, chemical state, and morphology of the catalysts.70 Xin et al.68 demonstrated that by constructing an SMSI state, it is possible to promote the formation of a defective MoO3−x overlayer surrounding Ru nanoparticles (Ru@MoO3−x) at a low temperature of 250 °C. This modification favors RWGS, achieving >99% CO selectivity, which could easily revert to CH4 formation upon O2 treatment. Therefore, constructing SMSI between active metals and support materials is one of the strategies that could be implemented to increase the higher alcohol selectivity, which also operates under low-temperature reaction conditions. For instance, Zhang et al.71 identified that only SiO2 and Si3N4 supports managed to stabilize the Co2C active sites by generating Si–O–Co bonds. In their study, the influence of different catalyst supports (Al2O3, ZnO, AC, TiO2, SiO2, and Si3N4) in ethanol synthesis over supported Na-promoted cobalt catalysts was investigated, as illustrated in Fig. 7. Na–Co/Al2O3, Na–Co/ZnO, Na–Co/AC and Na–Co/TiO2 catalysts exhibited very low alcohol selectivity of less than 1%. In comparison, Na–Co/SiO2 and Na–Co/Si3N4 catalysts exhibited a total alcohol selectivity of 9%. The experimental result was explained by the formation of the Co2C active phase on Na–Co/SiO2 and Na–Co/Si3N4 catalysts, which remained stable even after the reaction. Although the Co2C active phase was also detected on Na–Co/AC and Na–Co/TiO2 catalysts, the Co2C active phase was almost fully decomposed during the reaction condition due to a lack of SMSI. In contrast, no Co2C active phase was found on Na–Co/Al2O3 and Na–Co/ZnO catalysts. The authors inferred that Co2C is the active phase in CO2 hydrogenation, with its stability attributed to SMSI facilitated by the formation of Si–O–Co bonds, which also depends on the nature of the supports utilized. | ||
| Fig. 7 Schematic of Co phase transformation during CO reduction and CO2 hydrogenation reaction on Na–Co catalysts supported on different supports. Reprinted with permission from ref. 71. Copyright 2020 Elsevier. | ||
An et al.41 exploited the SMSI between La4Ga2O9 and cobalt particles, facilitating the formation of active Co0–Co2+ pairs. Perovskite oxides (PTO), with highly tunable composition and structure, can regulate oxygen vacancies through their bulk and surface properties. Co/La4Ga2O9 catalyst was synthesized using perovskite type LaCo1−xGaxO3 support and investigated for CO2 hydrogenation reaction. Ethanol selectivity of 34.7% was obtained over Co/La4Ga2O9 catalyst under reaction conditions of 270 °C, 3.5 MPa and GHSV = 3000 mL gcat−1 h−1. Co nanoparticles loaded on La4Ga2O9 were formed by reducing the LaCo0.5Ga0.5O3 precursor. SMSI between Co and La4Ga2O9 are postulated, resulting in electron donation from cobalt to La4Ga2O9, thus forming Co0–Co2+ pairs as active sites. CO2 is adsorbed and activated on the La4Ga2O9 surface, which favors RWGS in producing CO intermediates. Then, the produced CO intermediates migrate to Co0–Co2+ sites to promote ethanol synthesis. However, it is unfortunate that ethanol yield decreased when reaction time increased. This can be attributed to the increase in the Co0/Co2+ ratio, which caused an imbalance in the formation of intermediates over longer periods. Apart from that, SMSI between Co3O4 particles and isolated silanols, resulting in the formation of Si–O–Co chemical bonds on Co/S-1 catalysts, had also been observed by Ding et al.42 The SMSI was supported by XPS results, which demonstrated an increased number of electronic defects in Co3O4 on Co/S-1 and a higher electron density of Si–O bonds on S-1. This further consolidates the electron transfers from Co3O4 to Si–O bonds, contributing to the presence of Si–O–Co chemical bonds. The active sites of the Co/S-1 catalyst can be tuned through Si–O–Co chemical bonds, which stabilize the co-existing phases of Co0 and CoO (CoOx sites) by preventing the complete reduction of Co3O4. DFT calculations revealed the roles of Co0 and CoO sites in CO2 hydrogenation to ethanol, respectively. Specifically, Co0 sites favor the COOH* species, while CoO sites promote the formation of HCOO* species. The COOH* and HCOO* species are then coupled with 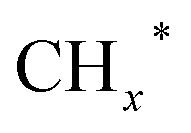 species on CoOx sites to produce ethanol.
species on CoOx sites to produce ethanol.
Moreover, a few studies have investigated the confinement effect of supports to achieve catalyst stability in CO2 hydrogenation. Wang et al.72 recently postulated that the confinement effect in In2O3–TiO2 catalysts promotes the dispersion of the In2O3 guest catalyst onto the TiO2 host surface, thereby improving performance in CO2 hydrogenation to CO. The formation of In–O–Ti interfacial bonds was crucial in inducing In2O3 dispersion and stabilizing the metastable InOx layers. Their findings revealed that the confinement effect at oxide/oxide interfaces benefits CO2 hydrogenation. For CO2 hydrogenation in HAS, Ding et al.35 synthesized the Cu@Na-beta catalyst, embedding the Cu nanoparticles in the beta molecular sieve. The authors highlighted that the synergistic interaction between copper nanoparticles and the beta zeolite framework is essential for HAS catalytic performance as this synergy suppresses the CO2 adsorption site and prevents the over-reduction of the catalyst. Na-beta zeolite as support in Cu@Na-eta catalyst demonstrates a superior confinement effect that shapes the copper nanoparticles into unique configurations with surface sites and limits the reactants along the catalyst surface, thus successfully preventing the formation of by-products such as methanol, formic acid, or acetic acid. In another study, Cu nanoparticles were embedded into BEA and MFI (S-1) zeolites, and their mesoporous equivalents (m-zeolites) were obtained via the carbon templating method.73 Based on the catalytic test result, mesoporous zeolites exhibited a higher CO2 conversion and ethanol selectivity than their non-mesoporous counterparts. Besides, the introduction of mesopores facilitated the formation of isopropanol. Cu-based mesoporous S-1 zeolite (Cu@m-S1), which underwent an additional recrystallization procedure, achieved a higher isopropanol selectivity and yielded 20.51 mmol gcat−1 h−1. The authors claimed that the mesoporous effect of zeolites positively affects the production of alcohol by facilitating the accessibility of reactant access to Cu active sites and improving the SMSI to accelerate alcohol formation. Tran et al.52 developed a Na–Rh–FeOx/ZSM-5 catalyst, which exhibited a higher ethanol selectivity than a Na–Rh–FeOx catalyst without zeolite support. The high specific surface area, smaller pore size, and decrease in iron particle size can be attributed to the excellent synergy between the Na–Rh–FeOx and ZSM-5 support. The ZSM-5-supported catalyst showed a suppression effect on hydrocarbon formation. However, the CO2 conversion did not increase despite the higher density of active sites due to higher surface area. This indicates that the zeolite support can modify the properties of the active sites.
Critical roles of oxygen vacancies
Various catalytic materials (e.g., Rh, Co, Fe, Cu, and Au) are active sites to catalyze CO2 hydrogenation and produce higher alcohols.16,29,74,75 However, monometallic catalysts have limitations and restrictions for HAS from CO2, such as low conversion and selectivity. Thus, metal oxides (e.g., ZrO2, TiO2, and CeO2) are often used as supports or promoters due to their inherent ability to generate abundant oxygen vacancies. The oxygen vacancy is typically recognized as the active site for CO2 adsorption and activation, facilitating the formation of intermediates and promoting SMSI, hence assisting in accelerating CO2 hydrogenation.41,44,76,77 Yang et al.16 synthesized the N-doped CuFeZn catalyst and added an organic ligand, 2,6-pyridine dicarboxylic acid. Accordingly, adding PDA to the CuFeZn catalyst increases the generation of oxygen vacancies, which regulates the electron structure of the catalyst. The oxygen vacancy captures CO2 by trapping O, and the nearby Zn or Cu reacts with the captured CO2 to form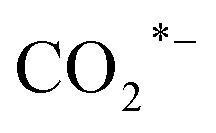 or CO* species. This is most likely because O in CO2 recharges the oxygen vacancy and strengthens the interaction between O and the active metals of the catalyst, assisting in establishing the pathway for electron transfer, facilitating the activation, dissociation, and reduction of CO2 molecules, and thus promoting the formation of higher alcohols. Xi et al.44 reported the K-promoted Fe–In/Ce–ZrO2 catalyst in which the existence of reduced InxOx phases with oxygen vacancies is of great importance for CO2 activation; thereby, these oxygen vacancies will assist CO2 activation in the formation of key reaction intermediates such as HCOO*, H2COO*, and H2CO* species. This study demonstrated that in situ pretreatment with CO at 350 °C for 3 h boosted the oxygen vacancy by 348.6% compared to air-calcination treatment. Moreover, Li et al.17 reported tailoring the Fermi level of a Cu-doped Zn catalyst synthesized using the urea homogeneous hydrolysis technique, suggesting that a lower Fermi level promotes better carbon chain growth. The electron trap effect of oxygen vacancy regulates the Fermi level of semiconductors. It influences their catalytic performance via charge transfer between the surface and adsorbed substances with an imbalanced electron structure, resulting in fast electron transfer. Thus, oxygen vacancies could boost the formation of higher alcohols. Besides, surface oxygen vacancies also play a key role in promoting CO2 adsorption and activation. In the Ir1–In2O3 SAC catalyst, the oxygen vacancies combine with monoatomic Ir to form Lewis acid–base pairs, which serve as active sites for CO2 adsorption and activation into CO* intermediates. The Irδ+–CO* intermediates interact with the CH3O*–Ov intermediates, thus enhancing ethanol selectivity.46
or CO* species. This is most likely because O in CO2 recharges the oxygen vacancy and strengthens the interaction between O and the active metals of the catalyst, assisting in establishing the pathway for electron transfer, facilitating the activation, dissociation, and reduction of CO2 molecules, and thus promoting the formation of higher alcohols. Xi et al.44 reported the K-promoted Fe–In/Ce–ZrO2 catalyst in which the existence of reduced InxOx phases with oxygen vacancies is of great importance for CO2 activation; thereby, these oxygen vacancies will assist CO2 activation in the formation of key reaction intermediates such as HCOO*, H2COO*, and H2CO* species. This study demonstrated that in situ pretreatment with CO at 350 °C for 3 h boosted the oxygen vacancy by 348.6% compared to air-calcination treatment. Moreover, Li et al.17 reported tailoring the Fermi level of a Cu-doped Zn catalyst synthesized using the urea homogeneous hydrolysis technique, suggesting that a lower Fermi level promotes better carbon chain growth. The electron trap effect of oxygen vacancy regulates the Fermi level of semiconductors. It influences their catalytic performance via charge transfer between the surface and adsorbed substances with an imbalanced electron structure, resulting in fast electron transfer. Thus, oxygen vacancies could boost the formation of higher alcohols. Besides, surface oxygen vacancies also play a key role in promoting CO2 adsorption and activation. In the Ir1–In2O3 SAC catalyst, the oxygen vacancies combine with monoatomic Ir to form Lewis acid–base pairs, which serve as active sites for CO2 adsorption and activation into CO* intermediates. The Irδ+–CO* intermediates interact with the CH3O*–Ov intermediates, thus enhancing ethanol selectivity.46
Reaction mechanisms of CO2 hydrogenation for HAS
The reaction mechanisms for the synthesis of higher alcohols from CO2 hydrogenation typically involve the following steps: the initial stage for the activation of CO2 and H2, resulting in the formation of C1 intermediates including CO*,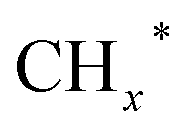 , and HCOO* species; and the second stage for the production of ethanol or higher alcohols with a longer carbon chain (C2+–C4+) through C–C coupling of
, and HCOO* species; and the second stage for the production of ethanol or higher alcohols with a longer carbon chain (C2+–C4+) through C–C coupling of 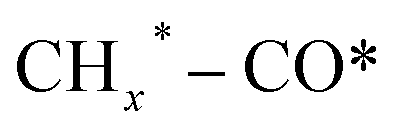 (a CO-mediated pathway) or
(a CO-mediated pathway) or 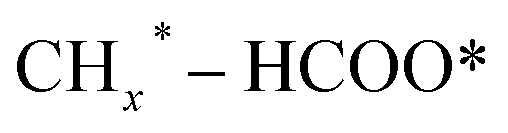 (a formate-mediated pathway).17,29,37,44,71,78 Yang et al.16 proposed reaction mechanisms for HAS from CO2 hydrogenation over N-doped CuZnFe, as illustrated in Fig. 8. The active sites (e.g., Cu0, ZnO, Fe3O4, and Fe5C2) adsorb the reactant molecules. Through capturing O from CO2, the oxygen vacancies generated on the surface of ZnO facilitate the adsorption and activation of CO2 molecules. The weak base of doped N also contributes to activating CO2 molecules. This leads to the generation of
(a formate-mediated pathway).17,29,37,44,71,78 Yang et al.16 proposed reaction mechanisms for HAS from CO2 hydrogenation over N-doped CuZnFe, as illustrated in Fig. 8. The active sites (e.g., Cu0, ZnO, Fe3O4, and Fe5C2) adsorb the reactant molecules. Through capturing O from CO2, the oxygen vacancies generated on the surface of ZnO facilitate the adsorption and activation of CO2 molecules. The weak base of doped N also contributes to activating CO2 molecules. This leads to the generation of 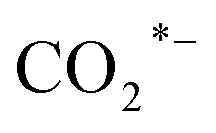 species as CO2 captures electrons over the surface of ZnO or N. At the interface of Cu–ZnO, H2 is dissociated and activated due to the vacant s orbital of the Cu species. Through the RWGS reaction, at this interface of Cu–ZnO, CO2 is dissociated to form CO* species. Subsequently, CO* species may undergo hydrogenation to form methanol or desorb to produce CO. Furthermore, at the Cu–FeC2 interface, a considerable portion of CO* dissociates, resulting in C* and O* radicals. The C* radical is further hydrogenated to generate the alkyl species
species as CO2 captures electrons over the surface of ZnO or N. At the interface of Cu–ZnO, H2 is dissociated and activated due to the vacant s orbital of the Cu species. Through the RWGS reaction, at this interface of Cu–ZnO, CO2 is dissociated to form CO* species. Subsequently, CO* species may undergo hydrogenation to form methanol or desorb to produce CO. Furthermore, at the Cu–FeC2 interface, a considerable portion of CO* dissociates, resulting in C* and O* radicals. The C* radical is further hydrogenated to generate the alkyl species 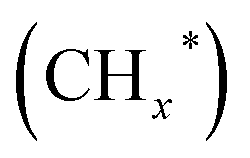 , and carbon chain growth occurs through continuously inserting
, and carbon chain growth occurs through continuously inserting 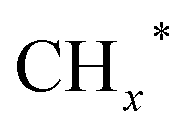 to form hydrocarbons (CnHm). Subsequently, CO* is inserted into CnHm on Cu sites to form higher alcohols (C2+).
to form hydrocarbons (CnHm). Subsequently, CO* is inserted into CnHm on Cu sites to form higher alcohols (C2+).
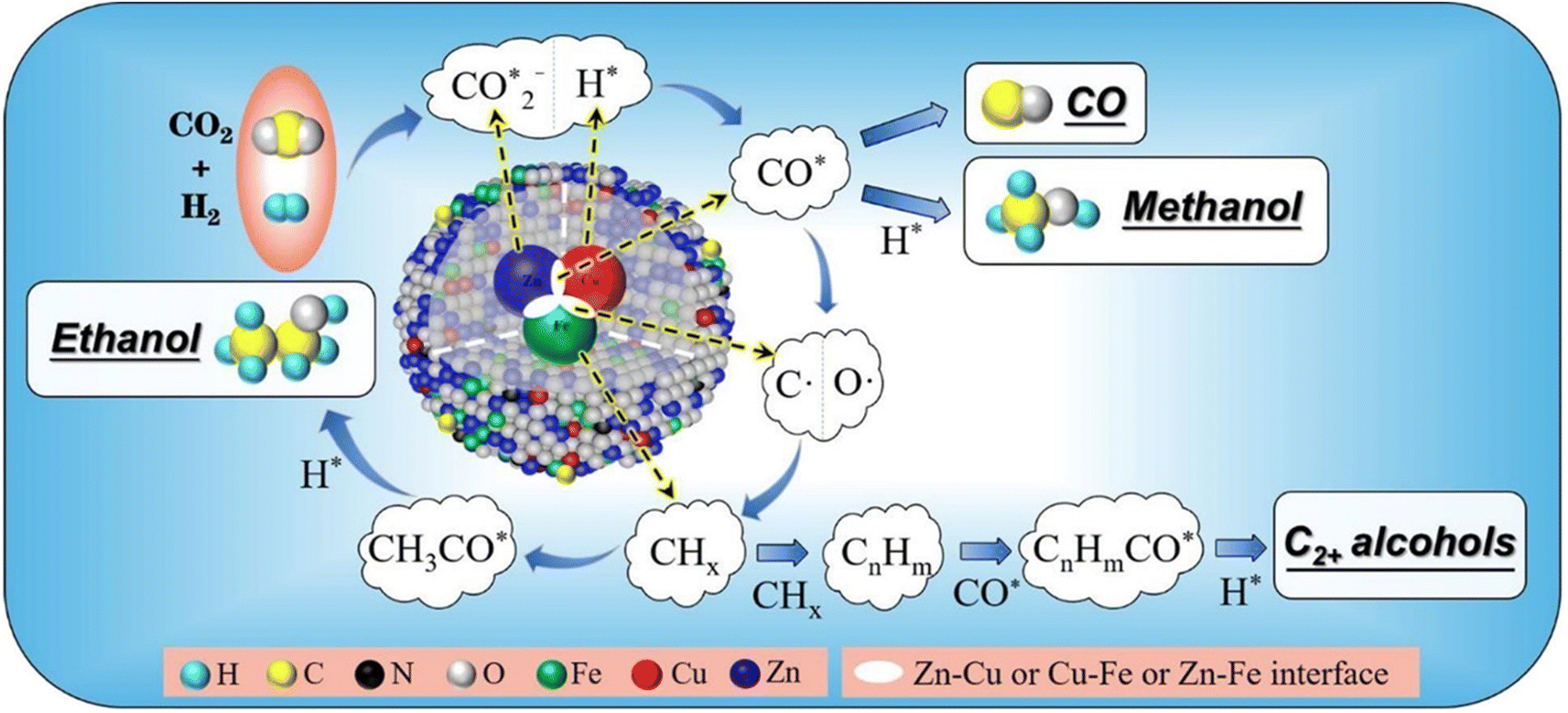 | ||
Fig. 8 The weak base of doped N promotes the generation of 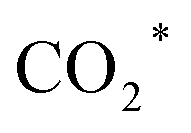 species through CO2 activation, which is further regulated by the CuZnFe catalyst to form intermediates including CO*, CHx, and HCOO* species to form higher alcohols through CO* insertion into CnHm. Reprinted with permission from ref. 16. Copyright 2022 Elsevier. species through CO2 activation, which is further regulated by the CuZnFe catalyst to form intermediates including CO*, CHx, and HCOO* species to form higher alcohols through CO* insertion into CnHm. Reprinted with permission from ref. 16. Copyright 2022 Elsevier. | ||
Zhou et al.26 proposed a reaction pathway for HAS from CO2 hydrogenation over a 0.3K–1Pd/Fe2O3 catalyst, as illustrated in Fig. 9. The authors investigated the reaction intermediates (e.g., CO, C2H5O*, CH3CHO*, CH3CH2O*, and CH4) after injecting CO2/H2 over the surface of the catalyst by in situ DRIFTS. The introduction of highly dispersed K (0.3 wt%) restricts the hydrogenation of the 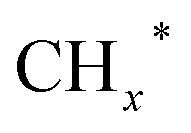 species. It enhances the ratio of
species. It enhances the ratio of 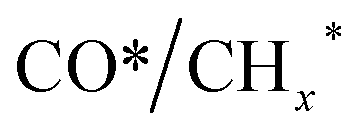 , promoting the insertion of CO, while introducing 1% Pd increases the formation rate of
, promoting the insertion of CO, while introducing 1% Pd increases the formation rate of 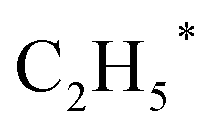 species. The synergy of K and PdFe alloys on a 0.3K–1Pd/Fe2O3 catalyst can regulate the ratio of
species. The synergy of K and PdFe alloys on a 0.3K–1Pd/Fe2O3 catalyst can regulate the ratio of 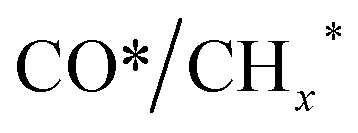 on the surface of the catalyst. The observed key reaction intermediates by in situ DRIFTS suggest the reaction pathway following the CO-mediated reaction mechanism by the insertion of CO* into
on the surface of the catalyst. The observed key reaction intermediates by in situ DRIFTS suggest the reaction pathway following the CO-mediated reaction mechanism by the insertion of CO* into 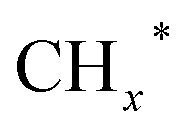 species to form intermediates (CHxCO*) through C–C coupling, which may instantly conjugate CO* and
species to form intermediates (CHxCO*) through C–C coupling, which may instantly conjugate CO* and 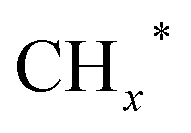 species, resulting in the formation of higher alcohols. Additionally, the study performed CO-TPSR-MS experiments to investigate the influence of K and Pd loading on Fe2O3 towards the behavior of the dissociative and non-dissociative activations of CO by measuring the ratio of CO2/CH4 from the desorption peak area. The signals CH4 and CO2 signify the dissociative activation of CO and the non-dissociative activation of CO, respectively. Fe2O3 shows a strong dissociative activation of CO. The addition of 1% Pd to 1Pd/Fe2O3 slightly enhances the non-dissociative activation of CO, which is attributed to the formation of PdFe alloys. However, adding 0.3% K to 0.3K/Fe2O3 shows a significant non-dissociative activation of CO. The CO-TPSR-MS experiments indicate that the synergy of K and PdFe alloys along with Fe7C3 regulates a favorable ratio of
species, resulting in the formation of higher alcohols. Additionally, the study performed CO-TPSR-MS experiments to investigate the influence of K and Pd loading on Fe2O3 towards the behavior of the dissociative and non-dissociative activations of CO by measuring the ratio of CO2/CH4 from the desorption peak area. The signals CH4 and CO2 signify the dissociative activation of CO and the non-dissociative activation of CO, respectively. Fe2O3 shows a strong dissociative activation of CO. The addition of 1% Pd to 1Pd/Fe2O3 slightly enhances the non-dissociative activation of CO, which is attributed to the formation of PdFe alloys. However, adding 0.3% K to 0.3K/Fe2O3 shows a significant non-dissociative activation of CO. The CO-TPSR-MS experiments indicate that the synergy of K and PdFe alloys along with Fe7C3 regulates a favorable ratio of 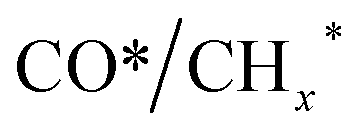 on the surface of the 0.3K–1Pd/Fe2O3 by balancing the dissociative and non-dissociative activations of CO This facilitates the C–C coupling of CO* and
on the surface of the 0.3K–1Pd/Fe2O3 by balancing the dissociative and non-dissociative activations of CO This facilitates the C–C coupling of CO* and 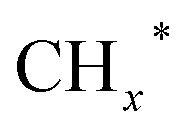 species to produce higher alcohols.
species to produce higher alcohols.
 | ||
| Fig. 9 Proposed reaction pathway that shows the non-dissociative activation of CO regulated by K and Pd to balance the dissociative activation of CO regulated by Fe2O3 catalyst for promoting the C–C coupling mechanism. Reprinted with permission from ref. 26. Copyright 2024 American Chemical Society. | ||
Xu et al.37 proposed an integrated reaction pathway of CO2 hydrogenation over a Cs–CuFeZn catalyst with a favorable balance of synergistic sites (Cu–ZnO and copper–iron carbide interfaces), as illustrated in Fig. 10. At the CuZnO interfaces, methanol formation proceeds directly from CO2 through HCOO* intermediates following the formate pathway. A proper proportion of the Cs promoter reduces the rate of methanol formation, which is favorable to HAS. Furthermore, at high temperatures, particularly above 300 °C, thermodynamics limits the reaction equilibrium and causes methanol to convert back to CO2 and H2, while the formation of CO is considerably accelerated. However, at high temperatures, the formation of higher alcohols is more favorable because Cu–ZnO produces CO through the RWGS reaction. At the same time, copper–iron carbide facilitates the insertion of C(H)O* species to surface hydrocarbon moieties to produce C2+OH at a similar rate. Furthermore, the Cs promoter is essential for driving the CO insertion reaction during the CO2-to-HA process by regulating the hydrogenation reaction over the CuFeZn catalyst. Since it still does not reach its thermodynamic equilibrium, the rate of C2+OH formation increases as temperature increases. Likewise, a large C2+OH/ROH proportion is produced at high temperatures because of the comparatively slow rate of methanol synthesis in conjunction with the increased rate of C2+OH formation.
 | ||
| Fig. 10 Proposed reaction mechanism of CO2 hydrogenation for higher alcohols through C(H)O* insertion and CO insertion into hydrocarbons over Cs–CuFeZn catalysts. Reprinted with permission from ref. 37. Copyright 2020 American Chemical Society. | ||
Li et al.17 proposed the reaction pathway for HAS from CO2 hydrogenation over an electronically modified ZnO catalyst, as illustrated in Fig. 11. According to the analysis of the characterization and the activity of an electronically modified ZnO catalyst, the synergistic effect between ZnO and Cu drives the catalytic activity of CO2 hydrogenation, which helps to propose the reaction mechanism for HAS. ZnO plays a critical role in facilitating the activation of CO and H2 and promoting the dissociation of the C–O bond. Moreover, the oxygen deficiency species (Znx+, 0 < x < 2) promotes carbon chain growth in ZnO. The non-dissociative hydrogenation of CO to generate methoxy species is facilitated by Cu embedded on the ZnO surface. Furthermore, Cu controls the regulation of the electronically modified Fermi level within the ZnO lattice and thus accelerates the electron transfer, facilitating the formation of surface alkyl species (CHx) by direct C–O dissociation or H-assisted C–O dissociation. As a result, the mechanism of CO insertion with CHx–C(Hx)O coupling produces higher alcohols (C2+).
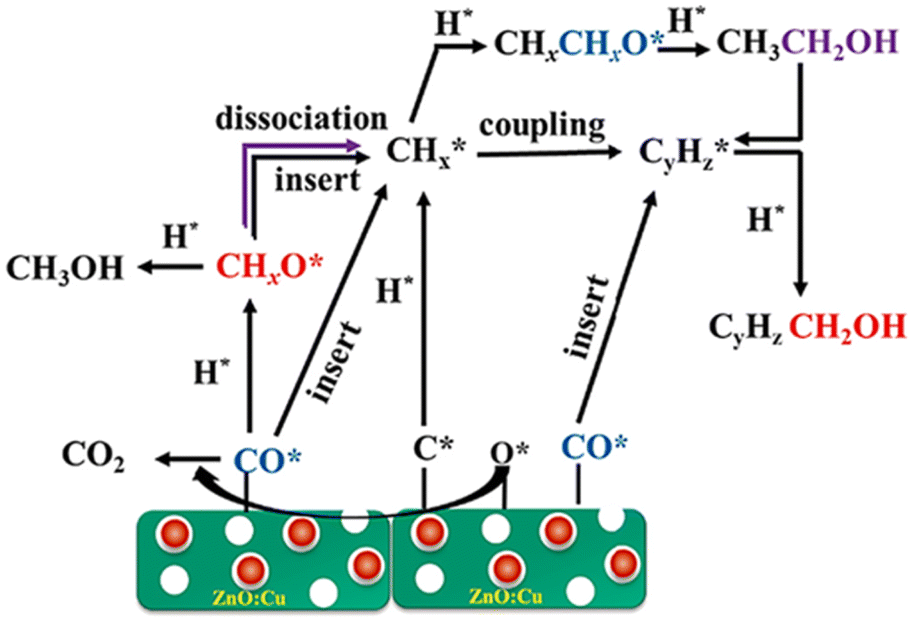 | ||
| Fig. 11 Electronically modified Fermi level facilitates the formation of alkyl species to promote CO insertion with CHx–C(Hx)O coupling for higher alcohols. Reprinted with permission from ref. 17. Copyright 2023 Elsevier. | ||
Ye et al.53 proposed the reaction pathway for ethanol production from CO2 hydrogenation over the βMo2C nanowires with a single Rh atom and an alkali metal K (K0.2Rh0.2/βMo2C), as illustrated in Fig. 12. An unmodified βMo2C catalyst could not convert CO2 into ethanol (or limit the conversion to only methanol). Rh loading on the βMo2C catalyst promoted the dissociative adsorption of CO2, and the CO* was adsorbed on the monoatomic Rhδ+. Moreover, hydrogen was more likely to be adsorbed and activated on Rh, which helped to prevent the production of methanol and methane. Additionally, Moδ+–CO, Mo2+–CO, and Mo4+–CO exhibited IR peaks at 2094, 2108, and 2129 cm−1, respectively. The peaks at 1081, 1053, and 1033 cm−1 were assigned to strongly adsorbed carbonate species (CO3δ−) formed on the Rh1.0/βMo2C. Thus, the bifunctional catalyst of monoatomic Rh on βMo2C was tailored for promoting the C–C coupling. However, achieving high ethanol selectivity by merely adjusting the Rh content was challenging, as increased Rh led to changes in its dispersion and enhanced oxidation of the carrier, resulting in dominant methanol production under mild conditions. Effective C–C coupling relied on SAC bifunctional active centers, where optimized synergy between the two catalytic pathways was essential for efficient carbon chain growth. To further improve the selectivity of ethanol, introducing K assisted in the adsorption and activation of CO2 on the βMo2C surface by generating more carbonate species. Furthermore, adding an appropriate amount of K assisted in regulating the carrier properties by controlling the H2 activation, which could effectively reduce the formation of methanol, yielding a higher selectivity of ethanol. In addition, the nature of single atom Rh was retained to promote the C–C coupling, resulting in ethanol formation.
 | ||
| Fig. 12 Proposed reaction mechanism of CO2 hydrogenation to ethanol on the β-Mo2C nanowires with a single Rh atom for promoting the C–C coupling mechanism and an introduction of an alkali metal K to regulate the balanced performance of the two active centers for improving the selectivity of ethanol. Reprinted with permission from ref. 53. Copyright 2022 Elsevier. | ||
Conclusions and future perspectives
CO2 hydrogenation to higher alcohols is a novel strategy to mitigate CO2 emissions. Efforts have been made to improve the CO2 conversion and resolve the unsatisfactory selectivity towards higher alcohols. The stability of the catalyst is also a key issue in CO2 hydrogenation due to catalyst deactivation. The construction of catalysts with various combinations of metals and metal oxides has garnered attention and emerged as a rising trend in enhancing HAS. The introduction of second active metal sites has been proven to hydrogenate the reaction to higher alcohols instead of C1 product formation, such as methane and methanol. However, designing an ideal catalyst is not easy to achieve. It requires precise control of interplay between active metal sites, supports, and promoters, which could assist in enhanced dispersion of the active metal sites, modify electronic properties, assist in the adsorption and activation of CO2 on the surface of catalysts, control the activation of H2, and promote the carbon chain growth, which could suppress the formation of methane and methanol and thus enhancing the activity and selectivity towards higher alcohols. These factors induce a synergistic interaction among the active metal, the support, and the promoter to facilitate a higher conversion of CO2 with enhanced selectivity for higher alcohols, as discussed in detail in the case-by-case studies presented in this review. Our understanding remains limited despite significant efforts over the past few years to study catalyst design for product selectivity control and mechanistic investigation in HAS. Unlike the well-established and commercially industrialized C1 production, such as methanol synthesis, the complexity of these reaction pathways and the challenges in catalyst design (requiring a balance among product selectivity, catalyst stability, and catalytic activity) pose substantial hurdles. Therefore, in-depth investigations of the nature of active sites and reaction pathways through combining advanced characterization (e.g., in situ XPS, in situ DRIFTS, and CO-TPSR-MS) with computational catalysts (e.g., DFT calculations and machine learning) are strongly recommended to develop highly efficient and cost-effective catalysts. After all, future trends in catalyst design can focus on tailoring the interaction between the active metal sites and the support through the formation of strong metal support interaction (SMSI) to optimize the catalytic activity of catalysts, adjusting the appropriate amount of promoter to enhance the carbon chain growth, prevent the formation of C1 products, and boost the selectivity towards higher alcohols. Additionally, promoting the confinement effect of supports to reduce particle size and understanding the reaction mechanism to predict the catalytic reaction pathway are essential for achieving a higher conversion of CO2 and greater selectivity for higher alcohols.Data availability
The data supporting the findings in this study are sourced from existing literature. Citations are given throughout the paper. Readers may contact the corresponding author, A. C. K. Y., to inquire about this review paper.Author contributions
A. T.: methodology, investigation, data curation, formal analysis, writing – original draft, writing – review & editing; M. I.: methodology, investigation, data curation, formal analysis, writing – original draft, writing – review & editing; M. P.: formal analysis, writing – original draft, writing – review & editing, funding acquisition; A. C. K. Y.: methodology, investigation, data curation, formal analysis, writing – original draft, writing – review & editing, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the Second Century Fund (C2F), Chulalongkorn University.References
- J. Zhong, X. Yang, Z. Wu, B. Liang, Y. Huang and T. Zhang, Chem. Soc. Rev., 2020, 49, 1385–1413 RSC.
- J. Rogelj, D. Huppmann, V. Krey, K. Riahi, L. Clarke, M. Gidden, Z. Nicholls and M. Meinshausen, Nature, 2019, 573, 357–363 CrossRef CAS.
- Y. Gao, X. Gao and X. Zhang, Engineering, 2017, 3, 272–278 CrossRef.
- G. Yergaziyeva, Z. Kuspanov, M. Mambetova, N. Khudaibergenov, N. Makayeva and C. Daulbayev, J. CO2 Util., 2024, 80, 102682 CrossRef CAS.
- R.-P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell, Q. Li, M. Fan and Y.-G. Yao, Nat. Commun., 2019, 10, 5698 CrossRef CAS.
- Y. Jiang, K. Wang, Y. Wang, Z. Liu, X. Gao, J. Zhang, Q. Ma, S. Fan, T.-S. Zhao and M. Yao, J. CO2 Util., 2023, 67, 102321 CrossRef CAS.
- S. Aslam, S. Rani, K. Lal, M. Fatima, T. Hardwick, B. Shirinfar and N. Ahmed, Green Chem., 2023, 25, 9543–9573 RSC.
- I. Dincer, Int. J. Hydrogen Energy, 2012, 37, 1954–1971 CrossRef CAS.
- A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Chem. Rev., 2017, 117, 9804–9838 CrossRef PubMed.
- P. Gao, L. Zhang, S. Li, Z. Zhou and Y. Sun, ACS Cent. Sci., 2020, 6, 1657–1670 CrossRef CAS PubMed.
- F. Dalena, A. Senatore, M. Basile, S. Knani, A. Basile and A. Iulianelli, Membranes, 2018, 8, 98 CrossRef.
- M. A. Ratcliff, J. Luecke, A. Williams, E. Christensen, J. Yanowitz, A. Reek and R. L. McCormick, Environ. Sci. Technol., 2013, 47, 13865–13872 CrossRef CAS PubMed.
- V. F. Andersen, J. E. Anderson, T. J. Wallington, S. A. Mueller and O. J. Nielsen, Energy Fuels, 2010, 24, 3647–3654 CrossRef CAS.
- D. Xu, Y. Wang, M. Ding, X. Hong, G. Liu and S. C. E. Tsang, Chem, 2021, 7, 849–881 CAS.
- J. M. Spero, B. DeVito and L. Theodore, Regulatory Chemicals Handbook, CRC Press, 1st edn, 2000, DOI:10.1201/9781482270389.
- C. Yang, B. Wang, Y. Wen, M. Fan, Y. Jia, S. Zhou and W. Huang, Fuel, 2022, 327, 125055 CrossRef CAS.
- F. Li, P. Jia, Q. Zhang, Y. Liu, V. A. Vinokurov and W. Huang, Fuel Process. Technol., 2023, 241, 107600 CrossRef CAS.
- G. Cui, Y. Lou, M. Zhou, Y. Li, G. Jiang and C. Xu, Catalysts, 2024, 14, 232 CrossRef CAS.
- A. I. Latsiou, N. D. Charisiou, Z. Frontistis, A. Bansode and M. A. Goula, Catal. Today, 2023, 420, 114179 CrossRef CAS.
- D. Xu, M. Ding, X. Hong and G. Liu, ACS Catal., 2020, 10, 14516–14526 CrossRef CAS.
- B. An, Z. Li, Y. Song, J. Zhang, L. Zeng, C. Wang and W. Lin, Nat. Catal., 2019, 2, 709–717 CrossRef CAS.
- J. Nebel, S. Schmidt, Q. Pan, K. Lotz, S. Kaluza and M. Muhler, Chin. J. Catal., 2019, 40, 1731–1740 CrossRef.
- R. Yao, J. Wei, Q. Ge, J. Xu, Y. Han, Q. Ma, H. Xu and J. Sun, Appl. Catal., B, 2021, 298, 120556 CrossRef CAS.
- R. Yao, B. Wu, Y. Yu, N. Liu, Q. Niu, C. Li, J. Wei and Q. Ge, Appl. Catal., B, 2024, 355, 124159 CrossRef CAS.
- Y. Wang, K. Wang, B. Zhang, X. Peng, X. Gao, G. Yang, H. Hu, M. Wu and N. Tsubaki, ACS Catal., 2021, 11, 11742–11753 CrossRef CAS.
- Y. Zhou, Y. Wang, H. Lu, T. Liu, X. Hong and G. Liu, ACS Sustain. Chem. Eng., 2024, 12, 3322–3330 CrossRef CAS.
- L. Wang, L. Wang, J. Zhang, X. Liu, H. Wang, W. Zhang, Q. Yang, J. Ma, X. Dong, S. J. Yoo, J. G. Kim, X. Meng and F. S. Xiao, Angew Chem. Int. Ed. Engl., 2018, 57, 6104–6108 CrossRef CAS PubMed.
- J. Graciani, D. C. Grinter, P. J. Ramírez, R. M. Palomino, F. Xu, I. Waluyo, D. Stacchiola, J. Fdez Sanz, S. D. Senanayake and J. A. Rodriguez, ACS Catal., 2022, 12, 15097–15109 CrossRef CAS.
- G. Wang, R. Luo, C. Yang, J. Song, C. Xiong, H. Tian, Z.-J. Zhao, R. Mu and J. Gong, Sci. China: Chem., 2019, 62, 1710–1719 CrossRef CAS.
- B. Ouyang, S. Xiong, Y. Zhang, B. Liu and J. Li, Appl. Catal., A, 2017, 543, 189–195 CrossRef CAS.
- D. Wang, Q. Bi, G. Yin, W. Zhao, F. Huang, X. Xie and M. Jiang, Chem. Commun., 2016, 52, 14226–14229 RSC.
- M. Kishida, K. Yamada, H. Nagata and K. Wakabayashi, Chem. Lett., 2006, 23, 555–556 CrossRef.
- B. Kang, S. Qi, X. Wang, F. Bai, N. Wang, R. Hu, Y. Zhang and H. Su, Catal. Commun., 2020, 137, 105945 CrossRef CAS.
- S. Liu, H. Zhou, Q. Song and Z. Ma, J. Taiwan Inst. Chem. Eng., 2017, 76, 18–26 CrossRef CAS.
- L. Ding, T. Shi, J. Gu, Y. Cui, Z. Zhang, C. Yang, T. Chen, M. Lin, P. Wang, N. Xue, L. Peng, X. Guo, Y. Zhu, Z. Chen and W. Ding, Chem, 2020, 6, 2673–2689 CAS.
- M. Irshad, H.-J. Chun, M. K. Khan, H. Jo, S. K. Kim and J. Kim, Appl. Catal., B, 2024, 340, 123201 CrossRef CAS.
- D. Xu, M. Ding, X. Hong, G. Liu and S. C. E. Tsang, ACS Catal., 2020, 10, 5250–5260 CrossRef CAS.
- D. Xu, H. Yang, X. Hong, G. Liu and S. C. Edman Tsang, ACS Catal., 2021, 11, 8978–8984 CrossRef CAS.
- Q. Zhang, S. Wang, R. Geng, P. Wang, M. Dong, J. Wang and W. Fan, Appl. Catal., B, 2023, 337, 123013 CrossRef CAS.
- S. Zhang, Z. Wu, X. Liu, Z. Shao, L. Xia, L. Zhong, H. Wang and Y. Sun, Appl. Catal., B, 2021, 293, 120207 CrossRef CAS.
- K. An, S. Zhang, J. Wang, Q. Liu, Z. Zhang and Y. Liu, J. Energy Chem., 2021, 56, 486–495 CrossRef CAS.
- X. Ding, J. Fu, Y. Lyu, L. Ma, Y. Xu and X. Liu, Chem. Eng. J., 2024, 494, 152923 CrossRef CAS.
- T. Witoon, T. Numpilai, S. Nijpanich, N. Chanlek, P. Kidkhunthod, C. K. Cheng, K. H. Ng, D.-V. N. Vo, S. Ittisanronnachai, C. Wattanakit, M. Chareonpanich and J. Limtrakul, Chem. Eng. J., 2022, 431, 133211 CrossRef CAS.
- X. Xi, F. Zeng, H. Zhang, X. Wu, J. Ren, T. Bisswanger, C. Stampfer, J. P. Hofmann, R. Palkovits and H. J. Heeres, ACS Sustain. Chem. Eng., 2021, 9, 6235–6249 CrossRef CAS.
- H. Yang, Z. Wei, J. Zhang, Y. Dang, S. Li, X. Bu, Z. Zhou, C. Gong, H. Wang, J. Li, Y. Liu, Y. Yang, T. Xiao, C. Liu, Y. Sun and P. Gao, Chem, 2024, 10, 2245–2265 Search PubMed.
- X. Ye, C. Yang, X. Pan, J. Ma, Y. Zhang, Y. Ren, X. Liu, L. Li and Y. Huang, J. Am. Chem. Soc., 2020, 142, 19001–19005 CrossRef CAS PubMed.
- Y. Lou, F. jiang, W. Zhu, L. Wang, T. Yao, S. Wang, B. Yang, B. Yang, Y. Zhu and X. Liu, Appl. Catal., B, 2021, 291, 120122 CrossRef CAS.
- S. Bai, Q. Shao, P. Wang, Q. Dai, X. Wang and X. Huang, J. Am. Chem. Soc., 2017, 139, 6827–6830 CrossRef CAS PubMed.
- Y. Wang, Y. Zhou, X. Zhang, M. Wang, T. Liu, J. Wei, G. Zhang, X. Hong and G. Liu, Appl. Catal., B, 2024, 345, 123691 CrossRef CAS.
- F. Zhang, W. Zhou, X. Xiong, Y. Wang, K. Cheng, J. Kang, Q. Zhang and Y. Wang, J. Phys. Chem. C, 2021, 125, 24429–24439 CrossRef CAS.
- S. Ji, F. Hong, D. Mao, Q. Guo and J. Yu, Chem. Eng. J., 2024, 495, 153633 CrossRef CAS.
- C.-C. Tran and S. Kaliaguine, Chem. Eng. J., 2024, 496, 153636 CrossRef CAS.
- X. Ye, J. Ma, W. Yu, X. Pan, C. Yang, C. Wang, Q. Liu and Y. Huang, J. Energy Chem., 2022, 67, 184–192 CrossRef CAS.
- H. Lei, R. Nie, G. Wu and Z. Hou, Fuel, 2015, 154, 161–166 CrossRef CAS.
- H. Lei, Z. Hou and J. Xie, Fuel, 2016, 164, 191–198 CrossRef CAS.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703 RSC.
- Y. Suo, Y. Yao, Y. Zhang, S. Xing and Z.-Y. Yuan, J. Ind. Eng. Chem., 2022, 115, 92–119 CrossRef CAS.
- J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek, J. F. Sanz and J. A. Rodriguez, Science, 2014, 345, 546–550 CrossRef CAS PubMed.
- C. Yang, S. Liu, Y. Wang, J. Song, G. Wang, S. Wang, Z.-J. Zhao, R. Mu and J. Gong, Angew. Chem., Int. Ed., 2019, 58, 11242–11247 CrossRef CAS PubMed.
- M. Zhang, R. Yao, H. Jiang, G. Li and Y. Chen, Appl. Surf. Sci., 2017, 412, 342–349 CrossRef CAS.
- S. Liu, C. Yang, S. Zha, D. Sharapa, F. Studt, Z.-J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2022, 61, e202109027 CrossRef CAS PubMed.
- G. Prieto, ChemSusChem, 2017, 10, 1056–1070 CrossRef CAS.
- N. A. Sholeha, H. Holilah, H. Bahruji, A. Ayub, N. Widiastuti, R. Ediati, A. A. Jalil, M. Ulfa, N. Masruchin, R. E. Nugraha and D. Prasetyoko, S. Afr. J. Chem. Eng., 2023, 44, 14–30 Search PubMed.
- M. Wang, P. Wang, G. Zhang, Z. Cheng, M. Zhang, Y. Liu, R. Li, J. Zhu, J. Wang, K. Bian, Y. Liu, F. Ding, T. P. Senftle, X. Nie, Q. Fu, C. Song and X. Guo, Sci. Adv., 2023, 9, eadg0167 CrossRef CAS PubMed.
- H. Kusama, K. Okabe, K. Sayama and H. Arakawa, Catal. Today, 1996, 28, 261–266 CrossRef CAS.
- K. Kitamura Bando, K. Soga, K. Kunimori and H. Arakawa, Appl. Catal., A, 1998, 175, 67–81 CrossRef CAS.
- X. Li, M. Song, Y. Zhou, P. Zhou, D. Xu, T. Liu and X. Hong, ChemCatChem, 2024, 16, e202301577 CrossRef CAS.
- H. Xin, L. Lin, R. Li, D. Li, T. Song, R. Mu, Q. Fu and X. Bao, J. Am. Chem. Soc., 2022, 144, 4874–4882 CrossRef CAS.
- W. Zhang, H. Lin, Y. Wei, X. Zhou, Y. An, Y. Dai, Q. Niu, T. Lin and L. Zhong, ACS Catal., 2024, 14, 2409–2417 CrossRef CAS.
- M. Ren, Y. Zhang, X. Wang and H. Qiu, Catalysts, 2022, 12, 403 CrossRef CAS.
- S. Zhang, X. Liu, Z. Shao, H. Wang and Y. Sun, J. Catal., 2020, 382, 86–96 CrossRef CAS.
- J. Wang, R. Li, G. Zhang, C. Dong, Y. Fan, S. Yang, M. Chen, X. Guo, R. Mu, Y. Ning, M. Li, Q. Fu and X. Bao, J. Am. Chem. Soc., 2024, 146, 5523–5531 CrossRef CAS PubMed.
- D. Iltsiou, J. Mielby and S. Kegnaes, Chempluschem, 2024, 89, e202300313 CrossRef CAS PubMed.
- B. Liu, B. Ouyang, Y. Zhang, K. Lv, Q. Li, Y. Ding and J. Li, J. Catal., 2018, 366, 91–97 CrossRef CAS.
- S. Zhang, K. An, J. Xin, Y. Jiang, M. Niu, P. Song, C. Wu, H. Wang and Y. Liu, Chem. Eng. J., 2024, 486, 150334 CrossRef CAS.
- C. Zhang, L. Wang, U. J. Etim, Y. Song, O. M. Gazit and Z. Zhong, J. Catal., 2022, 413, 284–296 CrossRef CAS.
- T. Liu, D. Xu, M. Song, X. Hong and G. Liu, ACS Catal., 2023, 13, 4667–4674 CrossRef CAS.
- S. Liu, Y. He, W. Fu, J. Chen, J. Ren, L. Liao, R. Sun, Z. Tang, C. Mebrahtu and F. Zeng, J. CO2 Util., 2023, 67, 102322 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |