
Graphene oxide offers precise molecular sieving, structural integrity, microplastic removal, and closed-loop circularity in water-remediating membranes through a covalent adaptable network†
Ria
Sen Gupta†
 ,
Samir
Mandal‡
,
Amit
Malakar‡
,
Siddhesh
Rege
,
Sk. Safikul
Islam
,
Ketaki
Samanta
,
Ashok
Misra
and
Suryasarathi
Bose
*
,
Samir
Mandal‡
,
Amit
Malakar‡
,
Siddhesh
Rege
,
Sk. Safikul
Islam
,
Ketaki
Samanta
,
Ashok
Misra
and
Suryasarathi
Bose
*
Department of Materials Engineering, Indian Institute of Science, Bangalore, 560012, India. E-mail: sbose@iisc.ac.in; Fax: +91-(0)80-2360 0472
First published on 20th November 2023
Abstract
Herein, a scalable method was adopted for hosting membrane reusability via a technique in which a covalent adaptable network was installed in the membrane by covalently anchoring an interpenetrating polymeric network membrane with a Diels–Alder adduct. The membrane was then used for water treatment applications. The presence of the laterally large GO sheets in the membrane matrix helped to control the pore size and contributed towards significant separation performance. The fabricated membrane was characterized by high water flux and nearly 97% rejection of stringent contaminants, including dyes and ions. The hydrophilic membrane surface helped in exhibiting antifouling characteristics and assisted in hitting the age-old selectivity–permeability offset. The cytocompatibility of the designed membranes added required leverage in terms of real-time deployment. The recyclability of the membranes enabled by the dynamic bonds augmented the retention of mechanical properties and satisfactory water-remediating features. This study has the potential to buffer the ill effects of microplastic pollution generated by the deplorable management of membrane disposal and provide a quick remedy in terms of cleaner and greener membranes.
Introduction
Statistical evaluation by the United Nations roughly estimates that nearly two-thirds of the global population face severe water scarcity issues for at least a month every year and by the end of 2030, nearly half of the world population will have little or no access to safe and potable drinking water.1–3 To remediate this crisis, steps should be taken as a priority to come up with sustainable solutions of water purification, even from non-conventional sources. Membrane separations, although they fit all the criteria mentioned above ,4 suffer heavily from fouling and scaling issues which greatly impair basic membrane performance.5 In general, PVDF is the most widely used polymer for the fabrication of membranes owing to its chemical inertness, significantly high mechanical strength, robustness, thermal integrity and stability, as well as aging resistance. Moreover, PVDF can be easily processed into a variety of configurations, be it flat sheets, tubular structures, or hollow fiber membranes. Thus, state-of-the-art membranes are usually made of polymers that are non-recyclable or non-biodegradable, and hence most of the membranes, after their use, end up in landfill or are completely burned.6,7 This adds to the global carbon footprint and cuts down on the sustainability aspect of the membranes.8Many studies have already been reported on membranes from biodegradable polymers, but research concerning their reusability is still in its nascency.9 Thus, it is of utmost importance to design membranes with circular life regimes and enhanced antifouling characteristics while maintaining their robust separation performance. This initiative also calls for a significant research focus on managing the end-use fate of the membranes.10,11 These steps would ensure augmented sustainability along with proper management in terms of membrane disposal.
From this perspective, covalent adaptable networks (CANs) can offer numerous advantages.12–16 The dynamic covalent bonds generated can endow the membranes with advantageous properties in terms of high chemical and thermal stability. They can also help to realize the recyclability aspect of used and fouled membranes, thus contributing towards a decline in the carbon footprint generated by membrane incineration. However, in order to increase the mechanical robustness of the membrane, strategies should be integrated which not only enable the formation of dynamic covalent bonds but also retain the mechanical durability of the membranes.
To this end, integrating engineering aspects of covalent adaptable networks and interpenetrating polymeric networks can have numerous advantages. The synergism of both strategies leads to the development of a system with enhanced mechanical properties coupled with the desired recyclability. It also helps in tailoring the pore sizes of membranes which in turn is highly important for target-specific applications.17,18
Herein, we have thoughtfully screened a pair of diene and dienophile for the successful formation of thermos-reversible CANs via a Diels–Alder (DA) reaction pathway. Diels–Alder was selected since the adduct can be formed at room temperature without posing any risk to the porous structure of the membrane.17,19 The DA precursors and the IPN components were chosen such that they could have specific interactions, thus facilitating the formation of a robust and reusable membrane enabled by in situ generated CANs.
GO and BMI were involved in the Diels–Alder reaction, and the mussel-inspired IPN matrix was composed of PVDF and dopamine monomers.20 BMI could undergo specific covalent interaction with dopamine and later react with GO to form the desired DA adduct.21 The molar ratios of dopamine and GO-BMI were chosen such that even after the successful reaction between these components, sufficient provision for polymerization of dopamine still exists. The successful polymerization of the tagged dopamine was effectively probed with XPS spectra (Fig. S1†).
During IPN formation, the sequential in situ polymerization of the GO-BMI-tagged dopamine ensures homogeneity in terms of the distribution of the exchangeable bonds throughout the membrane matrix. Additionally, the presence of GO in the membrane architecture would inherently aid in the increase in thermal and mechanical properties of the final designed membrane. Furthermore, the GO-BMI moieties would act as charge-carrying centres, thus enhancing separation performance.
In this work, we report the facile fabrication of an easily recyclable IPN membrane via the NIPS technique and installed dynamic covalent bonds during the fabrication process itself, thus helping the membranes to fill in the gaps present in the circular economy.22 The CAN-enabled IPN membranes exhibit alternative pathways for introducing sustainable strategies for recycling and reusing fouled membranes in a practicable manner. The membrane demonstrated significantly high rejection performance and its biocompatibility was ascertained by carrying out in vitro cytotoxicity studies in accordance with an MTT assay. The novel combination of covalent adaptable networks and IPNs can be representative of new-generation membranes with robust and stable performance while maintaining the sustainability factor. This advance could be inspirational for the fabrication of high-performing green and sustainable membranes, with an effective reduction in membrane waste.
Experimental section
Materials and methods
PVDF (Kynar 761 grade, Mw = 440![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) was provided by Arkema. Sodium meta per-iodate (NaIO4, ≥99.8%), dopamine hydrochloride monomer ((DA, 99%), and bismaleimides (BMI, Mw = 97.07 g mol−1) were obtained from sigma Aldrich. Buffer Tris(hydroxymethyl)aminomethane (Tris, 99.8–100%) was supplied by Sisco Research Laboratories Pvt Ltd. Sodium hydroxide pellets (NaOH, 97.0%), graphene oxide (GO), propan-2-ol (IPA, 99.5%), hydrochloric acid (HCl, 35–38%), and N,N-dimethylformamide (DMF, 99.0%) were obtained from SD Fine Chemicals Ltd. All the dyes, viz. Methyl Orange (MO, 327.33 Da), Rhodamine B (RB, 479.02 Da), Methylene Blue (MB), Methyl Red (MR), Congo Red (CR), Amido Black (AB), and Acridine Orange (AO), were supplied by a local vendor. Tetracycline (Mw = 444.43 g mol−1), oxytetracycline (Mw = 460.43 g mol−1), and Sudan (Mw = 248.28 g mol−1) were obtained from Sigma Aldrich, whereas Vit-B12 (Mw = 1355.38 g mol−1) and azithromycin (Mw: 785 g mol−1) were obtained from a local pharmacist.
000 g mol−1) was provided by Arkema. Sodium meta per-iodate (NaIO4, ≥99.8%), dopamine hydrochloride monomer ((DA, 99%), and bismaleimides (BMI, Mw = 97.07 g mol−1) were obtained from sigma Aldrich. Buffer Tris(hydroxymethyl)aminomethane (Tris, 99.8–100%) was supplied by Sisco Research Laboratories Pvt Ltd. Sodium hydroxide pellets (NaOH, 97.0%), graphene oxide (GO), propan-2-ol (IPA, 99.5%), hydrochloric acid (HCl, 35–38%), and N,N-dimethylformamide (DMF, 99.0%) were obtained from SD Fine Chemicals Ltd. All the dyes, viz. Methyl Orange (MO, 327.33 Da), Rhodamine B (RB, 479.02 Da), Methylene Blue (MB), Methyl Red (MR), Congo Red (CR), Amido Black (AB), and Acridine Orange (AO), were supplied by a local vendor. Tetracycline (Mw = 444.43 g mol−1), oxytetracycline (Mw = 460.43 g mol−1), and Sudan (Mw = 248.28 g mol−1) were obtained from Sigma Aldrich, whereas Vit-B12 (Mw = 1355.38 g mol−1) and azithromycin (Mw: 785 g mol−1) were obtained from a local pharmacist.
Membrane characterization techniques
The prepared m-IPN membrane was thoroughly characterized via different analytical techniques. Fourier transformation IR spectroscopy (FTIR) in the mid-IR range was used to ascertain the different functional groups. (FTIR spectra were recorded on a PerkinElmer spectrometer using an ATR accessory. The ATR accessory contained a diamond flat plate crystal at a nominal incident angle of 45°, and an average penetration depth of 2 μm. Each spectrum presented is the result of 8 accumulated scans obtained with a resolution of 4 cm−1 over a range of 650–4000 cm−1 with air as the background.) A UV-visible spectrophotometer from PerkinElmer was used to study the absorbance spectra generated by the feed and permeate dye and microplastic solutions. The particle size of the microplastic feed and permeate were studied with dynamic light scattering (DLS) using a ZetaPALS zeta potential analyzer. Crystallinity studies of the membranes were done with a PANalytical X'pert PRO. XPS from Axis Ultra where Al (1.486 KeV) was the monochromatic source, was carried out for both the IPN and m-IPN. A study of the morphology was done with a Karl Zeiss Ultra55 FE-SEM scanning electron microscope with inbuilt EDAX, which ultimately helped to detect the elements present in the membrane. Membrane pore size was evaluated from molecular weight cut-off studies via the rejection of a range of neutral solutes and from N2 adsorption–desorption experiments (BET). A TA Q500 performed the thermogravimetric analysis. A TA Q800 was used to test the mechanical integrity of some membranes. The measurements were performed in tension mode with a tension force of 0.01 N from room temperature to 140 °C at a heating rate of 10 °C min−1. Micro UTM was also used for studying the mechanical properties using a load cell of 25 N and a loading rate of 10 mm min−1. The hydrophilicity of the membranes was assessed with the contact angle measurement technique with water as the solvent. Using an adjustable cell with a gap height of 100 μm and at pH 7, a Surpass 3 instrument from Anton Paar was used to measure the surface charge of the membranes.Fabrication of IPN membrane
Dopamine monomer and PVDF were combined in 10 ml of DMF to synthesize dope solutions (20 wt%). The reaction mixture was heated to 70 °C and vigorously stirred to create a homogeneous solution. The final solution underwent a rigorous degassing process to remove any remaining bubbles. The membranes were then cast onto a glass plate using a doctor blade at a speed of 7–8 cm s−1 and a thickness of 300 μm. The film was precipitated in a coagulation bath containing a solution of Tris buffer (10 mM, pH = 8.5) and NaIO4 (5 mM) at a temperature of 4 °C. For optimum reduction in pore wall size, the membranes were kept submerged for a maximum of 7 days.18 Before testing and assessments started, the ready membranes were washed properly with ultrapure water and designated as IPN.Fabrication of PVDF/DOPA/GO/BMI (m-IPN) membrane
In a facile process, the first 20% dope solution was prepared by mixing 3 g of PVDF in DMF (7 ml) along with another solution mixture consisting of 2 g of bismaleimide and 1 g of dopamine hydrochloride monomer in DMF (3 ml). After mixing, both solutions were stirred for 3–4 h at 80 °C to obtain a homogenous mixture. In the meantime, 75 mg of GO was added to DMF (2 ml) and thoroughly sonicated first by bath sonication followed by probe sonication. The deep black GO suspension was poured on the dope solution and allowed to stir for another 12 h at the same temperature. Degassing was done for 10–15 minutes to remove any residual bubbles in the dope solution. Next, a doctor blade with a thickness of 300 μm and a speed 8–9 cm s−1 was used to cast the membrane on the glass plate. The cast films were then precipitated in a coagulation bath comprising NaIO4 (5 mM) and Tris buffer (pH = 8.5, 10 mM) where the temperature was maintained at 0–5 °C. The fabricated membranes were kept in the buffer solution for an optimized 7 days to achieve maximum reduction in the pore wall. The fabricated membranes were designated as m-IPN membranes. Before starting any assessment and testing, the as-prepared membranes were thoroughly washed with deionized water.Pure water flux and antifouling studies
An important requirement for a filtration module is the water flux derived from membranes. For typical commercial membranes, the common trade-off between membrane permeability and selectivity frequently presents a bottleneck. A crossflow setup is utilized to determine the flux for pure water. Before starting off the experiment, token membrane coupons (45 mm in diameter) were carefully placed into the test cell and compacted for 30 minutes at 10 psi (about 0.7 bar). Next, the transmembrane pressure was gradually increased to 150 psi (about 10.5 bar) and the following formula was applied to calculate the flux (Jw):| Jw = V/(A × t) (lm−2 h−1) | (1) |
All tests were performed in triplicate to ensure repeatability. These membranes should have stable and repeatable performance over time. In order to verify the membrane stability, a transmembrane pressure of 100 psi (7 bar) was maintained continuously for 21 days. Here, V represents volume in litres, A stands for effective membrane area and t stands for the time in hours.
The antifouling performance of the membranes was studied using 1000 ppm BSA (bovine serum albumin) solution as the protein model foulant. With prior knowledge of the pure water flux (Jw) generated by the membranes, flux values with BSA solution were subsequently recorded at 100 psi for 1 h. 0.9 wt% of NaCl solution was then used to thoroughly backflush the membrane. Then pure water flux values were again recorded (Jw1). The flux recovery ratio was then calculated to gauge the antifouling performance of the membrane.
| FRR (flux recovery ratio) = Jw1/Jw × 100 | (2) |
Dye rejection studies
To study dye rejection, some commonly used dyes, viz. Acridine Orange (AO), Amido Black (AB), Congo Red (CR), Methyl Red (MR), Methylene Blue (MB), Rhodamine B (RB), and Methyl Orange (MO), were used. Next, the in-house cross-flow setup was properly fitted with a 45 mm diameter coupon membrane. For each dye rejection cycle, 2000 ml (20 ppm) feed solutions were studied. The efficiency of dye rejection was calculated with the following equation, where the concentration of permeate was evaluated with a UV-vis spectrophotometer.| (%) rejection = [1 − (Cp/Cf)] × 100 | (3) |
Salt rejection studies
The salt rejection performance was evaluated by taking a 2000 ppm solution for NaCl. The same crossflow setup (used for dye rejection) was used for the study. Before applying the transmembrane pressure 100 psi (7 bar) for salt rejection, the membrane was compacted at 60 psi (4.2 bar) for 30 minutes. Permeate concentrations were measured at regular intervals with a TDS (total dissolved solute) meter. All the measurements were performed in triplicate and the equation for dye rejection efficiency was used to determine the salt rejection efficiency. To evaluate the long-term performance of the membrane, the experiment was run for an additional 21 days.Microplastic removal
The study regarding the ability of the membranes to remove microplastics consists of the following stages. To begin with, a small part of PVC pipe used for sanitary work was rubbed with emery paper (mesh size 800) to obtain PVC pipe powder. The powders were added to DI water and centrifuged for 10 minutes at 14000–15000 rpm resulting in a colloidal suspension containing microplastics. The supernatant was carefully collected. Next, the supernatant solution (feed) was passed through the m-IPN membrane through a dead-end setup and the permeate was duly collected. Dynamic light scattering (DLS) was performed to measure the particle size in both the feed and the permeate. A UV-vis spectroscopic method was introduced to ascertain the removal of microplastics by observing the characteristic peak of PVC in both the permeate and the feed microplastic solutions.
Chlorine tolerance evaluation
The variation in efficiency for salt rejection before and after exposure to high concentrations of sodium hypochlorite solution is typically used to measure the chlorine tolerance of membranes. The level of tolerance of a manufactured membrane to chlorine increases with decreasing deviation. The membrane was initially submerged for over 4 hours in a 2000 ppm (pH = 10) solution of sodium hypochlorite. The membranes were then thoroughly washed with DI water and their ability to reject 2000 ppm of NaCl solutions set forth in a manner comparable to the salt rejection studies that were previously mentioned.In vitro cytotoxicity measurements and recyclability studies
An MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] cytotoxicity assay is commonly performed to ascertain the cytocompatibility of samples, where cell viability is indicated by metabolic activity. In the presence of live cells, water-soluble yellow MTT is metabolically converted to blue-violet insoluble formazan. The photometric assessment of colour intensity following the dissolution of formazan in DMSO reveals a direct correlation between the number of viable cells and the colour intensity. Cell line L929 (NCCS/1469) was used for this experiment. The cells were properly grown and maintained in Minimum Essential Media (MEM) with fetal bovine serum (FBS) (10%) before the examination. Then, 1 g of the sample (IPN membrane) was sterilized at 121 °C for 15 minutes in order to prepare the sample extract. Complete MEM medium (10 ml) was added to the extract and incubated at 37 °C (in a 5% CO2 atmosphere) for 24 hours. A concentration range of 10 to 100% was used for the assay, where 100% denotes the neat extract.In a 96-well plate, 10000 cells per 100 μl MEM culture per well were seeded and carefully maintained for 24 h to successfully fabricate a semi-confluent layer. In the above-mentioned concentration range (10–100%) the layer was exposed for 24 h. At the end of the stipulated time limit, formazan formation in a growth control and concentration-treated sample were fully collated via an optical density (O.D.) study at 570 nm. According to the following formula, the viability of the cells was used to compute the percentage of inhibition of proliferation for each treated concentration:
| % viability = (O.D for extract/O.D for blank) × 100 | (4) |
To evaluate the closed-loop recyclability of the membranes, all the membranes used for dye and salt rejection were put in a 100 ml round bottom flask filled with DMF and kept for heating at 140 °C for 10–15 minutes. DCM and water were added to the blackish solution mixture for a successful workup through the liquid extraction method. Based on contrasting solubility, DMF, salt, and dyes were extracted. The solvents could be evaporated to get the polymeric components, and the membrane was reconstructed using the NIPS approach, as previously mentioned.
Results and discussion
IPN and CAN work in tandem toward the integrity of the membranes
m-IPN membranes were fabricated by integrating the concepts of IPNs and CANs. Bismaleimide (BMI) was made to react selectively with one of the IPN components, i.e., dopamine, in a particular stoichiometric ratio. Following this, the dopamine-anchored BMI was exposed to the presence of GO, which facilitated the formation of a Diels–Alder adduct between the diene (GO) and the dienophile (BMI). This was added to PVDF dope solution and finally membranes were cast via the non-solvent induced phase separation (NIPS) technique in a coagulation bath of cold Tris buffer solution which helped in the in situ sequential polymerization of GO-BMI-anchored dopamine, which in turn led to the formation of a jammed 3-system network. Dopamine helped in dispersing the DA adducts throughout the membrane matrix and made these exchangeable bonds an integral part of the entire system. Scheme 1 illustrates the chemical reaction involved in the synthesis procedure and the membrane fabrication technique. The membranes were endowed with sufficient charge-carrying centres and small pore sizes, caused by the presence of the large GO sheets and the sequential polymerization of dopamine. Such characteristics augmented better separation performance and the fouled membranes after their usage could be easily recycled via a facile and easy scalable pathway. This recyclability was enabled again by the dynamic bonds present in the system, which also helped in retaining the mechanical integrity and rejection performance even after 3 recycling phases.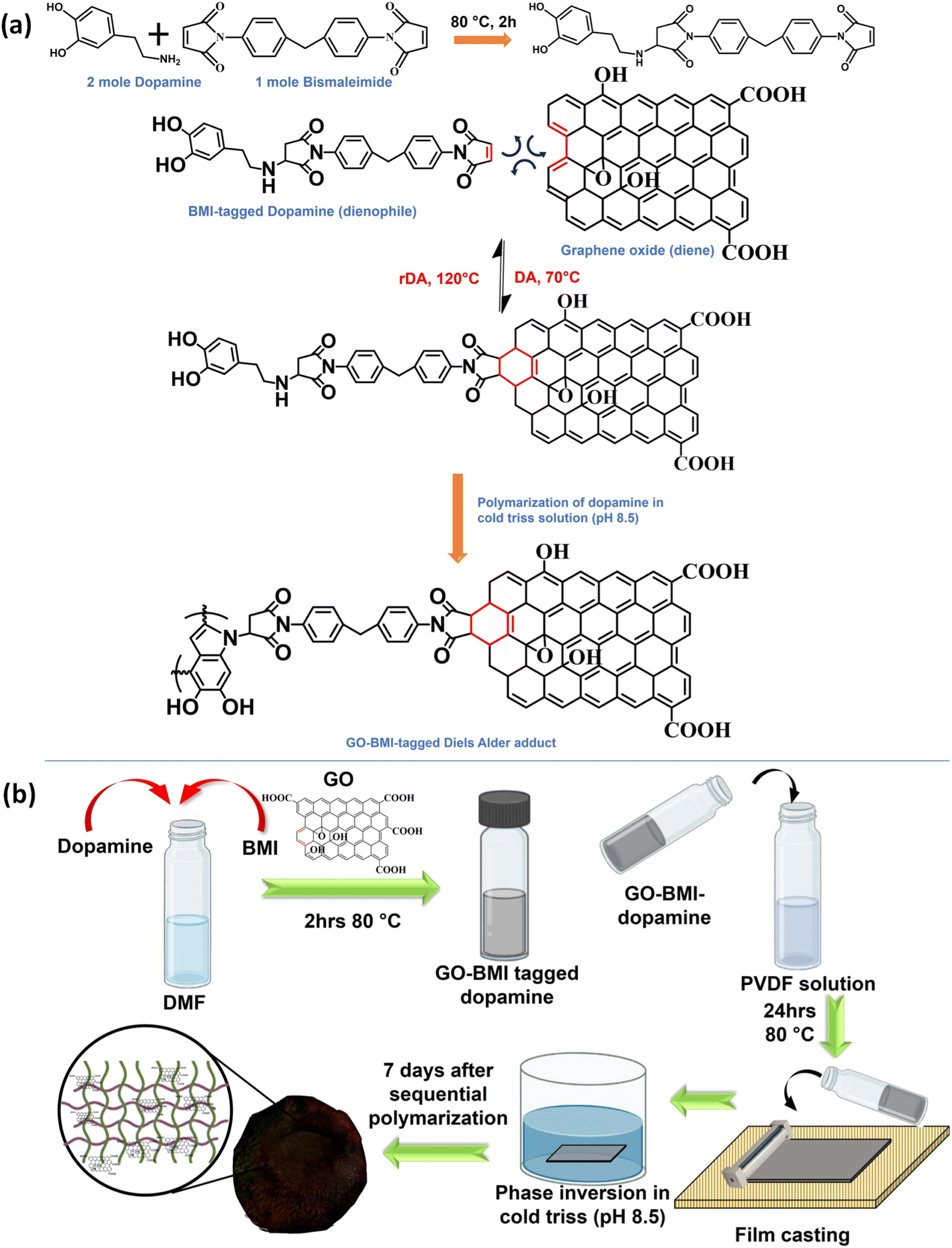 | ||
| Scheme 1 (a) Chemical reaction involved during the formation of a Diels–Alder adduct between graphene oxide (GO) and bismaleimide (BMI). (b) A cartoon depicting the sequential IPN membrane fabrication process. | ||
Chemical and crystalline environment of the fabricated membranes
A series of characterizations were carried out to ascertain the successful fabrication of the m-IPN membranes. To assess the chemical environment of the m-IPN membranes, FTIR analysis was performed (Fig. 1a). Apart from the regular absorption peak that usually arises from PVDF/PDA IPN membranes,18 a new peak at 1715 cm−1 could be ascribed to the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O imide bond in BMI.23 Additionally, the strategic absence of the N–H bending peak at 1635 cm−1 in m-IPN indicates the successful incorporation of BMI. The increment in the absorption peak intensity at 1512 cm−1 could be due to the increment in CC bonds due to the presence of GO.24
O imide bond in BMI.23 Additionally, the strategic absence of the N–H bending peak at 1635 cm−1 in m-IPN indicates the successful incorporation of BMI. The increment in the absorption peak intensity at 1512 cm−1 could be due to the increment in CC bonds due to the presence of GO.24
 | ||
| Fig. 1 (a) FTIR spectra, (b) XPS wide spectra and (c) XRD spectra for neat IPN and m-IPN membranes. | ||
The FTIR analysis was further supported with the XPS spectra. The spectra of IPN and m-IPM demonstrated the presence of carbon, nitrogen, oxygen, and fluorine elements (Fig. S1†).3 The wide XPS spectra provided valuable insights into the composition of these fabricated membranes (Fig. 1b). Through deconvolution of the 1s C core spectra of the m-IPN membrane, five distinct peaks were identified at approximately 284.72, 285.92, 287.12, 288.52, and 291.02 eV. These peaks corresponded to sp2/sp3 hybridized carbon, carbonyl C–O, CO bonds, –COOH, and –C–F bonds, respectively. Furthermore, the deconvoluted 1s N spectra of the m-IPN membrane exhibited peaks at 399.03, 400.43, and 402.23 eV, which can be attributed to the N–, –N–H, and N–C bonds, respectively.25 The deconvolution of the 1s O spectra revealed peaks at 530.63, 532.13, and 533.63 eV, providing additional evidence for the presence of CO, C–O, and C–O–C bonds on the membrane surface.4 Finally, the m-IPN membrane exhibited a prominent presence of 1s F, which originated from the PVDF component. This observation further confirmed the presence of –C–F bonds in the membrane structure.
The crystallinity aspect of the fabricated membranes was characterized via an XRD pattern. The broad peak at around 23° could be ascribed to the diffractions coming from the amorphous structures of PDA (Fig. 1c).24,26 The diffraction peak at 2θ = 20.2° ((110) plane) could possibly come from the semi-crystalline polymer PVDF present in the neat IPN membranes. In general, PVDF is a semicrystalline polymer (50–60%) that shows polymorphic phases (α, β, and γ); therefore a diffraction peak was observed corresponding to the γ-phase.27,28 However, in m-IPN apart from the peaks mentioned above, an additional peak was observed at 2θ = 10.4°, which corresponded to the reflection from the (002) plane of GO.29,30 The presence of this characteristic XRD peak confirmed the successful impregnation of GO in the m-IPN matrix.
Mechanical integrity of the membranes
The m-IPN membranes were found to be extremely robust in terms of mechanical strength. To ascertain this, DMA and UTM analyses were performed. A uniaxial tensile test was carried out on a micro UTM. Tensile samples of both neat IPN and m-IPN membranes were prepared according to the ASTM D882. Then, 10 mm min−1 was set as the loading rate during testing. It was observed (Fig. 2b and c) that the IPN membrane exhibited greater elongation before the final fracture, indicating its toughness. However, the mere addition of 75 mg of GO in the neat IPN matrix (m-IPN) resulted in enhanced stiffness and considerably reduced toughness.31,32 Subsequently, the elastic modulus was calculated from the slope of the Hookian region of the stress–strain plot. Here too, similar results were observed in which the presence of GO enhanced the modulus and strength compared to the bare IPN matrix. This observation could possibly be explained by the fact that GO being a nanofiller could effectively fill the free volume present in the neat IPN matrix, thus increasing chain rigidity. Additionally, the secondary polar–polar interactions between the functional groups of the GO and IPN chains increased the chain stiffness, thus enhancing the strength and modulus.33 | ||
| Fig. 2 (a) DMA profile, (b and c) tensile stress–strain curve and bar-graph of tensile strength and elastic modulus, (d) TGA profile of IPN and m-IPN membrane and (e) DSC thermogram of m-IPN membrane. | ||
To further corroborate the UTM findings, DMA studies were performed (Fig. 2a). It was observed that the incorporation of the GO-BMI moieties helped make a drastic improvement in the storage modulus of the IPN matrix. From 140 MPa for neat IPN membranes, the m-IPN ones recorded a storage modulus of 172 MPa. This increment can be attributed solely to the presence of GO, which is known for its high structural integrity.34
Thermal stability of the membranes: TGA and DSC analysis
Thermogravimetric (TGA) was carried out to probe the thermal stability of the IPN and m-IPN membranes and the evaluations were done using a TA Q500 (Fig. 2d). The control IPN membranes showed two-step degradation, the first occurring due to the degradation of catechol moieties around 150–300 °C and the second degradation around 300–500 °C occurring due to the flexible alkyl side chain.18 On the other hand, the m-IPN membranes also demonstrated a double-degradation thermal profile. The first degradation step at 250–350 °C corresponded to the degradation of the catechol group of dopamine and oxygen functional groups of GO, and the second one at 400–500 °C corresponded to the degradation of the flexible alkyl spacers. However, on closer inspection above 500 °C, it was observed that the neat IPN membrane degraded nearly 20% more than the m-IPN membranes. This significant enhancement in thermal stability could be attributed to the augmented rigidity in m-IPN due to the inclusion of pendent BMI over PDA and GO.To further substantiate the thermal stability, industrial scalability and finally the presence of the exchangeable bonds, DSC was carried out. Fig. 2e displays the differential scanning calorimetry (DSC) of the m-IPN membrane. On heating the membrane above 100 °C an endothermic peak corresponding to the retro-DA (rDA) reaction was observed at 112 °C,22 which was much higher than the boiling point of water. Therefore, we could conclude with an assertion that this dynamic bond-based membrane can operate at elevated temperatures. Additionally, the formation of the DA exchangeable bonds was further established by the DSC results.
Membrane pore size analysis: molecular weight cut-off (MWCO) and nitrogen adsorption–desorption studies
Membrane separations are driven by a synergism between pore size and surface charge. The pore sizes of the m-IPN membranes were calculated using MWCO studies (Fig. 3a). For the evaluation, a range of neutral solute molecules was taken into account. The molecular weights of the solutes ranged between 300 and 1400 Da. Being neutral in nature, the removal mechanism of these molecules can only stem from the pore size of the membranes. It was observed that the m-IPN membranes could successfully reject 94% of Giemsa stain, roughly 96% of Sudan IV, close to 97% of tetracycline and oxytetracycline, 98% of azithromycin, and just about 99% of vitamin B12. Thus, the pore size of the membranes lies in the nanofiltration regime, and this observation was further corroborated by BET experiments. | ||
| Fig. 3 (a) Molecular weight cut-off analysis of the m-IPN membrane using a range of neutral solutes and (b) pore size analysis from the N2 adsorption–desorption isotherm. | ||
To evaluate the permanent porosity of the m-IPN membrane, N2 adsorption–desorption experiments were performed at a temperature of 77.35 K. Fig. 3b shows the obtained N2 adsorption–desorption isotherms for the m-IPN. The resulting isotherm exhibited a type IV pattern according to IUPAC nomenclature and displayed a distinctive hysteresis loop in the multilayer range.35 This hysteresis loop was attributed to capillary condensation occurring within the mesoporous structures present in the membrane. Non-local density functional theory (NLDFT) was employed to determine the pore size distributions, and the pore size distributions were derived from the isotherms. The application of NLDFT allowed for precise analysis and characterization of the pore size distribution of the membrane. Interestingly, the experiments revealed that the m-IPN membrane had an average pore diameter of 21.4 nm. The impressive porous structure of the m-IPN membrane, its π-conjugated skeletons, substantial pore size, and permanent porosity hold exceptional potential for advancing next-generation molecular sieving membranes. The results demonstrated the excellent pore size control achieved during fabrication and highlighted the suitability of the membrane for various applications requiring precise molecular separation and filtration.36,37
Membrane surface properties: hydrophilicity, surface charge and morphology
The key variables determining the final performance of polymeric water filtration membranes are their hydrophilicity and surface charge. The more hydrophilic a membrane is, the more resistant it is to foulant attack. Antifouling characteristics mark one of the most critical demands of the membrane industry. In general, hydrophilic surfaces promptly promote the formation of a thick layer of hydration which the protein molecules find it difficult to penetrate. Measurement of water contact angle is the easiest method for determining the hydrophilicity or hydrophobicity of a particular membrane surface.38 When the absolute value of the water contact angle drops below 90°, a hydrophilic surface is hinted at. Similarly, a value above 90°, indicates a shift to the hydrophobic spectrum, thus signalling the membrane's aversion to water molecules. The static water contact angle values of the fabricated m-IPN and the neat IPN membranes were evaluated with the help of a goniometer (Fig. 4a). The neat IPN membranes had a WCA of 68°, whereas the m-IPN membranes had a WCA of 62°. The decline in the value can be well explained by the introduction of a greater number of hydrophilic groups (–OH, –COOH) due to the inclusion of GO in the membrane architecture. The hydrophilic nature of the membranes can be further ascertained from the water uptake measurements, where the uptake % rose from 85% for IPN membranes to 88% for the m-IPN membranes. Furthermore, from the AFM data, it was observed that the RMS (Sq) roughness values rose to 238 nm upon the introduction of GO-BMI linkages in the neat IPN matrix (RMS (Sq) roughness = 114 nm) (Fig. 4c and S2†). This increased roughness, in turn, suggests greater wettability for a hydrophilic surface, taking its cue from the Wenzel model theory.39 | ||
| Fig. 4 (a) Water contact angle, (b) zeta potential and AFM images of (c) neat IPN and (d) m-IPN membranes. | ||
Apart from surface roughness, the surface charge is the next major player in terms of deciding the surface features and hence the performance of the membranes. With the help of the Helmholtz–Smoluchowski equation, the zeta potential values of the membrane were calculated. Zeta potential is a direct representative of the interfacial interactions that come into play when a material (here a membrane) surface meets a liquid. The zeta potential of the fabricated m-IPN membrane was found to be around −38 mV, which was drastically greater than that of the neat IPN membranes (ca. −20 mV) (Fig. 4b). This increment in the surface charge values can be attributed to the introduction of a greater number of polar groups coming from GO and BMI. Thus, the Diels–Alder (DA) adduct could act as a charge-carrying center and hence help in significantly increasing the surface charge of the m-IPN membranes, which in turn would significantly improve the separation performance.
With the surface charge and nature in place, the surface and pore morphology were duly evaluated from SEM analysis (Fig. 5). The aimed-for reduction in pore size and variation in surface morphology brought about by the incorporation of DA bonds were probed via SEM and EDAX. It was observed that the membrane surface did not exhibit much of a difference compared to the neat IPN membranes. However, the m-IPN membrane displayed more surface coverage, which could be due to the polymerized PDA moieties as well as due to the presence of large GO sheets. As estimated from ImageJ, it was observed that there was a drastic decline in pore size, from a whooping 300–500 nm for the neat IPNs to 20–25 nm for the m-IPN membranes. Here again, the presence of GO sheets with high lateral dimensions could play a major role in targeted pore size engineering. From the cross-section morphology, a combination of macro-voids and network-like architecture (since it is very difficult to achieve precise control over pore size distribution via the NIPS method) was observed, which is a typical characteristic of common IPNs. The EDAX spectra confirmed the inclusion of the GO-BMI adducts since there was an increment in terms of carbon, nitrogen, and oxygen intensity in the EDAX spectra for the m-IPN membranes compared to the neat IPN membranes.
 | ||
| Fig. 5 SEM micrographs of (a–c) surface, cross-section and EDAX spectra of the neat IPN membrane and (d–f) surface, cross-section and EDAX spectra of m-IPN membranes. | ||
Membrane performance: water transport, ion rejection, chlorine tolerance, antifouling properties and microplastic removal
A thin, mechanically strong, and selective polymeric membrane can result in ideal filtration performance. The uniform distribution of the GO-BMI moieties anchored in the IPN matrix results in the establishment of a framework for the development of promising systems which offer improved water permeation and excellent rejection characteristics.Water permeation or flux obtained from the membranes is a key player in deciding the usability of a system in real-life conditions. Most polymeric membranes are plagued by the trade-off between permeability and selectivity; thus, quantification of pure water flux is of utmost importance. The assessment of pure water flux was performed by employing a spectrum of transmembrane pressure ranging from 25 psi to 150 psi (Fig. S4a†). A steady increment in the flux values was observed upon a linear increase in pressure across the membrane (134 LMH at 25 psi, 210 LMH at 50 psi, 251 LMH at 75 psi, 275 LMH at 100 psi, and about 290 LMH at 125 psi and 310 LMH at 150 psi). The linearity of the flux measurements could possibly stem from the fact that the pore architecture of the membrane developed changes upon an increase in transmembrane pressure. The flux values obtained qualify the membranes to be in the ultrafiltration to nanofiltration range. The flux values, however, were greatly reduced when compared to the neat IPN membrane (900 LMH at 50 psi). A plausible explanation could be that the reduction in pore sizes was brought about by the simultaneous in situ polymerization of the BMI-tagged dopamine molecules and the large GO sheets inside the membrane matrix, leading to the creation of a tortuous path for water permeation. The water transport via these membranes can be effectively explicated by a combination of pores and GO nanochannels.
The flux experiments were carried out for a continuous period of 3 weeks (100 psi) to determine the long-term stability of the fabricated membranes. The deviation in the flux values was found to be insignificant, thus corroborating its longevity (Fig. 6a).
 | ||
| Fig. 6 (a) Pure water flux stability of the m-IPN membranes under a transmembrane pressure of 100 psi and flux recovery obtained after performing 5 consecutive cycles of DA and retro-DA phases. (b) Stable NaCl rejection for a period of 21 days and subsequent chlorine tolerance property. (c) Effective cationic and anionic dye rejection by the m-IPN membranes. (d) Antifouling performance of the membranes. (e) Cytocompatibility of the m-IPN membranes as evaluated from an MTT assay using L929 cell lines. (f) Microplastic removal efficiency determined via UV-vis spectroscopic studies. | ||
Additionally, to see the effect of the in situ generated exchangeable bonds (Diels–Alder, DA), five consecutive cycles of DA and retro-DA were performed in accordance with the reported literature.21 In general, the membranes were heated at a temperature of 110 °C for a period of 6 h. This process initiated a retro-Diels–Alder process, leading to breakage of the Diels–Alder bonds. Following this, the membranes were cooled for a period of 6 h, which again facilitated the Diels–Alder reaction when the temperature reached 60–70 °C, leading to refurbishment of the bonds. The total duration of 12 h involving DA and retro-DA reactions constituted one cycle. After carrying out five such cycles, pure water flux measurements for the m-IPN membrane were again carried out. It was observed that the m-IPN membranes, after undergoing 5 such cycles, yielded 80% recovery of pure water flux (at 100 psi). This significant recovery could be attributed to the tuneable pore architecture of the fabricated membranes enabled via the exchangeable DA bonds. Here too the studies were continued for a period of 3 weeks, where negligible variance was observed in the results (Fig. 6a). Also, the m-IPN membranes were kept in water for a period of 1 month to observe whether the GO sheets present inside the membrane matrix undergo any delamination or not. However, it was seen from the XRD of the wet membrane (Fig. S4b†) that no change occurred in the d-spacing of the sheets, which corroborated the stability of the infused GO-BMI moieties. In other words, IPN formation helped in retaining the d-spacing of the sheets.
The presence of dopamine-grafted BMI and subsequent formation of DA adduct in the presence of GO sheets inside the IPN matrix helped in generating huge amounts of charge-carrying centers. This observation was further evidenced by the zeta potential values. The GO nanochannels, along with the highly negative membrane surface, were instrumental towards significant ion rejection efficiencies of the membrane. With satisfactory water flux values, the m-IPN membranes were consequently subjected to salt rejection experiments with 2000 ppm of feed salt solutions. Monovalent NaCl was discreetly chosen as the model salt foulant. Membranes that are capable of removing NaCl salt can invariably remove both divalent and trivalent salts since Na+ and Cl− exhibit the smallest hydrated radius. The m-IPN membranes could effectively remove 98.9% of NaCl salt. The rejection studies were continued for a period of 3 weeks, after which the rejection percentage was reduced to 96.8%. Such rejection values originated from the high negative charges present inside the membrane pores and the surface, along with the narrow nanochannels of GO sheets which effectively filtered the monovalent salt species. The Cl− ions suffered repulsion from these negative charge centers, and the Na+ ions faced cation–π interactions that occurred between the monovalent ions and the sp2 hybridized domains of the GO-BMI moieties.40,41 In general, a host of factors, including reduced pore size and substantial electrostatic interactions, played major roles in augmenting the rejection efficacies of the membrane while maintaining electronic balance on either side of the membrane.
The same membranes were exposed to chlorine (NaOCl) and were again deployed to reject NaCl salt. The lower the divergence in the salt rejection values, the more chlorine tolerant the membranes are. Chlorine tolerance marks an important feature of water-remediation systems since membranes during their lifetimes come into close contact with chlorine molecules since sodium hypochlorite solutions are often used as cleaning solutions. Membranes that disintegrate upon chlorine attack require frequent replenishment, which impede their usability as a long-term stable water filtration system. The m-IPN membranes could successfully reject high percentages of NaCl salt even after chlorine exposure (Fig. 6b). The membranes showed a decline of just 2–3% in salt rejection values. Such high values signified the robustness of the fabricated membrane, and the negligible decline can be attributed to the remaining –NH groups of PDA.
Furthermore, the charge-based ion-sieving property of the m-IPN membranes was assessed by selecting a gamut of cationic and anionic dyes. First, 20 ppm dye solutions were prepared, and the rejection efficiency was recorded at regular intervals using a UV-vis spectrophotometer. The membranes could effectively reject 99% of the cationic dyes (viz. Rhodamine B, Methylene Blue and Acridine Orange), and the rejection recorded for the anionic dyes (viz. Methyl Orange, Methyl Red, Congo Red and Amido Black) was about 96–98%. Although the dye rejection mechanism would mainly consist of Donnan exclusion and size-based sieving, the selectivity towards cationic dyes can be explained by the fact that the membranes were endowed with a great number of negatively charged species which aid in the removal of cationic dyes via electrostatic attraction which strongly adsorbs the dyes onto its surface. In the case of anionic dyes, heavy repulsion from the co-charged membrane surface and steric hindrance account for the rejection process. The UV-vis spectra for the various dye solutions demonstrated the absence of the characteristic peaks of the dyes in the permeates obtained (Fig. S3†). This observation further ratified the ability of the membranes in effective dye removal applications (Fig. 6c). The membranes could reject high volumes of dye in each cycle of the rejection study and could be easily refurbished by water backflushing for repeated usage for up to 10 cycles with no significant changes or decrease in efficiency.
Since polymeric membranes are plagued with fouling issues, which ultimately lead to performance failure, the antifouling characteristics of the m-IPN membranes were judged using BSA (bovine serum albumin). BSA was chosen as a model protein foulant, and a feed solution of the same helped to gauge the level of fouling tolerance of the membranes. Antifouling membranes, in general, are deemed to demonstrate less deviation in pure water flux values after passing runs of BSA feed solutions and subsequent cleaning (Fig. 6d). This quantity is usually evaluated from a ratio of flux recovery. The flux recovery ratio of the m-IPN membranes was found to be around 95%, which was increased greatly compared to the neat IPN membranes (85%).3 Such a high flux recovery ratio could stem from a combination of the membrane surface charge and hydrophilicity. The negatively charged surface could electrostatically repel the BSA molecules, further aided by the formation of a thick layer of hydration owing to the hydrophilic nature of the membrane (water contact angle = 62°). The hydration layer prevented the BSA molecules from coming into close contact with the membrane, inhibiting the chances of membrane fouling.
The ever-increasing water pollution caused by anthropogenic activities calls for measures to be taken on a war footing to diffuse this global crisis. Apart from the contaminants mentioned above, microplastic pollution poses serious health hazards for the entire ecosystem. Ordinary consumer products are the main source of microplastics, and they enter water resources via a variety of pathways including surface runoff and industrial and wastewater effluent. The size of these microplastics ranges between 1 and 1000 μm. Thus, using polymeric membranes with pore sizes less than the size of these microplastics is supposed to be effective for their efficient elimination from water streams. Water samples spiked with PVC powder obtained via powdering PVC sanitary pipelines (FTIR, S5) were used as a model microplastic-populated solution. The microplastic removal efficiency of the membranes was gauged using a dead-end setup. The size of the microplastics in the feed and permeate water samples were evaluated using the dynamic light scattering (DLS) method (Fig. S6†). The designed m-IPN membranes with pore sizes in the range 20–25 nm could effectively remove the larger microplastics via a size exclusion sieving mechanism. The removal was further confirmed using UV-vis spectroscopy techniques. The characteristic peak of PVC (at around 280 nm)42 was strategically absent from the permeate solutions, which further confirmed the removal of microplastic particles (Fig. 6f).
Risk evaluation and recyclability
Membranes intended for water treatment applications must abide by the requirements laid out for materials that meet mammals or mammalian cell lines. Materials that are characterized by inherent toxicity cannot be considered for water purification applications. Hence cytotoxic evaluations with mammalian cell lines is a major yardstick for evaluating the applicability of the designed system and gaining a comprehensive understanding of the associated risks when deploying the fabricated membrane. To assess the intrinsic cytocompatibility/cytotoxicity of the membranes, an MTT assay was performed using L929 (NCCS/1469) cell lines. The mathematical quantity used to measure the cytotoxic potential was the viability percentage of the cell lines. A considerably low viability percentage (<70% in the 100% test sample) would suggest higher cytotoxicity. The m-IPN membranes demonstrated a percent viability of 80% for the test sample with 100% of the membrane, and the values increased linearly for lower concentrations (almost 95% for the 10% test sample) (Fig. 6e). Higher values of cell viability percentage hinted at the presence of active cells in terms of metabolism. This result further indirectly implied the fact that no GO had leached from the system, since that would have increased the cytotoxicity of the system with mammalian cells. This cytocompatible characteristic further confirms the successful application of the B-COF@IPN membranes in water decontamination processes.The life cycle of most polymeric membranes is usually linear, and once fouled, it becomes a cumbersome and challenging task to restore them to their original state. Strong electrostatic forces that coexist between the foulants and membrane surface promote effective adhesion, which reduces membrane performance. To address this problem, recyclable membranes have attracted huge research interest and they allow the adoption of an easy technique for the separation of foulants before refabrication and reuse. The recyclability of the fouled m-IPN membranes was manifested by dissolving fouled membranes (membranes used for dye and salt rejection) in DMF solvent at 140 °C, and Fig. 7 exhibits the presence of two distinct phases in the reaction vial, one aqueous and the other organic, upon working up the dissolved membrane mixture with DCM and DI water. The top aqueous phase remained populated with foulants such as salts and dyes. And the bottom part was occupied by the polymeric components along with the DA precursors (no DA adduct since they break open during high-temperature dissolution in DMF by following the retro-DA pathway). Therefore, the foulants could be easily separated via the solvent extraction method. However, the polymeric part was recovered by solvent evaporation and refabricated using the abovementioned technique. The chemical structure of this regenerated membrane was analyzed by FTIR spectroscopy (Fig. 7b), and from the result, we can say the chemical integrity of the membrane was maintained even after three cycles of repeated recycling. Additionally, the negligible increase in pure water flux and retention in dye rejection performance corroborated the fact the recycling process did not hamper the basic architecture of the membrane (Fig. 7c and d). Furthermore, it was observed that the recycled membrane could reject Congo Red dye molecules more effectively than Methylene Blue, owing to its larger size. The recycled m-IPN membrane was also tested for its retention of thermal (Fig. 7a) and mechanical properties (Fig. 7e). It was observed that over the 3 recycling cycles, the peak location in the DSC curve for the occurrence of retro-DA remained unaltered. This indicated the excellent consistency maintained by the dynamic bond enabled membranes. To gauge the deviations in mechanical integrity between the m-IPN and recycled m-IPN membranes, tensile testing was conducted. It was observed that with each phase of membrane recycling, the tensile strength increased along with elongation at break compared with the original m-IPN, which indicated the onset of a stiffer and hence tougher membrane during recycling operations. This is a general trend observed during recycling and reprocessing since stiffness is enhanced due to the oxidation reaction of benzene rings and double bonds, and toughness enhancement facilitated the destruction of the interpenetrating networks of the m-IPN membrane.
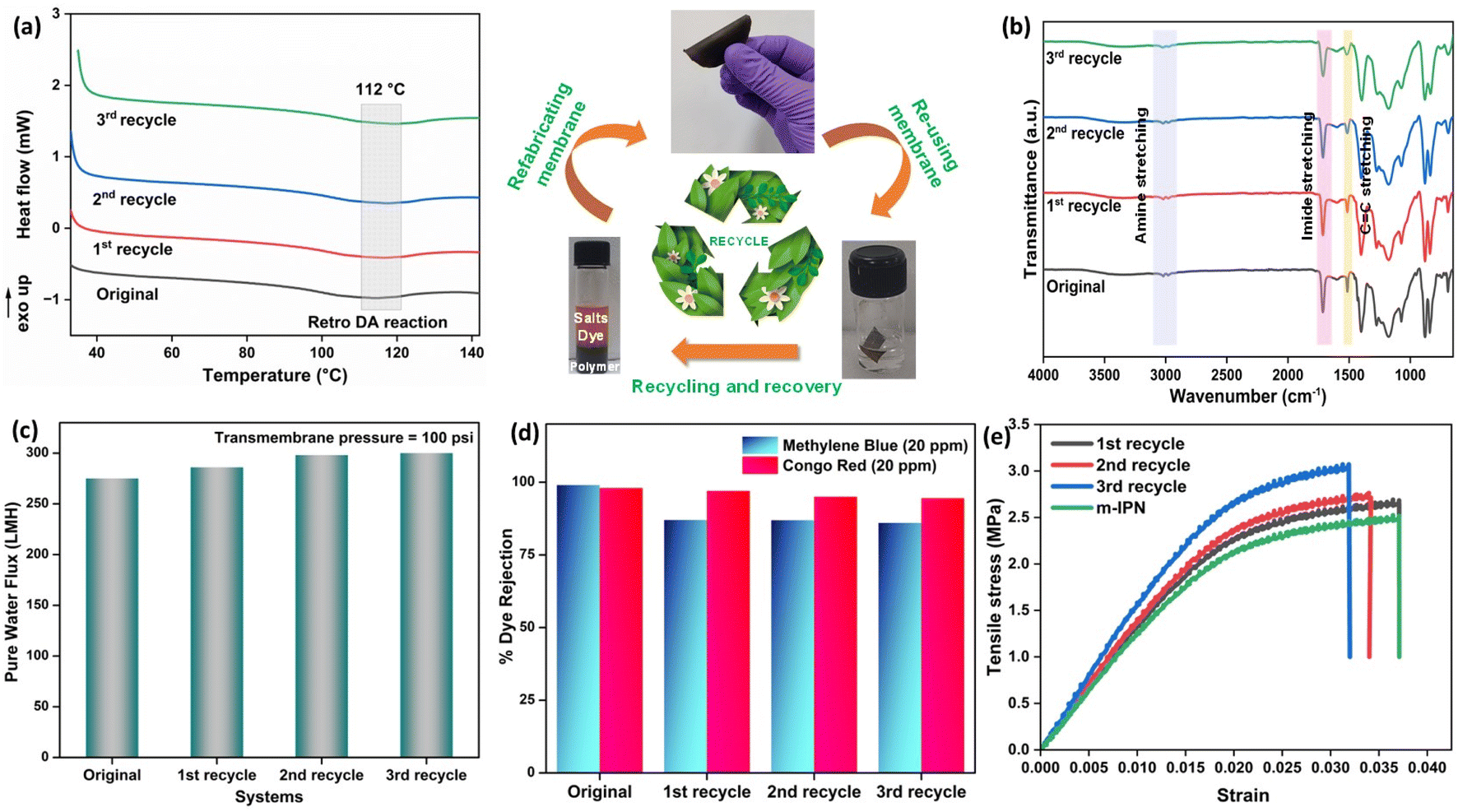 | ||
| Fig. 7 (a) DSC thermograms of the original and recycled m-IPN membranes. (b) FTIR spectra of the membranes before and after recycling. (c) Retention of pure water flux after recycling. (d) Dye rejection performance after recycling and reuse. (e) Mechanical properties of the neat m-IPN and recycled membranes. | ||
To gain a holistic understanding of the recycling abilities of the fabricated m-IPN membranes, the same procedure was repeated using a commercial PVDF membrane. Here too the commercial membranes were dissolved in DMF at similar temperatures and were re-cast. It was observed that the membranes thus formed had corrugated surfaces and the rejection efficiency decreased manyfold. Similar observations were reported in terms of the mechanical integrity of the recycled membrane, wherein the commercial membranes could not be recycled after the 1st cycle, owing to deterioration in mechanical features, which hindered further processing and casting of the membrane (Fig. 8).
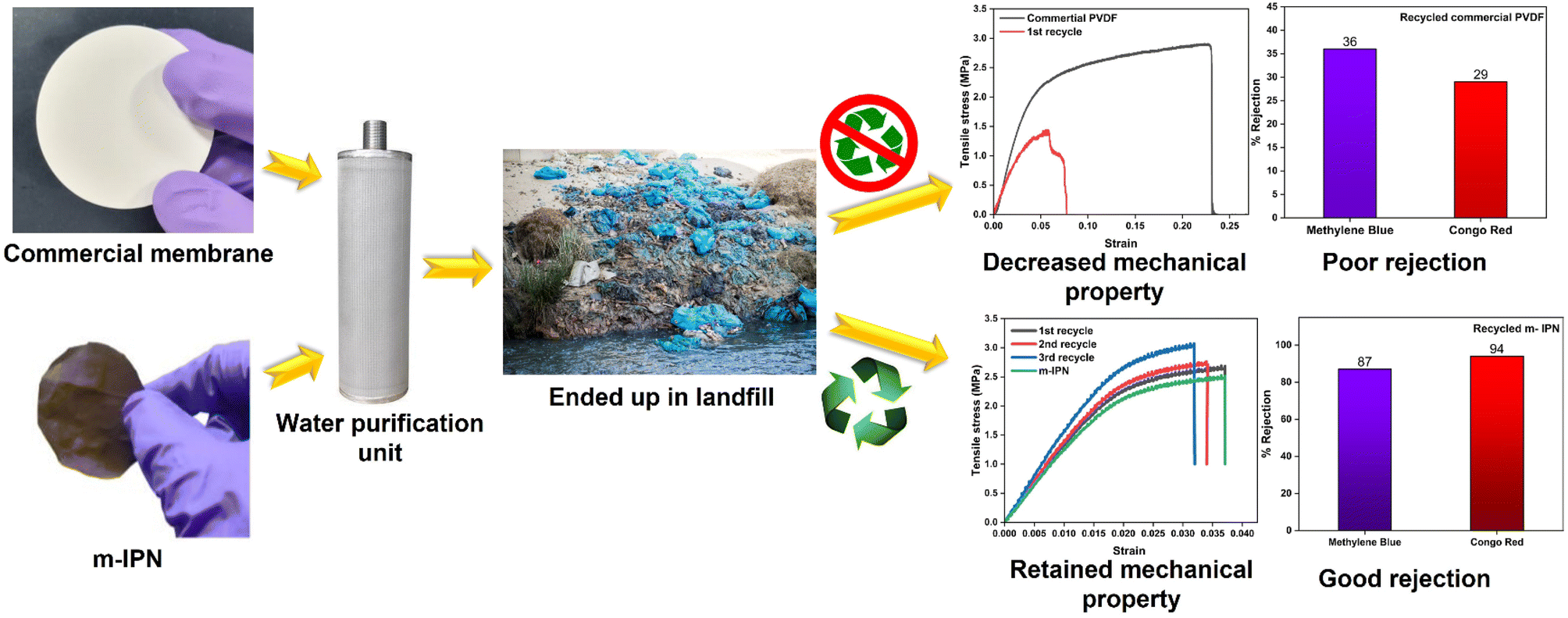 | ||
| Fig. 8 Recycling of commercial PVDF and fabricated m-IPN membranes. The commercial membranes as well as the m-IPN ones were recycled after deployment in water purification. The m-IPN membranes maintained rejection efficiency and mechanical integrity compared to commercial membranes. | ||
Conclusions
This study deals with the facile design of a recyclable membrane which can be reused multiple times via dissolution and refabrication, thus promoting a circular economy. Here the IPN (PVDF/PDA) membrane was tweaked by impregnating it with a selected diene–dienophile pair of GO and BMI moieties, which served as Diels–Alder precursors. The presence of the DA adducts (mainly GO) helped in enhancing the thermal and chemical stability of the membranes and also aided strategic pore engineering by effective reduction of pore sizes and also enhanced the surface charge of the designed membranes, thus facilitating rejection performance. Being covalently soldered to the IPN matrix, problems arising from GO leaching were ruled out in this case. This observation was further ascertained from cytotoxicity studies where the sheer cytocompatibility of the membranes towards mammalian cell lines was proof enough of its non-leaching properties. Also, being anchored to the IPN component dopamine and, eventually, its in situ sequential polymerization led to the formation of a jammed network system consisting of PVDF and GO-BMI-tagged PDA. In the designed membrane module, a synergism of electrostatic repulsion and surface hydrophilicity drove the separation efficacy. The membranes demonstrated high stability in terms of steady pure water flux (275 LMH at 100 psi) for a sustained duration of 3 weeks. Simultaneously, the membranes could retain their dye and salt rejection performance (>97%) for several operational cycles. In the process, the membranes did not lose their antifouling and chlorine-tolerant characteristics. Most importantly, these fouled membranes, owing to the presence of several dynamic covalent bonds throughout the matrix, could be easily reused via a facile recycling pathway, with preserved mechanical integrity over three recycling cycles. In a nutshell, these membranes have immense potential to revolutionize the membrane industry by introducing facile and practicable regeneration pathways, thus leading to a swift transposition from linear to circular life regimes.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors (SB) would like to acknowledge DST/SERB (Swarnajayanti fellowship) for the funding, RS and AM would like to acknowledge MHRD for Prime Minister's Research Fellowship (PMRF). SM and SR would like to thank MHRD for the institute fellowship. SSI would like to express his thankfulness for the funding from the National Postdoctoral Fellowship (PDF/2021/000629). The authors would like to express their heartfelt gratitude towards CENSE, IPC, and AFMM (IISc Bangalore) for characterization support and Biotech Testing Services, Mumbai for helping with biological studies.References
- S. L. Postel, Ecol. Appl., 2000, 10, 941–948 CrossRef
.
- R. Sen Gupta, N. Padmavathy and S. Bose, Adv. Sustainable Syst., 2021, 5, 2100213 CrossRef CAS
- R. S. Gupta, S. Mandal, S. Arya, S. Dutta, K. Manna, S. S. Islam, S. Pathan and S. Bose, Chem. Eng. J., 2023, 461, 141949 CrossRef
- S. Pathan, S. S. Islam, R. Sen Gupta, B. Maity, P. R. Reddy, S. Mandal, K. Anki Reddy and S. Bose, ACS Nano, 2023, 17(8), 7272–7284 CrossRef CAS
- D. M. Warsinger, S. Chakraborty, E. W. Tow, M. H. Plumlee, C. Bellona, S. Loutatidou, L. Karimi, A. M. Mikelonis, A. Achilli and A. Ghassemi, Prog. Polym. Sci., 2018, 81, 209–237 CrossRef CAS
- P. Bhol, S. Yadav, A. Altaee, M. Saxena, P. K. Misra and A. K. Samal, ACS Appl. Nano Mater., 2021, 4, 3274–3293 CrossRef CAS
- M. A. Shannon, P. W. Bohn, M. Elimelech, J. G. Georgiadis, B. J. Mariñas and A. M. Mayes, Nature, 2008, 452, 301–310 CrossRef CAS
- W. Xie, T. Li, A. Tiraferri, E. Drioli, A. Figoli, J. C. Crittenden and B. Liu, ACS Sustain. Chem. Eng., 2020, 9, 50–75 CrossRef
- P. K. Samantaray, A. Little, D. M. Haddleton, T. McNally, B. Tan, Z. Sun, W. Huang, Y. Ji and C. Wan, Green Chem., 2020, 22, 4055–4081 RSC
- S. P. Nunes, P. Z. Culfaz-Emecen, G. Z. Ramon, T. Visser, G. H. Koops, W. Jin and M. Ulbricht, J. Membr. Sci., 2020, 598, 117761 CrossRef CAS
- W. Lawler, Z. Bradford-Hartke, M. J. Cran, M. Duke, G. Leslie, B. P. Ladewig and P. Le-Clech, Desalination, 2012, 299, 103–112 CrossRef CAS
- P. R. Christensen, A. M. Scheuermann, K. E. Loeffler and B. A. Helms, Nat. Chem., 2019, 11, 442–448 CrossRef CAS PubMed
- T. E. Long, Science, 2014, 344, 706–707 CrossRef CAS PubMed
- P. Shieh, W. Zhang, K. E. Husted, S. L. Kristufek, B. Xiong, D. J. Lundberg, J. Lem, D. Veysset, Y. Sun and K. A. Nelson, Nature, 2020, 583, 542–547 CrossRef CAS
- M. Podgórski, B. D. Fairbanks, B. E. Kirkpatrick, M. McBride, A. Martinez, A. Dobson, N. J. Bongiardina and C. N. Bowman, Adv. Mater., 2020, 32, 1906876 CrossRef
- S. Billiet, K. De Bruycker, F. Driessen, H. Goossens, V. Van Speybroeck, J. M. Winne and F. E. Du Prez, Nat. Chem., 2014, 6, 815–821 CrossRef CAS
- J. M. Winne, L. Leibler and F. E. Du Prez, Polym. Chem., 2019, 10, 6091–6108 RSC
- R. S. Gupta, N. Padmavathy, P. Agarwal and S. Bose, Chem. Eng. J., 2022, 136997 CrossRef
- B. Briou, B. Améduri and B. Boutevin, Chem. Soc. Rev., 2021, 50, 11055–11097 RSC
- P. Banerjee, S. Kumar and S. Bose, ACS Appl. Nano Mater., 2021, 4, 6821–6831 CrossRef CAS
- W. Zhang, J. Duchet and J.-F. Gérard, J. Colloid Interface Sci., 2014, 430, 61–68 CrossRef CAS PubMed
- B. Li, S. Wang, X. J. Loh, Z. Li and T.-S. Chung, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2301009120 CrossRef CAS PubMed
- S. Chandran M, M. Krishna and K. Rai, Int. J. Polym. Sci., 2010, 2010, 987357 Search PubMed
- H. Luo, C. Gu, W. Zheng, F. Dai, X. Wang and Z. Zheng, RSC Adv., 2015, 5, 13470–13477 RSC
- M. Wang, M. Cui, W. Liu, X. Liu and B. Xu, Electroanalysis, 2018, 30, 842–851 CrossRef CAS
- F. Wang, R. Han, G. Liu, H. Chen, T. Ren, H. Yang and Y. Wen, J. Electroanal. Chem., 2013, 706, 102–107 CrossRef CAS
- X. Cai, T. Lei, D. Sun and L. Lin, RSC Adv., 2017, 7, 15382–15389 RSC
- A. Popa, D. Toloman, M. Stan, M. Stefan, T. Radu, G. Vlad, S. Ulinici, G. Baisan, S. Macavei and L. Barbu-Tudoran, J. Inorg. Organomet. Polym. Mater., 2021, 31, 1642–1652 CrossRef CAS
- X. Jiao, Y. Qiu, L. Zhang and X. Zhang, RSC Adv., 2017, 7, 52337–52344 RSC
- H.-H. Huang, K. K. H. De Silva, G. Kumara and M. Yoshimura, Sci. Rep., 2018, 8, 6849 CrossRef
- Y. Wang, R. Ou, H. Wang and T. Xu, J. Membr. Sci., 2015, 475, 281–289 CrossRef CAS
- Z. Wang, H. Yu, J. Xia, F. Zhang, F. Li, Y. Xia and Y. Li, Desalination, 2012, 299, 50–54 CrossRef CAS
- R. Sabater i Serra, J. Molina-Mateo, C. Torregrosa-Cabanilles, A. Andrio-Balado, J. M. Meseguer Dueñas and Á. Serrano-Aroca, Polymers, 2020, 12, 702 CrossRef
- Y. Zhang, Z. Xu, T. Zhang, Q. Meng, X. Zhang, C. Shen, Y. Lu, G. Zhang and C. Gao, J. Mater. Chem. A, 2020, 8, 16985–16993 RSC
- S. S. Islam, N. Salam, R. A. Molla, S. Riyajuddin, N. Yasmin, D. Das, K. Ghosh and S. M. Islam, Mol. Catal., 2019, 477, 110541 CrossRef
- Y. Lu, W. Liu, K. Wang and S. Zhang, J. Mater. Chem. A, 2022, 10, 20101–20110 RSC
- L. Cseri, R. Hardian, S. Anan, H. Vovusha, U. Schwingenschlögl, P. M. Budd, K. Sada, K. Kokado and G. Szekely, J. Mater. Chem. A, 2021, 9, 23793–23801 RSC
- P. K. Samantaray, R. Sen Gupta and S. Bose, Adv. Sustainable Syst., 2023, 7, 2200385 CrossRef CAS
- T. Le, E. Jamshidi, M. Beidaghi and M. R. Esfahani, ACS Appl. Mater. Interfaces, 2022, 14(22), 25397–25408 CrossRef CAS PubMed
- Z. Liu, Z. Xu, X. Hu and C. Gao, Macromolecules, 2013, 46, 6931–6941 CrossRef CAS
- P. Sun, F. Zheng, M. Zhu, Z. Song, K. Wang, M. Zhong, D. Wu, R. B. Little, Z. Xu and H. Zhu, ACS Nano, 2014, 8, 850–859 CrossRef CAS
- M. Hasan, R. Kumar, M. Barakat and M. Lee, RSC Adv., 2015, 5, 14393–14399 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta04539k |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |