
The Mott–Schottky Co2P/Co heterocatalyst encapsulated by N,P-doped graphene/carbon nanotubes as high-efficiency trifunctional electrocatalysts for cable-type flexible Zn–air batteries and water splitting†
Quynh Phuong
Ngo
 a,
Thanh Tuan
Nguyen
a,
Manjinder
Singh
a,
Nam Hoon
Kim
*ac and
Joong Hee
Lee
*abc
a,
Thanh Tuan
Nguyen
a,
Manjinder
Singh
a,
Nam Hoon
Kim
*ac and
Joong Hee
Lee
*abc
aDepartment of Nano Convergence Engineering (BK21 FOUR), Jeonbuk National University, Jeonju, Jeonbuk 54896, Republic of Korea. E-mail: jhl@jbnu.ac.kr; nhk@jbnu.ac.kr; Fax: +82 832702341; Tel: +82 832702342
bCarbon Composite Research Centre, Department of Polymer – Nano Science and Technology, Jeonbuk National University, Jeonju, Jeonbuk 54896, Republic of Korea
cAHES Co., 445 Techno Valley-ro, Bongdong-eup, Wanju-gun, Jeonbuk 55314, Republic of Korea
First published on 12th December 2023
Abstract
The preparation of highly efficient and low-priced multi-functional electrocatalysts for the hydrogen evolution reaction (HER), oxygen reduction reaction (ORR), and oxygen evolution reaction (OER) is crucial for robust zinc–air batteries (ZABs) and water electrolyzers. Herein, we report the preparation of novel Mott–Schottky Co2P/Co heterostructures encapsulated by N,P co-doped graphene and carbon nanotubes (Co2P/Co@N-CNT/NPG) using an in situ pyrolysis strategy, achieving superior trifunctional catalyst performance for the ORR, OER, and HER. The theoretical calculation indicates that the synergistic effect of the Mott–Schottky catalyst could increase the electron transport, trigger the active sites, and enhance the performance toward the ORR/OER. The Co2P/Co@N-CNT/NPG-based ZAB displays a considerable peak power density of 145 mW cm−2, and an outstanding cycle-life of 800 h. Furthermore, the flexible ZAB delivers superior mechanical properties with high flexibility, demonstrating its potential feasibility for practical application. Additionally, the water electrolysis device constructed using Co2P/Co@N-CNT/NPG electrodes requires a small cell voltage of 1.66 V at 10 mA cm−2, indicating the impressive ability to apply the catalyst for commercial energy storage and harvesting devices.
Introduction
Recently, the overuse of fossil fuels has led to environmental concerns, such as climate change and carbon emission trouble.1–3 The emergence of energy storage and harvesting devices, including water electrolyzers and zinc–air batteries (ZABs), could provide good solutions for future clean energy.4–7 However, the main reaction at the cathode (hydrogen evolution reaction, HER) and anode (oxygen evolution reaction, OER) during the water electrolysis process, and oxygen reactions (OER and the oxygen reduction reaction, ORR) at the ZAB cathode are the main challenges, because of their sluggish nature and poor durability.8–10 So far, the platinum group metals (PGMs) have been demonstrated as the benchmark electrocatalysts, such as Pt-based for the HER and ORR, and Ru-/Ir-based for the OER.11–13 But, their rarity, high cost, and singular functional properties have limited their large-scale application in energy storage and conversion devices.14 In addition, poor long-term stability has hindered the use of PGMs.15 Therefore, the development of cheap, high-performance, and long-term stable multi-functional catalysts is urgent for future energy industries. However, the design of nanostructures is still troublesome because of complicated nanostructure synthesis, multi-functional properties, or enriched active sites and interfaces.The transition metal compound-based electrocatalysts are illustrated as potential alternatives to PGMs due to their excellent activities, abundant supply, and inexpensive nature. In particular, metal phosphides have attracted intense attention in academia because of their outstanding catalytic activity and tunable nanostructures. For example, Liu et al. constructed Co2P implanted in Co metal, in combination with N,P dual-doped carbon, which exhibited good interaction of Co2P active sites and multi-element doped carbon, and promoted the catalyst ability for the ORR, OER, and HER.16 Parra-Puerto et al. investigated a variety of carbon-supported metal phosphides (MxPy) for the HER, ORR, and OER under various pH conditions, which could be comparable to the benchmark catalysts, such as Pt/C and IrO2.17 However, only a few types of such trifunctional electrocatalysts have been reported, and deep understanding of their mechanism is lacking.
On the other hand, the development of heterostructure catalysts is a favorable approach to boost the active sites, reduce the reaction energy barrier, facilitate charge transfer, and synergistically enhance electrocatalytic performance. These benefits were obtained from the optimal design of the multiple crystalline components or various phases to achieve the heterostructures.18 For example, Diao et al. developed W2N/WC heterostructure catalysts with abundant interfaces for water electrolyzer and zinc–air battery application. The as-prepared electrocatalysts presented a considerable half-wave potential of 0.81 V for the ORR, and required a small overpotential of 320 mV (OER) and 148.5 mV (HER) to attain a current density of 10 mA cm−2.19 Moreover, the electrocatalysts with the Mott–Schottky effect, which could spontaneously drive electrons to flow across the heterointerfaces and improve the electrochemical activities, were hindered by poor interaction with the active center.20 As a result, the chemical adsorption/desorption of intermediate species to the substrate could be improved, which accelerates the reaction conversion.21 Xue et al. prepared Janus Co/CoP nanoparticles that showed outstanding HER and OER performance in different concentrations and pH ranges.22 Nevertheless, the preparation of trifunctional catalysts with exceptional activities and excellent durability for ZABs and full water electrolyzers remains a great challenge.23 Therefore, an optimal approach that could prepare trifunctional and low-cost electrocatalysts with a facile, large-scalable process, and numerous active sites is an urgent task to satisfy the industrial application.24
Herein, we describe an in situ fabrication step that controlled the formation and generated the heterostructure of Co2P/Co encapsulated with nitrogen-doped carbon nanotubes (N-CNTs) and N,P co-doped graphene sheets (Co2P/Co@N-CNT/NPG). The Co2P/Co@N-CNT/NPG heterostructures provide a unique nanostructure with numerous active sites. One of the distinctive features of our work is the successful synthesis of an hierarchical nanostructure with an optimal ratio of Co2P/Co nanoparticles homogeneously encapsulated by N-CNTs, and interconnected with heteroatom N,P-co-doped porous graphene sheets. This configuration could generate the Mott–Schottky effect, associate the intrinsic potential and electric field at the catalyst interface by rearrangement of the electron cloud density and increase the adsorption/desorption of reactive species; therefore, it tolerates electrical charge through the active region and external integrated circuit. This composite structure possesses a unique combination of multiple ingredients within a single framework and significantly enhances electrocatalytic activity. In addition, the extreme surface area, large pore volume, and excellent conductivity of the N-CNT and NPG with an effective synergistic effect could promote the catalytic activities for the HER, OER, and ORR and were successfully applied for zinc–air batteries and water splitting application, making it a noteworthy advancement in the field. The Co2P/Co@N-CNT/NPG catalysts showed a small overpotential of 98 and 309 mV for the HER and OER to accomplish a current density of 10 mA cm−2. For the ORR process, the catalysts showed a high half-wave voltage of 0.91 V with an onset voltage of 1.05 V. The exceptional performance of Co2P/Co@N-CNT/NPG is superior to that of Pt/C or RuO2 electrocatalysts. Likewise, the water-splitting devices that were assembled with the same Co2P/Co@N-CNT/NPG electrodes needed a small voltage cell of 1.66 V to obtain a current density of 10 mA cm−2 with high stability for 40 h. When the Co2P/Co@N-CNT/NPG catalysts serve as the air-cathode, the as-fabricated ZAB showed a maximum power density of 145 mW cm−2, and an excellent stability of 800 h without degradation. Moreover, the solid-state flexible rechargeable ZAB presented superior mechanical properties with different bending and twisting states.
Experimental section
Synthesis of Co2P/Co@N-CNT/NPG
Graphene oxide (GO) was prepared using the previous procedure.25 50 mg GO was dispersed in 50 mL of DI water in a glass vial under tip sonication for 90 min. Then, 0.3 mmol cobalt(II) acetate (Co(ac)2), 2 mmol dicyandiamide (DCDA), and 1 mmol triphenylphosphine (TPP) were dissolved in GO solution under stirring. After that, the solution was dried using a freeze-drying machine to collect a dark grey powder. The resultant material was pyrolyzed at 800 °C under Ar/H2 (100 sccm) flow and held for 2 h. For comparison, Co@N-CNT/NG, Co2P/Co@PG, and NPG were also synthesized by a similar process, without adding TPP, DCDA, or Co(ac)2, respectively.Material characterization
The morphologies were observed using a field emission scanning electron microscope (FE-SEM) (JEOL, JSM-6701F, Japan) which is installed at the Center for University-wide Research Facilities (CURF), Jeonbuk National University (JBNU). The internal nanostructure was recorded by using a transmission electron microscope (TEM) (ARM200F, JEOL, Japan) at Korea Basic Science Institute (KBSI), Jeonju. The Raman spectra were recorded using a Nanofinder-30 machine (Tokyo Ins. Co., Japan) using a laser wavelength of 532 nm. The X-ray diffraction pattern (XRD) was recorded with a Rigaku Ultima IV (Japan). The N2-sorption isotherms and pore volume distributions were obtained using a Micromeritics ASAP 2020 at ≈77 K. The elemental components were accomplished by EDAX (SUPRA 40 VP; Germany). The X-ray photoelectron spectroscopy (XPS) data were collected by using a Theta probe instrument (Thermo Fisher Scientific, UK) at the Korea Basic Science Institute (KBSI), Jeonju.Electrochemical characterization
The electrochemical measurement was performed with a CHI potentiostat (CHI 760E, CH Ins. Inc., USA). The as-prepared catalyst-coated nickel foam (NF), graphite, and Hg/HgO were employed as the working, auxiliary, and reference electrodes. The obtained linear sweep voltammetry (LSV) results were corrected by 100% iR compensation to investigate the OER and HER performance. A rotating disk electrode (RDE) was employed to investigate the ORR performance, with the ORR polarization curves being corrected for the contribution of the double-layer capacitance. The EIS was performed in the frequency range 105 to 10−2 Hz at constant potentials in the catalytic turnover regions where all the studied catalytic interfaces possessed appreciable OER activity (in this instance, at 1.65 V vs. RHE). The as-obtained potential was switched to reversible hydrogen electrode (RHE) potentials using the formula:| ERHE = EHg/HgO + 0.098 + 0.059 pH | (1) |
The catalyst mixture was formulated by blending 2 mg catalyst and 5 μL 0.5% Nafion in 1 mL water + ethanol (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and ultrasonicated for 30 min. After that, 7 μL of the mixture was dropped onto the electrode and kept for drying.
:1) and ultrasonicated for 30 min. After that, 7 μL of the mixture was dropped onto the electrode and kept for drying.
The K–L plots are derived from plots of id−1 and the inverse of the square root of the rotation speed:
| 1/J = 1/JL + 1/JK = 1/Bω1/2 + 1/JK | (2) |
| B = 0.62nFC0D02/3ν−1/6 | (3) |
The H2O2 percentage and the number of electrons transferred (n) were projected by the rotating ring-disk electrode (RRDE) approach, and determined from the equations below:
| H2O2 (%) = 200 × (IR/N)/(IR/N + ID) | (4) |
| n = 4 × ID/(ID + IR/N) | (5) |
ZAB testing
Fabrication of aqueous ZABs: rechargeable ZABs were assembled in a homemade cell. A catalyst loading of 2 mg cm−2 was applied to a gas diffusion carbon cloth (W1S1010, Cetech, Taiwan) using a spatula. The anode and electrolyte consisted of a zinc metal plate and 6 M KOH with 0.2 M zinc acetate. Charge–discharge profiles were collected using a Wonatech multichannel battery testing system (WMPG1000S, WonATech Co., Ltd., Korea).Fabrication of the cable-type flexible all-solid state ZAB (CF-ZAB): a plastic tube was punched with holes and employed as the cover for the CF-ZAB. The air cathode prepared above was 0.5 cm in width and 15 cm in height. A zinc rod was employed as the anode. A PVA hydrogel film with 6 M KOH was fabricated and exploited as the quasi-solid electrolyte and separator. First, the zinc rod was placed in a template of 0.6 cm diameter and 8 cm length. Then the polymer electrolyte solution was poured into the template and cooled in a freezer for 6 h to obtain the CF-ZAB.
Theoretical calculations
The density functional theory (DFT) calculations were accomplished using the CASTEP code.26,27 The model structures were built with Co@NG, Co2P/Co@PG, and Co2P/Co@NPG with a k-point mesh of 3 × 3 × 1. The vacuum layer was set at 20 Å along the z-axis for slabs. The self-consistent field (SCF) tolerance is 1.0 × 10−5 eV per atom. The maximum force for geometry optimization convergence tolerance was set at 0.02 eV Å−1. Generalized gradient approximation (GGA) and Perdew–Burke–Ernzerhof (PBE) were employed for electron exchange and correlation energy.28,29 The plane-wave energy cut-off was set at 300 eV.30,31Results and discussion
Material characterization
The Co2P/Co@N-CNT/NPG nanostructure was fabricated using an in situ controlled pyrolysis route, as shown in Fig. 1.32 First, GO, metal sources, DCDA, and TPP were mixed and stirred vigorously for 40 min to obtain a well-dispersed mixture. After that, it was freeze-dried and annealed under Ar gas protection. During the high-temperature treatment process, the DCDA and TPP were decomposed, to provide the initial source for the formation of Co2P/Co@N-CNT/NPG. Subsequently, the N and P doping into the graphene framework could tune the electronic properties and increase the porosity and active sites to improve the electrochemical efficiency.33 Furthermore, Co metal could be obtained from the reduction process with the appearance of H2 gas and the phosphorous source, provided by the decomposition of TPP to form metal phosphide (Co2P) and partial reduction to Co metal. The synergistic effect generated from the Mott–Schottky catalysts of Co2P/Co could increase the interfacial charge transfer, tune the charge interface, and restrain the adsorption of intermediate species; therefore, it could significantly enhance the electrocatalyst performance.34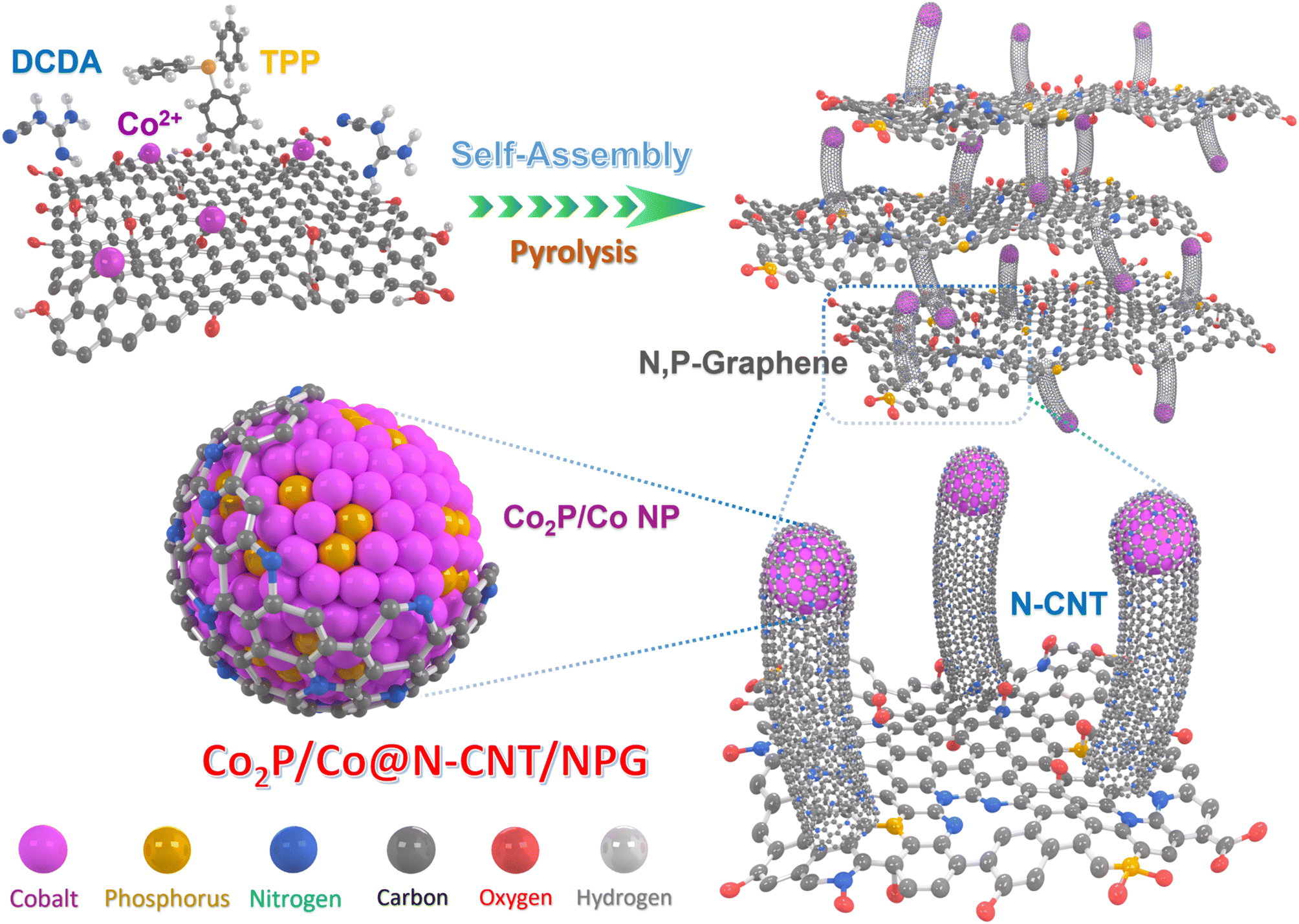 | ||
| Fig. 1 Schematic of the fabrication procedure for the Co2P/Co@N-CNT/NPG nanohybrid. | ||
The surface morphologies and internal nanostructures of nanostructures were evaluated by FE-SEM and TEM. Fig. 2a–c reveal the numerous heterogenous Co2P/Co NPs with a diameter of 24.5 ± 4.3 nm that were encapsulated by CNTs and uniformly distributed on the N,P-doped graphene sheets. The plentiful hierarchical architecture observed by the conjugation of 1D N-CNT and 2D NPG is expected to favor both mass transfer and electronic conductivity during the electrocatalytic reactions (Fig. S1†). The combination of 1D and 2D hierarchical nanostructures could provide outstanding physicochemical properties, and effectively inhibit agglomeration of nanoparticles; therefore, it could increase the exposed active sites of the catalyst.35 The N-doped CNTs are uniformly grown with an average diameter of 20 ± 5 nm to provide an efficient channel for electron transport.36 In comparison, the N-CNT could not be formed without the appearance of DCDA sources or Co catalysts (Fig. S2 and S3c, f†). In contrast, the morphology of Co@N-CNT/NG without the addition of TPP showed random size and distribution (Fig. S3a and d†), illustrating the critical role of TPP during the formation of heterocatalysts. Fig. 2c and e show that the Co2P/Co heterostructure NPs on the bamboo-like N-CNT were perfectly sealed by several layered carbon shells with a lattice spacing of 3.52 Å, related to the d-spacing of the (002) facet of graphitic carbon.37 The high-resolution transmission electron microscopy (HR-TEM) images of Co2P/Co heterostructure NPs clearly show the different regions of Co2P and Co with lattice fringes of 2.07 and 2.14 Å that imply the (111) and (121) facets of cobalt and cobalt phosphide, respectively (Fig. 2d).38 The formation of the Mott–Schottky heterojunction between the semiconductor Co2P and the metallic Co successfully induces the redistribution of electrons and holes, leading to the alignment of work functions on both sides.39 This could synergistically enhance the kinetic activities of multifunctional catalysts for the ORR, OER, and HER. Moreover, the scanning transmission electron microscope (STEM) images and energy dispersive spectrometry (EDS) color mapping reveal that Co and P are mainly located inside the carbon layers, while the C and N elements are consistently distributed in the Co2P/Co@N-CNT/NPG (Fig. 2f). Moreover, according to the findings from inductively coupled plasma atomic emission spectroscopy (ICP-AES), the Co element content in the Co@N-CNT/NG, Co2P/Co@N-CNT/NPG, and Co2P/Co@PG nanostructures is determined to be 9.31, 11.93, and 13.14 wt%, respectively (Fig. S4†). In addition, the encapsulated metallic nanoparticles could synergistically activate and further boost the electrochemical activities for the HER, OER, and ORR of the surrounding nitrogen-doped carbon layers/carbon nanotubes.40 In particular, the emerged synergy between the N and P co-doped carbon framework and the metal elements led to the bonding of cation and anion (M-N,P) segments, which could properly serve as the active sites, efficiently tailoring the electronic properties and chemical reactivity.41 Moreover, the carbon substrate provides an open and highly accessible site for oxygen intermediate species, which could be beneficial for improving the sluggish oxygen reactions.42
 | ||
| Fig. 2 Morphological analysis of the Co2P/Co@N-CNT/NPG nanohybrid: (a) overview of the SEM image, (b and c) TEM images (inset (c) shows the particle size distribution), (d and e) HR-TEM images, and (f) HAADF-STEM image with EDS color mapping of Co, P, N, C, and O. | ||
The crystallinity of the as-synthesized nanostructures was characterized by XRD. Fig. 3a reveals broad peaks at 2θ ≈ 26.1°, indicating the graphene and CNT signals.15 Moreover, the observed peaks at 2θ ≈ 44, 51, and 76° indicate Co metal (ICDD#00-015-0806), which demonstrated the successful formation after the reduction process.43 In addition, the XRD pattern of Co2P/Co@N-CNT/NPG and Co2P/Co@PG presented the diffraction peaks at 2θ ≈ 40.7, 40.9, and 43°, which were assigned to the (101), (201), and (211) facets of Co2P, respectively (ICDD#00-032-0306).44 These results verified the appearance of both Co metal and Co2P phases in the Co2P/Co@N-CNT/NPG heterostructure with the controlled synthesis protocol. The Raman spectra of all the as-prepared materials exhibit three typical bands at 1350, 1592, and 2682 cm−1 (Fig. 3b and S5†), related to the D, G, and 2D bands. The ID/IG ratio is a powerful tool to evaluate the defect and graphical contribution in carbon nanostructures.45 The ID/IG ratio displays a value of 1.35 for Co2P/Co@N-CNT/NPG, which is higher than that for Co@N-CNT/NG, Co2P/Co@PG, NPG, and GO of 1.20, 1.12, 1.25, and 0.95, respectively. These results suggest that the formation of the metal/metal phosphide heterostructure could release more defects from the graphene sheets, and therefore could be beneficial to the electrocatalytic activities. To examine the specific surface area and further prove its effect on electrochemical performance, we investigated the N2 sorption isotherms. The Brunauer–Emmett–Teller (BET) measurement results of the as-prepared samples show the representative type-IV isotherm (Fig. 3c), suggesting the mesoporous nanostructure.46Fig. 3c shows that the BET specific surface area of Co2P/Co@N-CNT/NPG is 493.6 m2 g−1, which is higher than that of Co@N-CNT/NG, Co2P/Co@PG, and NPG of 368.7, 300.2, and 403.1 m2 g−1, respectively. Fig. S6† shows that the pore diameter of the as-synthesized materials is mainly contributed at ≈3.5 nm, a little increased compared to that of pure NPG, due to the formation of the Co2P/Co heterostructure. The porous architecture with the excellent specific surface area of Co2P/Co@N-CNT/NPG could increase the contact sites with the electrolyte, promote ion/electron transport, and improve electrochemical performance.47
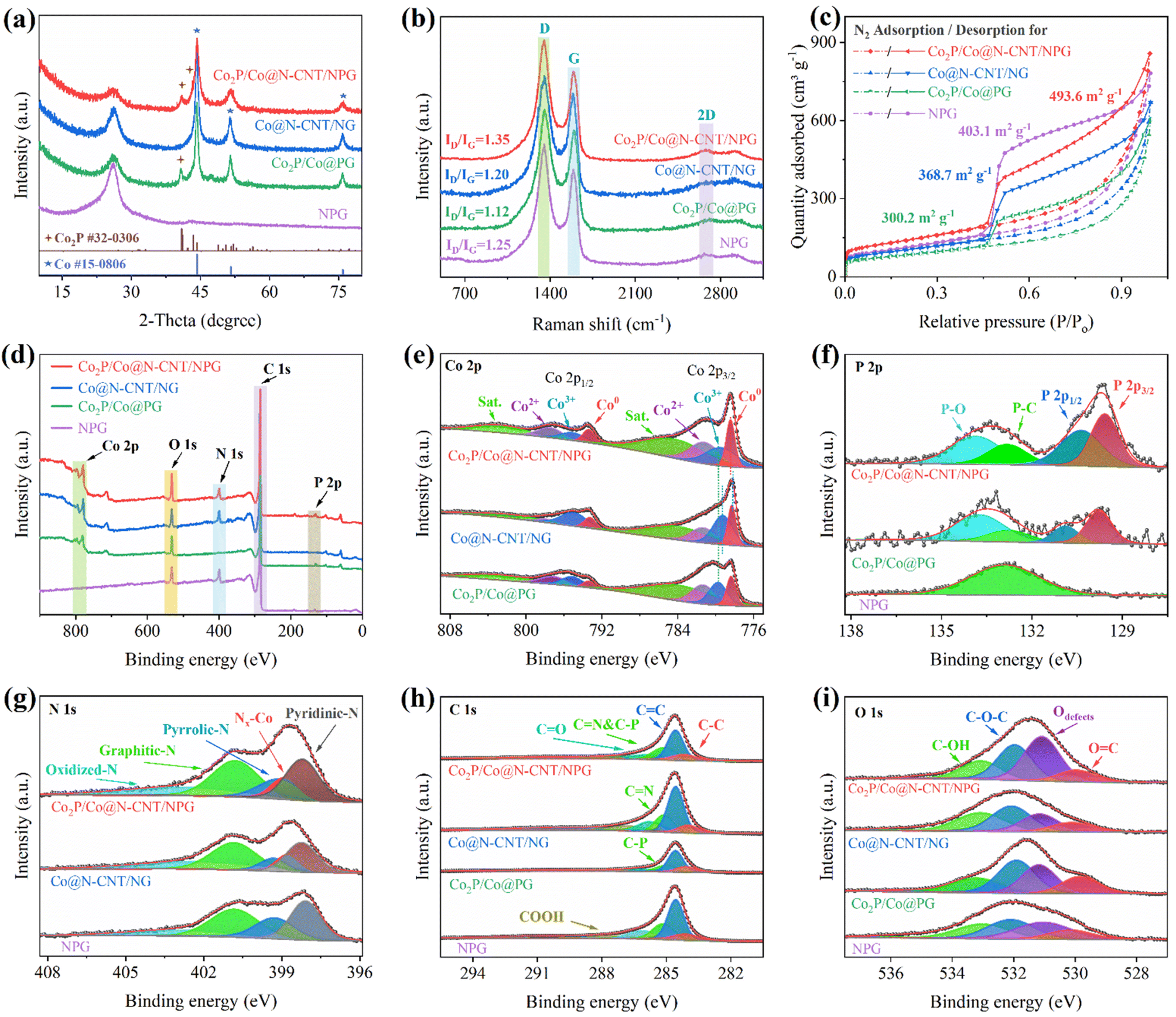 | ||
| Fig. 3 Structural investigations of the synthesized materials: (a) XRD patterns, (b) Raman spectrum, (c) N2 sorption isotherm, (d) XPS survey spectrum with high-resolution XPS spectra for the (e) Co 2p, (f) P 2p, (g) N 1s, (h) C 1s, and (i) O 1s of the Co2P/Co@N-CNT/NPG, Co@N-CNT/NG, Co2P/Co@PG nanohybrids, and NPG. | ||
To study the oxidation state and chemical bonding of the as-prepared materials, we performed XPS. The survey XPS of the Co2P/Co@N-CNT/NPG showed the existence of Co, O, N, C, and P elements (Fig. 3d and Table S1†). The XPS of Co 2p could be deconvoluted by the spin-orbiting couples with different oxidation states at 793.4, 795.2, and 797.5 eV for Co 2p1/2 and 778.4, 779.5, and 781.3 eV for Co 2p3/2, respectively (Fig. 3e). Moreover, two peaks positioned at binding energies of 784.8 and 802.9 eV represented satellites. The formation of Co0, Co3+, and Co2+ species could provide highly efficient active sites to improve the catalytical properties for the ORR, OER, and HER, which is consistent with the previous reports in the literature.7 In compared to Co@N-CNT/NG, the electron binding energy of the Co 2p in Co2P/Co@N-CNT/NPG and Co2P/Co@PG shows a positive shift to higher binding energy, demonstrating the electron transport from Co2P to Co metal through the Mott–Schottky interface (Fig. 3e).39 This result underscores the primary role of the Mott–Schottky interaction in significantly enhancing their performance in both the HER and OER processes.22 The deconvolution of P 2p showed two separated peaks at the positions of 129.6 and 130.1 eV, related to P 2p3/2 and P 2p1/2. Moreover, the two peaks at 133.7 and 132.6 eV could indicate unavoidable oxidized P on the surface of Co2P due to contact with air and P–C in NPG, respectively (Fig. 3f).48 The N 1s showed the main peaks at binding energies of 389.9, 398.2, 399.4, 400.9, and 403.5 eV, which represented the pyridinic-, Co-, pyrrolic-, graphitic-, and oxidized-N, respectively (Fig. 3g).49 The dominance of pyridinic- and pyrrolic-N in NPG and the N-CNT nanostructure due to the high thermodynamic stability at the edges of the graphene lattice can create an n-type material, and play an important role as high-efficiency catalytic sites for electrochemical reactions.50 The C 1s spectrum (Fig. 3h) could fit well the typical peaks for the C–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CN/C–P, CO, and COOH bonds at 283.9, 284.6, 285.1, 285.9, and 287.1 eV, respectively.51 The O 1s XPS spectra (Fig. 3i) display four peaks at binding energies of 529.9, 531.0, 532.0, and 533.1 eV, corresponding to OC, defective oxygen, C–O–C, and C–OH, respectively.52 The presence of defective oxygen could form the O–O bridge with the oxygen-containing species, and enhance the catalytic activities.53
C, CN/C–P, CO, and COOH bonds at 283.9, 284.6, 285.1, 285.9, and 287.1 eV, respectively.51 The O 1s XPS spectra (Fig. 3i) display four peaks at binding energies of 529.9, 531.0, 532.0, and 533.1 eV, corresponding to OC, defective oxygen, C–O–C, and C–OH, respectively.52 The presence of defective oxygen could form the O–O bridge with the oxygen-containing species, and enhance the catalytic activities.53
X-ray absorption spectroscopy (XAS) was systematically conducted at the Co K-edge to unravel intricate details concerning the local coordination environment of Co atoms. As shown in Fig. S7a,† X-ray absorption near-edge structure (XANES) showed the white line peak in the XANES spectra of Co2P/Co@N-CNT/NPG with Co foil and Co3O4 reference that demonstrated the existence of both Co metal and Co–P embedded in the N-CNT/NPG framework.9,10,54
Furthermore, the extended X-ray adsorption fine structure (EXAFS) spectra at the Co K-edge of Co2P/Co@N-CNT/NPG display a prominent broad peak. This peak is likely attributed to the coexistence of Co–P (1.79 Å) and Co–Co (2.14 Å) bonds, which is well-consistent with Co foil and Co3O4 reference.55 Additionally, the absence of Co–O and Co–O–Co bonds suggests that the phosphide compound was successfully formed in the Co2P/Co@N-CNT/NPG nanostructure, corroborating the results obtained from the XRD analysis (Fig. S7b†).
Electrochemical measurement
The oxygen reactions, including the ORR and OER, mainly drive the electrochemical performance of the ZAB. To successfully utilize the as-prepared electrodes for zinc–air battery application, their performance was first investigated for the ORR and OER. The cyclic voltammetry (CV) profiles of all the as-synthesized catalysts were acquired at a fixed scan rate of 10 mV s−1 in 0.1 M KOH O2-saturated electrolyte (Fig. 4a). The CV profile of the Co2P/Co@N-CNT/NPG revealed a sharp peak at 0.84 V, superior to other counterparts of Co@N-CNT/NG (0.82 V), Co2P/Co@PG (0.69 V), and NPG (0.77 V), suggesting a high-efficiency electrocatalytic activity for the ORR. In contrast, the CV profiles of all catalysts showed pseudo capacitance properties in N2-saturated solution, confirming that the corresponding peak revealed the oxygen reactions. The catalytic activities of all catalysts for the ORR were examined by the LSV technique under steady state polarization at 1600 rpm. The LSV curve of the Co2P/Co@N-CNT/NPG catalyst possessed an excellent onset potential of 1.04 V and a half-wave potential of 0.91 V. This performance is superior to that of benchmark Pt/C of 1.02 and 0.89 V, and outstanding compared to that of Co@N-CNT/NG, Co2P/Co@PG, and NPG of 0.97 and 0.86 V, 0.90 and 0.76 V, and 0.94 and 0.83 V, respectively (Fig. 4b, c and S8†). Moreover, the limiting current density illustrated the maximum diffusion current through the electrolyte during the ORR. Fig. 4b shows the tremendous limiting current density of 5.36 mA cm−2 for Co2P/Co@N-CNT/NPG, which is greater than that of other comparative catalysts of Co@N-CNT/NG, Co2P/Co@PG, NPG, and the benchmarked Pt/C of 4.72, 3.57, 4.39 and 5.19 mA cm−2, respectively. The Tafel slope is a critical factor that indicates the electron transport kinetics and reveals the electrocatalyst performance during the ORR. Fig. 4d exhibits the smallest Tafel slope of 66.3 mV dec−1 for the Co2P/Co@N-CNT/NPG catalyst, confirming the outstanding kinetics for the ORR. The electrochemical performance of the Co2P/Co@N-CNT/NPG catalyst was shown to be superior to that of the other non-precious electrocatalysts for the ORR that were recently reported in the literature (Table S2†). The excellent catalytical ability of the Co2P/Co@N-CNT/NPG catalyst originates from the enhanced active sites of Co2P/Co on the N,P dual-doped graphene and CNT hierarchical nanostructures. The LSV profiles at various rotational speeds from 400 to 2500 rpm were collected to investigate the reaction kinetics of the Co2P/Co@N-CNT/NPG catalyst (Fig. 4e and S9†). The limiting-current density regularly increases with increasing rotational speeds, demonstrating high mass transport and the kinetic-controlled nature of the ORR.56 The Koutecky–Levich (K–L) plots derived from the LSV profiles of the Co2P/Co@N-CNT/NPG exhibited excellent linearity, and validated the reaction kinetics toward the oxygen dissolution (Fig. 4f).57 The number of electrons transferred (n) was determined as 3.97, suggesting that the ORR followed the four-electron reaction procedure. The electrocatalytic durability of Co2P/Co@N-CNT/NPG was investigated by the chronoamperometric method in a three-electrode configuration. Fig. 4g reveals the outstanding stability of the Co2P/Co@N-CNT/NPG electrocatalyst with a retained current of about 94% after 40 h, which is better performance than that of Pt/C catalysts that retained 73% after 17 h. The LSV curve collected after long-term durability revealed a negligible shift, compared to the initial catalysts, illustrating enhanced durability for the ORR (Fig. S10†). Furthermore, we analyzed the Co2P/Co@N-CNT/NPG catalyst after long-term ORR performance. As shown in Fig. S11,† the SEM, TEM, XRD, Raman, and XPS data of the Co2P/Co@N-CNT/NPG catalyst after the ORR revealed a similar structure to the initial state, confirming the superior stability of the catalyst. We performed the LSV measurement using the RRDE to further examine the reaction mechanism of the as-obtained catalysts for the ORR (Fig. 4h). The LSV of all catalysts showed good consistency with the RDE results. Moreover, the Co2P/Co@N-CNT/NPG catalyst showed very low hydrogen peroxide yields of less than 5%, and an electron transfer number (n) value of around 4 (Fig. 4i), which shows good consistency with the K–L plot approach. Methanol tolerance was employed to verify the selectivity of the catalysts during chronoamperometry measurement. As shown in Fig. S12,† the Co2P/Co@N-CNT/NPG electrode displays a small change of current after adding 3 mL of methanol at 750 s. In contrast, a major change in current occurs for the Pt/C catalyst, due to the catalyst poisoning by methanol addition. This result confirms the good electrochemical performance of Co2P/Co@N-CNT/NPG for the ORR and suggests the high possibility for real industrial application.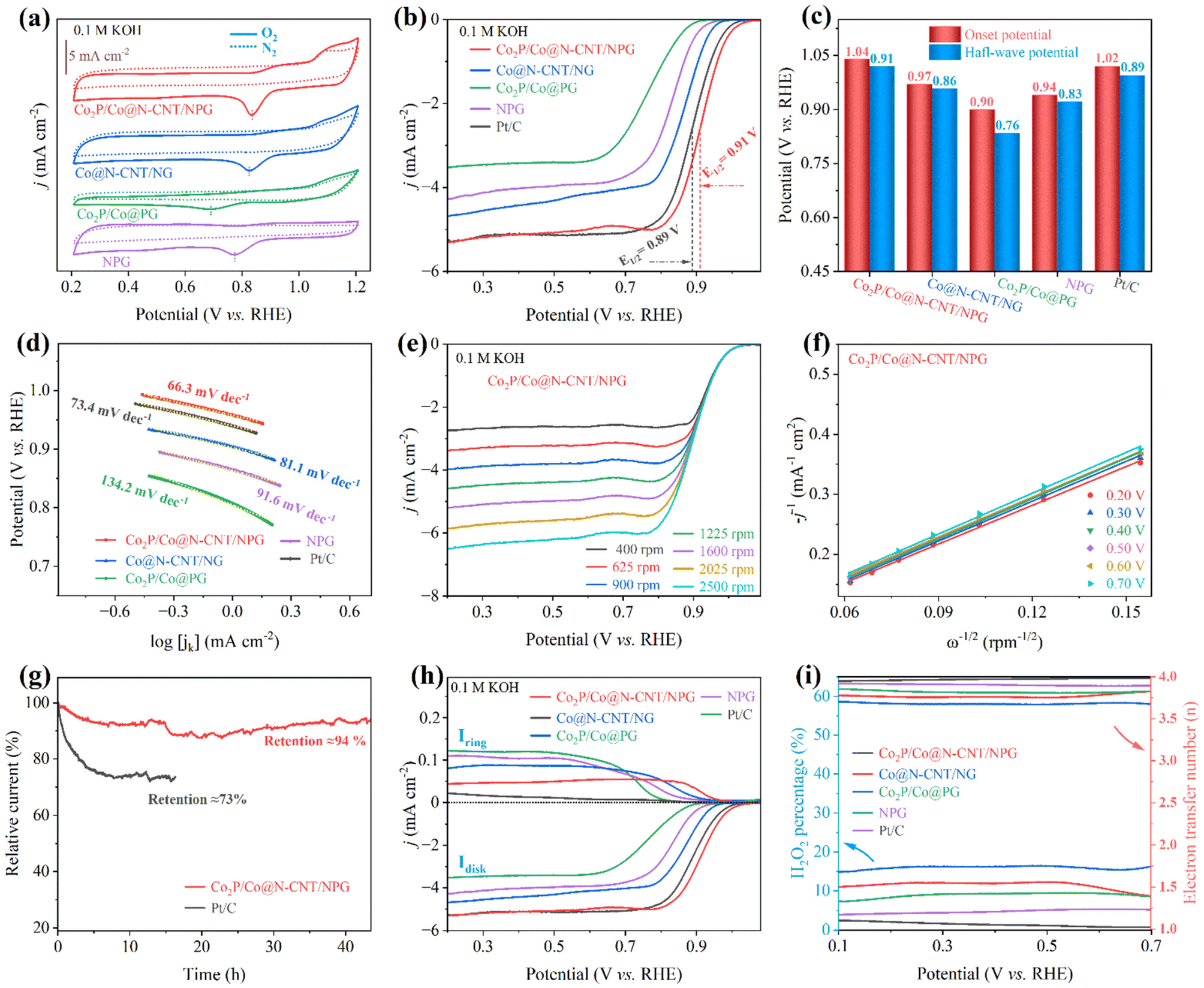 | ||
| Fig. 4 Electrocatalytic characterization of the synthesized materials in 0.1 M KOH toward the ORR: (a) the CV profiles of the synthesized materials at a sweep rate of 10 mV s−1 in N2 and O2 saturated electrolyte solution, (b) LSV polarization curves at 1600 rpm, which display the jdisk (disk current), and the corresponding (c) comparison of the as-prepared catalysts, (d) Tafel plots, and (e) LSV responses at various electrode rotational speeds. (f) K–L plots of Co2P/Co@N-CNT/NPG. (g) Chronoamperometric stability of the Co2P/Co@N-CNT/NPG and Pt/C catalysts after continuous operation, (h) LSV displayed jdisk and jring (ring current), and (i) H2O2 yield and electron transfer number (n) of Co2P/Co@N-CNT/NPG, Co@N-CNT/NG, Co2P/Co@PG, NPG, and Pt/C. | ||
The OER performance plays a vital role in the charging process of the ZAB. We carried out the OER measurement with a three-electrode configuration in 1 M KOH solution. Fig. 5a exhibits the LSV plots of the as-synthesized electrocatalysts. The Co2P/Co@N-CNT/NPG catalyst required a low overpotential of 309 mV to obtain a current density of 10 mA cm−2, which is exceptional, compared to that of benchmark RuO2 (341 mV), and the counterpart electrocatalysts of Co@N-CNT/NG, Co2P/Co@PG, and NPG of 356, 363, and 402 mV, respectively (Fig. S13†). Moreover, the Co2P/Co@N-CNT/NPG catalyst is outstanding when compared with other recent reports of transition metal-based catalysts for the OER performance (Table S3†). The Tafel slope was calculated from LSV plots of the Co2P/Co@N-CNT/NPG catalyst and exhibited a low value of 60.5 mV dec−1, which is even better than that of the benchmark RuO2 (74.2 mV dec−1), and superior to that of the counterparts of Co2P/Co@PG, Co@N-CNT/NG and NPG of 70.6, 71.7, and 102.6 mV dec−1, respectively (Fig. 5b). This result suggests that the synergistic effect between Co2P and metallic Co could improve the catalyst effective sites and accelerate electron transport. The long-term stability of the Co2P/Co@N-CNT/NPG catalyst was examined by collecting the LSV profile after 10000 continuous CV cycles. Fig. S14† reveals excellent electrochemical stability with a slight shift in LSV curves after 10000 cycling tests. In addition, the chronopotentiometry profile at different current densities of the Co2P/Co@N-CNT/NPG catalyst before and after 10000 CV cycles in Fig. 5c verifies the possibility of the catalyst at high current and long-term stability. The chronoamperometry was performed at a fixed potential, and Fig. S15† shows the result. The Co2P/Co@N-CNT/NPG catalyst presented outstanding durability with a current retention of 87% after 40 h, which is better than the benchmarked RuO2 catalyst of 69% current retention. The SEM, TEM, XRD, Raman, and XPS of the Co2P/Co@N-CNT/NPG catalyst were further performed after the OER measurement. The morphology and crystalline feature shown in Fig. S16† demonstrate the superior performance of the Co2P/Co@N-CNT/NPG catalyst for long-term OER stability. For practical application of water electrolysis, the OER performance was investigated at a higher KOH concentration of 3 M at 80 °C. The Co2P/Co@N-CNT/NPG catalyst needed a low overpotential of 183 and 213 mV at 10 and 50 mA cm—2, the values being smaller than those of Co@N-CNT/NG, Co2P/Co@PG, NPG, and benchmark RuO2 of 186 and 218 mV, 196 and 229 mV, 207 and 251 mV, and 191 and 227 mV, respectively (Fig. S17†). To estimate the electrochemical performance of the Co2P/Co@N-CNT/NPG bifunctional catalyst for oxygen reactions, we performed the LSV for both the ORR and OER at the steady state potential ranging from 0.1 to 2.0 V (Fig. 5d). The Co2P/Co@N-CNT/NPG catalyst exhibits exceptional ORR/OER catalyst activity with a slight voltage difference (ΔE = 0.82 V). The excellent catalytic activities and outstanding durability of the Co2P/Co@N-CNT/NPG catalysts demonstrate their great potential for use as non-precious metal catalysts for industrial ZAB and water electrolyzer applications (Table S4†).58
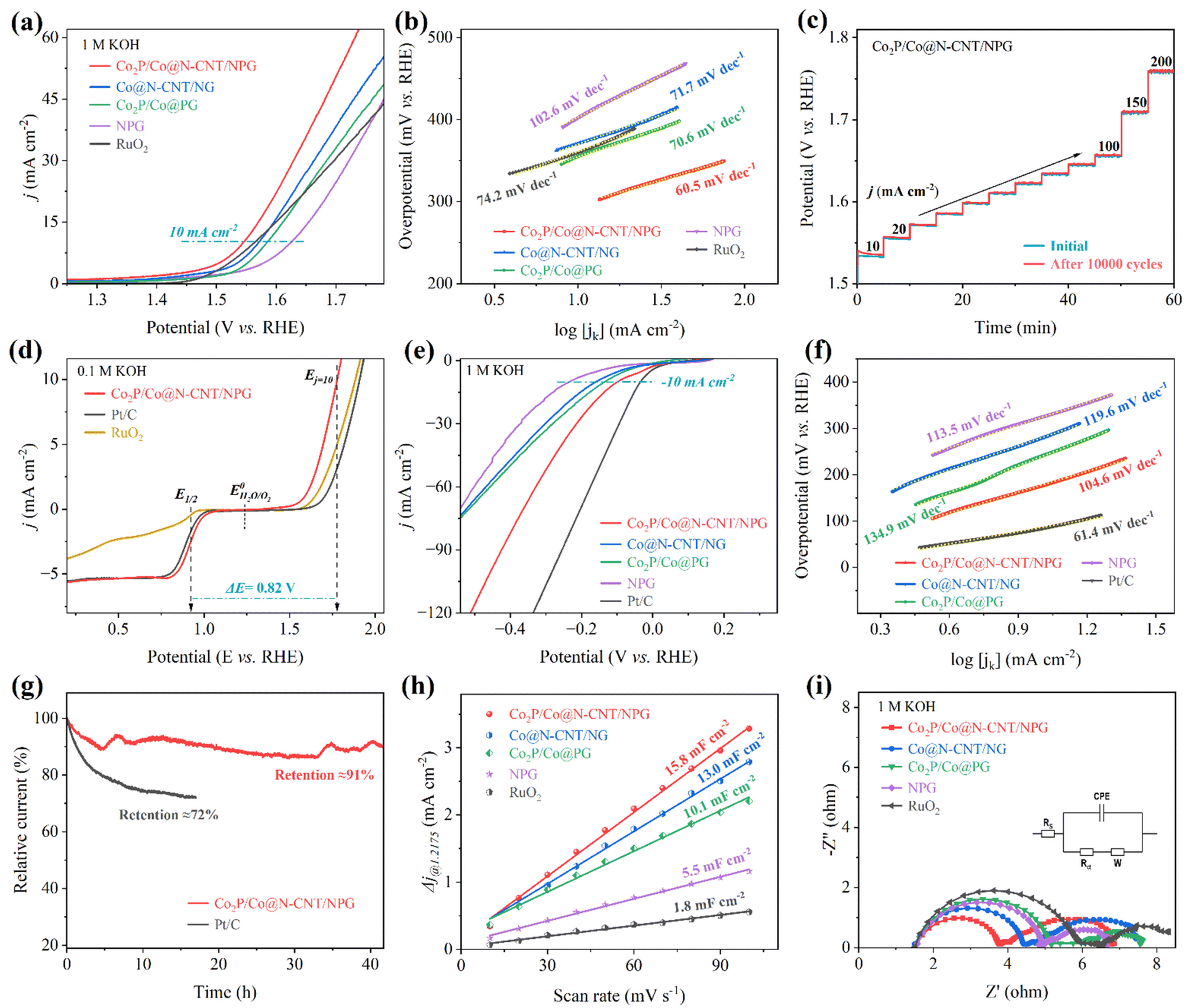 | ||
| Fig. 5 Electrocatalytic characterization of the synthesized materials toward the OER and HER in 1 M KOH: (a) LSV polarization curves for the OER. (b) Tafel plots of Co2P/Co@N-CNT/NPG, Co@N-CNT/NG, Co2P/Co@PG, NPG, and RuO2, (c) cycling stability of Co2P/Co@N-CNT/NPG before and after 10000 cycles at various currents, (d) the LSV curves of Co2P/Co@N-CNT/NPG, Pt/C, and RuO2 for both the OER and ORR in 0.1 M KOH, (e) HER LSV polarization curves, (f) Tafel plots of the as-prepared catalysts, (g) stability testing, (h) Cdl values, and (i) EIS analyses of the different synthesized catalysts. | ||
To further apply the as-synthesized catalysts for water electrolysis application, we examined the electrocatalytic characteristics for the HER in 1 M KOH solution. The LSV curves show that the Co2P/Co@N-CNT/NPG hybrid needs a minimal overpotential of 98 mV to reach a current density of 10 mA cm−2, prominently overtaking that of the Co2P/Co@PG, Co@N-CNT/NG, and NPG of 135, 163, and 236 mV, respectively (Fig. 5e and S13†). Under highly alkaline concentration conditions of 3 M KOH at 80 °C, the Co2P/Co@N-CNT/NPG catalyst required a small overpotential of 86 and 221 mV at 10 and 50 mA cm−2, respectively, which is equivalent to that of Pt/C at 10 and 50 mV, and exceptional when compared with that of the counterpart catalysts (Fig. S18†). The Tafel slope of the Co2P/Co@N-CNT/NPG catalyst calculated from the corresponding LSV profile showed a low value of 104.6 mV dec−1, which is better than that of Co2P/Co@PG (134.9 mV dec−1), Co@N-CNT/NG (119.6 mV dec−1), NPG (113.5 mV dec−1), and similar to that of Pt/C (61.4 mV dec−1) catalysts (Fig. 5f). Co2P/Co@N-CNT/NPG showed outstanding HER catalytic activities compared to the recent reports of transition metal and carbon-based catalysts in the literature (Table S5†). The long-term working ability is a crucial factor for commercialization. We performed the chronoamperometry at a fixed potential of −0.1 V vs. RHE to evaluate the stability of the Co2P/Co@N-CNT/NPG catalyst (Fig. 5g). The current density degradation of the Co2P/Co@N-CNT/NPG catalyst was about 91% after 40 h; meanwhile, the benchmarked RuO2 catalyst retained only 72% current density after 16 h. Moreover, the LSV polarization curves of Co2P/Co@N-CNT/NPG were accumulated after 10000 CV cycles. As shown in Fig. S19,† a negligible shift was obtained, illustrating the good durability of the catalyst for the HER. Additionally, the morphology and structure of the Co2P/Co@N-CNT/NPG sample after the HER were also examined through SEM, TEM, XRD, Raman, and XPS analyses (Fig. S20†). The microstructure and crystalline features of the Co2P/Co nanoparticles remain well-preserved even after prolonged HER operation. This preservation suggests that their stable structure results from the protective effect of carbon layers. The electrochemical surface area (ECSA) of electrocatalysts illustrates real active sites that could be investigated by using the CV measurement at various scan rates in a non-redox potential range (Fig. S21†). The double-layer capacitance (Cdl) is associated with ECSA behavior. Fig. S22† shows the CV profile of all catalysts at a constant rate of 10 mV s−1, which exposes the greatest area for Co2P/Co@N-CNT/NPG, compared to the other catalysts, demonstrating the exposure area of the catalyst to electrolyte. The Cdl value of Co2P/Co@N-CNT/NPG is high at 15.8 mF cm−2, which is greater than that of Co@N-CNT/NG, Co2P/Co@PG, NPG, and RuO2 of 13.0, 10.1, 5.5, and 1.8 mF cm−2, respectively, suggesting that Co2P/Co@N-CNT/NPG has numerous active sites for outstanding ORR, OER, and HER performance (Fig. 5h). Since the ECSA value is related to Cdl, it could be anticipated that Co2P/Co@N-CNT/NPG possessed a better ECSA than the other catalysts. The intrinsic activity of the catalysts was assessed based on normalized-ECSA.59 The LSV response was normalized using ECSA to diminish the influence of ECSA variations on catalytic activity. As depicted in Fig. S23,† the Co2P/Co@N-CNT/NPG catalyst showed outstanding performance for both the OER and HER when compared to other counterparts of Co@N-CNT/NG, Co2P/Co@PG, and NPG. This result illustrated the excellent intrinsic activity of Co2P/Co@N-CNT/NPG among the synthesized materials. In addition, the electrochemical turnover frequency (TOF) of each active site was examined to provide further insights into the intrinsic activity of the catalysts. To determine the number of active sites, we employed the CV method at a sweep rate of 50 mV s−1 in a PBS solution.60 Subsequently, we calculated the TOF value of the as-synthesized catalyst for the HER and OER and presented in Fig. S24.† The notably higher TOF values of Co2P/Co@N-CNT/NPG across the entire surveyed potential range unequivocally affirm its exceptional intrinsic catalytic activity for both the HER and OER in an alkaline medium. Furthermore, electrochemical impedance spectroscopy (EIS) characterization was employed to examine the charge-transfer resistance (Rct) of the as-prepared catalysts (Fig. 5i).61 The Co2P/Co@N-CNT/NPG catalyst exhibits a low Rct value of 2.24 Ω, which is smaller than that of Co@N-CNT/NG, Co2P/Co@PG, NPG, and RuO2 of 2.92, 3.63, 3.43, and 4.43 Ω, respectively, proposing the good charge transfer ability, and therefore it could improve the catalytic reaction kinetics. The tremendous catalytic activities of the Co2P/Co@N-CNT/NPG catalyst toward the ORR, OER, and HER showed promise for ZAB and water electrolysis applications and could be caused by: (i) firstly, the N,P dual-doping could enhance the conductivity and active sites of the graphene and CNT substrate, while the excellent conductivity framework could provide strong interfacial coupling with the Co2P/Co heterostructure, and enhance the electron mobility. (ii) Secondly, the complete encapsulation of graphene and CNTs over the Co2P/Co heterostructure could both improve the durability and ensure a huge loading amount of catalyst, thereby delivering more active sites and high-accessibility surface of catalysts to electrolyte. (iii) Thirdly, the synergistic effect between Co2P and metallic Co in the heterostructure could accelerate the ORR, OER, and HER. (iv) Last, but not least, the 3D hierarchical structure interrelated carbon framework is favorable for electron/ion transport and promoting the adsorption of intermediary species for the ORR/OER/HER during the redox reaction process, accelerating the prolonged reaction.
Theoretical calculation
The electronic tuning and acceleration of electron transport in the Mott–Schottky Co2P/Co@N-CNT/NPG catalyst was investigated using the first principles density functional theory (DFT) calculation. The models of different nanostructure were constructed as shown in Fig. 6a–c. The projected density of states (PDOS) for as-built models presented in Fig. 6d–f exhibits the improvement of the density of states (DOS) at the Fermi level for Co2P/Co@NPG when compared to other comparative structures such as Co2P/Co@PG and Co@NG, respectively.26,28–30,62,63 Moreover, the band structure of models is presented in Fig. S25,† illustrating the electronic properties of materials. On the other hand, to prove the advantage of Mott–Schottky interface formation, we have built and determined the charge density difference of Co2P/Co model and the result is shown in Fig. S26.† The cyan and yellow color display the accumulation and depletion of electrons that demonstrated the movement of electrons from Co to Co2P after Mott–Schottky interface formation.64 In addition, the work function calculation demonstrated the higher value for p-type semiconductor Co2P of 4.106 eV, which is higher than that of metal Co (3.719 eV).65,66 The interfacial metal/semiconductor was formed and generated the electric field that drove the electron movement from Co to Co2P side until the Fermi level reached equilibrium (Fig. S27†). Constantly, the Schottky barrier was established and reconstructed the heterogeneous interface which could optimize the electrochemical performance of the catalyst. The modelled Co/Co2P exhibited a lower work function of 3.605 eV that produces an easier pathway for electron transport between the electrolyte and electrode and therefore it enhances the electrocatalytic activities. These results further confirmed the synergistic mechanism and tuning of the electronic transport with the formation of the Mott–Schottky catalyst; it could speed up charge transfer and be beneficial for electrochemical performance at the catalyst interface.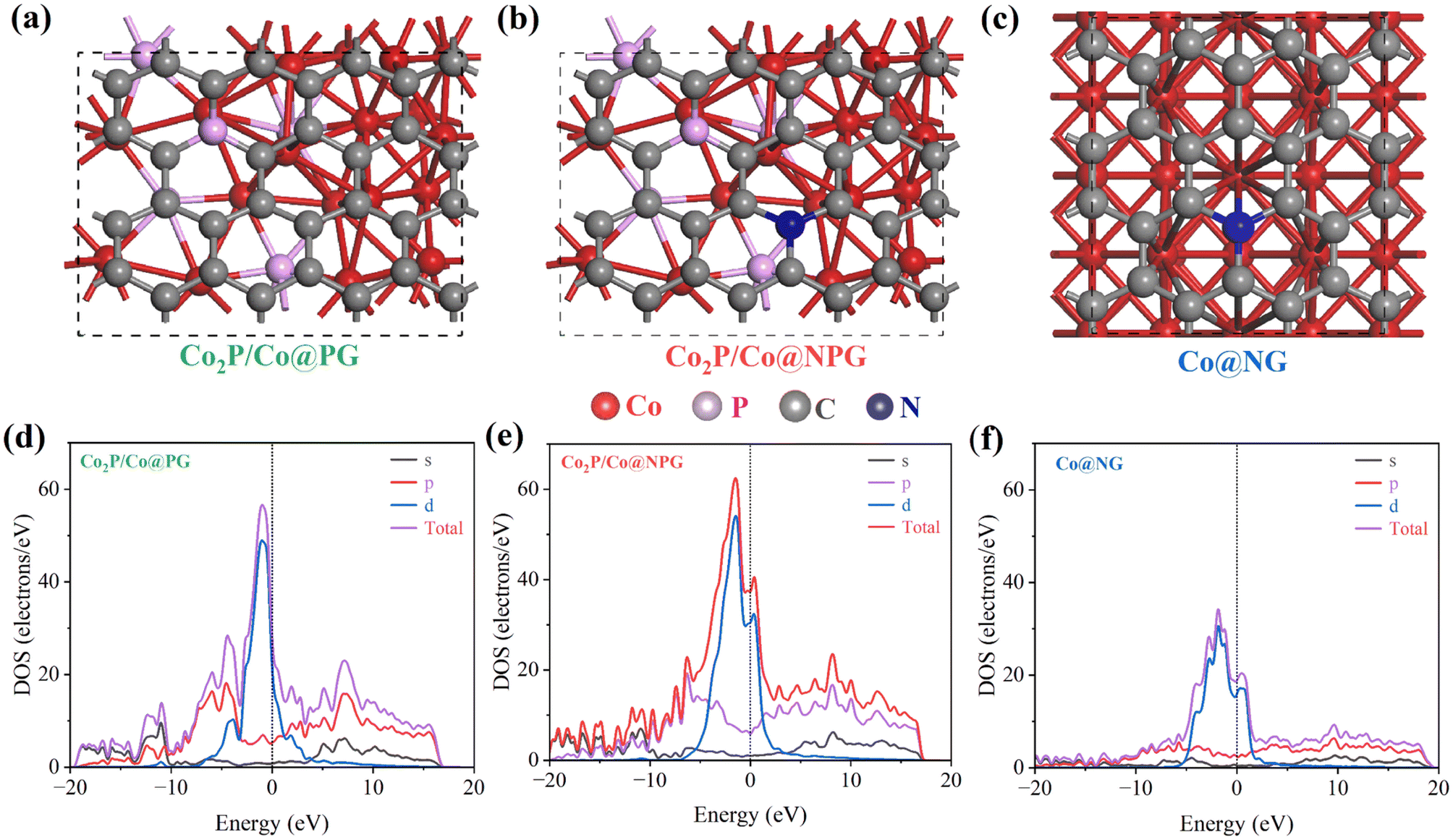 | ||
| Fig. 6 Structural models of (a) Co2P@PG, (b) Co2P/Co@NPG, and (c) Co@NG, and (d–f) corresponding PDOS calculation. | ||
Performance of the rechargeable ZAB and overall water splitting
The excellent multi-functional catalytic activity of Co2P/Co@N-CNT/NPG for the OER and ORR could offer high potential for application as the cathode for ZABs. The ZAB assembly process is illustrated in Fig. 7a with the Co2P/Co@N-CNT/NPG catalyst, Zn metal, and KOH + Zn(ac)2 as the air-cathode, anode, and electrolyte. The blend of Pt/C + RuO2 was prepared, and the performance examined for comparison. The as-constructed ZAB with the Co2P/Co@N-CNT/NPG cathode displayed a high open circuit potential of 1.458 V, which is better than that of Pt/C + RuO2 ZAB (1.411 V) (Fig. 7b and S28†). To explore the energy storage ability of the ZAB, the i–v polarization plots were collected (Fig. 7c). The ZAB with the Co2P/Co@N-CNT/NPG-based air-cathode showed a maximum current density of 145 mW cm−2, which outperforms the ZAB with the Pt/C + RuO2 cathode of 98 mW cm−2. In addition, the ZAB with the Co2P/Co@N-CNT/NPG-based air-cathode showed high discharge voltages of 1.36, 1.34, 1.31, 1.28, and 1.2 V at the various current densities of 1.0, 2.0, 5.0, 10.0, and 20.0 mA cm−2, which confirms the high-rate performance of the ZAB. When the current was set back to 1 mA cm−2, the discharge potential salvaged was 1.36 V, implying the good reversibility of the Co2P/Co@N-CNT/NPG cathode (Fig. S28†). The galvanostatic discharge tests were implemented to determine the capacity of the Co2P/Co@N-CNT/NPG cathode-based ZAB presenting a high value of 781 mA h g−1, which is more outstanding than that of Pt/C + RuO2 based ZAB at 608 mA h g−1 (Fig. 7d). The two as-assembled ZABs can supply power for a series of light-emitting diodes (LEDs), revealing its feasible practical application (inset of Fig. 7d). The charge–discharge profile exhibited small polarization of the Co2P/Co@N-CNT/NPG ZAB compared to that of the Pt/C + RuO2 ZAB, that benefits from the excellent performance of the Co2P/Co@N-CNT/NPG catalyst for the OER and ORR that is mainly driven during the charge–discharge process (Fig. 7e). The long-term durability of the Co2P/Co@N-CNT/NPG-based ZAB was estimated by continuous charge–discharge at 5 mA cm−2. The Co2P/Co@N-CNT/NPG cathode-based ZAB showed a minimal potential gap of 0.781 V, which is smaller than that of the Pt/C + RuO2 cathode-based ZAB (0.843 V) (Fig. 7f). After consecutive cycling for 800 h, the Co2P/Co@N-CNT/NPG cathode-based ZAB exhibited a minimal change in the potential gap of 0.798 V, suggesting a slight polarization behavior, compared to the Pt/C + RuO2-based ZAB. Even at a high current of 20 mA cm−2, the ZAB with the Co2P/Co@N-CNT/NPG-based air-cathode revealed exceptional long-term durability, with a slight change in voltage after 280 h (Fig. S29†). This excellent performance of our ZAB outperformed that of the recent reports for ZABs based on transition metal-based cathodes in the literature (Table S6†).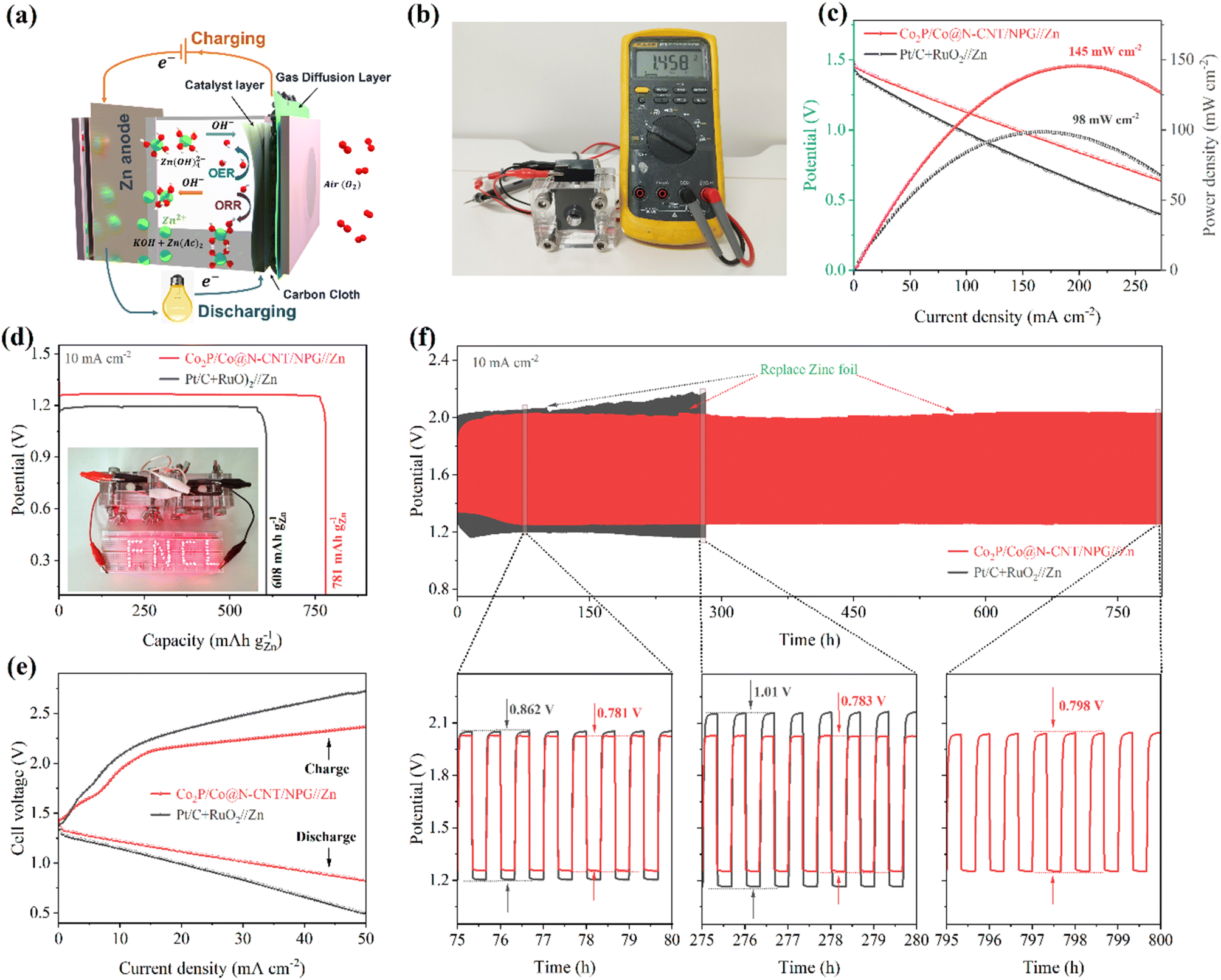 | ||
| Fig. 7 (a) Modelling of liquid-based ZABs with the Co2P/Co@N-CNT/NPG air cathode, (b) the as-obtained open-circuit voltage, (c) the i–v polarization and power density curves of the ZAB, (d) specific capacity of the ZAB at 10 mA cm−2, and inset showing the photo of the powered LEDs with configuration of the series-connected rechargeable ZAB, (e) charge and discharge polarization curves of the rechargeable ZAB, and (f) cycling stabilities of the rechargeable ZAB with Co2P/Co@N-CNT/NPG and Pt/C + RuO2 air cathodes. | ||
To sustain the demand for energy storage for different practical applications, the newly developed CF-ZAB was constructed. The rechargeable CF-ZAB was assembled with the Co2P/Co@N-CNT/NPG as the cathode, zinc as the anode, and PVA gel film with 6 M KOH as electrolyte (Fig. 8a). The CF-ZAB with the Co2P/Co@N-CNT/NPG-based cathode exhibits superior long-term stability for 205 h after continuous charge–discharge for 615 cycles with negligible voltage change. In contrast, the CF-ZAB with the Pt/C + RuO2-based cathode showed significant deterioration after 70 h (Fig. 8b). Moreover, the CF-ZAB with the Co2P/Co@N-CNT/NPG-based cathode presented a high open circuit potential of 1.415 V, and two series-connected batteries could steadily power numerous LEDs, as shown in Fig. 8c and e, suggesting great potential for practical application. To evaluate the flexibility performance of batteries at different states, we performed the charge–discharge measurement at various bending angles (Fig. 8d). The CF-ZAB with the Co2P/Co@N-CNT/NPG-based cathode showed high durability charge/discharge performance at various bending states. The outstanding electrochemical performance of the CF-ZAB could be compared to other reports of the solid-state ZABs in the literature (Table S7†). Movie S1† demonstrates the ability to maintain a steady power supply in the continuously bent state of the rechargeable CF−ZAB-based Co2P/Co@N−CNT/NPG cathode. These results suggest that Co2P/Co@N−CNT/NPG could be an applicable candidate to serve as the cathode for CF-ZABs and various wearable devices.
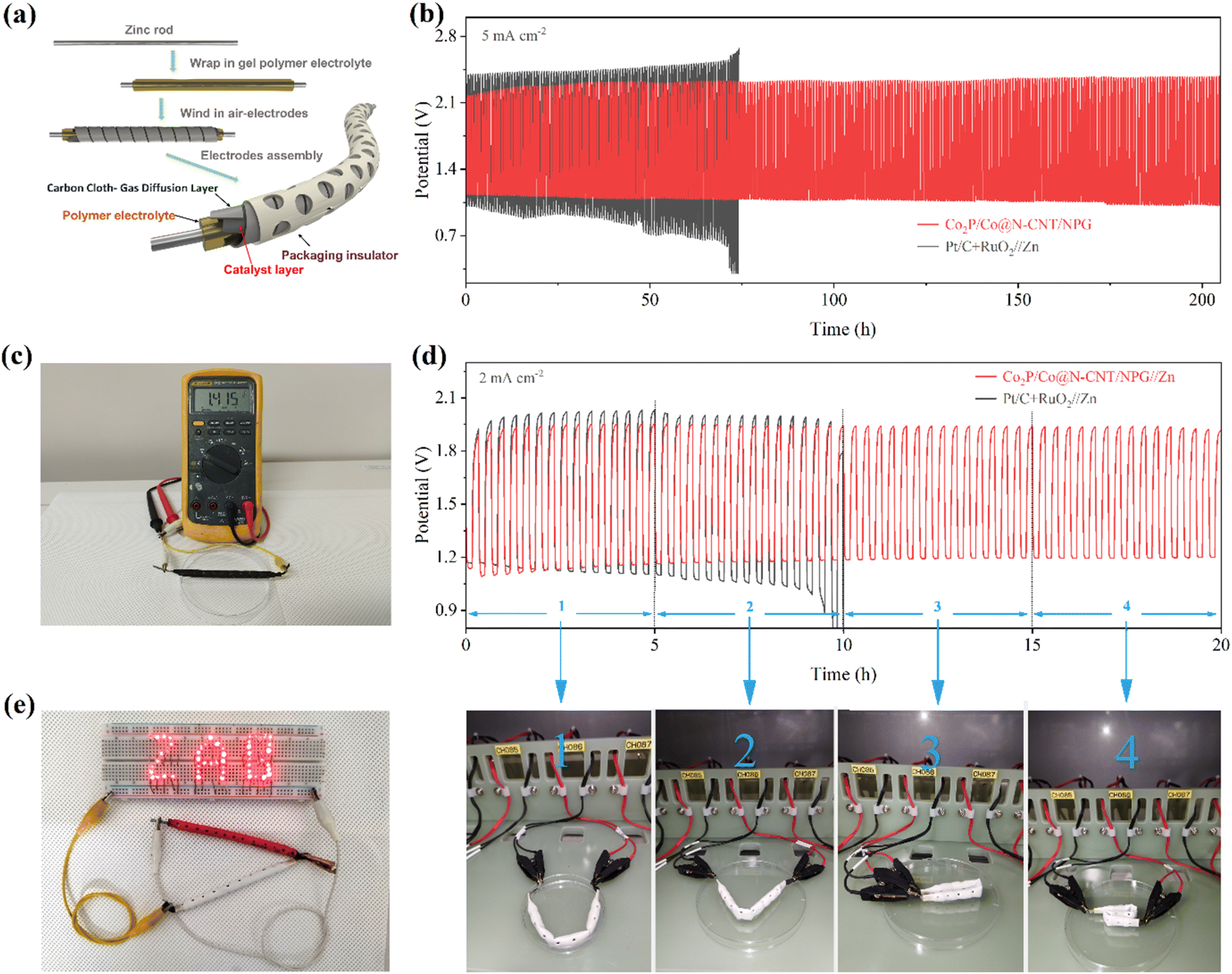 | ||
| Fig. 8 (a) Schematic of the assembled rechargeable CF-ZAB, (b) cycling stability of the CF-ZAB with Co2P/Co@N-CNT/NPG and Pt/C + RuO2 cathodes at 5 mA cm−2, (c) the OCV CF-ZAB, (d) the charge–discharge curves of the rechargeable CF-ZAB with different bending and twisting states, (e) the CF-ZAB supplies power to the LEDs. | ||
In addition, the excellent HER and OER catalytic activities could reveal the possibility of the Co2P/Co@N-CNT/NPG catalyst for water electrolysis. To investigate the water electrolysis performance, we constructed an electrolyzer using Co2P/Co@N-CNT/NPG as both the anode and cathode (Fig. 9a). Remarkably, the Co2P/Co@N-CNT/NPG(+,−) electrolyzer required small cell voltages of 1.66 and 1.89 V to achieve 10 and 100 mA cm−2 (Fig. 9b), verifying the outstanding overall water splitting performance. Moreover, the as-assembled electrolyzer presented superior long-term durability after 40 h with a high retention of 84% in current density, which is exceptional, compared with the Pt/C(−)//RuO2(+) electrolyzer (retaining 62% current density after 15 h) (Fig. 9c). To combine the utilization of the ZAB and electrolyzer, we constructed a self-powered system using two series-connected ZABs to supply the power for the water electrolysis process. Fig. 9d clearly shows the numerous H2 and O2 bubbles at the cathode and anode. At a high concentration of alkaline electrolyte of 3 M KOH at 80 °C, the electrolyzer needs a small cell potential of 1.43 and 1.63 V to attain the current density of 10 and 100 mA cm−2, the values being close to those of the benchmark Pt/C(−)//RuO2(+) based electrolyzer of 1.43 and 1.52 V (Fig. 9e). Moreover, the durability of the Co2P/Co@N-CNT/NPG(+,−) electrolyzer showed a small current loss of 18% after 40 h, demonstrating outstanding long-term durability, compared to Pt/C(−)//RuO2(+) (with degradation of 42% after 10 h) (Fig. 9f). The catalytic activity of the electrode was significantly enhanced upon increasing the pH and temperature of the working media. This enhancement can be attributed to the superior conductivity of the 3 M KOH electrolyte at 80 °C and the excellent charge transfer ability of the electrode in such an environment (Fig. 9g). The excellent water splitting performance of the Co2P/Co@N-CNT/NPG(+,−) electrolyzer is superior to that of other reports of transition metal catalysts in the literature (Fig. 9h and Table S8†). Our finding suggests that the unique hierarchical interactions of Co2P/Co@N-CNT/NPG to form the Mott–Schottky type catalyst could significantly improve the electrocatalytic properties for the water electrolysis system. This could be obtained from the synergistic effect of Co2P and Co and the encapsulation of N,P-doped graphene and N-doped CNTs. The self-power-driven water electrolysis system could offer promise for further application to the renewable energy complex.
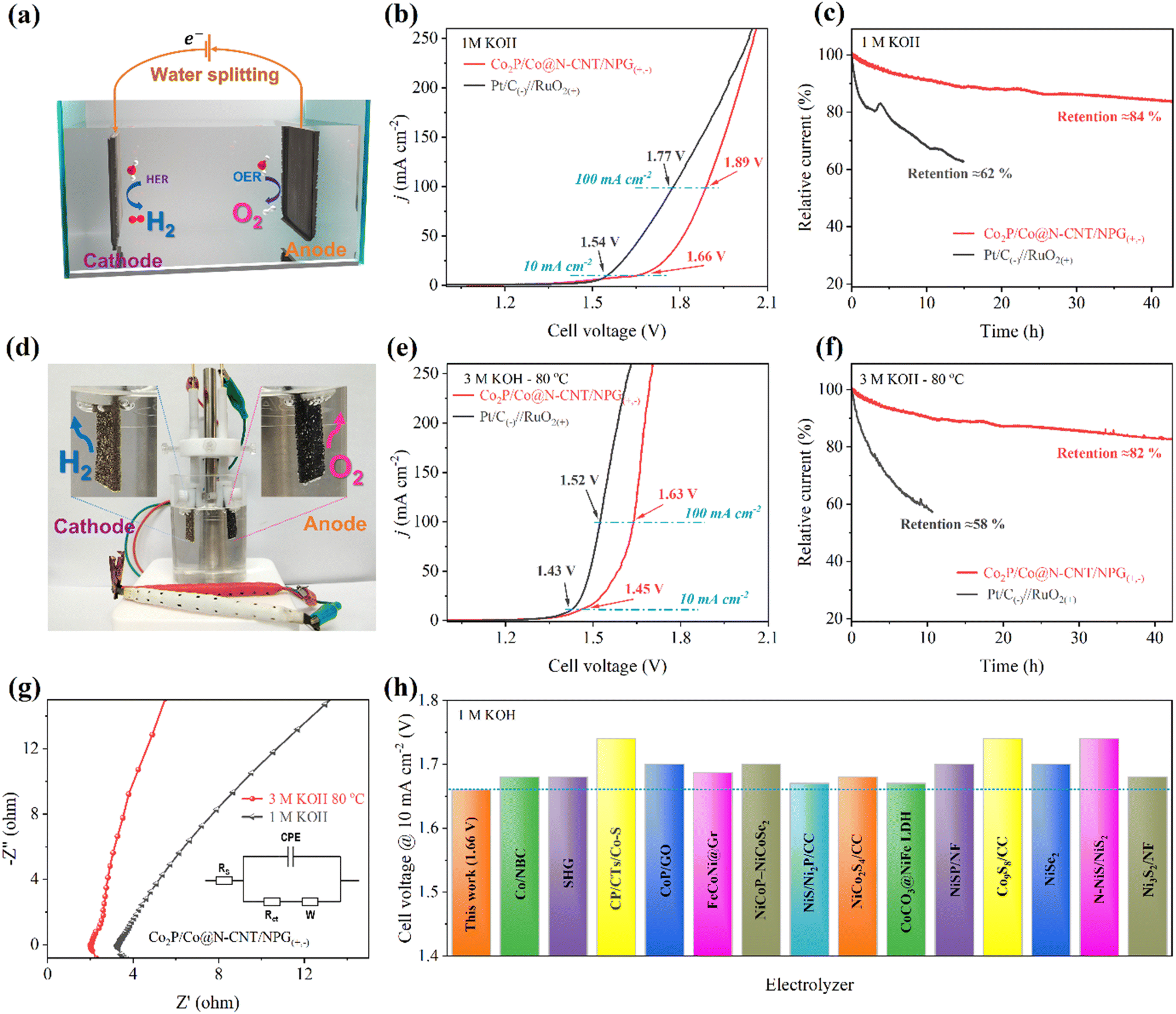 | ||
| Fig. 9 Electrochemical performances of the water splitting cells: (a) schematic of the assembled water electrolysis cells, (b) polarization curves for overall water splitting, (c) durability test for Co2P/Co@N-CNT/NPG(+,−) and Pt/C(−)‖RuO2(+) in 1 M KOH, (d) photograph showing the generation of H2 and O2 bubbles powered by CF-ZABs, (e) polarization curves for overall water splitting, (f) stability test, (g) EIS analyses for Co2P/Co@N-CNT/NPG(+,−) in 3 M KOH at 80 °C and 1 M KOH, and (h) performances for water splitting of some recently reported materials, specific values are listed in Table S8.† | ||
Conclusions
In summary, we have demonstrated a tunable coupling, followed by an in situ pyrolysis strategy to synthesize the novel Mott–Schottky trifunctional electrocatalyst of Co2P/Co@N-CNT/NPG. The enriched heterojunction Co2P and metallic Co encapsulated by N,P co-doped graphene and the CNT matrix exhibit superior catalytic properties for the ORR, OER, and HER. More importantly, the ZAB constructed from the Co2P/Co@N-CNT/NPG air-cathode demonstrates superior electrochemical performance with high peak power density (145 mW cm−2) and excellent stability for 800 h. The CF-ZAB presents tremendous mechanical stability, with almost unchanged performance. Furthermore, when serving as the bifunctional electrode, the Co2P/Co@N-CNT/NPG(+,−) electrolysis device requires a small cell potential of 1.66 and 1.45 V to acquire a current density of 10 mA cm−2 in 1 and 3 M KOH electrolyte, respectively. Undoubtedly, this material development suggests a novel strategy to prepare low-cost multi-functional electrocatalysts for numerous applications in next-generation energy-related devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Regional Leading Research Center Program (2019R1A5A8080326) through the National Research Foundation funded by the Ministry of Science and ICT of the Republic of Korea. And was supported by the Technology Innovation Program (20020872, Development of high crystalline graphitic structure-based high-efficiency and high-performance catalyst for water splitting) funded by the Ministry of Trade, Industry & Energy (MOTIE, Korea).Notes and references
- S. S. Shinde, J. Y. Jung, N. K. Wagh, C. H. Lee, D.-H. Kim, S.-H. Kim, S. U. Lee and J.-H. Lee, Nat. Energy, 2021, 6, 592–604 CrossRef CAS.
- M. Li, H. Li, X. Jiang, M. Jiang, X. Zhan, G. Fu, J.-M. Lee and Y. Tang, J. Mater. Chem. A, 2021, 9, 2999–3006 RSC.
- M. Li, X. Wang, K. Liu, Z. Zhu, H. Guo, M. Li, H. Du, D. Sun, H. Li, K. Huang, Y. Tang and G. Fu, Adv. Energy Mater., 2023, 13, 2301162 CrossRef CAS.
- N. K. Wagh, S. S. Shinde, C. H. Lee, S.-H. Kim, D.-H. Kim, H.-D. Um, S. U. Lee and J.-H. Lee, Nano-Micro Lett., 2022, 14, 190 CrossRef CAS PubMed.
- M. Li, X. Wang, K. Liu, H. Sun, D. Sun, K. Huang, Y. Tang, W. Xing, H. Li and G. Fu, Adv. Mater., 2023, 35, 2302462 CrossRef CAS PubMed.
- A. P. Grosvenor, S. D. Wik, R. G. Cavell and A. Mar, Inorg. Chem., 2005, 44, 8988–8998 CrossRef CAS PubMed.
- S. Ye, W. Xiong, P. Liao, L. Zheng, X. Ren, C. He, Q. Zhang and J. Liu, J. Mater. Chem. A, 2020, 8, 11246–11254 RSC.
- H. W. Go, T. T. Nguyen, Q. P. Ngo, R. Chu, N. H. Kim and J. H. Lee, Small, 2023, 19, 2206341 CrossRef CAS PubMed.
- S. Ye, Z. Chen, G. Zhang, W. Chen, C. Peng, X. Yang, L. Zheng, Y. Li, X. Ren, H. Cao, D. Xue, J. Qiu, Q. Zhang and J. Liu, Energy Environ. Sci., 2022, 15, 760–770 RSC.
- S. Ye, Y. Lei, T. Xu, L. Zheng, Z. Chen, X. Yang, X. Ren, Y. Li, Q. Zhang and J. Liu, Appl. Catal. B., 2022, 304, 120986 CrossRef CAS.
- J. Shi, X. Guo, S. Liu, Y. Sun, J. Zhang, Y. Liu, X. Zheng and Q. Kong, Compos. B Eng., 2022, 231, 109589 CrossRef CAS.
- T. T. Nguyen, J. Balamurugan, D. H. Kim, N. H. Kim and J. H. Lee, Small, 2020, 16, e2004661 CrossRef PubMed.
- M. Singh, T. T. Nguyen, P. Muthu Austeria, Q. P. Ngo, D. H. Kim, N. H. Kim and J. H. Lee, Small, 2023, 19, 2206726 CrossRef CAS PubMed.
- H. Li, B. Ren, W. Liu, L. Jing, R. Y. Tay, S. H. Tsang, L. Ricardez-Sandoval, A. Yu and E. H. T. Teo, Nano Energy, 2021, 88, 106246 CrossRef CAS.
- Q. P. Ngo, T. T. Nguyen, M. Singh, R. Balaji, N. H. Kim and J. H. Lee, Appl. Catal. B Environ., 2023, 331, 122674 CrossRef CAS.
- H. Liu, J. Guan, S. Yang, Y. Yu, R. Shao, Z. Zhang, M. Dou, F. Wang and Q. Xu, Adv. Mater., 2020, 32, 2003649 CrossRef CAS PubMed.
- A. Parra-Puerto, K. L. Ng, K. Fahy, A. E. Goode, M. P. Ryan and A. Kucernak, ACS Catal., 2019, 9, 11515–11529 CrossRef CAS.
- Q. Zheng, Y. Xiong, K. Tang, M. Wu, H. Hu, T. Zhou, Y. Wu, Z. Cao, J. Sun, X. Yu and C. Wu, Nano Energy, 2022, 92, 106750 CrossRef CAS.
- J. Diao, Y. Qiu, S. Liu, W. Wang, K. Chen, H. Li, W. Yuan, Y. Qu and X. Guo, Adv. Mater., 2020, 32, 1905679 CrossRef CAS PubMed.
- L. Peng, L. Su, X. Yu, R. Wang, X. Cui, H. Tian, S. Cao, B. Y. Xia and J. Shi, Appl. Catal. B Environ., 2022, 308, 121229 CrossRef CAS.
- Q. Dong, G. Li, F. Liu, J. Ren, H. Wang and R. Wang, Appl. Catal. B Environ., 2023, 326, 122415 CrossRef CAS.
- Z.-H. Xue, H. Su, Q.-Y. Yu, B. Zhang, H.-H. Wang, X.-H. Li and J.-S. Chen, Adv. Energy Mater., 2017, 7, 1602355 CrossRef.
- L. Zhou, T. Yang, Z. Fang, J. Zhou, Y. Zheng, C. Guo, L. Zhu, E. Wang, X. Hou, K.-C. Chou and Z. L. Wang, Nano Energy, 2022, 104, 107876 CrossRef CAS.
- J. Yu, J. Li, C.-Y. Xu, Q. Li, Q. Liu, J. Liu, R. Chen, J. Zhu and J. Wang, Nano Energy, 2022, 98, 107266 CrossRef CAS.
- T. T. Nguyen, P. Bandyopadhyay, X. Li, N. H. Kim and J. H. Lee, J. Membr. Sci., 2017, 540, 108–119 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Zeitschrift für Kristallographie - Crystalline Materials, 2005, 220, 567–570 CrossRef CAS.
- G. P. Francis and M. C. Payne, J. Phys.: Condens. Matter, 1990, 2, 4395–4404 CrossRef.
- M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulos, Rev. Mod. Phys., 1992, 64, 1045–1097 CrossRef CAS.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- B. G. Pfrommer, M. Cote, S. G. Louie and M. L. Cohen, J. Comput. Phys., 1997, 131, 233–240 CrossRef CAS.
- T. T. Nguyen, J. Balamurugan, H. W. Go, Q. P. Ngo, N. H. Kim and J. H. Lee, Chem. Eng. J., 2022, 427, 131774 CrossRef CAS.
- H. Yin, C. Z. Zhang, F. Liu and Y. L. Hou, Adv. Funct. Mater., 2014, 24, 2930–2937 CrossRef CAS.
- R. Saroha, J. H. Oh, J. S. Lee, Y. C. Kang, S. M. Jeong, D.-W. Kang, C. Cho and J. S. Cho, J. Chem. Eng., 2021, 426, 130805 CrossRef CAS.
- G. Zhou, G. Liu, X. Liu, Q. Yu, H. Mao, Z. Xiao and L. Wang, Adv. Funct. Mater., 2022, 32, 2107608 CrossRef CAS.
- Y. Guo, P. Yuan, J. Zhang, H. Xia, F. Cheng, M. Zhou, J. Li, Y. Qiao, S. Mu and Q. Xu, Adv. Funct. Mater., 2018, 28, 1805641 CrossRef.
- O.-K. Park, N. H. Kim and J. H. Lee, Carbon, 2022, 187, 330–337 CrossRef CAS.
- K. Kawashima, K. Shin, B. R. Wygant, J.-H. Kim, C. L. Cao, J. Lin, Y. J. Son, Y. Liu, G. Henkelman and C. B. Mullins, ACS Appl. Energy Mater., 2020, 3, 3909–3918 CrossRef CAS.
- X. Wang, M. Li, P. Wang, D. Sun, L. Ding, H. Li, Y. Tang and G. Fu, Small Methods, 2023, 7, 2300100 CrossRef CAS PubMed.
- X. Cui, P. Ren, D. Deng, J. Deng and X. Bao, Energy Environ. Sci., 2016, 9, 123–129 RSC.
- H. J. Son, Y. R. Cho, Y.-E. Park and S. H. Ahn, Appl. Catal. B Environ., 2022, 304, 120977 CrossRef CAS.
- X. Wang, Y. Liu and P. Wu, J. Chem. Eng., 2017, 328, 417–427 CrossRef CAS.
- X. Peng, L. Wang, L. Hu, Y. Li, B. Gao, H. Song, C. Huang, X. Zhang, J. Fu, K. Huo and P. K. Chu, Nano Energy, 2017, 34, 1–7 CrossRef CAS.
- K. Xu, H. Ding, M. Zhang, M. Chen, Z. Hao, L. Zhang, C. Wu and Y. Xie, Adv. Mater., 2017, 29, 1606980 CrossRef PubMed.
- T. T. Nguyen, J. Balamurugan, K. T. Lau, N. H. Kim and J. H. Lee, J. Mater. Chem. A, 2021, 9, 9092–9104 RSC.
- D. Jang, Y. Lee, Y. Shin, S. Park, C. Jo, Y.-H. Kim and S. Park, Appl. Catal. B Environ., 2020, 263, 118337 CrossRef CAS.
- S. Kim, S. Ji, H. Yang, H. Son, H. Choi, J. Kang and O. L. Li, Appl. Catal. B Environ., 2022, 310, 121361 CrossRef CAS.
- P. Jiang, Q. Liu, C. Ge, W. Cui, Z. Pu, A. M. Asiri and X. Sun, J. Mater. Chem. A, 2014, 2, 14634–14640 RSC.
- C. Zhang, L. Fu, N. Liu, M. Liu, Y. Wang and Z. Liu, Adv. Mater., 2011, 23, 1020–1024 CrossRef CAS PubMed.
- X. Wang, X. Li, L. Zhang, Y. Yoon, P. K. Weber, H. Wang, J. Guo and H. Dai, Science, 2009, 324, 768–771 CrossRef CAS PubMed.
- A. Ananthanarayanan, Y. Wang, P. Routh, M. A. Sk, A. Than, M. Lin, J. Zhang, J. Chen, H. Sun and P. Chen, Nanoscale, 2015, 7, 8159–8165 RSC.
- O. Ovdat, Y. Don and E. Akkermans, Phys. Rev. B, 2020, 102, 075109 CrossRef CAS.
- Z. Li, Y. Zhang, Y. Feng, C.-Q. Cheng, K.-W. Qiu, C.-K. Dong, H. Liu and X.-W. Du, Adv. Funct. Mater., 2019, 29, 1903444 CrossRef.
- H. Song, M. Wu, Z. Tang, J. S. Tse, B. Yang and S. Lu, Angew. Chem., Int. Ed., 2021, 60, 7234–7244 CrossRef CAS PubMed.
- Z. Chen, X. Zeng, X. Li, Z. Lv, J. Li and Y. Zhang, Adv. Mater., 2022, 34, 2106724 CrossRef CAS PubMed.
- S. Yasuda, A. Furuya, Y. Uchibori, J. Kim and K. Murakoshi, Adv. Funct. Mater., 2016, 26, 738–744 CrossRef CAS.
- G. A. Ferrero, K. Preuss, A. Marinovic, A. B. Jorge, N. Mansor, D. J. L. Brett, A. B. Fuertes, M. Sevilla and M.-M. Titirici, ACS Nano, 2016, 10, 5922–5932 CrossRef CAS PubMed.
- K. R. Park, D. T. Tran, T. T. Nguyen, N. H. Kim and J. H. Lee, J. Chem. Eng., 2021, 422, 130048 CrossRef CAS.
- Q. P. Ngo, T. T. Nguyen, Q. T. T. Le, J. H. Lee and N. H. Kim, Adv. Energy Mater., 2023, 13, 2301841 CrossRef CAS.
- D. Merki, S. Fierro, H. Vrubel and X. Hu, Chem. Sci., 2011, 2, 1262–1267 RSC.
- S. Anantharaj and S. Noda, ChemElectroChem, 2020, 7, 2297–2308 CrossRef CAS.
- G. P. Francis and M. C. Payne, J. Phys.: Condens. Matter, 1990, 2, 4395 CrossRef.
- B. G. Pfrommer, M. Côté, S. G. Louie and M. L. Cohen, J. Comput. Phys., 1997, 131, 233–240 CrossRef CAS.
- H. Yang, B. Wang, S. Kou, G. Lu and Z. Liu, Chem. Eng. J., 2021, 425, 131589 CrossRef CAS.
- R. Liang, Y. Wang, C. Qin, X. Chen, Z. Ye and L. Zhu, Langmuir, 2021, 37, 3321–3330 CrossRef CAS PubMed.
- S. Yang, G. Chen, A. G. Ricciardulli, P. Zhang, Z. Zhang, H. Shi, J. Ma, J. Zhang, P. W. M. Blom and X. Feng, Angew. Chem., Int. Ed., 2020, 59, 465–470 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta05029g |
| This journal is © The Royal Society of Chemistry 2024 |