
Fe-based dual-atom catalysts for the oxygen reduction reaction
Wuyi
Zhang
 a,
Shiyuan
Yi
a,
Yihong
Yu
a,
Hui
Liu
bc,
Anthony
Kucernak
d,
Jun
Wu
*bc and
Song
Li
*ae
a,
Shiyuan
Yi
a,
Yihong
Yu
a,
Hui
Liu
bc,
Anthony
Kucernak
d,
Jun
Wu
*bc and
Song
Li
*ae
aKey Lab for Anisotropy and Texture of Materials (MoE), School of Materials Science and Engineering, Northeastern University, Shenyang, 110819, China. E-mail: lis@atm.neu.edu.cn
bSchool of Metallurgy and Environment, Central South University, Changsha, 410083, China. E-mail: wujun18229914766@163.com
cChinese National Engineering Research Center for Control and Treatment of Heavy Metal Pollution, Changsha, 410083, China
dMolecular Science Research Hub, Imperial College London, White City Campus, London, W12 7SL, UK
eInstitute for Frontier Technologies of Low-Carbon Steelmaking, Shenyang, 110819, China
First published on 27th November 2023
Abstract
The oxygen reduction reaction (ORR) is widely employed at the cathode of next-generation energy devices such as fuel cells and metal–air batteries to accommodate electrons produced by anode reactions. The development of highly efficient and durable electrocatalysts for the ORR has been constrained by the involvement of multiple oxygen-containing intermediates and their scaling relations. Recently, dual-atom catalysts (DACs) supported on carbon materials have been intensively studied as ORR electrocatalysts due to their potential to precisely tune the adsorption/reactive performance of each metal site. In particular, Fe-based DACs exhibit outstanding ORR activities, holding great promise as substitutes for state-of-the-art Pt-based catalysts. However, the adjustment of the microenvironment of metal sites, loading density, scaling relation limitation, and excessively strong adsorption energy pose limitations on the practical applications of Fe-based DACs. To promote studies of Fe-based DACs, we summarize the current research status in this review by focusing on (1) the fundamental of the ORR and effects of Fe-based DACs, (2) common synthesis strategies of Fe-based DACs, and (3) ORR performance evaluations of Fe-based DACs. Additionally, this review provides our viewpoint on future directions and possible strategies to design catalysts for further optimization of the ORR.
 Jun Wu | Dr Jun Wu is now working in the School of Metallurgy and Environment at Central South University, China. He received his Bachelor's degree in Materials Science and Engineering from Central South University (2016) and his Master's (2017) and Ph.D. degrees (2022) in Chemistry from Imperial College London, UK. His research interests focus on developing novel materials for efficient energy conversion and storage (e.g., PEMFCs and ZABs) and air pollution control during non-ferrous metal smelting (e.g., CO, SO2 and NOx). |
 Song Li | Song Li is a professor of Materials Science and Engineering at Northeastern University, China. He received his Ph.D. in materials physics from the University of Lorraine in Metz and Northeastern University in Shenyang (2009). He was a visiting scholar at Boston College from 2015 to 2016. Li's recent research interests focus on developing novel materials for industrial decarbonization and electrification of chemical processes. |
1. Introduction
The world is rapidly shifting its energy structure from fossil-based to one dominated by renewable resources such as sunlight and wind.1,2 Due to uneven distribution in space and time, these renewables require efficient storage in chemical bonds and conversion via electrochemical routes.3–5 Due to their high energy density, fuel cells and metal–air batteries are recognized as promising next-generation energy devices.6–8 Although different fuels or metals are used, both devices generate electricity through fuel oxidation at the anode, which is paired by the oxygen reduction reaction (ORR) at the cathode.9–11 Compared to fast anodic reactions, the cathodic ORR suffers from sluggish kinetics, as it involves complex and multistep proton-coupled electron transfer processes. Highly efficient ORR electrocatalysts should be employed to resolve significant limits on the energy efficiency of the two devices.Currently, Pt-based materials are identified as the benchmark ORR electrocatalyst to lower the overpotential and accelerate the ORR kinetics, but their large-scale applications are hindered by their high cost and low abundance.12–15 In recent decades, tremendous efforts have been devoted to engineering nonprecious metal catalysts as alternatives, including alloys and oxides of various nanostructures, molecular catalysts, and single-atom catalysts (SACs).16–19 Among these catalysts, SACs have attracted much attention due to the well-defined configuration of metal sites, nearly 100% metal utilization, and ease of tailoring coordination/electronic structures.20–22 Typical SACs with ORR activity are composed of transition metal atoms anchored on nitrogen-doped carbon (M–N/C, M = Fe, Co, Ni, etc.). As the ORR activities of M–N/C follow the order of Fe > Co > Ni,23–25 Fe–N–C SACs have been considered the most potential and effective substitutes for Pt-based catalysts. Although great activity has been achieved, an enormous difference in the onset potentials of the ORR and OER (oxygen evolution reaction) exists, which is widely attributed to scaling relation limitations (SRL), especially on the single active site. The detailed mechanism will be discussed in the following sections.
To extend the engineering space of active sites, dual atom catalysts (DACs), which contain bimetallic atoms in isolated active sites or contain two different metal monoatomic sites, have been constructed and investigated for the ORR. Fe-based DACs improve the intrinsic ORR activity by introducing a second adjacent metal atom to modulate the electronic structure of the original active site. A synergistic effect is therefore observed, which may change the adsorption pattern of oxygen-containing species to break the SRLs and exploit the activity of the OER, achieving bifunctionality to satisfy the requirements of relevant devices.
In the past five years, various Fe-based DACs have been successfully developed with high performance in the ORR and relevant electrochemical devices, including Fe–Co, Fe–Ni, Fe–Mn, Fe–Cu, and Fe–Fe. However, the mechanism of enhanced ORR activity in Fe-based DACs remains unclear.
Given the increasing research interests, it is essential to provide up-to-date progress on Fe-based DACs promptly. In this review, the ORR reaction principles and related electrochemical devices are first introduced. Then, the main synthetic strategies of Fe-based DACs are presented categorically. Finally, the current progress of Fe-based DACs and the research work on the basis of Fe-based DACs are summarized. Previous reviews in this area mainly discuss the effect of catalysing electrochemical reactions with DACs. We systematically and carefully compiled and reviewed the profiles and research works on ORR-containing electrochemical energy devices, the shortcomings of utilizing SACs and how to overcome them with DACs, as well as in situ characterization and recent work on the basis of Fe-based DACs. This comprehensive review will provide insights into the strength of Fe-based DACs and recommendations for their further design and optimization.
2. Electrochemical devices and oxygen reduction reaction
2.1 Proton exchange membrane fuel cells (PEMFCs) and Zn–air batteries
The fuel cell is the next-generation energy device that generates electrical power from chemical energy in fuels through redox reactions on separate electrodes. Among various fuel cells, PEMFCs have attracted much attention due to their high power density, excellent stability, low toxicity, high energy conversion efficiency (theoretically more than 80%), and suitability in electric vehicles at low temperatures.26 The membrane electrode assembly (MEA) is the most important part of PEMFCs and includes the proton exchange membrane (PEM), porous gas diffusion layers (GDL), anode catalyst layer (ACL), and cathode catalyst layer (CCL).27–29 PEMs play various roles in transmitting protons (H+), preventing electron transfer, and separating the cathodic reaction. During operation, H2 as the fuel arrives at the surface of the anode catalyst via diffusion and breaks into H+ and e−. Protons arrive at the cathode through the PEM, while electrons flow along the external circuit to the cathode (eqn (1)). O2 reaches the cathodic catalyst surface through diffusion and is reduced to water via the ORR (eqn (2)). The overall reaction shows that H2 is oxidized by O2 into H2O (eqn (3)). The increasing popularity of various types of automobiles is further expected to boost the demand for PEM fuel cells. Although PEMFCs show many advantages and great potential, precious Pt-based catalysts are needed to reduce the overpotential and accelerate the sluggish kinetics of the ORR, resulting in the high cost of PEMFCs. Therefore, developing nonprecious metal catalysts is necessary, as there are still large activity and stability gaps between nonprecious metal catalysts and Pt-based catalysts.Cathodic reaction:
| O2 + 4H+ + 4e− → 2H2O | (1) |
Anodic reaction:
| 2H2 → 4H+ + 4e− | (2) |
Overall reaction:
| 2H2 + O2 → 2H2O | (3) |
Meanwhile, metal–air batteries, especially Zn–air batteries (ZABs), have also been considered promising next-generation energy storage systems due to their long-term stability, environmental friendliness, and extremely high energy density (1350 W h kg−1, which is five times higher than that of lithium-ion batteries). ZABs are mainly composed of four parts: the air electrode, zinc electrode, electrolyte, and separator. During the discharge process, O2 is reduced at the air electrode, while the Zn electrode is oxidized (eqn (4)–(6)). During the charging process, ZnO is reduced to Zn, while the oxygen evolution reaction (OER) occurs at the air electrode (eqn (7)–(9)). However, ZABs still suffer from low energy conversion efficiency, poor long-cycle stability, and low charge/discharge cycle current density. Bifunctional catalysts are designed to drive both the ORR and the OER, leading to bidirectional operation of Zn–air batteries. Pt-based catalysts and Ir/Ru oxides have been regarded as the best-performing ORR and OER catalysts to achieve bifunction in Zn–air batteries, but their high cost and poor battery performance restrict their large-scale application. On the other hand, Fe-based SACs, as a promising alternative to Pt-based catalysts, were designed and optimized for the ORR. However, since the catalysts for the OER need to provide extra active sites to accelerate the reaction, Fe–N/C SACs were inadequate to meet these specific requirements for the OER due to the slow O–O coupling process, excessive adsorption of OER intermediates, and invalid desorption of the final product (O2) and hence cannot be used as excellent bifunctional catalysts for Zn–air batteries.
Recently, Fe-based DACs have been considered as promising materials to introduce extra OER active sites and modulate the OER activity of Fe sites to achieve bifunctional ORR/OER activity.22,30,31 For example, Sun et al. found that interaction between Fe and Ni atoms in DACs could help to produce a high ORR performance (E1/2 of 0.85 V) and a low OER overpotential of 467 mV at ∼10 mA cm−2. A combined theoretical investigation on the OER activity supported that the modulation of Ni atoms in the FeN3–NiN3 structure reduces the adsorption of OH* intermediates to lower the OER overpotential on Fe atoms.
Air electrodes (discharge):
| 4OH− → O2 + 2H2O + 4e− | (4) |
Zn electrode (discharge):
| Zn + 2OH− → ZnO + H2O + 2e− | (5) |
The discharge process:
| 2Zn + O2 → 2ZnO (Eeq = 1.66 V vs. SHE) | (6) |
Air electrodes (charge):
| O2 + 2H2O + 4e− → 4OH− | (7) |
Zn electrode (charge):
| ZnO + H2O + 2e− → Zn + 2OH− | (8) |
The charge process:
| 2ZnO → 2Zn + O2 (Eeq = −1.66 V vs. SHE) | (9) |
2.2 Oxygen reduction reaction
As discussed in the previous section, the large-scale implementation and commercialization of PEMFCs and ZABs are limited by the slow ORR.32–34 The main processes of the ORR include (1) adsorption of O2 on the catalyst surface, (2) reduction of the adsorbed O2 by accepting electrons from the external circuit, and (3) desorption of the reduction products.35,36 There are three different modes of O2 adsorption on the surface of the catalyst, mainly the Griffiths mode, Bridge mode, and Pauling mode (Fig. 1a). In the Griffiths mode, two oxygen atoms are laterally adsorbed on the same active site. The intense interaction between O orbitals and the empty orbital of the active center weakens the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and makes it easier to break it. In the Bridge mode, two oxygen atoms are laterally adsorbed to two sites, and the synergy of the paired sites promotes OO bond breaking. In the Pauling mode, one oxygen atom is adsorbed on the active site, and the active site has a weak effect on the OO bond, which makes it difficult to break the OO bond and tends to produce H2O2.
O bond and makes it easier to break it. In the Bridge mode, two oxygen atoms are laterally adsorbed to two sites, and the synergy of the paired sites promotes OO bond breaking. In the Pauling mode, one oxygen atom is adsorbed on the active site, and the active site has a weak effect on the OO bond, which makes it difficult to break the OO bond and tends to produce H2O2.
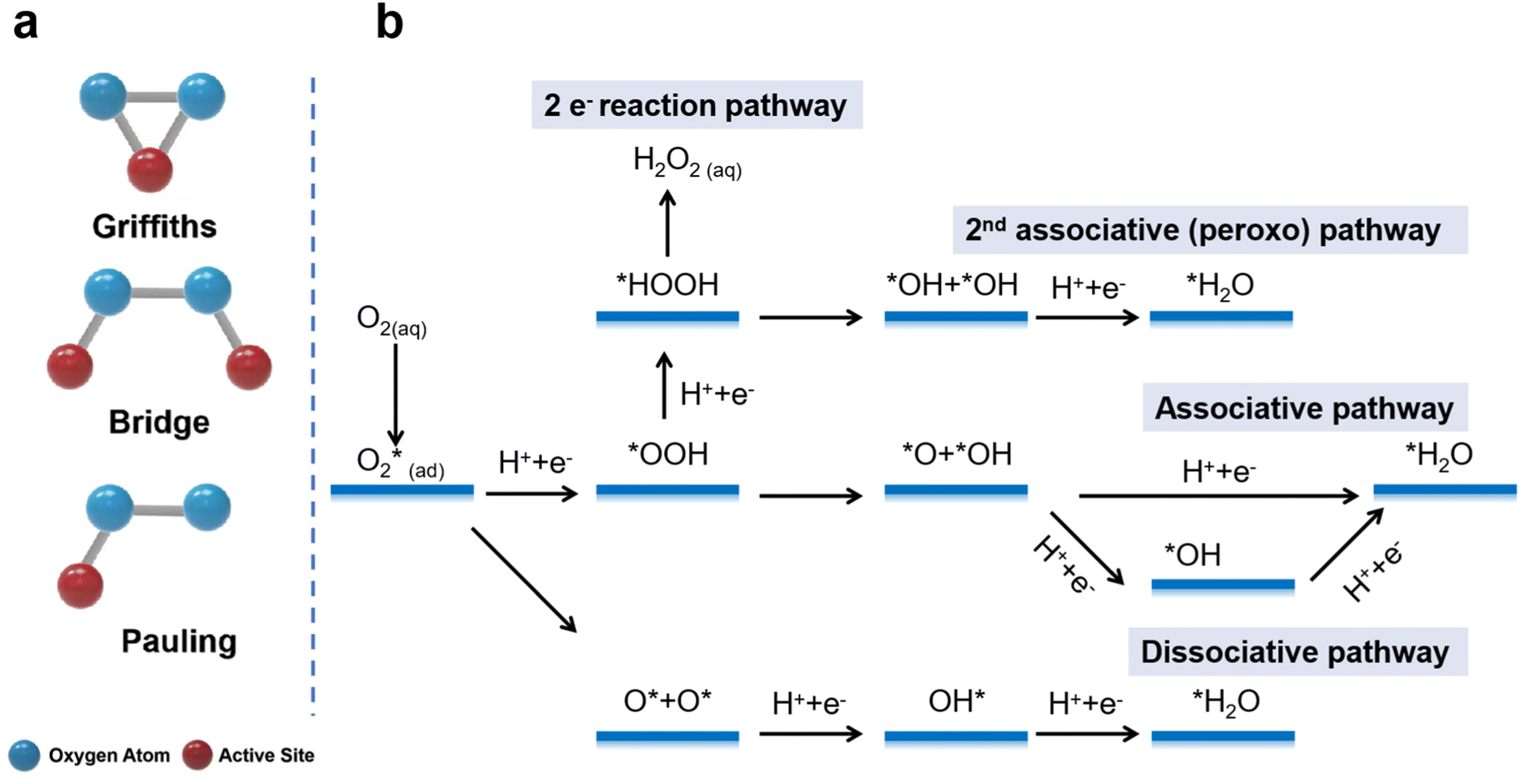 | ||
| Fig. 1 (a) Different modes of O2 adsorption on the catalyst surface. (b) Reaction pathways of the ORR. | ||
The ORR follows two different mechanisms of the multielectron transfer process: one is through the four-electron pathway to create H2O (eqn (10) and (13)), and the other is through the two-electron pathway to produce H2O2 (Fig. 1b), which can subsequently combine two electrons to produce H2O (eqn (11), (12), (14) and (15)).37–40 As the 4e− pathway exhibits high energy-conversion efficiency and the hydroxyl groups derived from H2O2 in the 2e− pathway can attack the PEMs, the direct 4e− pathway is more favorable for the ORR-related devices.
The 4e− pathway in alkaline electrolytes:
| O2 + H2O + 4e− → 4OH−, E° = 0.40 V vs. SHE | (10) |
The 2e− pathway in alkaline electrolytes:
| O2 + H2O + 2e− → OH− + HO2−, E° = 0.06 V vs. SHE | (11) |
| HO2− + H2O + 2e− → 3OH−, E° = 0.86 V vs. SHE | (12) |
The 4e− pathway in acidic electrolytes:
| O2 + 4H+ + 4e− → 2H2O, E° = 1.23 V vs. SHE | (13) |
The 2e− pathway in acidic electrolytes:
| O2 + 2H+ + 2e− → H2O2, E° = 0.70 V vs. SHE | (14) |
| H2O2 + 2H+ + 2e− → 2H2O, E° = 1.76 V vs. SHE | (15) |
The detailed reduction of the adsorbed O2* to H2O can be divided into three pathways: the dissociative pathway, the associative pathway, and the 2nd associative (peroxo) pathway (details shown in Fig. 1b).
2nd associative (peroxo) pathway:
| O2* + H+ + e− → OOH* | (16) |
| OOH* + H+ + e− → HOOH* | (17) |
| HOOH* → OH* + OH* | (18) |
| OH* + H+ + e− → H2O* | (19) |
In the 2nd associative (peroxo) pathway (Fig. 1b), the adsorbed O2 forms OOH* intermediates with H+ and electrons and there is further protonation of OOH* to HOOH*. Then, HOOH* breaks into 2 OH*. In the final step, OH* forms the final product H2O* with one H+ and one electron.
Dissociative pathway:
| O2* → O* + O* | (20) |
| O* + H+ + e− → OH* | (21) |
| OH* + H+ + e− → H2O* | (22) |
In the dissociative pathway (Fig. 1b), the adsorbed oxygen molecules dissociate into O intermediates. Then, 2 O* intermediates form 2 OH* intermediates with 2 H+ and 2 electrons; finally, 2 OH* intermediates obtain two electrons to form the final product 2 H2O with 2 H+.
Associative pathway:
| O2* + H+ + e− → OOH* | (23) |
| OOH* → O* + OH* | (24) |
| O* + H+ + e− → OH* | (25) |
| OH* + H+ + e− → H2O* | (26) |
In the associative pathway (Fig. 1b), the adsorbed oxygen molecules do not dissociate but undergo protonation to form OOH* intermediates. The OOH* intermediates break down into O* intermediates and OH* intermediates. Then, the O* intermediates are protonated to OH*. In the final step, the OH* forms the final product H2O* with one H+ and one electron.
Even though Fe–N/C SACs are promising substitutes for Pt-based catalysts, Fe–N/C SACs contain only one metal center. Thus, the reactants or intermediates have to adsorb on the single site via O atoms without the synergy of neighboring sites, which leads to principal limitations known as the scaling relationship limit.41,42 In catalysis, the scaling relationship correlates the binding energies of various intermediates (Fig. 2a). In terms of ORR, this relationship describes the trade-off between the binding strength of different oxygen intermediates (e.g., O*, OH*, OOH*) to the catalyst surface. Fe-based SACs typically contain active sites with symmetric coordination environments, such as Fe–N4 centers. These sites have uniform binding strengths for different ORR intermediates. The scaling relationship can be described mathematically as ΔGOH* = aΔGO* + b, where ΔGOH* is the binding energy of OH*, ΔGO* is the binding energy of O*, and “a” and “b” are coefficients determined by the catalyst structure. The “a” coefficient represents the extent to which the changes in O* binding energy would affect the OH* binding energy. When “a” is fixed due to the symmetric nature of the active sites in Fe-based SACs, the corresponding scaling behavior makes it challenging to independently optimize the OH* binding without affecting the binding of the other two intermediates. Therefore, the scaling relation limits the optimization of Fe-based SACs for all intermediates.
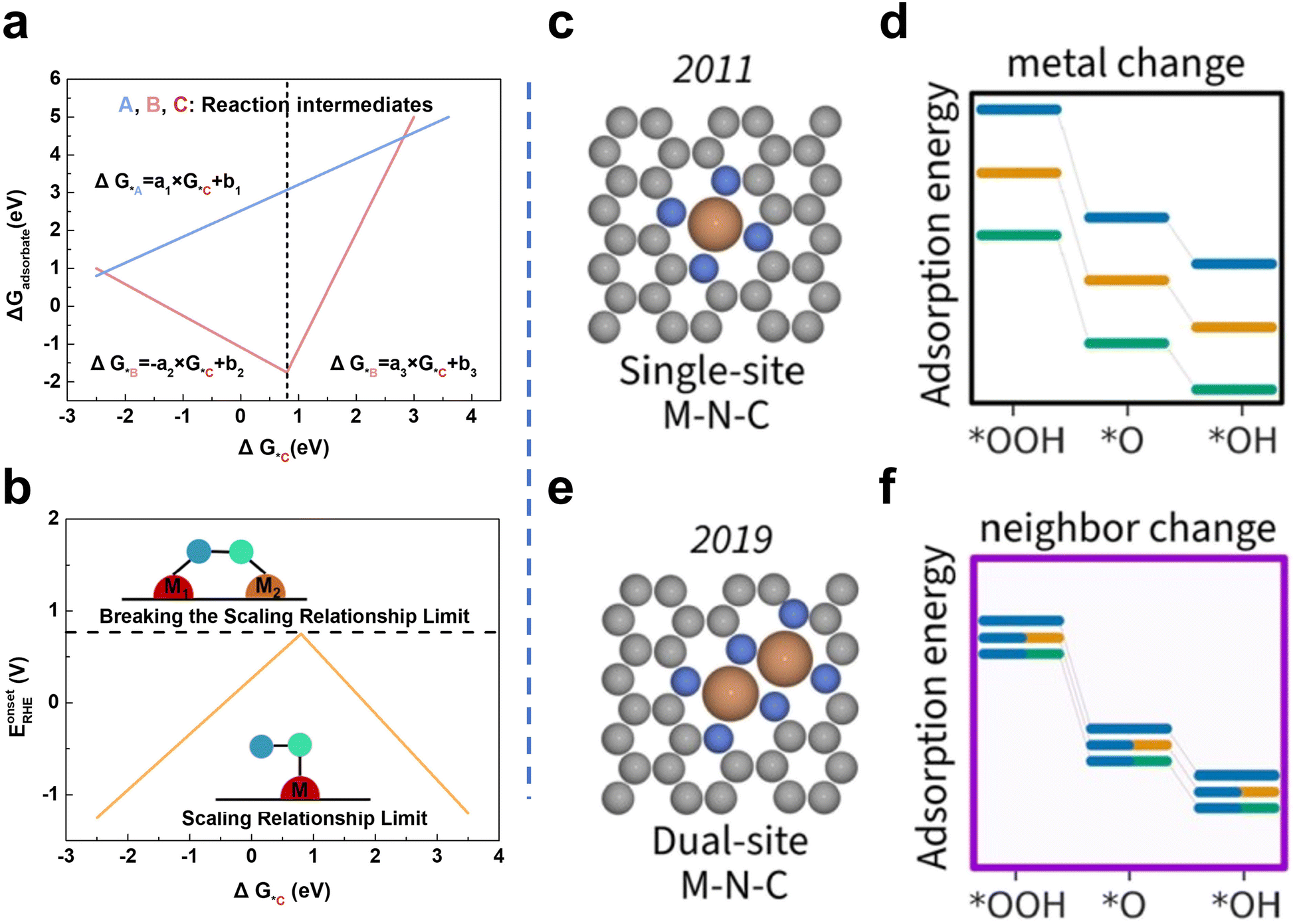 | ||
| Fig. 2 (a) Linear relationships among the adsorption free energy of intermediates for SACs. (b) Diagram of breaking the scaling relationship limit: from SACs to DACs41 (reproduced with permission from ref. 41 copyright 2022, ACS). (c) Configuration of MN4. (d) The adsorption energies of intermediates vary with changing M in the flat single-atom site model. (e) Configuration of M2N6. (f) The adsorption energies of intermediates vary with changing the second M in the flat dual-atom site model42 (reproduced with permission from ref. 42 copyright 2023, ACS). | ||
The M1–M2 configuration shows that the changing adsorption model can break the SRL (Fig. 2b). Taking the configuration of MN4 embedded in graphene as a model (Fig. 2c), the adsorption energies of OH* and OOH* (the vital intermediates in the ORR) would simultaneously and proportionally vary when changing the metal center (Fig. 2d). Thus, since the adsorption energies of reaction intermediates on individual metal sites are linearly correlated, the adsorption of all intermediates cannot be in the optimal state simultaneously, and the optimal reaction performance and the minimum theoretical overpotential cannot be obtained.41 By building the M2N6 model (Fig. 2e), it can be found that the combined variation of these metal sites can provide fine control of the adsorption energy, which could break the strong scaling relationship limit (Fig. 2f)42. From the aspect of the Sabatier principle, the active site should have a suitable binding energy to the reaction intermediate. However, Fe–N/C shows a strong M–O bond, which hinders the desorption of the final products. The introduction of the second metal provides different active sites and regulates the electronic structure of the Fe active site, thus effectively controlling the O2 adsorption strength and OO bond dissociation. Metal coupling in Fe-based DACs can lead to asymmetric charge distribution around the active site, thereby tuning the charge distribution and the adsorption/desorption properties of the intermediates.43,44 Yu et al. reported that adjacent Ni sites can modulate the electronic structure of Fe sites in DACs and further lower the energy barrier of the rate-determining step in the 4 e− ORR process based on DFT calculations. Accordingly, the prepared Fe–Ni DACs (FeNi SAs/NC) exhibited excellent ORR activity (E1/2 of 0.84 V vs. RHE and Eonset of 0.98 V vs. RHE).
2.3 Effects of Fe-based DACs
To address the drawbacks of SACs, Fe-based DACs were designed and employed as ORR catalysts. In this part, recent research work achieving various effects is discussed in detail. The effects of Fe-based DACs are summarized in Fig. 3 and further discussed below: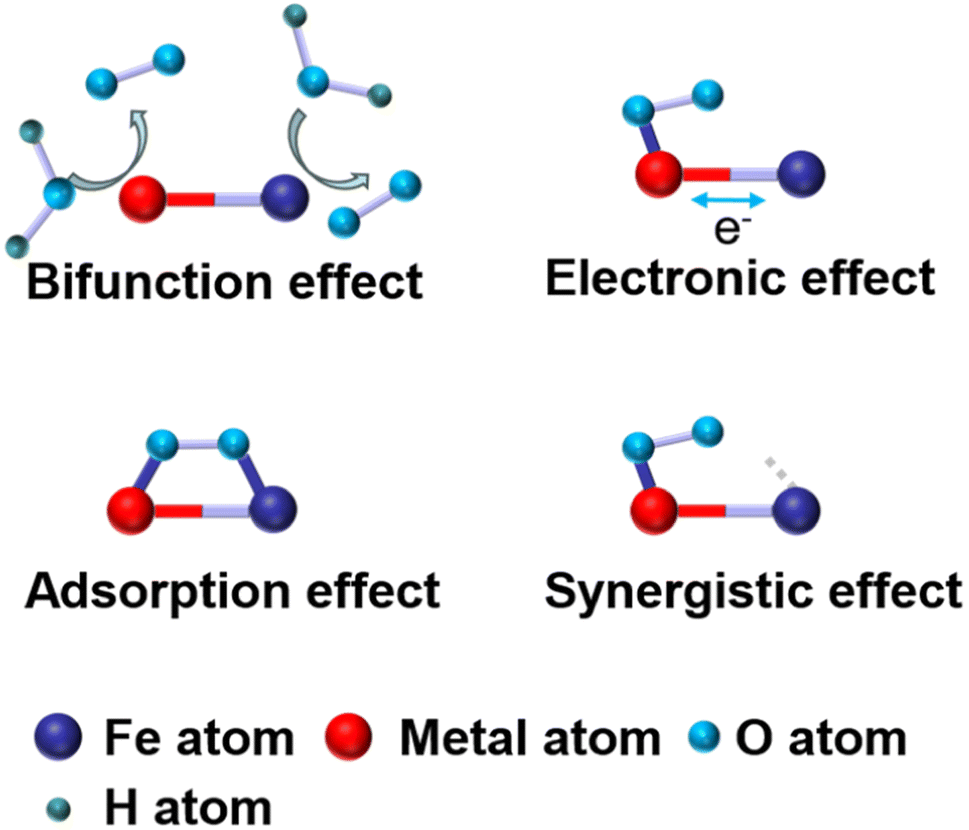 | ||
| Fig. 3 Fe-based dual atom catalysts' effect. | ||
(a) Introducing extra OER active sites into Fe–N/C. Liu et al. reported a bimetallic iridium–iron diatomic catalyst (IrFe–N–C) with an IrFeN6 configuration. Ir and Fe are coordinated with four N atoms, with two N atoms shared between IrN4 and FeN4 moieties (Fig. 4a). The Fe site in IrFeN6 shows a lower energy barrier (0.56 eV) for the rate determining step (RDS) than that of the Ir site (1.24 eV) towards the ORR. In terms of the OER process, where O* + OH− → OOH* + e− was regarded as the RDS of the reaction, the Fe site in IrFeN6 displays a higher energy barrier (0.67 eV) than the Ir site (only 0.43 eV) (Fig. 4b and c). Through the introduction of Ir sites for the OER, Fe-based DACs were successfully designed as bifunctional catalysts.45
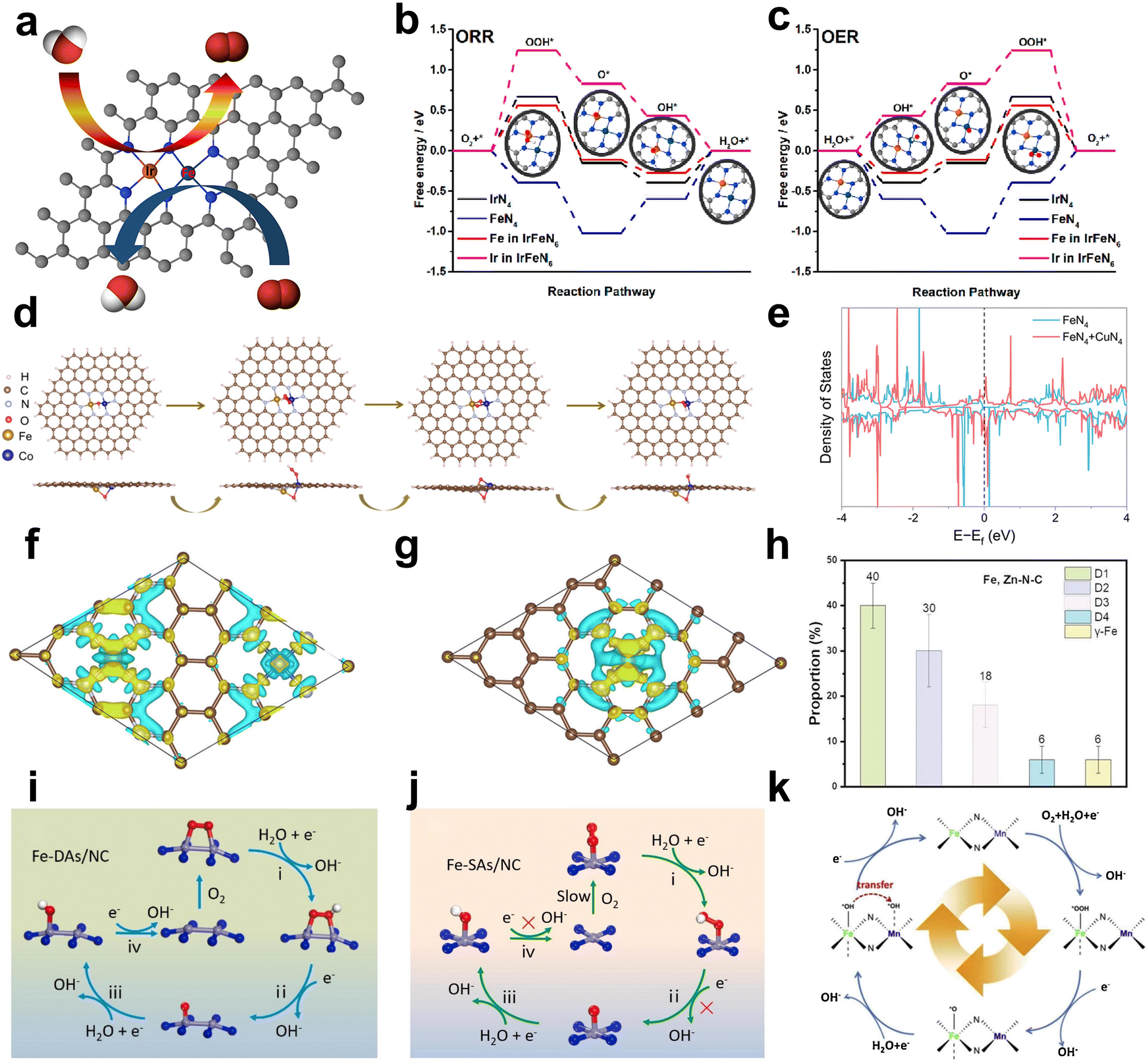 | ||
| Fig. 4 (a) Schematic diagram of bifunctional catalysis. Computational analysis of the IrFeN6 diatomic sites for the ORR and OER. Gibbs free-energy diagrams for (b) the ORR and (c) OER45 (reproduced with permission from ref. 45 copyright 2022, ACS). (d) Top view and side view of the initial structures after adsorption of OOH*, O*, and OH* on the FeCo–OH–NC model46 (reproduced with permission from ref. 46 copyright 2022, ACS). (e) Density of states for FeN4 + CuN4 and FeN4. (f) Electron density difference analysis of FeCu-SAC. (g) Electron density difference analysis of Fe-SAC. Density of states for FeN4 + CuN4 and FeN447 (reproduced with permission from ref. 47 copyright 2023, RSC). (h) The specific proportion of the chemical state of the Fe species in Fe, Zn–N–C49 (reproduced with permission from ref. 49 copyright 2023, Wiley). The proposed ORR mechanism on (i) Fe2–N6 moiety of Fe-DAs/NC and (j) Fe–N4 of Fe–SAs/NC51 (reproduced with permission from ref. 51 copyright 2023, Elsevier). (k) Scheme of the FeNx site and MnNx site cascade mechanism on Fe/Mnx–N–C52 (reproduced with permission from ref. 52 copyright 2021, Elsevier). | ||
(b) Constructing the FeMN6 site to exhibit both OER activity and ORR activity. The Fe–M dual atom sites show the potential to achieve the bifunction of ORR and OER. For instance, Wu prepared an atomically dispersed FeCo–NC catalyst that exhibits excellent bifunctional catalytic activity. Density functional theory (DFT) calculations demonstrate that the intermediates (*OH, *O, and *OH) exhibit a preference to adsorb on the top site of the metal atom (either Fe or Co) within the Fe–NC and Co–NC catalysts. Consequently, the ORR activity is predominantly governed by the electronic interactions between the intermediates and the metal atom rather than geometric factors. In contrast, these intermediates in the FeCo–OH–NC catalyst bond to the Fe site and tend toward Co, favoring adsorption at the bridge site between Fe and Co (Fig. 4d). Hence, the ORR activity depends on a combination of electronic interactions with the metal site and geometric effects. The Gibbs free energy change further suggests that the FeCo–N6 species in the FeCo–NC catalyst can simultaneously serve as the primary active sites for both the ORR and OER.46
(a) Charge transfer. The introduction of a second metal atom can lead to charge transfer between the two metal atoms, especially when one site is occupied by an adsorbed intermediate.47,48 Depending on the electronegativity and electron affinity, electrons can flow between the two metals, resulting in changes in oxidation states and charge redistribution. For instance, Yang et al. constructed FeN4 and CuN4 sites with varied neighboring distances on carbon black via a ligand-mediated method. Cu sites with higher electron density (d10) act as electron donors, which increases the electron density of the Fe atomic site, as confirmed by DFT calculation (Fig. 4e–g). The improved electron density optimizes the adsorption/desorption of intermediates, resulting in a lower energy barrier for the rate-determining step and enhanced ORR activity.47
(b) Spin state change. The spin state refers to the arrangement of electron spins within atomic or molecular orbitals. It has two main components: high spin and low spin. The interaction with the second metal may change the spin state of the Fe atom, which further affects the reactivity and electronic properties of the catalyst. For example, Zhang et al. developed Fe, Zn–N–C with excellent ORR activity (E1/2 = 0.867 V). The Mössbauer spectra reveal that the Fe, Zn–N–C half-metallic diatomic catalyst has approximately 90% low spin FeII and mid spin FeII. The large proportion of the D2 site (ferrous iron with a medium spin state) is approximately 30%, which was regarded as a stable and active site (Fig. 4h).49
(c) D-Band coupling. The electronic structure of transition metals is usually characterized by d-band electronic states, and when metal pairs are formed, coupling of the d-bands affects the energy levels and electronic properties of the atoms. For example, Li et al. prepared Fe–Mo bimetallic sites to tailor the Fe–Nx site. With the introduction of Mo atoms into the FeNx site, the Fe d band center shifted downwards. This modification enhances the adsorption and desorption characteristics of ORR intermediates on the FeMoN6 active site, ultimately leading to improved catalytic performance.50
3. Synthesis methods and strategies of Fe-based double-atom catalysts
Recently, Fe-based DACs have been developed to improve the performance of Fe–N/C SACs. However, Fe-based DACs with a relatively large surface energy tend to form metal agglomerates or nanoparticles during preparation, and thus requiring a strong anchor from the substrate via the introduction of N atoms. Additionally, the specific surface area and structure of the carbon matrix determine the electrical conductivity and mass transport as well as the number of sites that can be exposed, which is critical for the catalytic performance. Therefore, synthesis methods are essential to avoid agglomeration and control the structure and catalytic performance of Fe-based DACs. The developed synthesis methods mainly include the host–guest strategy (Fig. 5a), template-assisted strategy (Fig. 5b–d), and wet chemistry approach (Fig. 5e). In the synthesis process, high-temperature pyrolysis is often involved, and various methods may be combined for use.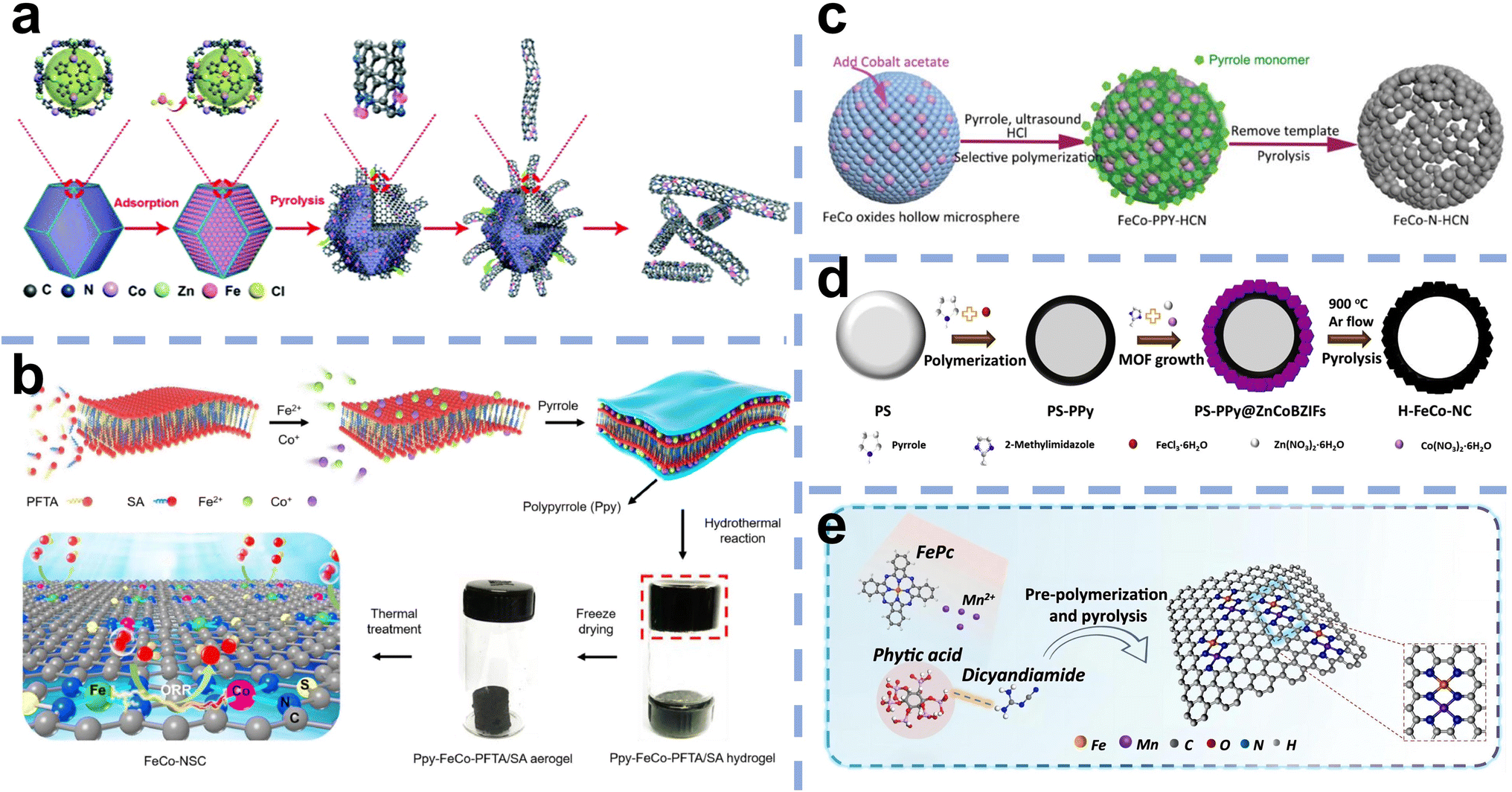 | ||
| Fig. 5 (a) Host–guest strategy: schematic illustration of the synthesis procedure for (Fe,Co)/CNT55 (reproduced with permission from ref. 55 copyright 2018, RSC). Template-assisted strategy: (b) schematic illustration of the synthesis procedure for FeCo-NSC56 (reproduced with permission from ref. 56 copyright 2022, Elsevier). (c) Schematic illustration of the formation process of FeCo–N–HCN from the FeCo oxide template57 (reproduced with permission from ref. 57 copyright 2021, Wiley). (d) Schematic of the synthesis procedure for the H–FeCo–NC catalyst from the PS sphere template58 (reproduced with permission from ref. 58 copyright 2022, Elsevier). Wet chemistry approach: (e) sol–gel method as a typical wet method to prepare Fe,Mn–N/C59 (reproduced with permission from ref. 59 copyright 2021, Nature Publishing Group). | ||
3.1 Host–guest strategy
The “host–guest strategy” is widely applied in constructing DACs, which involves adding another metal element (guest) to a precursor already containing the first metal (host). The key factors to build dual-atom catalysts are controlling the precise amount of metal and constructing the bond between two metals. Wang et al. first synthesized Zn/Co bimetallic MOFs (BMOFs) as the host material. Based on the precise control of binding between the Fe and Co nodes, FeCl3 was added to the Zn/Co BMOFs as the guest metal source. Then, Zn evaporates during the pyrolysis process due to its low boiling point, and Fe–Co dual sites inserted on porous N-doped carbon were prepared (Fig. 6a).60 Electron microscopy techniques confirm that the small bright double dots in a uniformly distributed porous carbon matrix are attributed to the FeCo dual sites (Fig. 6b–g).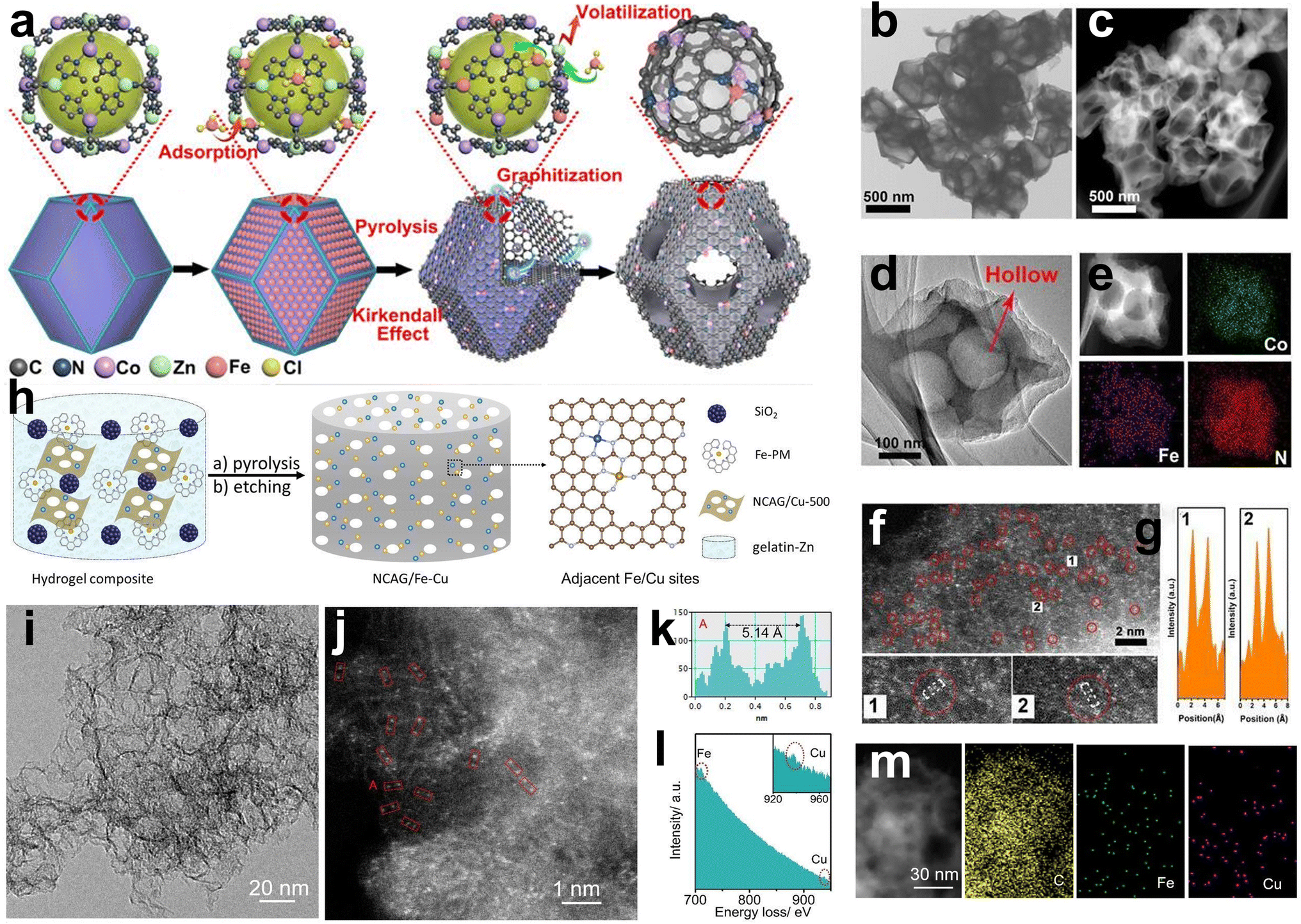 | ||
| Fig. 6 (a) Preparation of (Fe,Co)/N–C. (b) TEM, (c) HAADF-STEM, and (d) HRTEM of (Fe,Co)/N–C. (e) Corresponding EELS mapping of Co, Fe, and N. (f) Magnified HAADF-STEM of (Fe,Co)/N–C, showing Fe–Co dual sites dominant in (Fe,Co)/N–C. (g) Corresponding intensity profiles obtained on the zoomed-in areas in panel E60 (reproduced with permission from ref. 60 copyright 2017, ACS). (h) Schematic illustration of the preparation of NCAG/Fe Cu carbon aerogels. (i) TEM and (j) STEM images of NCAG/Fe Cu. (k) The intensity profile and (l) EELS spectrum of the red box from figure (j). Inset to (l) is the zoom-in of the Cu signal. (m) TEM image and the corresponding elemental maps of NCAG/Fe Cu22 (reproduced with permission from ref. 22 copyright 2022, Wiley). | ||
However, the utilization of MOF precursors suffers from their high cost and sophisticated preparation. In addition to BMOFs, other host materials have also been explored. For example, He et al. synthesized carbon aerogels doped with Cu named NCAG/Cu through controllable biomass gel template pyrolysis, which can be used as host materials to anchor Fe ions.22 The prepared NCAG/Cu aerogels were then dispersed into gelatin with Fe(II)-phenanthroline and SiO2 nanoparticles. The mixture was then pyrolyzed for the second time, followed by HF etching to obtain Fe, Cu-codoped carbon aerogels (Fig. 6h). STEM exhibited small bright dual dots in the carbon aerogel, and the EELS results showed that the pair of dots consisted of an Fe atom and a Cu atom (Fig. 6i, j and m). Elemental mapping analysis confirmed the uniform dispersion of Fe and Cu atoms (Fig. 6k and l).
3.2 Wet chemistry approach
Wet chemistry approaches have attracted increasing attention due to their low cost, simple operation steps, and mild reaction conditions. For example, Yang et al. employed dicyandiamide as the source of C and N, FePc, and Mn(NO3)2 as metal precursors to construct Fe, Mn/N–C catalysts with dual-metal atomic dispersion (Fig. 5e).59 All chemicals were dissolved in water under stirring and dried in an oven at 80 °C for 24 h to achieve prepolymerization for further pyrolysis. The dispersion of the dual sites in the N-doped defective carbon may be due to the adsorption of Mn salts and then binding to the adjacent Fe–N4 centers in iron phthalocyanine molecules. Fe, Mn/N–C showed outstanding ORR performance and favorable stability in alkaline and acidic electrolytes. SEM and TEM showed a graphene-like porous carbon sheet, while EDS elemental mapping, EELS, and comparison between HAADF-STEM of Fe, Mn–N/C and control samples confirmed the coexistence of Fe–Mn in an atomic pair rather than Fe–Fe pairs and Mn–Mn pairs (Fig. 7a–f). The theoretical Fe K-edge spectra of XANES based on the Fe, Mn–N6-1 moieties model calculated by DFT fit the experimental Fe K-edge XANES spectra well, which excludes other structures and moieties (Fig. 7g–j). Li et al. first precarbonized chitosan in a muffle furnace at 200 °C and then preoxidized it with strong acid.61 Chitosan, as a natural polymer with strong metal coordination ability and a rigid chain structure, has been regarded as an ideal matrix to host metals in a uniformly dispersed manner and form atomic sites. Chitosan with the above pretreatment, ammonium chloride, zinc chloride, and ferric chloride hydrate were used as the carbon, nitrogen, and metal sources, respectively. After stirring, evaporation of the solvent, and pyrolysis in N2, Fe/Zn–N–C was obtained as a durable half-metallic diatomic catalyst. The introduction of Zn not only significantly enhanced the ORR activity but also improved the stability of the catalyst by stabilizing the FeN4 site. Moreover, the wet chemistry approach has also been used to prepare hydrogel and aerogel catalysts. Chen et al. reported that Cu sites were introduced into Fe–N/C aerogels with adjacent FeN4 and Cu–N4 sites. First, the hydrogel composite was prepared via the polymerization of gelatin and the introduction of Cu(Ac)2·H2O. After freeze-drying and pyrolysis, carbon aerogels doped with Cu were constructed. Then the as-prepared copper-containing aerogels were mixed with gelatin and Fe salts to make hydrogel polymers, and SiO2 nanoparticles were utilized as templates. Porous aerogel catalysts containing Fe atomic sites and Cu atomic sites were successfully prepared by freeze-drying, pyrolysis and HF etching.22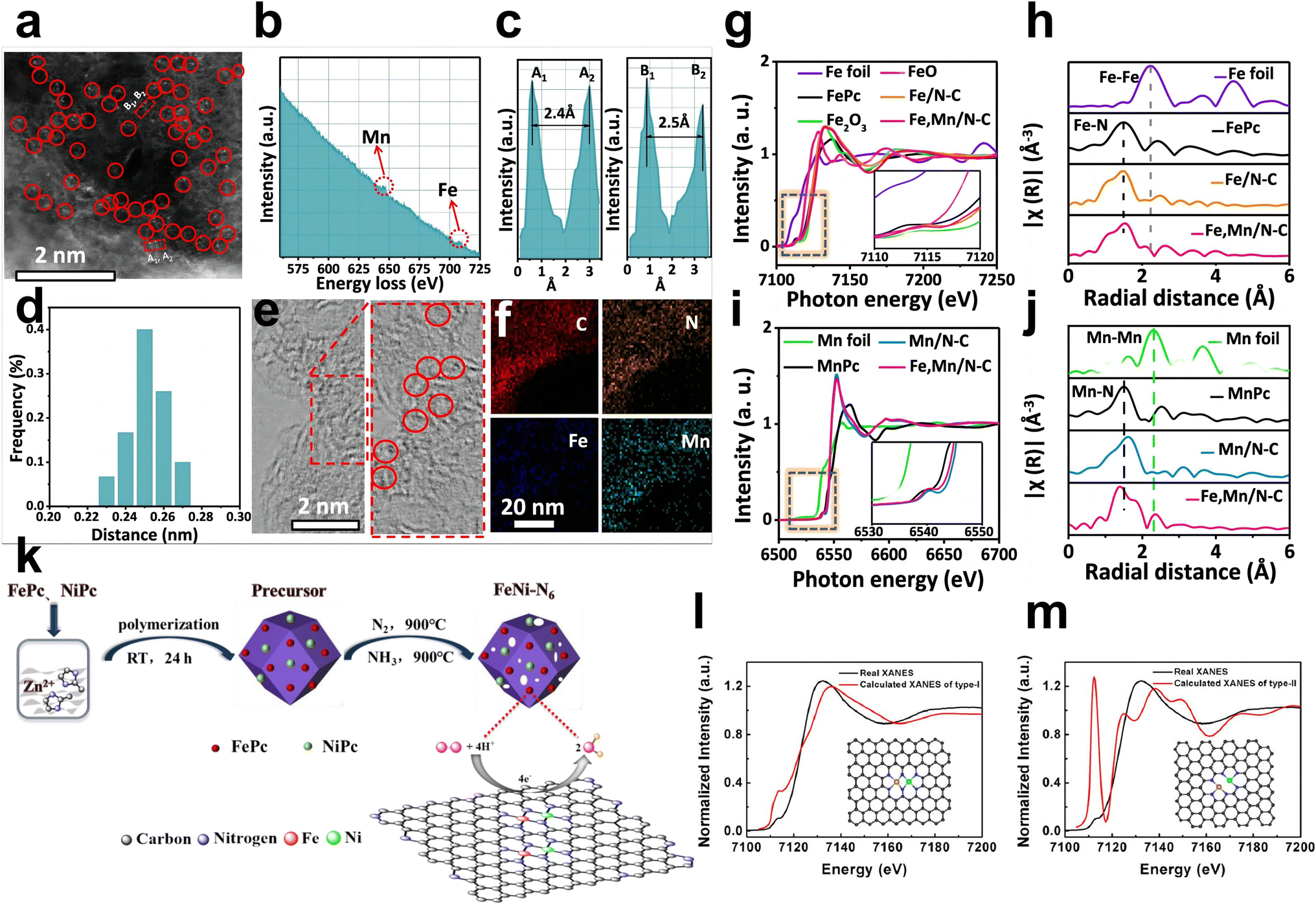 | ||
| Fig. 7 (a) Aberration-corrected HAADF-STEM image and some bimetallic Fe/Mn sites are highlighted by larger red circles. (b) Fe,Mn/N–C structure analyzed by EELS. (c) The intensity profiles obtained on two bimetallic Fe–Mn sites. (d) Statistical Fe–Mn distance in the observed diatomic pairs. (e) HR-TEM of Fe,Mn/N–C, in which some lattice distortions are highlighted by red circles. (f) HAADF-STEM image of Mn, Fe/N–C with mappings of individual elements (C, N, Fe, and Mn). (g) Fe K-edge XANES and (h) Fourier transform EXAFS spectra of Fe,Mn/N–C and reference samples. (i) Mn K-edge XANES and (j) Fourier transform EXAFS spectra of Fe,Mn/N–C and reference samples59 (reproduced with permission from ref. 59 copyright 2021, Nature Publishing Group). (k) Synthetic diagram of the FeNi–N6 catalyst. (l) Fe K-edge fitting curves of FeNi–N6 and the structural model (insets) of FeNi–N6 (type I) and (m) FeNi–N6 (type II)62 (reproduced with permission from ref. 62 copyright 2020, ACS). | ||
Zhou et al. utilized a modified wet chemical carbonization method to synthesize N-coordinated Fe, Ni dual-doped carbon with outstanding ORR performance under acidic conditions (Fig. 7k).62 Zn(NO3)2·6H2O and 2-methylimidazole were first dissolved in methanol, and liquid silica containing FePc and NiPc was added when white suspensions appeared. Liquid silica can inhibit the aggregation of metals and increase porosity. After strong stirring and polymerization, the precursor is pyrolyzed under nitrogen to obtain the final FeNi–N6 species embedded on a porous carbon catalyst with atomic dispersion. Powder X-ray diffraction, N2 adsorption–desorption isotherms, and high-resolution TEM confirmed only characteristic carbon peaks, a high specific surface area, and the absence of metal nanoparticles, respectively. HAADF-STEM measurements showed the uniform dispersion of Fe and Ni in the carbon matrix with a dual dot form. The k3-weighted Fourier transformed EXAFS (R space) result revealed no signal of Fe–metal binding and Ni–metal binding in FeNi–N6. Their XANES revealed that each metal atom coordinates with four nitrogen atoms, two of which coordinate with Fe and Ni simultaneously (Fig. 7l and m).
3.3 Template-assisted strategy
Template-assisted strategies also exhibit wide application in the synthesis of Fe-based DACs. Various templates are used depending on the synthesis pathway and the final catalyst structure, including silica, zinc oxide, and molten salts. Templates play an important role in constructing Fe-based DACs with different specific structures, such as core–shell structures, sphere structures, and nanorod structures. These specific structures show many advantages, including high specific surface areas, abundant micro/nanopores, improved mass-transport ability, enhanced exposure of catalytic sites, etc.Among these templates, silicon dioxide is the most commonly used hard template for preparing atomically dispersed catalysts due to its good thermal and chemical stability and ability to be etched by hot alkaline solutions and hydrofluoric acid. Wang et al. synthesized Fe/Co loaded nitrogen-doped hollow carbon sphere catalysts using dopamine as the C and N source, Fe(NO3)3·9H2O and Co(NO3)2·6H2O as the metal sources, and SiO2 spheres as the template.63 In this study, Fe3+/Co2+ ions were incorporated into polydopamine during dopamine polymerization on the surface of SiO2 spheres. After subsequent pyrolytic carbonization and removal of SiO2 by 10% HF, NHCS–FeCo was obtained. NHCS–FeCo exhibited a hollow carbon sphere structure with increased porosity and high specific surface area.
Li et al. first prepared porous CoFe2O4 hollow microspheres by a hydrothermal method, which can act as the template to prepare N-doped carbon nanocages and the metal source of Fe and Co (Fig. 5c).57 The pyrrole monomer was adsorbed on the CoFe2O4 surface as C and N sources. Then, HCl solution was added to dissolve the CoFe2O4 nanospheres to release Fe3+ and Co2+ ions. The difference in diffusion rate and chemical properties of Fe3+ and Co2+ ions (Fe3+ ions can trigger the polymerization of pyrrole monomers, while Co2+ ions cannot initiate polymerization) was used to selectively form polypyrrole nanocages on the surface of the nanospheres. In this case, the polypyrrole nanocage captures a small amount of Fe3+ and Co2+ ions as single-atom centers due to coordination with the N atoms in pyrrole. Finally, Fe and Co monotonically dispersed on N-doped carbon nanocages (FeCo–N–HCN) were obtained by controlled heat treatment in N2. TEM images exhibited an obvious carbon nanocage structure with a diameter of approximately 200 nm (Fig. 8a). HAADF-STEM images presented no concentrated bright particles but abundant bright dots with a distance of 0.5 nm between two adjacent metal atoms (Fig. 8b and c). Elemental mapping images indicated the uniform dispersion of Fe and Co atoms rather than a nanocrystalline structure (Fig. 8d and e). The HAADF-STEM measurement and corresponding tiny area of the 1 nm2 EELS spectrum of the catalyst showed adjacent Fe and Co atoms (Fig. 8f and g). The XAS results showed the absence of Fe–Fe and Co–Co signals, which were well fitted with the calculated models (Fig. 8h–m).
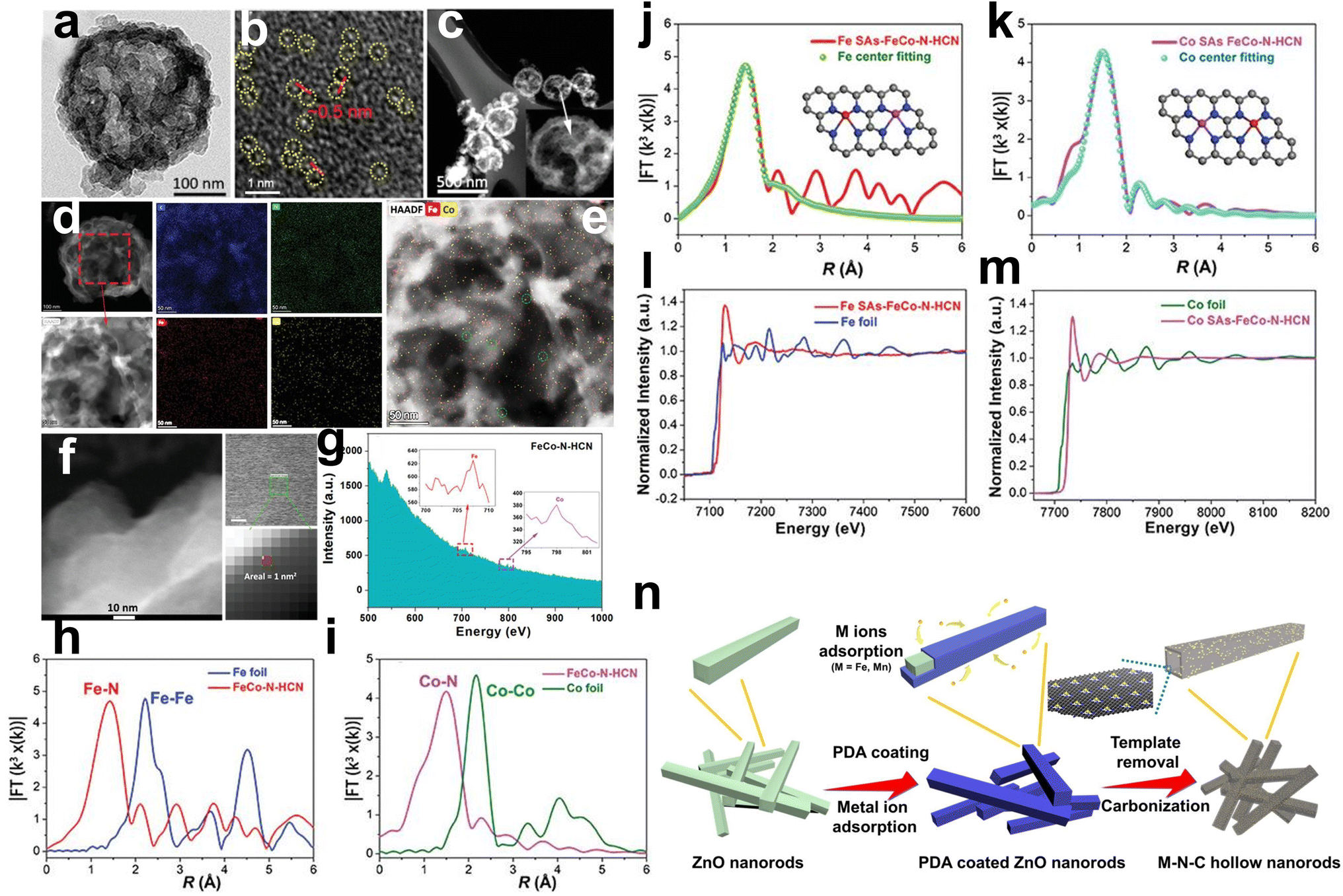 | ||
| Fig. 8 (a) TEM images of FeCo–N–HCN. (b and c) Aberration-corrected HAADF-STEM images of FeCo–N–HCN. (d) Typical TEM image and corresponding mapping images of FeCo–N–HCN. (e) Combined STEM-EDS elemental distribution of Fe and Co; bimetallic single atoms are highlighted in green circles. (f) The selection of a 1 nm2 small area in FeCo–N–HCN and (g) the corresponding EELS spectrum. (h) Fe K-edge XANES spectra of Fe foil and FeCo–N–HCN. (i) Co K-edge XANES spectra of Co foil and FeCo–N–HCN. Fourier transformed (FT) k3-weighted χ(k)-function of the EXAFS spectra for (j) Fe K-edge in Fe foil and FeCo–N–HCN and (k) Co K-edge in Co foil and FeCo–N–HCN. (l) Corresponding Fe K-edge EXAFS fitting with Fe centered neighboring Fe–N4–C and Co–N4–C model and (m) Co K-edge EXAFS fitting with Co centered neighboring Fe–N4–C and Co–N4–C model57 (reproduced with permission from ref. 57 copyright 2021, Wiley). (n) Schematic illustration of the formation process of Fe/Mn–Nx–C hollow nanorods from the ZnO nanorod template52 (reproduced with permission from ref. 52 copyright 2021, Elsevier). | ||
In addition to SiO2, ZnO has also been studied as a hard template due to its ability to display abundant various-dimensional nanostructures. Chen et al. reported that Fe/Mn–Nx–C bimetallic sites with atomic dispersion inserted in N-doped carbon catalysts were prepared by the simple construction of ZnO@PDA-FeMn precursors after pyrolysis (Fig. 8n). ZnO nanorods were used as templates to fabricate Fe/Mn–N–C hollow nanorods. High-quality single-crystal ZnO nanorods were fabricated based on the directional attachment of previously formed quasispherical ZnO nanoparticles. At the same time, the adsorption between metal ions (Fe3+ and Mn2+ derived from FeCl3 and MnCl2) and polydopamine (PDA) can be precisely tuned for electronic affiliation. In the carbonization process of PDA, the already adsorbed metal ions are immobilized at the defects of graphitized carbon through the formation of a typical M–Nx–C. After removing the template ZnO by HCl, the obtained M–Nx–C hollow nanorod samples exhibited a well-defined hollow structure.52
Nguyen et al. prepared hollow Fe, Co, and N codoped carbon catalysts (H–FeCo–NC) using polystyrene spheres as a template. H–FeCo–NC was constructed by using polypyrrole (PPy)-coated polystyrene (PS) spheres as the core, while Zn, Co bimetallic-ZIFs (ZnCoBZIFs) were used as the shell of core–shell particles after carbonization. PS spheres of 200 nm diameter were generated by emulsion polymerization and subsequently decomposed in the carbonization stage to remove the template leaving the hollow structure, while pyrrole was utilized as the C and N source. ZnCoBZIFs were grown from 2-methylimidazole, Zn(NO3)2·6H2O and Co(NO3)2·6H2O on the surface of PPy-coated PS spheres. Iron(III) chloride hexahydrate was used as a source of Fe which can also catalyze the polymerization of PPy on PS spheres. H–FeCo–NC catalysts possessed a high specific surface area of 324.08 m2 g−1 and uniform Fe and Co site distribution.58
Apart from the hard template, Wu et al. demonstrated a soft template-oriented route for the interlayer-constrained construction of Fe–Co DACs.56 Two amphiphiles, perfluorotetradecanoic acid (PFTA) and stearic acid (SA), self-assembled into lamellar micelles to form two-dimensional soft templates. Fe and Co ions were restricted between the two-dimensional soft template layer and the subsequently introduced polypyrrole (PPy) layer. A polypyrrole (PPy) layer was then coated. After pyrolysis, Fe–Co DACs (denoted as Fe–Co-NSC) were prepared, while Fe and Co monoatoms were separated on the two-dimensional carbon nanosheets.
In summary, although there are various methods to construct Fe-based DACs, the following challenges still need to be solved.
(1) The commonly used MOF is expensive, and the synthesis method is cumbersome, which is not conducive to large-scale production.
(2) Current common synthesis methods usually involve pyrolysis processes that cause metal atoms to aggregate easily and make it difficult to maintain the diatom configuration, thus limiting atomic site loading.
(3) The addition of various templates has caused a couple of problems, including increased costs, template removal, and separation of templates from catalysts.
4. Electrocatalytic performance of Fe-based DACs
In this section, we focus specifically on the different types of Fe-based DACs, in which Fe and the other metal atoms (e.g., Fe, Ni, Mn, Co, Cu) served as the active centers. We introduce the performance of catalysts with various metals and configurations, as well as the promoting effect of constructing Fe-based DACs.4.1 FeFe–N/C
First, the Fe2N6 structure was designed and prepared to investigate the effect of the adjacent Fe atom on the original Fe atom. Wu et al. synthesized Fe2N6 with planar-like structures for efficient ORR in PEMFCs by thermally migrating isolated FeN4 sites to a highly graphitized carbon substrate and then coupled to each other the form the Fe2N6 structure.64 The planar-like Fe2N6 structure showed an E1/2 of 0.84 V versus RHE in acidic electrolyte better than that of the FeN4 site (E1/2 = 0.76 V vs. RHE) and exhibited improved peak power densities of PEMFCs up to 845 mW cm−2 higher than the FeN4 site of approximately 300 mW cm−2 (Fig. 9a and b). The catalytic process of the planar Fe2N6 structure during the ORR was systematically investigated using in situ XAS measurements (Fig. 9c). The fitted oxidation state of the sample at the open circuit is +2.79, which is lower than the valence state of the oxygen atoms adsorbed on the individual FeN4 sites, +3.32. Thus, some Fe atoms in the planar Fe2N6 structure retain +2 under aqueous conditions, suggesting the formation of an O2–Fe3+–Fe2+ structure. The normalized Fe K-edge XANES spectra recorded at different potentials show that the absorption edge shifts to lower energies as the applied potential decreases, while the white line peak broadens. At 1.0 VRHE, an increase in the intensity of the edge peak indicated that the ORR had occurred. The difference in absorption intensity became more pronounced as the applied potential decreased, suggesting that more iron atoms were involved in the oxygen reduction process at low potentials. These results indicate that the planar-like Fe2N6 structure has high intrinsic activity and accelerated kinetics at positive onset potentials (Fig. 9d). The changes in the local coordination environment during the oxygen reduction process were investigated by the corresponding Fe K-edge FT-EXAFS oscillations. The Fe–N/O and Fe–Fe peaks can be clearly observed, and the intensity of the Fe–N/O shells is significantly reduced at low potentials, which suggests that the oxygen-containing adsorbates are desorbed at the Fe atoms. During the ORR process, oxygen adsorption, intermediate reactions and product desorption on two neighboring Fe atoms cause the stretching or compression of Fe–Fe coordination bonds, which ultimately leads to the dynamic change of Fe–Fe shells (Fig. 9e). The calculated contraction of the Fe–Fe bond length at 1.0 V versus RHE compared with that at the open circuit voltage indicates that oxygen intermediates were bonded with the adjacent Fe atoms (Fig. 9f). Dual-side adsorption was formed via this bond structure, which offered a strong OO bond breaking force to ameliorate the dissociation of the oxygenated intermediate and benefit the 4e− pathway. The absorption edge change of XANES showed that planar Fe2N6 follows a unique ORR redox transition from the initial state of Ox–Fe3+–Fe2+ to the final state Fe2+–Fe2+, which can be used to deduce the ORR catalytic cycle (Fig. 9g and h).
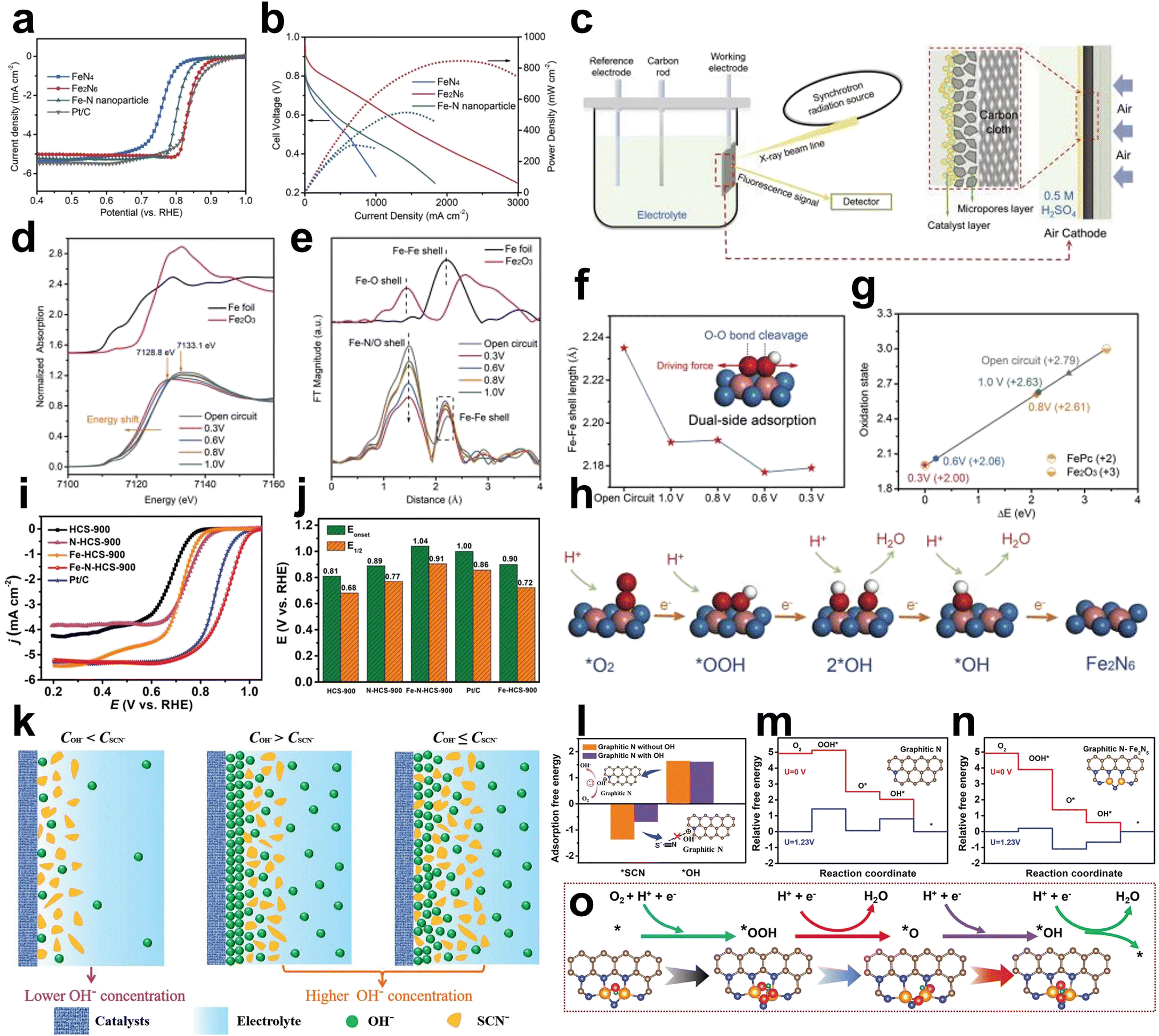 | ||
| Fig. 9 (a) ORR polarization curves of isolated FeN4, planar-like Fe2N6 structure and Fe–N nanoparticle catalysts in O2-saturated 0.5 M H2SO4 solution. Pt/C tested in O2-saturated 0.1 M HClO4 was used as a reference. (b) Polarization and power density curves of isolated FeN4, planar-like Fe2N6 structure and Fe–N nanoparticle-based membrane electrode assemblies in PEMFCs. Cell temperature: 80 °C; relative humidity (RH): 100%; H2/O2: 200 kPa. (c) Schematic of the operando XAS setup combined with XAS measurement and a three-electrode half-cell. (d) Operando XANES spectra and (e) corresponding FTs of Fe K-edge EXAFS oscillations of planar-like Fe2N6 structure at different applied potentials in 0.5 M H2SO4. (f) Fe–Fe shell length calculated from operando EXAFS spectra. Inset: deductive oxygenated intermediate adsorption state on the planar-like Fe2N6 structure. (g) Fitted average oxidation state of Fe in the planar-like Fe2N6 structure according to operando XANES spectra. (h) Proposed ORR reaction pathways on the planar-like Fe2N6 structure. The balls in blue, pink, red, and white represent N, Fe, O, and H atoms, respectively64 (reproduced with permission from ref. 64 copyright 2020, Wiley). (i) LSV curves, (j) corresponding Eonset and E1/2. (k) Schematic illustration of the SCN− poisoning mechanism on the catalyst under the different microenvironments. (l) DFT-calculated adsorption free energies of *SCN on the graphitic N species with or without OH*. (m and n) The free energy diagram for the ORR on the graphitic N structure and biactive graphitic N–Fe2N5 structure. (o) The proposed mechanism for the ORR on the biactive graphitic N and Fe2N5 structure65 (reproduced with permission from ref. 65 copyright 2022, Wiley). | ||
Apart from the Fe2N6 structure, Wang et al. constructed an edge-adjacent Fe2N5 structure and graphitic N species as the biactive ORR sites supported on hollow carbon spheres (Fe-N-HCS-900) via a continuous two-step synthetic strategy. Fe-N-HCS-900 exhibited better ORR performance (Eonset of 1.04 V, E1/2 of 0.91 V in 0.1 M KOH) than the control sample (Fe-HCS-900 with an Eonset of 0.9 V and E1/2 of 0.72 V) (Fig. 9i and j).65 SCN− anions were used in poisoning experiments and confirmed that the edge-adjacent Fe2N5 structure and graphitic N species performed as the biactive ORR sites in Fe-N-HCS-900 (Fig. 9k). When using DFT calculations to study the influence of microenvironments on poisoning the graphitic N species and Fe2N5 structure, hydroxyl groups were found to protect graphitic N from SCN− due to the higher adsorption free energy of SCN− and showed no obvious influence on the adsorption of OH* (Fig. 9l). Fig. 9m and n illustrate the adsorption free energy of the two models constructed by DFT, showing that graphitic N and Fe2N5 sites exhibit a more favorable ORR performance. Based on poisoning experiments and DFT calculations, the ORR mechanism is revealed in Fig. 9o. In detail, it can be divided into 4 steps, in which * is regarded as the active site. Step 1: * + O2 + e− + H+ → OOH*; step 2: OOH* + e− + H+ → O* + H2O; step 3: O* + e− + H+ → OH*; step 4: OH* + e− + H+ → * + H2O.
4.2 FeNi–N/C
Fe–Ni dual metal binding could modulate the charge distribution to ameliorate the adsorption of intermediates. Lu et al. prepared nitrogen-doped carbon hollow spheres containing atomically dispersed Fe–Ni bimetallic pairs (Fe–NiNC) with Fe–Ni metal binding.31 The optimized Fe–NiNC catalyst showed improved ORR and OER activity in alkaline solution (E1/2 = 0.85 V, η10 = 340 mV) compared to FeNC (E1/2 = 0.84 V, merely 5 mA cm−1 at 1.77 V) and NiNC (E1/2 = 0.78 V, η10 = 430 mV) counterparts (Fig. 10a and b). Compared to control samples, the charge distribution analysis shows that the metal bonding of Fe–Ni results in an increased positive charge and charge redistribution around the active site, which may improve the catalytic activity by encouraging the adsorption of intermediates on Fe and Ni sites.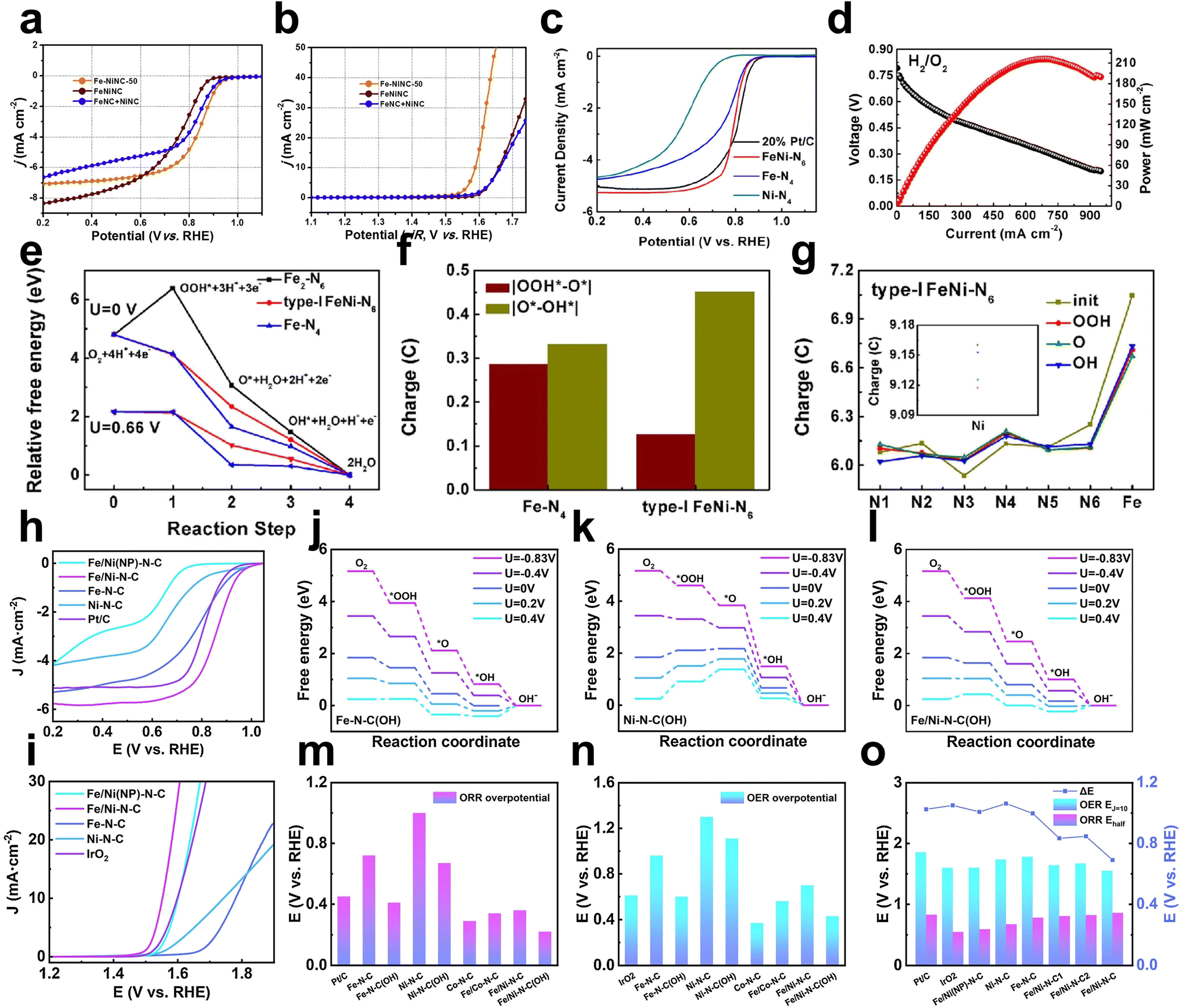 | ||
| Fig. 10 ORR (a) and OER (b) performance of Fe–NiNC-50 together with FeNiNC made via a codoping process and physically mixed FeNC + NiNC sample31 (reproduced with permission from ref. 31 copyright 2020, Elsevier). (c) ORR polarization curves of FeNi–N6, Fe–N4, Ni–N4, and 20 wt% Pt/C in 0.1 M HClO4 electrolyte at 1600 rpm. (d) H2/O2 PEMFC performance of FeNi–N6. (e) Free energy diagrams for the ORR on FeNi–N6 (type I), Fe–N4, and Fe2–N6. (f) Bader charge graph of O for Fe–N4 and FeNi–N6 (type I). (g) Bader charge graph of N, Fe, and Ni (internal graph) for FeNi–N6 (type I)62 (reproduced with permission from ref. 62 copyright 2020, ACS). The free energy diagrams at different electrode potentials for (h) Fe–N–C(OH), (i) Ni–N–C(OH) and (j) Fe/Ni–N–C(OH). (l) ORR overpotential, (m) OER overpotential and (n) ΔE for the bimetal catalyst and comparisons. LSV curves of Fe/Ni–N–C, Fe/Ni–N–C(NP), Ni–N–C, Fe–N–C and Pt/C (IrO2) for (k) the ORR and (o) OER in O2-saturated 0.1 M KOH solution44 (reproduced with permission from ref. 44 copyright 2021, Elsevier). | ||
In addition to Fe, Ni dual atoms with a binding structure, Fe, Ni dual atoms without metal binding were also investigated. Zhou et al. constructed a pair of Fe, Ni dual atoms anchored in porous N-doped carbon (named FeNi–N6) via a modified wet-chemistry carbonization method. FeNi–N6 exhibited better ORR activity (E1/2 is 28 mV larger than that of the control Fe–N4 sample) in HClO4 and excellent PEMFC performance (max power density ≈220 mW cm−2) (Fig. 10c and d). DFT calculations revealed the ambiguous structure of FeNi–N6 (type I: each metal atom is coordinated with four N atoms; type II: each metal atom is coordinated with three N atoms). The bimetallic structure in FeNiN6 is type I, as evidenced by the type I structure's predicted XANES's proximity to experimental spectra and its substantially lower calculated total energy (approximately 1.44 eV) than type II. The oxygen molecule is more favorably absorbed on the Fe atom in FeNiN6 (type I) than on the Ni atom, suggesting that the Fe atom is the active site for producing intermediates. FeNiN6 has the lowest absolute value of ΔG during the transition from OO to OOH*, which determines the overpotential of approximately 0.02 V which is lower than that of FeN4. The third step's ΔG of FeNiN6 is 0.47 eV, which is higher than FeN4's ΔG of 0.03 eV, indicating that FeNiN6 has stronger catalytic activity than FeN4 at this step (Fig. 10e). It can be observed that the entire FeNi–N6 structure is involved in the ORR and boosts the reaction activity by contrasting the Bader charge of O on FeNi–N6 and Fe–N4 at various stages of the ORR reaction and the fluctuation of the Bader charge of Fe, Ni, and N (Fig. 10f and g).62
In addition to Fe–Ni bonding, which affects the charge distribution on the reaction sites, the spin polarization of electrons has also received attention in the design of catalysts. A bifunctional catalyst with atomic pairs (Fe/Ni–N–C) for high-performance ORR and OER in 0.1 M KOH, reported by Li et al., had a minimal potential difference (ΔE = EOER − EORR = 1.552 V − 0.861 V) of 0.691 V, better than that of the Pt/C‖IrO2 system (Fig. 10h and i).44 Fe/Ni–N–C reached Eonset and E1/2 values of 1.005 V and 0.861 V in the ORR, respectively, better than those of the control samples. The researchers first predicted that the Fe–Ni diatomic active center had softened spin-polarized conduction electrons by DFT calculations. The active center Fe 3d in Fe–N–C is highly spin-polarized, and the metal in N–C exhibits paramagnetic properties with nearly zero spin magnetic moment. The spin coupling between the two metals weakened the degree of Fe 3d electron spin polarization by constructing Fe–Ni atomic pairs, which reduced the spin magnetic moment value from 1.88 μB to 1.48 μB. In the early stage of the ORR process, the spin polarization of the transition metals will produce a nonuniform heterodyne field that induces paramagnetic O2 molecules. Meanwhile, coupling with Ni 3d reduces the charge localization of Fe 3d. Hydroxyl modification can further enhance the charge nonlocalization of the active center and improve the charge transport ability. Additionally, the presence of conduction electrons will help to boost the charge transfer process between the catalyst and important reaction intermediates, which will benefit the catalytic activity. From DFT calculations, Fe/Ni–N–C shows superior bifunctional catalytic performance (ηORR = 0.22 V and ηOER = 0.43 V) with the self-binding of the OH ligand (Fig. 10j–o).
Long-range coupling allows the Fe and Ni sites, which are individually anchored with carbon, to modify their electronic structures and coordination environments. Atomically dispersed Fe and Ni in nitrogen-doped carbon (Fe/Ni-Nx/OC) with separated Fe–N4 and Ni–N4 sites were formed by combining the pyrolysis of templates with wet chemical metal ion impregnation. Due to the synergistic effects of the coexisting Fe–N4 and Ni–N4 sites, Fe/Ni–Nx/OC showed excellent ORR activity (E1/2 = 0.84 V in 0.1 M HClO4), outperforming Ni–Nx/OC (E1/2 = 0.505 V) and Fe–Nx/OC (E1/2 = 0.776 V).54
4.3 FeCo–N/C
Due to the synergetic effects between the highly dispersed FeNx and CoNx active sites, building FeCo DACs has received increasing interest. To construct Fe–Co dual sites imbedded on N-doped porous carbon called (Fe,Co)/N–C, Wang et al. established a host–guest technique based on Co nodes in Zn/Co MOFs binding to adsorbed Fe ions.60 Based on the same strategy, they further constructed Fe–Co double sites in N-doped carbon nanotubes ((Fe,Co)/CNT).55 (Fe,Co)/CNT exhibited admirable ORR performance with good stability and an Eonset of 1.15 V and E1/2 of 0.954 V in alkaline electrolytes, superior to Co/CNT-C (E1/2 = ∼0.74 V) and Fe/CNT-C (E1/2 = ∼0.73 V) (Fig. 11a and b). Experimental and DFT results showed that the OO bonds are weakened by the Fe–Co dual sites, which is advantageous for oxygen activation (Fig. 11c and d). (Fe,Co)/CNT has the potential to replace Pt/C due to its high current density of 178 mA cm−2 and maximum output power density of 260 mW cm−2 at the voltage of 1.0 V in Zn–air battery (Fig. 11e).
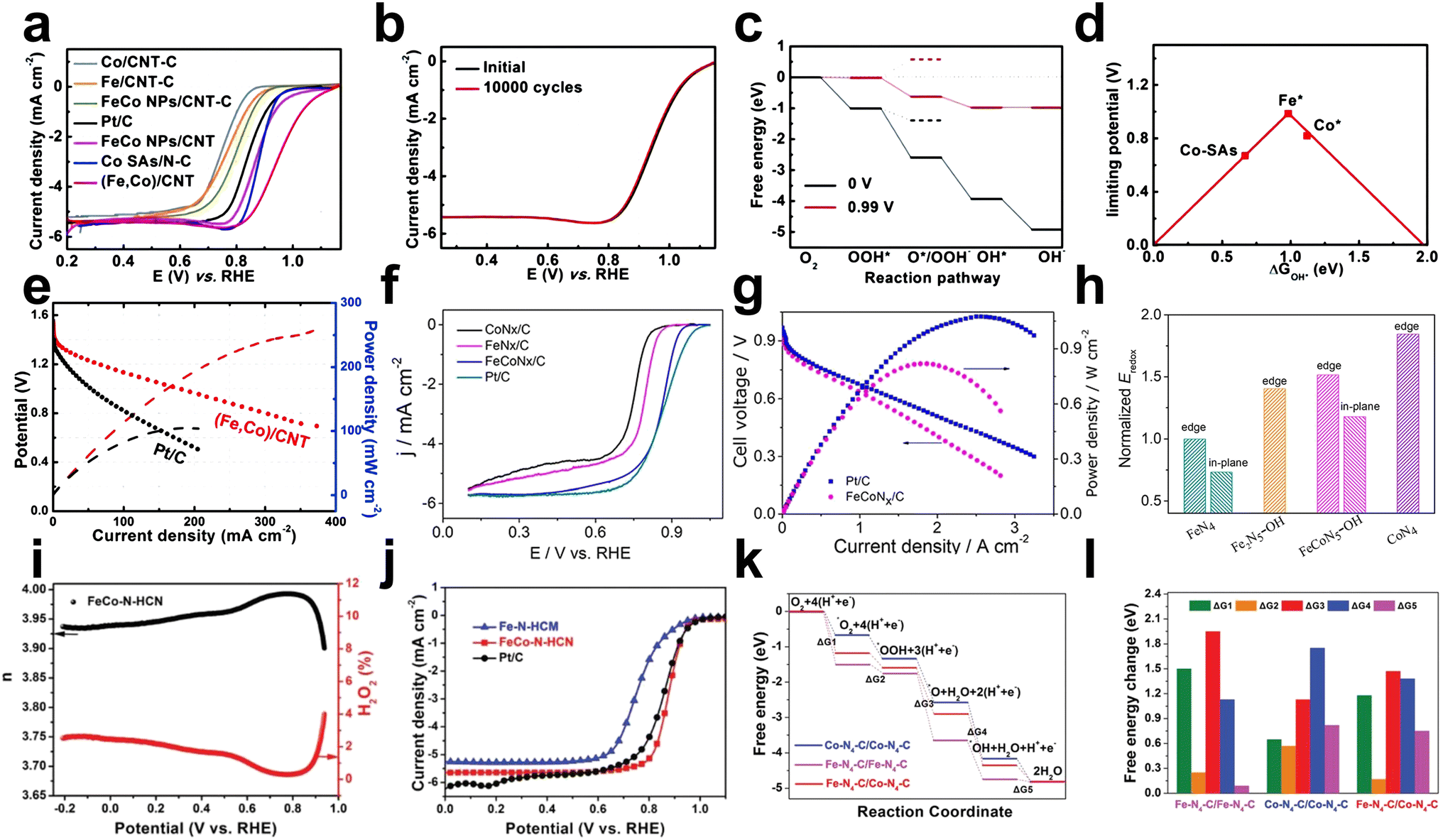 | ||
Fig. 11 (a) RDE polarization curves of samples in O2-saturated 0.1 KOH at a sweep rate of 10 mV s−1 and a rotation rate of 1600 rpm. (b) RDE polarization curves of (Fe,Co)/CNT before and after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 potential cycles ranging from 0.75 to 1.15 V (vs. RHE) in O2-saturated 0.1 M KOH. (c) Free energy diagram for the ORR on the Fe site of OH*-anchored (Fe,Co)/CNT at 0 V and 0.99 V. The energy profile also includes the 2e− pathway represented by dashed lines. C, N, Fe, Co, O and H atoms are presented by gray, blue, orange, green, red and white spheres, respectively. (d) Volcano plot of the ORR activity as a function of DGOH*, where Fe*, Co*, and Co–SAs represent Fe and Co sites on the OH*-anchored (Fe,Co)/CNT and the Co site on bare Co SAs/N–C, respectively. (e) Discharge polarization curves and the corresponding power density of Zn–air batteries with (Fe,Co)/CNT and Pt/C as cathode catalysts55 (reproduced with permission from ref. 55 copyright 2018, RSC). (f) ORR polarization curves with a scanning rate of 5 mV s−1 for the synthesized catalysts in 0.1 M HClO4 solution at 1600 rpm. (g) Polarization curves and corresponding power densities of membrane electrode assemblies fabricated with FeCoNx/C and Pt/C cathode catalysts. (h) Calculated M(II/III) (M = Fe or Co) redox potential of FeN4 (edge and in-plane sites), Fe2N5–OH, FeCoN5–OH (edge and in-plane sites), and CoN4, normalized with the Eredox of FeN4 at the edge66 (reproduced with permission from ref. 66 copyright 2019, ACS). (i) The calculated electron transfer number and H2O2 yield based on RRDE for FeCo–N–HCN. (j) LSV of Fe–N–HCN, Pt/C, and FeCo–N–HCN in 0.5 M H2SO4 at 1600 rpm. (k) Proposed mechanism for the ORR on the Fe–N4–C and Co–N4–C dual active centers. (l) Schematic energy profiles for the ORR pathway on different active sites in acid solution57 (reproduced with permission from ref. 57 copyright 2021, Wiley). 000 potential cycles ranging from 0.75 to 1.15 V (vs. RHE) in O2-saturated 0.1 M KOH. (c) Free energy diagram for the ORR on the Fe site of OH*-anchored (Fe,Co)/CNT at 0 V and 0.99 V. The energy profile also includes the 2e− pathway represented by dashed lines. C, N, Fe, Co, O and H atoms are presented by gray, blue, orange, green, red and white spheres, respectively. (d) Volcano plot of the ORR activity as a function of DGOH*, where Fe*, Co*, and Co–SAs represent Fe and Co sites on the OH*-anchored (Fe,Co)/CNT and the Co site on bare Co SAs/N–C, respectively. (e) Discharge polarization curves and the corresponding power density of Zn–air batteries with (Fe,Co)/CNT and Pt/C as cathode catalysts55 (reproduced with permission from ref. 55 copyright 2018, RSC). (f) ORR polarization curves with a scanning rate of 5 mV s−1 for the synthesized catalysts in 0.1 M HClO4 solution at 1600 rpm. (g) Polarization curves and corresponding power densities of membrane electrode assemblies fabricated with FeCoNx/C and Pt/C cathode catalysts. (h) Calculated M(II/III) (M = Fe or Co) redox potential of FeN4 (edge and in-plane sites), Fe2N5–OH, FeCoN5–OH (edge and in-plane sites), and CoN4, normalized with the Eredox of FeN4 at the edge66 (reproduced with permission from ref. 66 copyright 2019, ACS). (i) The calculated electron transfer number and H2O2 yield based on RRDE for FeCo–N–HCN. (j) LSV of Fe–N–HCN, Pt/C, and FeCo–N–HCN in 0.5 M H2SO4 at 1600 rpm. (k) Proposed mechanism for the ORR on the Fe–N4–C and Co–N4–C dual active centers. (l) Schematic energy profiles for the ORR pathway on different active sites in acid solution57 (reproduced with permission from ref. 57 copyright 2021, Wiley). | ||
Previous studies have indicated that M–N–C catalysts follow the Sabatier principle (suitable M–O bond strength contributes to better catalytic performance), while Fe–N–C catalysts exhibit excessive binding energy to O, hindering ORR performance. For this reason, Xiao et al. designed and constructed an FeCoN5 dual-atom site catalyst without metal bonds, where water spontaneously dissociates at the center to generate a new FeCoN5OH stabilization site, which can modulate the d-band energy of Fe and reduce the M–O binding energy. FeCoN5–OH exhibited excellent ORR performance with an Eonset of 1.02 V and E1/2 = 0.86 V in acidic media (Fig. 11f and g). Two adjacent metal atoms with a distance of 2.2–2.3 Å were confirmed by the bright paired dots in HAADF-STEM, and EELS spectra revealed that the Co and Fe atoms are located next to the N atoms (FeCoNx coordination). The combination of DFT calculations and in situ XANES confirmed that the Fe(III)/Fe(II) redox potential on this new active site was enhanced, demonstrating decreases in Fe–O binding energy and thus significant improvements in ORR performance (Fig. 11h).66
The FeCoN6 configuration demonstrated that the distinct vicinal structure can improve the electrochemical durability in ORR catalysis compared to the FeCoN5 configuration. Chen et al. constructed the dual-metal site catalyst FeCo–NC with the FeCoN6 configuration via pyrolysis of ZIF-8 containing Fe and Co.67 FeCo–NC demonstrated outstanding ORR activity with an E1/2 of 0.842 V in acidic electrolyte outperforming Fe–NC (E1/2 = 0.803 V) and exceptional durability with only an 11 mV decline in E1/2 in the 10000 cycle durability test. Fourier transform EXAFS curves indicated that metallic Fe–Fe and Co–Co scattering paths were not present. The electron density around the Fe center is increased by the vicinal effect of Co atoms on the Fe–N4 active site. Theoretical calculations suggest that the neighboring Co atoms improve ORR resistance and activity by preventing the formation of the *O(OH) intermediate and promoting *OH desorption from Fe–N4.
In addition to Fe–Co atomic pair types, M–N–C materials with adjacent single-atom dual-active centers have also been developed. Li et al. reported Fe–Co–N–HCN carbon nanocages with adjacent Fe–N4 and Co–N4 structures as dual active centers using a selective polymerization method.57 Electrochemical tests in 0.1 M KOH solution showed below 1.8% H2O2 yield and E1/2 of 0.86 V and Eonset of 0.98 V for FeCo–N–HCN, exceeding the commercial Pt/C (E1/2 = 0.85 V; Eonset = 1.03 V) and Fe–N-HCM (Eonset = 0.96 V; E1/2 = 0.76 V) (Fig. 11i and j). FeCo–N–HCN outperformed the Pt/C catalyst in Al–air batteries, displaying a maximum power density of 192.8 mW cm−2 at a current density of 368.1 mA cm−2. According to DFT calculations, the Fe–N4–C and Co–N4–C dual active centers first enhanced O2 splitting by activating the O–O bonds in the adsorbed O2. This prevented the generation of intermediate H2O2 and catalyzed the ORR through the 4 e− pathway. The ORR reaction energy barrier was significantly lowered by the dual active centers, which also facilitated the transfer of *OH intermediates from the strongly adsorbed Fe atomic centers to the Co atomic centers. This reduced the difficulty of desorption from the catalyst surface and lowered the overpotential (Fig. 11k and l).
4.4 FeCu–N/C
Many efforts have been devoted to achieving excellent ORR activity in Cu/Fe DACs.22,68,69As the rate-determining step during the ORR could be influenced by Cu atoms, Xiao et al. prepared Fe and Cu dual atoms inserted on N-doped porous carbon (FeCu–NC), which contained FeN4 and CuN4 dual active sites via a pyrolyzed FeCu–ZIF precursor under N2.69 FeCu–NC exhibited excellent ORR performance with an E1/2 of 0.882 V in alkaline media (0.1 M KOH), which was approximately 24 mV higher than that of the control sample Fe–N–C SAC (without the addition of copper acetylacetonate) (Fig. 12a). Furthermore, FeCu–NC showed exceptional durability and a higher peak power density of 0.91 W cm−2 than Pt/C (Fig. 12b). The free energy of different configurations showed that the RDS of Fe–N4 was OH*→H2O due to strong OH* adsorption, while the RDS of FeN4–CuN4-1 was O* → OH*, as the introduced adjacent CuN4 species can weaken OH* adsorption. Based on the d-band center theory, CuN4 species resulted in the shift of the Fe d-band center to a lower energy level and weaker adsorption ability, which increased the ORR activity (Fig. 12c–f).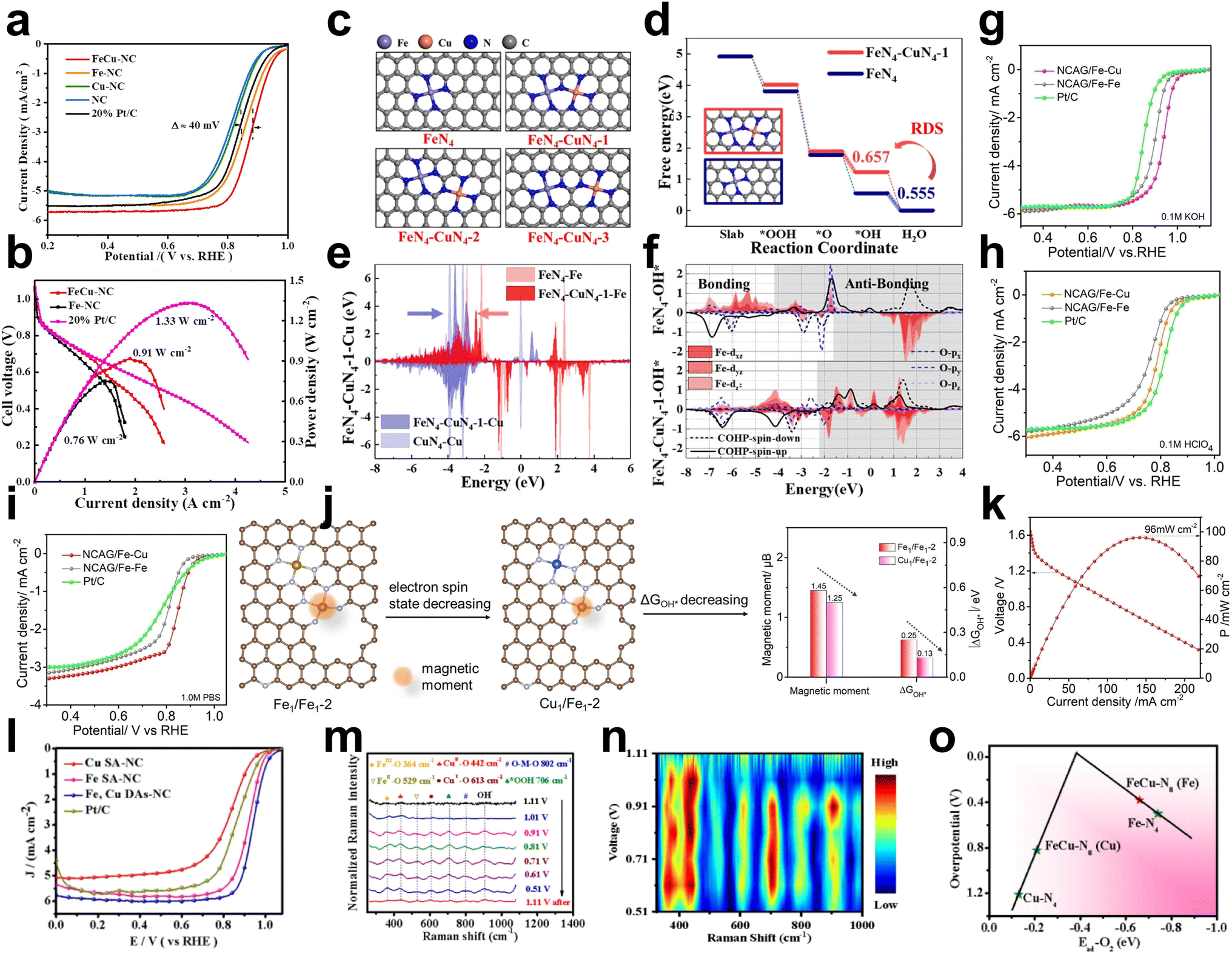 | ||
| Fig. 12 (a) ORR polarization curves of FeCu–NC, Fe–NC, Cu–NC, NC and 20% Pt/C in alkaline media. (b) Polarization curve and power density curve of FeCu–NC, Fe–NC and commercial Pt/C in H2–O2 HEMFC. Test conditions: anode loading 0.4 mg PtRu per cm2 (60% PtRu HISPEC10000), cathode loading: 1.0 mg cm−2, 5 cm2, 15 μm PAP-TP-85 membrane, 80 °C, 100% RH, 2.5 bar H2–O2 @ flow rate of 1.0–1.5 l min−1. (c) Configuration diagrams of FeN4, FeN4–CuN4-1, FeN4–CuN4-2 and FeN4–CuN4-3. (d) The free energy diagrams of FeN4 and FN4–CuN4-1, where the RDS denotes the rate-determining step. (e) The d-band changes of Fe and Cu between FeN4, CuN4 and FeN4–CuN4-1. (f) Suborbital PDOS of the Fe and O orbitals combined with the bonding state shown by COHP (removing the Fe dxy and dx2−y2 orbitals)69 (reproduced with permission from ref. 69 copyright 2022, Elsevier). (g) LSV curves of NCAG/Fe Cu, NCAG/Fe Fe and commercial Pt/C in 0.1 M KOH. (h) LSV curves of NCAG/Fe Cu, NCAG/Fe Fe and commercial Pt/C in 0.1 M HClO4. (i) LSV curves of NCAG/Fe Cu, NCAG/Fe Fe and commercial Pt/C in 1.0 M PBS. (j) Schematic illustration of the moderating effect of magnetic moment on ΔGOH* for the bimetal sites at the nanopore.22 (k) Discharge polarization and corresponding power density curves of a neutral/liquid Al–air battery assembled with NCAG/Fe Cu. (l) LSV polarization curves in O2-saturated 0.1 M KOH electrolyte. (m) In situ Raman spectra of Fe, Cu DAs-NC recorded at varied potentials in O2-saturated 0.1 M KOH electrolyte. (n) The corresponding 2D Raman intensity plot of voltage converted from (m). (o) The volcano plot of overpotential versus Ead-O2 for the different structures70 (reproduced with permission from ref. 70 copyright 2023, Elsevier). | ||
In addition to the modulated Fe d-band center, the magnetic moment of Fe could also be changed by Cu atoms. He et al. investigated the effect of a second metal atom on Fe–N/C using theoretical calculations. First-principles calculations revealed a positive linear correlation between the ORR key reaction step energy barrier and the magnetic moment of the Fe atom. DFT calculations showed that introducing Cu atoms could enhance the dz2 orbital of the Fe atoms with reverse spin, thus reducing the magnetic moment and the adiabatic step potential barrier of Fe (Fig. 12j). Subsequently, it was experimentally found that the magnetic moment of the Fe catalytic sites could be reduced by modifying Cu single atoms on Fe–N–C aerogel complexes (named NCAG/Fe–Cu). Thus, the ORR performance was significantly enhanced over a wide pH interval (0–14) (Fig. 12g–i). Neutral/solid Al–air cells and alkaline/solid Zn–air cells were assembled to achieve excellent open-circuit voltages of 2.00 V and 1.51 V and power densities of 130 mA cm−2 and 186 mA cm−2, respectively (Fig. 12k).22
Shi et al. reported a heterodiatomic catalyst decorated with N-bridged FeCu–N8 sites (Fe, Cu DAs-NC) with excellent ORR activity (E1/2 = 0.94 V in 0.1 M KOH) (Fig. 12l). Potential-dependent in situ Raman measurements were performed to monitor the structural changes of the intermediates on the Fe, Cu diatomic sites (Fig. 12m and n). At the initial state of 1.11 V, no obvious Raman peaks were observed because no reaction occurred. When the potential decreased to 1.01 V, two peaks appeared at 442 and 529 cm−1 attributed to CuII–OH and FeII–OH, respectively. When the potential was further shifted from 1.01 V to 0.51 V, the Raman peaks at 442 and 529 cm−1 became weaker, and two additional peaks at 613 and 364 cm−1 appeared, which were related to the CuI–O and FeIII–O oscillations, respectively, indicating that CuII/FeII was converted to CuI/FeIII during the ORR. In addition, the band observed at 802 cm−1 is characterized by O–M–O oscillations in Cu–O–Cu and Fe–O–Fe species, which implies that Fe and Cu species are the main active sites for the ORR. The new peak observed at 706 cm−1 belongs to the O–O stretching vibration of *OOH, the intensity of which increases as the potential shifts from 1.11 V to 0.51 V, indicating a large accumulation of oxygen intermediates. The peak centered at 991 cm−1 can be attributed to the OH deformation mode due to the production of OH. The volcano plot of overpotential versus Ead-O2 was obtained from DFT results, showing that the FeN4 sites in the N-bridged FeCu–N8 have the highest ORR activity within the lowest overpotential (Fig. 12o).70
4.5 FeMn–N/C
The introduction of Mn could change the electronic structure of Fe to improve the ORR performance. Gong et al. synthesized atomically dispersed Fe and Mn embedded on N-doped porous carbon showing excellent ORR performance (Fe,Mn–N/C-900) by pyrolyzing ZIF-8 precursors containing Fe and Mn ions.43 The XANES spectra and DFT calculations show that the addition of Mn can change the electronic structure of Fe in the Fe–Nx active site and that the energy barrier for the ORR process can be lowered by the synergistic interaction between Fe and Mn. Fe, Mn–N/C needs less energy than Fe–N/C to protonate O* to OH* during the ORR process.The strategy of Fe spin state modulated by Mn atoms has also been utilized to design FeMn DACs. Yang et al. reported atomically dispersed Fe and Mn on N-doped carbon (Fe,Mn/N–C) via pyrolysis processes.59 Fe,Mn/N–C exhibited excellent ORR performance (E1/2 = 0.928 V in 0.1 M KOH, E1/2 = 0.804 V in 0.1 M HClO4) (Fig. 13a and b) with OER activity, resulting in a lower difference between the OER and ORR metrics (E1/2 − Ej=10) than Pt/C and RuO2 (Fig. 13c). Furthermore, the negligible difference between Ej=10 and E1/2 of Fe,Mn/N–C leads to superior durability comparing to commercial Pt/C and Zn–air battery performance with long-term stability (Fig. 13d and e). DFT calculations suggest that the adjacent Mn–N can activate the Fe(III) site effectively, enabling Fe(III) to achieve single electron filling (t42ge1g) in the FeN4 site, which is more favorable for electrons to enter the antibonding orbital of O. Meanwhile, Fe and Mn/N–C possess appropriate bond lengths and binding energies with oxygen intermediates, accelerating the reaction kinetics (Fig. 13f–h and k). The optimized atomic structures for the main process of the ORR reveal that Fe,Mn/N–C can adsorb O2 in Bridge mode and construct Mn–O–Fe, while Fe–N/C adsorbs O2 in Griffiths mode (Fig. 13i and j).
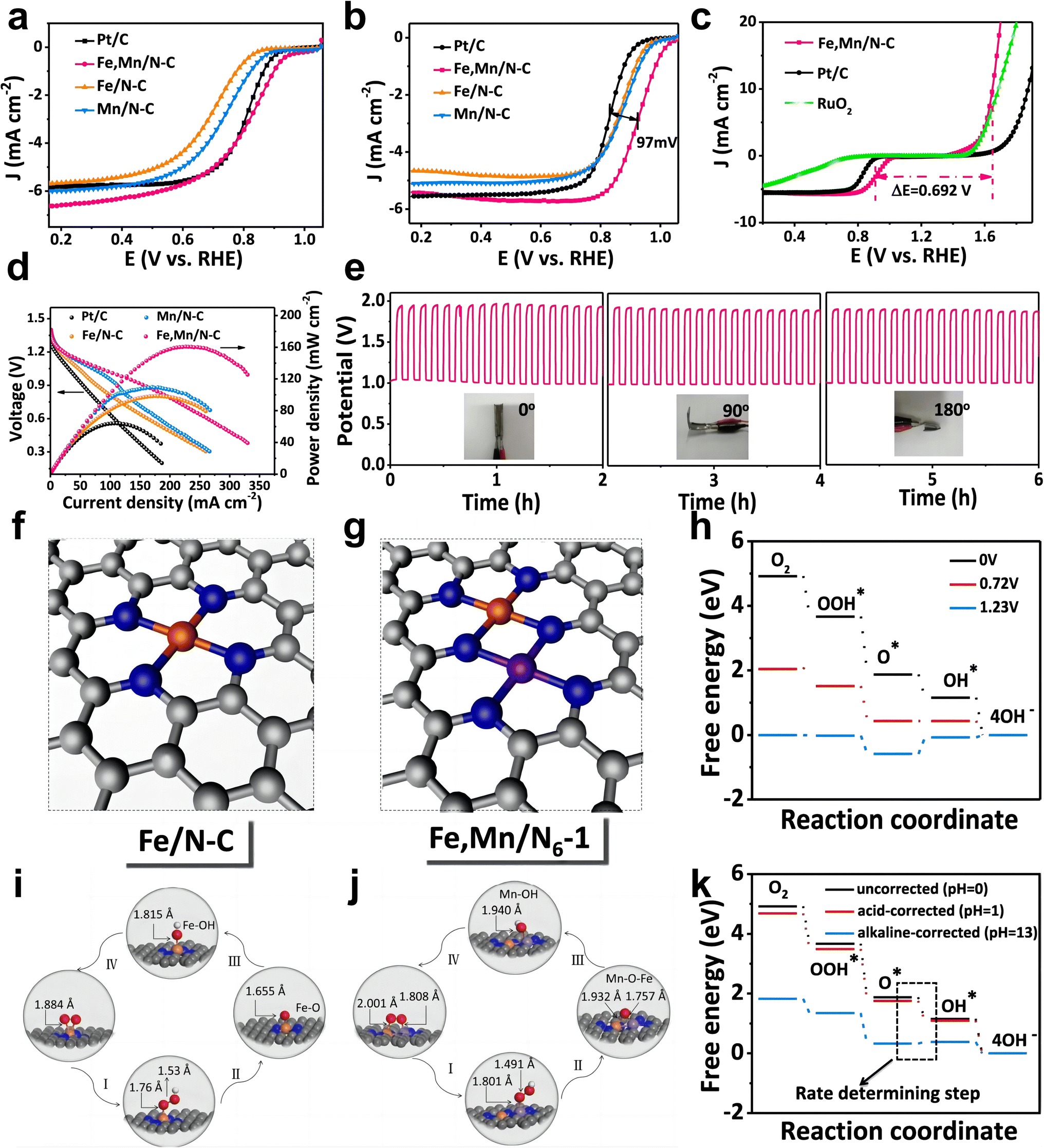 | ||
| Fig. 13 (a) LSV curves of Fe, Mn/N–C, Fe/N–C, Mn/N–C and Pt/C catalysts in O2-saturated 0.1 M HClO4 solution. (b) LSV curves of Fe, Mn/N–C, Fe/N–C, Mn/N–C, and Pt/C catalysts in O2-saturated 0.1 M KOH solution. (c) LSV curves of Fe,Mn/N–C, commercial Pt/C, and RuO2 catalysts on an RDE in 0.10 M KOH, indicating the bifunctional activities toward both ORR and OER. (d) Polarization and power density curves of the primary Zn–air batteries of the Fe,Mn/N–C, Fe/N–C, Mn/N–C and Pt/C catalysts in O2-saturated 6 M KOH solution. (e) Galvanostatic discharge–charge cycling curve at 1 mA cm−2 for the all-solid-state rechargeable ZAB, applying bending strain (as depicted by the inset images) every 2 h. DFT calculations of the ORR activity on Fe,Mn/N–C and Fe/N–C catalysts: the optimized structure of (f) Fe/N–C and (g) Fe,Mn/N–C. (h) The pathways for Fe,Mn/N–C are summarized at U = 0 V, 0.72 V, and 1.23 V, respectively. Optimized atomic structures for the main process of an ORR: (i) Fe/N–C and (j) Fe,Mn/N–C. (k) pH-corrected free energy diagram of Fe,Mn/N6-1 59 (reproduced with permission from ref. 59 copyright 2021, Nature Publishing Group). | ||
Mn–O π donation and Fe–O π–π repulsion assist the adsorption of O2 and desorption of products. By a facile one-step strategy, Subhajit Sarkar et al. reported binary (Fe–Mn) active sites in Fe-, Mn-, and N-doped carbon with hierarchically porous nanostructures (Fe, Mn, N-FGC). Fe, Mn, and N-FGC outperformed the commercial Pt/C catalyst in ORR and rechargeable Zn–air batteries.53 In detail, Fe, Mn, N-FGC showed good ORR performance in alkaline media with an Eonset of 1.03 V and E1/2 of 0.89 V. The performance of the rechargeable Zn–air cell exhibited an open-circuit voltage of 1.41 V, a power density of 220 mW cm−2 at a current density of 260 mA cm−2, and a constant charge–discharge voltage at high current densities (Fig. 14a–c and f). The XANES curves exhibited a peak at approximately 2.25 Å, which is different from the Fe–Fe bond (2.30 Å) in the Fe foil and the Mn–Mn bond (2.32 Å) in the Mn foil, suggesting the presence of the Fe–Mn bond. Following the proposed mechanism of dioxygen adsorption on the bimetallic sites, 3C–2e bonds are created in the electrocatalytic reaction (Fig. 14d and e). The inner sphere electron transfer pathway, which includes Mn–O π donation and Fe–O π–π repulsion, is responsible for the transfer of electron density from the bimetallic center to the adsorbed O2 during bond formation. Consequently, π–π repulsion and π donation can be boosted when Fe2+ and Mn2+ are coupled. This assists in the adsorption of O2 on the bimetal active site and stabilizes the 3C–2e bond. Fe and Mn will return to their ground state and cancel the positive charges that have been produced on them as a result of electron donation. Due to this tendency, the bimetal–O2 connection can be easily broken, releasing the reduction products. By weakening the OO bond, the synergistic action of the Fe–Mn bimetallic core facilitates the ORR kinetics.
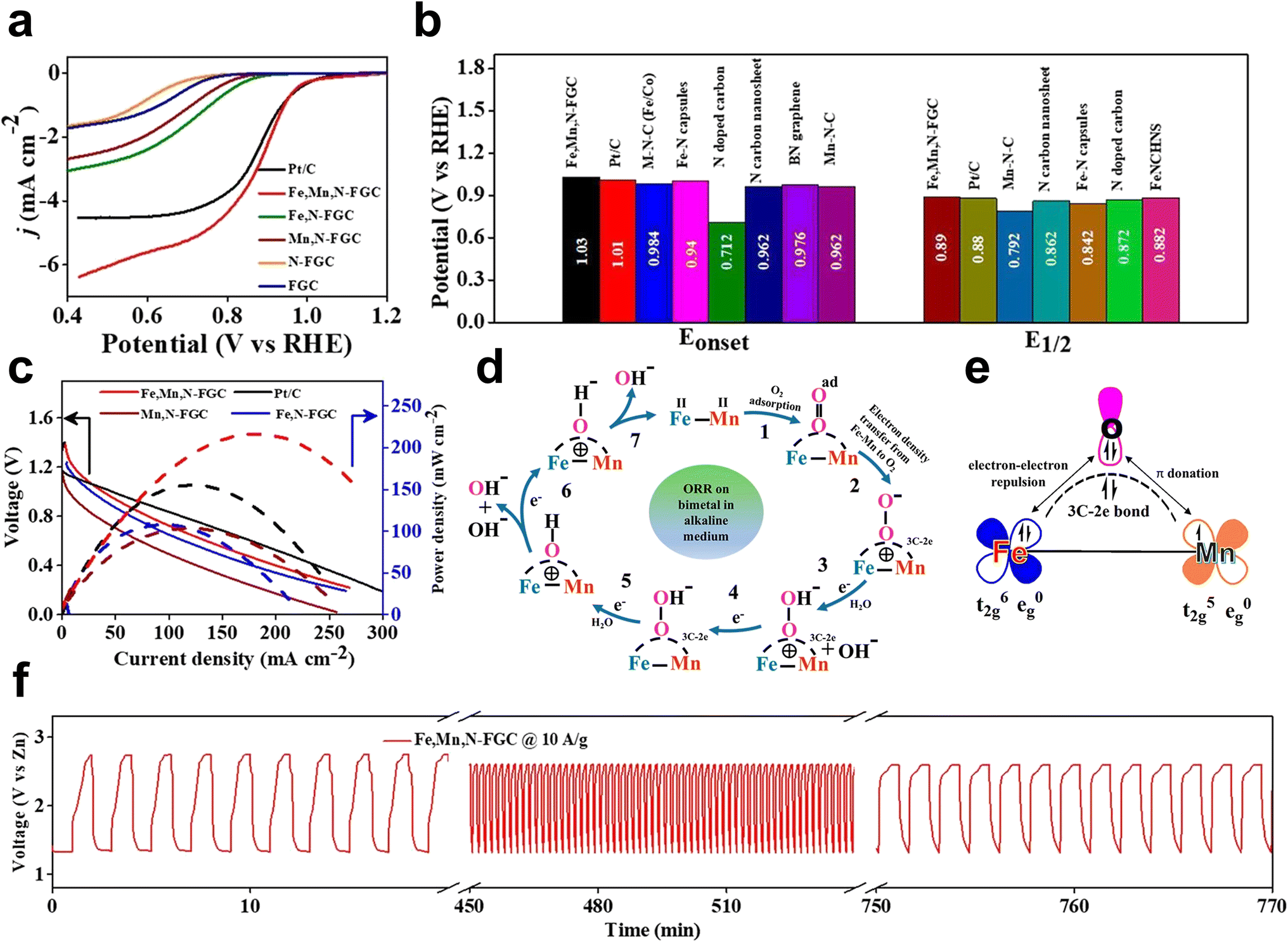 | ||
| Fig. 14 (a) LSV curves of Fe,Mn,N-FGC (0.3:0.3:1:1), Fe,N-FGC, Mn,N-FGC, N-FGC, and FGC catalyst at 1600 rpm in O2-saturated 0.1 M KOH medium; scan rate 10 mV s−1. (b) Comparison plot of Eonset and E1/2 for the Fe,Mn,N-FGC (0.3:0.3:1:1) catalyst with literature and commercial Pt/C. CV response of Fe,Mn,N-FGC catalyst in Ar (argon) (black) and O2 saturated (red). (c) Comparison based on polarization and power density curves of the Zn–air battery with Fe,Mn,N-FGC, Fe,N-FGC, Mn,N-FGC, and Pt/C catalysts. (d) ORR mechanism on bimetal (Fe–Mn) in alkaline medium and (e) corresponding orbital representation of Fe–Mn coupling and O2 adsorption in Fe,Mn,N-FGC (0.3:0.3:1:1) catalyst. (f) Continuous charge–discharge cycle of the battery with the Fe,Mn,N-FGC cathode catalyst at 10 A g−1 for 770 min 53 (reproduced with permission from ref. 53 copyright 2020, ACS). | ||
5. Conclusions and perspectives
In recent decades, significant achievements have been made in synthesizing Fe-based DACs with unique electronic structures and exceptional properties. This review summarizes recent progress on Fe-based DACs for the ORR and applications in PEM fuel cells and Zn–air batteries (details shown in Tables 1 and 2). The fundamentals of the ORR and electrochemical devices involving the ORR process are briefly introduced. Primary strategies for building Fe-based DACs are categorized and discussed with examples, including host–guest, wet chemistry, and template-assisted strategies. Finally, recent progress on Fe-based DACs, such as FeCo–N/C, FeNi–N/C, FeCu–N/C, and FeMn–N/C, was discussed based on the different metal and structural configurations.| Material | E onset | E 1/2 | Electrolyte | Maximum power density | Current density [mA cm−2] | Stability | Ref. |
|---|---|---|---|---|---|---|---|
| FeCo–NC | 0.982 V | 0.842 V | 0.1 M HClO4 | 800 mW cm−2 | ∼800 at 0.6 V | 100 h @ 400 mA cm−2 | 67 |
| (Fe,Co)/N–C | 1.06 V | 0.863 V | 0.1 M HClO4 | 505 mW cm−2 | 550 at 0.6 V | 100 h @ 600 mA cm−2 | 60 |
| FeCoNx/C | 1.02 V | 0.86 V | 0.1 M HClO4 | 819 mW cm−2 | ∼850 at 0.6 V | 5000 cycles CV | 66 |
| FeCu–NC | 0.96 V | 0.882 V | 0.1 M KOH | 590 mW cm−2 | — | — | 69 |
| Planar-like Fe2N6 | 0.84 V | 0.5 M H2SO4 | 845 mW cm−2 | 2112 at 0.4 V | — | 64 |
| Material | E onset | E 1/2 | Overpotential (η10) | ΔE | Open-circuit voltage | Peak power density | Stability | Ref. |
|---|---|---|---|---|---|---|---|---|
| (Fe,Co)/CNT | 1.15 V | 0.954 V | — | — | 1.63 V | 260 mW cm−2 | 6.7 h @ 20 mA cm−2 | 55 |
| NCAG/Fe Cu | 1.07 V | 0.94 V | 376 mV | 0.67 V | 1.51 V | 186 mW cm−2 | 800 cycles @ 5 mA cm−2 | 22 |
| Cu@Fe–N–C | 1.01 V | 0.892 V | — | — | 1.48 V | 92 mW cm−2 | 50000 s @ 20 mA cm−2 |
68 |
| FeMn-DSAC | 1.04 V | 0.922 V | 405 mV | 0.713 V | 1.45 V | 184 mW cm−2 | 80 h @ 2 mA cm−2 | 22 |
| Fe, Mn–N/C | — | 0.904 V | — | — | 1.45 V | 140 mW cm−2 | 23000 s @ 20 mA cm−2 |
43 |
| Fe,Mn,N-FGC | 1.03 V | 0.89 V | — | — | 1.41 V | 220 mW cm−2 | 770 min charge–discharge cycle | 53 |
| Fe,Mn/N–C | — | 0.928 V | 285 mV | 0.692 V | 1.33 V | 160.8 mW cm−2 | 81 h @ 5 mA cm−2 | 59 |
| Fe/Ni–N–C | 1.005 V | 0.861 V | 322 mV | 0.691 V | — | — | 720 min @ 5 mA cm−2 | 44 |
| Fe–NiNC-50 | 1.0 V | 0.85 V | 340 mV | 0.73 V | 1.41 V | ∼220 mW cm−2 | 100 h charge discharge cycle | 31 |
| Fe/Ni–Nx/OC | — | 0.938 V | — | — | 1.525 V | 148 mW cm−2 | 300 charge discharge cycle @ 20 mA cm−2 | 54 |
Fe-based DACs originate from the requirements of further improving the ORR performances of Fe-based SACs. However, Fe-based DACs should be considered as a transition stage and new direction rather than the final step of optimizing Fe-based catalysts. Apart from the above sections, various research works including Fe-based DACs have been developed and achieved, especially the following aspects.
5.1 Metal clusters and nanoparticles can also be introduced in Fe-based DACs
Inspired by the current work on the synergistic catalysis of clusters, nanoparticles and atomic sites, the use of clusters and nanoparticles to enhance the activity and stability of diatomic catalysts has also become a possible strategy.71,72 Li et al. fabricated uniformly dispersed Fe and Co codoped with nitrogen/oxygen into a three-dimensional highly porous tungsten–carbon matrix in aqueous solvents by a simple method (Fig. 15a). The enhanced electrocatalytic performance can be attributed to the synergistic effects of highly exposed Fe, Co–N/Ox and Fe/Co nanoparticles of the active substance tungsten–carbon structure, optimized electronic structure, self-generated carbon nanotubes, and enriched hierarchical porous interfaces.72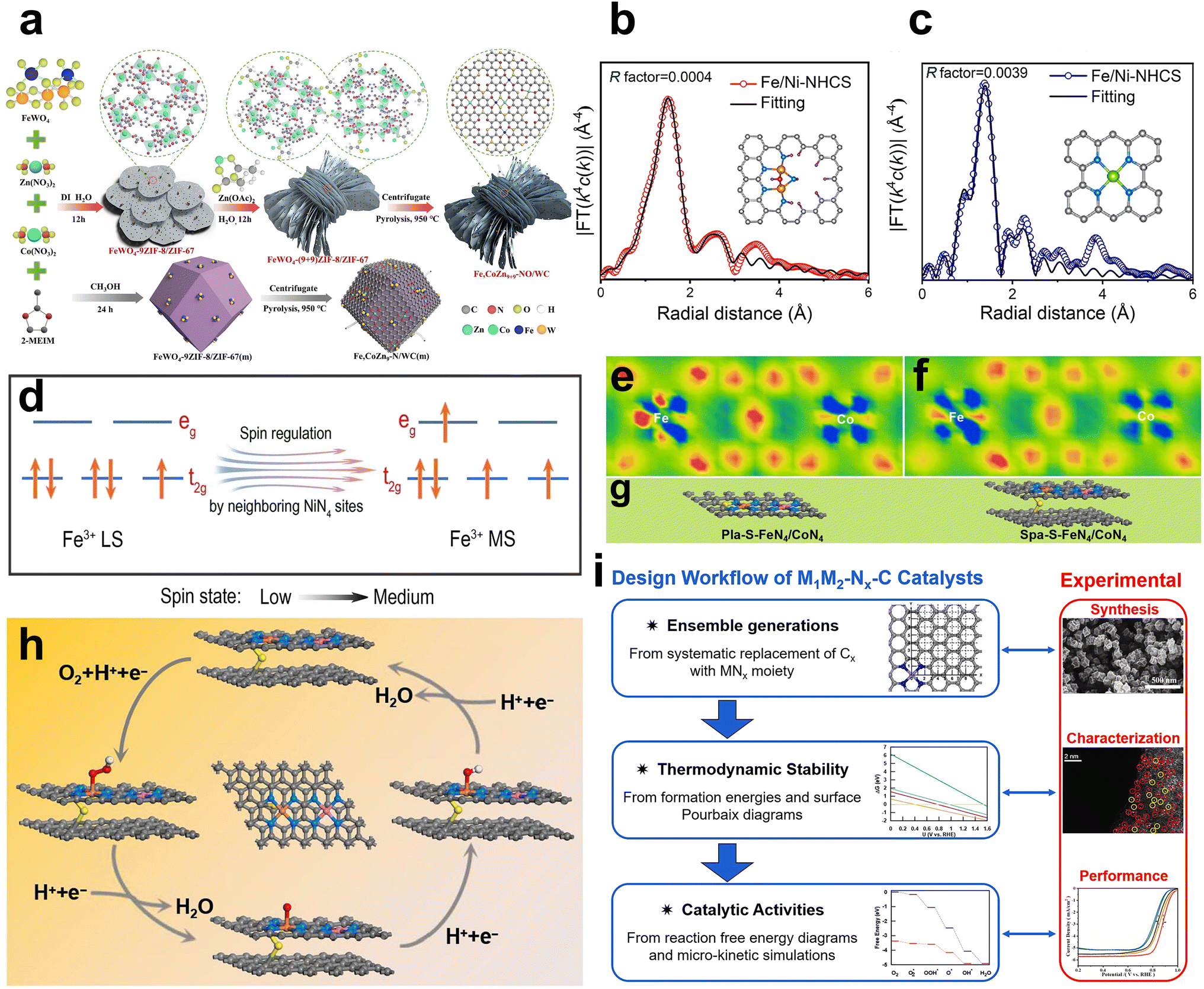 | ||
| Fig. 15 (a) Schematic diagram for the synthetic procedure of the Fe,CoZn9+9-NO/WC catalyst72 (reproduced with permission from ref. 72 copyright 2023, Elsevier). (b) FT-EXAFS fitting curves of Fe/Ni–NHCS at the Fe K-edge. (c) FT-EXAFS fitting curves of Fe/Ni–NHCS at the Ni K-edge. (d) The calculated number of unpaired electrons in the Fe-3d orbitals of Fe–NHCS and Fe/Ni–NHCS73 (reproduced with permission from ref. 73 copyright 2023, Elsevier). Calculated charge density differences for (e) Pla-S-FeN4/CoN4 and (f) Spa-S-FeN4/CoN4. (g) Model structures for calculations. (h) Proposed ORR mechanism on the Spa-S-FeN4/CoN4 site74 (reproduced with permission from ref. 74 copyright 2023, ACS). (i) Computational framework used to systematically study DMSCs for their catalytic activity toward ORR76 (reproduced with permission from ref. 76 copyright 2023, ACS). | ||
5.2 Combination of SACs and DACs
Based on Fe-based DACs, triatomic active sites have also been studied and developed to achieve better performance.73 Huang et al. reported that spin-state modulation of iron atom pairs by neighboring nickel sites improves ORR activity and zinc–air battery performance. N-doped hollow carbon spheres (Fe/Ni-NHCS) accommodated with adjacent NiN4 monometallic and Fe2N5 bimetallic atoms were constructed using hollow polymer spheres, which were deduced by XAS fitting (Fig. 15b and c). Both theoretical calculations and magnetic measurements indicate that the introduction of NiN4 plays an important role in optimizing the electronic spin state of Fe2N5 and lowering the energy barrier for the adsorption and conversion of oxygen-containing intermediates (Fig. 15d).735.3 Heteroatom (e.g., S, P and O) doping in Fe-based DACs
Similar to doping S, P and O in Fe-based SACs to achieve optimal modulation of the electronic structure, this strategy has also been researched and used for tailoring Fe-based DACs.72,74,75 For instance, Zeng et al. constructed highly active Fe2N5P sites on P-doped hollow carbon supports. Theoretical calculations suggest that the presence of P atoms on the Fe2N5P sites is favorable for the formation of OOH* and consequently enhances their activity.74 Meanwhile, Dai et al. reported S-bridge ligands to modulate the coordination environment of the Fe–Co dual active center (Spa-S-FeN4/CoN4) with excellent PEMFC performance. DFT calculations indicated that the adjacent S ligand and Co–N4 sites can synergistically regulate the d-orbital electronic structure of the Fe–N4 site (Fig. 15e–g). Combined with the DFT calculations and experimental results, the ORR mechanism on Spa-S-FeN4/CoN4 was proposed, as shown in Fig. 15h.745.4 Theoretical understanding in the design of Fe-based DACs
Theoretical understanding plays an essential role in guiding the design and development of Fe-based DACs. By utilizing theoretical methods, researchers can gain deeper insights into the electronic and structural properties of these catalysts, as well as predict their catalytic performance.76,77 For example, Hu et al. established a computational framework that incorporates configuration generation, phase diagram construction, and reaction free energy calculations to develop deeper insights into the active site structures and catalytic mechanisms on DACs (Fig. 15i). Thirty-one configurations of the Fe–Cu model were generated by populating the hexagonal graphene lattice and scrutinizing the atomic arrangements under reaction conditions. Experimental observations indicate that the *OH intermediates preferentially cover the Fe sites while leaving the Cu sites unoccupied over a wide potential window. Investigations confirmed that the ORR on Fe–Cu DACs follows a bimolecular pathway, with the desorption of *OH serving as the RDS. Furthermore, in-depth analysis unveiled a direct relationship between the limiting potential on Fe and the magnetic moment, suggesting that a shorter separation between the two metal sites contributes to improved catalytic performance.76To further enhance the activity and durability of Fe-based DACs for practical applications, several perspectives are proposed as follows:
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Fundamental Research Funds for the Central Universities (N2202012).References
- D. Zhao, Z. W. Zhuang, X. Cao, C. Zhang, Q. Peng, C. Chen and Y. D. Li, Atomic site electrocatalysts for water splitting, oxygen reduction and selective oxidation, Chem. Soc. Rev., 2020, 49(7), 2215–2264 RSC.
- J. W. Hu, W. Liu, C. C. Xin, J. Y. Guo, X. S. Cheng, J. Z. Wei, C. Hao, G. F. Zhang and Y. T. Shi, Carbon-based single atom catalysts for tailoring the ORR pathway: a concise review, J. Mater. Chem. A, 2021, 9(44), 24803–24829 RSC.
- J. L. Qiao, Y. Y. Liu, F. Hong and J. J. Zhang, A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels, Chem. Soc. Rev., 2014, 43(2), 631–675 RSC.
- V. Balzani, A. Credi and M. Venturi, Photochemical conversion of solar energy, ChemSusChem, 2008, 1(1–2), 26–58 CrossRef CAS PubMed.
- M. Hereher and A. M. El Kenawy, Exploring the potential of solar, tidal, and wind energy resources in Oman using an integrated climatic-socioeconomic approach, Renewable Energy, 2020, 161, 662–675 CrossRef.
- Y. Zhang, G. Wang, F. Yu, G. Xu, Z. Li, M. Zhu, Z. Yue, M. Wu, H.-K. Liu, S.-X. Dou and C. Wu, Highly reversible and dendrite-free Zn electrodeposition enabled by a thin metallic interfacial layer in aqueous batteries, Chem. Eng. J., 2021, 416, 128062 CrossRef CAS.
- H. Y. Li, W. J. Wan, X. J. Liu, H. X. Liu, S. B. Shen, F. Iv and J. Luo, Poplar-Catkin-Derived N, P Co-doped Carbon Microtubes as Efficient Oxygen Electrocatalysts for Zn-Air Batteries, ChemElectroChem, 2018, 5(7), 1113–1119 CrossRef CAS.
- J. F. Xie and Y. B. Wang, Recent Development of CO2 Electrochemistry from Li-CO2 Batteries to Zn-CO2 Batteries, Acc. Chem. Res., 2019, 52(6), 1721–1729 CrossRef CAS PubMed.
- K. Q. Zhong, M. Li, Y. Yang, H. G. Zhang, B. P. Zhang, J. F. Tang, J. Yan, M. H. Su and Z. Q. Yang, Nitrogen-doped biochar derived from watermelon rind as oxygen reduction catalyst in air cathode microbial fuel cells, Appl. Energy, 2019, 242, 516–525 CrossRef CAS.
- X. F. Lu, B. Y. Xia, S. Q. Zang and X. W. Lou, Metal-Organic Frameworks Based Electrocatalysts for the Oxygen Reduction Reaction, Angew. Chem., Int. Ed., 2020, 59(12), 4634–4650 CrossRef CAS PubMed.
- L. Du, G. X. Zhang, X. H. Liu, A. Hassanpour, M. Dubois, A. C. Tavares and S. H. Sun, Biomass-derived nonprecious metal catalysts for oxygen reduction reaction: The demand-oriented engineering of active sites and structures, Carbon Energy, 2020, 2(4), 561–581 CrossRef CAS.
- P. Lang, N. Yuan, Q. Jiang, Y. Zhang and J. Tang, Recent Advances and Prospects of Metal-Based Catalysts for Oxygen Reduction Reaction, Energy Technol., 2020, 8(3), 1900984 CrossRef CAS.
- M. Liu, Z. Zhao, X. Duan and Y. Huang, Nanoscale Structure Design for High-Performance Pt-Based ORR Catalysts, Adv. Mater., 2019, 31(6), 1802234 CrossRef PubMed.
- Z. Fang, P. Li and G. Yu, Gel Electrocatalysts: An Emerging Material Platform for Electrochemical Energy Conversion, Adv. Mater., 2020, 32(39), 2003191 CrossRef CAS PubMed.
- H. M. Xu, S. Q. Ci, Y. C. Ding, G. X. Wang and Z. H. Wen, Recent advances in precious metal-free bifunctional catalysts for electrochemical conversion systems, J. Mater. Chem. A, 2019, 7(14), 8006–8029 RSC.
- A. Han, Z. Zhang, J. Yang, D. Wang and Y. Li, Carbon-Supported Single-Atom Catalysts for Formic Acid Oxidation and Oxygen Reduction Reactions, Small, 2021, 17(16), 2004500 CrossRef CAS PubMed.
- L. Liu, Platinum group metal free nano-catalysts for proton exchange membrane water electrolysis, Curr. Opin. Chem. Eng., 2021, 34, 100743 CrossRef.
- H. Y. Kim, J. M. Kim, Y. Ha, J. Woo, A. Byun, T. J. Shin, K. H. Park, H. Y. Jeong, H. Kim, J. Y. Kim and S. H. Joo, Activity Origin and Multifunctionality of Pt-Based Intermetallic Nanostructures for Efficient Electrocatalysis, ACS Catal., 2019, 9(12), 11242–11254 CrossRef CAS.
- S. T. Hunt, M. Milina, A. C. Alba-Rubio, C. H. Hendon, J. A. Dumesic and Y. Roman-Leshkov, Self-assembly of noble metal monolayers on transition metal carbide nanoparticle catalysts, Science, 2016, 352(6288), 974–978 CrossRef CAS PubMed.
- Y. Wang, D. Wang and Y. Li, Rational Design of Single-Atom Site Electrocatalysts: From Theoretical Understandings to Practical Applications, Adv. Mater., 2021, 33(34), 2008151 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Single-atom catalysis of CO oxidation using Pt1/FeOx, Nat. Chem., 2011, 3(8), 634–641 CrossRef CAS PubMed.
- T. He, Y. Chen, Q. M. Liu, X. W. Song, H. T. Liu, M. Liu, Y. N. Liu, Y. Zhang, X. P. Quyang and S. W. Chen, Theory-Guided Regulation of FeN4 Spin State by Neighboring Cu Atoms for Enhanced Oxygen Reduction Electrocatalysis in Flexible Metal-Air Batteries, Angew. Chem., Int. Ed., 2022, 61(27), e202201007 CrossRef CAS PubMed.
- H. Q. Yang, Z. W. Chen, S. Q. Kou, G. L. Lu, D. H. Chen and Z. N. Liu, Carbon-supported catalysts with atomically dispersed metal sites for oxygen electroreduction: present and future perspectives, J. Mater. Chem. A, 2021, 9(29), 15919–15936 RSC.
- O. Yadorao Bisen, A. Kumar Yadav, B. Pavithra and K. Kar Nanda, Electronic structure modulation of molybdenum-iron double-atom catalyst for bifunctional oxygen electrochemistry, Chem. Eng. J., 2022, 449, 137705 CrossRef CAS.
- X. Wei, X. Luo, H. Wang, W. Gu, W. Cai, Y. Lin and C. Zhu, Highly-defective Fe-N-C catalysts towards pH-Universal oxygen reduction reaction, Appl. Catal., B, 2020, 263, 118347 CrossRef CAS.
- T. K. Maiti, J. Singh, P. Dixit, J. Majhi, S. Bhushan, A. Bandyopadhyay and S. Chattopadhyay, Advances in perfluorosulfonic acid-based proton exchange membranes for fuel cell applications: A review, Chem. Eng. J. Adv., 2022, 12, 100372 CrossRef CAS.
- V. Das, S. Padmanaban, K. Venkitusamy, R. Selvamuthukumaran, F. Blaabjerg and P. Siano, Recent advances and challenges of fuel cell based power system architectures and control - A review, Renewable Sustainable Energy Rev., 2017, 73, 10–18 CrossRef CAS.
- M. K. Debe, Electrocatalyst approaches and challenges for automotive fuel cells, Nature, 2012, 486(7401), 43–51 CrossRef CAS PubMed.
- W. Gao, Q. Yin, X. Zhang, C. Zhang, Y. Lei and C. Wang, Low-platinum dissymmetric membrane electrode assemblies for fuel cells suitable for a variety of external humidification conditions, J. Power Sources, 2022, 547, 232013 CrossRef CAS.
- T. He, Y. Chen, Q. Liu, B. Lu, X. Song, H. Liu, M. Liu, Y.-N. Liu, Y. Zhang, X. Ouyang and S. Chen, Theory-Guided Regulation of FeN4 Spin State by Neighboring Cu Atoms for Enhanced Oxygen Reduction Electrocatalysis in Flexible Metal–Air Batteries, Angew. Chem., Int. Ed., 2022, 61(27), e202201007 CrossRef CAS PubMed.
- X. Zhu, D. Zhang, C.-J. Chen, Q. Zhang, R.-S. Liu, Z. Xia, L. Dai, R. Amal and X. Lu, Harnessing the interplay of Fe–Ni atom pairs embedded in nitrogen-doped carbon for bifunctional oxygen electrocatalysis, Nano Energy, 2020, 71, 104597 CrossRef CAS.
- J. T. Zhang, L. T. Qu, G. Q. Shi, J. Y. Liu, J. F. Chen and L. M. Dai, N,P-Codoped Carbon Networks as Efficient Metal-free Bifunctional Catalysts for Oxygen Reduction and Hydrogen Evolution Reactions, Angew. Chem., Int. Ed., 2016, 55(6), 2230–2234 CrossRef CAS PubMed.
- A. Morozan, P. Jegou, B. Jousselme and S. Palacin, Electrochemical performance of annealed cobalt-benzotriazole/CNTs catalysts towards the oxygen reduction reaction, Phys. Chem. Chem. Phys., 2011, 13(48), 21600–21607 RSC.
- J. Liu, M. Jiao, L. Lu, H. M. Barkholtz, Y. Li, Y. Wang, L. Jiang, Z. Wu, D.-j. Liu, L. Zhuang, C. Ma, J. Zeng, B. Zhang, D. Su, P. Song, W. Xing, W. Xu, Y. Wang, Z. Jiang and G. Sun, High performance platinum single atom electrocatalyst for oxygen reduction reaction, Nat. Commun., 2017, 8(1), 15938 CrossRef CAS PubMed.
- J. H. Zagal and M. T. M. Koper, Reactivity Descriptors for the Activity of Molecular MN4 Catalysts for the Oxygen Reduction Reaction, Angew. Chem., Int. Ed., 2016, 55(47), 14510–14521 CrossRef CAS PubMed.
- M. M. Tong, F. F. Sun, Y. Xie, Y. Wang, Y. Q. Yang, C. G. Tian, L. Wang and H. G. Fu, Operando Cooperated Catalytic Mechanism of Atomically Dispersed Cu-N-4 and Zn-N-4 for Promoting Oxygen Reduction Reaction, Angew. Chem., Int. Ed., 2021, 60(25), 14005–14012 CrossRef CAS PubMed.
- F. Xiao, Y.-C. Wang, Z.-P. Wu, G. Chen, F. Yang, S. Zhu, K. Siddharth, Z. Kong, A. Lu, J.-C. Li, C.-J. Zhong, Z.-Y. Zhou and M. Shao, Recent advances in electrocatalysts for proton exchange membrane fuel cells and alkaline membrane fuel cells, Adv. Mater., 2021, 33(50), 2006292 CrossRef CAS PubMed.
- C. Wang, X. Zhang and Y. Liu, Promotion of multi-electron transfer for enhanced photocatalysis: A review focused on oxygen reduction reaction, Appl. Surf. Sci., 2015, 358, 28–45 CrossRef CAS.
- Y. Y. Pang, H. Xie, Y. Sun, M. M. Titirici and G. L. Chai, Electrochemical oxygen reduction for H2O2 production: catalysts, pH effects and mechanisms, J. Mater. Chem. A, 2020, 8(47), 24996–25016 RSC.
- B. B. Blizanac, P. N. Ross and N. M. Markovic, Oxygen electroreduction on Ag(111): The pH effect, Electrochim. Acta, 2007, 52(6), 2264–2271 CrossRef CAS.
- L. B. Li, K. Yuan and Y. W. Chen, Breaking the scaling relationship limit: from single-atom to dual-atom catalysts, Acc. Mater. Res., 2022, 3(6), 584–596 CrossRef CAS.
- R. Cepitis, N. Kongi, J. Rossmeisl and V. Ivanistsev, Surface curvature effect on dual-atom site oxygen electrocatalysis, ACS Energy Lett., 2023, 8(3), 1330–1335 CrossRef CAS PubMed.
- S. P. Gong, C. L. Wang, P. Jiang, L. Hu, H. Lei and Q. W. Chen, Designing highly efficient dual-metal single-atom electrocatalysts for the oxygen reduction reaction inspired by biological enzyme systems, J. Mater. Chem. A, 2018, 6(27), 13254–13262 RSC.
- H. Li, J. Wang, R. Qi, Y. Hu, J. Zhang, H. Zhao, J. Zhang and Y. Zhao, Enhanced Fe 3d delocalization and moderate spin polarization in FeNi atomic pairs for bifunctional ORR and OER electrocatalysis, Appl. Catal., B, 2021, 285, 119778 CrossRef CAS.
- Z. P. Yu, C. W. Si, A. P. LaGrow, Z. X. Tai, W. A. Caliebe, A. Tayal, M. J. Sampaio, J. P. S. Sousa, I. Amorim, A. Araujo, L. J. Meng, J. L. Faria, J. Y. Xu, B. Li and L. F. Liu, Iridium-iron diatomic active sites for efficient bifunctional oxygen electrocatalysis, ACS Catal., 2022, 12(15), 9397–9409 CrossRef CAS.
- Y. He, X. Yang, Y. Li, L. Liu, S. Guo, C. Shu, F. Liu, Y. Liu, Q. Tan and G. Wu, Atomically dispersed Fe–Co dual metal sites as bifunctional oxygen electrocatalysts for rechargeable and flexible Zn–air batteries, ACS Catal., 2022, 12(2), 1216–1227 CrossRef CAS.
- H. Z. Yang, H. Huang, Q. Wang, L. Shang, T. R. Zhang and S. G. Wang, Fe, Cu dual-metal single atom catalyst on commercial carbon black for efficient oxygen reduction reaction, J. Mater. Chem. A, 2023, 11(12), 6191–6197 RSC.
- Y. Rao, Y. Wu, X. Dai, Y.-W. Zhang, G. Qin, W. Qi and S. Li, A Tale of two sites: neighboring atomically dispersed pt sites cooperatively remove trace H2 in CO-rich stream, Small, 2022, 18(51), 2204611 CrossRef CAS PubMed.
- Y. Yao, T. Jiang, S. Y. Lim, C. Frandsen, Z. Li, Y. Dou, F. Wu, J. Qin, J. Zou, E. Stamate and W. Zhang, Universal synthesis of half-metallic diatomic catalysts for efficient oxygen reduction electrocatalysis, Small, 2023, 2304655 CrossRef CAS PubMed.
- P. Zhu, X. Xiong, X. L. Wang, C. L. Ye, J. Z. Li, W. M. Sun, X. H. Sun, J. J. Jiang, Z. B. Zhuang, D. S. Wang and Y. D. Li, Regulating the FeN4 moiety by constructing Fe-Mo dual-metal atom sites for efficient electrochemical oxygen reduction, Nano Lett., 2022, 22(23), 9507–9515 CrossRef CAS PubMed.
- R. Liu, R. Tang, J. Feng and T. Meng, Mechanistic insight into the Fe-atom-pairs for breaking the scaling relation in ORR, Chem. Eng. J., 2023, 470, 144261 CrossRef CAS.
- Z. Chen, X. Liao, C. Sun, K. Zhao, D. Ye, J. Li, G. Wu, J. Fang, H. Zhao and J. Zhang, Enhanced performance of atomically dispersed dual-site Fe-Mn electrocatalysts through cascade reaction mechanism, Appl. Catal., B, 2021, 288, 120021 CrossRef CAS.
- S. Sarkar, A. Biswas, T. Purkait, M. Das, N. Kamboj and R. S. Dey, Unravelling the role of Fe–Mn binary active sites electrocatalyst for efficient oxygen reduction reaction and rechargeable Zn-air batteries, Inorg. Chem., 2020, 59(7), 5194–5205 CrossRef CAS PubMed.
- Z. Zhu, H. Yin, Y. Wang, C.-H. Chuang, L. Xing, M. Dong, Y.-R. Lu, G. Casillas-Garcia, Y. Zheng, S. Chen, Y. Dou, P. Liu, Q. Cheng and H. Zhao, Coexisting single-atomic Fe and Ni sites on hierarchically ordered porous carbon as a highly efficient ORR electrocatalyst, Adv. Mater., 2020, 32(42), 2004670 CrossRef CAS PubMed.
- J. Wang, W. Liu, G. Luo, Z. J. Li, C. Zhao, H. R. Zhang, M. Z. Zhu, Q. Xu, X. Q. Wang, C. M. Zhao, Y. T. Qu, Z. K. Yang, T. Yao, Y. F. Li, Y. Lin, Y. Wu and Y. D. Li, Synergistic effect of well-defined dual sites boosting the oxygen reduction reaction, Energy Environ. Sci., 2018, 11(12), 3375–3379 RSC.
- Y. Wu, C. Ye, L. Yu, Y. Liu, J. Huang, J. Bi, L. Xue, J. Sun, J. Yang, W. Zhang, X. Wang, P. Xiong and J. Zhu, Soft template-directed interlayer confinement synthesis of a Fe-Co dual single-atom catalyst for Zn-air batteries, Energy Storage Mater., 2022, 45, 805–813 CrossRef.
- H. Li, Y. Wen, M. Jiang, Y. Yao, H. Zhou, Z. Huang, J. Li, S. Jiao, Y. Kuang and S. Luo, Understanding of neighboring Fe-N4-C and Co-N4-C dual active centers for oxygen reduction reaction, Adv. Funct. Mater., 2021, 31(22), 2011289 CrossRef CAS.
- Q. H. Nguyen, K. Im and J. Kim, Synthesis of hollow Fe, Co, and N-doped carbon catalysts from conducting polymer-metal-organic-frameworks core-shell particles for their application in an oxygen reduction reaction, Int. J. Hydrogen Energy, 2022, 47(57), 24169–24178 CrossRef CAS.
- G. Yang, J. Zhu, P. Yuan, Y. Hu, G. Qu, B.-A. Lu, X. Xue, H. Yin, W. Cheng, J. Cheng, W. Xu, J. Li, J. Hu, S. Mu and J.-N. Zhang, Regulating Fe-spin state by atomically dispersed Mn-N in Fe-N-C catalysts with high oxygen reduction activity, Nat. Commun., 2021, 12(1), 1734 CrossRef CAS PubMed.
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li, W. Chen, C. Jia, T. Yao, S. Wei, Y. Wu and Y. Li, Design of N-coordinated dual-metal sites: a stable and active Pt-free catalyst for acidic oxygen reduction reaction, J. Am. Chem. Soc., 2017, 139(48), 17281–17284 CrossRef CAS PubMed.
- H. Li, S. Di, P. Niu, S. Wang, J. Wang and L. Li, A durable half-metallic diatomic catalyst for efficient oxygen reduction, Energy Environ. Sci., 2022, 15(4), 1601–1610 RSC.
- Y. D. Zhou, W. Yang, W. Utetiwabo, Y. M. Lian, X. Yin, L. Zhou, P. W. Yu, R. J. Chen and S. R. Sun, Revealing of active sites and catalytic mechanism in N-coordinated Fe, Ni dual-doped carbon with superior acidic oxygen reduction than single-atom catalyst, J. Phys. Chem. Lett., 2020, 11(4), 1404–1410 CrossRef CAS PubMed.
- Y. B. Wang, D. P. Yang, A. L. Chen and Q. F. Yi, Fe/Co-loaded hollow carbon sphere nanocomposites as excellent cathodic catalysts of Zn-air battery, J. Electrochem. Soc., 2021, 168(9), 090512 CrossRef CAS.
- N. Zhang, T. Zhou, J. Ge, Y. Lin, Z. Du, C. a. Zhong, W. Wang, Q. Jiao, R. Yuan, Y. Tian, W. Chu, C. Wu and Y. Xie, High-density planar-like Fe2N6 structure catalyzes efficient oxygen reduction, Matter, 2020, 3(2), 509–521 CrossRef.
- X. Wang, Z. Chen, Z. Han, H. Gai, J. Zhou, Y. Wang, P. Cui, J. Ge, W. Xing, X. Zheng, M. Huang and H. Jiang, Manipulation of new married edge-adjacent Fe2N5 catalysts and identification of active species for oxygen reduction in wide pH range, Adv. Funct. Mater., 2022, 32(18), 2111835 CrossRef CAS.
- M. Xiao, Y. Chen, J. Zhu, H. Zhang, X. Zhao, L. Gao, X. Wang, J. Zhao, J. Ge, Z. Jiang, S. Chen, C. Liu and W. Xing, Climbing the apex of the ORR Volcano plot via binuclear site construction: electronic and geometric engineering, J. Am. Chem. Soc., 2019, 141(44), 17763–17770 CrossRef CAS PubMed.
- Y. B. Chen, J. J. Li, Y. P. Zhu, J. Zou, H. Zhao, C. Chen, Q. Q. Cheng, B. Yang, L. L. Zou, Z. Q. Zou and H. Yang, Vicinal Co atom-coordinated Fe-N-C catalysts to boost the oxygen reduction reaction, J. Mater. Chem. A, 2022, 10(18), 9886–9891 RSC.
- Z. Wang, H. Jin, T. Meng, K. Liao, W. Meng, J. Yang, D. He, Y. Xiong and S. Mu, Fe, Cu-coordinated ZIF-derived carbon framework for efficient oxygen reduction reaction and zinc–air batteries, Adv. Funct. Mater., 2018, 28(39), 1802596 CrossRef.
- Z. Xiao, P. Sun, Z. Qiao, K. Qiao, H. Xu, S. Wang and D. Cao, Atomically dispersed Fe-Cu dual-site catalysts synergistically boosting oxygen reduction for hydrogen fuel cells, Chem. Eng. J., 2022, 446, 137112 CrossRef CAS.
- F. Kong, M. Wang, Y. Huang, G. Meng, M. Chen, H. Tian, Y. Chen, C. Chen, Z. Chang, X. Cui and J. Shi, Cu-N-bridged Fe-3d electron state regulations for boosted oxygen reduction in flexible battery and PEMFC, Energy Storage Mater., 2023, 54, 533–542 CrossRef.
- Q. Xiong, J. Zheng, B. Liu, Y. Liu, H. Li and M. Yang, In situ self-templating construction of FeNi/N co-doped 3D porous carbon from bimetallic ions-coordinated porous organic polymer for rechargeable zinc-air batteries, Appl. Catal., B, 2023, 321, 122067 CrossRef CAS.
- J. Xue, Z. Liu, Y. Fan, R. Wang and Y. Li, ZIF-derived Fe,Co coordinated N/O-codoped three-dimensional tungsten–carbon matrix for the performance-enhanced zinc-air flow battery and water splitting, Chem. Eng. J., 2023, 146502 CrossRef CAS.
- C. Zhang, X. Wang, Z. Ma, H. Yao, H. Liu, C. Li, J. Zhou, R. Xu, X. Zheng, H. Wang, Q. Li, M. Gu, H. Jiang and M. Huang, Spin state modulation on dual Fe center by adjacent Ni sites enabling the boosted activities and ultra-long stability in Zn-air batteries, Sci. Bull., 2023, 68(18), 2042–2053 CrossRef CAS.
- Q. Miao, Z. Chen, X. Li, M. Liu, G. Liu, X. Yang, Z. Guo, C. Yu, Q. Xu and G. Zeng, Construction of Catalytic Fe2N5P Sites in Covalent Organic Framework-Derived Carbon for Catalyzing the Oxygen Reduction Reaction, ACS Catal., 2023, 13(16), 11127–11135 CrossRef CAS.
- F. Liu, L. Shi, X. Lin, B. Zhang, Y. Long, F. Ye, R. Yan, R. Cheng, C. Hu, D. Liu, J. Qiu and L. Dai, Fe/Co dual metal catalysts modulated by S-ligands for efficient acidic oxygen reduction in PEMFC, Sci. Adv., 2023, 9(23), eadg0366 CrossRef CAS PubMed.
- C. Brea and G. Hu, Mechanistic insight into dual-metal-site catalysts for the oxygen reduction reaction, ACS Catal., 2023, 13(7), 4992–4999 CrossRef CAS.
- Y. L. Sun, J. Wang, Q. Liu, M. R. Xia, Y. F. Tang, F. M. Gao, Y. L. Hou, J. Tse and Y. F. Zhao, Itinerant ferromagnetic half metallic cobalt-iron couples: promising bifunctional electrocatalysts for ORR and OER, J. Mater. Chem. A, 2019, 7(47), 27175–27185 RSC.
| This journal is © The Royal Society of Chemistry 2024 |