
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
p-block doped semi-metallic xenes as highly selective and efficient transition-metal free single atom catalysts for electrochemical CO reduction†
Huong T. D.
Bui
and
Tore
Brinck
 *
*
Department of Chemistry, CBH, KTH Royal Institute of Technology, Stockholm SE-100 44, Sweden. E-mail: tore@kth.se
First published on 15th December 2023
Abstract
The development of robust and inexpensive catalysts for the electrochemical CO reduction reaction (CORR) is key for sustainable production of valuable chemicals, yet it remains a long-standing challenge. Herein, we conduct a systematic theoretical investigation on p-block doping of semi-metallic xene monolayers to afford transition metal free catalysts for the CORR. Silicene (Si) and germanene (Ge) are suitable platforms for capturing the dopant (B/Al) to ensure high stability. Our single atom catalysts (SACs) are promising candidates for CORR due to their favorable initial CO adsorption and the selectivity of CO reduction over H2 evolution. B@Si, Al@Si, Al@Ge and B@Ge exhibit superior CORR catalytic activity with a limiting potential UL of 0.04, −0.39, −0.40, and −0.40 V, respectively. Notably, B@Si is identified as the best CORR electrocatalyst with an overpotential of less than 0.1 V. B@Si, Al@Si, Al@Ge exhibit high CORR selectivity towards CH3OH production, whereas B@Ge is predicted to form mainly CH4. The fundamental principles behind the outstanding CORR catalytic enhancement are disclosed by analyzing the structural and electronic configurations of two key intermediates, CO* and CHO*. CO* binds the dopant with moderate strength through a combination of σ-donation and π-backdonation unique for a transition metal free catalyst, whereas CHO* adsorbs strongly to the surface by the simultaneous binding to two neighboring atomic sites; consequently, the binding of the two intermediates breaks the scaling relation that limits the CORR activity of conventional catalysts. The optimal adsorption behaviors are attributed to the surface charge modulation induced by the substitutional doping. Hence, these findings may facilitate rational design of xene-based SACs for CORR and advance other catalytic applications.
1. Introduction
Given the advancement of industrialized society, the rampant consumption of carbon-based fossil fuels (coal, oil, and natural gas) has led to excessive release of carbon dioxide (CO2) into the atmosphere.1 Electrochemical conversions of CO2 (CO2RR) and CO (CORR) into sustainable fuels and chemicals, powered by renewable electricity, have emerged as promising long-term solutions to maintain a global carbon balance and address energy and environmental issues.1,2 While CO2RR towards more-than-2e− (>2e−) products still has poor energy efficiency and low selectivity, CORR has recently gained attention owing to its exceptional selectivity towards desired products.2–5 Nevertheless, the search for efficient, selective, robust, stable, and economical electrocatalysts for CO electroreduction in ambient aqueous operation conditions is not aligned with its potential. Additionally, considering CO* as a key intermediate of CO2RR, there are common reaction pathways between CO2RR and CORR.3–5 From a broader perspective, new catalyst design strategies and mechanistic insights for direct CORR can greatly benefit the enhancement and application of the CO2RR.Since the successful exfoliation of graphene and the subsequent exploration of its unique physical and chemical attributes, the discovery of crystalline two-dimensional (2D) materials has been at the forefront of the developments in catalysis and other applied fields.6–8 Very recently, silicene (Si) and germanene (Ge), two members of the 2D semi-metallic xene family (“x” referring to main group IV in this study), have been successfully fabricated and theoretically investigated.9–13 Of particular interest is their markedly enhanced chemical reactivities in comparison to graphene, positioning them as potential alternatives to graphene.14–18 This distinct advantage originates from their buckled honeycomb structures, where Si atoms (silicene) and Ge atoms (germanene) exhibit mixed sp2–sp3 hybridization, in contrast to the stable sp2 hybridization of C atoms (graphene), resulting in lowered aromaticity.
To date, incorporating foreign atoms into the 2D materials through doping engineering has been identified as a promising strategy to enhance catalytic efficiency.19–26 Single atom catalysts (SACs), a novel subgroup of heterogeneous catalysts, has aroused considerable attention due to their high specific areas, maximum atom utilization efficiency, unique electronic structure, coordination unsaturated surface atoms, etc., which boosts catalytic activities.27–29 Several previous theoretical reports have highlighted Si and Ge monolayers as appropriate matrices for anchoring adatoms and the resulting Si/Ge-based SACs often exhibit distinguished catalytic performances.18,30–32 Specifically, P doping can promote H2 evolution reaction (HER) performance of both Si and Ge.18 Fe/Ru on single-layer Ge show efficacy in removing CO contamination in H2.31 Pt/Au-decorated Si are found to be high-activity catalysts for CO oxidation.33,34 Notably, we have found that the integration of the non-transition metals (non-TM) B and Al into Si enables CO chemisorption,35 which is in contrast to CO physisorption on pristine Si.36,37 This stimulated us to extensively investigate further CO reduction on various xene-based SACs. Our observation is pivotal as CO adsorption is regarded as a preliminary step, influencing catalytic CORR efficiency. Moreover, to the best of our knowledge, no prior endeavors have explored CORR on potential Si/Ge-related SACs.
Here, for the first time, we thoroughly examine the catalytic proficiency of single p-block non-TM atom doped semi-metallic xenes (Si and Ge) for CORR. Two doping heteroatoms (B/Al) from the third main groups (IIIA) can be deposited stably into monovalent Si/Ge to build four different p-type materials with no remaining lone-pair electron on the dopants. Inspired by the unique combination of σ-donation (C to B) and π-backdonation (B to C) that we previously observed in induced pyramidal B@(Si-/Ge-)3 motifs for CO adsorption,35 we find that our new systems are capable of inert CO binding and activation without transition metal (TM) participation. All B/Al@xene structures not only effectively hamper the competitive HER, but also demonstrate superior CORR efficiency and high selectivity towards CH3OH via thermodynamically favored pathways with ultra-low limiting potentials. B@Si is anticipated to be the most efficient and robust SAC for CORR, remarkably, with overpotential less than 0.1 V. By probing into geometric and electronic properties, the underlying origin behind optimal adsorptions of two crucial species, CO* and CHO* on four B/Al@xenes is deduced, which sheds light on the enhancement of CORR catalysis. The incorporation of dopants serves dual functions: modifying the charge redistribution in xene substrates and providing effective active sites for optimizing adsorbed species, thereby boosting CORR performance. Thus, these findings open a new avenue for non-TM doping into the heavier analogs of graphene, known as xenes, thereby broadening the scope of xene-based SACs for multi-electron catalytic reactions.
2. Computational methods
Spin-polarized DFT calculations were performed using the projector augmented wave (PAW) potentials,38,39 as implemented in VASP.40–42 We employed the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional43 to define the electron interactions. The van der Waals (vdW) correction was described by applying semiempirical DFT-D3 method of Grimme.44 An energy cutoff of 500 eV was adopted for the plane-wave basis set. The convergence threshold for electronic structure iterations and for residual force were 1 × 10−5 eV and 0.02 eV Å−1, respectively. The Brillouin zone was sampled by using Monkhorst–Pack with 3 × 3 × 1 and 5 × 5 × 1 k-point grids for structural optimization and electronic calculations, respectively. A 20 Å vacuum space length was set along the z direction for all structure models to avoid the interaction between the periodic images. The solvation effects were corrected by using VASPsol model as implemented by Mathew and Henning.45 For model structures, one dopant (B/Al) was substituted into 5 × 5 supercell of xene (silicene/germanene), creating four B/Al@xene systems with 49 Si/Ge and 1 B/Al. Bader charge analysis was deployed to compute charge population.46 The projected crystal orbital hamilton populations (pCOHP) calculations were analyzed by the LOBSTER package.47 Vaspkit was used for data post-processing of PDOS and wavefunction visualization.48Ab initio molecular dynamics (AIMD) simulation was carried out under the NVT ensemble at 500 K and a time step of 2 fs to investigate the thermal stability of the structures.49 The search for transition states and energy barriers were calculated using the climbing image nudged elastic band (CI-NEB) method.50The computational hydrogen electrode model (CHE) proposed by Norskov's and co-workers51 was employed to obtain the thermodynamics of electrochemical reactions. The change of Gibbs free energy (ΔG) can be calculated via: ΔG = ΔE + ΔEZPE − TΔS + ΔGU + ΔGpH where ΔE, ΔEZPE, and ΔS correspond to the electronic energy difference, the zero-point energy change and the entropy change; ΔGU = −eU (e and U are the number of transferred electrons and the applied electrode potential, respectively) and ΔGpH = 0 for acidic medium. Here, the zero-point energy was computed from the vibrational frequencies via the equation:  and entropy was determined through the equation:
and entropy was determined through the equation:  where εi is the vibrational energy of mode i, N is the number of atoms in the adsorbates, kB is Boltzmann constant (1.38 × 10−23 J K−1),T = 298.15 K. The entropies of free gas molecules were taken from the National Institute of Standards and Technology (NIST) database. The limiting potential (UL) of the reaction was calculated as UL = −ΔGmax/e, in which ΔGmax is the free energy change of potential determining step and e is the corresponding number of transferred electrons. The overpotential is calculated as η = Ue − UL where Ue= −ΔGEq/e is the equilibrium potential (or standard potential) and ΔGEq is the standard free energy change of the overall reaction according to the computational hydrogen electrode model. The computed value of −0.13 eV is used for ΔGEq.
where εi is the vibrational energy of mode i, N is the number of atoms in the adsorbates, kB is Boltzmann constant (1.38 × 10−23 J K−1),T = 298.15 K. The entropies of free gas molecules were taken from the National Institute of Standards and Technology (NIST) database. The limiting potential (UL) of the reaction was calculated as UL = −ΔGmax/e, in which ΔGmax is the free energy change of potential determining step and e is the corresponding number of transferred electrons. The overpotential is calculated as η = Ue − UL where Ue= −ΔGEq/e is the equilibrium potential (or standard potential) and ΔGEq is the standard free energy change of the overall reaction according to the computational hydrogen electrode model. The computed value of −0.13 eV is used for ΔGEq.
3. Results and discussion
3.1 Geometry and stability
The structural properties of two pristine silicene (Si) and germanene (Ge) nanosheets, are initially investigated. The 5 × 5 supercells of Si and Ge monolayers, comprising 50 atoms each, are constructed and optimized as illustrated in Fig. S1.† The calculated lattice constants (3.87 Å and 4.06 Å) and bond lengths (dSi–Si = 2.28 Å and dGe–Ge = 2.44 Å) for Si and Ge structures, respectively, are in good agreement with previous theoretical works,12,13,16 thereby validating the accuracy of our computational models.In this study, two representative elements in the group IIIA, B and Al, are employed as dopants within two analogs of graphene, silicene (Si) and germanene (Ge). For the design strategy of impurity doping into the xene lattice, one host atom (Si/Ge) from the pristine structures is replaced by one dopant (B/Al) to form a single atom catalyst (SAC). As presented in Fig. 1a, the doped xene geometries are not significantly altered compared to the pristine Si/Ge ones. Specifically, in B-substituted xenes, the average bond lengths of B and three host substrate atoms (Si/Ge) nearest to the B, denoted as dB–Si and dB–Ge, are 1.97 and 2.05 Å, respectively. In contrast, in Al-substituted xenes, the average dAl–Si and dAl–Ge bond lengths are 2.35 and 2.45 Å, respectively. Thus, the presence of B is more likely to induce a planar conformation in proximity to the dopant, while the Al dopant tends to slightly protrude from the xene lattice. The observed variations in geometry upon substitution can be attributed to the differing covalent radii of B, Al, Si, and Ge atoms (0.81, 1.43, 1.18 and 1.22 Å, respectively). Indeed, the lengths of the bonds between B and Si/Ge are in almost perfect agreement with the sums of the covalent radii of the elements, which indicate the bonds to be strong covalent bonds.
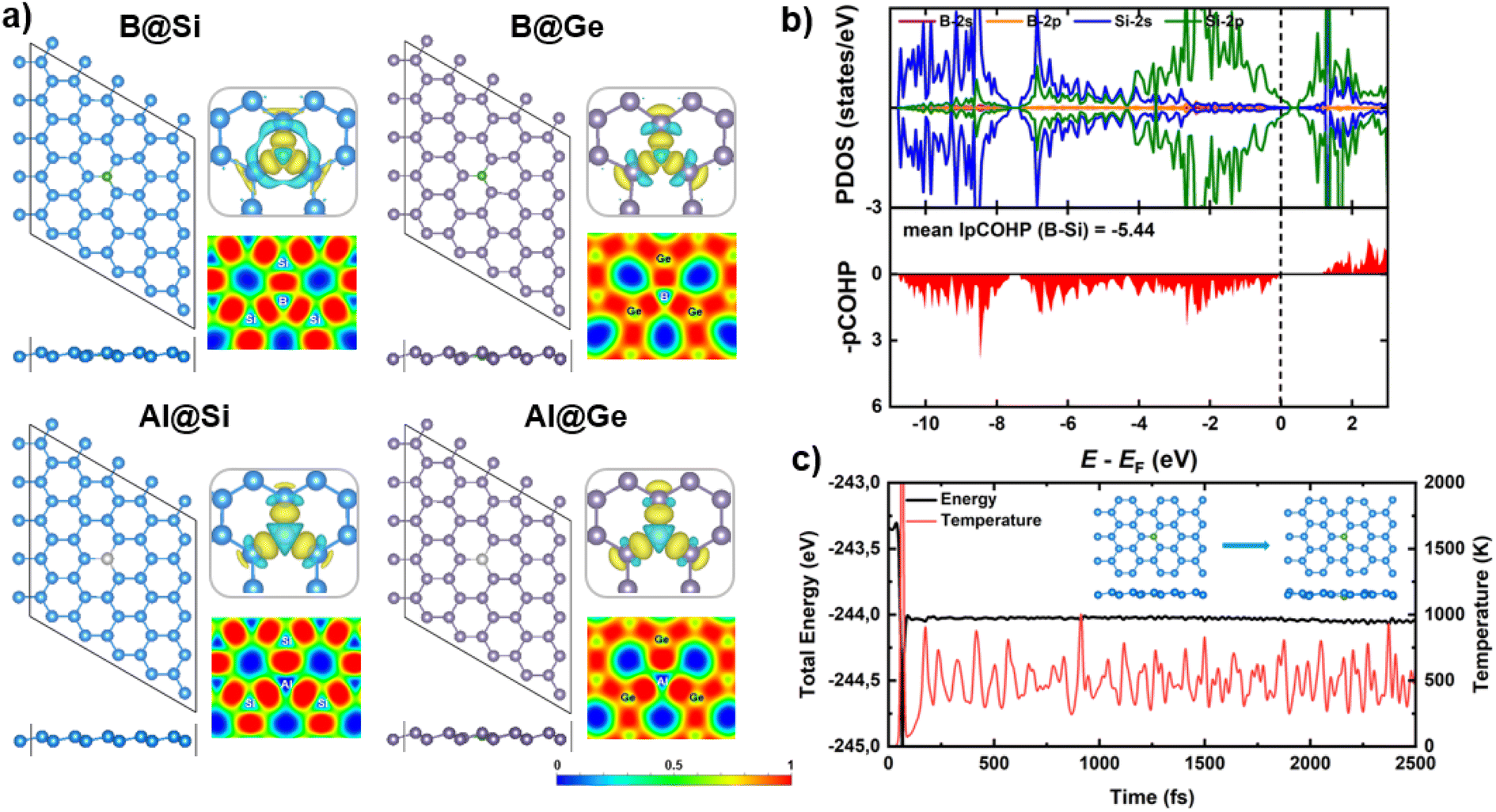 | ||
| Fig. 1 (a) Schematic representations of four boron/aluminum-embedded silicene/germanene monolayers (B/Al@Si/Ge) from top and side views, corresponding charge density difference and electron localization function (ELF) maps. The isosurface value is 0.0035 e Å−3, and the yellow (cyan) region represents electron gain (loss). Color code: B – green, Al – silver, Si – light blue, Ge – violet. (b) Partial density of states (PDOS) projected on the s and p orbitals of dopants and host substrates in B@Si; projected crystal orbital hamilton populations (pCOHP) of the B–Si bond. The pCOHP of the spin-down component is masked by that of the spin-up. The Fermi energy is set at zero. (c) Ab initio molecular dynamics (AIMD) simulation of B@Si at 500 K with the initial and final structures. | ||
It is widely acknowledged that structural stability stands as a paramount criterion for the selection of potential catalysts. Especially for SACs, diffusion and aggregation of single atoms may undermine catalytic performance as well as the stability of the catalytic cycles, i.e. the turnover number. To examine the stability aspect, the binding energy (Eb) of dopant atom into defected xene monolayers is calculated via Eb = Edoped-xene − ESV-xene − Edopant, in which Edoped-xene, ESV-xene and Edopant are the total energies for the optimized equilibrium configuration of doped xene structures, single vacancy (SV) xenes, and isolated single B atom in its ground state, respectively. The more negative Eb values are, the more stable the doped structures are. B exhibits a strong binding affinity with Si and Ge, possessing Eb values of −9.00 and −7.23 eV, respectively, which are much stronger than the cohesive energy of elemental B of −5.81 eV.52,53 The computed Eb values of Al in Si and Ge are −5.95 and −5.08 eV, respectively, thereby surpassing the cohesive energy of Al of −3.39 eV per atom.53 These results affirm the high thermodynamic stability of our substitutional systems. The silicene monolayer presents a more promising host for accommodating dopants compared to germanene, with more negative Eb quantities. In addition, a common trend is discernible across each xene system, wherein the interatomic distance of dopant (B/Al) and host atom decreases as the magnitude of the binding energy increases. This indicates that B is more stably incorporated into both xene substrates compared to Al.
Electronic structure calculations and chemical bonding analysis have been performed to unveil the underlying origins of the structural stability. Charge density difference plots (displayed in the top right panels of Fig. 1a) reveal electron density accumulation at three bond regions, verifying the strong interaction between dopants and their neighboring atoms of the SV-xene substrates. Interestingly, the electron density of the Al adatom appears to be reduced, in contrast to the significant electron localization in the vicinity of B. The charge distribution aligns well with the Bader charge analysis. To be clear, there is charge transferred from the substrate to B (resulting in a slight negative charge on B), whereas the reverse phenomenon is observed for Al (inducing a positive charge on Al). This behavior stems from the electronegativity hierarchy as follows B (2.04) ∼ Ge (2.01) > Si (1.90) > Al (1.61). The strong covalent bonds between dopants (B/Al) and adjacent (Si/Ge) atoms are also verified by the electron localization function (ELF) maps (presented in the bottom right panels of Fig. 1a), which show high electron localization between dopants and the three nearest host atoms. Spin-polarized partial density of states (PDOS) and projected crystal orbital hamilton populations (pCOHP) for B/Al-doped xenes are characterized as shown in Fig. 1b and S2.† One can be seen that the Fermi level (EF) resides inside the valence band (VB) in all structures since both B and Al substituents have three valence electrons, one less than Si and Ge, resulting in p-type doing of the doped xene systems. More importantly, there are apparent PDOS overlaps between dopants (B/Al) and neighbors (Si/Ge) near the EF, which signifies the substantial electron transfer ascribed by the formation of strong dopant-host atom bonds within the doped systems. Particularly, the significant overlap between B orbitals (primarily p orbitals) and Si/Ge-(s + p) orbitals is discernible in B-doped structures. In contrast, a modest overlap is observed between Al-p states and Si/Ge-(s + p) states, accompanied by the considerable engagement of Al-s states near the EF. Note that the greater contribution of B-p states at the band edges strengthen the interaction with xene substrates compared to Al substituent. This electronic trait also holds promise for the formation of the later CORR intermediates in B-doped materials, as discussed later. The high degree of covalency of the interactions between dopants and neighboring host atoms is also confirmed by the pCOHP result, which shows that all B/Al–Si/Ge bonding fall into the bonding regions around the EF. Moreover, the B–Si/Ge show higher covalent bonding strength than the Al–Si/Ge counterparts, with the IpCOHP values per bond (mean IpCOHP) for B–Si/Ge (−5.44/−5.50) being more negative than those of Al–Si/Ge (−4.41/−4.40). As a result, the high structural stability of our B/Al-doped xenes can primarily be attributed to the strong covalent bonds between B/Al and Si/Ge.
In addition, the thermal stability of B/Al-doped xenes has been assessed by means of AIMD simulation, as depicted in Fig. 1c and S3.† Energy and temperature fluctuate within a marginal range; and the structural geometry remains intact over the simulation time at 500 K. In fact, the dopants (B/Al) consistently occupy their preferred anchoring sites and maintain strong bonds with their three adjacent Si/Ge atoms, indicating high thermal stability and structural integrity of the catalysts.
3.2 Favorability of CORR over HER
Hydrogen evolution reaction (HER) is the main competitive side reaction during the CORR process. Indeed, the first protonation step of CORR to form CHO* or COH* competes with the first step of HER to form H*. Hence, it is necessary to evaluate the catalytic selectivity of CORR over HER by comparing the changes in reaction free energy for the adsorbed CHO*/COH* and H* species (ΔGCHO*/COH*vs. ΔGH*). In this assessment, the formation of the COH* intermediate is omitted due to its structural instability with a high ΔGCOH* value as well as its lack of involvement in favored CORR pathways, as elaborated in the subsequent section. From Fig. 2a, the CHO* formation of all catalysts is below a bisecting line, which implies a thermodynamic propensity towards CHO* over H*, leading to enhancement of the CORR selectivity.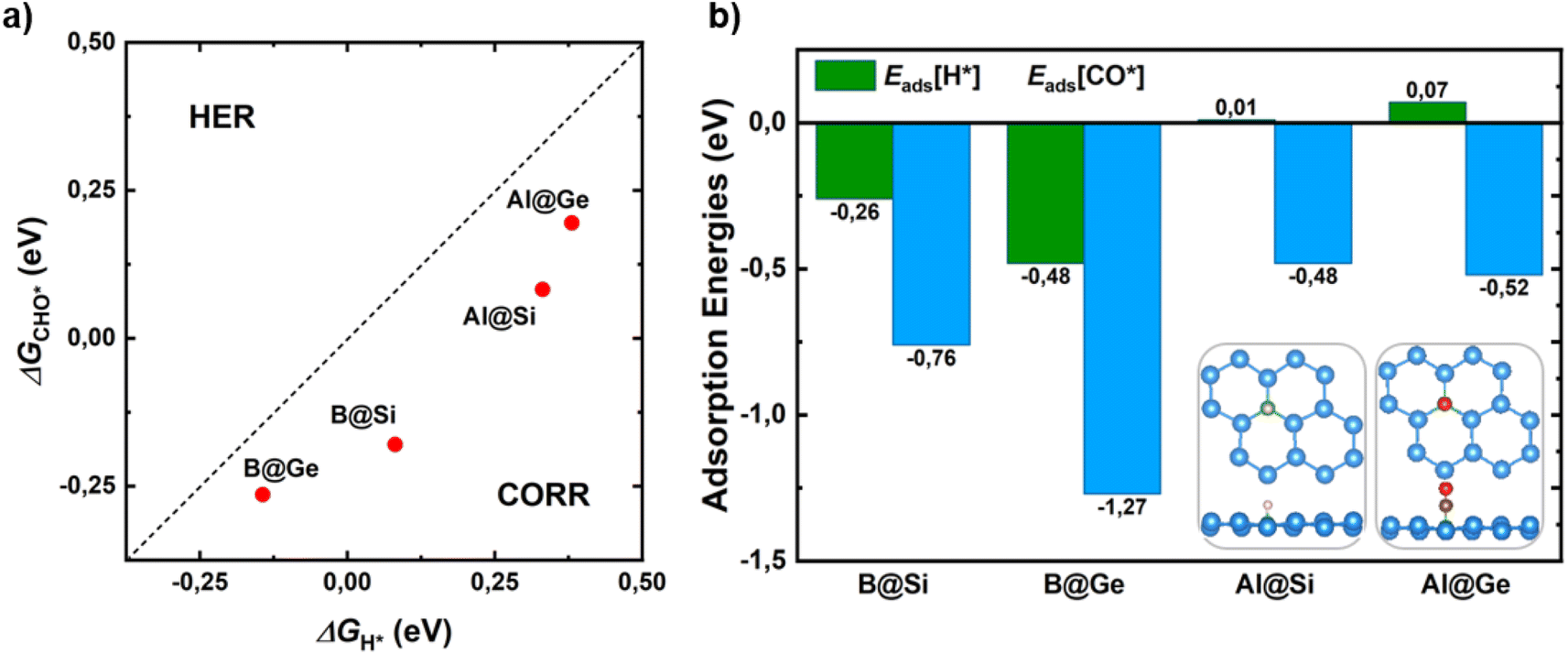 | ||
| Fig. 2 (a) First protonation free energy change ΔGCHO (CORR) vs. ΔGH (HER). The catalysts below the dashed parity line indicate the preference of CORR over HER. (b) Adsorption energies of the most stable H*/CO*-adsorbed species on four different doped systems. Top and side views of corresponding optimized configurations in downright panels. Take B@Si as an example; green, light blue, red, brown, and light pink spheres represent B, Si, O, C and H, respectively. | ||
In addition, a proficient initial CO activation is regarded as a fundamental requisite for a functioning CORR catalyst. Strong CO adsorption on the catalyst surface can prevent H from obstructing active sites for CORR, thereby impeding HER and promoting enhanced CORR activity and selectivity. In this manner, the catalysts with a lower adsorption energy of CO* than that of H* demonstrate a selectivity of CORR over HER (Eads[CO*] < Eads[H*]). A meticulous investigation of all possible adsorption scenarios for CO* and H* species on the surfaces, along with their corresponding adsorption energies, reveals the most stable configurations. In Fig. 2b and S4,† the CO molecule shows a clear energetic preference for vertical chemisorption onto the B/Al dopant, orienting its C end towards the surfaces. This is characterized by moderate-to-strong adsorption energy values ranging from −0.48 to −1.27 eV. As for H, it tends to anchor at the top site of B in B@Si/Ge, and at the bridge site formed by an Al dopant and its neighbor (Si*/Ge*) in Al@Si/Ge. Fig. 2b unveils that B-substituted systems possess stronger CO and H adsorptions than Al-substituted counterparts. More importantly, CO exhibits tighter binding than H to all catalysts, as evidenced by more negative Eads values. This finding highlights the inherent capability of our catalysts to effectively suppress the HER, thereby minimizing the HER adverse impact on the CORR.
3.3 CORR activity and selectivity
We establish all possible CO reduction pathways towards various C1 products, including hydrocarbons and oxygenates, on four non-transition metal (non-TM) xenes. As aforementioned, our models meet the requirements for efficient CO adsorption and selectivity of CORR over HER. Detailed reaction free energy diagrams that clarify their CORR activity and selectivity are shown in Fig. 3, while Fig. 4 presents optimized geometries of corresponding intermediates involved in the CORR process.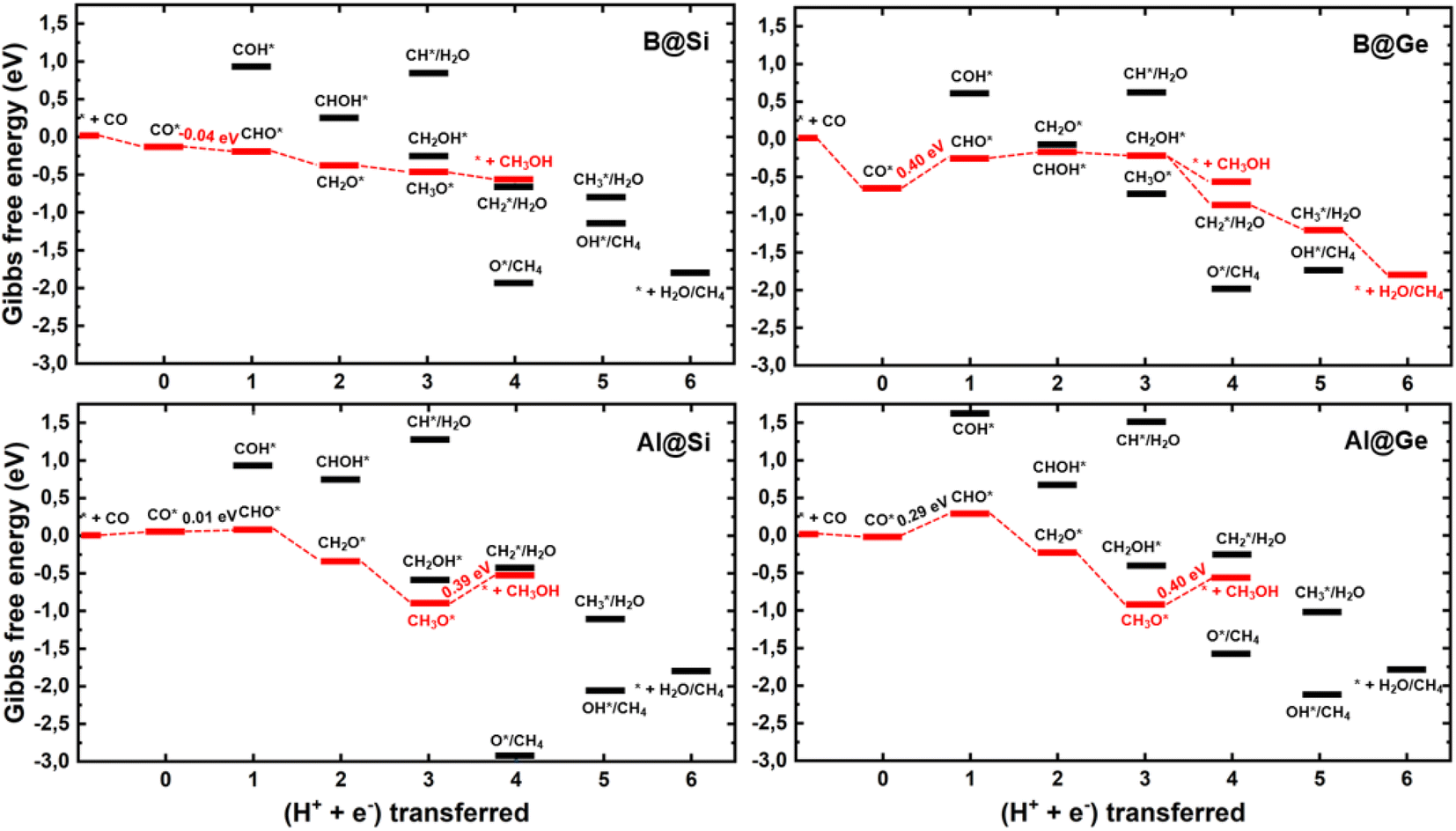 | ||
| Fig. 3 Free-energy profiles for CO reduction on four model systems. The red line displays the most favorable overall pathway, and the potential determining step is labeled with the corresponding free energy change. | ||
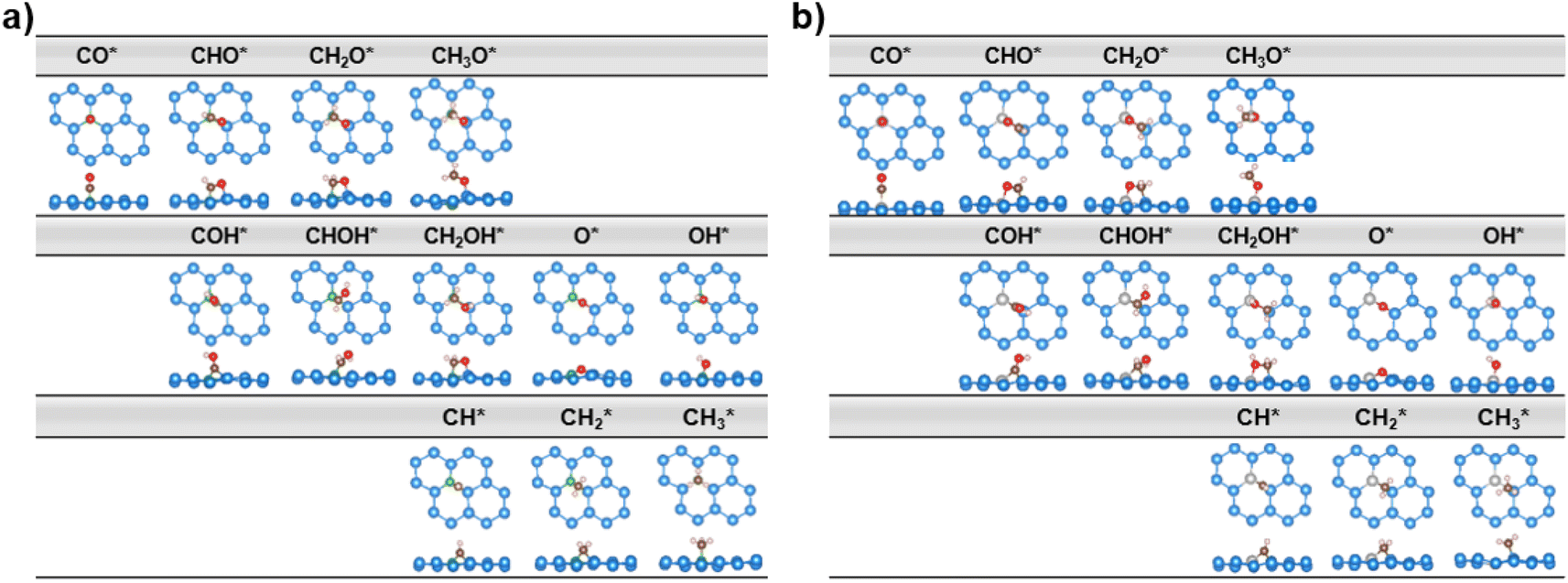 | ||
| Fig. 4 Top and side views of the optimized lowest-energy intermediate geometries along all possible CORR pathways of two systems, (a) B@Si and (b) Al@Si. Color code: B – green, Al – silver, Si – light blue, C – brown, O – red, H – light pink. | ||
The first hydrogenation step of CO* leads to the generation of either CHO* or COH*. It is obvious that the creation of CHO* demands lower energy than that of COH* in all systems owing to their binding modes. As depicted in Fig. 4, CHO* tends to connect to the catalytic surface through two-site adsorption mode, resulting in two distinct bonds: C–B and O–Si/Ge in B-doped xenes, and C–Si/Ge and O–Al in Al-doped xenes. Intriguingly, the Al dopant due to its metallic nature binds to O of the CHO* species, which is in contrast to the B–C bond with the B dopant. This suggests that Al-substituted structures bind less tightly to CHO* than their B-substituted analogs. In terms of the COH* intermediates, C prefers to bind to the bridge site of the dopant (B/Al) and one adjacent host atom (Si/Ge), rendering COH* less stable than CHO*. Thereby, the thermodynamically favorable reaction pathway predominantly involves the key CHO* intermediate. While the hydrogenation step of CO* to CHO* is slightly endergonic in most cases, B@Si emerges as an exception with an exergonic step and thereby positioning it as the most promising CORR catalyst. The second protonation at either the C or O site of CHO* can create CH2O* or CHOH* intermediates, which shares analogous binding modes to CHO* and COH*, respectively. The formation of CH2O* is also found to be more favorable in energy than that of CHOH*, except for B@Ge. The third hydrogenation step gives rise to three possible intermediates, CH2OH*, CH3O* or CH* together with the liberation of H2O (CH* + H2O). While the protonation of CHOH* to CH* and H2O requires a large energy cost, the generation of CH2OH* or CH3O* releases a significant amount of energy. CH3O* tends to bind tightly to the top site of the dopant (B/Al) via O, whereas CH2OH* maintains a comparable adsorption arrangement as CH2O*, indicating a stronger adsorption of CH3O* than CH2OH* on the catalytic surface. As a result, most systems are inclined to produce CH3O* rather than CH2OH*, except for the B@Ge structure, which produces CH2OH* from CHOH*. Besides the CH3OH product, the fourth protonation could potentially lead to the creation of O* along with the release of CH4 from CH3O*. This pathway would reduce the catalytic efficiency as O* is strongly adsorbed and the subsequent protonation to form OH* is endergonic (except for Al@Ge). However, a detailed analysis of the B@Si system indicates that O*/CH4 is not formed by protonation of CH3O* in a single electrochemical transformation. Instead, our analysis shows that CH3OH* is formed in the initial electrochemical step, and that the subsequent chemical transformation to form O*/CH4 has an energy barrier of 1.2 eV. The adsorption energy of CH3OH* is −0.26 eV and thus the release of CH3OH is kinetically favored over the formation of O*/CH4, see Fig. S5† for the details of the analysis. B@Ge has the potential to form a mixture of CH3OH and CH4 as both pathways are exergonic and diverging after the protonation of CH2OH*. In the latter more favorable pathway, the free energy changes for the consecutive hydrogenation steps via the CH2* → CH3* → * + CH4 path are downhill. In brevity, according to the lowest-free energy change of each elementary step, and considering that there is a kinetic barrier for forming O*/CH4, our catalysts generally prefer to produce CH3OH over CH4 through the CO* → CHO* → CH2O* → CH2OH*/CH3O* → * + CH3OH route (shown in red in Fig. 3). Remarkably, CO reduction on B@Si is exergonic with a limiting potential of 0.04 V and an overpotential of less than 0.1 V. Moreover, the Al@Si, B@Ge and Al@Ge materials exhibit relatively high CORR performance with low limiting potentials of around −0.40 V. The CO* → CHO* step is the potential determining step for the B catalysts whereas the CH3O* → * + CH3OH step has the corresponding role for the Al catalysts. The effect on the free energy diagram of each system of applying a potential corresponding to the limiting potential is illustrated in Fig. S6.† The limiting potential is the highest potential where all electrochemical steps are exergonic, and in the case of B@Si it corresponds to a slightly positive potential (0.04 V).
A comparison between the limiting potentials and the free energy diagrams obtained with VaspSol with the corresponding vacuum results suggests that the aqueous environment has only a limited effect on the overall CORR activity and selectivity of the single-atom catalysts (SACs), compare Fig. 3 and S7.† The limiting potentials with VaspSol and in vacuum (value in parentheses) for B@Si, Al@Si, B@Ge and Al@Ge are 0.04 (−0.02), −0.39 (−0.46), −0.40 (−0.49) and −0.40 (−0.45 V). Thus, the solvation correction in no case has a larger effect than 0.09 V on the limiting potential.
3.4 Origin of CORR enhancement
To gain deeper understanding of the CORR catalytic activity, we calculate the adsorption energies of two crucial intermediates, Eads[CO*] and Eads[CHO*], which not only involves the potential determining step (PDS) in most non-TM@xenes but also plays a decisive role in the catalytic efficiency. It is generally known that the linear scaling relation between CO* and CHO* results in a high limiting potential and reduced catalytic activity.54–56 This is because strong adsorption of CO is closely linked to a binding enhancement for the CHO species, leading to a high reaction free energy change of the CO* → CHO* step. From Fig. 5 it is evident that our four SACs significantly deviate from the linear relationships between Eb[CO*] and Eb[CHO*] that exist for the (111) and (211) transition metal surfaces, resulting in enhanced catalytic activity for our models as compared to the crystalline transition metal catalysts. Especially, B@Si with moderate CO* adsorption and strong CHO* adsorption has a much lower free energy change of the PDS (CO* → CHO*), resulting in the excellent CORR activity. The breaking of the traditional linear scaling relation leads to the enhancement of the CORR performance.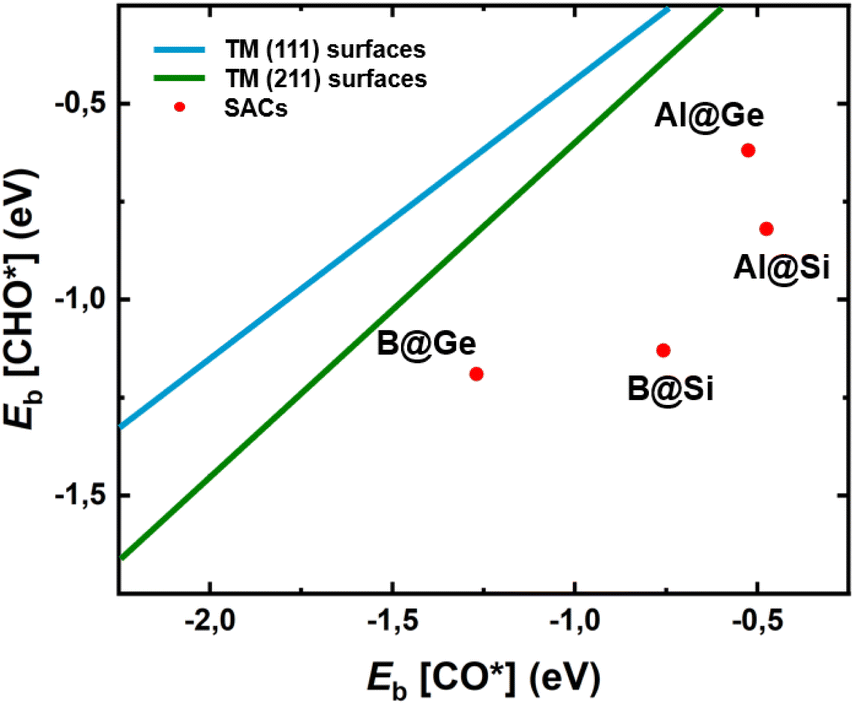 | ||
| Fig. 5 Relationship between adsorption energies of two intermediates CO* and CHO*, Eb[CO*] and Eb[CHO*], on four model systems. The linear scaling relations of transition-metal (TM) (111) and (211) facets are plotted in dark blue and green lines from ref. 57, respectively. SACs with the deviation from the traditional scaling lines of TMs exhibit high CORR activity. | ||
The mechanism for the enhanced CO and CHO adsorption is further comprehensively elucidated by their electronic and geometric characteristics. Bader charge variation of CO* and CHO* adsorption on four structures is displayed in Fig. 6. Both CO* and CHO* show negative charge; however, the CHO* species accept more electrons than CO* in each system, which can be attributed to an extraordinary two-way electron transfer within the CO* adsorption configurations, as discussed later. The host substrate, SV-Si/Ge, represented by green lines, acts as electron donor with the doping of B substituent and conversely as electron acceptor with the doping of Al counterpart in both CO* and CHO* intermediates. The electronegativity order of atoms (B ∼ Ge > Si > Al) may account for the varying amount of charge transfer. Consequently, the B dopant is slightly negatively charged, whereas Al is positively charged within the CO* and CHO* systems. Intriguingly, the more negatively charged the dopant center the more likely it is to stabilize the CO* and CHO* intermediates. For example, in the CO*/CHO* state, the B adatom in germanene has the most negative charge among all systems, and consequently the B@Ge structure has the lowest Eads[CO*] and Eads[CHO*] values among the four substrates. This implies that the dopant serves as a critical electron modulator as well as the key active site for CO reduction.
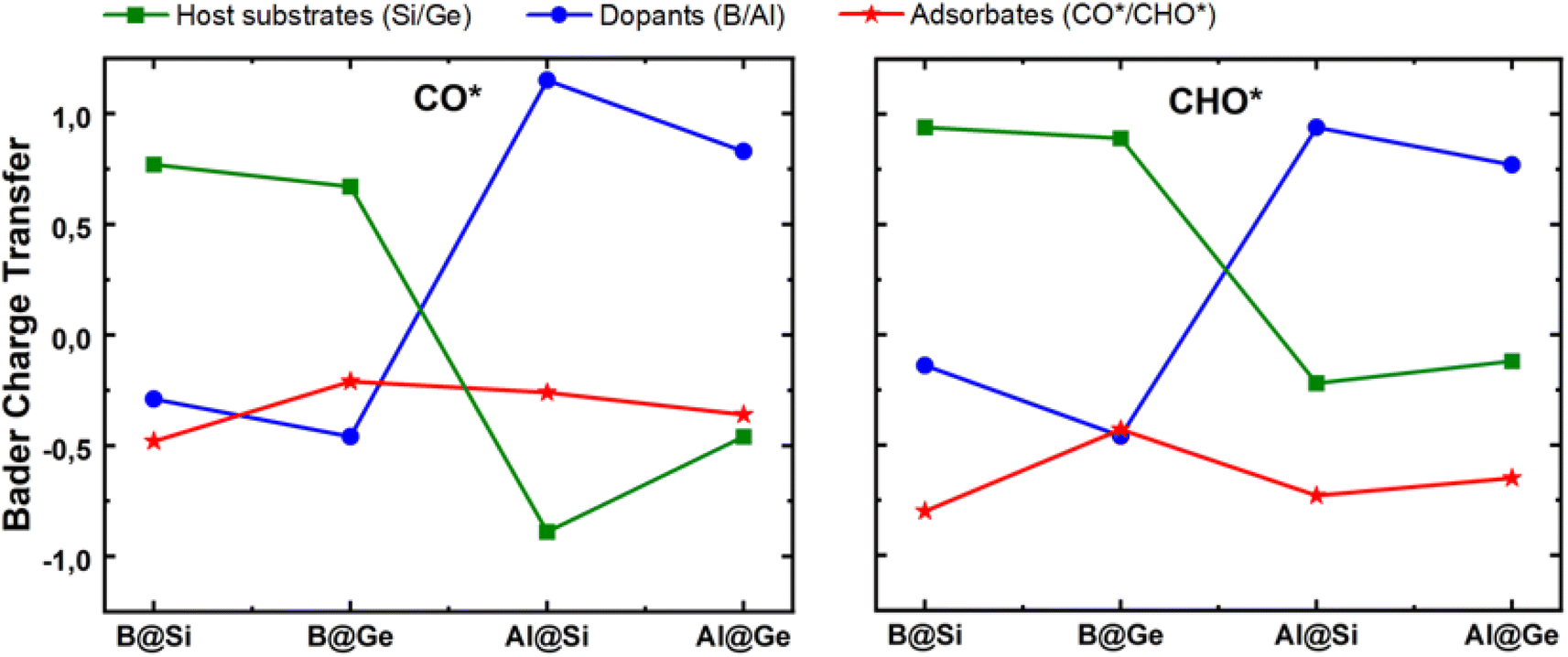 | ||
| Fig. 6 Bader charge variation of three inspected moieties (single vacancy host substrate, dopant and adsorbate) within CO* and CHO* intermediates. Unit: |e|. | ||
Spin-polarized partial density of states (PDOS), charge density differences and molecular orbitals of CO absorbed on the four structures are presented in Fig. 7. The comparison of PDOS between the B-doped and Al-doped structures unveils a much greater overlap with the CO molecule for B than for Al, providing a reasonable explanation for why CO binds more strongly on B-doped catalysts than on Al-doped ones. Specifically, in B-doped systems, there is a pronounced interaction between CO 4σ/5σ and B 2pz orbitals, with minor DOS contribution of adjacent Si/Ge 2p orbitals (refer to Fig. S8† for further details). In contrast, the 2pz orbitals of Al have a modest interaction with the 4σ/5σ CO orbitals. The more significant overlap in B-doped systems suggests a stronger B–C σ-bonding than Al–C σ-bonding, leading to shorter B–C than B–Al bond lengths (Fig. S9†). This stronger bonding is a consequence of the higher electron attraction capacity of the B dopant with its empty 2pz orbital, which withdraws more electrons from the CO molecule compared to its Al counterpart. Besides, owing to the occupied orbitals of the dopant in distorted pyramidal B/Al-doped xenes geometries, a constructive π-type backbonding arises from a noticeable overlap between B/Al 2px/y orbitals and C 2px/y orbitals (denoted as CO 1π), shortening the distance between CO and the catalytic framework. Briefly, the 4σ, 5σ and 1π CO orbitals significantly engage in the chemical bonding with the materials, thereby improving CO adsorption on the catalytic surface. Of particular importance, there is an overlap between CO 2π* and B/Al 2p orbitals over a broad energy window close to the EF, which results in the elongation of C–O bond length (dC–O). In agreement with our earlier molecular study,35 the states of the B dopant are more delocalized and interact more strongly with the CO 2π* orbitals than the corresponding Al states, accounting for the more elongated dC–O in B@Si/Ge than in Al@Si/Ge (1.17/1.18 Å vs. 1.15/1.16 Å, respectively). The partial occupancy of anti-bonding CO 2π* orbitals is also verified by the Bader charge analysis, in which the extra electron density residing on CO* adsorbate varies from −0.21 to −0.48 |e|. These charges that reflect partial occupation of the 2π* orbitals due to π-backdonation are congruent with a weakened C–O triple bond and furthered CO activation. Furthermore, interaction is observed between the Si/Ge 2p and C 2p orbitals close to the EF, promoting the charge transfer upon CO adsorption (Fig. S8†). The electron density difference (Fig. 7a) also highlights the bidirectional charge transfer between CO and B@Si/Ge, which is in contrast to Al@Si/Ge, and in perfect accordance with the above PDOS observation. To sum up, the combination of σ-donation from C to B and π-backdonation from B to C plays a pivotal role in the binding and activation of CO, thereby enhancing the propensity for CO reduction on B/Al-substituted xenes.
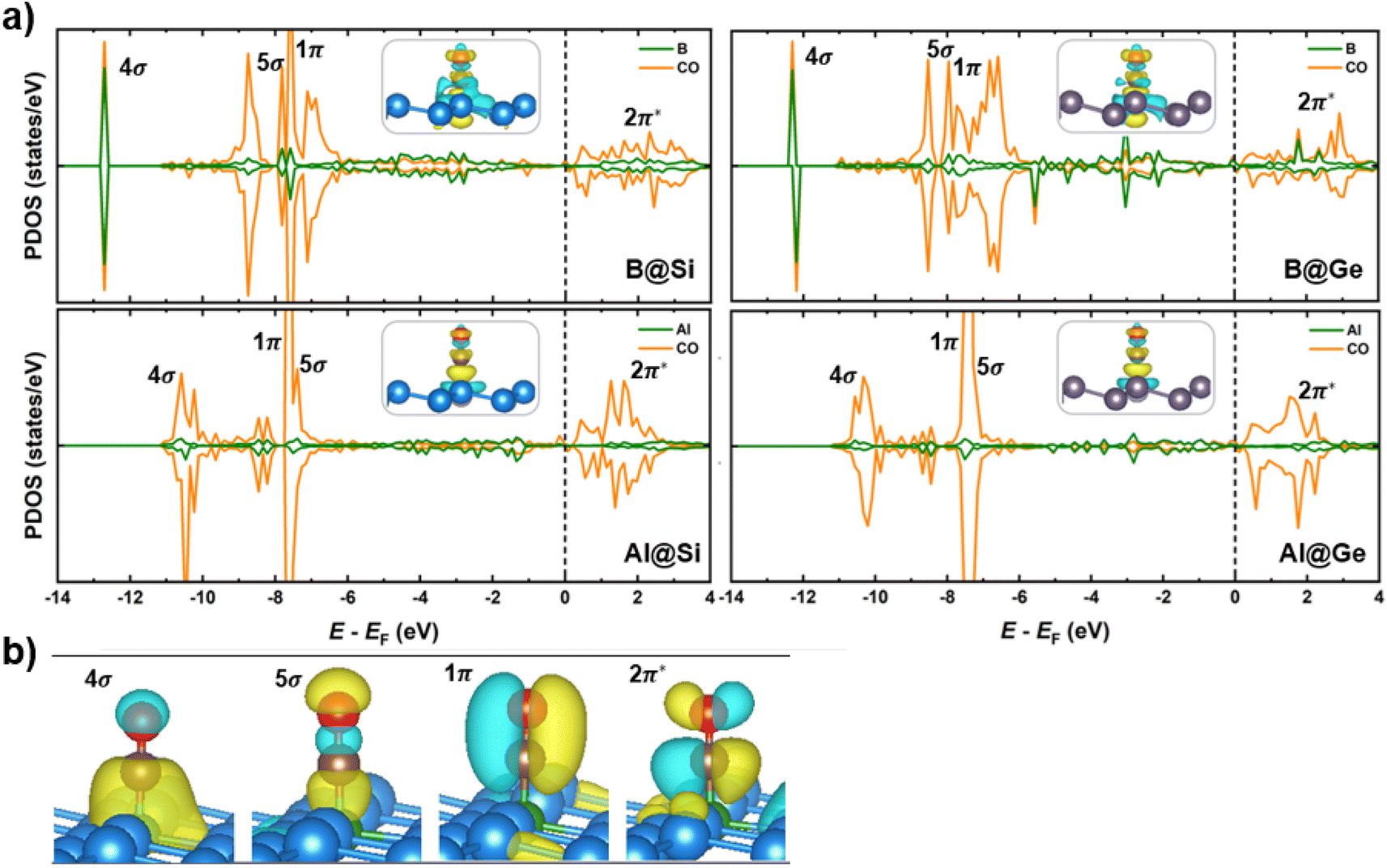 | ||
| Fig. 7 (a) Partial density of states (PDOS) of CO and dopants (B/Al) of four structures upon CO adsorption. The Fermi level shown in dashed line is set to zero. Selected bands are labeled according to the labeling of the corresponding canonical orbitals of bare CO. The insets show their corresponding charge density difference. Color code: yellow – charge accumulation, cyan – charge depletion. (b) Charge density of 4σ, 5σ, 1π, and 2π* orbitals of adsorbed CO molecule on B-doped silicene (B@Si) from side view. | ||
When it comes to CHO* adsorption, the combination of atoms and their configuration determine the binding strength. As mentioned earlier, the dopant together with one of three adjacent Si/Ge atoms on the substrate (referred to as Si*/Ge*) constitute two heteronuclear active sites acting as C* and O* binding sites. Specifically, B prefers to interact with C, and the Si*/Ge* atom interacts with O. In contrast, Al tends to bind to O, and Si*/Ge* binds to C. Such binding modes allow the CHO*-adsorbed geometries to become stabilized with negative adsorption energy values (Table S1†). The PDOS and electron density differences (Fig. 8a) suggest significant orbital mixing between the B/Al@xenes and the CHO* group, confirming the strong CHO* binding. The two most noteworthy features are the four interaction regions below the EF, and that the structures sharing the common dopant exhibit similar overlap patterns. To be specific, the sharp peak (denoted as (i)) around −12 eV corresponds to the strong σ-bonding interaction between C and B-2pz in B-embedded systems, and C and Si*/Ge*-2pz in Al-embedded ones (see Fig. S10† for more details), which is mainly responsible for the strong binding of CHO* to the catalytic surfaces. The second and third zones represent the overlap of C and O (CHO*) with the two active surface atoms (one B/Al dopant and one substrate atom Si*/Ge*). In addition, a weak PDOS overlap between C and its favored anchoring partner of the surface is observed in the range of −3 to −1 eV (iv), strengthening the binding between the CHO* group and B/Al@xenes. Notably, the PDOS of B-integrated systems displays a higher degree of interaction with CHO* compared to their Al-integrated counterparts. This phenomenon is apparent through the denser electron clouds in the interaction regions of B-doped structures as illustrated in Fig. 8b. Consequently, the average binding distances of C–B and O–Si*/Ge* in B-doped catalysts are shorter than those of C–Si*/Ge* and O–Al in Al-doped ones (see Fig. S11†). Furthermore, concerning the Al-integrated systems, Al@Si adsorbs CHO* stronger than Al@Ge does, which is due to the higher DOS contribution from Si* compared to Ge* in the primary interaction peaks. This leads to a lower reaction free energy change in the protonation step to form CHO* intermediate in Al@Si compared to Al@Ge (0.01 vs. 0.29 eV, respectively), resulting in the overall thermodynamically more favorable CO reduction on the Al-doped silicene.
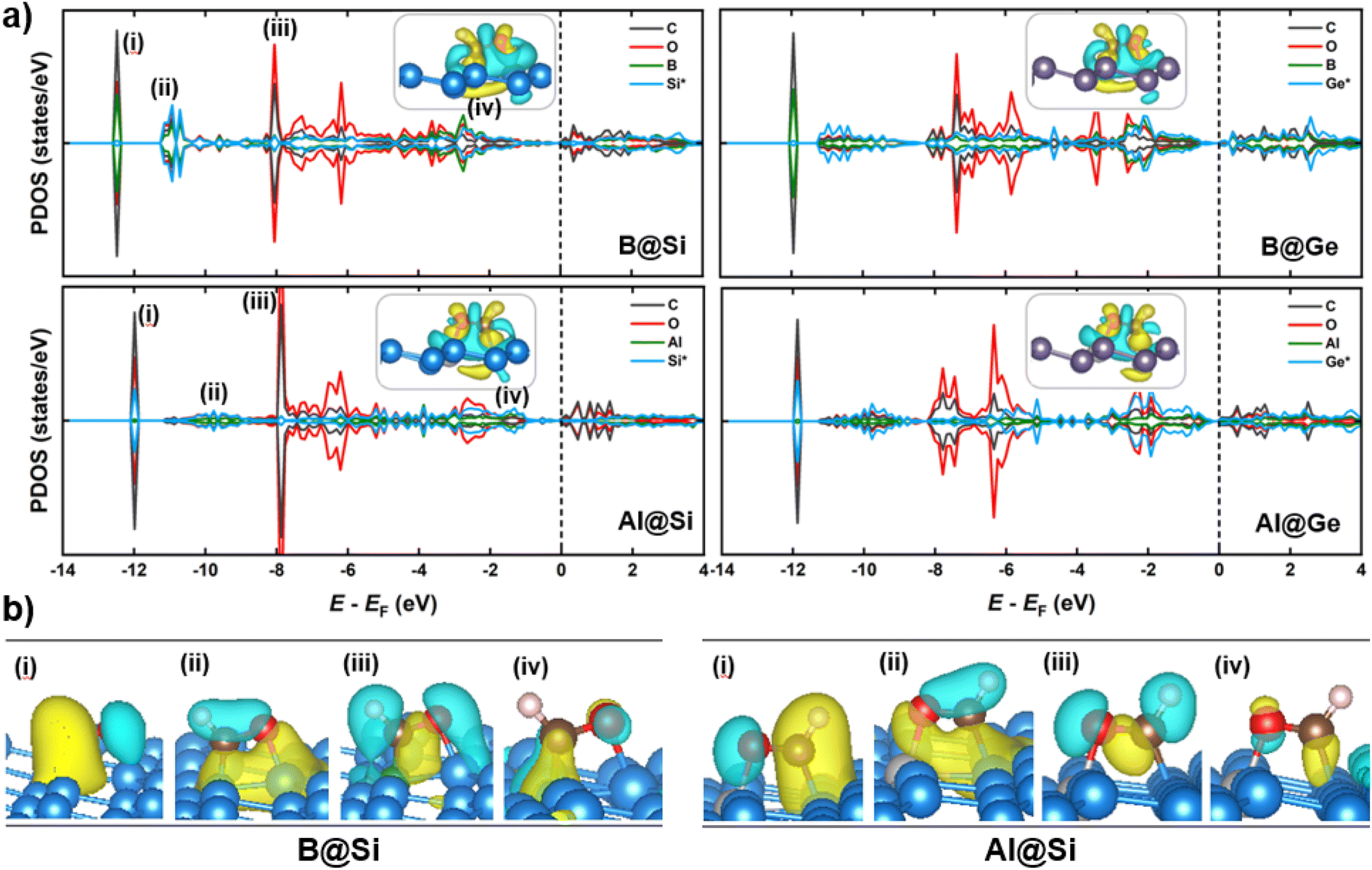 | ||
| Fig. 8 (a) Partial density of states (PDOS) of C and O atoms in CHO*, dopant (B/Al) and one substrate atom (Si*/Ge*) of CHO* species adsorption on four structures. Asterisk (*) means the substrate atom directly bonding CHO* adsorbate. The Fermi level shown in dashed line is set to zero. The insets show their corresponding charge density difference. Color code: yellow – charge accumulation, cyan – charge depletion. (b) Charge density isosurfaces of orbitals/bands at the energy level of CHO* adsorbed on B@Si and Al@Si from side view. | ||
4. Conclusion
In summary, by means of DFT computations combined with a CHE model, we carry out a thorough investigation into the viability of xene monolayers (silicene and germanene) doped with two p-block atoms (B/Al) as electrocatalysts for CORR. Our results reveal that dopant forms three localized bonds with three neighboring host atoms, preserving the pristine xene configuration, with negative binding energy (Eb), offering high structural stability. All four systems are put forth as potential candidates for CORR based on favorable initial CO adsorption and preference of CORR over HER. Calculated adsorption energies of vital adsorbed species of CO* versus CHO* break the traditional scaling relations, lowering the limiting potential for CO reduction. As a result, B@Si, Al@Si, Al@Ge and B@Ge exhibit excellent CORR performance with a limiting potential UL value of 0.04, −0.39, −0.39, and −0.40 V respectively. B@Si shows the best CORR catalytic activity with an overpotential of less than 0.1 V. B@Si, Al@Si, Al@Ge have a high CORR selectivity towards the CH3OH product, following the preferred pathway: CO* → CHO* → CH2O* → CH3O* → * + CH3OH. B@Ge may form a mixture of CH3OH and CH4, but the formation of CH4 is energetically preferred via the pathway CO* → CHO* → CHOH*→ CH2OH*→ CH2*→ CH3*→ * + CH4.We uncover the fundamental factors driving the catalytic CORR enhancement based on structural and electronic features of the CO* and CHO* intermediates. It is found that CO moderately chemisorbs on top of dopants via the intriguing “C to B σ-donation – B to C π-backdonation” scenario, whereas the strengthened adsorption of the CHO* species on dual atomic sites of the surface with distinct bonding orientations is guided by the substituents. The introduction of dopants modifies the surface charge distribution, and it thereby allows for fine-tuning of the binding strength of adsorbed species and optimization of the CORR performance. Hence, our study not only offers theoretical insights into the enhanced CORR catalytic mechanism, but also paves the way for designing high-performance heterogenous SACs for other various applications.
Author contributions
Huong T. D. Bui: conceptualization, data curation, formal analysis, methodology, investigation, and writing. Tore Brinck: conceptualization, investigation, and writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swedish Research Council (VR) grant number 2021-05881. The computations were enabled by resources provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) at the National Supercomputer Centre in Linköping University (NSC) and the PDC Centre for High Performance Computing (PDC-HPC) partially funded by the Swedish Research Council through grant agreement no. 2022-06725.References
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef.
- A. Bagger, L. Arnarson, M. H. Hansen, E. Spohr and J. Rossmeisl, J. Am. Chem. Soc., 2019, 141, 1506–1514 CrossRef CAS PubMed.
- H. Du, J. Fu, L.-X. Liu, S. Ding, Z. Lyu, Y.-C. Chang, X. Jin, F. O. Kengara, B. Song, Q. Min, J.-J. Zhu, D. Du, C. Gu, Y. Lin, J.-S. Hu and W. Zhu, Mater. Today, 2022, 59, 182–199 CrossRef CAS.
- B. Ruqia, G. M. Tomboc, T. Kwon, J. Kundu, J. Y. Kim, K. Lee and S.-I. Choi, Chem Catal., 2022, 2, 1961–1988 CrossRef CAS.
- L. Wang, S. A. Nitopi, E. Bertheussen, M. Orazov, C. G. Morales-Guio, X. Liu, D. C. Higgins, K. Chan, J. K. Nørskov, C. Hahn and T. F. Jaramillo, ACS Catal., 2018, 8, 7445–7454 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- S. Z. Butler, S. M. Hollen, L. Cao, Y. Cui, J. A. Gupta, H. R. Gutiérrez, T. F. Heinz, S. S. Hong, J. Huang, A. F. Ismach, E. Johnston-Halperin, M. Kuno, V. V. Plashnitsa, R. D. Robinson, R. S. Ruoff, S. Salahuddin, J. Shan, L. Shi, M. G. Spencer, M. Terrones, W. Windl and J. E. Goldberger, ACS Nano, 2013, 7, 2898–2926 CrossRef CAS.
- D. Deng, K. S. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218–230 CrossRef CAS PubMed.
- L. Li, S. Lu, J. Pan, Z. Qin, Y. Wang, Y. Wang, G. Cao, S. Du and H.-J. Gao, Adv. Mater., 2014, 26, 4820–4824 CrossRef CAS.
- B. Aufray, A. Kara, S. Vizzini, H. Oughaddou, C. Léandri, B. Ealet and G. Le Lay, Appl. Phys. Lett., 2010, 96, 183102 CrossRef.
- S. Cahangirov, M. Topsakal, E. Aktürk, H. Şahin and S. Ciraci, Phys. Rev. Lett., 2009, 102, 236804 CrossRef CAS.
- J. C. Garcia, D. B. De Lima, L. V. C. Assali and J. F. Justo, J. Phys. Chem. C, 2011, 115, 13242–13246 CrossRef CAS.
- S. Lebègue and O. Eriksson, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 115409 CrossRef.
- X. Lin and J. Ni, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 075440 CrossRef.
- J. Feng, Y. Liu, H. Wang, J. Zhao, Q. Cai and X. Wang, Comput. Mater. Sci., 2014, 87, 218–226 CrossRef CAS.
- W. Xia, W. Hu, Z. Li and J. Yang, Phys. Chem. Chem. Phys., 2014, 16, 22495–22498 RSC.
- S. S. Raya, A. S. Ansari and B. Shong, Surf. Interfaces, 2021, 24, 101054 CrossRef CAS.
- W. Liao, G. Yu, L. Zhao, H. Zhu and W. Chen, Nanoscale, 2022, 14, 10918–10928 RSC.
- A. Kumar, V. Q. Bui, J. Lee, L. Wang, A. R. Jadhav, X. Liu, X. Shao, Y. Liu, J. Yu and Y. Hwang, Nat. Commun., 2021, 12, 6766 CrossRef CAS.
- T. He, K. Reuter and A. Du, J. Mater. Chem. A, 2020, 8, 599–606 RSC.
- H. T. D. Bui, V. Q. Bui, X. Shao, A. Kumar, S.-G. Kim, H. M. Le, Y. Kawazoe and H. Lee, J. Phys. Chem. C, 2021, 125, 13176–13184 CrossRef CAS.
- S. Ajmal, H. T. D. Bui, V. Q. Bui, T. Yang, X. Shao, A. Kumar, S.-G. Kim and H. Lee, Chem. Eng. J., 2022, 429, 132282 CrossRef CAS.
- H. T. D. Bui, V. Q. Bui, S.-G. Kim, Y. Kawazoe and H. Lee, Phys. Chem. Chem. Phys., 2021, 23, 25143–25151 RSC.
- Y. Li and W. An, J. Energy Chem., 2023, 80, 350–360 CrossRef CAS.
- S. Tang, Q. Dang, T. Liu, S. Zhang, Z. Zhou, X. Li, X. Wang, E. Sharman, Y. Luo and J. Jiang, J. Am. Chem. Soc., 2020, 142, 19308–19315 CrossRef CAS.
- Y. Zhang, T. Liu, X. Wang, Q. Dang, M. Zhang, S. Zhang, X. Li, S. Tang and J. Jiang, ACS Appl. Mater. Interfaces, 2022, 14, 9073–9083 CrossRef CAS.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- Y. Wang, J. Mao, X. Meng, L. Yu, D. Deng and X. Bao, Chem. Rev., 2019, 119, 1806–1854 CrossRef CAS.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- W.-Z. Li, M.-Y. Liu, L. Gong, M.-L. Zhang, C. Cao and Y. He, Appl. Surf. Sci., 2021, 560, 150041 CrossRef CAS.
- Z.-Z. Lin and X. Chen, Mater. Des., 2016, 107, 82–89 CrossRef CAS.
- W.-Z. Li, Y. He, Y. Mao and K. Xiong, ACS Omega, 2022, 7, 33156–33166 CrossRef CAS.
- F. Ersan, Ö. Arslanalp, G. Gökoğlu and E. Aktürk, Appl. Surf. Sci., 2016, 371, 314–321 CrossRef CAS.
- C. Li, S. Yang, S.-S. Li, J.-B. Xia and J. Li, J. Phys. Chem. C, 2013, 117, 483–488 CrossRef CAS.
- T. Brinck and S. K. Sahoo, Phys. Chem. Chem. Phys., 2023, 25, 21006–21019 RSC.
- J. Prasongkit, R. G. Amorim, S. Chakraborty, R. Ahuja, R. H. Scheicher and V. Amornkitbamrung, J. Phys. Chem. C, 2015, 119, 16934–16940 CrossRef CAS.
- D. K. Nguyen, D.-Q. Hoang and D. M. Hoat, RSC Adv., 2022, 12, 9828–9835 RSC.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140, 084106 CrossRef.
- W. Tang, E. Sanville and G. Henkelman, J. Phys. Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Comput. Chem., 2016, 37, 1030–1035 CrossRef CAS PubMed.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- D. M. Bylander and L. Kleinman, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 13756–13761 CrossRef.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- D. L. V. K. Prasad and E. D. Jemmis, Phys. Rev. Lett., 2008, 100, 165504 CrossRef.
- Cohesive Energy | The Elements Handbook at KnowledgeDoor, https://www.knowledgedoor.com/2/elements_handbook/cohesive_energy.html, accessed August 4, 2023 Search PubMed.
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- B. Ruqia, G. M. Tomboc, T. Kwon, J. Kundu, J. Y. Kim, K. Lee and S.-I. Choi, Chem Catal., 2022, 2, 1961–1988 CrossRef CAS.
- Y. Ouyang, L. Shi, X. Bai, Q. Li and J. Wang, Chem. Sci., 2020, 11, 1807–1813 RSC.
- C. Shi, H. A. Hansen, A. C. Lausche and J. K. Nørskov, Phys. Chem. Chem. Phys., 2014, 16, 4720–4727 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Optimized structures of pristine silicene and germanene monolayers, optimized geometries of CO* and H* potential intermediates, partial density of states of CO* and CHO* adsorption on SACs, AIMD simulation results, kinetic analysis of CH3OH* analysis, free energy profiles with applied potentials and in vacuum, optimized configurations of CO* and CHO* adsorption on four systems and their corresponding bond lengths, coordinates of optimized structures of lowest-energy intermediates. See DOI: https://doi.org/10.1039/d3ta05155b |
| This journal is © The Royal Society of Chemistry 2024 |