
Solvation structure regulation of an organic small molecule additive for dendrite-free aqueous zinc-ion batteries†
Xiaomin
Li‡
a,
Jinwei
Miao‡
b,
Fulong
Hu
a,
Kang
Yan
b,
Lin
Song
 a,
Huiqing
Fan
b,
Longtao
Ma
c and
Weijia
Wang
*b
a,
Huiqing
Fan
b,
Longtao
Ma
c and
Weijia
Wang
*b
aFrontiers Science Center for Flexible Electronics, Institute of Flexible Electronics, Northwestern Polytechnical University, Xi'an 710072, P. R. China
bState Key Laboratory of Solidification Processing, School of Materials Science and Engineering, Northwestern Polytechnical University, Xi'an 710072, P. R. China. E-mail: weijia.wang@nwpu.edu.cn
cSchool of Materials Science and Engineering, Guangdong Provincial Key Laboratory of Advanced Energy Storage Materials, South China University of Technology, Guangzhou 510641, China
First published on 27th November 2023
Abstract
The aqueous zinc-ion battery is a potential energy storage device due to its environmental sustainability and cost-effectiveness. Nonetheless, the reduced reversibility of the Zn anode arising from continuous parasitic reactions (hydrogen evolution reaction (HER) and corrosion) and random dendrite growth has a significant impact on its life and performance. Herein, organic small-molecule formamide (FA) is proposed as an additive to the aqueous ZnSO4 electrolyte and realizes a dendrite-free and highly reversible Zn anode by regulating the solvation structure. Through theoretical calculation, as well as physicochemical and electrochemical characterization, it is evidenced that FA additive shows the following characteristics: (1) it replaces some of the H2O molecules in the solvation structure to participate in the solvation structure of Zn2+, and significantly inhibit the HER, (2) breaks the original hydrogen bonding network, resulting in the improvement of the low temperature performance of the battery, (3) preferentially adsorbs on the Zn surface to regulate the charge distribution at the Zn anode/electrolyte interface and inhibit Zn corrosion during the initial cycling process, and (3) contributes to the formation of an inorganic–organic double-layer solid electrolyte interface (SEI) for dendrite-free uniform zinc deposition. Benefiting from these advantages, Zn‖Cu asymmetric batteries assembled with this electrolyte show more than 1700 highly reversible zinc plating/stripping cycles, and the average coulombic efficiency reaches 99.24%. Zn‖Zn symmetric batteries survive more than 1600 h of stable cycling at 1 mA cm−2 and 0.5 mA h cm−2. As a proof of concept, the Zn‖MnO2 full cells provide excellent cycle performance over 2000 cycles at the current density of 4 A g−1, and maintain a high capacity retention of 85.1%.
1. Introduction
Aqueous zinc-ion batteries have the advantages of high safety, low cost and non-toxicity,1–4 while their continuous electrochemical corrosion induced by the aqueous electrolyte and the resulting low reversibility of the Zn anode encumber their further applications.5–9 Specifically, when the battery is at rest or in operation, the solvated Zn2+ near the electric double-layer (EDL) structure needs to overcome an energy barrier to desolvate and to release a large number of electrochemically reactive H2O molecules. The reactive H2O molecules lead to the evolution of H+ into H2 and the variation of local pH values, which will attack the metallic Zn and trigger parasitic side reactions. As a results, a heterogeneous zinc surface and a porous structure is formed, which inevitably exacerbate the chemical corrosion of the Zn anode during repeated plating/stripping processes.10–12 Meanwhile, loose Zn particles with irregular morphology lead to the loss of electrical contact between the deposited Zn and the substrate, further deteriorating the reversibility of the Zn anode.13 To make matters worse, irregularly shaped zinc dendrite particles tend to puncture the separator, resulting in a short circuit in the battery.14–16 To alleviate the above problems, researchers have proposed strategies such as constructing artificial interfacial layers,17 modifying collectors,18 optimizing the internal structure of the zinc anode,19 modifying the separator,20 and increasing the salt concentration21 to protect the Zn anode from the deleterious reactions associated with aqueous electrolyte. While these methods provide new insights into zinc metal protection, they are difficult to implement from the perspective of manufacturing process and production cost.22 Considering the above difficulties, it would be more practical to explore suitable electrolyte additives to inhibit the formation of dendrites and to improve the reversibility of the Zn anode.23–26 Currently, the commonly used electrolyte additives for aqueous zinc-ion batteries can be categorized into four types, including ionic, organic, inorganic, and metallic additives.27,28 Among them, organic additives have attracted wide attention due to their high solubility in aqueous solution and unique regulation mechanisms.11,24,29Most electrolyte additive engineering strategies are based on regulating the solvation structure of the Zn2+ hydration layer [Zn(H2O)62+], which reduces the solvation effect between Zn2+ and H2O molecules, thus inhibiting the side reactions. In addition, the regulation of solvation structure can affect the desolvation and nucleation processes of Zn2+, leading to a more uniform deposition of zinc.30 These organic solvents are mixed with aqueous solvents to form a mixed electrolyte that retains the inherent advantages of each system, namely non-flammability and non-toxicity of the aqueous system and the improved electrochemical stability of the non-aqueous system.31 Using organic solvents successfully resolves the conflict between performance, cost, interface chemistry, sensitivity to ambient humidity, and environmental friendliness. In mixed electrolytes, it is necessary to make a reasonable selection of organic molecules.32 Generally speaking, organic molecules need to have a strong polarity (usually containing N, O, F, and their lone pair electrons can be coordinated with metal ions); be miscible with water and form strong hydrogen bonds and reduce the activity of water; contain functional groups that can participate in chemical reactions and be reduced to form a dense solid electrolyte interface (SEI). As an example, Wang et al. reported a ZnCl2/H2O-DMSO electrolyte by introducing the organic additive DMSO into the ZnCl2 aqueous electrolyte.33 Since the donor number of DMSO (29.8) is much higher than that of H2O (18), the solvation structure of Zn2+ preferentially combines with DMSO and therefore alters the hydrated Zn2+ solvation structure. During zinc deposition, DMSO is more likely reduced to generate a dense Zn12(SO4)3Cl3(OH)15–5H2O–ZnSO3–ZnS SEI, which inhibits the decomposition of solvated H2O. In addition, the activation of H2O is greatly reduced, due to the enhanced interaction between H2O and DMSO. All the phenomena above demonstrate that DMSO plays the role of inhibiting the passivation of the zinc surface, impeding the formation of zinc dendrites, and reducing the decomposition of H2O. Triethyl phosphate (TEP) is also considered as a cosolvent with a strong solvation ability in aqueous/nonaqueous electrolytes. TEP has a higher donor number (26 kcal mol−1) than H2O (18 kcal mol−1) and tends to form an inner solvation structure occupied by TEP around Zn2+ with a strong interaction of H-bonds. The coordination structure of TEP can reduce water activity to inhibit cathode dissolution and form a solid polymer–inorganic interphase with composition of poly-ZnP2O6 and ZnF2 to effectively prevent Zn dendrite growth and parasitic HER. Meanwhile, organic solvents such as TEP and trimethyl phosphate (TMP) can still maintain the safety of aqueous electrolytes and improve the reversibility of Zn.34 Acrylamide (AM) was introduced into a high-voltage aqueous electrolyte for aqueous Zn-ion batteries, in which small dipole molecules replace part of the water molecules and participate in the solvation structure of Zn2+, thus reducing the activity of water and expanding the ESW. In addition, these dipole molecules can form hydrogen bonds with water molecules and disrupt the hydrogen bond network, thereby reducing parasitic reactions associated with water molecules.35 It is of great importance to develop easier-to-handle and environmentally friendly organic additives with excellent zincophilicity. Regulating the solvation structure can provide experience in electrolyte development engineering, as well as guidance for the rational design of a stable and reversible Zn anode.24
In this work, a simple electrolyte additive engineering strategy is developed by introducing formamide (FA) into ordinary ZnSO4 electrolytes to modulate the surface chemistry of the Zn anode and bulk electrolyte structure. For the unprotected Zn anode, Zn2+ tends to nucleate at protrusions with low nucleation potentials, which inevitably leads to deterioration of dendrites. Due to the strong interaction between the carbonyl group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of the FA molecule and the zinc atoms on the surface of the Zn anode, a hydrophobic interface is formed, which reduces the reactivity of H2O with the Zn anode. This unique metal–molecule interface acts as a corrosion inhibitor against water-induced side reactions, resulting in a corrosion-free Zn anode that avoids worsening of the tip effect. Meanwhile, the lone pair electrons on the O element make FA zincophilic and provide rich sites to coordinate with Zn2+, which fundamentally reduces the activity of H2O and inhibits the HER, thus improving the quality of zinc deposition. In addition, FA molecules are preferentially reduced over H2O molecules, resulting in the in situ generation of an SEI, which realizes a good protective effect on the Zn anode.
O) of the FA molecule and the zinc atoms on the surface of the Zn anode, a hydrophobic interface is formed, which reduces the reactivity of H2O with the Zn anode. This unique metal–molecule interface acts as a corrosion inhibitor against water-induced side reactions, resulting in a corrosion-free Zn anode that avoids worsening of the tip effect. Meanwhile, the lone pair electrons on the O element make FA zincophilic and provide rich sites to coordinate with Zn2+, which fundamentally reduces the activity of H2O and inhibits the HER, thus improving the quality of zinc deposition. In addition, FA molecules are preferentially reduced over H2O molecules, resulting in the in situ generation of an SEI, which realizes a good protective effect on the Zn anode.
2. Experimental
MnO2 cathode materials are prepared by the hydrothermal method. 2.67 mmol (0.403 g) of MnSO4 and 16 mmol (2.529 g) of KMnO4 are added into 70 ml of deionised water and stirred until the reactants are completely dissolved. The solution is transferred into a hydrothermal kettle and reacted at 160 °C for 12 h. The suspension is collected and washed with deionized water and anhydrous ethanol several times to remove impurities, finally the MnO2 product is obtained by drying at 60 °C.36 The physical characterization of the zinc foils, by-products and SEI is mainly carried out by X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS). The coordination state of Zn2+ in aqueous solution is detected by Raman spectroscopy and Fourier transform infrared spectroscopy (FTIR). The surface morphology of the zinc foils is observed using a Field Emission Scanning Electron Microscope (FESEM), and the SEI is observed by transmission electron microscopy (TEM). The manufactured CR2032 coin cells are subjected to galvanostatic charge–discharge (GCD) tests using battery test systems. The electrochemical tests are all performed using an electrochemical workstation via techniques such as linear scanning voltammetry (LSV), Tafel, chronoamperometry (CA), cyclic voltammetry (CV), and electrochemical impedance spectroscopy (EIS).3. Results and discussion
FA, as an excellent organic solvent, has the advantages of high dielectric constant, strong polarity, low viscosity and fast ion diffusion. Herein, 2 M ZnSO4 is chosen as the base electrolyte and 30% FA electrolyte is prepared by adjusting the volume ratio of FA to H2O (volume ratio of FA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 3:7). Similarly, electrolytes containing 10% FA, 20% FA, 40% FA and 50% FA are also prepared in the same way. As shown in Fig. S1,† the electrolyte with 50% volume fraction of FA crystallizes out after standing for one month, while others show a uniform dissolved state without significant change.
:H2O = 3:7). Similarly, electrolytes containing 10% FA, 20% FA, 40% FA and 50% FA are also prepared in the same way. As shown in Fig. S1,† the electrolyte with 50% volume fraction of FA crystallizes out after standing for one month, while others show a uniform dissolved state without significant change.
3.1 Zn2+ solvation structure and physicochemical properties of electrolytes
Firstly, the role of FA in regulating the solvation structure of Zn2+ is demonstrated by theoretical calculation.37Fig. 1a shows the electrostatic potential diagrams of the FA molecule and H2O molecule. Obviously, similar to the O atom in the H2O molecule, the carbonyl group (CO) in FA has a negative potential, indicating that there is a strong electrostatic potential between Zn2+ and FA, which makes them attract each other. The evolution of the solvation structure of Zn2+ is investigated by molecular dynamics (MD) simulation, and the electrolyte with FA volume fraction of 30% is chosen for demonstration (denoted as ZnSO4/FA). Fig. 1b shows the snapshot of the MD simulation of the pure ZnSO4 electrolyte and the typical solvation structure, where Zn2+ is found to coordinate with H2O molecules and SO42− anions to form the [Zn(H2O)6−x(SO4)x]2+ solvation structure. As presented in the snapshot of the MD simulation of ZnSO4/FA electrolyte (Fig. 1c), a solvation structure consisting of Zn2+ coordinated with four H2O molecules and two FA molecules is observed, indicating that FA molecules coordinate with Zn2+ and break the original solvation structure, i.e., FA molecules replace some H2O molecules to participate in the solvation structure of Zn2+. The distribution of the nearest-neighbor molecules is investigated by the radial distribution function (RDF), and the differences between the solvation structure of Zn2+ in pure ZnSO4 electrolyte and ZnSO4/FA electrolyte are discussed quantitatively. As displayed in Fig. 1d and S2a,† for the pure ZnSO4 electrolyte, Zn2+ is coordinated with 5.17 H2O molecules and 0.83 SO42− anions to form the [Zn(H2O)5.17(SO4)0.83]2+ solvation structure. Due to the introduction of FA additive, the peak at 2.05 Å can be attributed to Zn2+–O(FA) (Fig. 1e), demonstrating the participation of FA molecules in the solvation structure of Zn2+. To be precise, the Zn2+ primary solvation structure in the ZnSO4/FA electrolyte consists of about 4.22 H2O molecules, 0.53 FA molecules and 1.25 SO42− anions (Fig. S2b†), expressed as [Zn(H2O)4.22(FA)0.53(SO42−)1.25]2+. In addition, it is found that the amount of SO42− coordinated with Zn2+ increased with the addition of FA, indicating a transformation of the electrolyte structure from the solvent separated ion pair (SSIP) to contact ion pair (CIP). The transformation of the average solvation structure of Zn2+ indicates a greatly reduced amount of solvated H2O, thus mitigating the HER and hopefully protecting the Zn anode from surface corrosion. As shown in the linear scanning voltammetry (LSV) (Fig. S3†), the addition of FA leads to a lower HER potential as compared with pure ZnSO4 electrolyte (from −0.139 V to −0.181 V vs. Zn2+/Zn).
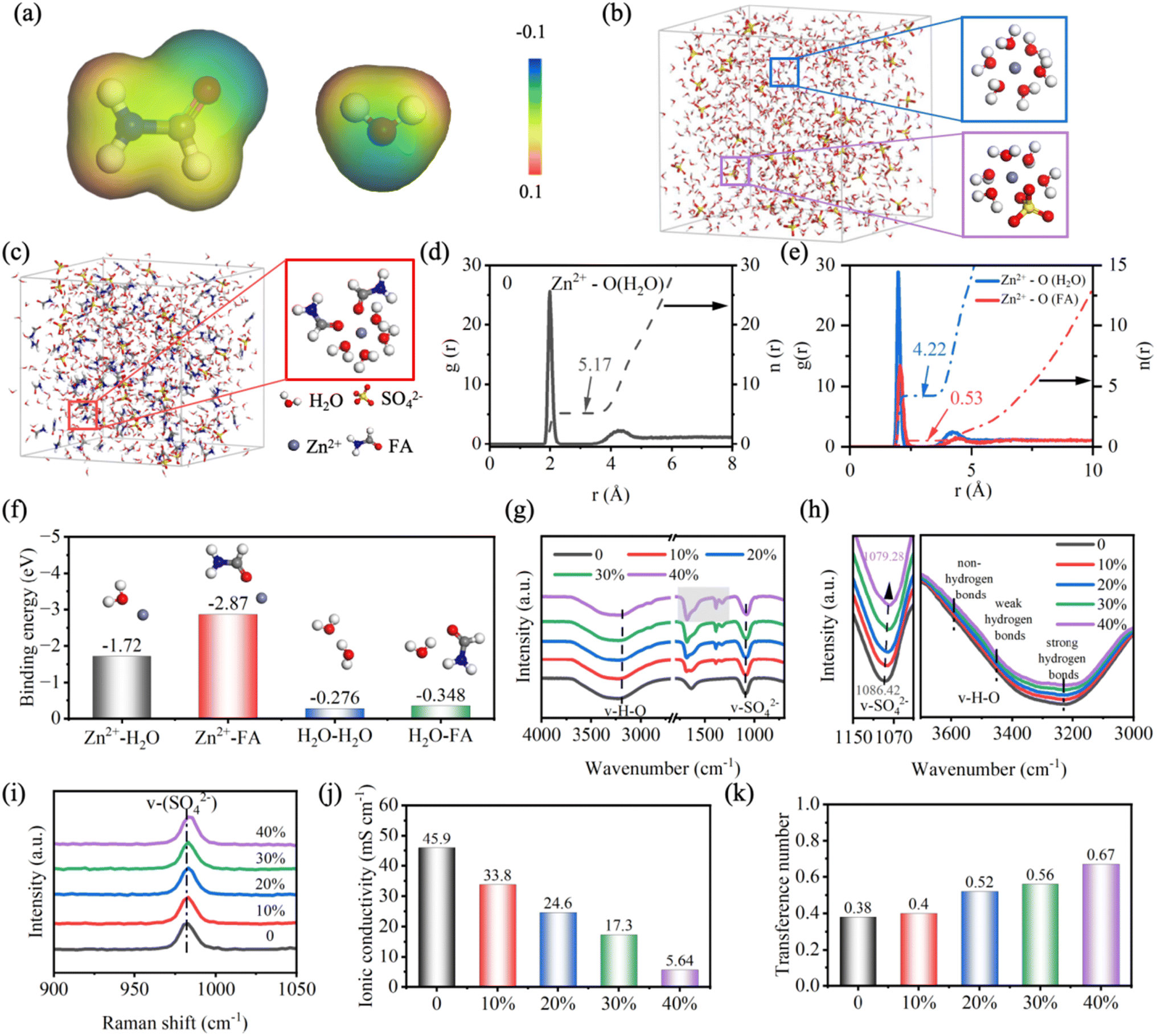 | ||
| Fig. 1 (a) Electrostatic potential diagram of formamide (FA) and H2O molecules. Molecular dynamics (MD) simulation snapshots and typical Zn2+ solvation structure of (b) pure ZnSO4 electrolyte and (c) ZnSO4/FA electrolyte. Radial distribution function (RDF) plots of Zn2+–O of (d) pure ZnSO4 electrolyte and (e) ZnSO4/FA electrolyte. (f) Binding energy of Zn2+–H2O, Zn2+–FA, H2O–H2O, and H2O–FA. Infrared spectra (g) and the corresponding enlarged view of v-SO4 and v-H–O (h) of ZnSO4 electrolytes without and with various concentrations of FA. (i) Raman spectra of ZnSO4 electrolytes without and with various concentrations of FA. (j) Ionic conductivity and (k) Zn2+ transference number of ZnSO4 electrolytes without and with various concentrations of FA. | ||
Density functional theory (DFT) calculation also illustrates the influence of FA additive on the primary solvation structure of Zn2+. As displayed in Fig. 1f, the binding energy of the FA molecule with Zn2+ is stronger than that of H2O (−2.87 eV vs. −1.72 eV), indicating that FA is more likely to replace the H2O in the solvation structure.38 Meanwhile, the binding energy between FA and H2O (−0.35 eV) is higher than that between H2O and H2O (−0.28 eV), suggesting that H2O inclines to bind with FA thereby damaging the H-bond network in the native electrolyte, thus lowering the freezing point and facilitating the operation of the batteries at low temperatures.
In order to better understand the interaction of FA with the components of the original ZnSO4 electrolyte, Raman and Fourier transform infrared (FT-IR) tests are performed. Raman spectra of different electrolytes are shown in Fig. 1i. The peaks occurring near 980 cm−1 can be assigned to the v-SO42− band. Based on the classical Eigen–Tamm (ET) mechanism, these strong peaks can be divided into contact ion pairs (CIP) and solvent-separated ion pairs (SSIP).39,40 With the addition of FA, the peak of the v-SO42− band shifts to higher frequencies, and the proportion of CIPs gradually increases, which proves the transformation of solvation structure from SSIP to CIP. The detailed results are displayed in Fig. S4.† The influence of FA molecules on the interaction between Zn2+ and SO42− is further confirmed by FT-IR results (Fig. 1g and h). In addition to the FA molecule (1677.45, 1387.64, 1323.41 cm−1), the stretching vibration of v-SO42− (ZnSO4 1086.42 cm−1, ZnSO4/FA 1079.28 cm−1) shows a redshift, suggesting an enhancement of the electrostatic coupling between Zn2+ and SO42−, which further indicates the increase of the proportion of CIP.40,41 In addition, a distinct wide peak at 3000–3700 cm−1 can be attributed to the O–H stretching vibration of H2O, which can be further divided into three peaks, corresponding to strong hydrogen bonds (∼3230 cm−1), weak hydrogen bonds (∼3450 cm−1), and non-hydrogen bonds (∼3620 cm−1), respectively.42,43 For pure ZnSO4 electrolyte, strong hydrogen bonds account for the highest proportion in the hydrogen bond environment, but with the addition of FA, the strong hydrogen bonds attenuated more significantly, and Fig. S5† shows the results of the three hydrogen bonds. We can see that with the increase of FA, the proportion of strong hydrogen bonds decreases from 83.03% to 80.03%, while the proportion of weak hydrogen bonds increases from 13.73% to 18.24%, and the proportion of non-hydrogen bonds is small and has little change. This indicates that the original H-bond network in water is weakened, resulting in the inhibition of the HER, but at the same time the ionic conductivity of the electrolyte may be affected. The local environment of 1H from H2O in different electrolyte solutions was further investigated by nuclear magnetic resonance (1H NMR) measurements, as shown in Fig. S6.†44 The 1H chemical shift peak moves to a higher value after adding FA, which is mainly because the strong interaction between FA and H2O molecules weakens the electron density around the protons in the H2O molecules, which also demonstrates the changes in solvation structure and hydrogen bonding.
In order to investigate the effect of FA on ion transport, ionic conductivity and transference number are tested. As shown in Fig. 1j, the ionic conductivity in pure ZnSO4 electrolyte is 45.9 mS cm−1, which decreases to 33.8, 24.6, 17.3, and 5.64 mS cm−1 with the increase of FA with a volume ratio of 10%, 20%, 30%, and 40%, respectively. This may be ascribed to the slow migration of cations due to elevated electrolyte viscosity with the addition of additives. The detailed results are shown in Fig. S7,† which is consistent with FT-IR results. As presented in Fig. 1k, the transference number of Zn2+ increases from 0.38 to 0.67 as FA increases (detailed results are shown in Fig. S8†). This suggests that addition of FA promotes mass transfer at the electrolyte/anode interface, thereby facilitating the deposition of Zn. Ionic conductivity is influenced by factors such as zinc salt, composition of solvent, temperature, etc. When zinc salt content and temperature are consistent, the composition of solvent plays a significant role. In general, the higher the solvent viscosity, the lower the ionic conductivity. The viscosity of ZnSO4 electrolytes without and with various concentrations of FA is shown in Fig. S9.† As the FA volume fraction increases from 0 to 40%, the viscosity increases, resulting in a decrease in ionic conductivity. Regarding transfer numbers, the anion migration is greatly reduced due to the formation of a unique solvation structure, while the cation is less affected, therefore the transfer number of Zn2+ increases with the increase of FA volume fraction.33 As a result, the ionic conductivity and transfer numbers exhibit an opposite trend with the increase in concentration. In order to balance the adverse effects of the reduction of ion conductivity caused by FA additive, 30% addition is a relatively better choice. It possesses a high Zn2+ transference number and appropriate conductivity, so the electrolyte with FA volume ratio of 30% and pure ZnSO4 electrolyte are investigated, denoted as ZnSO4/FA and ZnSO4, respectively.
3.2 Adsorption of FA and optimized electrolyte/anode interface characteristics
The adsorption energies of H2O and FA molecules on the surface of Zn are analyzed by density functional theory (DFT) (Fig. 2a). Specifically, the adsorption energy of FA molecules on the Zn (002) (−0.77 eV) and (100) (−0.56 eV) planes is much lower than that of H2O molecules on the Zn (002) (−1.45 eV) and (100) (−0.46 eV) planes. The adsorption energy calculation results demonstrate that the ZnSO4/FA electrolyte has zincophilic properties. FA molecules replace the adsorbed H2O dipoles at the anode/electrolyte interface in the aqueous system, which significantly reduces the amount of free water and leads to a poor H2O metal/molecule interface. To verify the zincophilic properties of FA molecules, the contact angle of the electrolytes on the surface of zinc foil is tested (Fig. 2b). It is found that the contact angle of ZnSO4/FA electrolyte (99.11°) is smaller than that of pure ZnSO4 electrolyte (102.12°), indicating that the addition of FA improves the zincophilic properties of the electrolyte, which is consistent with the calculation of adsorption energy. The effect of the metal–molecule interface on zinc metal corrosion is quantified by a corrosion test (Fig. 2c).45–47 Compared with pure ZnSO4 electrolyte, the Zn anode in ZnSO4/FA electrolytes has a lower corrosion current density (from 39.62 mA cm−2 to 5.28 mA cm−2), and the corrosion potential is more positive, indicating that the metal–molecule interface enhances the corrosion resistance of the Zn anode in ZnSO4/FA electrolyte.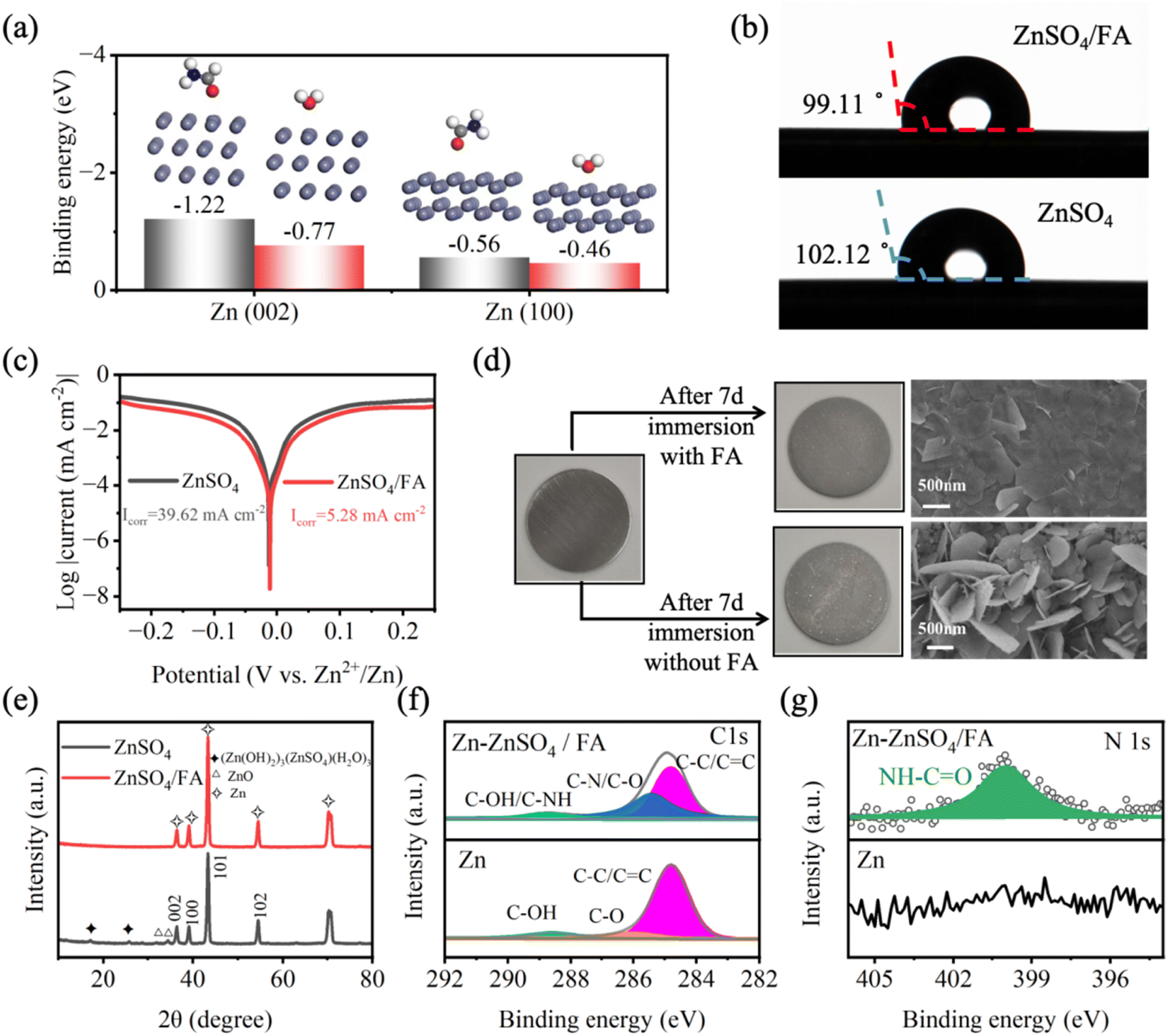 | ||
| Fig. 2 (a) The adsorption energy of H2O and FA molecules on Zn (002) and Zn (100) surfaces, respectively. (b) Contact angles of pure ZnSO4 electrolyte and ZnSO4/FA electrolyte. (c) Linear polarization curves of the Zn anode in the pure ZnSO4 electrolyte and ZnSO4/FA electrolyte. (d) Optical photographs and SEM images of the pristine Zn anode and Zn anode after immersion. (e) XRD patterns of the Zn anode after immersion. XPS spectra of (f) C 1s and (g) N 1s for the Zn foil before and after immersion. | ||
Furthermore, self-corrosion tests are carried out to verify the protective effect of the metal–molecule interface during the battery rest by soaking the zinc metal in electrolytes for 7 days. In general, due to the thermodynamic instability of zinc in aqueous solution, self-corrosion occurs spontaneously when the zinc metal is immersed in the aqueous electrolyte, resulting in the loss of the zinc anode and the formation of loose and irregular sheets of corrosion by-products on the surface.48 Compared with pure ZnSO4 electrolyte, the surface of zinc metal immersed in ZnSO4/FA electrolyte for 7 days is more homogeneous and smoother (Fig. 2d), indicating that the adsorbed FA layer can effectively block the side reactions in aqueous electrolyte, which is also confirmed by XRD results (Fig. 2e). In the XRD pattern of the zinc foil after cycling in the pure ZnSO4 electrolyte, it can be observed that peaks corresponding to ZnO and (Zn(OH)2)3(ZnSO4)(H2O)3 by-products appear in the range of 0° to 35°. However, in the XRD pattern of the zinc foil after cycling in the ZnSO4/FA electrolyte, no obvious by-product peaks are observed, indicating that the FA additive has an inhibition effect on side reactions. Subsequently, the binding structure between adsorbed FA and the Zn anode is studied by X-ray photoelectron spectroscopy (XPS) (Fig. 2f and g). In the C 1s spectrum of the zinc foil soaked in ZnSO4/FA electrolyte, a new peak of C–NH appears, which is correlated with the FA additive. Similarly, in the N 1s spectrum, it can be found that there is no peak related to N in the pure ZnSO4 electrolyte. However, a peak of NH–CO appears in the ZnSO4/FA electrolyte, which provides evidence of the adsorption of FA molecules on the surface of zinc metal. In addition, in order to further illustrate the inhibition effect of metal–molecule interface on side reactions, the dynamic measurement of Zn‖Zn symmetric batteries is carried out by alternating cycling and resting measurement methods, that is, 30 h of resting after every 30 h of cycling at a current density of 1 mA cm−2 and a capacity of 0.5 mA h cm−2. As shown in Fig. S10,† the voltage distribution of the Zn‖Zn symmetric battery containing pure ZnSO4 electrolyte shows a serious fluctuation after a 123 h test, while the Zn‖Zn symmetric battery containing the FA electrolyte shows stable voltage hysteresis throughout the 900 h test, demonstrating the ability of the metal–molecule interface to stabilize the Zn anode.
3.3 SEI characteristic and homogeneous zinc deposition
The energy levels of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of FA and H2O molecules are calculated to clarify the redox stability (Fig. S11†). It is found that FA has a higher HOMO energy level and a lower LUMO energy level than H2O (−5.877 eV vs. −6.748 eV, −0.599 eV vs. 1.061 eV), which means that FA molecules easily gain electrons, and are reduced to SEI before H2O reduction, thereby inhibiting side reactions on the zinc surface and reducing the production of by-products. In order to obtain more accurate results, zinc was deposited directly on a blank TEM copper grid using ZnSO4/FA electrolyte, and TEM was performed. As shown in Fig. S12,† the lattice fringes of ZnO (002) and ZnS (111) are identified in the SEI layer, which is formed in the first few charge and discharge cycles. In the energy dispersive X-ray spectroscopy (EDS) mapping, it can be observed that N, Zn, O, and S elements are uniformly dispersed on the whole particle surface within the Zn deposits.49 In addition, the surface was further investigated by XPS with different sputtering depths. Combined with peak fitting (Fig. 3a–d), it is evidenced that the component at the top surface is primarily composed of organic components, represented by C–O, CO and C–C. The signal of inorganic components ZnS and ZnO becomes stronger when sputtered to the depth of 10 nm. Additionally, the non-sputtered electrode shows the weakest signal in the Zn 2p spectrum due to the overcoating of the organic component. The analysis indicates that the upper surface of SEI is primarily composed of organic components, while the inner part of SEI is composed of inorganic substances like ZnS and ZnO. The SEI layer not only blocks the contact between the zinc metal and the water and inhibits the side reactions, but also provides the diffusion path of Zn2+ and homogenizes the Zn2+ flux.50 According to previous reports, ZnS and ZnO play an important role in improving the stability and reversibility of zinc anodes.51,52 In order to further understand the positive effects of the in situ constructed SEI on stabilizing the Zn anode and on regulating deposition behavior, the morphology of deposited Zn and the electrochemical properties of ZnSO4 and ZnSO4/FA electrolytes are further explored. Fig. 3e and f are the SEM images of the surface of the zinc foils after one cycle in the two electrolyte system. It can be seen that the surface of the zinc foil using pure ZnSO4 electrolyte has a typical dendrite morphology, while the surface of the zinc foil using ZnSO4/FA electrolyte is flat. According to the SEM images of the cross sections of zinc foils after cycling in the two electrolyte system as shown in Fig. 3g and h, the zinc foil with ZnSO4/FA electrolyte is smooth and dense, while the zinc foil with pure ZnSO4 electrolyte has obvious protrusions on the surface and disordered and loose deposition. Not surprisingly, the deposition thickness (32 μm) is larger than that in the ZnSO4/FA electrolyte (28 μm), demonstrating that FA can promote uniform and dense zinc deposition.
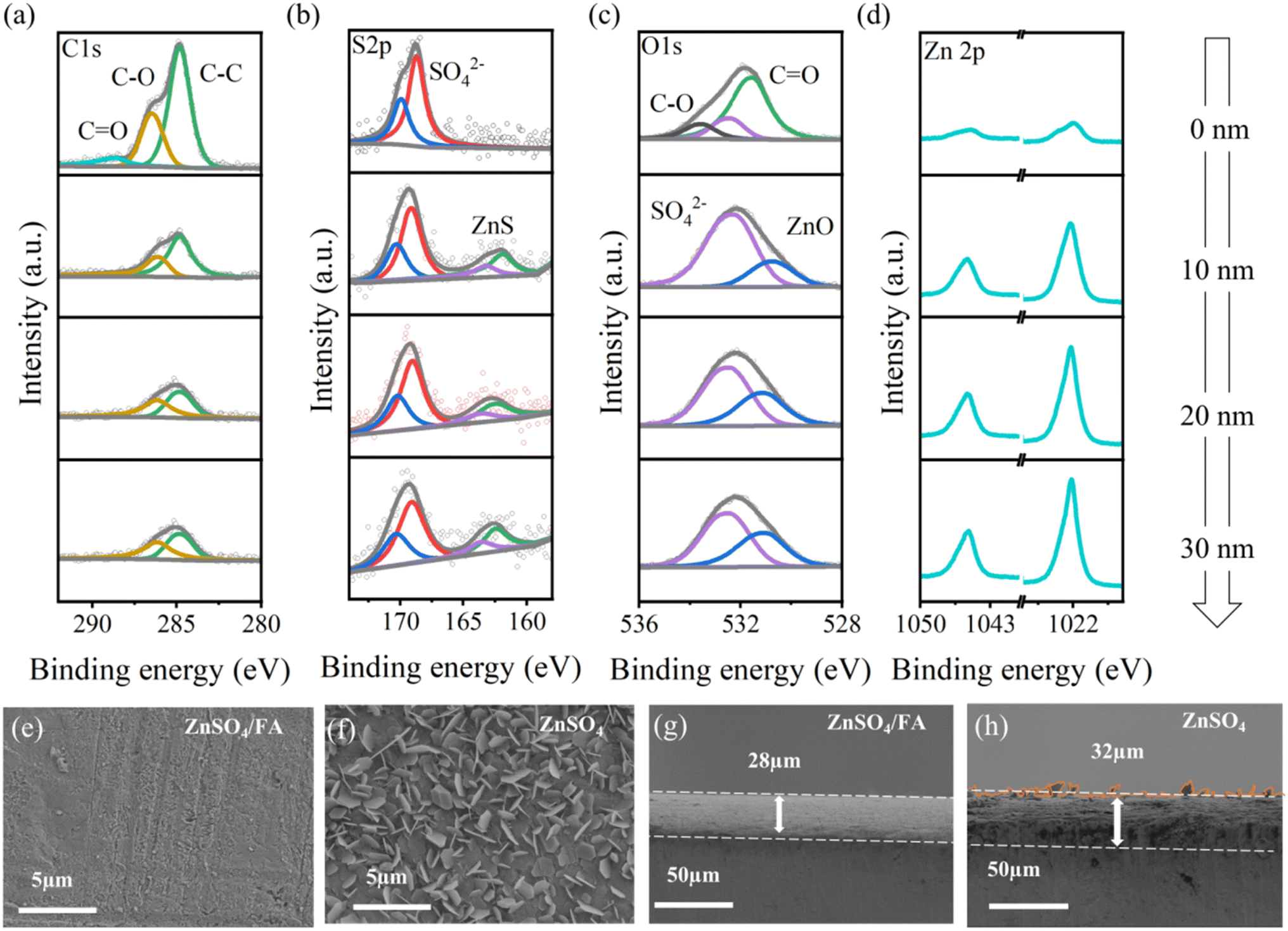 | ||
| Fig. 3 (a–d) XPS depth profile of the Zn anode after 25 plating/stripping cycles in ZnSO4/FA electrolyte at a current density of 1 mA cm−2 with a capacity of 0.5 mA h cm−2. (e and f) The plan-view and (g and h) cross section SEM images of the Zn anode after a plating/stripping cycle in (e and g) ZnSO4/FA electrolyte and (f and h) pure ZnSO4 electrolyte at a current density of 1 mA cm−2 with a capacity of 0.5 mA h cm−2. | ||
CA is an electrochemical method for characterizing changes in the concentration of electroactive substances near a surface. Fig. S13† shows the change of current response determined by the nucleation center with time during deposition time of 300 s under −200 mV overpotential. For pure ZnSO4 electrolyte, the current density reaches its stable value (∼−125 mA cm−2) shortly after overpotential being applied, suggesting activation of all nucleation sites. In contrast, the activation time of the battery using ZnSO4/FA electrolyte is prolonged, and the current continues to increase slowly and maintains a smaller current value than that of the pure ZnSO4 electrolyte, which implies that the nucleation sites in the ZnSO4/FA electrolyte are slowly and gradually exposed, avoiding the rapid uneven growth of zinc nuclei and thus inhibiting the growth of zinc dendrites. The differences in the nucleation mechanism and the stable current density can be attributed to the adsorption of FA molecules on the surface of the Zn anode, thereby reducing the number of nucleation sites in the ZnSO4/FA electrolyte and slowing down the formation of nuclei. In addition, from the deposition behavior of zinc (Fig. S14†), it is suggested that the nucleation and growth overpotential in the ZnSO4/FA electrolyte are higher than those in pure ZnSO4 electrolyte. The higher nucleation overpotential usually requires a stronger driving force in the initial nucleation process, thereby depositing fine zinc particles and inhibiting zinc dendrite growth. The XRD pattern of zinc anode cycling for 20 cycles in pure ZnSO4 electrolyte and ZnSO4/FA electrolyte (Fig. S15†) shows that for the Zn anode cycling in pure ZnSO4 electrolyte a peak of Zn4(OH)6SO4·xH2O appears at about 9.8°, indicating that the addition of FA can inhibit the side reactions.
3.4 Highly reversible zinc deposition/stripping behavior
In order to further investigate the effect of FA additive on the stability and reversibility of aqueous zinc-ion batteries, Zn‖Zn symmetric batteries and Zn‖Cu asymmetric batteries are assembled and tested. According to the galvanostatic charge–discharge (GCD) curve (Fig. 4a), the symmetric battery using ZnSO4/FA electrolyte exhibits a constant plating/stripping voltage hysteresis of about 50 mV at a current density of 1 mA cm−2 and a cut-off capacity of 0.5 mA h cm−2, with stable cycling for more than 1600 hours. The Zn‖Zn symmetric battery using pure ZnSO4 electrolyte experiences a sudden decrease in polarization voltage only after 375 hours of cycling, which may be due to a short circuit caused by dendrite growth. For the Zn‖Zn symmetric battery with ZnSO4/FA electrolyte running at a current density of 2 mA cm−2, cut-off capacity of 0.5 mA h cm−2 and current density of 0.5 mA cm−2, cut-off capacity of 0.25 mA h cm−2 can stably cycle for 1400 h and 1800 h, respectively, which are much better than those of batteries using pure ZnSO4 electrolyte under the same conditions (Fig. S16 and S17†). As presented in Fig. 4b, in the rate performance of the Zn‖Zn symmetric battery, the symmetric battery using pure ZnSO4 electrolyte has a short circuit in the cycle at the current density of 15 mA cm−2, while the symmetric battery using ZnSO4/FA electrolyte has a stable performance, showing smooth voltage distribution and small voltage hysteresis at different current densities. The detailed results of voltage hysteresis are shown in Table S1.† This excellent stability is mainly attributed to the improved transport dynamics of Zn2+ by the FA-derived SEI. The low temperature characteristics of ZnSO4/FA electrolyte are also verified. Fig. 4c shows the performance of Zn‖Zn symmetric batteries at varying temperatures. Zn‖Zn symmetric batteries using ZnSO4/FA electrolyte in the temperature range from 30 °C to −30 °C exhibit a smooth voltage distribution and small voltage hysteresis. However, in the pure ZnSO4 electrolyte, a short circuit occurs at −10 °C. These results demonstrate that a much better low temperature performance is achieved with using ZnSO4/FA electrolyte as compared with pure ZnSO4 electrolyte. The Zn‖Zn symmetric battery using ZnSO4/FA electrolyte was then tested at a low temperature of −20 °C at 1 mA cm−2 and 0.5 mA h cm−2, as shown in Fig. S18.† It can be seen that the Zn‖Zn symmetric battery can cycle stably for more than 1800 h at a low temperature of −20 °C, which further proves the good low-temperature performance of the ZnSO4/FA electrolyte. The improved low-temperature performance of the ZnSO4/FA electrolyte is attributed to the disruption of the original hydrogen bonding network in the electrolyte. The results of binding energy calculation, and the alteration in the type of hydrogen bonding as indicated in the infrared spectra, demonstrate that the addition of FA breaks the original hydrogen bonding network. This leads to a reduction in the proportion of strong hydrogen bonds between H2O molecules, and an increase in the proportion of weak hydrogen bonds between FA and H2O. The freezing process involves a complex rearrangement of disordered water molecules into ordered ice. This rearrangement is driven by forming extra hydrogen bonds (H-bonds). It is a process in which the hydrogen bond changes from disorder to order. Consequently, the breaking of the hydrogen bonding network reduces the degree of order, resulting in a lower freezing point and thus better low-temperature properties.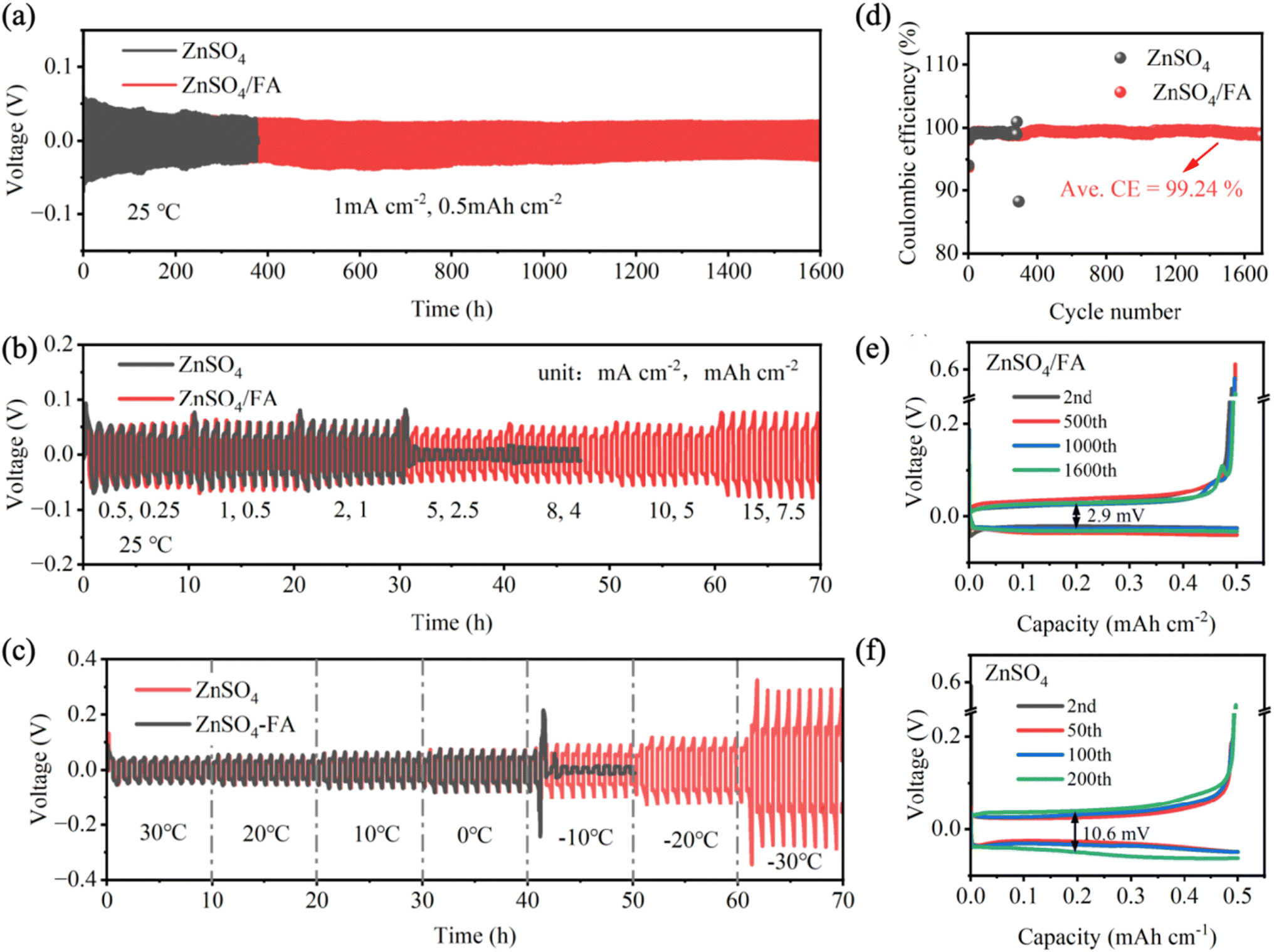 | ||
| Fig. 4 (a) Cycling performance of Zn‖Zn symmetric batteries at 1 mA cm−2 and 0.5 mA h cm−2. (b) Rate performance of the Zn‖Zn symmetric batteries. (c) Variable temperature performance of Zn‖Zn symmetric batteries in the temperature range from 30 °C to −30 °C. (d) The coulombic efficiency (CE) of the Zn‖Cu asymmetric batteries at 1 mA cm−2 and 0.5 mA h cm−2. The corresponding voltage–capacity curves for the selected cycles in (e) ZnSO4/FA electrolyte and (f) pure ZnSO4 electrolyte. | ||
In addition, the reversible zinc behavior of Zn‖Cu asymmetric batteries is investigated at the current density of 1 mA cm−2 and a cutoff capacity of 0.5 mA h cm−2 (Fig. 4d–f). The Zn‖Cu asymmetric battery using ZnSO4/FA electrolyte has a high initial coulombic efficiency of 93.71%, stable operation for more than 1700 hours, and an average coulombic efficiency of 99.24%, much better than cycling in pure ZnSO4 electrolyte (98.28% for 292 cycles). Additionally, the overpotential for the selected cycles of Zn‖Cu asymmetric batteries using ZnSO4/FA electrolyte is about 2.9 mV, which is lower than that of Zn‖Cu asymmetric batteries using pure ZnSO4 electrolyte (10.6 mV), and the voltage curve is more consistent, indicating that the Zn‖Cu asymmetric batteries using ZnSO4/FA electrolyte have better reversibility during the process of Zn stripping/plating at the electrode/electrolyte interface.
3.5 Performance of Zn‖MnO2 full cells
As a proof-of-concept, MnO2 is chosen as the cathode material for full cell assembly to verify the practicality of our strategy. Both CV curves show similar redox peaks (Fig. 5a), indicating that FA plays a negligible role in electrochemical redox processes. Meanwhile, the reduction of the redox peak in the CV curve for ZnSO4/FA may be due to the strong interaction between FA and Zn2+ and sluggish interfacial Zn2+ charge transfer kinetics.33 The Zn‖MnO2 full cells are tested for long cycle and rate performance. As shown in Fig. 5b, Zn‖MnO2 full cells with ZnSO4/FA electrolyte exhibit better long-cycle performance and maintain a discharge capacity of 160.5 mA h g−1 after 2000 cycles at a current density of 4 A g−1, with a capacity retention of 85.1%. Under the same cycling conditions, pure ZnSO4 electrolyte-based cells show significantly reduced capacity, and the retention rate is only 54.7% after 1616 cycles. Furthermore, the rate performance of the Zn‖MnO2 full cell is studied and shown in Fig. 5c–e. The Zn‖MnO2 full cell using ZnSO4 electrolyte exhibits greater capacity decay as the current increases, and an irreversible capacity decay occurs when the current density returns to 1 A g−1 (89.6% capacity retention compared to the average capacity of the 10 cycles at 1 A g−1). In contrast, the Zn‖MnO2 full cell using ZnSO4/FA electrolyte offers higher capacities at different current densities, and provides a superior capacity retention rate of 99.8% when current density returns to 1 A g−1, indicating that the reversibility of the Zn anode is improved by using FA electrolyte additive.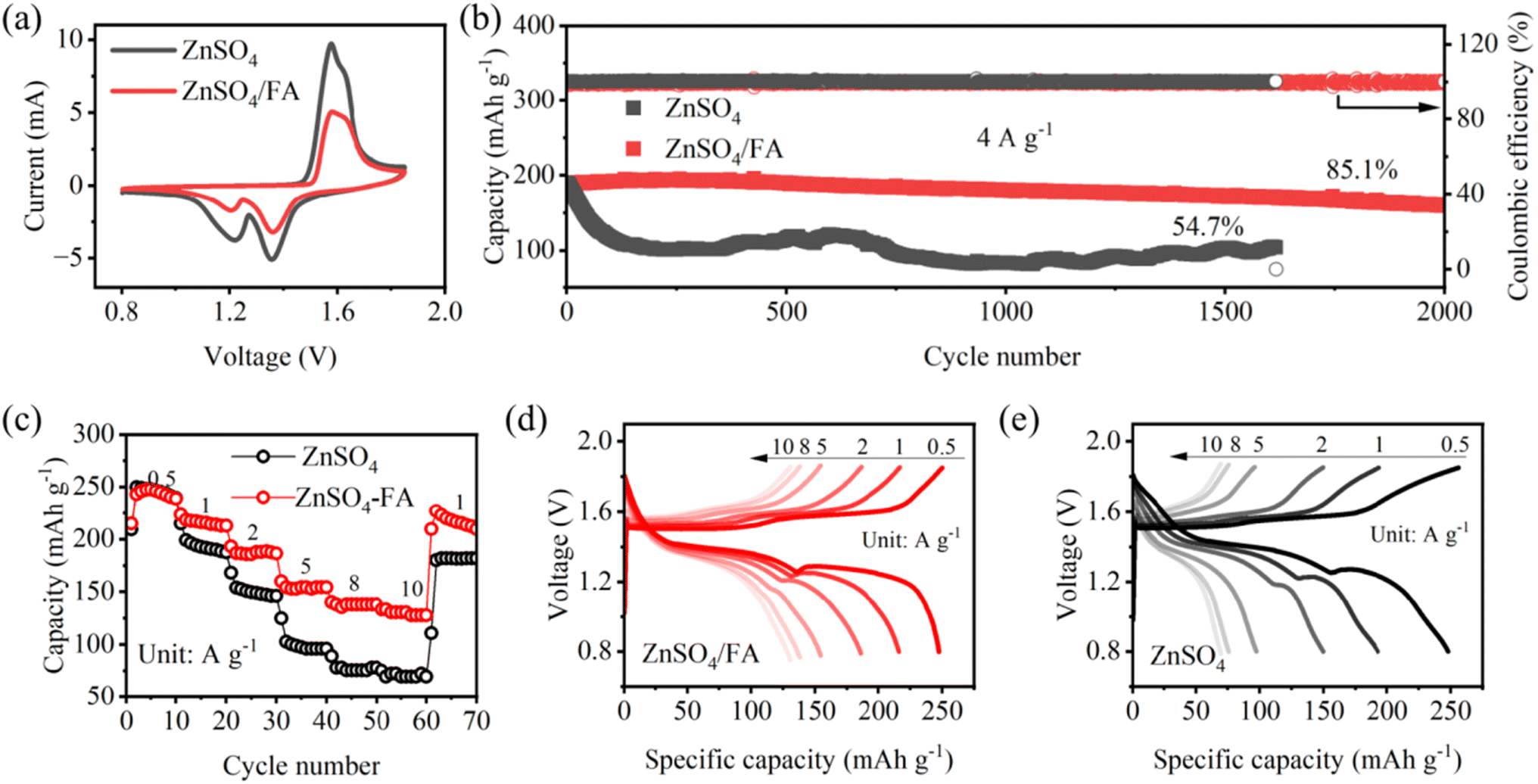 | ||
| Fig. 5 (a) The CV curves of the Zn‖MnO2 full cells in pure ZnSO4 electrolyte and ZnSO4/FA electrolyte. (b) The long-term cycling performance of the Zn‖MnO2 full cells in pure ZnSO4 electrolyte and ZnSO4/FA electrolyte. (c) The rate performance of the Zn‖MnO2 full cells in pure ZnSO4 electrolyte and ZnSO4/FA electrolyte and the corresponding charge–discharge profiles. Charging/discharging curves of Zn‖MnO2 full cells at different current densities in (d) ZnSO4/FA and (e) ZnSO4 electrolyte. | ||
4. Conclusions
In this work, formamide (FA) is introduced as an electrolyte additive to ZnSO4 aqueous electrolyte for the first time. Firstly, the addition of FA changes the solvation structure of Zn2+ and reduces the number of H2O molecules in the solvation structure. Moreover, FA interacts with the free H2O molecules to break the original hydrogen-bonding network, which radically reduces the activity of H2O molecules, inhibits the side reactions related to H2O molecules and improves the low temperature performance of the battery significantly. In addition, FA is found to be a good interfacial stabilizer. This is mainly due to the fact that FA has zinc affinity and prevents H2O from contacting the Zn anode surface by adsorbing on the surface of the Zn anode, forming a water-poor EDL structure, thus inhibiting side reactions at the interface. In the first cycle, the protective effect of adsorbed FA is dominant due to the incomplete formation of the SEI. With the charge–discharge cycles proceeding, the Zn surface will be completely covered by FA reduction products, and the dominant role of uniform zinc deposition is generated by the SEI. Benefiting from these positive effects, the Zn‖Zn symmetric battery using ZnSO4/FA electrolyte exhibits a constant plating/stripping voltage hysteresis of about 50 mV at a current density of 1 mA cm−2 and a cut-off capacity of 0.5 mA h cm−2, with stable cycling for more than 1600 hours. The Zn‖Cu asymmetric battery using ZnSO4/FA electrolyte has a high initial coulombic efficiency of 93.71% and stable operation for more than 1700 hours with an average coulombic efficiency of 99.24%. Additionally, the Zn‖MnO2 full batteries using ZnSO4/FA electrolyte have achieved excellent cycling performance with a capacity retention rate of 85.1% for more than 2000 cycles at a current density of 4 A g−1, demonstrating the great potential of the ZnSO4/FA electrolyte additive for future high-performance aqueous zinc-ion batteries.Author contributions
Xiaomin Li: conceptualization, methodology, investigation, formal analysis, data curation, writing – original draft, writing – review & editing. Jinwei Miao: formal analysis, methodology. Fulong Hu: formal analysis, writing – original draft, methodology. Kang Yan: formal analysis, methodology. Lin Song: writing – review & editing. Huiqing Fan: writing – review & editing. Longtao Ma: writing – review & editing. Weijia Wang: writing – review & editing, resources, supervision.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Xiaomin Li and Jinwei Miao contributed equally to this work. This study was sponsored by the National Natural Science Foundation of China (52272124), the Fundamental Research Funds for the Central Universities (D5000230071), and the Practice and Innovation Funds for Graduate Students of Northwestern Polytechnical University (PF2023150). This research performed SEM, TEM, XRD, Raman, and XPS tests in the Analytical & Testing Center of Northwestern Polytechnical University.References
- D. Wang, Q. Li, Y. Zhao, H. Hong, H. Li, Z. Huang, G. Liang, Q. Yang and C. Zhi, Adv. Energy Mater., 2022, 12, 2102707 CrossRef CAS.
- W. Chen, G. Li, A. Pei, Y. Li, L. Liao, H. Wang, J. Wan, Z. Liang, G. Chen, H. Zhang, J. Wang and Y. Cui, Nat. Energy, 2018, 3, 428–435 CrossRef CAS.
- J. Yan, E. H. Ang, Y. Yang, Y. Zhang, M. Ye, W. Du and C. C. Li, Adv. Funct. Mater., 2021, 31, 2010213 CrossRef CAS.
- J. Shi, T. Sun, J. Bao, S. Zheng, H. Du, L. Li, X. Yuan, T. Ma and Z. Tao, Adv. Funct. Mater., 2021, 31, 2102035 CrossRef CAS.
- J. Zhao, J. Zhang, W. Yang, B. Chen, Z. Zhao, H. Qiu, S. Dong, X. Zhou, G. Cui and L. Chen, Nano Energy, 2019, 57, 625–634 CrossRef CAS.
- J. Yang, H. Yan, H. Hao, Y. Song, Y. Li, Q. Liu and A. Tang, ACS Energy Lett., 2022, 7, 2331–2339 CrossRef CAS.
- L. Ma, M. A. Schroeder, O. Borodin, T. P. Pollard, M. S. Ding, C. Wang and K. Xu, Nat. Energy, 2020, 5, 743–749 CrossRef CAS.
- Y. Zhang, Z. Cao, S. Liu, Z. Du, Y. Cui, J. Gu, Y. Shi, B. Li and S. Yang, Adv. Energy Mater., 2022, 12, 2103979 CrossRef CAS.
- Z. Hou, Y. Gao, R. Zhou and B. Zhang, Adv. Funct. Mater., 2022, 32, 2107584 CrossRef CAS.
- Y. Lv, M. Zhao, Y. Du, Y. Kang, Y. Xiao and S. Chen, Energy Environ. Sci., 2022, 15, 4748–4760 RSC.
- Y. Liu, Y. An, L. Wu, J. Sun, F. Xiong, H. Tang, S. Chen, Y. Guo, L. Zhang, Q. An and L. Mai, ACS Nano, 2023, 17, 552–560 CrossRef CAS PubMed.
- S. H. Park, S. Y. Byeon, J.-H. Park and C. Kim, ACS Energy Lett., 2021, 6, 3078–3085 CrossRef CAS.
- Y. Yang, H. Yang, R. Zhu and H. Zhou, Energy Environ. Sci., 2023, 16, 2723–2731 RSC.
- S. Liu, J. Mao, W. K. Pang, J. Vongsvivut, X. Zeng, L. Thomsen, Y. Wang, J. Liu, D. Li and Z. Guo, Adv. Funct. Mater., 2021, 31, 2104281 CrossRef CAS.
- J. Zhu, Z. Bie, X. Cai, Z. Jiao, Z. Wang, J. Tao, W. Song and H. J. Fan, Adv. Mater., 2022, 34, 2207209 CrossRef CAS.
- J. Zhou, F. Wu, Y. Mei, Y. Hao, L. Li, M. Xie and R. Chen, Adv. Mater., 2022, 34, 2200782 CrossRef CAS PubMed.
- Q. Zhang, Y. Su, Z. Shi, X. Yang and J. Sun, Small, 2022, 18, 2203583 CrossRef CAS.
- Y. Mu, Z. Li, B.-k. Wu, H. Huang, F. Wu, Y. Chu, L. Zou, M. Yang, J. He, L. Ye, M. Han, T. Zhao and L. Zeng, Nat. Commun., 2023, 14, 4205 CrossRef CAS.
- C. Zhu, P. Li, G. Xu, H. Cheng and G. Gao, Coord. Chem. Rev., 2023, 485, 215142 CrossRef CAS.
- Y. Song, P. Ruan, C. Mao, Y. Chang, L. Wang, L. Dai, P. Zhou, B. Lu, J. Zhou and Z. He, Nano-Micro Lett., 2022, 14, 218 CrossRef CAS.
- L. Cao, D. Li, F. A. Soto, V. Ponce, B. Zhang, L. Ma, T. Deng, J. M. Seminario, E. Hu, X.-Q. Yang, P. B. Balbuena and C. Wang, Angew. Chem., Int. Ed., 2021, 60, 18845–18851 CrossRef CAS.
- C. Nie, G. Wang, D. Wang, M. Wang, X. Gao, Z. Bai, N. Wang, J. Yang, Z. Xing and S. Dou, Adv. Energy Mater., 2023, 13, 2300606 CrossRef CAS.
- J. Li, Z. Guo, J. Wu, Z. Zheng, Z. Yu, F. She, L. Lai, H. Li, Y. Chen and L. Wei, Adv. Energy Mater., 2023, 2301743 CrossRef CAS.
- Y. Geng, L. Pan, Z. Peng, Z. Sun, H. Lin, C. Mao, L. Wang, L. Dai, H. Liu, K. Pan, X. Wu, Q. Zhang and Z. He, Energy Storage Mater., 2022, 51, 733–755 CrossRef.
- T. Zhang, Y. Tang, S. Guo, X. Cao, A. Pan, G. Fang, J. Zhou and S. Liang, Energy Environ. Sci., 2020, 13, 4625–4665 RSC.
- G. Gao, G. Li, Y. Zhao, L. Ma and W. Huang, Matter, 2023, 6, 3732–3746 CrossRef CAS.
- C. Han, W. Li, H. K. Liu, S. Dou and J. Wang, Nano Energy, 2020, 74, 104880 CrossRef CAS.
- R. Qin, Y. Wang, M. Zhang, Y. Wang, S. Ding, A. Song, H. Yi, L. Yang, Y. Song, Y. Cui, J. Liu, Z. Wang, S. Li, Q. Zhao and F. Pan, Nano Energy, 2021, 80, 105478 CrossRef CAS.
- S. Guo, L. Qin, T. Zhang, M. Zhou, J. Zhou, G. Fang and S. Liang, Energy Storage Mater., 2021, 34, 545–562 CrossRef.
- X. Li, X. Wang, L. Ma and W. Huang, Adv. Energy Mater., 2022, 12, 2202068 CrossRef CAS.
- F. Wang, O. Borodin, M. S. Ding, M. Gobet, J. Vatamanu, X. Fan, T. Gao, N. Eidson, Y. Liang, W. Sun, S. Greenbaum, K. Xu and C. Wang, Joule, 2018, 2, 927–937 CrossRef CAS.
- N. Chang, T. Li, R. Li, S. Wang, Y. Yin, H. Zhang and X. Li, Energy Environ. Sci., 2020, 13, 3527–3535 RSC.
- L. Cao, D. Li, E. Hu, J. Xu, T. Deng, L. Ma, Y. Wang, X.-Q. Yang and C. Wang, J. Am. Chem. Soc., 2020, 142, 21404–21409 CrossRef CAS PubMed.
- S. Liu, J. Mao, W. K. Pang, J. Vongsvivut, X. Zeng, L. Thomsen, Y. Wang, J. Liu, D. Li and Z. Guo, Adv. Funct. Mater., 2021, 31, 2104281 CrossRef CAS.
- Z. Huang, T. Wang, X. Li, H. Cui, G. Liang, Q. Yang, Z. Chen, A. Chen, Y. Guo, J. Fan and C. Zhi, Adv. Mater., 2021, 34, e2106180 CrossRef PubMed.
- Y. Jin, L. Zou, L. Liu, M. H. Engelhard, R. L. Patel, Z. Nie, K. S. Han, Y. Shao, C. Wang, J. Zhu, H. Pan and J. Liu, Adv. Mater., 2019, 31, 1900567 CrossRef.
- K. Wang, F. Liu, Q. Li, J. Zhu, T. Qiu, X.-X. Liu and X. Sun, Chem. Eng. J., 2023, 452, 139577 CrossRef CAS.
- L. Zhou, F. Wang, F. Yang, X. Liu, Y. Yu, D. Zheng and X. Lu, Angew. Chem., Int. Ed., 2022, 61, e202208051 CrossRef CAS.
- W. W. Rudolph and C. C. Pye, Phys. Chem. Chem. Phys., 1999, 1, 4583–4593 RSC.
- A. C. Hayes, P. Kruus and W. A. Adams, J. Solution Chem., 1984, 13, 61–75 CrossRef CAS.
- W. W. Rudolph, M. H. Brooker and P. R. Tremaine, J. Solution Chem., 1999, 28, 621–630 CrossRef CAS.
- Q. Zhang, Y. Ma, Y. Lu, L. Li, F. Wan, K. Zhang and J. Chen, Nat. Commun., 2020, 11, 4463 CrossRef CAS PubMed.
- L. Miao, R. Wang, S. Di, Z. Qian, L. Zhang, W. Xin, M. Liu, Z. Zhu, S. Chu, Y. Du and N. Zhang, ACS Nano, 2022, 16, 9667–9678 CrossRef CAS PubMed.
- Y. Liu, X. Tao, Y. Wang, C. Jiang, C. Ma, O. Sheng, G. Lu and X. W. Lou, Science, 2022, 375, 739–745 CrossRef CAS.
- M. Yan, C. Xu, Y. Sun, H. Pan and H. Li, Nano Energy, 2021, 82, 105739 CrossRef CAS.
- X. Xie, S. Liang, J. Gao, S. Guo, J. Guo, C. Wang, G. Xu, X. Wu, G. Chen and J. Zhou, Energy Environ. Sci., 2020, 13, 503–510 RSC.
- Y. Jin, K. S. Han, Y. Shao, M. L. Sushko, J. Xiao, H. Pan and J. Liu, Adv. Funct. Mater., 2020, 30, 2003932 CrossRef CAS.
- R. Sun, D. Han, C. Cui, Z. Han, X. Guo, B. Zhang, Y. Guo, Y. Liu, Z. Weng and Q.-H. Yang, Angew. Chem., Int. Ed., 2023, 62, e202303557 CrossRef CAS PubMed.
- M. Chen, J. Zheng, Y. Liu, O. Sheng, Z. Ju, G. Lu, T. Liu, Y. Wang, J. Nai, Q. Wang and X. Tao, Adv. Funct. Mater., 2021, 31, 2102228 CrossRef CAS.
- C. Huang, X. Zhao, S. Liu, Y. Hao, Q. Tang, A. Hu, Z. Liu and X. Chen, Adv. Mater., 2021, 33, 2100445 CrossRef CAS.
- X. Wang, K. Feng, B. Sang, G. Li, Z. Zhang, G. Zhou, B. Xi, X. An and S. Xiong, Adv. Energy Mater., 2023, 2301670 CrossRef CAS.
- J. Feng, D. Ma, K. Ouyang, M. Yang, Y. Wang, J. Qiu, T. Chen, J. Zhao, B. Yong, Y. Xie, H. Mi, L. Sun, C. He and P. Zhang, Adv. Funct. Mater., 2022, 32, 2207909 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta05814j |
| ‡ Xiaomin Li and Jinwei Miao contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |