
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photochemical transformation of a perylene diimide derivative beneficial for the in situ formation of a molecular photocatalyst of the hydrogen evolution reaction†
Estera
Hoffman
 a,
Karol
Kozakiewicz
ab,
Małgorzata
Rybczyńska
ab,
Michał
Mońka
a,
Daria
Grzywacz
ab,
Beata
Liberek
b,
Piotr
Bojarski
a and
Illia E.
Serdiuk
*a
a,
Karol
Kozakiewicz
ab,
Małgorzata
Rybczyńska
ab,
Michał
Mońka
a,
Daria
Grzywacz
ab,
Beata
Liberek
b,
Piotr
Bojarski
a and
Illia E.
Serdiuk
*a
aFaculty of Mathematics, Physics and Informatics, University of Gdańsk, Wita Stwosza 57, 80-308 Gdańsk, Poland. E-mail: illia.serdiuk@ug.edu.pl; Tel: +48 58 523 22 44
bFaculty of Chemistry, University of Gdańsk, Wita Stwosza 63, 80-308 Gdańsk, Poland
First published on 31st January 2024
Abstract
The photocatalytic splitting of water gains more and more attention as one of the most promising environmentally friendly approaches for the production of green hydrogen fuels. In this article, we report a peculiar feature of a carbazole derivative of the perylene diimide dye Cbz-PDI which forms in the presence of platinum salt and sunlight an efficient molecular photocatalyst of the hydrogen evolution reaction (HER). Such a photocatalyst (Cbz-PDCA-Pt2) represents a unique group of organometallic platinum complexes with good sunlight absorption, satisfactory solubility, high stability and HER photoactivity. The in situ formation seems to be a convenient solution to the problems with the preparation, isolation and storage of such compounds. The best HER performance (in terms of the turnover number of 1900, decreased platinum usage and increased stability for longer than 3 days) was achieved when Cbz-PDCA-Pt2 formed a mixture with Cbz-PDI and the molar percentage of platinum in the mixture did not exceed 10%. In such a system, Cbz-PDI facilitates charge separation and the storage of excitation energy due to the long lifetimes of its excited states observed as a result of thermally activated delayed fluorescence (TADF). The discovered photoreactivity of the perylene diimide derivative can hopefully open a new direction in the organometallic materials for use in molecular (photo)catalysis.
Introduction
Research towards alternative fuels gains increasing attention and efforts lately due to threatening climate changes on the one hand and decreasing stability of oil and gas markets on the other hand, requiring a concerted effort from humanity, specifically the scientific community. There is an urgent need for environmentally friendly, abundant and efficient energy sources. In response to global warming and the requirements of reduction of CO2 emission, at least partial replacement of fossil fuels with hydrogen is regarded as one of the most promising solutions to be implemented in the near future.1,2 In fact, from the viewpoint of combustion process, among other fuels, hydrogen affords the highest energy capacitance due to its minimal molecular weight and zero CO2 emissions because the only product of the oxidation of pure H2 is water. The key problem is how to produce “green” H2, avoiding the use of fossil fuel energy at any stage of preparation, including electricity generated by the combustion of coal, oil or gas.The photocatalytic splitting of water seems to be an elegant solution representing the direct transformation of sunlight energy to transportable chemical fuels, including H2.3–5 The application of photocatalysis for hydrogen evolution reaction (HER) from water is a promising approach that is still in the early stages of development. A growing number of investigations aiming to improve the parameters of the photocatalytic HER are driven by its potential to considerably increase the efficiency and decrease the costs of converting solar energy into H2 as an alternative to the “green” H2 obtained by electrolysis using electricity from photovoltaic cells.6
The light-induced HER requires a photocatalytic system, which can be of two types: heterogeneous or homogeneous. In the first type, the key processes such as light absorption, charge transport, and water reduction usually occur on separate fragments of the photocatalytic system: the photosensitizer, mediator and (co)catalyst, respectively.7 These are the most intensively studied systems and have the highest state-of-the-art photocatalytic HER efficiencies and stabilities. In contrast, in a homogeneous system represented by a molecular photocatalyst, all these processes should occur in the same molecule. The achievement of a high efficiency of sunlight absorption and photocatalytic activity, together with long-term stability within a single molecular system, is extremely challenging, which explains the very limited number of molecular HER catalysts known to date, especially those applicable to photocatalysis. Nevertheless, the key advantage of homogeneous systems is their higher solubility and much lower aggregation in water. In the case of high stability, these factors can enable a sufficient number of active photocatalytic sites present in a limited solution volume, thus affording high power efficiency of photocatalytic HER and finally bringing us closer to the development of a respective “green” H2 technology.
As compared to the heterogeneous photocatalytic systems, the homogeneous catalysts offer higher atom economy and selectivity of photoreaction, while their mechanistic investigations and analysis of their structure–property relationships are generally much easier. For these reasons, research on molecular catalysts like metalorganic complexes is one of topical directions of material science.
An ideal metalorganic complex for the photocatalytic HER reaction should have (i) strong sunlight absorption, (ii) an appropriate LUMO level above −4.4 eV (vs. vacuum) to enable water reduction, (iii) balanced hydrogen adsorption/desorption thermodynamics to enable high H2 evolution and (iv) high photo- and mechanical stability. The organic ligand and its interactions with the metal have a crucial role in the modulation of parameters (i), (ii) and (iv). Recently developed molecular catalytic systems include various metal complexes with porphyrin and corrole ligands. Particularly, Co,8 Ni9 and Cu10 complexes applied to the surface of an electrode show high stability and efficiency in the electrocatalytic HER tests. Other efficient electrocatalytic Ni complexes include various diazadiphosphacyclooctane11 and azadiphosphacycloheptane12 derivatives as stable and strongly binding ligands.
From the point of view of HER thermodynamics, platinum has the most suitable electronic configuration and presents at the maximum of the volcano plot,13 binding neither too strongly nor too weakly to a substrate (H2O or H3O+) and product (H and H2). Its superior catalytic properties are usually realized in heterogeneous catalytic systems, which use it in the form of colloids, nanoparticles or alloys.14 Such approaches, however, waste considerable amounts of this precious metal beneath the active surface and have considerable problems with mechanical stability (e.g. the aggregation of (nano)particles), decreasing the active surface area. To form a homogeneous complex, Pt is chelated with various aliphatic amines and their derivatives as ligands.15
In spite of excellent electrocatalytic performance, the above-mentioned Pt complexes rarely catalyze the photoinduced HER. For the latter purpose, molecular catalysts are usually incorporated onto the surface of a (semi)conductor (e.g. TiO2,16 graphene derivatives17,18 or metal colloids19) to give a heterogeneous system. Naturally occurring [Fe–Fe]-hydrogenase20 can serve as one of the rare examples of a water-soluble HER photocatalyst with medium-to-low efficiency.
In spite of the fact that platinum complexes seem to be a route to the most efficient and relatively economic “green” H2 production, examples of their use in the role of photocatalyst itself are surprisingly scarce.21 A few reported examples include Pt complexes with Schiff bases, although there is evidence of considerable problems with photostability, HER efficiency22 and solubility in aqueous solutions.23 The nature of the ligand (e.g. its chelating ability and light absorptivity) seems to be important in functional metalorganic materials with Pt. Moreover, synthesis of such complexes requires specific equipment and storage conditions due to the sensitivity of Pt precursors towards light, oxidation and other reactions.
In response to the high demand for efficient photocatalysts of the HER, in this article we report the remarkable ability of a perylene diimide derivative to undergo photochemical activation to a ligand that forms a stable molecular platinum complex. This Pt complex can act as an efficient photocatalyst of the HER. As a result, a convenient method for the in situ preparation of a highly stable Pt complex Cbz-PDCA-Pt2 (Fig. 1) with photocatalytic activity is described. The ligand fragment responsible for the absorption of light is formed via the irradiation of 1,7-bis[4-(carbazol-9-yl)phenyl]-N,N-bis(2-ethylhexyl)perylene-3,4:9,10-tetracarboxylic acid diimide (Cbz-PDI, Fig. 1) with sunlight, which, in the presence of the platinum salt, forms Cbz-PDCA-Pt2. The molecular design of the ligand presented here applies the electron donor–acceptor molecular architecture, photoreactive sites and alkyl groups that prevent aggregation and improve solubility.
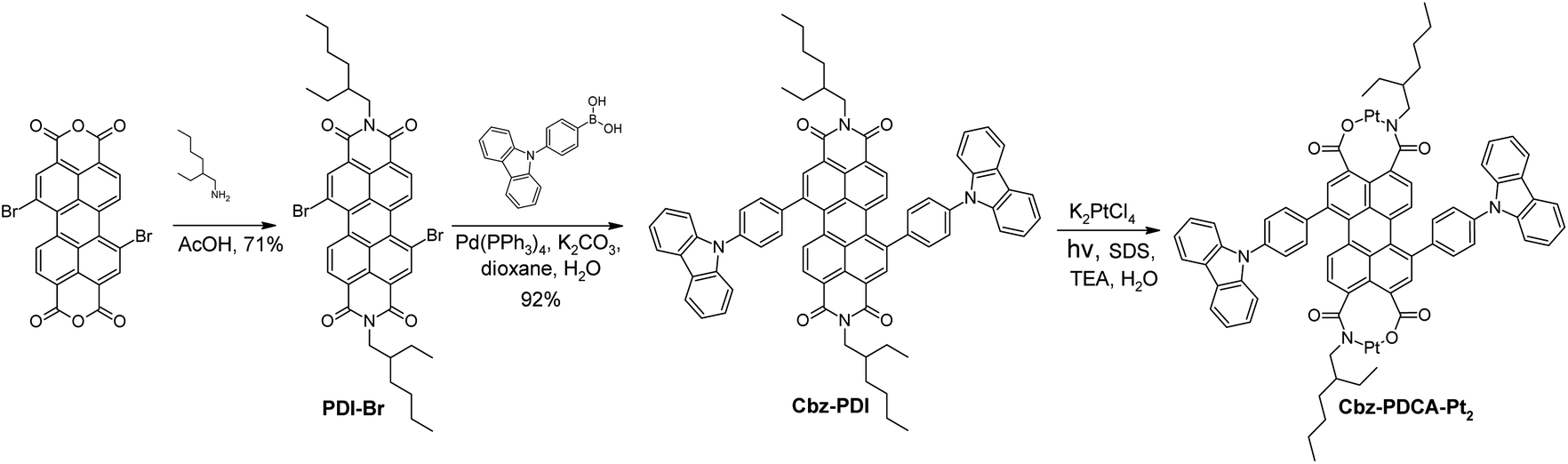 | ||
| Fig. 1 Synthesis of Cbz-PDI and Cbz-PDCA-Pt2. | ||
Perylene diimide was selected as an electron acceptor fragment with good sunlight absorption and which, as we discovered, forms complexes with Pt under irradiation. Moreover, perylene itself shows good semiconductor properties and a high (photo)chemical stability. Carbazole was selected as a (photo)chemically stable and bulky electron donor which enables the formation of a charge transfer (CT) state under excitation yet preserving reasonable light absorption, and provides partial separation of the frontier orbitals to facilitate charge separation. The designed Cbz-PDCA-Pt2 molecule undergoes more than 1000 turnovers of the HER in 24 h under sunlight irradiation in the presence of sacrificial electron donors and remains active for more than 3 days. The unique photochemical transformation of a perylene diimide fragment, affording a strong chelating ligand under the conditions of the HER, is a new approach in the development of homogeneous HER photocatalysts.
Results and discussion
Preparation of Cbz-PDI
The precursor of the ligand, Cbz-PDI, was prepared in a two-step reaction via a modified procedure.24 Briefly, 1,7-dibromo-perylene-3,4,9,10-tetracarboxylic dianhydride reacted with 2-ethylhexyl amine and further with 4-[(carbazol-9-yl)phenyl]boronic acid in a Suzuki coupling to give Cbz-PDI in a 65% yield. The details of the synthetic procedures and analyses are provided in the ESI (Fig. S1 and S2).†Spectral properties and changes during the formation of the Pt complex
Due to the specific interest in the light absorption properties and their changes during the photochemical formation of the Pt complex, the origin of the electronic transitions in Cbz-PDI and related perylene derivatives is discussed in detail.Cbz-PDI shows a broad absorption covering almost all of the visible region from 380 to 650 nm. This originates mainly from two key electronic transitions: a low-energy charge transfer (CT) transition involving an N-phenylcarbazole donor and a PDI acceptor and a locally excited (LE) transition within the PDI acceptor fragment (Fig. S3, ESI†). Typically, the CT transition appears as a broad band with absorption centered at 540 nm (Fig. 2A, Table 1). The respective CT fluorescence band is specifically sensitive to medium polarity, namely shifting by 0.23 eV (100 nm) from cyclohexane to chloroform solutions (Fig. 2B, Table 1). The LE transition has a higher energy: its absorption maximum appears near 500 nm in Cbz-PDI and free PDI acceptor. The respective LE fluorescence can be observed in solutions of PDI, with a maximum near 530 nm. In the view of the following discussion, it is important to mention that the LE energy increases when the cyclic amide fragment is absent. In fact in the perylene parent molecule, the absorption and fluorescence maxima appear at 440 nm and 445 nm, respectively. This indicates that the conjugation of the perylene core with the carboxyl amide groups regulates the stabilization of the LE transition and structural changes in this fragment are reflected in the electronic spectra.
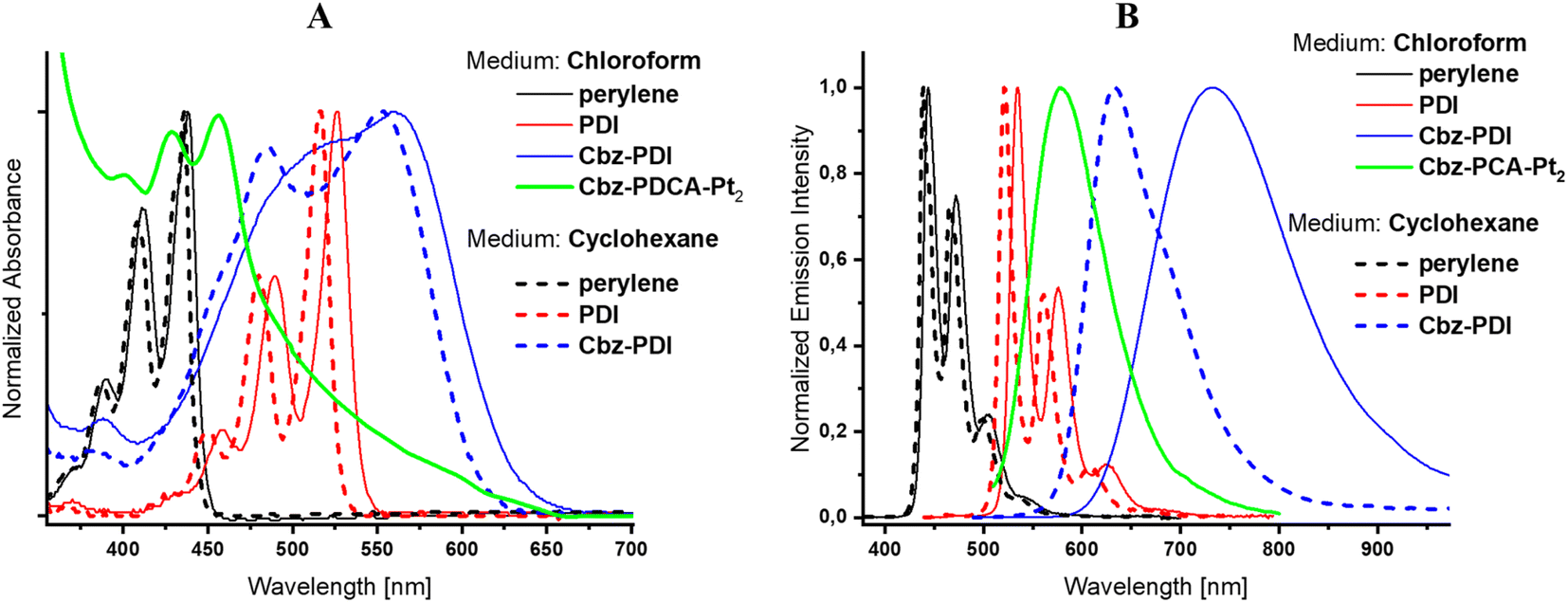 | ||
| Fig. 2 Absorption (A) and fluorescence (B) spectra of Cbz-PDI before and after activation, PDI, and perylene in chloroform and cyclohexane. | ||
| Compound | Medium | λ abs [nm] | λ em [nm] |
|---|---|---|---|
| Perylene | Cyclohexane | 435 | 440 |
| Chloroform | 440 | 445 | |
| PDI | Cyclohexane | 515 | 520 |
| Chloroform | 525 | 535 | |
| Cbz-PDI | Cyclohexane | 550 | 630 |
| Chloroform | 560 | 730 | |
| Cbz-PDCA-Pt2 | Chloroform | 450 | 580 |
When an aqueous solution of Cbz-PDI is irradiated by sunlight in the presence of triethylamine (TEA, 0.3 M), K2PtCl4 (50 μM) and sodium dodecyl sulphate (2.1 mM), referred further as activation, its color changes from bright violet to dark yellow and the absorption spectra undergo strong changes (Fig. 3A). Within the first hour of irradiation, the 545 nm band shifts to one at 500 nm with a complex structure and a band centered at 750 nm appears. Further irradiation leads to a shift of the 500 nm band to 450 nm. The 750 nm band disappears, most likely indicating the formation and deactivation of the radical anion species of Cbz-PDI, formed as a result of the quenching of its excited state by TEA. This mixture produces hydrogen, as shown in Fig. 3B. During the first hour of irradiation, minimal amounts of H2 are produced, but after 2–3 h, the TOF value reaches its maximum of 30 mol H2 per 1 mol of Pt for 1 h and then remains stable. Reaching the TOF maximum correlates with the maximum contribution of the 450 nm band in the absorption spectra. This indicates that species with this absorption are responsible for the HER activity of the investigated system.
 | ||
| Fig. 3 Changes in the absorption spectra (A) and contamination of H2 in the gaseous phase (B) during the activation of Cbz-PDI; the numbers on the plot indicate the TOF values; TON is calculated per mole of K2PtCl4. Fluorescence excitation (C) and emission (D) spectra, emission decay (E) and mass spectrum (F) of the activated mixture containing Cbz-PCA-Pt2. | ||
In an attempt to reveal the key activation steps and structural components of these species, we performed various HER tests under different conditions. It was found that under irradiation in the absence of K2PtCl4, the mixture did not produce H2, whereas the appearance of 450 nm band was not observed in the absorption spectra. The same lack of activation was observed when TEA was replaced by ascorbic acid (AA), an acidic sacrificial electron donor. The activated species were thus formed under basic conditions and contained Pt.
As compared to Cbz-PDI, the absorption of the activated mixture lacks the CT band centered at 540 nm, indicating a significant decrease in the strength of acceptor or donor. The LE absorption maximum is higher than that of the PDI acceptor, but lower than that of perylene. Considering the high stability of the carbazole donor under the applied conditions and importance of basic conditions for the activation process, we conclude that the irradiation of Cbz-PDI facilitated the gradual hydrolysis of the cyclic diimide of the PDI fragment, decreasing its electron acceptor properties. The nucleophilic attack of hydroxyl anions can lead to the opening of the diimide ring with the formation of a chelate fragment consisting of an N-(2-ethylhexyl)carboxamide group in the vicinity of a carboxylate anion. Such a chelate fragment can form a complex with Pt.
Analysis of the fluorescence excitation and emission spectra identified two main spectral components. For this analysis, the mixture was activated for a relatively long time of 24 h to ensure a lack of Cbz-PDI. After the removal of TEA by evaporation, the major component with the absorption and fluorescence excitation band at 450 nm showed a fluorescence emission with a pronounced vibronic structure centered at 500 nm (Fig. 3C and D). The second minor component appeared in the absorption spectrum as a low-intensity tail; its fluorescence excitation spectrum had broad bands with an apparent maximum at 480 nm. This second component could be individually excited with a 500–550 nm light, which gave rise to a broad, structureless emission band centered at 590 nm. This emission was detected for a relatively long time (>100 ns) (Fig. 3E, Table 2).
| Compound | c(TEA) [M] | λ abs [nm] | ε [M−1 cm−1] | λ em [nm] | τ PF [ns] | τ DF [μs] |
|---|---|---|---|---|---|---|
| a λ abs, λem – wavelength of absorption and emission maxima, respectively; ε – molar extinction coefficient; τPF, τDF – lifetimes of prompt (PF) and delayed (DF) fluorescence, respectively, monitored at the respective emission bands. | ||||||
| Cbz-PDI | 0 | 545 | 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 550 550 |
750 | 1.6 | 1.1 |
| 0.3 | 545 | 16550 |
750 | 0.7 | — | |
| Cbz-PDCA-Pt2 | 0 | 450 | 10250 |
695 | 4.2 | — |
| 0.3 | 450 | 10250 |
695 | 1.6 | — | |
As our attempts to isolate the activated species from the photocatalytic mixture using column chromatography and HPLC were unsuccessful, we further used LCMS analysis performed directly on the same mixture activated for 24 h. Two mass components were identified. In spite of the long activation time and lack of Cbz-PDI in the absorption spectra, a significant amount of a component with a mass peak of 1097.50 m/z, identical to the initial compound, was detected (Fig. S1 and S2, ESI†). This behavior can indicate the reverse transformation of the activated species back to Cbz-PDI, probably under the conditions of the chromatographic analysis. The second component had a peak at 1518.42 m/z corresponding to Cbz-PDCA-Pt2, the structure of which is shown in Fig. 3F. This shows that platinum atoms are directly incorporated into the structure of the activated species forming an organometallic complex, explaining the origin of the photocatalytic activity in the HER.
To understand the mechanism of formation of Cbz-PDCA-Pt2, we performed DFT calculations for the thermodynamics of the possible stages of transformation starting from Cbz-PDI, as depicted in Scheme 1. The opening of the first cyclic diamide starts with the nucleophilic attack of a hydroxyl anion on the carbonyl group, thus forming an anion localized on the amide nitrogen atom (from Cbz-PICA-aN). Such a species further transforms to a more stable tautomeric form with the charge localized on the oxygen atom (Cbz-PICA-aO) via intramolecular proton transfer, resulting in a decrease in the total Gibbs free energy of −301 kJ mol−1. Cbz-PICA-aO coexists in dynamic equilibrium with its neutral protolytic form Cbz-PICA-n.
 | ||
| Scheme 1 Proposed mechanisms for the Cbz-PDCA-Pt2 formation in the presense of TEA. The green arrows indicate the stepwise mechanism, the blue arrows indicate the synchronous mechanism and the gray arrows indicate reverse transformations in neutral or acidic media. | ||
There are two possibilities of further transformations of Cbz-PICA. According to the first, in the synchronous mechanism marked by blue arrows in Scheme 1, another nucleophilic attack of a hydroxyl anion on Cbz-PICA-aO and opening of the second ring occurs in a similar way as described above with the formation of the dianion Cbz-PDCA-daO and a −103 kJ mol−1 decrease in ΔG. Protolytic equilibrium affords the neutral form of Cbz-PDCA-n, which can react with two equivalents of K2PtCl4 to give Cbz-PDCA-Pt2 with a total ΔG of −442 kJ mol−1. Another possibility, marked by green arrows in Scheme 1, is the formation of a mononuclear complex Cbz-PICA-Ptvia the reaction of Cbz-PICA-n with one equivalent of K2PtCl4 (ΔG = −216 kJ mol−1), followed by basic hydrolysis of the cyclic diamide in Cbz-PICA-Pt (ΔG = −294 kJ mol−1) and further reaction with K2PtCl4 (ΔG = −226 kJ mol−1) to give Cbz-PDCA-Pt2.
Interestingly, the neutral forms with open ring(s) (Cbz-PICA-n, Cbz-PDCA-n, and Cbz-PDCA-Pt-n) easily lose water, transforming back to closed-ring species. This makes the hydrolysis stages reversible in the absence of platinum, especially in neutral and acidic media, where considerable amounts of such neutral forms are present. In fact, such a feature of the 3-carboxamide-4-carboxyl perylene derivatives was applied recently for the preparation of PDI derivatives under mild conditions at room temperature.25 The reversibility of the above mentioned hydrolysis stages explains the lack of activation of Cbz-PDI under irradiation in the absence of TEA and K2PtCl4. The Pt2+ dication(s) stabilize the ring-opened form(s), affording an organometallic complex. The reversibility of the hydrolysis of Cbz-PDI may be also one of the reasons for the difficulties in isolating Cbz-PDCA-Pt2. In such a case, the in situ formation of Cbz-PDCA-Pt2 described here seems to be the most optimal way of preparing such an HER-active molecular catalyst.
While both of the above mentioned pathways are thermodynamically allowed, the experimental data indirectly support the first synchronous mechanism. Taking into account the results of analyses described above, we assume that the main component of the activated solution is Cbz-PDCA with both diamide cycles cleaved. The absorption and emission of such species should have a maxima close to those of perylene and band shape with a pronounced vibronic structure. Reverse cyclization under the conditions of the LCMS spectrum should yield Cbz-PDI, as in fact was discussed above. Moreover, the frontier molecular orbitals of Cbz-PDCA are localized on perylene (Fig. S3, ESI†), explaining the vibronic structure of the electronic spectra of the observed band at 450 nm. The second component of the activated mixture is thus Cbz-PDCA-Pt2, with broad absorption and emission bands in the long-wavelength region due to the metal–ligand charge-transfer nature of the lowest energy transitions (Fig. S3, ESI†). It should be mentioned that more allowed electronic transitions responsible for the absorption of Cbz-PDCA-Pt2 have a similar energy to Cbz-PDCA as they are also localized on perylene (Fig. S3, ESI†).
HER performance
To optimize the micellar photocatalytic system, the influence of SDS concentration was analyzed first (Fig. S4A, ESI†). We found that, due to low solubility, Cbz-PDI is not activated without SDS or when its concentration is below 1 mM. The optimum SDS concentration was found to be close to 2.1 mM, while systems with higher values showed a lower TON for the HER.
The molar ratio of Cbz-PDI and Pt is the key parameter for the investigated system. Fortunately, from the point of view of attempts to minimize the use of precious platinum, the maximum TON values were achieved with a much smaller ratio than that suggested by the Cbz-PDCA-Pt2 stoichiometry (1:2). In contrast, the use of a Cbz-PDI concentration of 80 μM and c(Pt) = 5 μM in a 16:1 molar ratio forty times higher than that suggested by the stoichiometry of the complex facilitated the activation procedure in terms of reaching the maximum intensity of the 450 nm absorption band more rapidly (Fig. 4A and B) and enabled a stable TOF value of 35 TON/h for the first 8 hours of irradiation after the addition of AA (Fig. 4C). Such experiments with an increased concentration of Cbz-PDI revealed that it is not completely degraded under these activation conditions, even after hours of irradiation. In fact, some remaining amounts of Cbz-PDI repeatedly detected in the absorption spectra of the activated mixture correlated with high, stable TOF values (Fig. 4B) and an estimated TON24 of 800 (Table 3). The role of the remaining amounts of Cbz-PDI will be discussed further. Unfortunately, at such Cbz-PDI concentrations, the photocatalytic system is not stable due to the precipitation of large amounts of Cbz-PDI.
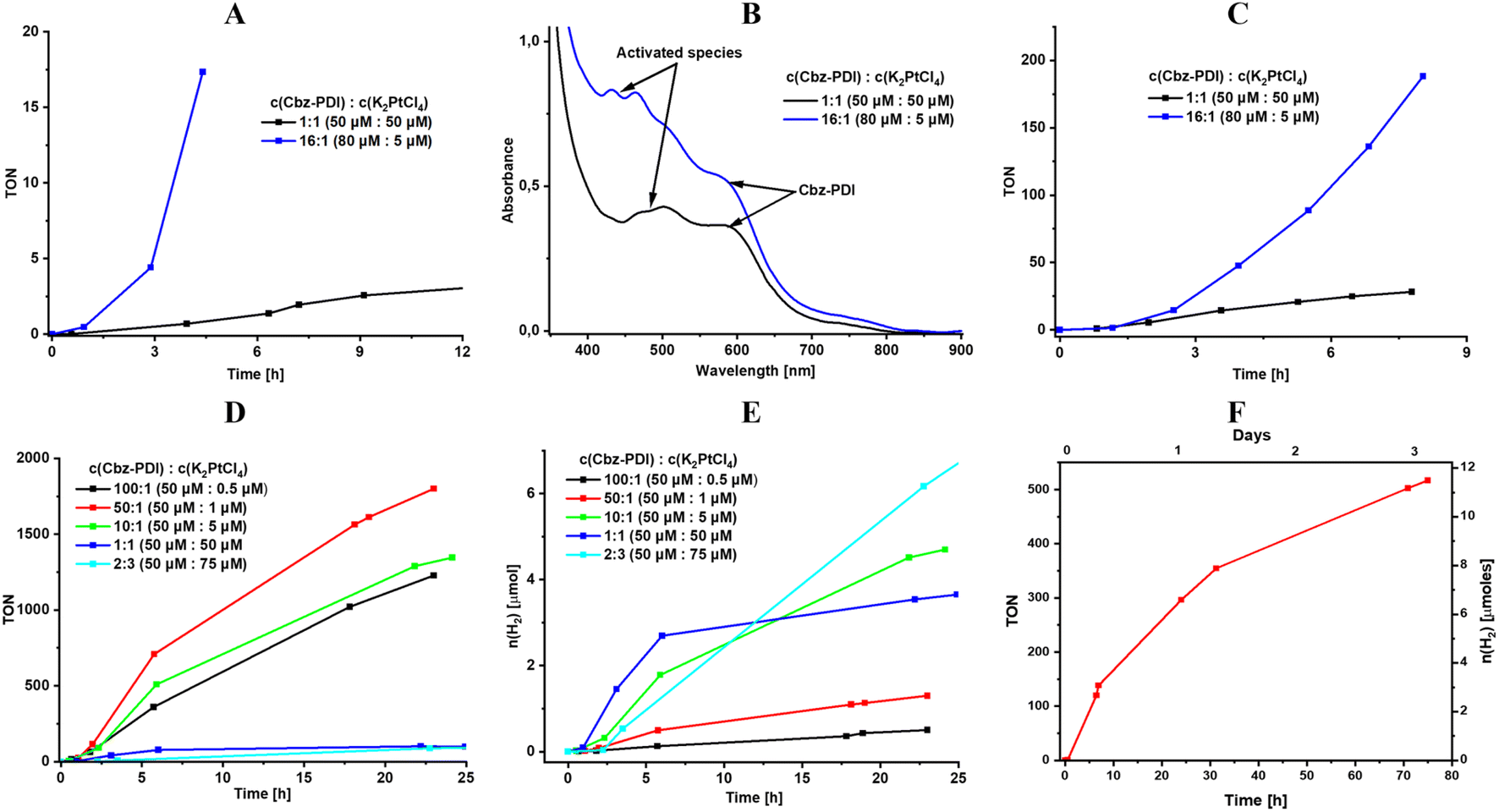 | ||
| Fig. 4 HER tests on 3 mL solutions at 1 sun irradiation. Changes in the TON values (A) during activation with an excess of Cbz-PDI in the presence of TEA (0.3 M); absorption spectra (B) and HER performance (C) of the activated mixture containing AA (0.7 M). The HER performance of activated mixtures with various ratios of Cbz-PDI and Pt measured in TON (D) and number of moles of H2 (E). HER stability test on 20 mL of solution (F). | ||
| Test | V sol. [mL] | c(Cbz-PDI) [μM] | c(K2PtCl4) [μM] |
c(Cbz-PDI):c(K2PtCl4) |
c(AA) [M] | n(H2)24h [μmol] | TON24h | TOFmax [1/h] |
|---|---|---|---|---|---|---|---|---|
| a Conditions: test 1 was performed in DFM with addition of 6.5% of H2O without SDS, c(TEA) = 0.3 M; tests 2–4 were performed in water, c(SDS) = 2.1 mM, c(TEA) = 0.3 M. | ||||||||
| 1 (DMF) | 3 | 50 | 0.5 | 100:1 |
0 | <0.004 | — | — |
| 3 | 50 | 50 | 1:1 |
0 | 0.05 | 14 | 0.7 | |
| 2 (H2O) | 3 | 50 | 50 | 1:1 |
0 | 0.1 (19 h) | 4 (19 h) | 0.7 |
| 0.7 | 0.8 (8 h) | 28 (8 h) | 6 | |||||
| 3 | 80 | 5 | 16:1 |
0 | 0.06 (4.5 h) | 9 (4.5 h) | 9 | |
| 0.7 | 4.3 | 800 | 43 | |||||
| 3 (H2O) | 3 | 50 | 0.5 | 100:1 |
0.7 | 0.6 | 1270 | 197 |
| 3 | 50 | 1 | 50:1 |
0.7 | 1.4 | 1900 | 157 | |
| 3 | 50 | 5 | 10:1 |
0.7 | 4.2 | 600 | 117 | |
| 3 | 50 | 50 | 1:1 |
0.7 | 3.4 | 100 | 18 | |
| 3 | 50 | 75 | 2:3 |
0.7 | 6.3 | 90 | 6 | |
| 4 (H2O) | 21 | 50 | 5 | 10:1 |
0 | 6.5 | 280 | 13 |
| 0.7 | 12.1 | 520 | 42 | |||||
To avoid deviations due to the uncontrolled precipitation of Cbz-PDI, we explored systems with a constant concentration of Cbz-PDI of 48 μM. The highest TON24 values, reaching 1900, were achieved in a mixture with an even larger molar ratio of 50:1 (Fig. 4D, Table 3), where the actual Pt concentration was 1 μM. Such a TON24 value calculated per mole of Cbz-PDCA-Pt2 is, however, underestimated because the Cbz-PDCA-Pt2 concentration is calculated assuming the complete complexation of Pt and not taking into account the losses during side-reactions. When the molar ratio was increased to 100:1 or decreased to 10:1, the TON24 value decreased to 1270 and 600, indicating an insufficient amount and less efficient utilization of Pt, respectively. The use of 1:1 and 2:3 molar ratios is almost one hundred times less efficient, indicating a high wastage of Pt, most likely due to the reduction and precipitation of metallic (nano)particles.
From the point of view of the actual amount of H2 produced by the photocatalytic system, a 10:1 ratio seems to be the most optimum (Fig. 4E). A 3 mL volume of such a solution produces 4.6 μmol of H2 in 24 h, which is only 1.4 times lower as compared to the mixture with a 1:2 ratio containing 20 times more Pt.
To estimate relatively long-term photoactivity of the Cbz-PDCA-Pt2 systems, an HER test was performed using an activated mixture of 50 μmol l−1Cbz-PDI, 5 μmol l−1 K2PtCl4, 2.1 mM SDS, 0.3 M TEA and 0.7 M AA. The photoreaction was carried out in a 43 mL reactor using 20 mL of the photocatalytic mixture. Under such conditions, more than 12 μmol of H2 were produced within 3 days and the photocatalytic activity remained as high as TOF72h = 7 h−1, which is a 43% decrease compared to that during the first 24 h (Fig. 4F). Cycle tests revealed gradual photobleaching and a three-fold decrease in the photocatalytic activity when irradiated with a full sunlight spectrum (Fig. S4C, ESI†). In contrast, when the UV light was optically filtered and only the >400 nm light was used for irradiation, the samples showed an impressively high quantitative stability of TOF = 5 h−1 for at least 3 days and significantly decreased photobleaching (Fig. S4C, ESI†).
In an attempt to analyze the activity of the Cbz-PDCA-Pt2 molecular catalyst in a soluble form without micellar support, we performed activation and HER tests on dimethylformamide with the addition of 6.5% (v/v) H2O and 0.3 M TEA as a sacrificial electron donor. The activated mixtures showed similar changes in the absorption spectra as described earlier for the water–SDS medium but much lower activity. The activated mixture with a 1:1 molar ratio of Cbz-PDI and Pt afforded 50 nmol of H2 in 24 h (TON24h = 14, Table 3), whereas the activity of the mixture with a 100:1 ratio was below the detection limit of the chromatographic method used (4 nmol H2). In contrast to the micellar nanoparticle medium where the reverse tendency was observed, this proved that the molecular catalyst was formed and was HER-active itself, but its performance in the dissolved form was much lower than in combination with a nanoparticle system, which correlates well with previous reports on Pt complexes.26
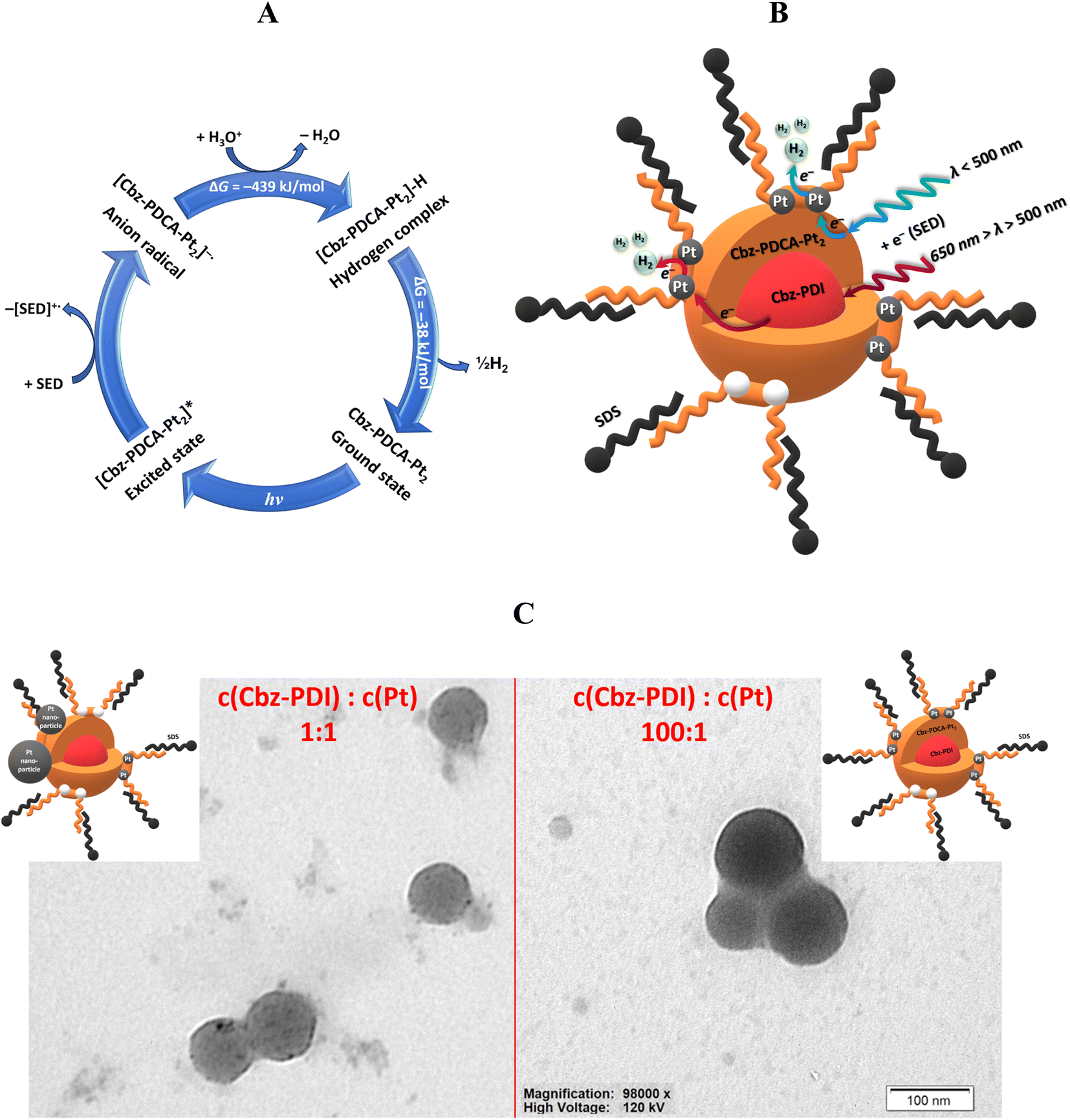 | ||
| Fig. 5 The photocatalytic cycle of the Cbz-PDCA-Pt2 catalyst during HER (A). The suggested optimum structure of a photocatalytically active nanoparticle consisting of a Cbz-PDI core and Cbz-PDCA-Pt2 surface in SDS–water solutions (B). Comparison of the TEM images of two samples activated with different amount of Pt and the suggested structures of the nanoparticles (C). | ||
The above mentioned improvement in the performance of the HER in the presence of residual Cbz-PDI indicates its role as an electron mediator, which facilitates the storage of the energy of sunlight and electrons before the reduction of water occurs on Cbz-PDCA-Pt2. In fact, in SDS-stabilized nanoparticles, this compound exhibits thermally activated delayed fluorescence (TADF) (Fig. S5, ESI†). This phenomenon occurs in molecular donor–acceptor systems when the energies of the excited singlet and triplet states are close. In the PL decay, such compounds show a region of delayed fluorescence (DF), which originates from the upconversion of T1 to S1 and further light emission. The duration of DF thus indicates the timescale of the stability of the T1 state and the ability to store excitons for such a period of time. The occurrence of TADF in Cbz-PDI is evidence for the relatively high stability of its excited states, whereas its DF lifetime of 1.1 μs (Table 2) indicates the storage of excitation energy for a few microseconds. According to the computational results, the HOMO energy of Cbz-PDI is −5.70 eV, which is appropriate for the reaction of excited Cbz-PDI with the SEDs used. In fact, as supported by the emission decay measurements, its excited state is effectively quenched by TEA with a rate constant of 7.2 × 108 s−1 (Fig. S5, ESI†), affording the anionic radical. Perylene diimides are also known for the relatively high stability of their anionic radical species28 and good electron transporting features.29 Like various inorganic semiconductors used as electron mediators in heterogeneous photocatalytic systems, Cbz-PDI acts as a reservoir of excitation and electronic energies. Taking into account the LUMO energy of Cbz-PDI of −3.83 eV, electron is further transferred to the molecular catalyst Cbz-PDCA-Pt2 with a sufficiently lower LUMO of −4.26 eV.
Last but not least, the broader absorption of Cbz-PDI enables more efficient harvesting of sunlight than Cbz-PDCA-Pt2 itself. The above-mentioned considerations are perfectly supported by the HER test performed under irradiation with a 550 nm longpass optical filter in which the light was only absorbed by Cbz-PDI (Fig. S4B, ESI†). When corrected by the difference in absorbance, the >550 nm light yielded only half the amount of H2 obtained with the full sunlight spectrum, even though the molecular catalyst was not excited directly. Therefore, as summarized in Fig. 5B, in the discussed systems, Cbz-PDI serves as a photosensitizer of the red portion of the spectrum and can store excitation and electronic energies.
The core–shell nanoparticle structure presented in Fig. 5B is supported by the analysis of TEM images. Due to low solubility, the initial solution consists of nanoparticles of Cbz-PDI in SDS micelles (Fig. S6, ESI†). During activation, only the surface of such nanoparticles can easily transform to Cbz-PDCA-Pt2, whereas the core retains Cbz-PDI, which, due to the lack of contact with solution, is hardly activated. In the case of the most efficient photocatalytic systems obtained under activation with a molar ratio of Cbz-PDI and Pt from 10:1 to 100:1, the recorded TEM images with distinct, well-defined, round-shaped nanoparticles with diameters of 10–100 nm (mode near to 37.7 nm) (Fig. 5C and S6, ESI†). Importantly, the uniform and relatively intense coloration of such nanoparticles supports the assumption on the metalorganic nature of their surface. Such observations indicate that Cbz-PDCA-Pt2 formation occurs on the surface of the nanoparticles, supporting the ideal situation presented in Fig. 5B. When the concentrations of Cbz-PDI and Pt were equimolar, the TEM images showed round-shaped nanoparticles with various degrees of coloration (Fig. 5C and S6, ESI†). The weaker coloration of most of the nanoparticles mainly indicates their organic nature, probably due to the less efficient formation of Cbz-PDCA-Pt2. However, their surfaces are covered with smaller black spots of metallic Pt nanoparticles, formed by the light-induced reduction of K2PtCl4 in the presence of TEA. In such a case, the platinum atoms of Cbz-PDCA-Pt2 probably serve as one of the centers of crystallization for the Pt nanoparticles. Such equimolar samples are thus inhomogeneous, with most of the platinum concentrated in separate or aggregated nanoparticles (Fig. 5C), which explains the above mentioned lower TON values calculated per mol of Pt.
According to the TEM images and absorption changes during activation experiments, the number of Cbz-PDI molecules and thus the size of the core varies depending on the initial concentration of Cbz-PDI. The Cbz-PDCA-Pt2 species represent the shell of the nanoparticles because they are formed on the surface in direct contact with the components of the solution. Depending on the amount of K2PtCl4 used, Cbz-PDCA-Pt2 can cover either all the surface of the nanoparticle, which is the ideal case or more realistically, only the part of the surface. The remaining ring-opened species Cbz-PDCA remain in a neutral or anionic form without platinum (Scheme 1). SDS molecules form a micelle around a core–shell nanoparticle. As mentioned above, very low concentrations of SDS are not enough to enable solubility of the photoactive nanoparticles, whereas high concentrations can block the access of the solution components to Cbz-PDCA-Pt2 on the surface and thus decrease the long-term photocatalytic efficiency.
Conclusions
In summary, the light-assisted cleavage of amide bonds in the perylene diimide derivative Cbz-PDI was discovered. In the presence of a platinum salt and triethylamine, the resulting bidentate ligand forms an organometallic complex Cbz-PDCA-Pt2 that acts as a photocatalyst in the hydrogen evolution reaction. The formation of this complex is accompanied by a blue shift in the absorption spectra due to a decrease in the electron-withdrawing strength of the perylene diimide acceptor. Taking into account the problematic isolation of Cbz-PDCA-Pt2 due to its low chemical stability under ambient conditions, the described in situ formation of this species seems to be the most convenient way of preparing this molecular catalyst. Analysis of the thermodynamics provided by the DFT calculations and experimental data indicated the formation of a bidentate ligand Cbz-PDCA, which, on further reaction with two equivalents of K2PtCl4, afforded Cbz-PDCA-Pt2, rather than a stepwise mechanism via a single-ring opening reaction and the formation of a mononuclear platinum complex. To the best of our knowledge, this is the first report of the light-induced formation of perylene-based organometallic compounds, opening a new direction for molecular photocatalysts in various applications.The use of a mixture of Cbz-PDCA-Pt2 and Cbz-PDI in the HER tests afforded photocatalytic systems with the TON24h exceeding 1900, economical use of platinum down to 10 molar percent, and retention of 67% of the photocatalytic stability after 3 days of HER performance. When calculated per unit mass of the active catalyst, one of the most efficient mixtures, containing 2 molar percent of Pt, afforded a hydrogen production rate of 30 mmol g−1 h−1 during the first 5 h and produced over 370 mmol g−1 H2 after 24 h. This is higher than the efficiencies reported for other perylene-based systems, like N-annulated perylene diimide with platinized polymeric carbon nitride catalyst (12 mmol g−1 h−1),30 perylene diimide on palladium quantum dots (29 mmol g−1 h−1)31 and self-assembled phosphoric acid substituted perylene diimide (11.7 mmol g−1 h−1).32 In contrast, a higher hydrogen production rate of 61 mmol g−1 h−1 was reported for nitrogen bay-annulated perylene diimide supramolecular photocatalysts,33 indicating a promising pathway for further improvements in Cbz-PDCA-Pt2 systems.
Our results also indicate that the stability and microsecond lifetime of the excited states of Cbz-PDI are beneficial for the photocatalytic activity of the system. In general terms, the organic materials showing thermally activated delayed fluorescence can serve as electron mediators and excitation energy storage materials for applications in (photo)catalysis. In terms of stability, Cbz-PDCA-Pt2 and Cbz-PDCA-Pt2/Cbz-PDI systems exceed the reported photocatalytic activity of other Pt(II) complexes more than 10 times,20 which usually lose their activity after 6–8 h. The TON values achieved are also very high, although comparison with previous reports is complicated due to the different conditions used and lack of TON parameters in most of the reports.
Undoubtedly, the described molecular photocatalyst and its heterogenous mixture with Cbz-PDI needs further improvement. Further attempts will focus on the complexation with different metals, improvement of sunlight absorption, and solubility.
Experimental and computational methods
Sample preparation for spectroscopic measurements
Absorption and emission spectra of Cbz-PDI and Cbz-PDCA-Pt2 molecules were investigated in water micelles and/or in organic solvents as mentioned above. Water micelles were prepared by the addition of 20 μM of a 1 mM THF solution of a compound in 2.5 mL of a 0.01 M SDS water solution under sonication. The UV-Vis absorption spectra were recorded using a Shimadzu UV-1900 spectrophotometer. Steady-state photoluminescence spectra were recorded using an FS5 spectrofluorometer (Edinburgh Instruments).Time-resolved emission spectra and emission decays were investigated using a customized system34 consisting of a pulsed YAG:Nd laser (PL2251A, EKSPLA) coupled with an optical parametric generator (PG 401/SH) as the excitation light source and 2501S grating spectrometer (Bruker Optics) combined with a streak camera system (C4334-01 Hamamatsu) as the detection unit. These measurements provided the time-wavelength-intensity 3D matrices from which the decay of the emission band(s) was analyzed (for more details, see ESI†).
Sample preparation for Cbz-PDI activation and HER tests
Activation and photocatalytic tests were performed under irradiation with a sun simulator SCIENCETECH SF-300-A equipped with an airmass filter AM1.5 G enabling a light intensity of 1 sun at the sample surface. Samples for the hydrogen evolution reaction were prepared in the following manner. First, an appropriate amount of the 1 mM Cbz-PDI in THF solution was added dropwise under sonication to the 0.01 M SDS solution in water and the mixture was sonicated for 5 min. Next, an appropriate amount of the 1 mM K2PtCl4 solution in water was added, followed by the addition of an appropriate amount of TEA to achieve a 0.3 M solution. The mixture was placed into a reactor with a silicon septum and air was replaced with argon. The mixture was irradiated by simulated sunlight at 35–40 °C with continuous stirring for the activation of Cbz-PDI and formation of Cbz-PDCA-Pt2. Depending on the amount of contamination of the solution and mixture, complete activation was achieved in 3 h to 24 h.For target HER tests, ascorbic acid was added to the activated mixture up to a concentration of 0.7 M. Air was replaced with argon and the mixture was irradiated with simulated sunlight.
Calculations of parameters of the photocatalytic reaction
The amount of hydrogen produced was determined using a gas chromatograph GC-2030 (Shimadzu) with a BID detector calibrated using a gas mixture containing 0.092% H2.The production of one H2 molecule requires two photocatalytic cycles. Therefore, unless otherwise stated, the turnover number (TON) describing the number of cycles that occurred on a catalyst is given by eqn (1):
| TON = 2 n(H2)/n(Cbz-PDCA-Pt2), | (1) |
The rate of change of TON in a period of time, (Δt) and the turnover frequency (TOF), is given by eqn (2):
| TOF = ΔTON/Δt. | (2) |
Quantum chemical calculations
Unconstrained geometry optimizations were carried out for individual species at ground state using the B3LYP35 hybrid functional and Lan-L2DZ basis set implemented in the Gaussian 16 program package.36 The energies of the molecular orbitals and electronic transitions were available from the single-point calculations calculated for optimal ground state geometries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support within the LIDER XI grant LIDER/47/0190/L-11/19/NCBR/2020 of the National Centre for Research and Development (NCBR), Poland is gratefully acknowledged. The quantum chemical calculations were performed on computers at the Wroclaw Centre for Networking and Supercomputing (WCSS), Poland.References
- Commission Delegated Regulation (EU) 2023/1185, Off. J. Eur. Union, L 157/20, 2023 Search PubMed.
- A. Wolf and N. Zander, Inter Econ., 2021, 56, 316 Search PubMed.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503 CrossRef CAS PubMed.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216 RSC.
- Y. Zhang, Y.-J. Heo, J.-W. Lee, J.-H. Lee, J. Bajgai, K.-J. Lee and S.-J. Park, Catalysts, 2018, 8, 655 CrossRef.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K.-Y. Wong, Chem. Rev., 2020, 120, 851 CrossRef CAS PubMed.
- K. Maeda, J. Photochem. Photobiol., C, 2011, 12, 237 CrossRef CAS.
- B. Mondal, K. Sengupta, A. Rana, A. Mahammed, M. Botoshansky, S. G. Dey, Z. Gross and A. Dey, Inorg. Chem., 2013, 52, 3381 CrossRef CAS PubMed.
- Y. Han, H. Fang, H. Jing, H. Sun, H. Lei, W. Lai and R. Cao, Angew. Chem., Int. Ed., 2016, 55, 5457 CrossRef CAS PubMed.
- H. Lei, H. Fang, Y. Han, W. Lai, X. Fu and R. Cao, ACS Catal., 2015, 5, 5145 CrossRef CAS.
- U. J. Kilgore, M. P. Stewart, M. L. Helm, W. G. Dougherty, W. Scott Kassel, M. Rakowski DuBois, D. L. DuBois and R. Morris Bullock, Inorg. Chem., 2011, 50, 10908 CrossRef CAS PubMed.
- M. P. Stewart, M.-H. Ho, S. Wiese, M. L. Lindstrom, C. E. Thogerson, S. Raugei, R. Morris Bullock and M. L. Helm, J. Am. Chem. Soc., 2013, 135, 6033 CrossRef CAS PubMed.
- B. E. Conway and G. Jerkiewicz, Electrochim. Acta, 2000, 45, 4075 CrossRef CAS.
- S. K. Lakhera, A. Rajan, T. P. Rugma and N. Bernaurdshaw, Renewable Sustainable Energy Rev., 2021, 152, 111694 CrossRef CAS.
- M. M. Quintanilha, B. A. Schimitd, A. M. F. Costa, D. H. Nakahata, D. de Alencar Simoni, J. C. T. Clavijo, D. H. Pereira, A. C. Massabni, W. R. Lustri and P. P. Corbi, J. Mol. Struct., 2021, 1236, 130316 CrossRef CAS.
- F. Lakadamyali and E. Reisner, Chem. Commun., 2011, 47, 1695 RSC.
- P. Tang, H. J. Lee, K. Hurlbutt, P.-Y. Huang, S. Narayanan, C. Wang, D. Gianolio, R. Arrigo, J. Chen, J. H. Warner and M. Pasta, ACS Catal., 2022, 12, 3173 CrossRef CAS PubMed.
- C. Li, Z. Chen, H. Yi, Y. Cao, L. Du, Y. Hu, F. Kong, R. K. Campen, Y. Gao, C. Du, G. Yin, I. Y. Zhang and Y. Tong, Angew. Chem., Int. Ed., 2020, 59, 15902 CrossRef CAS PubMed.
- P. Du, J. Schneider, P. Jarosz and R. Eisenberg, J. Am. Chem. Soc., 2006, 128, 7726 CrossRef CAS PubMed.
- F. Wang, W.-G. Wang, H.-Y. Wang, G. Si, C.-H. Tung and L.-Z. Wu, ACS Catal., 2012, 2, 407 CrossRef CAS.
- K. Sakai and H. Ozawa, Coord. Chem. Rev., 2007, 251, 2753 CrossRef CAS.
- C. Wang, Y. Chen and W.-F. Fu, Dalton Trans., 2015, 44, 14483 RSC.
- E. J. L. McInnes, R. D. Farley, C. C. Rowlands, A. J. Welch, L. Rovatti and L. J. Yellowlees, J. Chem. Soc., Dalton Trans., 1999, 4203 RSC.
- A. Keerthi and S. Valiyaveettil, J. Phys. Chem. B, 2012, 116, 4603 CrossRef CAS PubMed.
- M. C. Kwakernaak, M. Koel, P. J. L. van den Berg, E. M. Kelder and W. F. Jager, Org. Chem. Front., 2022, 9, 1090 RSC.
- G. Li, M. F. Mark, H. Lv, D. W. McCamant and R. Eisenberg, J. Am. Chem. Soc., 2018, 140, 2575 CrossRef CAS PubMed.
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283 CrossRef CAS.
- J. Cann, B. S. Gelfand and G. C. Welch, Mol. Syst. Des. Eng., 2020, 5, 1181 RSC.
- V. Sharma, J. D. B. Koenig and G. C. Welch, J. Mater. Chem. A, 2021, 9, 6775 RSC.
- F. Yu, Z. Wang, S. Zhang, K. Yun, H. Ye, X. Gong, J. Hua and H. Tian, Appl. Catal., B, 2018, 237, 32 CrossRef CAS.
- Y. Li, X. Zhang and D. Liu, J. Photochem. Photobiol., 2021, 48, 100436 CrossRef CAS.
- K. Kong, S. Zhang, Y. Chu, Y. Hu, F. Yu, H. Ye, H. Dinga and J. Hua, Chem. Commun., 2019, 55, 8090 RSC.
- H. Xu, Z. Wang, S. Feng, X. Liu, X. Gong and J. Hua, Int. J. Hydrogen Energy, 2023, 48, 8071 CrossRef CAS.
- A. A. Kubicki, P. Bojarski, M. Grinberg, M. Sadownik and B. Kukliński, Opt. Commun., 2006, 269, 275 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1371 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: It includes the synthetic procedures and results of analyses; the results of the DFT calculations, including the optimized structures, energies and molecular orbitals; and additional results from the spectroscopic measurements. See DOI: https://doi.org/10.1039/d3ta05930h |
| This journal is © The Royal Society of Chemistry 2024 |