
Modulation of intermolecular interactions in hole transporting materials for improvement of perovskite solar cell efficiency: a strategy of trifluoromethoxy isomerization†
Jiayi
Qi
a,
Ruiqin
Wang
a,
Xin
Chen
a,
Fei
Wu
*b,
Wei
Shen
 a,
Ming
Li
a,
Rongxing
He
*a and
Xiaorui
Liu
*a
a,
Ming
Li
a,
Rongxing
He
*a and
Xiaorui
Liu
*a
aChongqing Key Laboratory of Soft-Matter Material Chemistry and Function Manufacturing, School of Chemistry and Chemical Engineering, Southwest University, Chongqing 400715, P. R. China. E-mail: herx@swu.edu.cn; liuxiaorui@swu.edu.cn
bChongqing Key Laboratory for Advanced Materials and Technologies of Clean Energy, School of Materials and Energy, Southwest University, Chongqing 400715, P. R. China. E-mail: feiwu610@swu.edu.cn
First published on 4th January 2024
Abstract
Rational design of hole transport materials (HTMs) can significantly enhance the performance of perovskite solar cells (PSCs). In this work, in order to provide a strategy of molecular design, three HTMs (JY4–JY6) are designed through modulating the intermolecular interactions within carbazole-diphenylamine-based HTMs for enhancing the efficiency of PSCs. On the basis of the DFT, TD-DFT, Marcus theory, and MD simulations, though the designed HTMs exhibit similar electron structures, optical properties and solubility, trifluoromethoxy isomerization in the designed HTMs can yield different intermolecular interactions and surface adsorption on perovskite films. Specifically, the molecule of JY6 demonstrates an inter-molecular co-directional alignment, which can offer an additional C–H/π stacking pathway for promoting a strong electronic-coupling between the adjacent molecules. Related experiments further confirmed heightened hole mobility, smoother film morphology, enhanced hole extraction and reduced charge recombination at the perovskite/JY6 interface. Therefore, PSC devices based on JY6 achieve a champion PCE of 22.06% compared to the devices based on JY4 (PCE of 18.84%) and JY5 (PCE of 20.94%) under equivalent conditions. Through a combination of theoretical calculations and experimental exploration, we substantiate the feasibility of obtaining potential HTMs via trifluoromethoxy isomerization to modulate the intermolecular interactions for enhancing the efficiency of PSCs.
Introduction
In recent years, perovskite solar cells (PSCs) have garnered widespread research interest as an emerging photovoltaic technology, owing to their advantages of high efficiency and low cost. The PSC devices have achieved a certified power conversion efficiency (PCE) of up to 26.1%.1 The PSC devices typically employ a sandwich structure comprising a perovskite photoactive absorption layer, an electron transport layer (ETL), a hole transport layer (HTL) and a metal electrode (e.g. silver electrode).2,3 Given various processing methods that enable the fabrication of high-quality perovskite films and electron transport materials (ETMs), the design and development of organic small-molecule hole transport materials (HTMs) have become particularly crucial.4–6 HTMs as an integral part between the electrode and perovskite layer in PSC devices play a critical role in the transporting and extraction of photo-generated carriers, and the protection of the perovskite.7–9 Therefore, rational design of HTMs can significantly enhance the performance of PSCs.In the molecular design of HTMs for PSCs, the main strategies include adjusting molecular structures, designing isomeric forms, and introducing functional groups. For example, Tang et al.10 considered the structure–property relationship and designed and synthesized two HTMs (PQ and DB) with similar molecular structures. Among them, DB, which has a core benzoyl structure, due to its flexible molecular structure, effectively interacts with the underlying perovskite, thereby enhancing the overall performance and stability of the devices. Yang et al.11 developed two fluorinated isomeric HTMs (spiro-mF and spiro-oF) based on spiro-OMeTAD for assembling PSC devices. The presence of fluorine atoms inhibits the intrusion of oxygen and water for promoting the device stability. The meta position fluorine atom in spiro-mF induces non-covalent interactions and internal interactions, resulting in a higher hole mobility and improved performance of PSC devices based on spiro-mF. Kin et al.12 proposed an end-group trifluoromethoxy substitution strategy to enhance the long-term stability of PSCs. Given its high hole mobility and strong hydrophobicity, the PCE of PSCs based on SGT-405s (C2, CF3) increased from 19.31% to 20.14%. Modulation of the molecular structure is an effective way to develop efficient HTMs. However, for the development of HTMs, we should not only pay close attention to the change of the chemical structure, but also should have an insight into the influence of intermolecular interactions on the performance of HTMs in PSC applications.
In fact, the intermolecular interactions within HTMs play an important role in promoting the efficiency of PSC devices owing to their influence on electronic structure, charge transport properties, and interfacial characteristics between HTMs and the perovskite photoactive layer. For instance, Jen et al.13 reported two donor–π-linker–donor (D–π–D) HTMs (N01 and N02), where N01 self-assembled into an ordered layered structure through intermolecular interactions, exhibiting excellent hole mobility and interfacial properties. Cheng et al.14 investigated the enhanced intermolecular interactions in YT-MPF with the peripheral fluorine groups, which featured a highly polarized structure and exhibited strong electron density delocalization to achieve the improvement of hole mobility. In addition, density functional theory (DFT), time-dependent DFT (TD-DFT), and molecular dynamics (MD) simulation studies can provide a clearer understanding of molecular geometry, optoelectronic properties, and the optimization of PSC device performance.15–18 Wei et al.19 employed DFT calculations to reveal the superior isotropic coordination ability of the halogenated HTM with perovskites, passivating Pb2+ defects to a greater extent to improve device stability. Cheng et al.20 utilized a multifluorinated molecule (6FPPY) as an interface modifier. Theoretical calculations and experimental results demonstrated that the fluorine functional groups in 6FPPY along with its highly conjugated planar core strengthened interface interactions and reduced energy losses during charge recombination. In our previous work, DFT predictions were utilized to elucidate the theoretical characteristics of isomers RQ4–RQ6. The results of theoretical simulations and experimental findings indicated that the para-substituted RQ6 exhibited closely packed molecular stacking, which results in improving the interfacial interactions at the perovskite/HTM interface.21
On the basis of the molecular design of HTMs in PSC applications, we focus on the influence of intermolecular interactions on the material function and the performance of PSC devices. Starting from carbazole-diphenylamine-based HTMs, three HTMs of JY4–JY6 were designed with a trifluoromethoxy isomerization strategy to modulate intermolecular interactions. As depicted in Fig. 1a, trifluoromethoxy groups are positioned at the para, meta, and ortho positions of the end-chain phenyls, respectively. Employing DFT, TD-DFT, Marcus theory, and MD simulations, we have investigated their geometric structures, frontier molecular orbital energy levels, optical absorption, hole transport properties, intermolecular interactions, and the interface characteristics between the perovskite and HTMs. Theoretical simulation results reveal that the designed HTMs exhibit similar electron structures, optical properties and solubility. However, trifluoromethoxy isomerization in the designed HTMs can yield different intermolecular interactions and surface adsorption on perovskite films. For example, compared to JY4 and JY5, JY6 was demonstrated an intermolecular co-directional alignment, which can offer an additional C–H/π stacking pathway for promoting a strong electronic-coupling between the adjacent molecules. To validate the accuracy of the theoretical findings, JY4–JY6 were synthesized for the fabrication of PSC devices. Experimental characterization confirmed that when used as the HTM, JY6 exhibited a higher hole mobility, smoother film morphology, and more efficient hole extraction. Under identical conditions, PSC devices based on JY6 achieved a champion PCE of 22.06%, surpassing the performance of devices based on JY4 (PCE of 18.84%), JY5 (PCE of 20.94%) and spiro-OMeTAD (PCE of 21.53%). This work underscores the efficacy and reliability of coupling theoretical simulations with experimental investigations. Furthermore, an efficient strategy of trifluoromethoxy isomerization in carbazole-diphenylamine-based HTMs to modulate the intermolecular interactions for enhancing the efficiency of PSCs is feasible.
 | ||
| Fig. 1 (a) Chemical structures of corresponding isomeric HTMs. (b) Optimized geometries of the investigated molecules in this work as obtained using the B3P86/6-311G(d,p) method. | ||
Results and discussion
Theoretical simulations
In order to investigate the relationship between the molecular structure and molecular interactions for providing a strategy for promoting the material function, three isomers of HTMs (JY4, JY5, and JY6) were designed. Starting from carbazole-diphenylamine derivatives with biphenyl substitution, trifluoromethoxy groups in the designed JY4, JY5, and JY6 are introduced to replace the end phenyl rings in the para, meta, and ortho positions, respectively (Fig. 1a). According to our previous studies, the B3P86/6-311G (d,p) functional and basis set can give accurate results in line with the experimental values for carbazole-diphenylamine derivative based HTMs.22 Therefore, ground-state structural optimization for the designed molecules was performed on the basis of the B3P86/6-311G (d,p) functional and basis set. As depicted in Fig. 1b, the dihedral angles of JY4, JY5 and JY6 between the trifluoromethoxy-substituted benzene and benzene ring are 142.0°, 142.2° and 128.7°, respectively. Between the middle benzene ring and the carbazole-diphenylamine, the dihedral angles are about 129.2–129.8° for the designed molecules. It can be seen that the JY6 molecule exhibits a smaller dihedral angle and higher planarity than other molecules, which potentially enhance the intermolecular π–π interactions in favor of facilitating hole transport.23 In addition, in comparison with the ground-state dipole moments of JY4 (2.01D) and JY5 (3.77D) (Table 1), the ortho substitution of trifluoromethoxy in JY6 yields a significantly higher ground-state dipole moment (3.81D), which is favorable for strengthening the intermolecular interactions and promoting the hole extraction and transport.24,25| HOMO | LUMO | E g | μ | η | ΔG | Absorptiona | Emissiona | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| λ abs | f | Assignments | λ em | Shift | |||||||
| a The results in S0–S1 states are calculated from TD-PBE0/6-31G(d) levels in dichloromethane solvent. | |||||||||||
| JY4 | −5.14 | −2.00 | 3.14 | 2.01 | 2.41 | −0.47 | 402 | 0.05 | H → L + 1 (80%) | 446 | 44 |
| H → L (17%) | |||||||||||
| JY5 | −5.15 | −2.13 | 3.02 | 3.77 | 2.41 | −0.48 | 403 | 0.11 | H → L + 1 (71%) | 446 | 43 |
| H → L (26%) | |||||||||||
| JY6 | −5.10 | −1.95 | 3.15 | 3.81 | 2.46 | −0.48 | 401 | 0.04 | H → L + 1 (75%) | 446 | 45 |
| H → L (23%) | |||||||||||
The distribution of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) within the frontier molecular orbitals (FMO) reflects the electronic characteristics of the molecules.26 In fact, there are no significant differences of the FMO distributions for the JY4, JY5 and JY6, as shown in Fig. 2a. For the molecules, the electron density of the HOMO is mainly delocalized on the carbazole-diphenylamine moiety, while the LUMO is primarily distributed over the biphenyl units. The well-separated HOMO and LUMO indicate charge transfer characteristics, which can promote the transfer of charges within the molecules from the donor of the carbazole-diphenylamine part to the acceptor of trifluoromethoxy-substituted biphenyl.27 The alignment of energy levels for HTMs with the perovskite is crucial for enhancing the photovoltaic efficiency.28 A better alignment of energy levels not only improves hole extraction and transport but also minimizes energy losses.29 As shown in Table 1, employing the B3P86/6-311G(d,p) functional and basis set, the HOMO energy levels for JY4, JY5, and JY6 are calculated to be −5.14, −5.15, and −5.10 eV, respectively. These values are significantly higher than the valence band (VB) of the perovskite, which facilitates the enhancement of electron–hole separation efficiency and promotes the effective extraction and transport of holes.30 The LUMO energy levels for JY4, JY5, and JY6 are −2.00, −2.13, and −1.95 eV, respectively, all of which are higher than the conduction band (CB) of the perovskite. These higher energy levels than the CB of the perovskite can prevent the photogenerated electrons from flowing towards the metal electrode while obstructing non-radiative recombination between electrons and holes.31 To corroborate the findings from the FMO analysis, each molecule was segmented into four parts such as the carbazole as the donor, the terminal trifluoromethoxy-benzene as the acceptor, and the benzene ring serving as the π-bridge in the intermediate. Density of states (DOS) analysis (Fig. 2b–d) for the designed HTMs was performed. It can be observed that the HOMO of JY4, JY5, and JY6 is mainly contributed by diphenylamine (DPA) and the core, with percentages in the order JY6 (97.49%) > JY5 (97.37%) > JY4 (97.34%). This indicates that the carbazole-diphenylamine portion of these three molecules essentially acts as the donor. In contrast, the LUMO of all three molecules is mainly concentrated on the π-bridge and acceptor segments. Specifically, the contribution of the trifluoromethoxy–benzene moiety to the LUMO is greater for JY6 (49.98%) than for JY4 (43.27%) and JY5 (48.20%). This observation supports the results of the FMO analysis.26,32
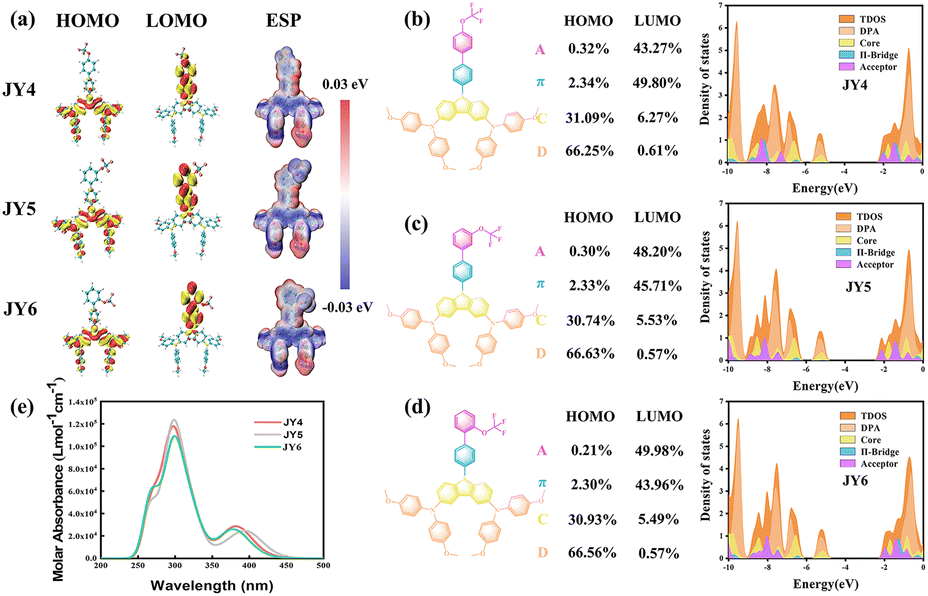 | ||
| Fig. 2 (a) Frontier molecular orbitals of all investigated molecules. (b–d) Graphical representation of the density of states (DOSs) of studied molecules (JY4–JY6), the waveforms of different colors represent the partial density of states (PDOSs) for various components within molecules. (e) Simulated absorption spectra of molecules JY4, JY5, and JY6 using the TD-PBE0/6-31G(d) functional and basis set in DCM. | ||
Electrostatic potential (ESP) maps illustrate the charge distribution within molecules, as shown in Fig. 2a. In the maps, red, white, and blue represent regions with positive charges, neutral regions, and regions of concentrated negative charges, respectively. It can be observed that the negative charges mainly gather around the carbazole center and the trifluoromethoxy group. This indicates that both of these components can act as Lewis bases to a greater extent, inhibiting the migration of lead ions and improving the quality of the perovskite film.33 Furthermore, good solubility of HTMs in organic solvents and stability in air are prerequisites for the preparation and application of PSCs. As shown in Table 1, JY4, JY5, and JY6 exhibit similar solvation free energies (ΔG) in chlorobenzene, which are approximately 0.47, 0.48, and 0.48 eV, respectively. This suggests that all molecules have good solubility in chlorobenzene, which is favorable for uniform film formation during spin-coating. In turn, it can induce uniform growth of the perovskite active layer on the top surface, promoting better interface contact between the perovskite film and HTMs.34 The molecular stability of HTMs can be assessed using the absolute hardness (η), where a higher η value indicates greater stability.35 As can be seen in Table 1, the η values for JY4, JY5, and JY6 are 2.41, 2.41, and 2.46 eV, respectively. The UV-vis absorption spectra of JY4–JY6 in dichloromethane solvent were predicted at the TD-PBE0/6-31G(d) level. As shown in Fig. 2e and Table 1, the designed JY4–JY6 exhibit similar optical properties. The maximum absorption wavelengths (λabs, max) for these molecules are 402 nm, 403 nm, and 401 nm, respectively, with corresponding orbital transitions being HOMO → LOMO + 1. The oscillator strengths (f) and stokes shifts are 0.05, 0.11, 0.04, and 44 nm, 43 nm, 45 nm, respectively.
The designed organic small molecules as HTMs were expected to exhibit high hole mobility for PSC applications. To further obtain insight into the hole transport capabilities, molecular dynamics simulations (MD) were performed using the GROMACS program. Hole mobility (μh) for the three molecules was calculated based on the Marcus electron transfer theory and the Einstein equation.36,37 One key parameter affecting the charge mobility is the reorganization energy (λ) which arises from geometric relaxation during charge transfer.38,39 The calculated hole reorganization energies (λh) for JY4, JY5, and JY6 are 0.194, 0.193, and 0.196 eV, respectively. Another important parameter of electron transfer integral (V) for the adjacent fragments was calculated.40V could be simulated from the PW91/TZP levels in the ADF program,41,42 which signifies the intermolecular electron coupling between two adjacent molecules in the structure. The values of V were affected by the orbital overlap and adjacent interactions. In order to obtain the models of molecular arrangement for the designed HTMs, we constructed a 50 × 50 × 50 Å box and placed 24 HTM molecules in it to perform molecular dynamics simulations of isolated HTMs. (see Fig. 3a). Simulations were carried out under NPT system conditions of 298.15 K, time step of 1 fs, Berendsen pressure coupling and velocity scale temperature coupling. According to the simulated results, the transporting pathways taking the central molecule with all neighboring molecules were constructed. The main pathways were extracted as shown in Fig. 3b. On the basis of the selected transporting pathways, the V of each pathway was calculated. The parameters for each pathway, as well as the calculated values of hole transfer integral (Vh), hole transport rate (kij), and hole mobility (μh), are presented in Table 2. The μh values for the three HTMs are as follows: JY6 (5.80 × 10−3 cm2 V−1 s−1) > JY5 (1.06 × 10−3 cm2 V−1 s−1) > JY4 (2.80 × 10−4 cm2 V−1 s−1).
 | ||
| Fig. 3 (a) Representative snapshot of molecular dynamics simulation. (b) Main charge-hopping pathways of JY4, JY5, and JY6. (c) Isodensity surface plots of the intermolecular HOMO overlaps of the pathways with the largest hole transfer integrals. The isodensity value is set at 1 × 10−6. | ||
| Molecules | Pathways | λ [eV] | ri [Å] | V [eV] | k ij [s−1] | μ h [cm2 V−1 s−1] |
|---|---|---|---|---|---|---|
| a Inner reorganization energy for holes. b Center-of-mass distance. c Hole coupling V (eV). d Hole transport rate kij. e Hole mobility μh. | ||||||
| JY4 | 1 | 0.194 | 12.707 | −3.13 × 10−4 | 5.72 × 108 | 2.80 × 10−4 |
| 2 | 14.719 | 4.71 × 10−4 | 1.30 × 109 | |||
| 3 | 13.508 | −7.79 × 10−4 | 3.55 × 109 | |||
| 4 | 11.181 | −4.80 × 10−4 | 1.35 × 109 | |||
| JY5 | 1 | 0.193 | 14.066 | 1.46 × 10−3 | 1.26 × 1010 | 1.06 × 10−3 |
| 2 | 9.060 | 1.56 × 10−3 | 1.43 × 1010 | |||
| 3 | 9.136 | 6.08 × 10−5 | 2.18 × 107 | |||
| 4 | 12.949 | 1.04 × 10−3 | 6.34 × 109 | |||
| JY6 | 1 | 0.196 | 11.082 | 8.78 × 10−4 | 4.38 × 109 | 5.80 × 10−3 |
| 2 | 13.875 | −1.05 × 10−3 | 6.30 × 109 | |||
| 3 | 15.336 | −4.12 × 10−4 | 9.64 × 108 | |||
| 4 | 12.499 | 3.42 × 10−3 | 6.66 × 1010 | |||
The higher hole mobility of JY6 is attributed to its larger electronic coupling. Visually, the orbital overlaps of the largest Vh for each HTM (JY4–JY6) were extracted. As can be seen in Fig. 3c, the regions enclosed in red and yellow indicate the areas of overlap between the HUMO orbitals of two adjacent molecules with the same and opposite phases, respectively. Under the same isosurface, JY6 exhibits a larger HOMO–HOMO overlap, which is more favorable for hole transport.17 Moreover, utilizing the independent gradient model (IGM) method, a systematic understanding of the intermolecular interactions of the designed molecules was attained.43,44 As depicted in Fig. 4a, each molecule was treated as an individual fragment. It is evident that all dimers exhibit a green region, which highlights a weak interaction with reduced electronic density, suggesting π–π van der Waals attractions between the dimers. Every dimer also showcases blue and red zones, indicative of localised attraction and repulsion forces, respectively.45 It is noteworthy that the intermolecular interactions within the JY4 dimer are relatively dispersed, whereas those in JY5 and JY6 present a more pronounced IGM isosurface area, reflecting their enhanced interactions. Contrary to the JY5 dimer, the JY6 dimer manifests a co-facial orientation, with the diphenylamine segments of the molecules interacting. This provides an additional avenue for C–H/π stacking, which is also instrumental in JY6 exhibiting an elevated hole mobility.45,46
 | ||
| Fig. 4 (a) IGM analysis based on the original molecular density shows the weak interaction of the designed HTM dimer. All iso-surfaces (isovalue = 0.001) are colored for the promolecular density range of −0.05 < ρ(r) < 0.05 a.u. (b) The distribution of HTMs on the perovskite surface after 10 ns is simulated by the MD method. (c–e) Adsorption energy of the perovskite surface to the HTM and electron charge density differences. | ||
The stacking and orientation of HTMs on the perovskite surface are crucial for enhancing the device performance. Therefore, we further conducted MD simulations to explore the deposition process of HTMs on the perovskite layer. The simulations were conducted under NVT ensemble conditions. At 10 ns, the interactions between HTMs and the perovskite surface are as follows: JY6/PVK (−1074.27 kJ mol−1) < JY5/PVK (−966.05 kJ mol−1) < JY4/PVK (−871.71 kJ mol−1), indicating a stronger interaction of JY6 with the perovskite. Additionally, we investigated the adsorption states of single HTMs on the perovskite surface. Based on configurations sampled from MD simulations, JY6 exhibited a flatter adsorption state compared to the randomly stacked JY4 and JY5. Utilizing first-principles calculations to comprehend molecular properties and behaviors, we simulated the charge density difference (CDD) between the perovskite surface and HTMs (JY4–JY6), as shown in Fig. 4c–e. Here, the yellow region represents electron accumulation, while the blue region represents electron depletion. The corresponding adsorption energies (Eads) of HTMs on the perovskite surface are JY6 (−3.27 eV) < JY5 (−3.11 eV) < JY4 (−3.07 eV). Clearly, JY6 demonstrates stronger interface contact.
The results from theoretical simulations indicate that the three newly designed HTMs possess appropriate energy levels and exhibit similar optical properties. However, by controlling the substitution positions of trifluoromethoxy groups, the adjacent-substituted JY6 molecule undergoes meaningful changes in its internal charge properties. Compared to JY4 and JY5, JY6 provides additional intermolecular stacking modes, demonstrating a higher hole mobility. It displays a distinct mechanism for enhancing interface interactions at the perovskite/HTMs interface. Therefore, JY6 holds promise as a candidate HTM.
Synthesis, characterization, and device performance
The synthesis routes for these HTMs are presented in Fig. S1.† The 1H NMR spectra, 13C NMR spectra, and HRMS data can be found in Fig. S2–S10.† Using Pd(PPh3)4 as a catalyst, 1,4-dioxane as the solvent, and a potassium carbonate solution to provide a basic environment, the three target compounds were successfully synthesized via Suzuki coupling reactions. Taking into account the filling effect of the molecular film, we characterized the HOMO energy levels of JY4–JY6 films using ultraviolet photoelectron spectroscopy (UPS). The cutoff energy region, onset energy region, and corresponding energy diagrams in the UPS spectra are shown in Fig. 5a and b. The work functions of JY4–JY6 films are −4.13, −4.05, and −4.15 eV, resulting in HOMO energy levels of −4.90, −4.88, and −4.83 eV for JY4, JY5, and JY6, respectively (as shown in Fig. 5b). The UV-visible absorption spectra of JY4, JY5, and JY6 are depicted in Fig. 5c, and the corresponding parameters are listed in Table 3. Three HTMs exhibit similar absorption features, with their absorption bands primarily located in the ultraviolet region. Notably, the absorption peaks are observed within the 250 to 500 nm range, with maximum absorption wavelengths (λabs) occurring at around 420 nm. The onset of absorption (λonset) is found to be 441, 443, and 440 nm for JY4, JY5, and JY6, respectively. Calculations based on the onset of absorption yield optical bandgaps (Eg) of 2.81 eV for JY4, 2.80 eV for JY5, and 2.81 eV for JY6. Based on UPS and UV experimental data, the energy level diagrams of JY4–JY6 as HTMs applied to PSC devices were obtained (Fig. 5d). Importantly, the HOMO and LUMO levels of the designed HTMs are higher than the VB and CB of the perovskite, indicating a good level alignment between HTMs and the perovskite. This alignment facilitates effective hole extraction and reverse recombination of electrons. The electrochemical performance of JY4, JY5, and JY6 was analyzed using cyclic voltammetry (CV), as shown in Fig. S11.† Ferrocene was used as an internal standard for these measurements. All three HTMs exhibit two distinct redox processes.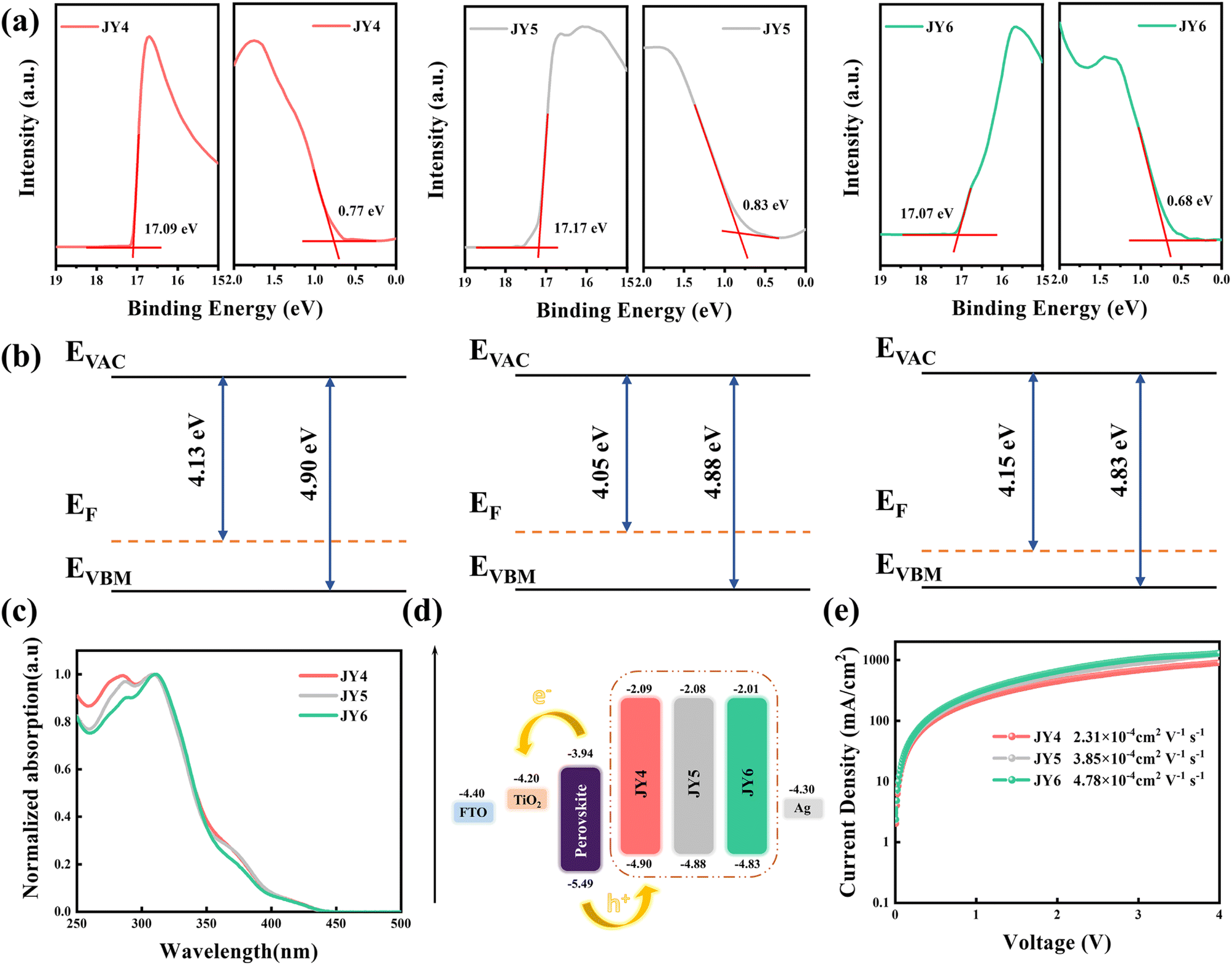 | ||
| Fig. 5 (a) UPS spectra of the JY4–JY6 films (b) energy diagram for JY4–JY6. (c) Measured absorption spectra of JY4, JY5, and JY6. (d) Energy levels of JY4, JY5, and JY6 in PSC devices. (e) Dark J–V curves of JY4, JY5, and JY6 for measurement of hole mobility. | ||
| λ max [nm] | λ onset [nm] | E ox [eV] | E g [eV] | E HOMO [eV] | E LUMO [eV] | Hole mobility [cm2 V−1 s−1] | |
|---|---|---|---|---|---|---|---|
| a Absorption spectra were measured in DCM solution. b Onset of oxidation potentials measured by cyclovoltammetry. c Optical band gap (Eg) obtained from the onset values of absorption (λonset). | |||||||
| JY4 | 417 | 441 | 0.37 | 2.81 | −4.90 | −2.09 | 2.31 × 10−4 |
| JY5 | 419 | 443 | 0.37 | 2.80 | −4.88 | −2.08 | 3.85 × 10−4 |
| JY6 | 420 | 440 | 0.38 | 2.82 | −4.83 | −2.01 | 4.78 × 10−4 |
The parameters of hole mobility for JY4, JY5, and JY6 were determined using the space charge limited current (SCLC) method with an ITO/PEDOT:PSS/HTMs/Ag configuration for hole-only devices.47 As shown in Fig. 5e, the hole mobilities of the HTMs are as follows: JY6 (4.78 × 10−4 cm2 V−1 s−1) > JY5 (3.85 × 10−4 cm2 V−1 s−1) > JY4 (2.31 × 10−4 cm2 V−1 s−1). This trend is consistent with the computational results. However, the higher hole mobility of JY6 is likely attributed to the enhanced dipole–dipole interactions between neighboring molecules due to the ortho-position substituted trifluoromethoxy groups, which promote close molecular packing.48
Through the grazing-incidence wide-angle X-ray scattering (GIWAXS) technique, we conducted a detailed investigation into the molecular stacking of JY4–JY6 films. Fig. 6 presents the two-dimensional (2D) GIWAXS patterns and corresponding one-dimensional (1D) film GIWAXS line cuts. As shown in Fig. 6a–c, it is observed that the molecules in films of JY4–JY6 exhibit random orientation. According to Fig. 6(d), the JY4, JY5, and JY6 films exhibit π–π diffraction peaks at 1.31, 1.33, and 1.37 Å–1, respectively, with corresponding π–π stacking distances of 4.79, 4.72, and 4.58 Å. Experimental results indicate that the substitution of adjacent trifluoromethoxy groups in the JY6 film contributes to a more compact molecular stacking. This positively influences the charge transport characteristics and device performance of JY6.48–50 To further investigate the surface morphology of the films for the designed HTMs, we conducted atomic force microscopy (AFM) measurements. The root mean square roughness (RMS) of the bare perovskite film surface was the highest at 27.7 nm (Fig. 7a). After spin-coating the designed HTMs onto the perovskite film, the RMS significantly decreased in the following order: perovskite/JY6 (9.64 nm) < perovskite/JY5 (9.82 nm) < perovskite/JY4 (12.1 nm). This suggests that when these three molecules are applied to PSC devices, JY6 exhibits better film-forming ability. A smoother film morphology leads to improvements in hole transport at the perovskite/JY6 interface and a reduction in charge recombination, which has a significant impact on enhancing the performance of PSC devices.51
 | ||
| Fig. 6 The GIWAXS images of (a) JY4, (b) JY5 and (c) JY6. (d) The corresponding line cuts from GIWAXS patterns for JY4–JY6. | ||
 | ||
| Fig. 7 (a) AFM image of the perovskite film. (b–d) AFM images of JY4, JY5, and JY6 films on the perovskite. | ||
Considering the suitable energy levels, excellent hole mobility, and outstanding film-forming ability of the designed HTMs, we fabricated PSCs with the structure of FTO/TiO2/Cs0.05FA0.85MA0.10Pb(I0.88Br0.04Cl0.08)3/HTMs/Ag (Fig. 8a) to explore the photovoltaic performance of JY4, JY5, and JY6. We optimized the thickness of the HTM films in PSC devices with JY4–JY6 at different concentrations (40, 50, and 60 mg mL−1). The relevant data are presented in Tables S5–S7.† The film thickness data of HTMs at different concentrations are detailed in Table S8.† In this work, the devices exhibited optimal performance when the concentration of HTMs was 50 mg mL−1. Fig. 8b illustrates the best J–V curves of PSCs based on JY4, JY5, and JY6 at the optimized concentration under AM 1.5G illumination, with parameters detailed in Table 4. It can be observed that the PSC based on JY6 exhibits a champion PCE of 22.06%, with a VOC of 1.10 V, JSC of 24.46 mA cm−2, and FF of 81.99%, significantly higher than those of the PSC based on JY4 (PCE of 18.84%) and the PSC based on JY5 (PCE of 20.94%). Through the above tests, we find that by tuning the substitution positions of the trifluoromethoxy groups, the VOC, JSC, and FF parameters of devices based on ortho, meta, and para HTMs gradually improve. The significant improvement in JSC and FF for JY6 can be attributed to its excellent control over the perovskite film quality and higher hole mobility, while the additional intermolecular interactions provided by JY6 and its stronger interaction with the perovskite improve the VOC. Furthermore, we conducted a comparative analysis of the performance of devices based on the classic Spiro-OMeTAD under identical conditions (as illustrated in Fig. S12 and Table S3†). The maximum PCE achieved for Spiro-OMeTAD-based devices was 21.53%, with a VOC of 1.114 V, JSC of 24.61 mA cm−2, and FF of 78.53%. Notably, PSC devices utilizing JY6 exhibited superior efficiency compared to those based on spiro-OMeTAD. Hysteresis has a significant impact on the performance and stability of PSC devices.52 Fig. S13 and Table S4† present the forward and reverse scan J–V curves, along with the corresponding photovoltaic parameters, for the optimal devices based on JY4–JY6 and spiro-OMeTAD. The hysteresis index (HI) for PSC devices is ranked as follows: spiro-OMeTAD (3.16%) < JY6 (3.17%) < JY5 (3.82%) < JY4 (5.10%). The HI of the PSC devices based on JY6 is comparable to that of PSC devices based on spiro-OMeTAD, indicating that by tuning the substitution positions of the trifluoromethoxy groups at the ortho, meta, and para positions, the charge accumulation at the perovskite/HTM interface has been reduced.53 Considering the reproducibility of device photovoltaic performance, we fabricated 18 devices for each HTM (Fig. S14†). It can be seen that compared to JY4, PSCs based on JY5, JY6 and spiro-OMeTAD exhibit better reliability, particularly JY6, which achieves a higher PCE due to its higher VOC, while devices based on JY4 show a lower PCE mainly because of the lower VOC and FF.
 | ||
| Fig. 8 (a) PSC device assembly diagram. (b) J−V curves of JY4, JY5, and JY6 based PSC devices. (c) J−V curves under reverse and forward scans for JY4, JY5, and JY6. (d) EQE and integrated JSC of the JY4–JY6 based PSCs. (e) Steady-state PL spectra of the perovskite films, and JY4, JY5, and JY6 films on the perovskites. (f) Time-resolved PL decay of the perovskite films, and JY4, JY5, and JY6 films on the perovskites. | ||
| HTMs | V OC (V) | J SC (mA cm−2) | FF (%) | PCE (%) |
|---|---|---|---|---|
| a The maximum value. b The average values were obtained from 18 devices. | ||||
| JY4 | 1.034 (1.002 ± 0.020) | 24.08 (24.14 ± 0.17) | 75.64 (73.47 ± 2.09) | 18.84a (17.78 ± 0.64)b |
| JY5 | 1.077 (1.061 ± 0.022) | 24.15 (24.14 ± 0.15) | 80.51 (79.89 ± 1.9) | 20.94 (20.45 ± 0.34) |
| JY6 | 1.100 (1.098 ± 0.023) | 24.46 (24.15 ± 0.14) | 81.99 (79.94 ± 1.13) | 22.06 (21.20 ± 0.47) |
Further investigation of the photoelectric conversion characteristics of the devices was conducted through external quantum efficiency (EQE) spectra, as shown in Fig. 8d, in conjunction with the integrated short-circuit current density curve. We observed strong EQEs for all devices in the range of 400 to 750 nm. The integrated current density values for JY4, JY5, and JY6 devices were 22.78, 22.85, and 23.22 mA cm−2, respectively, which are consistent with the J–V curve results.
Furthermore, we performed steady-state photoluminescence (PL) spectroscopy and time-resolved photoluminescence (TRPL) decay measurements of the perovskite films, as well as perovskite films deposited with HTMs, to analyze the carrier dynamics at the interface between the perovskite and HTM. As shown in Fig. 8e, compared to the pristine perovskite, there is a significant quenching of PL intensity at 787 nm when the HTM is deposited on the surface. The quenching intensity at the perovskite/HTM interface is ranked as JY6 > JY5 > JY4, with JY6 exhibiting the strongest quenching, indicating faster hole transfer at the perovskite/JY6 interface. By fitting the TRPL decay curves of the perovskite samples coated with the three HTMs using a double-exponential equation (Fig. 8f), it is evident that the lifetimes of all HTM-deposited perovskite films are significantly reduced compared to that of the pristine perovskite film, following the trend of JY6 < JY5 < JY4. The TRPL results indicate that among the designed HTMs, the highest hole extraction efficiency occurs between the perovskite and JY6. Furthermore, charge recombination at the perovskite/JY6 interface is effectively suppressed. Therefore, JY6 exhibits a pronounced photoluminescence quenching capability and a shorter luminescence decay lifetime, possibly due to enhanced molecular stacking facilitated by the co-directional alignment of the molecules.54
Conclusions
In summary, we provided a strategy of trifluoromethoxy isomerization, aiming to modulate the intermolecular interactions within carbazole-diphenylamine-based HTMs for enhancing the efficiency of PSCs. Employing DFT, TD-DFT, Marcus theory, and MD simulations, we have investigated their geometric structures, frontier molecular orbital energy levels, optical absorption, hole transport properties, intermolecular interactions, and the interface characteristics between the perovskite and HTMs. Though the designed HTMs exhibit similar electron structures, optical properties and solubility, trifluoromethoxy isomerization in the designed HTMs can yield different intermolecular interactions and surface adsorption on perovskite films. Specifically, the molecule of JY6 demonstrated an intra-molecular co-directional alignment, which can offer an additional C–H/π stacking pathway for promoting a strong electronic-coupling between the adjacent molecules. Related experiments further confirmed heightened hole mobility, smoother film morphology, enhanced hole extraction and reduced charge recombination at the interface/HTMs. Therefore, PSC devices based on JY6 achieved a champion PCE of 22.06% compared to the devices based on JY4 (PCE of 18.84%) and JY5 (PCE of 20.94%) under equivalent conditions. Our research underscores the efficacy and reliability of coupling theoretical simulations with experimental investigations and offers an efficient strategy of trifluoromethoxy isomerization to modulate the intermolecular interactions for enhancing the efficiency of PSCs.Author contributions
Jiayi Qi: investigation, data curation, formal analysis, and writing – original draft. Ruiqin Wang: investigation, visualization and validation. Xin Chen: validation. Fei Wu: supervision. Wei Shen: resources. Ming Li: resources. Rongxing He: supervision, resources. Xiaorui Liu: conceptualization, funding acquisition, project administration, resources, supervision, and writing – review and editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by supported by the Fundamental Research Funds for the Central Universities (Grant No. SWU-KT23009 and SWU-XDJH202314) and Natural Science Foundation of Chongqing (Grant No. cstc2020jcyj-msxmX0379).Notes and references
- Z. Wu, E. Bi, L. K. Ono, D. Li, O. M. Bakr, Y. Yan and Y. Qi, Nano Energy, 2023, 115, 108731 CrossRef CAS.
- L. M. Nhari, R. M. El-Shishtawy and A. M. Asiri, Dyes Pigm., 2021, 193, 109465 CrossRef CAS.
- P. Yan, D. Yang, H. Wang, S. Yang and Z. Ge, Energy Environ. Sci., 2022, 15, 3630–3669 RSC.
- A. Liu, K. Liu, H. Zhou, H. Li, X. Qiu, Y. Yang and M. Liu, Sci. Bull., 2018, 63, 1591–1596 CrossRef CAS PubMed.
- H. Sun, P. Dai, X. Li, J. Ning, S. Wang and Y. Qi, J. Energy Chem., 2021, 60, 300–333 CrossRef CAS.
- Y. Niu, C. Tian, J. Gao, F. Fan, Y. Zhang, Y. Mi, X. Ouyang, L. Li, J. Li, S. Chen, Y. Liu, H.-L. Lu, X. Zhao, L. Yang, H. Ju, Y. Yang, C.-F. Ding, M. Xu and Q. Xu, Nano Energy, 2021, 89, 106455 CrossRef CAS.
- P. Murugan, T. Hu, X. Hu and Y. Chen, J. Mater. Chem. A, 2022, 10, 5044–5081 RSC.
- C. Zhang, K. Wei, J. Hu, X. Cai, G. Du, J. Deng, Z. Luo, X. Zhang, Y. Wang, L. Yang and J. Zhang, Mater. Today, 2023, 67, 518–547 CrossRef CAS.
- Y. Fu, Y. Li, G. Xing and D. Cao, Mater. Today Adv., 2022, 16, 100300 CrossRef CAS.
- R. Tang, H. Liu, Y. Xu, K. Chen, J. Zhang, P. Zhang, C. Zhong, F. Wu and L. Zhu, Adv. Funct. Mater., 2023, 33, 2208859 CrossRef CAS.
- M. Jeong, I. W. Choi, E. M. Go, Y. Cho, M. Kim, B. Lee, S. Jeong, Y. Jo, H. W. Choi, J. Lee, J.-H. Bae, S. K. Kwak, D. S. Kim and C. Yang, Science, 2020, 369, 1615–1620 CrossRef CAS PubMed.
- C. Lu, M. Aftabuzzaman, C. Hoon Kim and H. Kyu Kim, Chem. Eng. J., 2022, 428, 131108 CrossRef CAS.
- N. Cai, F. Li, Y. Chen, R. Luo, T. Hu, F. Lin, S. Yiu, D. Liu, D. Lei, Z. Zhu and A. K. -Y. Jen, Angew. Chem., Int. Ed., 2021, 60, 20437–20442 CrossRef CAS PubMed.
- M. Zhai, A. Wang, C. Chen, F. Hao, H. Wang, L. Ding, X. Yang and M. Cheng, Chem. Eng. J., 2021, 425, 131675 CrossRef CAS.
- Y. Li, Y. Zhang, P. Heng, R. Shao, Y. Liu, W. Qiao, L. Wang and J. Zhang, Org. Electron., 2018, 54, 14–20 CrossRef CAS.
- J. Xia, P. Luizys, M. Daskeviciene, C. Xiao, K. Kantminiene, V. Jankauskas, K. Rakstys, G. Kreiza, X. Gao, H. Kanda, K. G. Brooks, I. R. Alwani, Q. U. Ain, J. Zou, G. Shao, R. Hu, Z. Qiu, A. Slonopas, A. M. Asiri, Y. Zhang, P. J. Dyson, V. Getautis and M. K. Nazeeruddin, Adv. Mater., 2023, 35, 2300720 CrossRef CAS PubMed.
- R. Wang, X. Chen, C. Wu, J. Qi, X. Jiang, F. Wu and X. Liu, Appl. Surf. Sci., 2023, 639, 158192 CrossRef CAS.
- K. Yang, Q. Liao, J. Huang, Z. Zhang, M. Su, Z. Chen, Z. Wu, D. Wang, Z. Lai, H. Y. Woo, Y. Cao, P. Gao and X. Guo, Angew. Chem., Int. Ed., 2022, 61, e202113749 CrossRef CAS PubMed.
- Z. Zhang, L. Shen, S. Wang, L. Zheng, D. Li, Z. Li, Y. Xing, K. Guo, L. Xie and Z. Wei, Adv. Energy Mater., 2023, 13, 2204362 CrossRef CAS.
- H. Wang, W. Zhang, B. Wang, Z. Yan, C. Chen, Y. Hua, T. Wu, L. Wang, H. Xu and M. Cheng, Nano Energy, 2023, 111, 108363 CrossRef CAS.
- R. Wang, C. Wu, J. Qi, W. Shen, F. Wu, M. Li, R. He and X. Liu, Adv. Funct. Mater., 2023, 2213843 Search PubMed.
- Q. Chen, H. Liu, R. Wang, C. Wu, F. Wu, X. Liu and X. Liu, Dyes Pigm., 2022, 206, 110604 CrossRef CAS.
- C. Igci, H. Kanda, S.-M. Yoo, A. A. Sutanto, O. A. Syzgantseva, M. A. Syzgantseva, V. Jankauskas, K. Rakstys, M. Mensi, H. Kim, A. M. Asiri and M. K. Nazeeruddin, Sol. RRL, 2022, 6, 2100667 CrossRef CAS.
- X. Ji, T. Wang, Q. Fu, D. Liu, Z. Wu, M. Zhang, H. Y. Woo and Y. Liu, Macromol. Rapid Commun., 2023, 2300213 CrossRef CAS PubMed.
- J. Xu, Q. Xiong, X. Huang, P. Sun, Q. Zhou, Y. Du, Z. Zhang and P. Gao, Small, 2022, 2206435 Search PubMed.
- R. Hussain, M. Adnan, K. Atiq, M. Usman Khan, Z. H. Farooqi, J. Iqbal and R. Begum, Sol. Energy, 2023, 253, 187–198 Search PubMed.
- Y. Yang, S. Un Ryu, F. Wu, H. Lu, K. Jia, C. Zhong, T. Park and L. Zhu, Chem. Eng. J., 2021, 424, 130396 CrossRef CAS.
- J. A. Hong, M. Jeong, S. Park, A. Lee, H. S. Kim, S. Jeong, D. W. Kim, S. Cho, C. Yang and M. H. Song, Adv. Sci., 2023, 10, 2205127 CrossRef CAS PubMed.
- J. Zhou, H. Li, L. Tan, Y. Liu, J. Yang, R. Hua and C. Yi, Angew. Chem., Int. Ed., 2023, e202300314 CAS.
- Z. Li, Y. Tong, J. Ren, Q. Sun, Y. Tian, Y. Cui, H. Wang, Y. Hao and C.-S. Lee, Chem. Eng. J., 2020, 402, 125923 CrossRef CAS.
- H. Lu, J. Xu, X. Liu, F. Wu and L. Zhu, Mater. Today Energy, 2021, 21, 100780 CrossRef CAS.
- S. J. Akram, J. Iqbal, M. Ans, Y. A. El-Badry, R. F. Mehmood and R. A. Khera, Sol. Energy, 2022, 237, 108–121 CrossRef CAS.
- B. Wu, Q. Fu, L. Sun, Y. Liu, Z. Sun, S. Xue, Y. Liu and M. Liang, ACS Energy Lett., 2022, 7, 2667–2676 CrossRef CAS.
- X. Gao, F. Wu, Y. Zeng, K. Chen, X. Liu and L. Zhu, J. Mater. Chem. C, 2023, 11, 11218–11224 RSC.
- M. Vatanparast and Z. Shariatinia, Sol. Energy, 2021, 230, 260–268 CrossRef CAS.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- R. A. Marcus, Annu. Rev. Phys. Chem., 1964, 15, 155–196 Search PubMed.
- A. Thomas, R. K. Chitumalla, A. L. Puyad, K. V. Mohan and J. Jang, Comput. Theor. Chem., 2016, 1089, 59–67 CrossRef CAS.
- J.-H. Pan, Y.-M. Chou, H.-L. Chiu and B.-C. Wang, J. Phys. Org. Chem., 2007, 20, 743–753 CrossRef CAS.
- E. F. Oliveira and F. C. Lavarda, Polymer, 2016, 99, 105–111 CrossRef CAS.
- G. Te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. Van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- C. Fonseca Guerra, J. G. Snijders, G. Te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 Search PubMed.
- C. Lefebvre, G. Rubez, H. Khartabil, J.-C. Boisson, J. Contreras-García and E. Hénon, Phys. Chem. Chem. Phys., 2017, 19, 17928–17936 RSC.
- T. Lu and Q. Chen, J. Comput. Chem., 2022, 43, 539–555 CrossRef CAS PubMed.
- Y. Jiang, E. J. Mattioli, M. Calvaresi and Z. Wang, Chem. Commun., 2020, 56, 11835–11838 RSC.
- Y. Tian, L. Wang, G. Fu, C. Zhang, R. Lu and X. Dong, Theor. Chem. Acc., 2020, 139, 78 Search PubMed.
- V. D. Mihailetchi, J. Wildeman and P. W. M. Blom, Phys. Rev. Lett., 2005, 94, 126602 CrossRef CAS PubMed.
- K. Lee, Y. Huang, W. Chiu, Y. Huang, G. Chen, G. B. Adugna, S. Li, F. Lin, S. Lu, H. Hsieh, K. Liau, C. Huang, Y. Tai, Y. Tao and Y. Lin, Adv. Funct. Mater., 2023, 2306367 CrossRef CAS.
- W. Li, C. Wu and X. Han, Molecules, 2023, 28, 3076 CrossRef CAS PubMed.
- H. Zhang, X. Yu, M. Li, Z. Zhang, Z. Song, X. Zong, G. Duan, W. Zhang, C. Chen, W. Zhang, Y. Liu and M. Liang, Angew. Chem., Int. Ed., 2023, 62, e202314270 Search PubMed.
- Y. Yao, C. Cheng, C. Zhang, H. Hu, K. Wang and S. De Wolf, Adv. Mater., 2022, 34, 2203794 Search PubMed.
- C. Yang, H. Wang, Y. Miao, C. Chen, M. Zhai, Q. Bao, X. Ding, X. Yang and M. Cheng, ACS Energy Lett., 2021, 6, 2690–2696 CrossRef CAS.
- J. Xia, V. Joseph, A. A. Sutanto, R. Balasaravanan, Y. Ezhumalai, Z.-X. Zhang, J.-S. Ni, S. T. Yogesh, S.-L. Yau, G. Shao, Z. Qiu, A. M. Asiri, M.-C. Chen and M. K. Nazeeruddin, Cell Rep. Phys. Sci., 2023, 4, 101312 CrossRef CAS.
- J. Yang, J. Huang, C. Zhang, H. Sun, B. Li, Y. Wang, K. Feng, Q. Liao, Q. Bai, L. Niu, H. Wang and X. Guo, Adv. Funct. Mater., 2022, 32, 2206311 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta06179e |
| This journal is © The Royal Society of Chemistry 2024 |