
Research progress of Prussian blue and its analogues for cathodes of aqueous zinc ion batteries
Jiayi
Liu
abc,
Zhongrong
Shen
 abc and
Can-Zhong
Lu
*abc
abc and
Can-Zhong
Lu
*abc
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, P. R. China. E-mail: czlu@fjirsm.ac.cn
bUniversity of the Chinese Academy of Sciences, Beijing 100049, P. R. China
cXiamen Key Laboratory of Rare Earth Photoelectric Functional Materials, Xiamen Institute of Rare Earth Materials, Haixi Institutes, Chinese Academy of Sciences, Xiamen 361021, P. R. China
First published on 1st January 2024
Abstract
Aqueous zinc-ion batteries (AZIBs) have garnered significant attention for large-scale applications due to their low cost, safety, and eco-friendliness. Among all cathode materials, Prussian blue and its analogues (PBAs) have garnered significant attention and been subjected to extensive research due to their diverse property-effectiveness, simple synthesis process, reversible Zn2+ insertion/removal capability, and other notable characteristics. However, numerous studies have revealed that the utilization of PBAs as a cathode material in AZIBs still presents certain challenges, including limited energy density and prolonged structural instability during cycling. In light of the aforementioned opportunities and challenges for PBAs, we conducted a comprehensive literature review encompassing synthesis, analysis of energy storage mechanisms, enhancement of electrolyte–electrode interfaces, and battery applications. The objective of this review is to encourage active engagement from researchers interested in AZIBs and PBA cathode materials, thereby fostering research and industrialization efforts that collectively contribute towards the advancement of safe and environmentally friendly energy storage.
 Jiayi Liu | Jiayi Liu is currently pursuing a Master of Engineering degree at the University of Chinese Academy, China. Her research interests focus on aqueous zinc-ion battery energy storage, particularly the application of Prussian blue materials in aqueous zinc-ion batteries and electrolyte engineering to enhance the anode interface. |
 Zhongrong Shen | Zhongrong Shen obtained his B.S. (2000), M.S. (2003) and PhD (2007) degrees from the University of Science and Technology of China (USTC), Institute of Chemistry, Chinese Academy of Sciences (ICCAS), and Japan Advanced Institute of Science and Technology (JAIST), respectively. After postdoctoral research at Japan Science and Technology Agency (JST), he worked as an assistant professor at the JAIST and Shinshu University, sequentially. He has been a professor at the Fujian Institute of Research on the Structure of Matter (FJIRSM), Chinese Academy of Sciences, since 2017. His research focuses on carbon hybrid materials and electrode materials in the fields of sodium ion batteries, zinc ion batteries, and hydrogen storage. |
 Can-Zhong Lu | Dr Can-Zhong Lu is a Professor at the Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, China. Prof. Lu received his B.S. in chemistry from Xiamen University in 1986 and PhD from the University of Essex, England, in 1999. Prof. Lu's research interests include polyoxides, coordination polymers, organic–inorganic composite materials, photoelectric conversion materials, etc., and he has made some important progress in the research of organic ligands or transition metal (rare earth) small molecules connecting polyoxides to construct organic–inorganic hybrid materials and host-guest materials, transition metal luminescent materials and their devices. |
1 Introduction
Due to the escalating environmental issues and the surging demand for sustainable energy, alternative novel energy sources have emerged as a prominent area of research. Owing to their exceptional attributes such as high energy density, wide voltage range, and prolonged cycle life, lithium-ion batteries (LIBs) have witnessed an exponential rise in popularity within portable electronic devices and commercial electric vehicle energy storage systems over the past few decades. However, the limited availability of lithium resources, the exorbitant cost of electrodes, and the significant safety hazards associated with the toxic and flammable nature of organic electrolytes diminish their competitiveness in addressing the forthcoming challenges of scalable energy devices.1–3 Therefore, the pursuit of novel generations of environmentally friendly battery materials has emerged as a primary objective in large-scale energy storage research. Diverse types of batteries, including monovalent Na+ and K+ and multivalent Mg2+, Zn2+ and Al3+ batteries, rapidly evolved into the next iteration of energy storage devices in the subsequent period.4–7 Among them, sodium and potassium ion batteries have been extensively investigated; however, the utilization of flammable and toxic organic electrolytes may still give rise to safety concerns.8 Aqueous zinc-ion batteries (AZIBs) are characterized by their exceptional properties, such as high safety, eco-friendliness, low redox potential, and good compatibility, which is closely related to the excellent properties of Zn metal (Table 1). AZIBs eliminate concerns regarding safety issues associated with flammable metals and toxic electrolytes, making them the optimal choice for large-scale storage applications of renewable energy.9,10 Furthermore, the substantial growth of literature in this field (Fig. 1a) attests to the significant research attention in recent years.11–14| Metal ions | Li+ | Na+ | K+ | Mg2+ | Zn2+ | Al3+ |
|---|---|---|---|---|---|---|
| Ionic radius [Å] | 0.76 | 1.02 | 1.38 | 0.72 | 0.740 | 0.535 |
| Theoretical specific capacity [mA g g−1] | 3862 | 1166 | 685 | 2205 | 820 | 2980 |
| Volumetric energy density [mA h cm−3] | 2061 | 1129 | 610 | 3834 | 5851 | 8064 |
| Standard potential (V vs. SHE) | −3.04 | −2.71 | −2.93 | −2.37 | −0.76 | −1.66 |
| Crustal abundance [ppm] | 18 | 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
21000 |
23000 |
79 | 82000 |
| Metal cost [$ per ton] | 165000 |
200 | 1000 | 4600 | 2570 | 2390 |
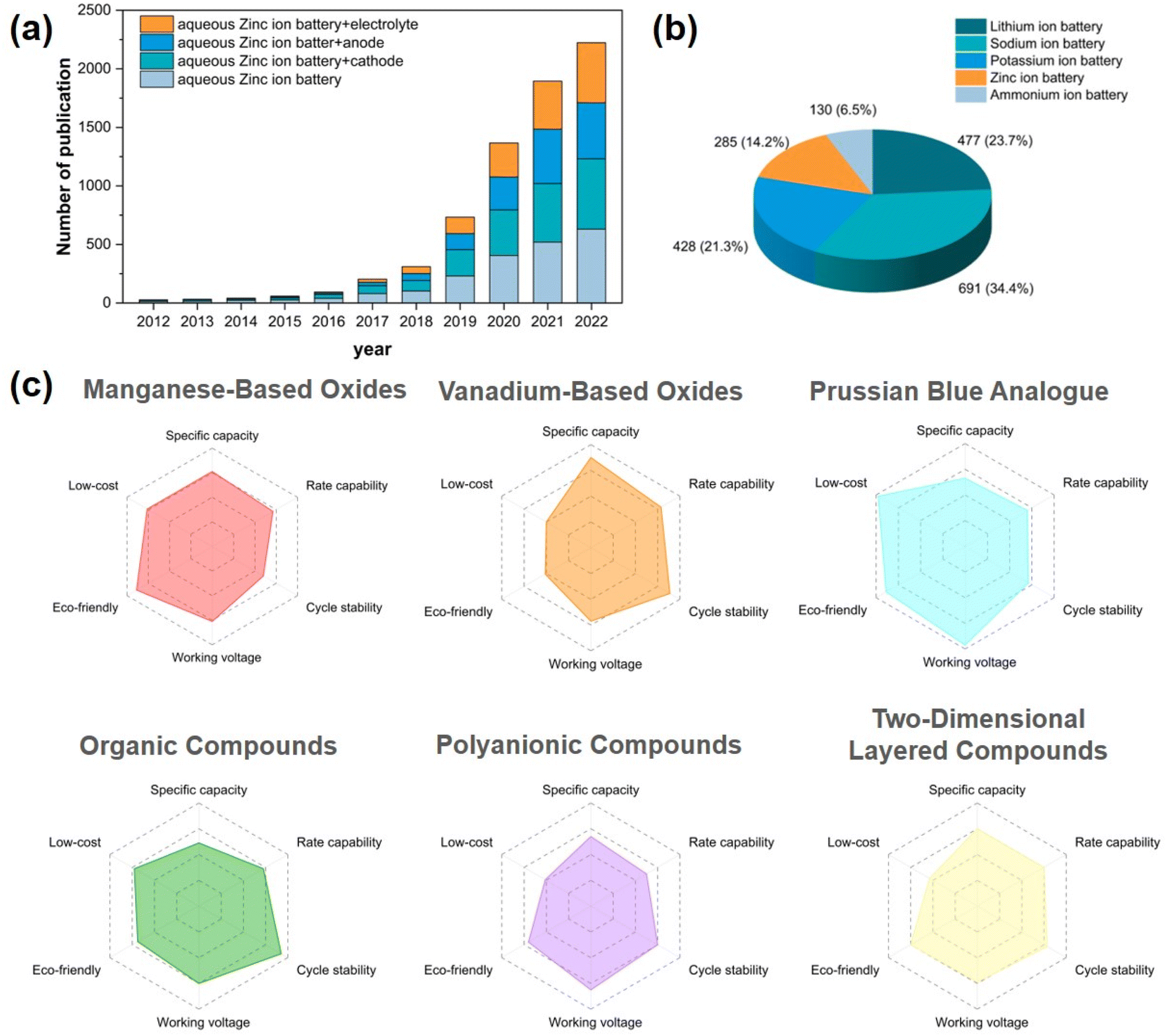 | ||
| Fig. 1 (a) Growth of publications on zinc ion batteries in the last decade. (b) The proportion of publication of PBAs in all kinds of battery fields. (c) Performance comparison radar diagram of different zinc ion battery cathode materials. | ||
The conventional aqueous zinc ion battery consists of a zinc metal anode, a near-neutral (or mildly acidic) aqueous electrolyte, and a cathode that facilitates the reversible intercalation (deintercalation) of Zn2+ ions. Among these, the design and modification of cathode materials is an essential stage in the development of AZIBs. Currently, Zn2+ can be reversibly inserted or removed from manganese-based oxides, vanadium-based oxides, Prussian blue analogues (PBAs), organic compounds, polyanionic compounds, and two-dimensional layered materials (Mxene, sulfides, and layered selenides), among others.11,15–23 We illustrate the performance characteristics of various materials with radar diagrams (Fig. 1c). Manganese-based materials often face challenges of rapid capacity decay due to structural instability caused by manganese dissolution during cycling, parasitic side reactions, and structural transitions.24–27 The stability and capacity of compounds based on the element V are enhanced compared to those of Mn-based cathodes due to their superior capacitance contribution and more robust crystal structure.
However, the average operating voltage of most V-based compounds compared to Zn/Zn2+ is approximately 0.75 V, which prevents the attainment of high energy density.28–31 Prussian blue and its analogues (PBAs) stand out as a class of high-voltage resistant materials. Their operating voltage range can reach 1.2 V, in addition to their simple synthesis process, low cost, and environmentally friendly characteristics.32,33
Among the 20000 species of MOFs, PBAs belong to a distinct subclass characterized by unique structural and electrochemical properties.34 The applications of PBAs span across various disciplines, encompassing hydrogen storage, biosensing, cancer therapy, and sea water desalination.35 In recent years, PBAs have been employed as precursors in the synthesis of a variety of functional materials. In the framework of PBA crystals, the substantial gap (∼4.6 Å) can effectively accommodate a wide range of alkali metal ions as well as multivalent metal ions. The success of monovalent ions in sodium ion and potassium ion batteries has prompted scientists to investigate the feasibility of incorporating bivalent ions into such materials (Fig. 1b).36–39 The original Prussian blue is ferric hexacyanoferrate (Fe4[Fe(CN)6]3). Early research focused primarily on the available Zn2+ storage host, where Fe atoms connected to N atoms can be replaced and interstitially modified with Cr, Zn, Mn, Co, Ni, Cu, and other elements to produce a series of metallic hexacyanoferrates (MHCFs) with a similar composition and crystal structure. These salts are known as Prussian blue analogues (PBAs), which have become an important electrode material in ZIBs due to their inexpensive raw materials, straightforward synthesis process, and working voltage of up to 1.5–1.8 V (vs. Zn2+/Zn) in aqueous electrolyte.
The investigation of PB and PBAs has led to an increasing number of papers reporting their utilization as cathode materials for zinc ion batteries in recent years. In this review, the author focuses on the current status and challenges associated with the use of PBAs in ZIB cathode materials (Fig. 2), by focusing on the structure and energy storage mechanism of Prussian blue and its analogues, as well as by referencing current academic research on the electrochemical energy storage of Prussian blue analogues. Here, various varieties of PBAs applied to ZIB cathode materials are described, along with their respective characteristics and current challenges.
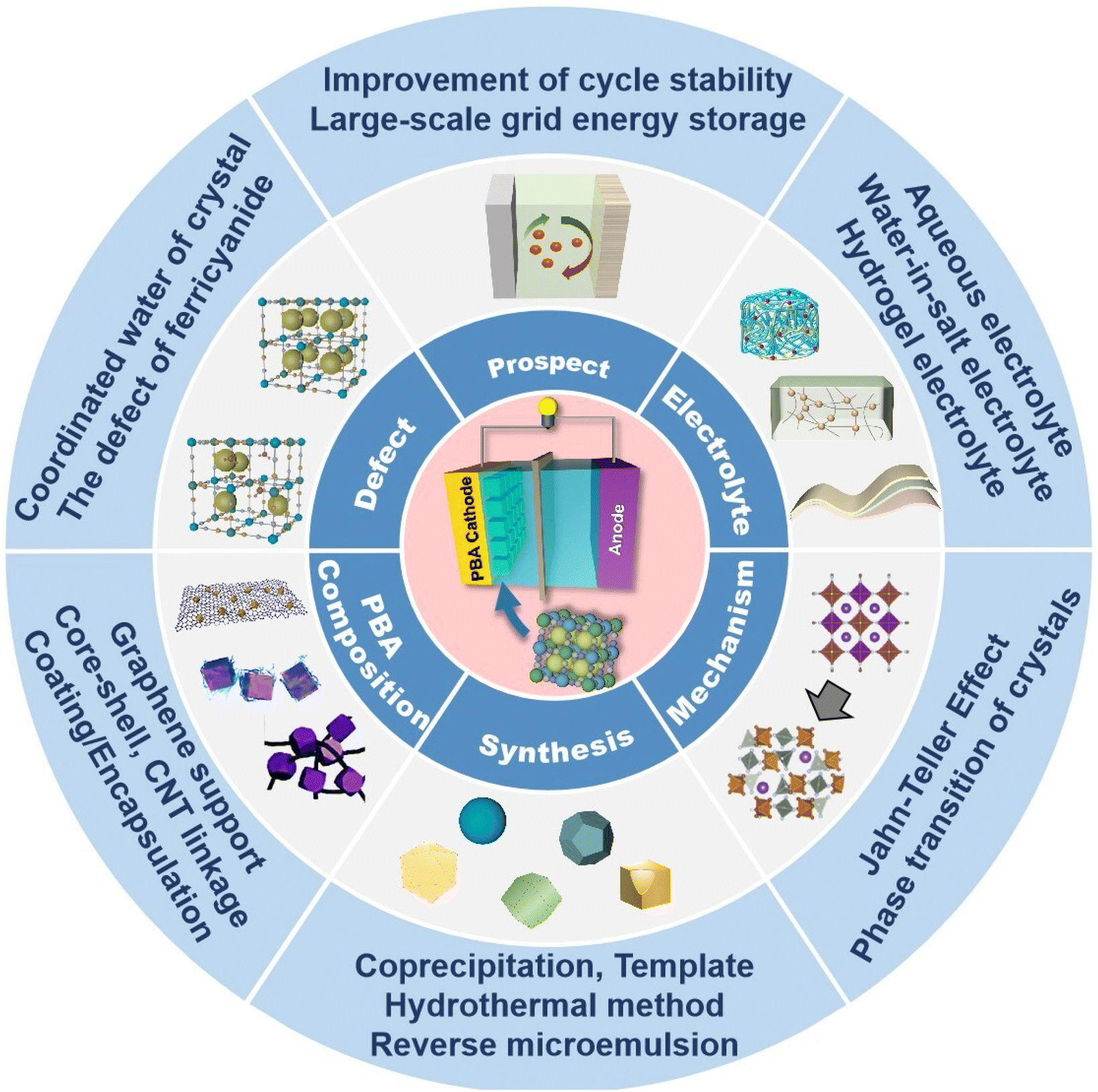 | ||
| Fig. 2 A summary of this review (reprinted with permission from ref. 88. Copyright 2022, Elsevier; reprinted with permission from ref. 50. Copyright 2021, John Wiley and Sons). | ||
2 PBA crystal structure and cathode mechanism
An ideal PBA is denoted using the formula A2MAMB(CN)6 (A = Li+, Na+, K+, zeolite water; MA = Fe, Ni, Mn, V, Mo, Cu, Co, Zn; MB = Fe, Co, Cr, Ru). Due to its high stability in aqueous solution, ferricyanide is the primary raw material for synthesis, so the preponderance of MB sites in the framework are occupied by Fe (Fig. 3a). PBA is a type of metal coordination compound with a body-centered cubic structure in which the nitrogen and carbon atoms of the CN ligand combine with the MB and MA atoms to form open ion channels and a large interstitial space (A site). Each individual framework channel (A site) is about 4.6 Å.40 Nonetheless, PBA typically possesses the defect vacancy of Fe(CN)6 whose N position is occupied and coordinated by water molecules (Fig. 3c), so the general formula is typically written as AxMA[MB(CN)6]1−Z□Z·nH2O (0 < X < 2; 0 < Z < 1), □ is a defect of Fe(CN)6. X is the occupied range of A ions, which is in the range of 0–2, and the existence of zeolite water will narrow this range. The defect part of the MB(CN)6 site is represented by Z in the general formula, and the range of Z is 0–1. As shown in Fig. 3a–c, the MA ions in the cubic lattice and the nitrogen atoms in the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N ligand form an MAN6 octahedron. MB ions and carbon atoms in the CN ligand form an MBC6 octahedron, a 3D open rigid frame face-centered cubic lattice structure that can accommodate metal ion insertion. MA and MB may be the same transition metal, distinguished by their valence states. Spin states are frequently determined by the concentration of the ligand surrounding the central ion. H2O in the general formula comprises zeolite water in the cavity gap as well as water coordinated with metal ions of MA, where H2O occupies the Fe[(CN)6]3− site in advance to coordinate with the metal ions of the MA site.35,41–43
N ligand form an MAN6 octahedron. MB ions and carbon atoms in the CN ligand form an MBC6 octahedron, a 3D open rigid frame face-centered cubic lattice structure that can accommodate metal ion insertion. MA and MB may be the same transition metal, distinguished by their valence states. Spin states are frequently determined by the concentration of the ligand surrounding the central ion. H2O in the general formula comprises zeolite water in the cavity gap as well as water coordinated with metal ions of MA, where H2O occupies the Fe[(CN)6]3− site in advance to coordinate with the metal ions of the MA site.35,41–43
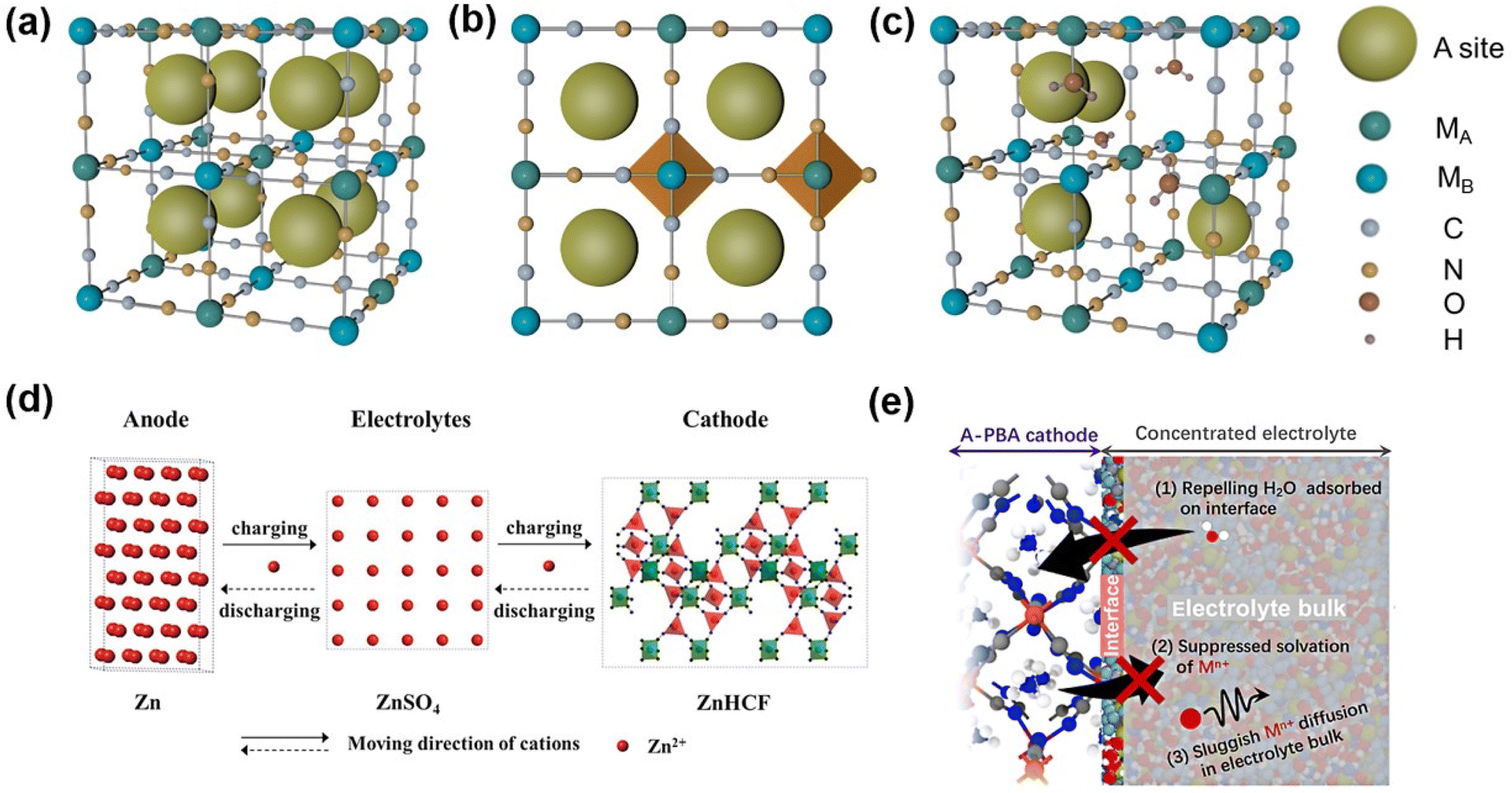 | ||
| Fig. 3 (a) Ideal Prussian blue analogue crystal structure. (b) Prussian blue ideal framework and its coordination form. (c) Prussian blue frame containing structural water. (d) Schematic diagram of a zinc ion battery based on a ZnHCF positive electrode. (Reprinted with permission from ref. 49. Copyright 2014, John Wiley and Sons). (e) Schematic of key factors in suppressing metal-ion dissolution of A-PBA in the concentrated electrolyte. (Reprinted with permission from ref. 54. Copyright 2022, American Chemical Society). | ||
2.1 The energy storage mechanism of PBAs
The 3D framework of MHCF has a large interstitial site (∼4.6 Å) surrounded by 12 CN−, which is larger than the radius of the majority of hydrated cations.44 At the same time, PBA possesses a special ion channel with a diameter of approximately 3.2 Å in the (100) direction.45,46 Large insertion sites and ion channels ensure that the majority of host ions with a small hydration radius have rapid reaction kinetics when inserting/extracting PBA, which ordinarily contributes to PBA's excellent cycle stability.47 According to the principle of charge conservation, MHCFs are oxidized and guest cations are liberated from the crystal skeleton during charging. During discharge, the reduction of MHCFs is concurrent with the reinsertion of metal cations into the skeleton. Chae et al.48 expressed the mechanism of Zn2+ insertion into NiHCF lattice vacancies as:| K0.86Ni[Fe(CN)6]0.954(H2O)0.766 + xZn2+ + 2xe− ↔ K0.86ZnxNi[Fe(CN))6]0.954(H2O)0.766 |
In 2014, Zhang et al.49 introduced zinc hexacyanoferrate (Zn3[Fe(CN)6]2, ZnHCF) as the positive electrode of an aqueous zinc ion battery, which demonstrates the removal of cations(Fig. 3d). The corresponding positive electrode electrochemical reaction shown in Fig. 3d can be expressed as:
Cathode:
| Zn2+ + 2xe− + Zn3[Fe(CN)6]2 ↔ Zn3+x[Fe(CN)6]2 |
The investigation of energy storage mechanisms necessitates careful attention to the structural transformation of PBAs during charge and discharge. Deng investigated the XRD analysis of a KMnHCF electrode under various cycling conditions and observed a monoclinic to cubic phase transition during the charging process, with both phases coexisting within a specific voltage range. Subsequently, the cubic phase persisted until complete charging of the electrode, followed by a reversion to the monoclinic phase during discharge engineering (Fig. 4a).50 Aili et al. observed a positive shift in the peak of KFCHCF (K1.66Fe0.25Co0.75[Fe(CN)6]·0.83H2O) in the X-ray diffraction pattern during the charging process, indicating contraction of the crystal structure after ion extraction, with a subsequent return to its original position upon discharge. This subtle structural change accounts for the high speed and recyclability exhibited by PBA materials (Fig. 4b and c).51
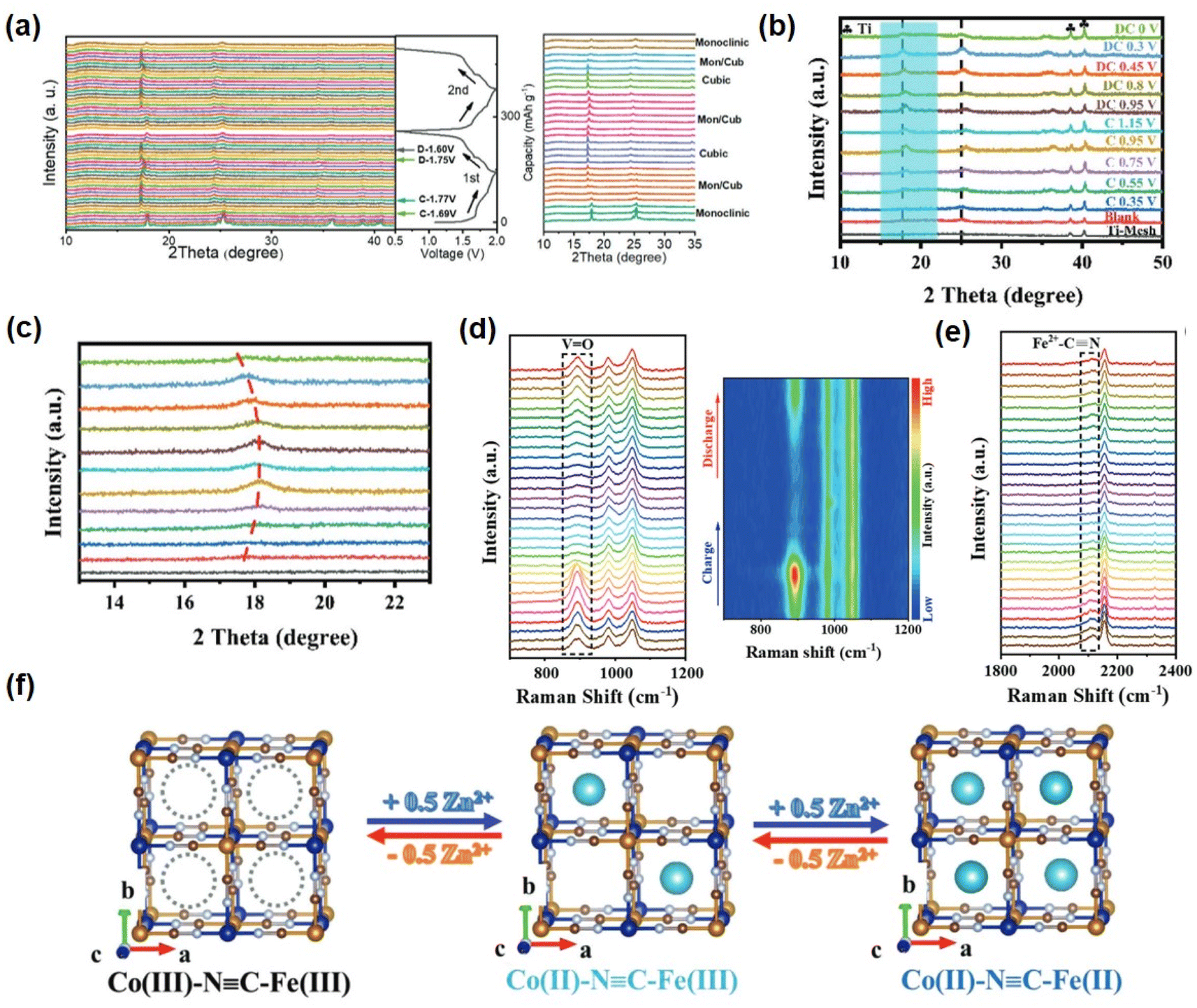 | ||
| Fig. 4 (a) In situ XRD patterns of a KMnHCF electrode. First two cycles to reveal the structure evolution during Zn2+ (de)insertion. (Reprinted with permission from ref. 50. Copyright 2021, John Wiley and Sons). (b) The ex situ XRD patterns and (c) the enlarged area of the KFCHCF sample at different charged and discharged states. (Reprinted with permission from ref. 51. Copyright 2023, John Wiley and Sons.). (d) and (e) In situ Raman spectroscopic analysis of the energy storage mechanism of VOHCF. (Reprinted with permission from ref. 53. Copyright 2023, John Wiley and Sons). (f) Schematic illustration of reversible Zn2+ intercalation/deintercalation in CoFe(CN)6 frameworks during the electrochemical process. (Reprinted with permission from ref. 60. Copyright 2019, John Wiley and Sons). | ||
The MB site in PBAs is the representative redox active site in this structure. In addition, the substitution of a different transition metal MA influences the reaction potential and capacity of PBAs. The MA and MB sites are occupied by distinct or identical metal atoms, resulting in distinct combination and reaction potentials.42,52 Fe, Mn, Co, and V are electrochemically active within the stable window of the organic electrolyte and aqueous system in PBAs. When these metal ions occupy the MA site, a double-electron redox reaction can occur. In situ analysis techniques were employed to investigate the PBAs at two REDOX sites. For the VOHCF electrode, the intensity of the V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 864–921 cm−1 gradually diminishes as the charge progresses, indicating a gradual ion insertion process. Upon discharge, there is a slow increase in the intensity of the VO peak, suggesting reversible ion ejection and embedding within VOHCF (Fig. 4d). In addition, the [Fe(CN)6]4− group gradually shifts to higher wavenumbers at around 2150 cm−1, followed by a reverse migration that corresponds to Fe2+ to Fe3+, thereby highlighting the pivotal role of [Fe(CN)6]4− as an additional REDOX active site (Fig. 4e).53 In addition, many PBAs have a reversible zinc ion intercalation/deintercalation reaction and high charge–discharge performance due to the coupling of multiple redox reactions and an ideal crystal structure.
O at 864–921 cm−1 gradually diminishes as the charge progresses, indicating a gradual ion insertion process. Upon discharge, there is a slow increase in the intensity of the VO peak, suggesting reversible ion ejection and embedding within VOHCF (Fig. 4d). In addition, the [Fe(CN)6]4− group gradually shifts to higher wavenumbers at around 2150 cm−1, followed by a reverse migration that corresponds to Fe2+ to Fe3+, thereby highlighting the pivotal role of [Fe(CN)6]4− as an additional REDOX active site (Fig. 4e).53 In addition, many PBAs have a reversible zinc ion intercalation/deintercalation reaction and high charge–discharge performance due to the coupling of multiple redox reactions and an ideal crystal structure.
In the process of a charge–discharge cycle, besides the crystal structure change induced by Zn2+ intercalation/deintercalation, PBAs as cathodes also show the dissolution of metal ions in the bulk.54 The bulk metal ion dissolution process undergoes the following main steps (Fig. 3e):(i) the metal ions in the material bulk diffuse to the surface of the solid electrode; (ii) metal ions are solvated by free solvation (free H2O molecules) at the solid–liquid interface; (iii) diffusion of solvation metal ions to electrolytic liquid phases.55–57 The dissolution of the electrode material in PBAs and its compatibility with the electrolyte will be comprehensively discussed in the section dedicated to electrolytes.
2.2 The alkaline metal ions
As the alkaline metal ion hexacyanoferrate is used as a source material during the synthesis process, the alkali metal ion will occupy the A site in advance.58 The distribution of K ions in the lattice vacancies is non-uniform, exhibiting a tendency to form clusters at specific locations within the compound lattice. Therefore, how to distribute gap vacancies in the skeleton in an electrochemical cycle also needs to be clarified by experimental study. You et al. disclosed the redox mechanism and phase transition of FeHCF during intercalation/deintercalation of sodium (Fig. 5a).59 In comparison to sodium-free crystals occupying Na vacancies, the XRD peak position shifts to a minor angle, indicating an increase in lattice parameters. Sodium-containing crystals appear diamond-shaped in comparison to cubic crystals lacking sodium. Recent research indicates that although alkaline metal ions occupy vacancies in advance, they will be removed prior to the intercalation of Zn ions. The research group of Professor Zhi produced KCoFe(CN)6 using a coordination compound and hexacyanoferrate ion at a controlled reaction rate.60 When Zn2+ is encapsulated in the KCoFe(CN)6 skeleton at constant current, one K+ is discharged from each skeleton in situ, as confirmed by XPS mapping. After the release of K+, it can still maintain a flawless 3D framework with only a minor volume reduction. Fig. 4f shows the insertion/deintercalation of Zn2+ during the electrochemical discharge/charge.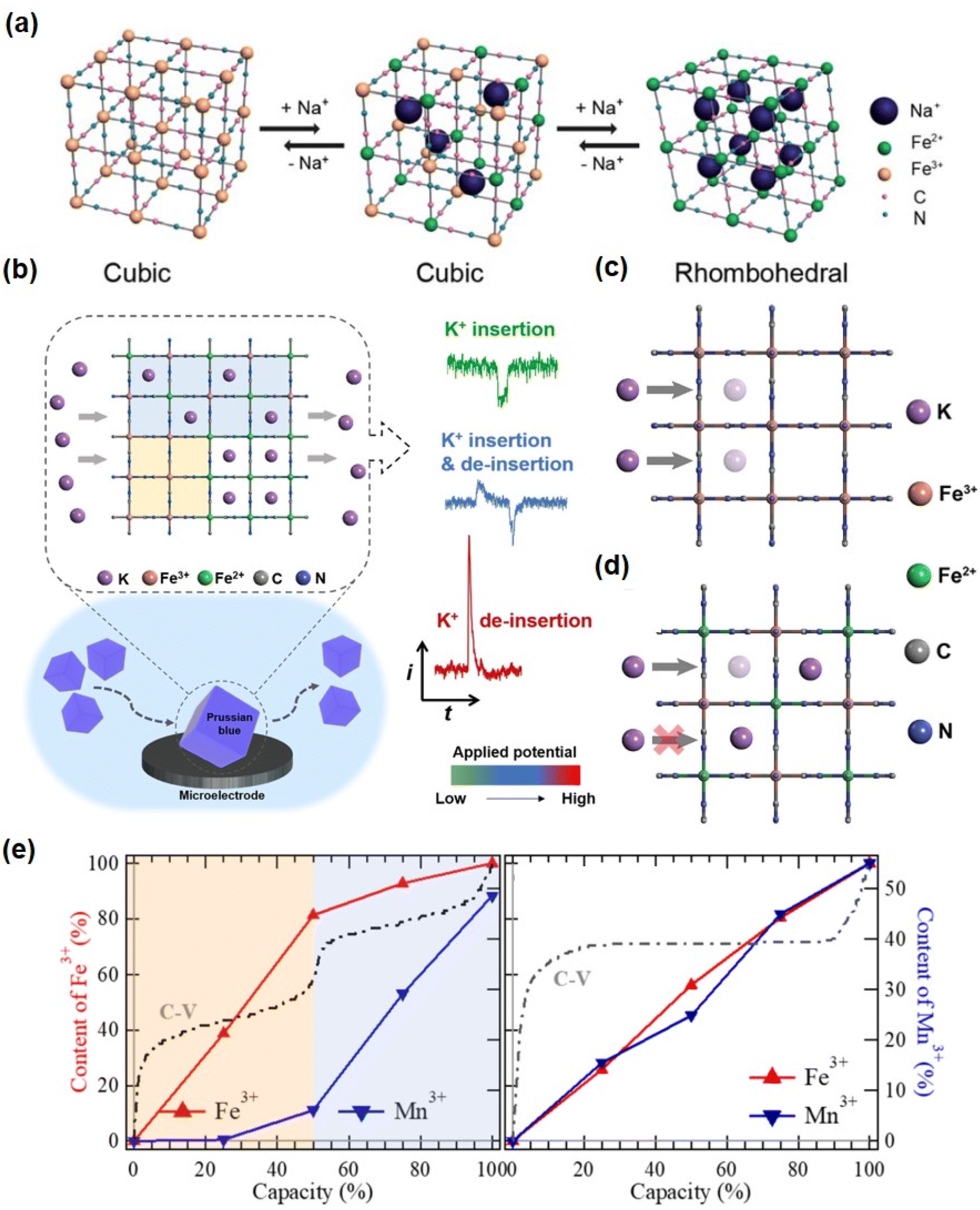 | ||
| Fig. 5 (a) Schematic illustration of the redox mechanism of HQ-NaFeHCF. (Reprinted with permission from ref. 59. Copyright 2014, Royal Society of Chemistry). (b) Schematic illustration of the nano-impact method for the kinetic study of K+ insertion/de-insertion in single PB particles. The ion channels at the interface of Fe[Fe(CN)6] (c) and KFe[Fe(CN)6] (d). (Reprinted with permission from ref. 63. Copyright 2019, John Wiley and Sons). (e) Concentration of the oxidation states of Fe3+ and Mn3+ upon electrochemical potentials. The percentage of oxidation states in hydrated and anhydrated samples reflects the REDOX sequence. (Reprinted with permission from ref. 69. Copyright 2017, American Chemical Society). | ||
In 2016, Ojwang et al.61 inserted the K+ portion into the CuHCF structure by reducing Fe(III) to Fe(II) in 0.1 M K2S2O3 aqueous electrolyte.61 The parameters of the CuHCF unit cell decrease as Fe(III) is converted to Fe(II) and as the quantity of K inserted increases. In their later work, which focused on the electrochemical-structural relationship of CuHCF cycling in 1 M ZnSO4, and it was demonstrated that the average cell voltage increased and a two-step plateau was observed during the initial CuHCF cycle.62 Consequently, the initiation (deinsertion) of zinc is associated with the nonlinear modification of CuHCF unit cell parameters during material operation. These results indicate that the reduction of CuHCF unit cells is greater in the presence of zinc ions compared to potassium ions.
Zhou et al.63 employed nano-impact electrochemistry (NIE), a novel technique, to reveal the intrinsic properties of the electrochemical processes that occur in PB nanoparticles. It was found that the KFHCF particle is not solely composed of KFe[Fe(CN)6] but also consists of K2Fe[Fe(CN)6] and Fe[Fe(CN)6]. Consequently, the electrochemical behavior of an individual PB particle enables the measurement of two distinct pairs of REDOX reactions within the same particle, irrespective of variations among different particles in the set. The feedback current signal exhibited by a single particle exhibits variation with respect to the applied potential, as depicted in Fig. 5b. When an electric potential is applied between two REDOX centers, both the oxidation and reduction current signals can be observed; however, the signal difference is related to the aggregation mode of K+.
| K2FeII[FeII(CN)6] ⇌ KFeIII[FeII(CN)6] ⇌ FeIII[FeIII(CN)6] |
The findings of this study demonstrate the kinetic influence of K+ on the ion embedding process. Specifically, the presence of K+ obstructs a portion of the ion transport channel, thereby restricting the reduction of KFe[Fe(CN)6] through slower interfacial transfer with K+, which is comparatively slower than the diffusion rate of K+ within the particle. In contrast, the reduction of Fe[Fe(CN)6] is constrained by the K+ within the particles, which is considered to exhibit slower kinetics compared to the transfer of K+ at the electrolyte composite particle interface (Fig. 5c and d).
2.3 Influence of two kinds of water on energy storage
Due to the rapid reaction rate of metal ions in combination with Fe(CN)63− during coprecipitation, the products frequently contain more iron-cyanide vacancies. In addition to occupying the crystal gap position, H2O also occupies the iron cyanide defect. On the one hand, the presence of [Fe(CN)6]3− vacancy defects reduces the structural stability of the material, and repeated intercalation of Zn2+ may cause the structure to collapse; on the other hand, water molecules will impede the transmission of Zn2+ in the crystal structure. Consequently, reducing vacancy defects and water content is advantageous for enhancing the cycling performance of electrode materials.64By means of neutron diffraction and X-ray single crystal diffraction, it is determined that there are two kinds of water molecules with different structural positions in the crystal structure of PBA.65,66 The first type is zeolite water, or interstitial water, which competes with inserted ions for site A, or vacancy in the framework. Fig. 3c depicts the second form of water, which is coordination water coordinated with the MA site ion at the hexacyanoferrate vacancy. The negatively charged oxygen atoms in these water molecules assist in shielding a large number of positive transition metal ion charges at the vacancies. There is a correlation between the quantity of vacancies and coordinated water in the lattice, with vacancy sites providing space for water molecules coordinated with MA ions and possibly providing sites for coordinated water. Doping MA sites in organic systems with nickel or electrochemically inert metals to preserve the structural stability advantages of vacancies and water is an active area of research. However, the interfacial charge transfer of an aqueous electrolyte is significantly superior to that of an organic electrolyte due to the fact that ion diffusion in an organic electrolyte requires solvation/desolvation and ion diffusion to the electrode surface. In an aqueous electrolyte, the activation energy required for only partial shedding of ion solvation spheres is less than that in an organic system; consequently, the kinetics are faster.67
However, the effect of water on ion conduction in a lattice is extremely complex and poorly understood. According to one theory, the movement of ion groups through crystal structure vacancies involves exchange with nearby water molecules and rotation around the surrounding ion hydration sheath. This ion migration assisted by rotation is called a “gear” or “paddle wheel” mechanism, which is used to move ions through channels and vacancies.68 It has been found that the presence of interstitial water in a Prussian blue lattice has an effect on the redox platform of transition metals.69 In particular, the traditional view holds that the redox potential is dependent on the ionization energy of a transition metal in a particular oxidation state. In this study, the standard ionization energy of Fe2+ is less than that of Mn2+, whereas the ligand field stability energy (LFSE) of Fe2+ (LS) is greater than that of Mn2+ (HS), resulting in an increase in the redox potential of Fe2+/3+. If the ionization energy and the competitive effect of LFSE are in equilibrium, the Fe2+ (LS) and Mn2+ (HS) redox potentials will overlap to form a single plateau (Fig. 5e). In a hydrated system, the interstitial water molecules dilute the ligand field in the FeC6 and MnN6 octahedra and disrupt the structure that defines the spin states. When the LFSE effect is unable to compete with the ionization energy, the redox potential gap between Fe2+/3+ and Mn2+/3+ reappears, resulting in a double plateau structure in the electrochemical morphology of the hydrated electrode.
Cappe et al. studied the effect of water on cation mobility in an A2Zn3[Fe(CN)6]2·xH2O skeleton by using impedance spectroscopy data, where A = Na, K, Rb, Cs.70 It was discovered that porous hexacyanometallates have low surface polarity and no partially exposed transition metal coordination centers, which reduces the possibility of H+ and OH− analogue formation. The presence of weakly bonded water molecules in the lattice increases the mobility impedance resistance. This inference can be explained by the fact that increased hydration of the solid leads to the formation of a denser network of water molecules within the porous framework, thereby reducing the number of available pathways for cation migration. When the number of water molecules in a cationic coordination environment increases, the water molecules become more closely associated with the solid, and the barrier to diffusion increases in energy. Therefore, further theoretical and targeted studies, such as nuclear magnetic resonance spectroscopy or isotope labeling studies, can more accurately distinguish the effects of zeolite water and coordination water on ion conduction.
3 Synthesis of PB/PBAs
3.1 Coprecipitation
According to the papers, there are numerous methods for preparing coordination compound powders containing two or more metal elements, among which the coprecipitation method is a significant method.71,72 Through various chemical reactions in solution, it is possible to directly obtain powder materials with a uniform chemical composition and distribution, and the process is simple and inexpensive. By mixing a metal salt with a salt containing [Fe(CN)6]4−/3− in an aqueous solution, nanoscale particle precipitation can be rapidly produced in the aqueous solution because the product has a very small Ksp (Ksp « 10−10).73,74 The crystal is formed by the coprecipitation method, whose growth can be regulated by using the concentration, temperature, stirring speed, surfactant, etc. (Fig. 6b and c). The analogues of Prussian blue synthesized by coprecipitation are summarized in Table 2. Sonochemistry can aid the coprecipitation technique in controlling the size of PBAs. Wu et al.75 reported the synthesis of various sizes of PB nanocubes in acidic media using a single reactant K4[Fe(CN)6] assisted by ultrasonic treatment (Fig. 6a). Moreover, the experiment demonstrated that the size distribution of the synthesized Prussian blue nanocubes was highly dependent on the reaction temperature and K4[Fe(CN)6] concentration. The average side length of nanocubes prepared at 40 degrees Celsius and 1 mmol L−1 K4[Fe(CN)6] was approximately 250 nanometers. However, when K4[Fe(CN)6] concentrations reach 10 and 100 mmol L−1 at the same temperature, the nanocube side lengths reach 300 and 500 nm, respectively. While the side length of the Prussian blue nanocube varies between 250 nm and 450 nm between 40 °C and 70 °C, the concentration of K4[Fe(CN)6] is maintained at 1 mmol L−1. | ||
| Fig. 6 (a) TEM images of Prussian blue nanocubes prepared at 40 °C using different concentrations of K4Fe(CN)6. (Reprinted with permission from ref. 75. Copyright 2006, American Chemical Society). (b) Schematic diagram of the nucleation and growth process of the ZnHCF particle shape at different drop rates of reactants. (c) SEM images of cubic octahedron ZnHCF, truncated angular octahedron ZnHCF and octahedron ZnHCF obtained at room temperature. (Reprinted with permission from ref. 83. Copyright 2015, Springer Nature). | ||
| Materials | Precursor | Chelating agent | T | Reaction time | Size | Ref. |
|---|---|---|---|---|---|---|
| K2Zn3[Fe(CN)6]2 | 0.1 M ZnSO4 + 0.05 M K3Fe(CN)6 | — | 60 °C | Several hours | 200 nm | 49 |
| K0.08Zn1.53[Fe(CN)6]·0.26H2O | 0.02 M ZnSO4 + 0.02 M K3Fe(CN)6 | — | Room temperature | 1 h | 500–800 nm | 81 |
| K1.88Zn2.88[Fe(CN)6]2(H2O)5 | 50 mM ZnSO4·7H2O + 50 mM K4Fe(CN)6·3H2O | — | 70 °C | 30 min | 70–120 nm | 82 |
| K0.07Zn[Fe(CN)6]0.69 | 0.1 M ZnSO4 + 0.05 M K3Fe(CN)6 | — | Room temperature | Immediately | 1.7 μm | 83 |
| K0.08Zn[Fe(CN)6]0.67 | 0.1 M ZnSO4 + 0.05 M K3Fe(CN)6 | — | Room temperature | 20 min | ∼3.2 μm | 83 |
| K0.07Zn[Fe(CN)6]0.68 | 0.1 M ZnSO4 + 0.05 M K3Fe(CN)6 | — | Room temperature | 2 h | ∼2.6 μm | 83 |
| Zn3[Fe(CN)6]2 | ZnSO4 + K3Fe(CN)6 | — | 70 °C | 1 h | 300 nm | 84 |
| CuHCF | Cu(NO3)2·3H2O + K3Fe(CN)6 | — | Room temperature | 30 min | 100 nm | 85 |
| K2x/3Cu[Fe(CN)6]2/3⋅nH2O | Cu(NO3)2·3H2O + K3Fe(CN)6 | — | Room temperature | 30 min | 40–100 nm | 86 |
| CuHCF (nano cube) | CuSO4 + K3Fe(CN)6 | — | Room temperature | 30 min | 50–100 nm | 43 |
| CuZnHCF | Cu(NO3)2·3H2O + Zn(NO3)2·6H2O + K3Fe(CN)6 | — | Room temperature | 30 min | 2–12 μm | 87 |
| NiHCF (nanocubes) | Ni(NO3)2·6H2O + K3Fe(CN)6 | Na3C6H5O7 | Room temperature | 10 min | 100 nm | 88 |
| NiFe(CN)6 | Ni(NO3)2 + Na4Fe(CN)6 | — | — | 2 h | 89 | |
| K1.51Ni[Fe(CN)6]0.954(H2O)0.766 | Ni(NO3)2·6H2O + K4[Fe(CN)6]·3H2O | — | Room temperature | 30 min | 5–25 nm | 48 |
| K1.6Mn[Fe(CN)6]0.94·0.63H2O | MnSO4 + K4Fe(CN)6·3H2O | Potassium citrate | Room temperature | 24 h | 500–600 nm | 50 |
| ZnMnFe-PBA | MnSO4·H2O + K3Fe(CN)6 + ZnSO4·7H2O | — | 80 °C | 1 h | 0.5–1 μm | 90 |
| K2MnFe(CN)6 | MnCl2·4H2O + K4Fe(CN)6 | 2 g PVP(K30) | 80 °C | 50 min | 2–4 μm | 91 |
| KCoFe(CN)6⋅5.5H2O | K3[Fe(CN)6] + Co(CH3COO)2·4H2O | — | Room temperature | 24 h | 500 nm | 60 |
| CoMn-PBA HSs | K4[Fe(CN)6]·3H2O + Mn(NO3)2·4H2O + Co(NO3)2·6H2O | — | 90 °C | 4 h | 1.2 μm | 92 |
| FeFe(CN)6 | K3[Fe(CN)6] + FeCl3 +30% HCl | — | 100 °C | 30 min | 300 μm | 33 |
| VHCF | VOPO4·2H2O + Na4Fe(CN)6⋅10H2O | — | 80 °C | 4 h | 23.6 nm | 93 |
| VHCF | VOSO4·xH2O + K3Fe(CN)6 | — | 60 °C | 4 h | 30 nm | 94 |
| VO-PBAs | VOSO4 + K3Fe(CN)6 | — | Room temperature | Immediately | 30–50 nm | 95 |
3.2 Hydrothermal method
The hydrothermal method is a well-established and straightforward synthesis technique capable of synthesizing thermally unstable materials close to their melting points. This method enables the production of large, high-quality crystals and allows for the control of particle size, agglomeration, and impurity pollution levels. The performance of the resulting products depends on both the formation mechanism and hydrothermal conditions.76 Yang et al.77 and Zheng et al.78 reported the synthesis of microcubes of Berlin green and Prussian blue, respectively. Since then, several studies have documented the utilization of the hydrothermal method for synthesizing a diverse range of Prussian blue-based materials. The reaction temperature will affect the morphology of PB/PBA, and high temperature will increase the dissolution or etching of PB/PBA.79 With the aid of organic additives, precise control over the morphology of Prussian blue particles can be achieved in hydrothermal systems, resulting in the formation of spherical nanocrystals.80 Zhang et al. reported that in hydrothermal systems, the introduction of bivalent cobalt into a potassium ferricyanide solution leads to the continuous reduction of trivalent iron and a cation exchange reaction between bivalent iron and cobalt, resulting in the gradual formation of uniform PBAs with an open cage structure.963.3 Reverse microemulsion method
This method has been gradually applied to the synthesis of nano-PB particles as a result of the reverse microemulsion technique. It has superior particle size control and leads to particle distribution uniformity. Polymer stabilizers can provide chemical and spatial constraints for the construction of nanoparticles, resulting in a distinct colloidal morphology and preventing agglomeration. Aucher et al.97 first reported the synthesis of nano-PB by inverse microemulsion control. The microemulsion has been prepared from sodium bis(2-ethylhexyl) sulfosuccinate (anionic surfactant) SOL (anionic surfactant (AOT) sol). In these processes, nanoarray growth is governed by a multi-step procedure involving the slow photoreduction of an iron oxalate precursor to form ferrous ions (FeII), which then react with the [Fe(CN)6]3− anion to form water-coated PB nuclei and clusters. Further exchange and fusion between microemulsion drops produces nanoparticles encapsulated in a shell of surface molecules. The highly hydrophobic surface properties of PB nanocubes encapsulated by AOT facilitate the fabrication of self-assembled nanoparticle arrays with two-dimensional and three-dimensional superlattice arrays. Zhang et al. prepared spherical PB nanoparticles with diameters ranging from 5 nm to 20 nm using biocompatible chitosan polymer as a protective agent (Fig. 7e and f).98 The surface of chitosan is positively charged due to the presence of protonated amines, which are good adsorbents for negative ions. [Fe(CN)6]3− interacts electrostatically with cationic macromolecules and subsequently reacts with FeCl2 (Fig. 6d). During the formation of chitosan PB, chitosan provides a template and spatial stability for the formation of PB nanoparticles. | ||
| Fig. 7 SEM images of PB particles prepared with different amounts of PVP added (a) 3 g, (b) 5 g, and (c) 10 g. (Reprinted with permission from ref. 106. Copyright 2012, Royal Society of Chemistry). TEM images of chitosan-PB nanoparticles prepared at [Fe2+], [Fe(CN)6]3− = 1 mM, [chitosan] = 3 mg mL−1 (d), 2 mg mL−1 (e), and 1 mg mL−1 (f). (Reprinted with permission from ref. 98. Copyright 2008, Elsevier). (g) Morphology evolution of Prussian blue from a cube to a star-like hexapod with increasing concentrations of HNO3. (Reprinted with permission from ref. 99. Copyright 2012, Korean Chemical Society). Field emission scanning electron microscope (FE-SEM) images of (h) PB-1 mL HCl, (i) PB-2 mL HCl, and (j) PB-3 mL HCl. (Reprinted with permission from ref. 100. Copyright 2017, John Wiley and Sons). | ||
3.4 Template method
In addition, PBA is a synthetic material that can be synthesized from templates and then evolved in an etching solution. The morphology of PBAs can also be altered and etched by using acids and bases. Selective chemical etching (acid–base etching): PB can be selectively etched in a hot acid solution if a polymer or surfactant is used as a covering. On the crystal surface, selective etching by a polymer or surfactant will be selectively adsorbed. The polymer or surfactant will selectively adsorb on the surface of the crystal, etching the corners and edges. Lee et al. demonstrated the evolution of PB morphology from a cube to a hexagonal polyhedron by adding nitric acid.99 Nitric acid concentration causes particles to evolve from a cube to a truncated cube, vertical body, truncated octahedron, hexapod with arms, and finally stellate hexapod. These findings indicate that the oxidation process begins at the nanocube's corner and reveal the distinct stability of the interal plane of the PB crystal structure (Fig. 7g). Liu et al. discovered that acids not only initiate chemical reactions but also serve as a morphology-modifying etching agent. In contrast to Lee's report that HNO3 corrodes from edges and corners, HCl solution tends to corrode from surfaces.100 As shown in Fig. 7h, when the amount of HCl added is 1 mL, the generated PB is micron sized cubic particles. After adding 2 mL HCl, the surface of PB cube in the fovea is very rough (Fig. 7i). Finally, when 3 mL HCl was added to the system, porous PB spheres (Fig. 7g) were obtained. Besides the external morphology can be etched by adding acid, and the internal hollow structure can also be etched by adding acid. Yamauchi et al.101 reported that hollow Prussian blue (PB) particles can be prepared by etching PB mesocrystals in HCl solution in the presence of polyvinylpyrrolidone (PVP). An inner cavity appeared in the center of the PB mesocrystals after 3.5 h of etching, and it grew larger when the etching duration was increased to 4 h. After 5 hours of etching, all of the PB nanocrystals dissolved. It should be noted that PVP plays an important role in the etching rate of a particle surface with hollow PB particles.3.5 Chelating agents and surfactants
Polyvinylpyrrolidone (PVP), sodium citrate, EDTA-2Na, SDS, etc. play an important role in determining the shape and particle size of PBAs, and control the morphology and particle size by regulating the nucleation rate and growth rate of the crystal. Irregular particles and non-uniform nanocube formation were observed in the absence of such directing agents. Kitagawa et al.102 were the first to report the formation of PB nanoparticles using PVP as a chelating agent. In the presence of PVP, by mixing Fe2+ and [Fe(CN)6]3− in water, the size of PB nanoparticles is adjusted between 12 and 27 nm, depending on the ratio of iron ions added to PVP and the initial concentration of iron ions. Chiang and co-workers precisely controlled the particle size in the range of 20 to 500 nm by varying the amount of sodium citrate added as a chelating agent and using different Fe sources.103 Additionally, their prior research demonstrated that sodium citrate could function as a chelating agent to interact with nickel ions, thereby influencing the crystallization rate of the final cyano-bridged coordination polymer.104 In general, the ultimate size of particles in a product is determined by the equilibrium between nucleation and crystal growth. In this system, stable release of free nickel ions from the nickel citrate complex leads to their initial reaction with ferricyanide(II) to form nuclei. The final product is formed by the interaction of these nuclei with liberated nickel ions and sodium(II) ferricyanide. In contrast, at low sodium citrate concentrations, the majority of nickel ions existed as free nickel ions, which reacted swiftly with sodium(II) ferricyanide. Consequently, there are numerous nuclei in the initial phases of the reaction, which expand rapidly to form small particles.Lou's Group controlled the amount of PVP and citrate to control the crystal growth kinetics of PBA. Frame-like nanostructures were found in the absence of additives, and nanocubes were formed with the concentration of additives.105 In another attempt, the influence of citrate and PVP concentration on the growth of a PBA crystal was discussed. High concentrations of PVP and citrate produce a surface closed cube with a truncated cube shape, while low concentrations of citrate result in an open cage structure with cavities at corners. Hu et al.106 developed a simple method to control the size and morphology of PB nanoparticles by systematically determining the pH value of the solution and the amount of K3[Fe(CN)6]. They developed three different PB nanoparticles: small (about 20 nm), medium (about 100 nm) and large (about 200 nm) (Fig. 7a–c). Currently, PVP is crucial in determining the final shape, crystallization process, size, and oxidation state of the metal skeleton. The pH of the solution, the concentration of K3[Fe(CN)6], and the amount of PVP control the size and morphology of the final product via a multistep mechanism. PVP is utilized primarily as a capping agent to stabilize the crystal nucleus in the initial stage. When the concentration of PVP is low, direct particle precipitation cannot expand. However, when the amount of PVP is sufficient, the nuclei first stabilize and grow in an orderly manner. The acid concentration regulates K3[Fe(CN)6] and produces trivalent Fe ions, which, along with the ions in [Fe(CN)6]4−, result in the formation of PB nanocubes. Therefore, a small amount of PVP is advantageous for products containing small nanoparticles, while a large amount of PVP is advantageous for products containing medium and large nanoparticles. It can be concluded that chelating agents and surfactants have a controllable effect on the crystallinity and morphology of synthesized PB/PBAs. The chelating agent regulates the release of precursor ions to facilitate crystal growth and the formation of large particles from the released ions. Surfactants inhibit the growth of particle size by acting directly on the surface of nanoparticles.
3.6 Design of PBA composites
The mechanism of PBAs as the cathode material of ZIBs is based on Zn2+ intercalation, while diffusion-controlled ion intercalation is usually affected by the rate performance. In order to improve the diffusion performance of ion intercalation, many studies have been devoted to the physical or chemical composite of PBAs and other materials to achieve unexpected improvements in the electrochemical performance of PBAs. Xue developed a new PBA-based ZIB composite cathode material for an aqueous system by loading nickel hexocyanoferrate (NiHCF) nanocubes onto sheets of reduced graphene oxide (RGO).88 In the NiHCF/RGO composite, the NiHCF nanoparticles are securely attached to the RGO layer, forming a conductive network (Fig. 8a–c). The strong synergistic interaction between NiHCF and the highly conductive RGO effectively increases the specific surface area, accelerates electron and ion transport, and inhibits the structural collapse of the NiHCF/RGO electrode during Zn2+ insertion/deintercalation. Benefiting from the above advantages, the NiHCF/RGO complex exhibited significantly improved electrochemical zinc storage properties, including an excellent reversible capacity of 94.5 mA h g−1 at a current density of 5 mA g−1 and 50.1 mA h g−1 at 200 mA g−1, and 80.3% volume retention after 1000 cycles at 200 mA g−1. This research presents a straightforward and efficient method for enhancing the electrochemical performance of PBA-based cathodes for aqueous ZIB applications.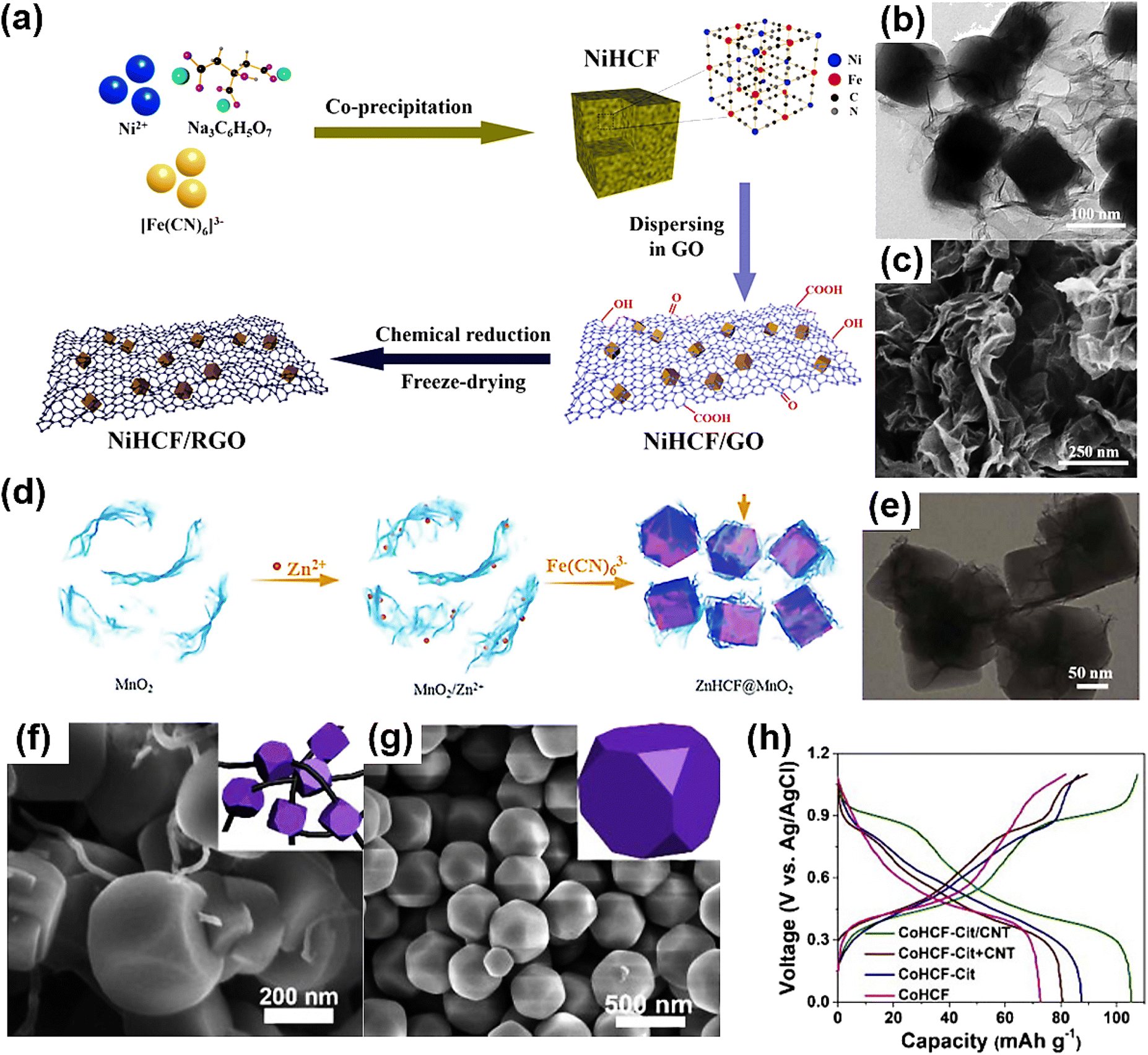 | ||
| Fig. 8 (a) Schematic illustration of the synthetic process of NiHCF/RGO. (b) TEM image of NiHCF/RGO. (c) SEM image of NiHCF/RGO. (Reprinted with permission from ref. 88. Copyright 2022, Elsevier). (d) Schematic diagram for the fabrication procedures of the ZnHCF@MnO2 composite, and (e) corresponding TEM morphology. (Reprinted with permission from ref. 107. Copyright 2017, Royal Society of Chemistry). SEM images of (f) CoHCF-Cit/CNT and (g) CoHCF. (h) Galvanostatic discharge/charge profiles. (Reprinted with permission from ref. 108. Copyright 2018, Elsevier). | ||
In 2013, Zhang et al.107 developed an in situ coprecipitation method to prepare manganese oxide-coated zinc hexocyanoferrate (ZnHCF) nanocubes (ZnHCF@MnO2). Zn2+ ions were adsorbed on the surface of two-dimensional MnO2 nanosheets via electrostatic interaction in an aqueous electrolyte, and then K3Fe(CN)6 was added dropwise during aging to form ZnHCF@MnO2 composites. The intercalated ZnHCF core is surrounded by a pseudocapacitive manganese oxide shell, and the 2D MnO2 nano-sheet serves as a buffer layer (Fig. 8d and e). ZnHCF was used as the central core, and 2D MnO2 nanosheets with a certain pseudocapacity served as the buffer layer. The re-embedded Zn2+ from ZnHCF after charging enters the peripheral MnO2 nanosheets, and Zn2+ from MnO2 remains embedded in ZnHCF during discharge, thereby reducing the diffusion control from the electrolyte to the electrode. In addition, the Zn2+ in the electrolyte contributes additional capacity to MnO2, thereby enhancing the cell's performance.
CNT and PB can be recombined to enhance PB's intrinsic structural vacancies and poor electronic conductivity. It is simple for carbon materials such as CNTs to aggregate and challenging for them to disseminate uniformly in PBA. In addition, the high surface energy of PBA facilitates aggregation during growth, preventing carbon materials from forming complete 3D conducting channels. Qian et al. synthesized a cobalt hexocyanoferrite truncated nanocube (CoHCF-Cit/CNT) with citrate and glycerol to address this problem (Fig. 8f and g).108 CoHCF-Cit/CNT was used as the cathode of a Na+/Zn2+ double ion battery for the first time. CNTs not only show effective internal resistance for CoHCF particles, but also show significant resistance between adjacent CoHCF particles, which significantly improves the electrochemical performance and cycle capacity of CoHCF-Cit/CNT (Fig. 8h). Additionally, research has produced PB-CNT composites that prevent PB particle agglomeration by penetrating single PB particles into carbon nanotubes.109 CNTs significantly enhance the composite's electron transport, resulting in four orders of magnitude greater electron conductivity at room temperature for PB/CNT than for PB alone. Due to the “metallic nature” of CNTs, the conductivity of the PB/CNT composite increased by 47.6% when cooled to −25 °C compared to the conductivity of the material without a CNT composite, indicating its potential in the construction of low-temperature batteries. The obtained materials exhibit outstanding electrochemical properties at low temperatures, and the synthesis strategy is applicable to the development of additional low temperature cathode materials.
All of the aforementioned composites improve the electrochemical performance of PBAs as a positive electrode material to some degree, and although the MnO2 coating introduces a new energy storage mechanism, it still has poor conductivity and excessive internal resistance, which will reduce the battery's lifespan. However, the CNT film coating had no negative impact on the internal resistance, but the capacity increase was insufficient. Moreover, during cycling, irreversible phase transition, transition metal dissolution, and structure collapse occur. Huang et al. coated PANI in ZnHCF by simple in situ polymerization to prepare ZnHCF/PANI positive electrodes.110 As the coating material of ZnHCF, PANI can effectively avoid the problems of high internal resistance and large cushioning stress in large deformation. PANI coating provides a new voltage platform of about 1.2 V for ZnHCF at 1.8 V. It can reach a high capacity of 150 mA h g−1 at 100 mA g−1, and the capacity rate is 75% after 350 cycles.
3.7 Optimization strategy of the PBA crystal structure by removing zeolite water
The weight loss below 200 °C corresponds to physical adsorption and removal of zeolite water, whereas the continuous weight loss between 200 and 300 °C is attributable to coordination water. This is because the coordination water is located exactly at the M(CN)6 vacancy in the PBA framework, which requires a higher temperature to remove.111 Currently, thermogravimetry (TGA) is utilized to quantify the water content of the PBA framework. In the literature on sodium ion batteries, however, it has been discovered that removing all of the water from the PBA's structure will also have a negative effect on its performance. Goodenough et al.112 studied a manganese-type PBA by XRD, which distorts into a rhombo-shaped geometry when severely sodiumized in a 1:1 NaClO4 electrolyte system containing 10% ethylene fluoride carbonate diethyl carbonate/ethylene carbonate. When all water is eliminated, it may be accompanied by a high concentration of small ions, which exacerbates the structural deformation. Wu et al. also found that the electrochemical capacity and recyclability of the PB skeleton have great influence on structural defects and water content in the lattice when searching for stable Na+ host materials in Na2SO4 electrolyte.113,114 In their later work, it was demonstrated that high capacity and cycle stability cathodes based on low cost PB compounds could be constructed by controlling structural defects and lattice water in the PB skeleton.115 This is also true for zinc-ion batteries, where it has been discovered that KMnHCF with a low water content performs better than those with a high water content.91
Particle size also has an effect on water adsorption, and smaller crystals may lead to stronger water adsorption.116 The sodium-rich Na1+xFeFe(CN)6 produced by Li et al. in a single step can increase the number of sodium ion skeletons with effective vacancies and coordination water.117 The amount of water in the MNHCF structure can be obtained by dewatering at high temperature under vacuum conditions.118,119 The vacancies and defect-rich PBA produced by dehydration are incapable of reaching their theoretical capacity due to the absence of Fe[(CN)6]3−/4− as a structural support during cation intercalation and disintercalation, resulting in a crystal structure that is fragile. By suppressing lattice defects, it is possible to increase the amount of water in the PB framework; the key to this strategy is to improve crystallinity.115,120,121 Reducing the crystallization rate is the key to improving PBA crystallization, and numerous researchers have proposed various solutions: (1) the chelator/surfactant-assisted coprecipitation method, where the crystallization rate can be slowed by using sodium citrate as a chelator to increase the crystallinity of PBA.114,122 (2) Decomposition of hexocyanate metal salts in acid: Guo and his colleagues59 fabricated high-quality PB nanocubic crystalline cubic crystal Na0.61Fe[Fe(CN)6]0.94 (HQ-NaFe) by a simple synthesis procedure using Na4Fe(CN)6 as the sole iron source. Due to the low water content of HQ-NaFe zeolite crystal growth, there is a small amount of [Fe(CN)6] vacancies in the crystal skeleton, which further enhances the ion storage capacity, enriches the transport path, and effectively preserves the crystal structure throughout the cycle.
The addition of a chelating agent has an significant effect on inhibiting the formation of a good crystal structure by adding Mn2+ in a K/Na-citrate solution (citric acid k/Na) chelating reactant to slow down the nucleation of KMHCF to achieve uniform and nearly chemical crystal growth.123 Compared with citrate as the chelating agent, EDTA-2K has stronger complexing ability to Mn2+ (Kstable[Mn(EDTA)]2− = 1013.8 » Kstable[Mn(citrate)]− = 103.67). The nucleation and growth of K2Mn[Fe(CN)6] can be greatly inhibited, and the defects and moisture of the synthesized sample (KMF-EDTA) are significantly reduced.124 In addition to the use of a chelating agent, the acid-assisted hydrothermal method can slow down the reaction rate of FeFe(CN)6 nanocrystals with low defects, which may be due to the acid-assisted dynamic balance between corrosion and growth. The precursor K3Fe(CN)6 slowly decomposes into Fe3+/Fe2+ in acid solution, and then reacts with residual [Fe(CN)6]3− to form FeFe(CN)6 nanocrystals with low defects.59,115 Therefore, some strategies to reduce water content in PBA can be summarized as follows: (a) coarsening PBA particles to reduce surface adsorption; (b) dehydration of PBA samples at higher temperatures under high vacuum; (c) introducing more basic ions into the skeleton to reduce the adsorption sites of zeolite water; (d) reduce the coordination sites of coordinating water by reducing the number of MB(CN)6 vacancies.
4 REDOX pairs in PBAs
As mentioned above, different metal ions occupying MA sites will produce one or two redox active sites in the PBA framework. In this section, according to the redox sites involved in the reaction, PB/PBA-based cathode materials can be divided into two categories: a single redox pair PBA and double redox pair PBA. The specific capacity voltage properties are shown in Fig. 9. PB/PBA containing a single active REDOX site exhibited a pair of anodic/cathodic react ion peaks, whereas PB/PBA containing two active REDOX sites typically exhibited two REDOX pairs. The redox potential of various metal ions is affected by both the ligand field and the standard redox potential.69 In certain exceptional circumstances, two redox pairs will combine into one. In the case of MnHCF, Fe2+/Fe3+ has a lower standard potential in comparison to Mn2+/Mn3+; however, CN− is a strong field ligand, and the low-spin Fe site coordinated with C has a strong ligand field, allowing for a higher Fe2+/Fe3+ REDOX potential. In the meantime, the weak ligand field of the high spin Mn site would reduce the redox potential of Mn2+/Mn3+. In this case, similar potentials would be generated at the Fe2+/Fe3+ and Mn2+/Mn3+ sites, resulting in a single pair of REDOX peaks. The specific capacity of ZnHC, FNiHCF, and CuHCF is typically limited to 80 mA h g−1 due to the electrically inert Zn2+, Ni2+ and Cu2+ ions, which can only provide a single active Fe2+/Fe3+ redox site. Single redox active sites and double redox active sites will be discussed with regard to PBAs containing various metals.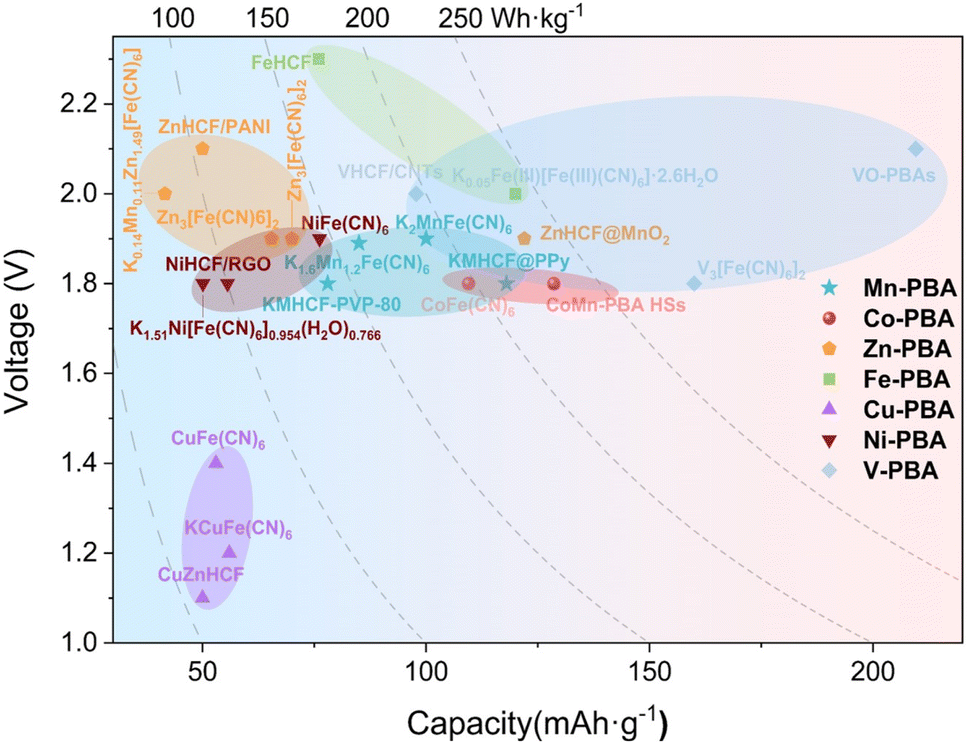 | ||
| Fig. 9 Comparison of Prussian blue applied to aqueous zinc-ion batteries. | ||
4.1 The single redox pair of PBAs
When Zn, Cu, or Ni ions occupy the MA site, ZIBs can exhibit a high voltage plateau, but their electrochemical performance is inactive; only Fe2+/Fe3+ can be used as a redox center. The number of sites occupied by intercalation ions determines the number of oxidizable Fe2+.125 Typically, these metal compound electrodes have a low capacity of approximately 60 mA h g−1 and a low high voltage stability (approximately 100 cycles).42 For example, ZnHCF and CuHCF are used as the positive electrode of ZIBs. Based on a ZnSO4 aqueous electrolyte system, the former exhibits an average operating voltage of 1.7 V and a capacity of 65.4 mA h g−1 for about 100 cycles at 60 mA g−1 current density,49 while the latter exhibits an average discharge potential of 1.73 V and provides 53 mA h g−1 at 60 mA g−1 current density, and its cycle life is similar to that of the former.84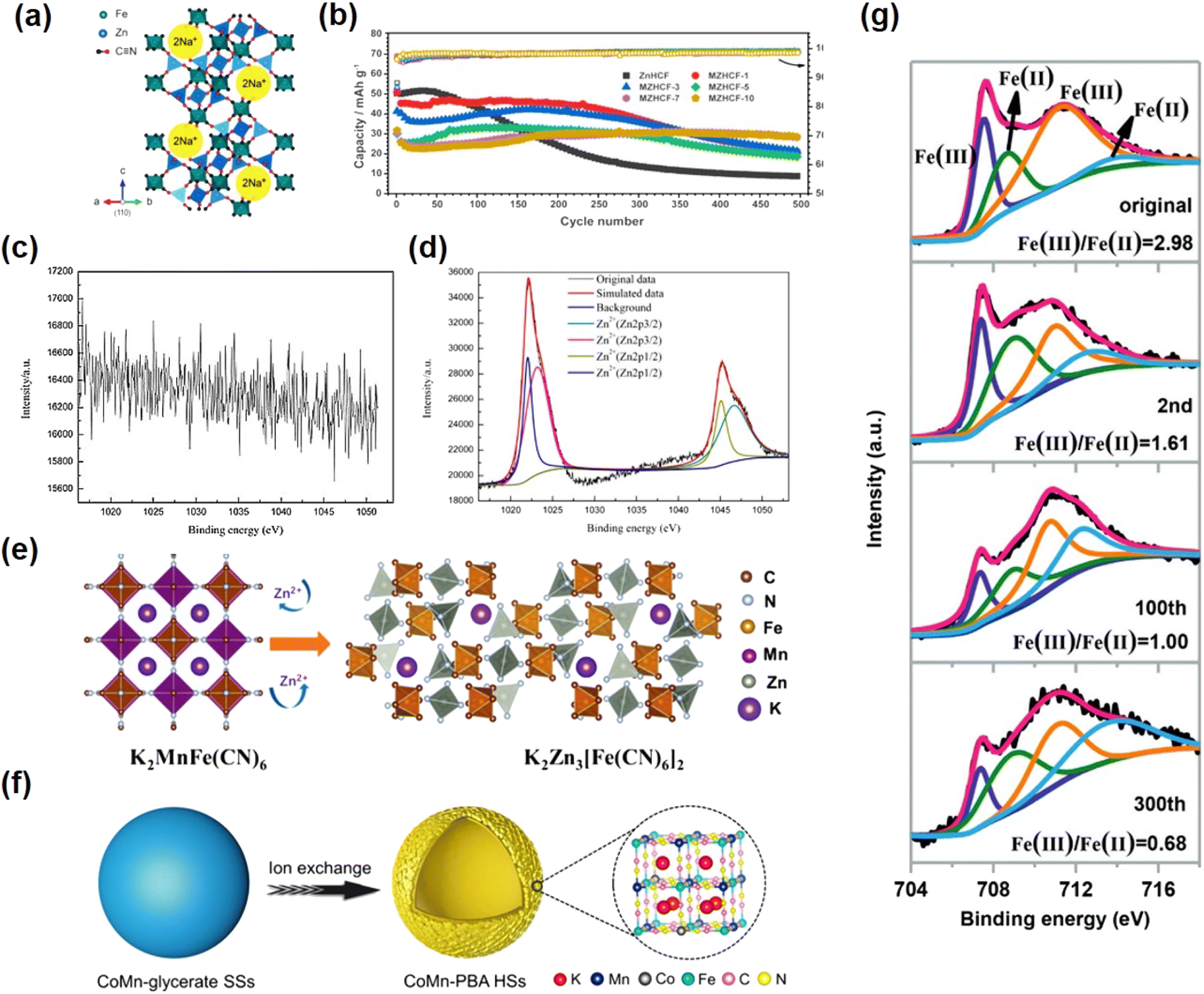 | ||
| Fig. 10 (a) Unit cell structure of NZH. (Reprinted with permission from ref. 36. Copyright 2012, Royal Society of Chemistry). (b) Cycling performance of ZnHCF and MZHCF at 250 mA g−1. (Reprinted with permission from ref. 81. Copyright 2021, American Chemical Society). XPS spectra of the original (c) and Zn-inserted CuHCF electrode (d). (Reprinted with permission from ref. 43. Copyright 2014, Elsevier). (e) The schematic diagram of the phase transformation for the KMnHCF electrode. (Reprinted with permission from ref. 50. Copyright 2021, John Wiley and Sons). (f) Synthesis process of CoMn-PBA HSs. (Reprinted with permission from ref. 92. Copyright 2021, John Wiley and Sons). (g) Ex situ XPS spectra of Fe 2p of FeHCF cathodes in the discharge state. (Reprinted with permission from ref. 138. Copyright 2019, John Wiley and Sons). | ||
In 2014, Zhang et al.49 reported for the first time that ZnHCF was used as a host material for divalent ion insertion on the basis of previous research studies. The experiments show the first use of ZnHCF as a cathode material for an aqueous zinc ion battery. When it is assembled with a zinc anode, the average operating voltage of ZnHCF can reach 1.7 V, which is a record operating voltage of aqueous zinc ion batteries. Based on the total active electrode materials, it also provides a specific energy density of 100 W h Kg−1 with a capacity retention rate of over 76% after 100 cycles. The electrode has longer cycle stability under ZnSO4 electrolyte than K2SO4 and Na2SO4 solutions. Although ZnHCF as a cathode material can significantly improve the working voltage of ZIBs, compared with Mn oxide cathode material, the lack of cycle performance of PBA analogues has always been a difficult problem that limits its large-scale maturity. Ni et al. introduced Mn ions into ZnHCF compounds by simple precipitation to study the effect of metals connected with N bonds on Zn intercalation chemistry and to improve electrochemical stability.81 A series of manganese-substituted ZnHCF materials (MZHCFs) were used as cathode materials for aqueous zinc ion batteries (ARZIBs), where an Mn content of 7% exhibited optimal cycling performance. Because the substitution of Mn ions inhibits the cuboidal to rhomboid phase transition, the solid solution mechanism becomes dominant, thus improving the structural stability during the insertion and extraction of Zn ions. Thus MZHCF exhibits a significantly improved capacity retention rate during the constant current cycle compared to the MnHCF and ZnHCF materials in ARZIBs (Fig. 10b).
In order to better understand the aging mechanism of CuHCF, Kasiri et al. studied the influence of electrolyte concentration and properties on the electrochemical performance of CuHCF.129 The results showed that the electrolyte had great influence on the degradation mechanism of CuHCF. In addition to the influence of the properties of different ions in the electrolyte, the ion concentration also affects the properties of the material (the higher the electrolyte concentration, the faster the aging of CuHCF). From the potential curves of CuHCF cycled in 100 mM ZnSO4, we can observe the formation of a two-step plateau and the change in average cell voltage at higher cycling concentrations. In particular, it is observed that higher electrolyte concentration and lower current rate lead to faster degradation of a positive electrode. The results show that the phase transition of CuHCF occurs when it is cycled in high concentration ZnSO4 and 100 mM Zn(ClO4)2 electrolyte, which negatively affects the aging of the active material. Kasiri et al. delayed the degradation mechanism by optimizing the structure of CuHCF by using Zn2+ in the synthesis.87 The experimental results show that the CuZnHCF mixture with a Cu:Zn ratio of 93:7 exhibits excellent specific charge and stability, and two different phases are produced during the charge–discharge cycle of CuHCF. The first phase (cubic) and the second phase (non-cubic) nanoparticles are found to increase the zinc-rich composition of the CuZnHCF (93:7) mixture as expected.
In 2014, Trócoli et al.85 synthesized Cu-HCF from Cu(NO3)2·3H2O and K3Fe(CN)6 with similar stoichiometric ratios, based on a 20 mM ZnSO4 aqueous solution (pH 6.0) electrolyte. The charge retention rate of the CuHCF–Zn cell after 100 cycles was 96.3% of the maximum charge (the 15th cycle). However, the stability of CuHCF in ZnSO4 is slightly worse than that of classical electrolytes KNO3 and HNO3 (pH = 2), and a phase transition occurs during cycling at higher electrolyte concentrations (100 mM). In order to improve the stability of CuHCF, the group added sodium salt 2 M NaClO4 into zinc-based electrolyte Zn(ClO4)2 to obtain a mixed ion battery.130 The results showed that after 500 cycles, the initial charge retention rate of the battery with NaClO4 was 73.3% higher than that without NaClO4 (58.1%). It is inferred that sodium can improve the electrochemical performance of the system by slowing down the phase transition. At the same time, the content of zinc in CuHCF is calculated to be about 1.5–2% that of sodium by mathematical treatment.
4.2 Double REDOX pair PBA
When the MA site is in Mn, Fe, Co, or V, two redox sites can participate in the reaction. Due to the presence of two redox centers, the cyanide salt composed of these three metals has a greater working voltage and capacity than ZuHCF, CuHCF, and NiHCF. Due to the limitation of the electrochemical stability window (ESW) on electrolyte operation and limited redox center activity, a potential high pressure platform of these materials has not yet been conclusively established.125As mentioned previously, the addition of a surfactant to the synthesis process can effectively reduce crystal defects. Cao et al. controlled the reaction process by adding PVP to the synthesis process and obtained a cubic granular KMHCF material with low water content and low defect uniformity.91 In this study, PVP can not only induce anisotropic growth through preferential adsorption to a single crystal plane, but also provide metastability through hydration-based spatial repulsion. The results show that the KMHCF-PVP-80 electrode also exhibits excellent long-cycle performance at other higher current densities. It has been reported that anorganic conductive polymer and Mn-PBA coated materials can also remedy this issue. Chen et al. coated polypyrrole (ppy) on the external surface of a KMnHCF cube, which not only effectively prolonged its cycle life but also led to excellent cycle performance.135 This is attributed to the limitation of Mn dissolution during electrochemical cycling by using ppy as a protective layer and its excellent electronic conductivity.
A recent study reported that the original K2MnFe(CN)6 cathode was gradually transformed into rhomboidic K2Zn3[Fe(CN)6]2 by the introduction of Zn2+ during the electrochemical cycle, and the introduction of Zn2+ caused a strong John-Teller effect on Mn3+, leading to strong lattice distortion (Fig. 10e).50 Together with the disproportionation reaction of manganese, the MnN6 octahedron is replaced by the ZnN4 tetrahedron, which eventually generates a new K2Zn3[Fe(CN)6]2 phase. The resulting solid structure of the K2Zn3[Fe(CN)6]2 phase contains wider channels for accommodating divalent ions, thereby enabling highly stable and reversible storage of Zn2+ ions. They also theoretically calculated different MnHCFs, in which the MnN6 octahedral surface exhibits a modest Jahn–Teller effect during K+ deintercalation from K2MnFe(CN)6 to MnFe(CN)6, and the crystal structure maintains a monoclinic skeleton without phase transition. In contrast, with Zn2+ insertion, some of the octahedral MnN6 are highly distorted and even become tetrahedral MnN4, indicating that Zn2+ induces a strong Jahn–Teller effect.
000 cycles (73% capacity retention).
Previous research has demonstrated that hollow structure construction is an effective method to reduce structural strain during ion insertion/ejection and enhance the stability of PBA.139,140 Based on this, Zeng et al.92 proposed an effective self-template strategy (Fig. 10f) to construct cobalt-rich substituted manganese-rich PBA hollow spheres (represented as CoMn-PBA HSs) by simple anion exchange. Due to the higher specific surface area, sufficient active sites, and low coordinated water content of CoMnPBA HSs, the electrochemical performance is better than that of Co-PBA HSs and Mn-PBA HS. In contrast, the Co-PBA HS electrode has low current density and poor electrochemical activity. The CoMn-PBA HS electrode has high current density and has a similar cyclic voltammetry (CV) curve to that of the Mn-PBA HS electrode. Capacitance recovery shows excellent magnification performance as the current density decreases back to 0.1 A g−1. In addition, after 1000 cycles, it exhibits a coulombic efficiency close to 100%.
N ligands can also delay vanadium ion dissolution in aqueous electrolytes.142 Consequently, vanadium-containing PBAs have a high specific capacity, a moderate operating voltage, and a high level of stability, but there are few reports on this subject. As reported by Zhang et al., they synthesized VHCF nanoparticles using a coprecipitation method and used them for the first time as an AZIB cathode material.93 In addition, their electrochemical properties and the mechanism of Zn2+ ion insertion/deintercalation were investigated. XPS revealed that three pairs of REDOX peaks and the corresponding three pairs of charging–discharge plateaus can be attributed to V3+/V4+ (I/I′), V4+/V5+ (II/II′), and [Fe(CN)6]4−/[Fe(CN)6]3− (III/III′) reversible REDOX processes. The specific capacity of the VHCF nanocathode was up to 172 mA h g−1, and 87.8% retention was obtained after 1000 cycles. Recently, Tian et al. used VO-PBAs in a WISE electrolytic liquid system of 21 m (LiTFSI) and 1 M Zn(CF3SO3)2 to effectively avoid side reactions and decomposition in the aqueous electrolyte, thus ensuring the high pressure of the system.95 However, this work significantly improves the capacity and coulombic efficiency of the positive electrode.
Despite the fact that numerous studies have focused on enhancing the capacity and cycling performance of V-PBA, the inherent low conductivity of V-PBA remains a limitation. Combining PBA with conductive agents to improve the conductivity of electrons is a viable remedy to this problem. Xue et al.94 designed a VHCF/CNT hybrid material in which vanadium hexanoate nanoparticles were grown on carbon nanotubes by in situ coprecipitation. On the one hand, the inherent 3D open framework of VHCF provides sufficient ion diffusion paths. On the other hand, since VHCF nanoparticles are connected through cross-linked CNTs, the conductive network formed by CNTs can accelerate the electron transfer between VHCF nanoparticles and promote the full utilization of active materials. However, considering that a single conductive carbon framework cannot provide adequate electrical conductivity, a double conductive carbon framework composed of internal and external conductive carbon components can provide higher electronic conductivity and better electrochemical properties. Wang et al. synthesized K1.14(VO)3.33[Fe(CN)6]2·6.8H2O cathode material, through the electron reaction of three different REDOX pairs V5+/4+, V4+/3+ and Fe3+/2+, and a dual conductive framework constructed from CNTs and Super P (SP).143 Benefiting from its structural and morphology engineering, the KVHCF@DCCF cathode material has a high specific capacity of 180 mA h g−1 at 400 mA g−1.
5 PB positive electrode-based aqueous electrolyte
The advantages of aqueous electrolytes, including low cost, high ionic conductivity, simple synthesis/processing, and benevolence, dominate research on electrolytes in ZIBs, and PB/PBAs as cathode materials can match aqueous electrolytes well. This section focuses primarily on the water system electrolyte as a PB/PBA complement in zinc ion batteries. Since dendrites and ZnO are more likely to form in alkaline electrolytes and strongly acidic electrolytes corrode the anode and collector, electrolytes that are neutral or weakly acidic are preferred. In addition to the hydrogen bond between water molecules and the interaction between Zn2+ and H2O, there are also interactions between positive–negative ions and negative ions–H2O in the aqueous zinc ion electrolyte system. The interaction between anions and H2O is stronger than the hydrogen bond between water molecules, which can contribute to the solvation of Zn2+via the network structure of hydrogen bonds between water molecules. Consequently, various anions have a significant impact on the electrochemical performance of the battery. Table 3 summarizes the electrolytes that complement the positive electrodes of PB/PBAs in ZIBs, including ZnSO4, Zn(CF3SO3)2, Zn(NO3)2, Zn(TFSI)2, Zn(CH3COO)2, Zn(ClO4)2, and ZnCl2. Among them, ZnSO4 and Zn(CF3SO3)2 are most commonly used because of their stability and compatibility with electrodes, and the corresponding ESW can reach 2.3 V and 2.4 V, respectively. However, Zn(CF3SO3)2 is significantly more expensive than ZnSO4, which may limit its commercial viability, while ZnSO4, despite its obvious price advantage, inevitably exihibits hydrogen evolution reactions and co-embedding with Zn2+/H+. A by-product (Zn4SO4(OH)6·4H2O) will be formed at the cathode material/electrolyte interface.144 In contrast, the bulky anion CF3SO3− in Zn(CF3SO3)2 can reduce the amount of free water around Zn2+, reduce solvation, and have higher reversibility and faster reaction kinetics.145There is a strong chemical/electrochemical interaction between polar water molecules and PB/PBAs in aqueous electrolyte, which has a negative impact on the stability of the electrode, and the operation will lead to inadequate dissolution of active substances. Due to the low water decomposition voltage of 1.23 V, the effective potential window of a water medium is also restricted. On the one hand, the minimal operating voltage increases the device's energy density. On the other hand, the narrow potential window does not permit the high operating voltage of the PB/PBA cathode, which may result in the hazard of water-induced side reactions (such as the hydrogen/oxygen evolution reaction (HER/OER)) and consequently the device performance. Approaches to expanding the electrochemical stability window include adjusting the pH of the electrolyte and utilizing a “water in salt” (WIS) electrolyte system. The most frequent reason for adjusting pH is to regulate the decomposition of aqueous electrolytes.146–148 Xia et al. inhibited hydrogen evolution by adjusting the pH value of the electrolyte to 13, and shifted the potential of hydrogen evolution from 0.43 V to 0.8 V. In this way, the electrochemical window will be shifted as a whole, while the entire electrochemical stability window remains the same width in the aqueous electrolytic liquid system.146
A new type of “water in salt” (WIS) electrolyte system can effectively inhibit the dissolution and side reactions of PB/PBAs due to its extremely low water content.149–151 Liu et al. developed an acetonitrile/water-in-salt (AWIS) mixed electrolyte. Compared with the aqueous electrolyte, the AWIS mixed electrolyte prolonged the life of a ZnZn battery from 150 hours to 2500 hours and increased the upper limit cut-off voltage of a Zn–MnO2 battery from 1.8 V to 2.2 V.150 In addition, it has been reported that the Zn-PB battery capacity, rate ability, and cycle stability are enhanced after high voltage scanning at 2.3 V (vs. Zn/Zn2+) in the HCZLE salt-covered water electrolyte of 21 M lithium trifluoromethane sulfonyl and 1 M zinc bis (trifluoromethane sulfonyl) imide. This improvement is a result of the activation of C-linked low FeHCF.138
The strong solvation effect of 30 M KFSI + 1 M Zn(CF3SO3)2 salt solution in water can significantly reduce the activity of water in aqueous solutions. This pronounced solvation effect is supported by Raman spectroscopy results (Fig. 11a). With increasing electrolyte concentration, the corresponding H–O stretching peak shifts to 3557 cm−1, indicating a robust coordination between water molecules and potassium ions. The XRD analysis of a single charge and discharge cycle also provides evidence that a high concentration electrolyte can effectively maintain the structural stability of the positive electrode KZnHCF (Fig. 11c). Specifically, no significant changes in the structure were observed at both the initial stage of charging and the final stage of discharging. Furthermore, the atomic ratio of the KZnHCF electrode pre- and post-cycling substantiates the incorporation of Zn2+ ions and underscores the structural stability of [Fe(CN)6]4− (Fig. 11b).153 Other studies have demonstrated a significant reduction in the concentration of Mn within the KMnHCF electrode when exposed to diluted electrolyte, with no detectable presence of manganese observed in the electrode after 100 cycles using a 30 M KFSI + 1 M Zn(CF3SO3)2 electrolyte (Fig. 11d).50 Li et al. demonstrated that diluted hydrolysates exhibit limited reversibility towards Zn2+ ions, and the presence of NiHCF in diluted electrolytes leads to the formation of novel phases. However, this phenomenon is effectively suppressed in highly concentrated 1 M Zn(TFSI)2 + 21 M LiTFSI electrolytes (Fig. 11e).154
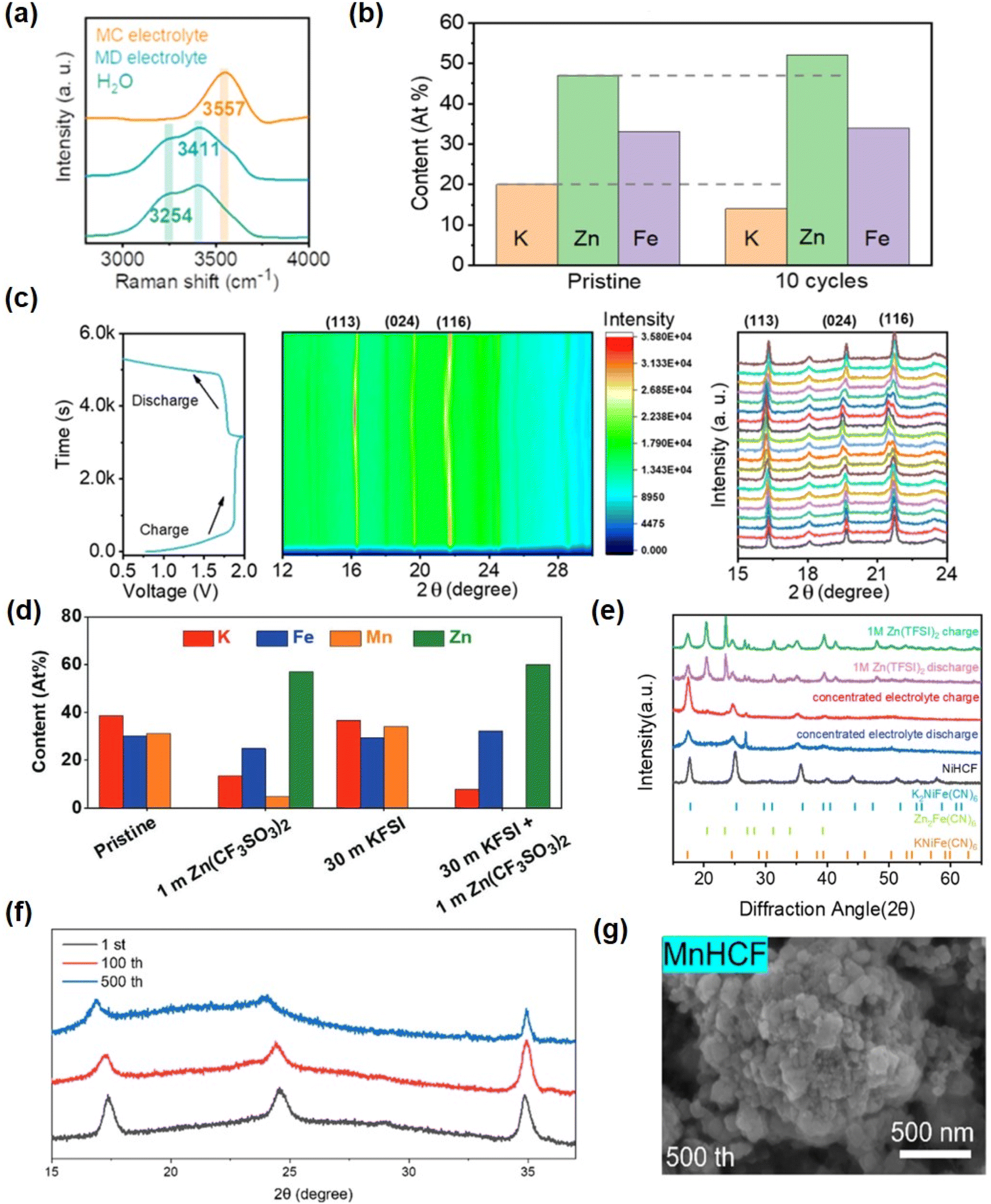 | ||
| Fig. 11 (a) Raman spectra of the two mixed electrolytes and pure water (H2O) observed in the range of 2500–4000 cm−1 corresponding to the O–H stretching modes of water molecules. (b) Atomic ratio of the KZnHCF cathode before and after cycles. (c) Structure stability analysis of KZnHCF. In situ XRD test of the KZnHCF/Zn cell in the MC electrolyte and the corresponding charge–discharge curve. (Reprinted with permission from ref. 153. Copyright 2022, American Chemical Society). (d) The EDS results corresponding to the KMnHCF electrode after 100 cycles in diluted electrolyte and WIS electrolyte at 0.3 A g−1. (Reprinted with permission from ref. 50. Copyright 2021, John Wiley and Sons). (e) XRD patterns for structure evolution of the NiHCF electrode over 50 cycles in different electrolytes. (Reprinted with permission from ref. 154. Copyright 2020, John Wiley and Sons). (f) XRD results of MnHCF after the 1st, 100th, and 500th cycles using gel-0.3. (g) The morphology of MnHCF after 200th cycles using gel-0.3. (Reprinted with permission from ref. 162. Copyright 2023, American Chemical Society). | ||
Despite the fact that the water-salt system can reduce the water content to a certain extent, batteries with a specific mechanical strength and shape deformation are required in certain exceptional circumstances. With the continuous miniaturization of electronic chips, the development of integrated electronic devices, such as implantable medical devices, wearable health monitoring systems, flexible displays, and smart clothing, has attracted the attention of scientists around the globe.155–157 The gel electrolyte has some mechanical strength and deformation capability, and the cross-linked polymer network can interact powerfully with polar water molecules and PB/PBAs. Typically, quasi-solid gel electrolytes are prepared by dissolving inorganic salts in a fluid polymer backbone, such as polyvinyl alcohol (PVA),158 polyacrylamide (PAM),138 sodium alginate (SA),159 and sodium carboxymethyl cellulose (CMC).160 After the solid/gel interface has been assembled into a device, it inhibits the dissolution of PB/PBAs and the interface water decomposition reaction effectively and has a lengthy service life. For instance, WIG electrolytes can attain robust properties due to SA's strong affinity for water. The formation of hydrogen bonds between water molecules and polar groups on the SA chain (including OH− and COO−) permits the storage of unbound water within the WIG electrolyte. The electrolyte enlarges the electrochemical window and reduces decomposition-induced water loss. NaCl/ZnSO4/sodium alginate electrolyte (NaCl/ZnSO4/SA) serves as the electrolyte to match the CuHCF cathode in order to obtain high performance in a Na–Zn hybrid battery.159
Zhang and his colleagues107 showed the practical application of zinc ion batteries in flexible wearable electronics. The flexible quasi-solid-state battery was prepared by coupling ZnHCF@MnO2 with Zn foil in ZnSO4/PVA gel electrolyte. The current change of the battery can be ignored under different bending angles, showing excellent flexibility. In addition, the discharge capacities of the flexible device at 100, 200, 400, 500 and 800 mA g−1 were 89, 78, 67, 58 and 53 mA h g−1, respectively. In addition to the higher discharge capacity, the quasi-solid state battery also has a higher rate capacity. When the current density was increased from 100 mA g−1 to 800 mA g−1, the discharge capacity could still exceed 49 mA h g−1 with a capacity retention of 55%. At the same time, the flexible battery can stably cycle for more than 500 times and still maintain 71% capacity. Even folding the flexible battery can power the LED bulb (drive voltage of 1.8 V).
Zhi et al. studied the effect of different factors on the electrochemical performance of the Zn/MnHCF battery with PVHF/MXene-g-PMA as the electrolyte.161 This solid state Zn/MnHCF cell can operate normally at −35, and the capacity retention rate is 36.4% compared with that at 25 °C. When the operating temperature was increased to 100 °C, a significant capacity of up to 143.2 mA h g−1 was achieved. After high or low temperature testing, the capacity of all-solid Zn/MnHCF full cells can be well recovered when the temperature is restored to 25 °C. These results demonstrate the excellent environmental adaptability of an all-solid Zn/MnHCF whole-cell and confirm the remarkable thermal and freezing resistance of this type of solid-state cell. Although PBA as a flexible zinc ion cathode material has high voltage and good zinc storage performance, its development is still limited by the generally low lifetime caused by dissolution in the electrochemical cycle. The hydrogel electrolyte can act as a physical barrier to the dissolution of the anode material, especially for Mn-PBAs and PBA-based cathode materials. Luo et al. employed gelatin as a quasi-solid electrolyte to effectively suppress the dissolution of Mn in MnHCF. Throughout the cycling process, XRD analysis revealed that the cathode of MnHCF maintained its monoclinic phase (Fig. 11f), and SEM imaging demonstrated the preserved cubic morphology of the overall block structure, thereby indicating excellent cycling stability of MnHCF in gel-0.3 electrolyte (Fig. 11g).162 Zhi's group60 introduced a sol–gel transition strategy by introducing a hydrogel as a flexible electrolyte. The assembled Zn/CoFe(CN)6 cell achieves 2200 cycles and an excellent performance of nearly 100% coulombic efficiency.
Adding inorganic163 and organic164 additives to aqueous electrolytes is considered to be an effective strategy to modify the interfacial properties and inhibit the dissolution of PB/PBAs. Generally speaking, an electrolyte additive is a substance that improves the electrode/electrolyte interface to improve the electrochemical performance of the battery without participating in the electrode reaction, for example, the common organic surfactant sodium dodecylsulfonate (SDS). SDS molecules face the electrode and the electrolyte with their hydrophilic groups and hydrophobic groups, respectively, and are adsorbed on the electrode surface. The adsorbed SDS layer is on the outer hydrophobic interface, which effectively prevents the passage of water molecules and expands the electrochemical stability window of the electrolyte to about 2.5 V. In addition, based on the density functional theory experiments and calculation results, it is shown that SDS can effectively inhibit the dissolution of Mn in the Na2MnFe(CN)6 cathode, and effectively improve the cycle life of the Na–Zn hybrid battery.165
6 Summary and outlook
This review offers a comprehensive overview of the crystal structure, energy storage mechanism, and synthesis methods of PBA materials, with the aim of enhancing readers' comprehension in this field. Importantly, PBAs not only demonstrates facile and cost-effective synthesis but also exhibits remarkable reversibility for embedding/detaching metal ions within its framework. The application of transition metal-containing Prussian blue analogues (PBAs) in zinc ion batteries is then comprehensively discussed. Despite the extensive research on PBAs over several decades, investigations into their potential as battery materials have only commenced within the past ten years. In conclusion, the majority of PBAs are synthesized using the coprecipitation method, and by controlling experimental parameters such as stirring speed and drop acceleration, and it is possible to obtain particles with diverse morphologies that significantly impact the properties of PBAs. Due to their open framework structure, which facilitates the reversible insertion of diverse metal ions, PBAs have undergone continuous modification and development since their initial use as for sodium and potassium ion batteries, now finding application as cathode materials for zinc ion batteries. This review discusses the impact of vacancy structure water and zeolite water on the framework and electrochemical properties of PBAs, based on their synthesis and structure. The comprehensive description encompasses the application of diverse cathode active materials in zinc ion batteries, including Zn-PBAs, Cu-PBAs, and Ni-PBAs within a single REDOX center, as well as Co-PBAs, Fe-PB, Mn-PBAs and V-P with a double REDOX center. Table 3 presents a comprehensive overview of the electrochemical characteristics exhibited by various types of PBAs as cathode materials for ZIBs. In terms of practical implementation, the primary concern lies in their limited volumetric capacity, which consequently results in reduced volume energy density and cycle stability. The particle morphology modification projects of certain PBAs are being considered as potential solutions to address the issue of low volumetric energy density. The potential areas for further investigation and advancement are summarized in Fig. 12. The diagram depicting the middle battery configuration represents the potential future of large-scale energy storage in PBAs as a cathode.| Material | Electrolyte | Working window [V] | Capacity [mA h g−1@mA g−1] | Cycle life [cycles, retention%@ mA g−1] | Ref. |
|---|---|---|---|---|---|
| Zn3[Fe(CN)6]2 | 3 M ZnSO4 | 0.8–1.9 | 66.5@60 | 200, 81@300 | 83 |
| Zn3[Fe(CN)6]2 | 1 M ZnSO4 | 0.8–1.9 | 65.4@60 | 200, 80@300 | 49 |
| K0.14Mn0.11Zn1.49[Fe(CN)6] | 1 M ZnSO4 | 1.0–2.0 | 41.6@50 | 500, 85.5@250 | 81 |
| CuFe(CN)6 | 0.02 M ZnSO4 | 0.5–1.4 | 53@60 | 100, 81@600 | 84 |
| KCuFe(CN)6 | 1 M ZnSO4 | 0.2–1.2 | 56@20 | 20, 77@20 | 43 |
| CuZnHCF | 0.02 M ZnSO4 | 0.2–1.1 | 50@85 | 1000, 88.74@85 | 87 |
| NiHCF/RGO | 2 M ZnSO4 | 0.7–1.8 | 50.1@200 | 1000, 80.3@200 | 88 |
| K1.51Ni[Fe(CN)6]0.954(H2O)0.766 | 0.5 M Zn(ClO4)2 | 0.6–1.8 | 55.6@0.2C | 20, 99.9@0.2C | 48 |
| NiFe(CN)6 | 0.5 M Na2SO4 + 0.05 M ZnSO4 | 0.9–1.9 | 76.2@100 | 1000, 81@500 | 152 |
| K1.6Mn1.2Fe(CN)6 | 2 M Zn(ClO4)2 in tetraethylene glycol dimethyl ether | 0.7–1.9 | 65@50 | 8500, 94@200 | 33 |
| K2MnFe(CN)6 | 30 M KFSI + 1 M Zn(CF3SO3)2 | 0.5–1.9 | 100@200 | 400, 72@200 | 50 |
| KMHCF-PVP-80 | 1 M ZnSO4 + 0.1 M MnSO4 | 0.4–1.8 | 78@200 | 400, ≈100@100 | 91 |
| KMHCF@PPy | 2 M ZnSO4 + 0.1 M MnSO4 | 0.4–1.8 | 110@100 | 100, 97.8@100 | 135 |
| K0.05Fe(III)[Fe(III)(CN)6]·2.6H2O | 1.0 M Zn(OAc)2/([Ch]OAc + 30 wt% water) | 0.8–2.0 | 120@10 | 50, 97@60 | 136 |
| Fe[Fe(CN)6]·2.5H2O | A bio-degradable ionic liquid-water mixture | 0.6–1.8 | 54@0.1 mA cm−3 | 50, 99.5@0.1 mA cm−3 | 137 |
| FeHCF | 21 M lithium LiTFSI + 1 M zinc bis(trifluoromethanesulfonyl)imide | 0–2.3 | 76@1000 | 10000, 73@3000 | 138 |
| CoFe(CN)6 | 4 M (Zn(OTf)2 | 0.7–1.8 | 109.5@6000 | 2200, ≈100@2000 | 60 |
| CoMn-PBA HSs | 2 M Zn(CF3SO3)2 | 0.5–1.8 | 128.6@50 | 300, 76.4@10000 |
92 |
| V3[Fe(CN)6]2 | 4 M Zn(CF3SO3)2 | 0.4–1.8 | 160@500 | 1000, 87.8@2000 | 93 |
| VHCF/CNTs | 2 M ZnSO4 | 0.3–2.0 | 97.8@50 | 1000, 98@3200 | 94 |
| VO-PBAs | 21 M (LiTFSI) + 1 M Zn(CF3SO3)2 | 0.3–2.1 | 209.6@100 | 2000, 95.5@1000 | 95 |
| ZnHCF@MnO2 | 0.5 M ZnSO4 | 1.4–1.9 | 120@100 | 1000, 77@100 | 107 |
| ZnHCF/PANI | 7.5 M ZnCl2 + 4 M NaCl | 0.8–2.1 | 50@50 | 350, 75@500 | 110 |
 | ||
| Fig. 12 Prospects for future research and application avenues of PBAs. | ||
The concentration of metal ions within the PBA framework exhibits a positive feedback effect on the capacity and cycling performance of sodium ion and potassium ion batteries, when used as cathode materials. However, the impact of the basic metal ion content within the framework of PBAs on the mechanism for Zn2+ removal remains insufficiently elucidated. Insufficient evidence suggests that the framework exerts a favorable influence on Zn2+ during synthesis. In addition, further investigation is variations in water content affect the crystal structure of Prussian blue. Notably, when subjected to vacuum drying, Prussian blue materials used in sodium ion batteries experience a reduction in water content which eliminates water molecules from the organic electrolyte system. In an aqueous-based zinc ion battery, it is unknown whether water will be continuously embedded in the lattice as the electrochemical reaction progresses, such that the amount of water molecules in the lattice will influence the transmission of metal ions and electrons. It is unclear, however, if the water molecules in the lattice will impact the transmission of metal ions and electrons. This is the distinction between an aqueous electrolyte and an organic electrolyte, so the discussion on this aspect is also a breakthrough in elucidating the mechanism. In summary, the research on high-performance PBAs should focus on effectively controlling the phase transition of defect water content and investigating the characteristics of lattice strain. For MnHCF, additional efforts are necessary to suppress the Jahn–Teller effect in order to enhance cycle stability.
In essence, the research on high-performance PBAs should primarily focus on effectively controlling the phase transition of defect water content and investigating the characteristics of lattice strain. Regarding MnHCF, additional endeavors are necessary to suppress the Jahn–Teller effect for enhancing cycle stability. From a practical application standpoint, the consideration of volume energy density becomes pivotal. Extensive research has been dedicated to the doping of transition metals and the development of composite materials thus far. Consequently, increasing novelty also involves enhancing ion utilization efficiency through the design of morphology and structure, such as incorporating hollow structures and core–shell structures. However, there is a dearth of research on modifying the cathode/electrolyte interface, excluding any modifications to the PBA material itself. The energy barrier for the embedding of completely dehydrated metal ions in a crystal lattice is distinctly different when they diffuse from the electrolyte to the electrode interface. Furthermore, the involvement of metal hydrated ions in intercalation forms at the electrode interface plays a pivotal role in battery dynamics. Moreover, exploring system and ion chemistry through simulation calculations and studying interfacial impedance represents a promising avenue for future research. It is worth noting that certain bimetallic PBAs exhibit substantial potential for research and development. In such a structure, one metal component acts as a supportive framework while the other imparts active sites. The diverse interactions among transition metals play a crucial role in mutually enhancing their electrochemical performance, thereby surpassing that of PBAs containing a single metal. Additionally, novel configurations or enhanced characteristics may be unveiled during the course of the investigation, and thus, the exploration of bimetallic PBAs holds potential for ground breaking outcomes. Increasing the specific surface area is a promising approach to enhance the electrochemical efficacy of PBAs. Exploring novel technologies for synthesizing particles with diverse morphologies and structures, as well as controlling various other factors, represents an intriguing research direction that will impact the electrochemical performance of manufactured goods. In conclusion, we anticipate that this study will offer researchers a comprehensive understanding of the current state of cathode materials based on Prussian blue analogues for zinc-ion batteries, while also inspiring them to identify and address challenges, as well as develop novel PBAs materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 52073286(Lu) and 22275185(Lu)), the XMIREM Autonomously Deployment Project (2023GG01(Lu)), and the Fujian Science& Technology Innovation Laboratory for Optoelectronic Information of China (2021ZZ115(Lu)).References
- L. Lu, X. Han, J. Li, J. Hua and M. Ouyang, J. Power Sources, 2013, 226, 272–288 CrossRef CAS.
- A. Kwade, W. Haselrieder, R. Leithoff, A. Modlinger, F. Dietrich and K. Droeder, Nat. Energy, 2018, 3, 290–300 CrossRef.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- J. Tarascon, Joule, 2020, 4, 1616–1620 CrossRef.
- Y. Liang, H. Dong, D. Aurbach and Y. Yao, Nat. Energy, 2020, 5, 646–656 CrossRef CAS.
- L. E. Blanc, D. Kundu and L. F. Nazar, Joule, 2020, 4, 771–799 CrossRef CAS.
- M. Wang, C. Jiang, S. Zhang, X. Song, Y. Tang and H. Cheng, Nat. Chem., 2018, 10, 667–672 CrossRef CAS PubMed.
- L. Jiang, Y. Lu, C. Zhao, L. Liu, J. Zhang, Q. Zhang, X. Shen, J. Zhao, X. Yu, H. Li, X. Huang, L. Chen and Y.-S. Hu, Nat. Energy, 2019, 4, 495–503 CrossRef CAS.
- J. Shin and J. W. Choi, Adv. Energy Mater., 2020, 10, 2001386 CrossRef CAS.
- K. Zhu, T. Wu, S. Sun, Y. Wen and K. Huang, ChemElectroChem, 2020, 7, 2714–2734 CrossRef CAS.
- L. Chen, Q. An and L. Mai, Adv. Mater. Interfaces, 2019, 6, 1900387 CrossRef.
- Y. Li, J. Fu, C. Zhong, T. Wu, Z. Chen, W. Hu, K. Amine and J. Lu, Adv. Energy Mater., 2019, 9, 1802605 CrossRef.
- T. C. Li, D. Fang, J. Zhang, M. E. Pam, Z. Y. Leong, J. Yu, X. L. Li, D. Yan and H. Y. Yang, J. Mater. Chem. A, 2021, 9, 6013–6028 RSC.
- H. Zhang, Q. Liu, D. Zheng, F. Yang, X. Liu and X. Lu, Nat. Commun., 2021, 12, 14 CrossRef CAS PubMed.
- G. Zampardi and F. La Mantia, Curr. Opin. Electrochem., 2020, 21, 84–92 CrossRef CAS.
- A. Konarov, N. Voronina, J. H. Jo, Z. Bakenov, Y.-K. Sun and S.-T. Myung, ACS Energy Lett., 2018, 3, 2620–2640 CrossRef CAS.
- J. Huang, Z. Guo, Y. Ma, D. Bin, Y. Wang and Y. Xia, Small Methods, 2019, 3, 1800272 CrossRef.
- X. Ma, X. Cao, Y. Zhou, S. Guo, X. Shi, G. Fang, A. Pan, B. Lu, J. Zhou and S. Liang, Nano Res., 2020, 13, 3330–3337 CrossRef CAS.
- K. W. Nam, S. S. Park, R. dos Reis, V. P. Dravid, H. Kim, C. A. Mirkin and J. F. Stoddart, Nat. Commun., 2019, 10, 4948 CrossRef PubMed.
- A. Khayum M, M. Ghosh, V. Vijayakumar, A. Halder, M. Nurhuda, S. Kumar, M. Addicoat, S. Kurungot and R. Banerjee, Chem. Sci., 2019, 10, 8889–8894 RSC.
- X. Li, M. Li, Q. Yang, H. Li, H. Xu, Z. Chai, K. Chen, Z. Liu, Z. Tang, L. Ma, Z. Huang, B. Dong, X. Yin, Q. Huang and C. Zhi, Acs Nano, 2020, 14, 541–551 CrossRef CAS PubMed.
- W. Shi, W. S. V. Lee and J. Xue, Chemsuschem, 2021, 14, 1634–1658 CrossRef CAS PubMed.
- F. Wan and Z. Niu, Angew. Chem., 2019, 131, 16508–16517 CrossRef.
- Y. Zhang, L. Tao, C. Xie, D. Wang, Y. Zou, R. Chen, Y. Wang, C. Jia and S. Wang, Adv. Mater., 2020, 32, 1905923 CrossRef CAS PubMed.
- Y. Shen and K. Kordesch, J. Power Sources, 2000, 87, 162–166 CrossRef CAS.
- B. Wu, G. Zhang, M. Yan, T. Xiong, P. He, L. He, X. Xu and L. Mai, Small, 2018, 14, 1703850 CrossRef PubMed.
- Y. Li, S. Wang, J. R. Salvador, J. Wu, B. Liu, W. Yang, J. Yang, W. Zhang, J. Liu and J. Yang, Chem. Mater., 2019, 31, 2036–2047 CrossRef CAS.
- Q. Sun, H. Cheng, W. Nie, X. Lu and H. Zhao, Chem. Asian J., 2022, 17, e202200067 CrossRef CAS PubMed.
- Y. Zhang, E. H. Ang, K. N. Dinh, K. Rui, H. Lin, J. Zhu and Q. Yan, Mater. Chem. Front., 2021, 5, 744–762 RSC.
- J. Ding, Z. Du, L. Gu, B. Li, L. Wang, S. Wang, Y. Gong and S. Yang, Adv. Mater., 2018, 30, 1800762 CrossRef PubMed.
- N. Zhang, Y. Dong, M. Jia, X. Bian, Y. Wang, M. Qiu, J. Xu, Y. Liu, L. Jiao and F. Cheng, ACS Energy Lett., 2018, 3, 1366–1372 CrossRef CAS.
- X. Zhang, T. Xiong, B. He, S. Feng, X. Wang, L. Wei and L. Mai, Energy Environ. Sci., 2022, 15, 3750–3774 RSC.
- Q. Li, K. Ma, G. Yang and C. Wang, Energy Storage Mater., 2020, 29, 246–253 CrossRef.
- A. Indra, T. Song and U. Paik, Adv. Mater., 2018, 30, 1705146 CrossRef PubMed.
- L. Ma, H. Cui, S. Chen, X. Li, B. Dong and C. Zhi, Nano Energy, 2021, 81, 105632 CrossRef CAS.
- H. Lee, Y.-I. Kim, J.-K. Park and J. W. Choi, Chem. Commun., 2012, 48, 8416–8418 RSC.
- Y. Lu, L. Wang, J. Cheng and J. B. Goodenough, Chem. Commun., 2012, 48, 6544–6546 RSC.
- C. D. Wessells, R. A. Huggins and Y. Cui, Nat. Commun., 2011, 2, 1–5 Search PubMed.
- D. Asakura, M. Okubo, Y. Mizuno, T. Kudo, H. Zhou, K. Ikedo, T. Mizokawa, A. Okazawa and N. Kojima, J. Phys. Chem. C, 2012, 116, 8364–8369 CrossRef CAS.
- C. Xu, Z. Yang, X. Zhang, M. Xia, H. Yan, J. Li, H. Yu, L. Zhang and J. Shu, Nano-Micro Lett., 2021, 13, 166 CrossRef CAS PubMed.
- A. Paolella, C. Faure, V. Timoshevskii, S. Marras, G. Bertoni, A. Guerfi, A. Vijh, M. Armand and K. Zaghib, J. Mater. Chem. A, 2017, 5, 18919–18932 RSC.
- K. Hurlbutt, S. Wheeler, I. Capone and M. Pasta, Joule, 2018, 2, 1950–1960 CrossRef CAS.
- Z. Jia, B. Wang and Y. Wang, Mater. Chem. Phys., 2015, 149, 601–606 CrossRef.
- H. Yi, R. Qin, S. Ding, Y. Wang, S. Li, Q. Zhao and F. Pan, Adv. Funct. Mater., 2021, 31, 2006970 CrossRef CAS.
- X. Guo, Z. Wang, Z. Deng, X. Li, B. Wang, X. Chen and S. P. Ong, Chem. Mater., 2019, 31, 5933–5942 CrossRef CAS.
- C. Gao, Y. Liu, L. Zheng, E. Feng, S. Sim, Y. Kim, J. Yun, M. Kim and S. W. Lee, J. Electrochem. Soc., 2019, 166, A1732 CrossRef CAS.
- S. Phadke, R. Mysyk and M. Anouti, J. Energy Chem., 2020, 40, 31–38 CrossRef.
- M. S. Chae, J. W. Heo, H. H. Kwak, H. Lee and S.-T. Hong, J. Power Sources, 2017, 337, 204–211 CrossRef CAS.
- L. Zhang, L. Chen, X. Zhou and Z. Liu, Adv. Energy Mater., 2015, 5, 1400930 CrossRef.
- W. Deng, Z. Li, Y. Ye, Z. Zhou, Y. Li, M. Zhang, X. Yuan, J. Hu, W. Zhao, Z. Huang, C. Li, H. Chen, J. Zheng and R. Li, Adv. Energy Mater., 2021, 11, 2003639 CrossRef CAS.
- U. Ali, B. Liu, H. Jia, Y. Li, Y. Li, Y. Hao, L. Zhang, S. Xing, L. Li and C. Wang, Small, 2023, 2305866 CrossRef PubMed.
- F. Scholz and A. Dostal, Angew Chem. Int. Ed. Engl., 1996, 34, 2685–2687 CrossRef.
- J. Yang, W. Hou, L. Ye, G. Hou, C. Yan and Y. Zhang, Small, 2023, 2305386 Search PubMed.
- L. Chen, W. Sun, K. Xu, Q. Dong, L. Zheng, J. Wang, D. Lu, Y. Shen, J. Zhang, F. Fu, H. Kong, J. Qin and H. Chen, ACS Energy Lett., 2022, 7, 1672–1678 CrossRef CAS.
- F. Wang, W. Sun, Z. Shadike, E. Hu, X. Ji, T. Gao, X.-Q. Yang, K. Xu and C. Wang, Angew. Chem., Int. Ed., 2018, 57, 11978–11981 CrossRef CAS.
- J. Yue, L. Lin, L. Jiang, Q. Zhang, Y. Tong, L. Suo, Y. Hu, H. Li, X. Huang and L. Chen, Adv. Energy Mater., 2020, 10, 2000665 CrossRef CAS.
- X. Dong, Y.-G. Wang and Y. Xia, Acc. Chem. Res., 2021, 54, 3883–3894 CrossRef CAS PubMed.
- Z. Ji, B. Han, H. Liang, C. Zhou, Q. Gao, K. Xia and J. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 33619–33625 CrossRef CAS PubMed.
- Y. You, X.-L. Wu, Y.-X. Yin and Y.-G. Guo, Energy Environ. Sci., 2014, 7, 1643–1647 RSC.
- L. Ma, S. Chen, C. Long, X. Li, Y. Zhao, Z. Liu, Z. Huang, B. Dong, J. A. Zapien and C. Zhi, Adv. Energy Mater., 2019, 9, 1902446 CrossRef CAS.
- D. O. Ojwang, J. Grins, D. Wardecki, M. Valvo, V. Renman, L. Häggström, T. Ericsson, T. Gustafsson, A. Mahmoud, R. P. Hermann and G. Svensson, Inorg. Chem., 2016, 55, 5924–5934 CrossRef CAS.
- V. Renman, D. O. Ojwang, M. Valvo, C. P. Gomez, T. Gustafsson and G. Svensson, J. Power Sources, 2017, 369, 146–153 CrossRef CAS.
- W. Xu, J.-J. Zheng, Y.-A. Li, X. Gao, X. Ji and Y.-G. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202306185 CrossRef CAS PubMed.
- Y. Shang, X. Li, J. Song, S. Huang, Z. Yang, Z. J. Xu and H. Y. Yang, Chem, 2020, 6, 1804–1818 CAS.
- G. W. Beall, W. O. Milligan, J. Korp and I. Bernal, Inorg. Chem., 1977, 16, 2715–2718 CrossRef CAS.
- F. Herren, P. Fischer, A. Ludi and W. Haelg, Inorg. Chem., 1980, 19, 956–959 CrossRef CAS.
- Y. Mizuno, M. Okubo, D. Asakura, T. Saito, E. Hosono, Y. Saito, K. Oh-ishi, T. Kudo and H. Zhou, Electrochim. Acta, 2012, 63, 139–145 CrossRef CAS.
- C. D. Wessells, S. V. Peddada, M. T. McDowell, R. A. Huggins and Y. Cui, J. Electrochem. Soc., 2011, 159, A98 CrossRef.
- J. Wu, J. Song, K. Dai, Z. Zhuo, L. A. Wray, G. Liu, Z. Shen, R. Zeng, Y. Lu and W. Yang, J. Am. Chem. Soc., 2017, 139, 18358–18364 CrossRef CAS PubMed.
- E. Perez-Cappe, M. Aguilar-Frutis, N. Chavez, J. Ribalta and E. Reguera, Microporous Mesoporous Mater., 2012, 163, 326–333 CrossRef CAS.
- G. Du and H. Pang, Energy Storage Mater., 2021, 36, 387–408 CrossRef.
- B. Singh and A. Indra, Mater. Today Energy, 2020, 16, 100404 CrossRef.
- M. B. Zakaria and T. Chikyow, Coord. Chem. Rev., 2017, 352, 328–345 CrossRef CAS.
- J. A. Dean, Lange’s Handbook of Chemistry, 1978, DOI:10.1080/10426919008953291.
- X. Wu, M. Cao, C. Hu and X. He, Cryst. Growth Des., 2006, 6, 26–28 CrossRef CAS.
- D. A. Peixoto, S. C. Silva, P. H. S. Borges, R. C. Lima and E. Nossol, J. Mater. Sci., 2023, 58, 2993–3024 CrossRef CAS.
- J. Yang, H. Wang, L. Lu, W. Shi and H. Zhang, Cryst. Growth Des., 2006, 6, 2438–2440 CrossRef CAS.
- X.-J. Zheng, Q. Kuang, T. Xu, Z.-Y. Jiang, S.-H. Zhang, Z.-X. Xie, R.-B. Huang and L.-S. Zheng, J. Phys. Chem. C, 2007, 111, 4499–4502 CrossRef CAS.
- S. Ying, C. Chen, J. Wang, C. Lu, T. Liu, Y. Kong and F.-Y. Yi, ChemPlusChem, 2021, 86, 1608–1622 CrossRef CAS PubMed.
- J. Gu, S. Fu, C. Jin, X. Liu, Y. Gao, J. Wu, Z. Bian, H. Tian, L. Wang and F. Gao, J. Solid State Chem., 2016, 239, 64–68 CrossRef CAS.
- G. Ni, X. Xu, Z. Hao, W. Wang, C. Li, Y. Yang, C. Zhou, L. Qin, W. Chen, X. Yao and J. Cai, ACS Appl. Energy Mater., 2021, 4, 602–610 CrossRef CAS.
- J. W. Heo, M. S. Chae, J. Hyoung and S.-T. Hong, Inorg. Chem., 2019, 58, 3065–3072 CrossRef CAS PubMed.
- L. Zhang, L. Chen, X. Zhou and Z. Liu, Sci. Rep., 2015, 5, 1–11 Search PubMed.
- R. Trócoli and F. La Mantia, ChemSusChem, 2015, 8, 481–485 CrossRef PubMed.
- R. Trocoli and F. La Mantia, ChemSusChem, 2015, 8, 481–485 CrossRef CAS PubMed.
- G. Zampardi, M. Warnecke, M. Tribbia, J. Glenneberg, C. Santos and F. La Mantia, Electrochem. Commun., 2021, 126, 107030 CrossRef CAS.
- G. Kasiri, J. Glenneberg, A. B. Hashemi, R. Kun and F. La Mantia, Energy Storage Mater., 2019, 19, 360–369 CrossRef.
- Y. Xue, Y. Chen, X. Shen, A. Zhong, Z. Ji, J. Cheng, L. Kong and A. Yuan, J. Colloid Interface Sci., 2022, 609, 297–306 CrossRef CAS PubMed.
- A. L. Lipson, S.-D. Han, S. Kim, B. Pan, N. Sa, C. Liao, T. T. Fister, A. K. Burrell, J. T. Vaughey and B. J. Ingram, J. Power Sources, 2016, 325, 646–652 CrossRef CAS.
- L. Fan, X. Guo, X. Hang and H. Pang, J. Colloid Interface Sci., 2022, 607, 1898–1907 CrossRef CAS PubMed.
- T. Cao, F. Zhang, M. Chen, T. Shao, Z. Li, Q. Xu, D. Cheng, H. Liu and Y. Xia, ACS Appl. Mater. Interfaces, 2021, 13, 26924–26935 CrossRef CAS PubMed.
- Y. Zeng, X. F. Lu, S. L. Zhang, D. Luan, S. Li and X. W. (David) Lou, Angew. Chem., Int. Ed., 2021, 60, 22189–22194 CrossRef CAS PubMed.
- Y. Zhang, Y. Wang, L. Lu, C. Sun and D. Y. W. Yu, J. Power Sources, 2021, 484, 229263 CrossRef CAS.
- Y. Xue, X. Shen, H. Zhou, J. Cao, J. Pu, Z. Ji, L. Kong and A. Yuan, Chem. Eng. J., 2022, 448, 137657 CrossRef CAS.
- Y. Tian, M. Ju, X. Bin, Y. Luo and W. Que, Chem. Eng. J., 2022, 430, 132864 CrossRef CAS.
- W. Zhang, H. Song, Y. Cheng, C. Liu, C. Wang, M. A. N. Khan, H. Zhang, J. Liu, C. Yu, L. Wang and J. Li, Advanced Science, 2019, 6, 1801901 CrossRef PubMed.
- S. Vaucher, M. Li and S. Mann, Angew. Chem., Int. Ed., 2000, 39, 1793–1796 CrossRef CAS PubMed.
- Q. Zhang, L. Zhang and J. Li, Electrochim. Acta, 2008, 53, 3050–3055 CrossRef CAS.
- S.-H. Lee and Y.-D. Huh, Bull. Korean Chem. Soc., 2012, 33, 1078–1080 CrossRef CAS.
- Y. Liu, G. Wei, M. Ma and Y. Qiao, Chem.–Euro. J., 2017, 23, 15991–15996 CrossRef CAS PubMed.
- M. Hu, S. Furukawa, R. Ohtani, H. Sukegawa, Y. Nemoto, J. Reboul, S. Kitagawa and Y. Yamauchi, Angew. Chem., Int. Ed., 2012, 51, 984–988 CrossRef CAS PubMed.
- T. Uemura and S. Kitagawa, J. Am. Chem. Soc., 2003, 125, 7814–7815 CrossRef CAS PubMed.
- Y.-D. Chiang, M. Hu, Y. Kamachi, S. Ishihara, K. Takai, Y. Tsujimoto, K. Ariga, K. C.-W. Wu and Y. Yamauchi, Eur. J. Inorg. Chem., 2013, 2013, 3141–3145 CrossRef CAS.
- M. Hu, S. Ishihara, K. Ariga, M. Imura and Y. Yamauchi, Chem.–Euro. J., 2013, 19, 1882–1885 CrossRef CAS PubMed.
- J. Nai, J. Zhang and X. W. D. Lou, Chem, 2018, 4, 1967–1982 CAS.
- H. Ming, N. L. K. Torad, Y.-D. Chiang, K. C.-W. Wu and Y. Yamauchi, CrystEngComm, 2012, 14, 3387–3396 RSC.
- K. Lu, B. Song, Y. Zhang, H. Ma and J. Zhang, J. Mater. Chem. A, 2017, 5, 23628–23633 RSC.
- D. Zhang, Z. Yang, J. Zhang, H. Mao, J. Yang and Y. Qian, J. Power Sources, 2018, 399, 1–7 CrossRef CAS.
- Y. You, H.-R. Yao, S. Xin, Y.-X. Yin, T.-T. Zuo, C.-P. Yang, Y.-G. Guo, Y. Cui, L.-J. Wan and J. B. Goodenough, Adv. Mater., 2016, 28, 7243–7248 CrossRef CAS PubMed.
- Q. Liu, Z. Ma, Z. Chen, M. Cui, H. Lei, J. Wang, J. Fei, N. He, Y. Liu, Q. Liu, W. Li and Y. Huang, Chem. Commun., 2022, 58, 8226–8229 RSC.
- C. Zhang, Y. Xu, M. Zhou, L. Liang, H. Dong, M. Wu, Y. Yang and Y. Lei, Adv. Funct. Mater., 2017, 27, 1604307 CrossRef.
- J. Song, L. Wang, Y. Lu, J. Liu, B. Guo, P. Xiao, J.-J. Lee, X.-Q. Yang, G. Henkelman and J. B. Goodenough, J. Am. Chem. Soc., 2015, 137, 2658–2664 CrossRef CAS PubMed.
- X. Wu, Y. Luo, M. Sun, J. Qian, Y. Cao, X. Ai and H. Yang, Nano Energy, 2015, 13, 117–123 CrossRef CAS.
- X. Wu, C. Wu, C. Wei, L. Hu, J. Qian, Y. Cao, X. Ai, J. Wang and H. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 5393–5399 CrossRef CAS PubMed.
- X. Wu, M. Shao, C. Wu, J. Qian, Y. Cao, X. Ai and H. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 23706–23712 CrossRef CAS PubMed.
- M. B. Zakaria, V. Malgras, T. Takei, C. Li and Y. Yamauchi, Chem. Commun., 2015, 51, 16409–16412 RSC.
- W.-J. Li, S.-L. Chou, J.-Z. Wang, Y.-M. Kang, J.-L. Wang, Y. Liu, Q.-F. Gu, H.-K. Liu and S.-X. Dou, Chem. Mater., 2015, 27, 1997–2003 CrossRef CAS.
- W. Li, C. Han, G. Cheng, S. Chou, H. Liu and S. Dou, Small, 2019, 15, 1900470 CrossRef PubMed.
- L. Wang, J. Song, R. Qiao, L. A. Wray, M. A. Hossain, Y.-D. Chuang, W. Yang, Y. Lu, D. Evans, J.-J. Lee, S. Vail, X. Zhao, M. Nishijima, S. Kakimoto and J. B. Goodenough, J. Am. Chem. Soc., 2015, 137, 2548–2554 CrossRef CAS PubMed.
- T. N. Vo, J. Hur and I. T. Kim, ACS Sustainable Chem. Eng., 2020, 8, 2596–2601 CrossRef CAS.
- G. Du, M. Tao, J. Li, T. Yang, W. Gao, J. Deng, Y. Qi, S.-J. Bao and M. Xu, Adv. Energy Mater., 2020, 10, 1903351 CrossRef CAS.
- Y. Liu, Y. Qiao, W. Zhang, Z. Li, X. Ji, L. Miao, L. Yuan, X. Hu and Y. Huang, Nano Energy, 2015, 12, 386–393 CrossRef CAS.
- A. Zhou, Z. Xu, H. Gao, L. Xue, J. Li and J. B. Goodenough, Small, 2019, 15, 1902420 CrossRef CAS PubMed.
- L. Deng, J. Qu, X. Niu, J. Liu, J. Zhang, Y. Hong, M. Feng, J. Wang, M. Hu, L. Zeng, Q. Zhang, L. Guo and Y. Zhu, Nat. Commun., 2021, 12, 2167 CrossRef CAS PubMed.
- J. Yan, E. H. Ang, Y. Yang, Y. Zhang, M. Ye, W. Du and C. C. Li, Adv. Funct. Mater., 2021, 31, 2010213 CrossRef CAS.
- G. Ni, B. Han, Q. Li, Z. Ji, B. Huang and C. Zhou, ChemElectroChem, 2016, 3, 798–804 CrossRef CAS.
- M. Oliver-Tolentino, G. Ramos-Sánchez, G. Guzmán, M. Avila, I. González and E. Reguera, Solid State Ionics, 2017, 312, 67–72 CrossRef CAS.
- C. Islas-Vargas, A. Guevara-García, M. Oliver-Tolentino, G. Ramos-Sánchez, I. González and M. Galván, J. Electrochem. Soc., 2018, 166, A5139 CrossRef.
- G. Kasiri, R. Trócoli, A. B. Hashemi and F. La Mantia, Electrochim. Acta, 2016, 222, 74–83 CrossRef CAS.
- R. Trócoli, G. Kasiri and F. L. Mantia, J. Power Sources, 2018, 400, 167–171 CrossRef.
- C. D. Wessells, S. V. Peddada, R. A. Huggins and Y. Cui, Nano Lett., 2011, 11, 5421–5425 CrossRef CAS PubMed.
- A. Mullaliu, J. Asenbauer, G. Aquilanti, S. Passerini and M. Giorgetti, Small Methods, 2020, 4, 1900529 CrossRef CAS.
- S. Wheeler, I. Capone, S. Day, C. Tang and M. Pasta, Chem. Mater., 2019, 31, 2619–2626 CrossRef CAS.
- G. Ni, Z. Hao, G. Zou, X. Xu, B. Hu, F. Cao and C. Zhou, Sustainable Energy Fuels, 2022, 6, 1353–1361 RSC.
- M. Chen, X. Li, Y. Yan, Y. Yang, Q. Xu, H. Liu and Y. Xia, ACS Appl. Mater. Interfaces, 2022, 14, 1092–1101 CrossRef CAS PubMed.
- Z. Liu, G. Pulletikurthi and F. Endres, ACS Appl. Mater. Interfaces, 2016, 8, 12158–12164 CrossRef CAS PubMed.
- Z. Liu, P. Bertram and F. Endres, J. Solid State Electrochem., 2017, 21, 2021–2027 CrossRef CAS.
- Q. Yang, F. Mo, Z. Liu, L. Ma, X. Li, D. Fang, S. Chen, S. Zhang and C. Zhi, Adv. Mater., 2019, 31, 1901521 CrossRef PubMed.
- J. Nai and X. W. Lou, Adv. Mater., 2019, 31, 1706825 CrossRef.
- L. Ren, J.-G. Wang, H. Liu, M. Shao and B. Wei, Electrochim. Acta, 2019, 321, 134671 CrossRef CAS.
- S. Liu, L. Kang, J. M. Kim, Y. T. Chun, J. Zhang and S. C. Jun, Adv. Energy Mater., 2020, 10, 2000477 CrossRef CAS.
- S. Husmann, A. J. Zarbin and R. A. Dryfe, Electrochim. Acta, 2020, 349, 136243 CrossRef CAS.
- F. Wang, Y. Li, W. Zhu, X. Ge, H. Cui, K. Feng, S. Liu and X. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 34468–34476 CrossRef CAS PubMed.
- F. Wan, L. Zhang, X. Dai, X. Wang, Z. Niu and J. Chen, Nat. Commun., 2018, 9, 1656 CrossRef PubMed.
- Q. Zhang, K. Xia, Y. Ma, Y. Lu, L. Li, J. Liang, S. Chou and J. Chen, ACS Energy Lett., 2021, 6, 2704–2712 CrossRef CAS.
- J.-Y. Luo, W.-J. Cui, P. He and Y.-Y. Xia, Nat. Chem., 2010, 2, 760–765 CrossRef CAS PubMed.
- L. Chen, Z. Guo, Y. Xia and Y. Wang, Chem. Commun., 2013, 49, 2204–2206 RSC.
- L. Chen, X. Dong, F. Wang, Y. Wang and Y. Xia, Chem. Commun., 2016, 52, 3147–3150 RSC.
- L. Suo, O. Borodin, W. Sun, X. Fan, C. Yang, F. Wang, T. Gao, Z. Ma, M. Schroeder, A. von Cresce, S. M. Russell, M. Armand, A. Angell, K. Xu and C. Wang, Angew. Chem., Int. Ed., 2016, 55, 7136–7141 CrossRef CAS PubMed.
- X. Song, H. He, M. H. Aboonasr Shiraz, H. Zhu, A. Khosrozadeh and J. Liu, Chem. Commun., 2021, 57, 1246–1249 RSC.
- L. Suo, Y.-S. Hu, H. Li, M. Armand and L. Chen, Nat. Commun., 2013, 4, 1481 CrossRef PubMed.
- K. Lu, B. Song, J. Zhang and H. Ma, J. Power Sources, 2016, 321, 257–263 CrossRef CAS.
- W. Deng, Z. Li, Y. Chen, N. Shen, M. Zhang, X. Yuan, J. Hu, J. Zhu, C. Huang, C. Li and R. Li, ACS Appl. Mater. Interfaces, 2022, 14, 35864–35872 CrossRef CAS PubMed.
- Z. Li, T. Liu, R. Meng, L. Gao, Y. Zou, P. Peng, Y. Shao and X. Liang, Energy Environ. Mater., 2021, 4, 111–116 CrossRef CAS.
- X. Li, Y. Tang, H. Lv, W. Wang, F. Mo, G. Liang, C. Zhi and H. Li, Nanoscale, 2019, 11, 17992–18008 RSC.
- P. Yu, Y. Zeng, H. Zhang, M. Yu, Y. Tong and X. Lu, Small, 2019, 15, 1804760 CrossRef.
- H. Dong, J. Li, J. Guo, F. Lai, F. Zhao, Y. Jiao, D. J. Brett, T. Liu, G. He and I. P. Parkin, Adv. Mater., 2021, 33, 2007548 CrossRef CAS.
- B. He, P. Man, Q. Zhang, C. Wang, Z. Zhou, C. Li, L. Wei and Y. Yao, Small, 2019, 15, 1905115 CrossRef CAS.
- W. Pan, Y. Wang, X. Zhao, Y. Zhao, X. Liu, J. Xuan, H. Wang and D. Y. C. Leung, Adv. Funct. Mater., 2021, 31, 2008783 CrossRef CAS.
- Q. Zhang, C. Li, Q. Li, Z. Pan, J. Sun, Z. Zhou, B. He, P. Man, L. Xie, L. Kang and others, Nano Lett., 2019, 19, 4035–4042 CrossRef CAS PubMed.
- Z. Chen, X. Li, D. Wang, Q. Yang, L. Ma, Z. Huang, G. Liang, A. Chen, Y. Guo, B. Dong and others, Energy Environ. Sci., 2021, 14, 3492–3501 RSC.
- L. Luo, Y. Liu, Z. Shen, Z. Wen, S. Chen and G. Hong, ACS Appl. Mater. Interfaces, 2023, 15, 29032–29041 CrossRef CAS.
- T. Shao, C. Li, C. Liu, W. Deng, W. Wang, M. Xue and R. Li, J. Mater. Chem. A, 2019, 7, 1749–1755 RSC.
- C. Liu, Y. Sun, J. Nie, D. Dong, J. Xie and X. Zhao, New J. Chem., 2020, 44, 4639–4646 RSC.
- Z. Hou, X. Zhang, X. Li, Y. Zhu, J. Liang and Y. Qian, J. Mater. Chem. A, 2017, 5, 730–738 RSC.
| This journal is © The Royal Society of Chemistry 2024 |