
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deciphering the role of Fe impurities in the electrolyte boosting the OER activity of LaNiO3†
Haritha
Cheraparambil
a,
Miquel
Vega-Paredes
 b,
Yue
Wang
a,
Harun
Tüysüz
a,
Christina
Scheu
b and
Claudia
Weidenthaler
*a
b,
Yue
Wang
a,
Harun
Tüysüz
a,
Christina
Scheu
b and
Claudia
Weidenthaler
*a
aMax-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, D-45470 Mülheim an der Ruhr, Germany. E-mail: weidenthaler@mpi-muelheim.mpg.de
bMax-Planck-Institut für Eisenforschung, Max-Planck-Straße 1, D-40237 Düsseldorf, Germany
First published on 29th January 2024
Abstract
Perovskites have emerged as potential catalysts for the alkaline oxygen evolution reaction. Iron impurities in the electrolyte play an important role in enhancing the catalytic activity of Ni centres, but the nature of active sites is elusive. In this article, we report a detailed study of iron incorporation dynamics and provide direct spectroscopic evidence for surface re-construction and dynamic active site evolution in LaNiO3 perovskite using identical location scanning transmission electron microscopy, electron energy loss spectroscopy and in situ electrochemical Raman spectroscopy. We demonstrate that the electrocatalytic activity is enhanced up to an amount of 7.5 ppm Fe traces in the electrolyte by lowering the Tafel slope from 115 to 49 mV dec−1. The iron impurities in the electrolyte enter the perovskite structure, leading to the dissolution of the A-site, vacancy formation, and amorphization of surface layers. The origin of activity arises from these amorphous layers which are ∼5 nm thick and rich in Ni oxyhydroxides, Fe oxyhydroxides, and a Ni–O–Fe coordinated environment. Together with pH-dependent studies, we confirm the lattice oxygen mechanism in the presence of Fe impurities. Our work provides new insights into the design and a deeper understanding of Ni–Fe synergetics in perovskite-based catalysts for alkaline OER.
Introduction
Electrochemical water splitting is an effective way to produce green hydrogen and has led to tremendous research efforts in the pursuit of carbon neutrality and sustainable energy production. Nevertheless, the overpotential of the process is high due to the sluggish kinetics of the anodic four-electron transfer oxygen evolution reaction (OER).1–3 Therefore, the development of an active, stable, and cost-effective OER catalyst is necessary. Even though the state-of-the-art catalysts, Ru- and Ir-based oxides, exhibit excellent OER performance, the scarcity and high costs impede their practical applications.4,5 Therefore, much of the ongoing research is focused on the preparation of alternative low-cost and highly active catalysts, of which transition-metal oxides such as perovskites (ABO3) have gained attention due to their mixed ionic–electronic conductivities, as well as structural and chemical tunability. By substituting the A or B site, perovskites can form myriad structures that can indirectly modulate physical, chemical, and catalytic properties.6–8Significant progress has been made in the rational design of perovskites by tailoring the valence, defects, and morphology to increase the number of accessible active sites.9–11 Numerous studies also direct towards surface reconstruction, stability, and degradation of the perovskite catalysts during OER.12–15 Most of the perovskites are identified to undergo a concerted electron–proton transfer, also known as the adsorbate evolution mechanism (AEM).6 Recently, this has been challenged by an alternate pathway known as the lattice oxygen mechanism (LOM), in which lattice oxygen from the oxides is also involved in the formation of O2, enhancing OER performance.16–18 It has also been shown that traces of Fe impurities from the electrolyte significantly enhance the OER performance of Ni-based oxides and perovskites.19–23
Despite these intensive studies, there remains a lot of ambiguity and unanswered questions on the iron incorporation dynamics of perovskite oxides. The extent to which Fe impurity from the electrolyte can enhance the OER activity and how it alters the surface layers of the catalyst are still unclear. It is important to study the surface alterations on atomic and local scales because such surface changes could result in the evolution of dynamic species that are different from the bulk nature of perovskite oxides.24 While the true nature of active sites in hydr(oxy)oxides is still under discussion,22,25–27 the structural complexity of perovskites makes it even harder to probe the short-lived dynamic species. In such instances, it is crucial to combine the observations from both in situ/operando and ex situ experiments to establish functional links between the surface reconstruction, and the catalytic mechanism.
Herein, we report a holistic study on lanthanum nickelate perovskite to understand the complex electrode–electrolyte interface at atomic and local scales. Systematic investigation of the effects of electrolyte impurities on the catalyst performance has shown that the presence of iron impurities enhances the electrocatalytic performance to some extent and has adverse effects beyond that. Using in situ electrochemical Raman spectroscopy, electron energy loss spectroscopy (EELS), and identical location scanning transmission electron microscopy (IL-STEM) reveals that the interplay between catalytically and dynamically evolving NiOOH and active Fe species from the electrolyte is crucial in favouring the LOM pathway. The studies establish functional links between the perovskite properties, the influence of electrolyte impurities, surface evolution, and the reaction mechanism.
Experimental section
Synthesis
Different perovskites were synthesized via the solution combustion method followed by calcination. In a typical reaction, stoichiometric amounts of nitrates [La(NO3)3·6H2O Alfa Aesar, CAS 10277-43-7, purity 99.99%, Ni(NO3)2·6H2O Alfa Aesar, CAS 13478-00-7, purity ≥ 97%] were dissolved in water. Glycerol (Sigma-Aldrich, CAS 56-81-5) was added as the fuel in an equal molar ratio concerning the metal nitrates. The mixture was stirred for 15 min on a magnetic stirrer and 3 mL nitric acid was added dropwise. The mixture was heated to 250 °C until the solvent was completely evaporated and an auto-combustion reaction took place. This resulted in the formation of a sponge-like powder which was further used as the precursor. The precursor was further calcined at 700 °C for 30 min to obtain the perovskite phase.Characterization
In identical location IL-STEM, the same region of the sample can be studied before and after an electrocatalytic reaction, and has been used for determining degradation mechanisms33,34 and the nature of active species35 of nanocatalysts. For the IL experiments, a 10 μL drop of a 0.3 mg mL−1 dispersion of LaNiO3 on deionized water was drop-cast on a hole carbon-coated Au TEM finder grid (Plano). After the initial characterization by STEM, the LaNiO3-containing grid was used as a working electrode in a three-electrode set-up. A Pt wire (Redoxme) and a reversible hydrogen electrode (Gaskatel) were used as counter and reference electrodes, respectively. For making electrical contact between the grid and the potentiostat instead of the classical glassy carbon–Teflon cap approach, a gold thread was used to avoid loss of current created by the evolved gas bubbles.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :isopropanol (1:1) and 50 μL Nafion 117 (Sigma-Aldrich) binder and further sonicating for 30 min to form a homogeneous ink. 5.25 μL of catalyst ink (catalyst loading of 0.12 mg cm−2) was drop cast onto the polished glassy carbon electrode and dried under argon atmosphere overnight. Cyclic voltammetry (CV) was performed at a scan rate of 50 mV s−1 within the 0.7 to 1.6 V vs. RHE potential window. Linear scan voltammetry (LSV) was measured after stabilizing the surface via CV in a potential window of 0.7 to 1.7 V vs. RHE at a scan rate of 10 mV s−1. Chronopotentiometry was performed at 10 mA cm−2 of geometric current density in 1 M KOH with 7.5 ppm Fe content to test the stability of the catalyst. Electrochemical impedance spectroscopy (EIS) was measured at 1.66 V vs. RHE and 5 mV of amplitude within the 100 mHz to 100 kHz frequency range and the obtained Nyquist plots were then fitted to the equivalent circuit model using the EC-Lab software. The IR drop was compensated at 85%.
:isopropanol (1:1) and 50 μL Nafion 117 (Sigma-Aldrich) binder and further sonicating for 30 min to form a homogeneous ink. 5.25 μL of catalyst ink (catalyst loading of 0.12 mg cm−2) was drop cast onto the polished glassy carbon electrode and dried under argon atmosphere overnight. Cyclic voltammetry (CV) was performed at a scan rate of 50 mV s−1 within the 0.7 to 1.6 V vs. RHE potential window. Linear scan voltammetry (LSV) was measured after stabilizing the surface via CV in a potential window of 0.7 to 1.7 V vs. RHE at a scan rate of 10 mV s−1. Chronopotentiometry was performed at 10 mA cm−2 of geometric current density in 1 M KOH with 7.5 ppm Fe content to test the stability of the catalyst. Electrochemical impedance spectroscopy (EIS) was measured at 1.66 V vs. RHE and 5 mV of amplitude within the 100 mHz to 100 kHz frequency range and the obtained Nyquist plots were then fitted to the equivalent circuit model using the EC-Lab software. The IR drop was compensated at 85%.
Results and discussion
Simple perovskite, LaNiO3 was synthesized by an optimized solution combustion method followed by calcination.37 Glycerol was used as the fuel, and the self-combustion occurred at 250 °C, resulting in the formation of spongy black precursors. A phase pure sample is obtained at 700 °C, as confirmed by Rietveld analysis (Fig. 1a). The compound crystallizes in a rhombohedral structure (space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c). The column length distribution was determined using the whole powder pattern modeling (WPPM) approach assuming spherical crystallites with a lognormal distribution of their column length.29 The volume-weighted crystallite size (LVol) was calculated to be 6–8 nm (Fig. S1†). Detailed analysis of the refined data reveals an asymmetric broadening of the reflections at 23° and 40° 2θ. In addition, the calculation of the Fourier difference map reveals the presence of residual electron density around the A site (La) in the direction of the c-axis and around the B site (Ni) around the a–b plane as depicted in Fig. 1b.
c). The column length distribution was determined using the whole powder pattern modeling (WPPM) approach assuming spherical crystallites with a lognormal distribution of their column length.29 The volume-weighted crystallite size (LVol) was calculated to be 6–8 nm (Fig. S1†). Detailed analysis of the refined data reveals an asymmetric broadening of the reflections at 23° and 40° 2θ. In addition, the calculation of the Fourier difference map reveals the presence of residual electron density around the A site (La) in the direction of the c-axis and around the B site (Ni) around the a–b plane as depicted in Fig. 1b.
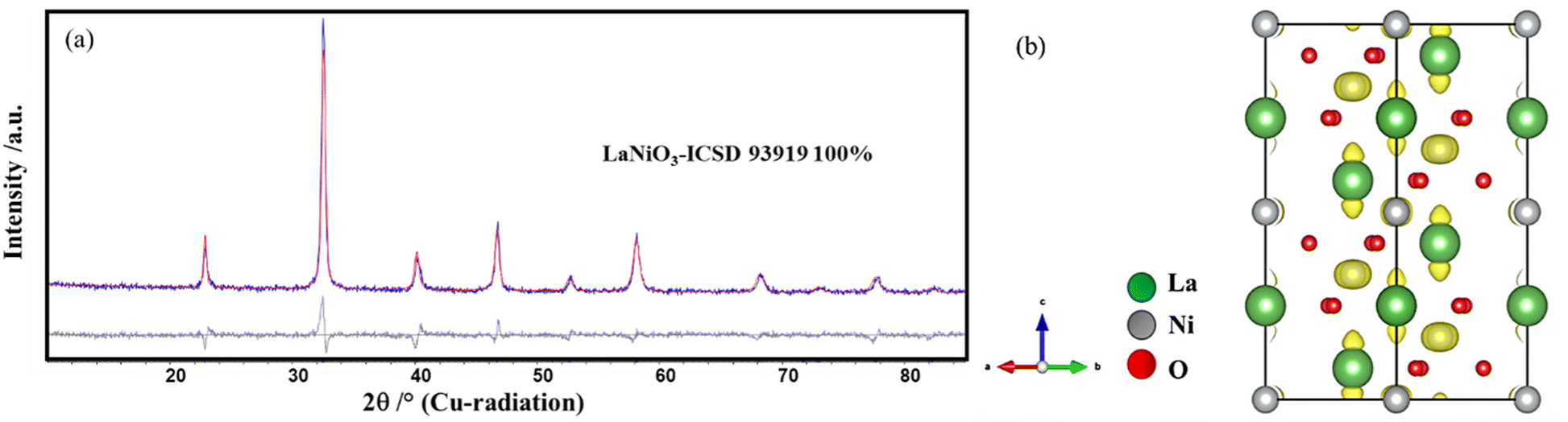 | ||
| Fig. 1 (a) Rietveld refinement plot of LaNiO3 obtained at 700 °C. (b) Fourier difference map showing the residual electron density (yellow in colour). | ||
To gain more information on the local structure, total scattering experiments were performed and the resulting PDFs were analyzed as shown in Fig. 2a. Total X-ray scattering data and subsequent pair distribution function (PDF) analysis have proven to be powerful tools for characterizing amorphous, distorted and crystalline structures not only in the short-range but also in the intermediate and long ranges.38,39 The phase crystallizes in a rhombohedral structure with space group Rc. In a perfect crystal system, the first pair correlation corresponds to the Ni–O distances in the NiO6 polyhedron which appears at 1.94 Å as shown in (i), and the next pair correlation is assigned to the La–O distances in a 9-fold coordinated polyhedron (ii). There are three shorter (2.43 Å) and six longer (2.71 Å) La–O bond lengths which result in a broadening of the pair correlation. However, in the present perovskite structure, the NiO6 polyhedra have shorter Ni–O bond lengths (1.85 Å) and La–O polyhedra have longer bond lengths (2.48 Å and 2.83 Å). These observations indicate the presence of disorder. The pair correlation at 3.8 Å (iii) includes contributions from the Ni–Ni distances and La–La distances in corner-sharing Ni–O and La–O polyhedra. La–O polyhedra share edges as well, where the La–La distances appear around 5.4 Å.
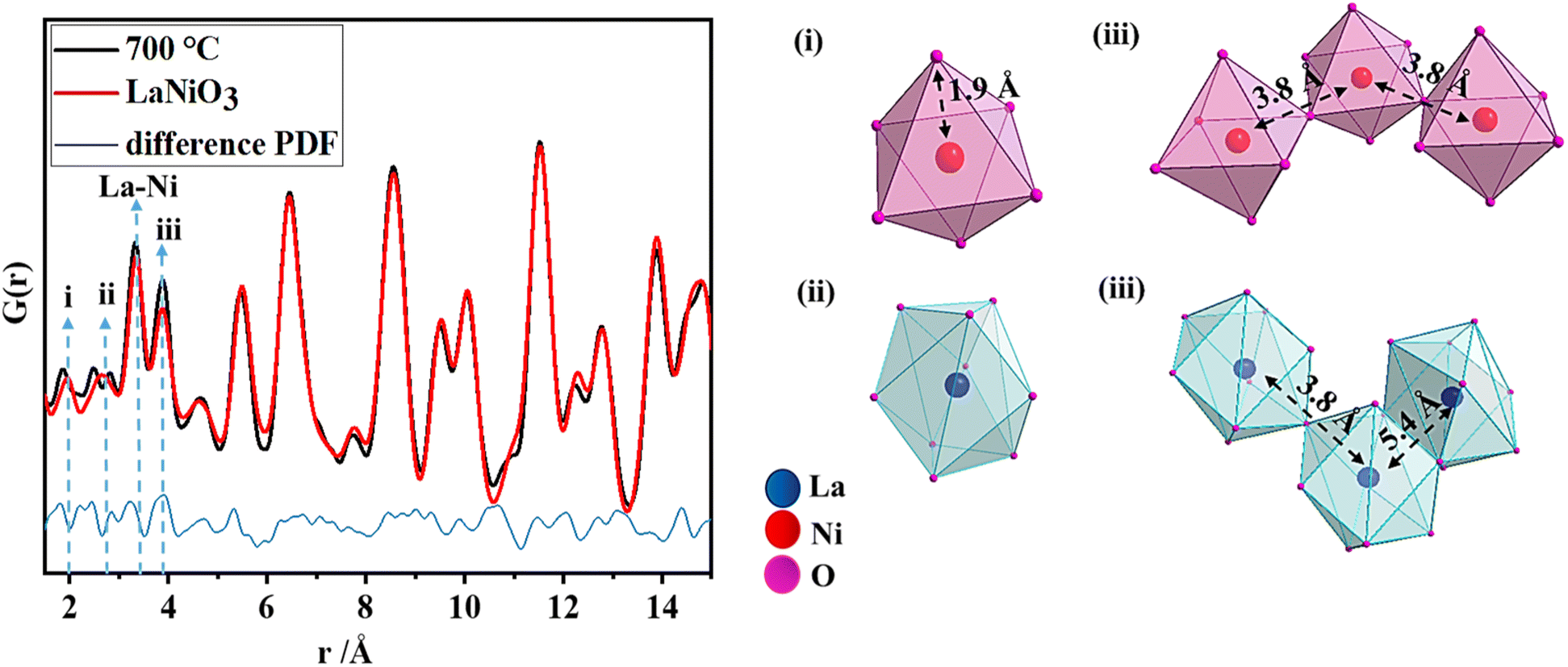 | ||
| Fig. 2 PDF refinement of LaNiO3 obtained at 700 °C, with the explanation of the nearest pair correlations (i–iii). | ||
Scanning electron microscopy (SEM) shows the surface topology of the sample (Fig. S2a and b†). The particles are densely packed with interparticle porosity and the BET surface area from physisorption experiments was calculated to be 10 m2 g−1 (Fig. S3†). Scanning transmission electron microscopy (STEM) probes the crystallinity of the material. It confirms the presence of stacking faults, as shown in Fig. 3a in the annular dark field (ADF) and Fig. 3b in the high-angle annular dark field (HAADF) images. This agrees well with the observations obtained from the Fourier difference map as well as from PDF analysis.
 | ||
| Fig. 3 (a) ADF image of LaNiO3, showing the presence of multiple planar defects. (b) High-resolution HAADF-STEM micrograph. The positions of the La (blue) and Ni (red) columns are indicated. | ||
Fe-dependent electrochemical performance
Once the structure of the catalyst was determined, the catalytic OER activity of the system was tested according to the adapted protocol proposed by Jaramillo and co-workers.40 The electrocatalysts can undergo various surface reconstructions in the presence of iron in the electrolyte.19,20 To systematically study the role of Fe, the electrolyte was first purified as reported in the literature.22,41 The iron concentration in the electrolyte was varied from 1 ppm to 10 ppm by the controlled addition of iron nitrate nonahydrate as the iron source. The concentration of Fe was further confirmed by inductively coupled plasma optical emission spectroscopy (ICP-OES) (Table S1†). As the Fe content in the electrolyte increases, the area under the redox peaks increases indicating that more Ni centres are getting activated (Fig. 4a). Along with the significant increase in the activation of the catalyst, there is also a shift in the redox behaviour corresponding to the electronic interaction between Fe and Ni centres, as shown in the inset of Fig. 4a.22,42 No redox behaviour of iron was observed because the oxidation of metallic Fe to Fe2+/Fe3+ oxides or hydroxides occurs in the potential range from −360 mV to −1.0 V vs. the reversible hydrogen electrode (RHE). This observation is also consistent with literature reports.43 However, the OER activity increases up to a maximum Fe content of 7.5 ppm in the electrolyte and then slowly decreases as shown in Fig. 4b. Even though iron species increase the activity of Ni centres, an excess amount can aggravate the charge transfer to water because of the insulating property of iron oxyhydroxides.41,42Fig. 4b and d show that the catalyst can deliver a maximum current density of 340 mA cm−2 with an overpotential of 300 mV vs. RHE, thus boosting the overall activity. The reaction kinetics are further analyzed using Tafel plots, which confirm the excellent performance of LaNiO3 in the presence of traces of Fe impurities, lowering the Tafel slope from 115 to 49 mV dec−1 as illustrated in Fig. 4c.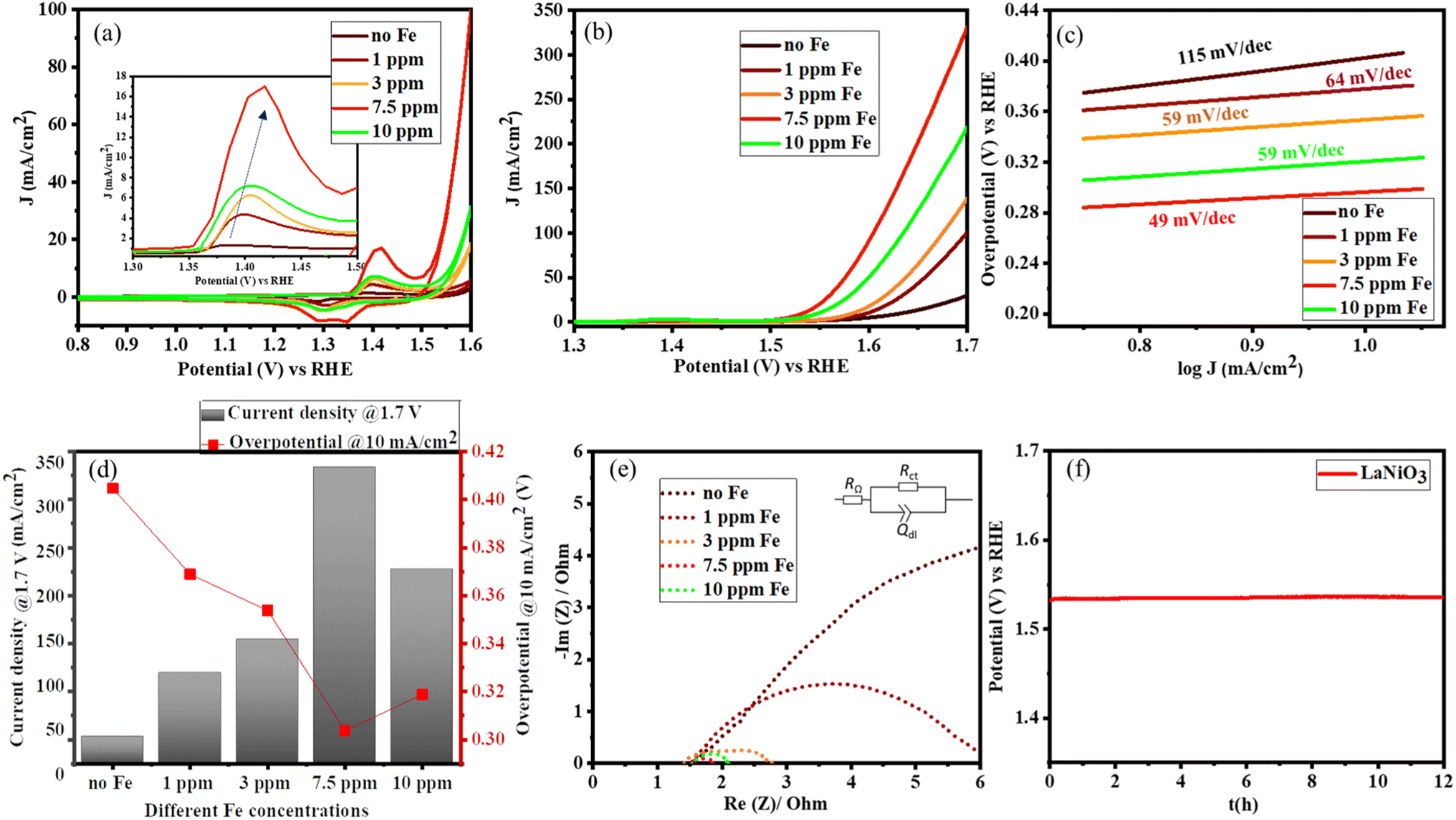 | ||
| Fig. 4 (a) Cyclic voltammetry after 50 cycles. The inset shows the shift in the oxidation peak of Ni in the presence of different Fe contents. (b) Linear sweep voltammetry (LSV) after 100 cycles of CV (c) Tafel plots (d) overview on the overpotential at 10 mA cm−2 and current density at 1.7 V vs. RHE (e) Nyquist plots from EIS measurements at 1.66 V vs. RHE on LaNiO3 in different concentrations of Fe in the electrolyte (1 M KOH) and (f) chronopotentiometry on LaNiO3 at 10 mA cm−2 for 12 h in 1 M KOH with 7.5 ppm of Fe impurity. | ||
Complementary electrochemical impedance spectroscopy (EIS) was used to probe the charge transfer behaviour in the presence of Fe impurities in the electrolyte. Fig. 4e depicts the Nyquist plots from which the charge transfer resistance was calculated using a simplified Randles model (RΩ)(RctQdl) as shown in the inset of Fig. 4e. The results are summarized in Table S2.† A significant decrease of the charge transfer resistance (Rct) in the presence of Fe impurities is observed for LaNiO3. This denotes the synergistic effects between Fe and the Ni centres that facilitate charge transfer. However, the Rct is lowest at a Fe concentration of 7.5 ppm in KOH, and at higher Fe concentrations, Rct increases, also indicating that the excess of Fe hinders the conductivity.
The chronopotentiometry (CP) measurements were performed on the catalyst in 1 M KOH with 7.5 ppm Fe impurities. This sample retains the activity at 10 mA cm−2 for up to 12 h (Fig. 4f). The high stability is also confirmed by the subsequently obtained TEM image, showing that the crystallinity is preserved (Fig. S4†).
Studying the surface reconstruction using IL-STEM
IL-STEM was used to follow the changes of the same particles on the atomic scale before and after the electrochemical cycles. After cycling LaNiO3 in 1 M KOH with Fe impurity (7.5 ppm), an amorphization of the surface is observed, while the crystallinity of LaNiO3 is preserved when cycled without Fe impurities (Fig. 5a–d). The amorphous layer was further studied as shown in Fig. 5e–h. STEM imaging and EDS analysis revealed that the Fe species are localized in the newly formed amorphous surface with a thickness of about 5 nm. | ||
| Fig. 5 Identical location ADF-STEM micrographs of LaNiO3 (a) before, (b) after OER in 1 M KOH, (c) before, and (d) after OER in 1 M KOH with Fe impurities. When Fe impurities are added to the electrolyte, an amorphous layer is formed (indicated by the red arrows in (d)). The structure and composition of the amorphous (oxy)hydroxide layer formed during OER are analyzed with (e) HAADF STEM (f) ADF STEM and (g) EDS composition maps. Corresponding intensity profiles (h) along the direction indicated by the arrow in (e) of a LaNiO3 cycled in 1 M KOH with Fe impurities. | ||
Identifying the nature of active sites
In situ Raman studies were performed to follow the structure reconstruction and the evolution of active species during OER. The vibration modes of LaNiO3 are presented in Table S3.† The characteristic band around 400 cm−1 corresponds to the vibrational modes of the NiO6 polyhedra.44 Once the catalyst contacts the KOH electrolyte, the band around 800 cm−1 appears, corresponding to the adsorbed hydroxide/oxyhydroxide species on the Au substrate.36,45 This band is observed in both samples and is very prominent during the onset of OER.In situ Raman spectra collected during changing potentials in purified KOH are shown in Fig. 6a. The vibrational mode at 400 cm−1 is preserved at all potentials, although its intensity decreases with the increase in the applied potentials. At a potential of 1.0 V, a broad Raman band appears between 450 cm−1 and 600 cm−1, which resolves into two separate bands at 1.3 V centred around 474 cm−1 and 555 cm−1. They correspond to the bending and stretching vibration modes of Ni–O in Ni-oxyhydroxide species. Due to resonance effects, these two vibrational modes are known to have a high Raman cross-section. From the relative intensities of the two bands (IEg/IA1g = 1.15), they are attributed to the presence of γ-NiOOH.46
 | ||
| Fig. 6 (a) In situ Raman spectra of LaNiO3 collected during the OER process (1.0–1.7 V vs. RHE) in 1 M purified KOH, (b) with the addition of Fe impurities. Comparison of EELS spectra collected for LaNiO3 in different conditions: before OER (black colour), after OER in 1 M purified KOH (green colour), nanoparticles after OER in 1 M KOH with Fe impurities (red colour), and amorphous layer (blue colour) formed after OER in 1 M KOH with Fe impurities. The energy loss region of the (c) O K-edge, (d) Fe L-edge, and (e) La M + Ni-L edge are shown. | ||
The in situ Raman spectra of LaNiO3 measured in the presence of Fe impurities are shown in Fig. 6b. The vibrational mode around 800 cm−1 is also observed here and becomes sharper at potentials between 1.35 and 1.45 V where Ni exhibits its classic redox behaviour (Ni3+ to Ni4+). When the potentials are applied, the vibrational mode at 400 cm−1 gradually disappears, hinting at surface distortion. The bands disappear completely around 1.45 V indicating local amorphization of the structure during OER in the presence of Fe. In addition, the broad Raman bands around 900–1150 cm−1 observed at potentials above 1.35 V, the region for Ni redox behaviour and onset of OER, are attributed to the formation of the active oxygen/superoxide species.47 The most dramatic effect of the Fe impurities is the early formation and change in absolute and relative intensities of the pair of NiOOH bands; both bands (Eg and A1g) are more intense and the relative intensity (IEg/IA1g = 1.41) increases with Fe incorporation, as shown in Fig. S5.† The high Raman cross-section of the NiOOH vibrations limits the amount of information that can be extracted from the Fe contribution with low Fe contents.19
The in situ Raman data were complemented by EELS experiments, which provide higher spatial resolution. HAADF-STEM micrographs of the areas where the EELS spectra were collected are provided in Fig. S6.† An amorphous layer is observed only when the electrolyte contains Fe impurities during OER, which is in good agreement with the IL STEM and Raman spectroscopy observations. Fig. 6c shows the O K-edge. It exhibits three prominent peaks: a pre-peak located around 527 eV related to electronic transitions from O 1s to O 2p orbitals hybridized with Ni 3d, followed by peaks around 534 eV and 540 eV corresponding to the long-range order of the crystal, as well as the density of La 5d and Ni 4s–O bond.35,48,49 The pre-peak changes significantly after OER in the presence of Fe impurities, and the intensity of the O K-edge pre-peak at ∼527 eV is reduced. This indicates that the local coordination and the valence state have changed and that lattice oxygen most likely takes part in the catalysis.35,49 For the amorphous layer, the main peak is shifted to higher energy losses and is at 540 eV. This could indicate the formation of Fe3+ species50 or Ni–O–Fe species since the O-K edges of α- and β-FeOOH and Ni-oxyhydroxide have similar features.51 Only minor changes in the O-K edge are observed for the system after OER in purified KOH.
The La M4,5 and Ni L2,3 edges are known as white lines, and their position and relative intensity value are known to be affected by the oxidation state of the element. The white lines are visible in Fig. 6e and appear for La M5 at an energy loss of 832 eV, while La M4 appears at 849 eV. For Ni, the white lines L3 and L2 possess energy losses of 852 eV and 872 eV. Due to the overlap between the La M4 and the Ni L3 white lines, the interpretation of the data is difficult, but some changes can be observed for the different samples. In the nanoparticle region measured after OER in the presence of Fe impurities, the intensity of the La M5 at 832 eV decreases compared to the M4 edge, but there is no shift in the edge position. This indicates that the oxidation state of La does not change and the La concentration decreases relative to Ni. This is also confirmed by ICP-OES measurements of the electrolyte after OER on LaNiO3 in the presence of Fe impurities where we detect La (0.04 ppm) dissolved into the electrolyte as shown in Table S4.† Since the peak at ∼850 eV contains white line contributions of La M4 and Ni L3, increasing the relative Ni concentration leads to a higher relative intensity of this peak. Similarly, the amorphous layer present after OER in KOH + Fe is also enriched in Ni. The L2,3-edges are caused by electronic excitations from 2p core levels to unoccupied 3d orbitals. This is directly reflected in the 3d electronic states and therefore provides information about the valence state. The peak of the La M4/Ni L3 white line is broadened and shifted to higher energy losses for the amorphous layer. In a first approximation, as the La M5 white line remains at the same energy loss, it is assumed that both are related to a change in the Ni ions. Accordingly, the shift of the Ni L3 white line to higher energy losses in the presence of Fe indicates that Fe promotes and stabilizes higher oxidation states of Ni.52 The broadness of the peaks shows the presence of different oxidation states.53
The Fe L-edge as shown in Fig. 6d displays a very broad peak centred around 708 eV, which becomes very prominent after OER in the presence of Fe impurities in the amorphous layer compared to the nanoparticle region. This is an indication that the amorphous layer is rich in Fe, complementing the EDS observations. The ICP-OES (Table S4†) data shows the reduction of the Fe content in the electrolyte before and after OER. It also confirms that Fe has entered the interface. The broad features could again indicate the presence of amorphous or disordered Fe oxyhydroxides or direct towards a mixed coordination environment with Ni. These observations are consistent with O K-edge and Ni L2,3-edges.
These results show that the origin of activity in LaNiO3 catalyst arises from NiOOH which interacts with Fe species from the electrolyte. This interaction leads to the dissolution of lanthanum and, the formation of oxygen vacancies, which subsequently result in the formation of amorphous and dynamically active oxyhydroxide layers of ca. 5 nm thick. The amorphous layers are identified to be a mixture of Ni-oxyhydroxides, Fe-oxyhydroxides, and a Ni–O–Fe coordinated environment. The presence of Fe not only promotes and stabilizes the higher oxidation state of Ni but also could act as a potential catalytic site.
Elucidating the OER reaction mechanism using pH-dependent studies
In addition, it was studied how the OER activity can be adjusted by varying the pH value. This affects the free energy of adsorbed intermediates, the concentration of OH−, or the surface coverage of the adsorbed *OH and *OOH intermediates.54 From theoretical calculations and O-isotope experiments on Sr-based perovskites, it was discussed that the pH-dependent OER activity experiments indicate the lattice oxygen mechanism.54,55 The OER activities of LaNiO3 were measured in alkaline electrolytes of different pH with (7.5 ppm Fe) and without Fe impurities. A high-purity semiconductor-grade KOH was used as the electrolyte.Cyclic voltammetry measurements show that the surface is activated with increasing pH, whether Fe impurities are present or not but the activation is pronounced in the presence of electrolyte impurities. However, the current density increases tenfold in the presence of Fe impurities (Fig. 7a), while the activation is boosted only threefold in the absence of Fe impurities (Fig. 7b) as the pH of the electrolyte increases from 12.5 to 14. A similar trend is observed for the OER overpotential in electrolytes with different pH values as summarized in Fig. 7c. Thus, both cyclic voltammetry and linear sweep voltammetry measurements show that the OER kinetics of LaNiO3 is pH-dependent and the dependence is particularly pronounced in the presence of Fe impurities, which increase the overall performance via the lattice oxygen mechanism.
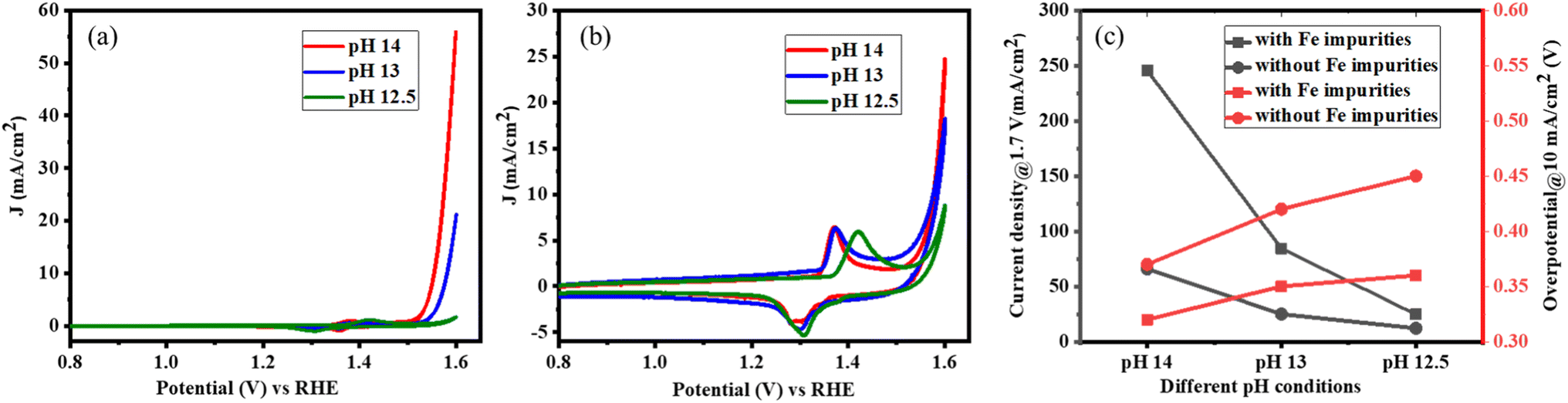 | ||
| Fig. 7 Cyclic voltammetry data after the 50th cycle was obtained for LaNiO3 under different pH conditions: (a) with Fe impurities, (b) without Fe impurities, (c) overview of the overpotential at 10 mA cm−2 and current density at 1.7 V vs. RHE at different pH conditions with and without Fe impurities in the electrolyte. | ||
As Fig. 8 depicts, when LaNiO3 comes in contact with the electrolyte, an electrochemical interface is established where the active Fe species interact with the perovskite surface resulting in surface alteration which includes La dissolution, formation of oxygen vacancies and the formation of an amorphous layer. However, the rate of ion dissolution and redeposition reaches an equilibrium state25 and establishes a stable interface over longer cycles. Additionally, the stable dynamic interface offers two active sites for OER, promoting the O2 production rate via the lattice oxygen mechanism.
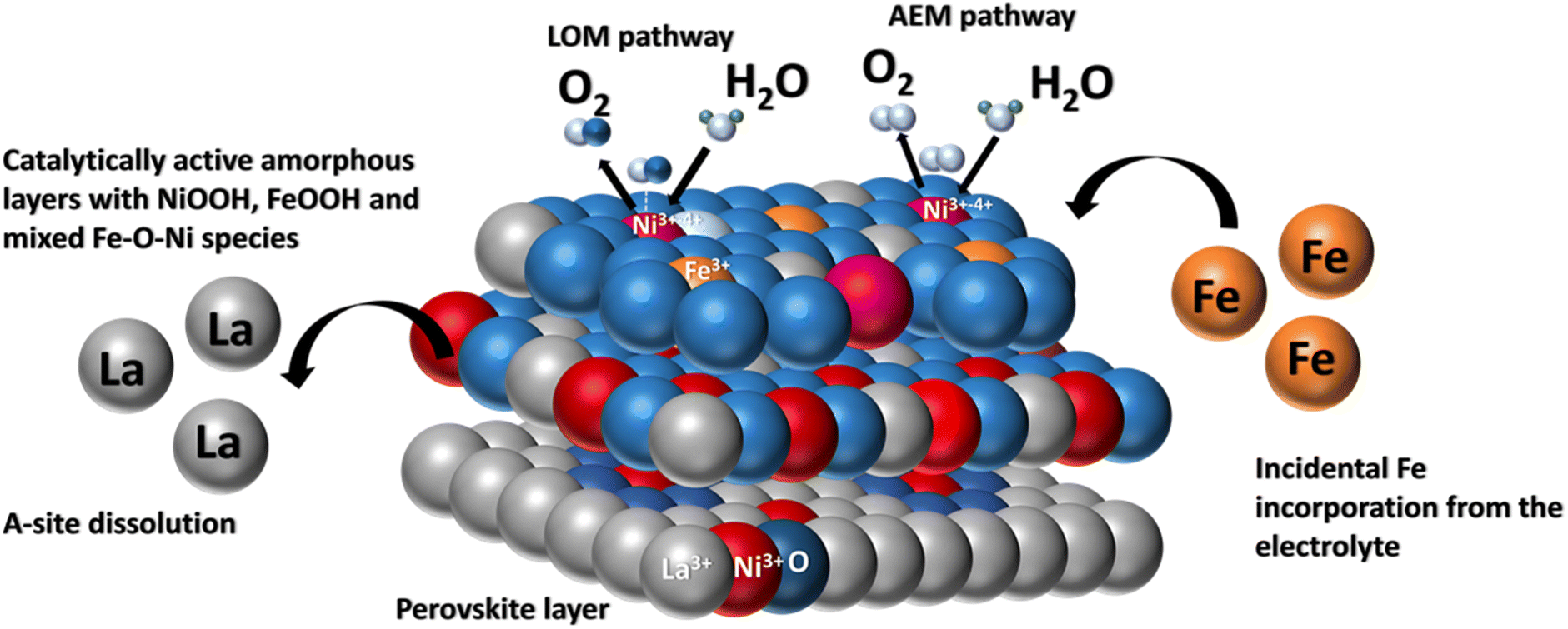 | ||
| Fig. 8 Pictorial representation of incidental Fe incorporation dynamics on a LaNiO3 system during OER. | ||
Conclusions
In summary, a combination of controlled perovskite synthesis, detailed structure analysis, atomic scale microscopy imaging, EELS, and in situ Raman spectroscopy demonstrated how the catalyst surface evolves and forms dynamic active sites during OER in the presence of Fe impurities in the electrolyte. Fe impurities in the electrolyte have been shown to favour the kinetics to some extent and, moreover, can hinder the OER activity of the catalyst. The Fe species from the electrolyte lead to significant surface modulation of the catalyst. They not only stabilize the higher oxidation state of Ni but also serve as a potential active site for OER. The presence of Fe impurity paves the way for the lattice oxygen mechanism boosting the O2 production rate. This work leads to a deeper mechanistic understanding of the dynamics of Fe incorporation and helps in the development of catalysts with better performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
HC and CW acknowledge the funding from the International Max Planck Research School for Interface Controlled Materials for Energy Conversion (IMPRS-SurMat) and Max Planck Society for the support. HT thanks the Volkswagen Foundation (96_742) and the Carbon2Chem consortium funded by the Bundesministerium für Bildung und Forschung (BMBF) and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) Projektnummer 388390466–TRR 247 within the Collaborative Research Centre/Transregio 247 “Heterogeneous Oxidation Catalysis in the Liquid Phase” for the funding. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at PETRA III, and we would like to thank PhD Alba San Jose Mendez for assistance in using beamline P02.1 (beamtime was allocated for proposal I-20210517). We would like to express our gratitude towards H. Petersen, P. Sharma, I. Kappel, and T. H. Ulucan for their support during in situ XRD beamtimes. We also thank H. Petersen, J. Ternieden, B. Florian, and E. Budiyanto for supporting microstructure analysis, XRD, ICP-OES, and electrocatalytic measurements.References
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef.
- B. M. Hunter, H. B. Gray and A. M. Muller, Chem. Rev., 2016, 116, 14120–14136 CrossRef PubMed.
- J. Rogelj, M. Schaeffer, M. Meinshausen, R. Knutti, J. Alcamo, K. Riahi and W. Hare, Environ. Res. Lett., 2015, 10, 105007 CrossRef.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef.
- S. Park, Y. Shao, J. Liu and Y. Wang, Energy Environ. Sci., 2012, 5, 9331–9344 RSC.
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Nat. Commun., 2016, 7, 11053 CrossRef PubMed.
- F. Song, L. Bai, A. Moysiadou, S. Lee, C. Hu, L. Liardet and X. Hu, J. Am. Chem. Soc., 2018, 140, 7748–7759 CrossRef PubMed.
- H. Wang, J. Qi, N. Yang, W. Cui, J. Wang, Q. Li, Q. Zhang, X. Yu, L. Gu, J. Li, R. Yu, K. Huang, S. Song, S. Feng and D. Wang, Angew Chem. Int. Ed. Engl., 2020, 59, 19691–19695 CrossRef PubMed.
- K. A. Stoerzinger, M. Risch, J. Suntivich, W. M. Lü, J. Zhou, M. D. Biegalski, H. M. Christen, Ariando, T. Venkatesan and Y. Shao-Horn, Energy Environ. Sci., 2013, 6, 1582–1588 RSC.
- J. Du, T. Zhang, F. Cheng, W. Chu, Z. Wu and J. Chen, Inorg. Chem., 2014, 53, 9106–9114 CrossRef PubMed.
- Z. Yan, H. Sun, X. Chen, X. Fu, C. Chen, F. Cheng and J. Chen, Nano Res., 2018, 11, 3282–3293 CrossRef.
- A. Grimaud, K. J. May, C. E. Carlton, Y. L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 2439 CrossRef PubMed.
- K. J. May, C. E. Carlton, K. A. Stoerzinger, M. Risch, J. Suntivich, Y.-L. Lee, A. Grimaud and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 3264–3270 CrossRef.
- E. Fabbri, M. Nachtegaal, T. Binninger, X. Cheng, B. J. Kim, J. Durst, F. Bozza, T. Graule, R. Schaublin, L. Wiles, M. Pertoso, N. Danilovic, K. E. Ayers and T. J. Schmidt, Nat. Mater., 2017, 16, 925–931 CrossRef PubMed.
- J.-W. Zhao, Z.-X. Shi, C.-F. Li, Q. Ren and G.-R. Li, ACS Mater. Lett., 2021, 3, 721–737 CrossRef.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y. L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef PubMed.
- C. Yang, M. Batuk, Q. Jacquet, G. Rousse, W. Yin, L. Zhang, J. Hadermann, A. M. Abakumov, G. Cibin, A. Chadwick, J.-M. Tarascon and A. Grimaud, ACS Energy Lett., 2018, 3, 2884–2890 CrossRef.
- J. S. Yoo, X. Rong, Y. Liu and A. M. Kolpak, ACS Catal., 2018, 8, 4628–4636 CrossRef.
- I. Spanos, J. Masa, A. Zeradjanin and R. Schlögl, Catal. Lett., 2020, 151, 1843–1856 CrossRef.
- S. Anantharaj, S. Kundu and S. Noda, Nano Energy, 2021, 80, 105514 CrossRef.
- R. Subbaraman, N. Danilovic, P. P. Lopes, D. Tripkovic, D. Strmcnik, V. R. Stamenkovic and N. M. Markovic, J. Phys. Chem. C, 2012, 116, 22231–22237 CrossRef.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef.
- H. Li, Y. Chen, J. Ge, X. Liu, A. C. Fisher, M. P. Sherburne, J. W. Ager and Z. J. Xu, JACS Au, 2021, 1, 108–115 CrossRef.
- R. Zhang, P. E. Pearce, Y. Duan, N. Dubouis, T. Marchandier and A. Grimaud, Chem. Mater., 2019, 31, 8248–8259 CrossRef.
- D. Y. Chung, P. P. Lopes, P. F. B. D. Martins, H. He, T. Kawaguchi, P. Zapol, H. You, D. Tripkovic, D. Strmcnik, Y. Zhu, S. Seifert, S. Lee, V. R. Stamenkovic and N. M. Markovic, Nat. Energy, 2020, 5, 222–230 CrossRef.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M. J. Cheng, D. Sokaras, T. C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Norskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef PubMed.
- S. Lee, L. Bai and X. Hu, Angew Chem. Int. Ed. Engl., 2020, 59, 8072–8077 CrossRef.
- A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef.
- P. Scardi and M. Leoni, J. Appl. Crystallogr., 2006, 39, 24–31 CrossRef.
- P. Scardi, Z. Kristallogr. - Cryst. Mater., 2002, 217, 420–421 CrossRef.
- P. Juhas, T. Davis, C. L. Farrow and S. J. L. Billinge, J. Appl. Crystallogr., 2013, 46, 560–566 CrossRef.
- X. Yang, P. Juhas, C. L. Farrow and S. J. L. Billinge, arXiv, 2015, preprint, arXiv:1402.3163v3, DOI:10.48550/arXiv.1402.3163.
- M. Vega-Paredes, R. Aymerich-Armengol, D. A. Esteban, S. Marti-Sanchez, S. Bals, C. Scheu and A. G. Manjon, ACS Nano, 2023, 17, 16943–16951 CrossRef.
- M. Vega-Paredes, C. Scheu and R. Aymerich-Armengol, ACS Appl. Mater. Interfaces, 2023, 15, 46895–46901 CrossRef.
- P. P. Lopes, D. Y. Chung, X. Rui, H. Zheng, H. He, P. F. B. D. Martins, D. Strmcnik, V. R. Stamenkovic, P. Zapol, J. F. Mitchell, R. F. Klie and N. M. Markovic, J. Am. Chem. Soc., 2021, 143, 2741–2750 CrossRef PubMed.
- E. Budiyanto, S. Salamon, Y. Wang, H. Wende and H. Tuysuz, JACS Au, 2022, 2, 697–710 CrossRef PubMed.
- P. Bera, Int. J. Self-Propag. High-Temp. Synth., 2019, 28, 77–109 CrossRef.
- S. J. Billinge and M. G. Kanatzidis, Chem. Commun., 2004, 749–760, 10.1039/b309577k.
- B. Ingham, Crystallogr. Rev., 2015, 21, 229–303 CrossRef.
- C. C. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef PubMed.
- G. H. Moon, M. Yu, C. K. Chan and H. Tuysuz, Angew Chem. Int. Ed. Engl., 2019, 58, 3491–3495 CrossRef.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef PubMed.
- A. Schober, J. Fowlie, M. Guennou, M. C. Weber, H. Zhao, J. Íñiguez, M. Gibert, J.-M. Triscone and J. Kreisel, APL Mater., 2020, 8, 061102 CrossRef.
- O. Diaz-Morales, F. Calle-Vallejo, C. de Munck and M. T. M. Koper, Chem. Sci., 2013, 4, 2269–2696 RSC.
- B. S. Yeo and A. T. Bell, J. Phys. Chem. C, 2012, 116, 8394–8400 CrossRef.
- B. J. Trzesniewski, O. Diaz-Morales, D. A. Vermaas, A. Longo, W. Bras, M. T. Koper and W. A. Smith, J. Am. Chem. Soc., 2015, 137, 15112–15121 CrossRef.
- N. Biškup, J. Salafranca, V. Mehta, M. P. Oxley, Y. Suzuki, S. J. Pennycook, S. T. Pantelides and M. Varela, Phys. Rev. Lett., 2014, 112, 087202 CrossRef.
- N. Gauquelin, E. Benckiser, M. K. Kinyanjui, M. Wu, Y. Lu, G. Christiani, G. Logvenov, H. U. Habermeier, U. Kaiser, B. Keimer and G. A. Botton, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 195140 CrossRef.
- S.-Y. Chen, A. Gloter, A. Zobelli, L. Wang, C.-H. Chen and C. Colliex, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 104103 CrossRef.
- X. Ren, C. Wei, Y. Sun, X. Liu, F. Meng, X. Meng, S. Sun, S. Xi, Y. Du, Z. Bi, G. Shang, A. C. Fisher, L. Gu and Z. J. Xu, Adv. Mater., 2020, 32, e2001292 CrossRef PubMed.
- N. Li, D. K. Bediako, R. G. Hadt, D. Hayes, T. J. Kempa, F. von Cube, D. C. Bell, L. X. Chen and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1486–1491 CrossRef.
- Y. Dou, C.-T. He, L. Zhang, M. Al-Mamun, H. Guo, W. Zhang, Q. Xia, J. Xu, L. Jiang, Y. Wang, P. Liu, X.-M. Chen, H. Yin and H. Zhao, Cell Rep. Phys. Sci., 2020, 1, 100077 CrossRef.
- Y. Pan, X. Xu, Y. Zhong, L. Ge, Y. Chen, J. M. Veder, D. Guan, R. O'Hayre, M. Li, G. Wang, H. Wang, W. Zhou and Z. Shao, Nat. Commun., 2020, 11, 2002 CrossRef.
- A. K. Tomar, U. N. Pan, N. H. Kim and J. H. Lee, ACS Energy Lett., 2022, 8, 565–573 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta06733e |
| This journal is © The Royal Society of Chemistry 2024 |