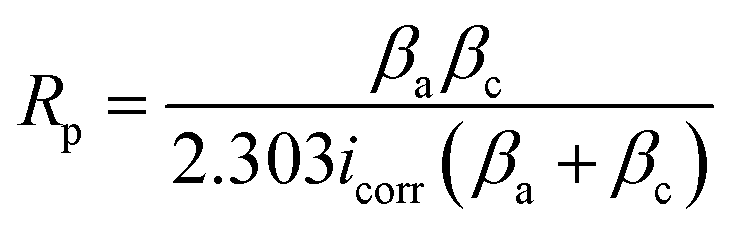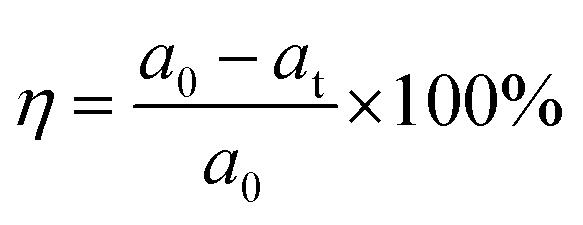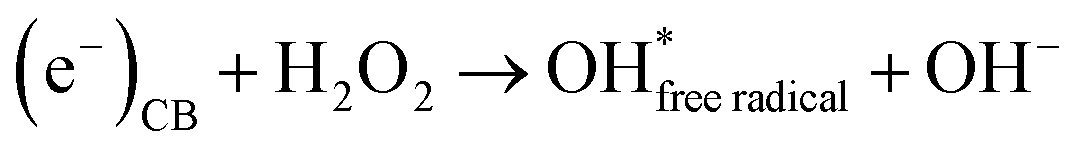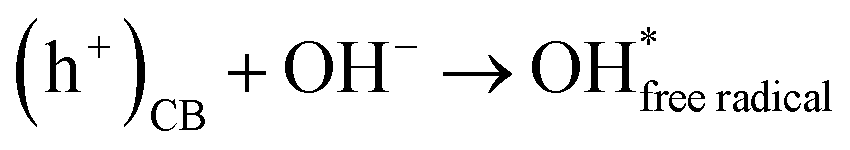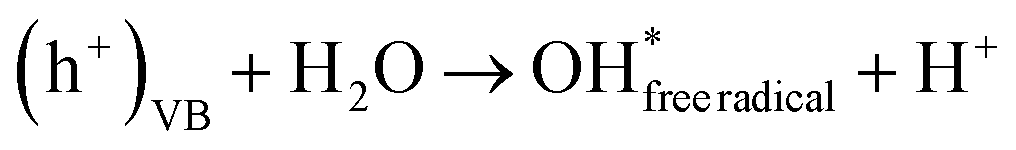DOI:
10.1039/D3TA06840D
(Paper)
J. Mater. Chem. A, 2024,
12, 3411-3433
Modulating chelation with pH sensitivity for controlled structural defects and enhanced electrochemical and photocatalytic activities of LDH films†
Received
8th November 2023
, Accepted 19th December 2023
First published on 11th January 2024
Abstract
The convergence of organic compounds and their incorporation into inorganic layers paves the way for pioneering organic–inorganic hybrid materials with finely tuned, precise functional properties, promising a new era of advanced applications that push the boundaries of current technology. This study employs a pH-responsive chelation approach to control defects in MgAl-LDH, aiming to enhance its electrochemical and photocatalytic properties. This involves the synthesis of MgAl-LDH on plasma-assisted oxidized substrates, followed by the sequential introduction of [EDTA]4− and [Zn(EDTA)]2− anions into the LDH galleries via an anion exchange route. The chelation of EDTA with the metallic ions is pH-dependent, where a higher pH value encourages the inclusion of [EDTA]4− anions, supported by the stability of LDH flakes, while a lower pH value facilitates the introduction of [Zn(EDTA)]2− anions. The results show that the integration of [Zn(EDTA)]2− anions leads to a more compact structure and a reduction in the size of LDH flakes, effectively sealing existing defects within the hybrid material. When exposed to a 3.5% NaCl solution, the LDH–EDTA–Zn sample exhibited superior electrochemical stability, characterized by a lower corrosion current density (2.12 × 10−9 A cm−2) and higher total polarization resistance (2.73 × 107 Ω cm2). Moreover, the LDH–EDTA–Zn sample demonstrated exceptional photocatalytic efficiency, effectively decomposing rhodamine dye in an aqueous solution, attaining an impressive photocatalytic efficiency of 99.7% within just 25 minutes, while maintaining stability throughout five consecutive cycles, surpassing the performance of previously reported LDH-based catalysts. The interaction mechanism between the chelated anions and the LDH surface was elucidated by applying density functional theory. This research presents a promising approach for developing advanced organic–inorganic hybrid materials with exceptional electrochemical and photocatalytic performance.
1. Introduction
Layered double hydroxides (LDHs), versatile and environmentally friendly materials, have gained significant attention in research and applications, owing to their multifunctional properties, which encompass their effectiveness as photocatalysts and their utility as corrosion-resistant coatings for valuable metals, attributes that arise from their adjustable band gap, exchangeable interlayer anions, substantial specific surface area, and impressive adsorption capacity, making them a promising choice for a wide range of eco-friendly solutions and innovative technologies.1–6 LDHs are composed of divalent metal ions (M2+) and trivalent metal ions (M3+), as well as anions that aid in charge balancing. These anions, together with water molecules, are accommodated between the layers of the LDH structure.7–10 The negatively charged anions residing in these interlayers can be replaced by specific anionic corrosion inhibitors, resulting in the formation of LDH films that incorporate these inhibitors.11–14 Extensive research has been conducted in the field of corrosion protection, investigating numerous inhibitors, such as 2-mercaptobenzothiazole,15 8-hydroxyquinoline,16 molybdate,17 benzoate,18 vanadate,19 aspartic acid,20 and carboxylate anions.21 For example, Jiang et al.18 reported the existence of a self-repairing ability combined with active corrosion protection for the AZ31 magnesium alloy. They achieved this by enhancing the LDH film, which had been intercalated with molybdate ions, through the use of fluoroalkyl silane and the infusion of perfluoropolyether treatments. Zeng et al.22 created a nanoplatelet-structured MgAl–MoO42− LDH coating, which effectively prevented corrosion on the AZ31 alloy, demonstrating its promising potential for corrosion protection in practical applications. In summary, the addition of corrosion inhibitor anions into the LDH interlayer of magnesium alloys notably enhances their corrosion resistance, marking a substantial advancement in corrosion prevention.
On the other hand, plasma electrolytic oxidation (PEO) is an emerging technique laying the groundwork for designing inorganic layers, providing structural protection against corrosion23 and wear,24 and enabling diverse functional applications such as in photocatalysis,25 electrocatalysis,26 and biomedicine.27 However, PEO can sometimes lead to porous coatings, increasing vulnerability to corrosion in extreme environments.28 Despite its potential drawbacks in terms of corrosion protection, porosity can actually be considered an advantageous feature for catalytic activity, as its high porosity offers a significant increase in surface area, promising enhanced catalytic activation, efficient dye impregnation, and accelerated degradation rates. Recently, there has been a considerable increase in research focusing on merging LDH coatings with PEO coatings. This method has gained interest because LDHs have great stability in keeping a flake-like structure in their films as well as the capacity to release corrosion inhibitors, as necessary.29 Numerous research groups have emphasized the potential use of LDH films as efficient sealants for the porous structure present in PEO coatings applied on lightweight metal alloys. For instance, Kuznetsov and his team found that effectively growing LDH films on an AA2024 aluminum alloy improved the alloy's corrosion inhibition properties. This improvement was most notable when they introduced vanadate anions into the LDH film.19 Similarly, another group reported that the synthesis of a Zn–Al LDH layer on the surface of the oxide layers considerably increased the corrosion resistance of a 2024 aluminum alloy.30 However, the presence of structural flaws in the LDH film, positioned perpendicularly to the coating surface, creates openings for corrosive substances to compromise both the coating and the substrate beneath it. This indicates that establishing a dependable and durable corrosion protection system cannot be simply achieved by using an LDH layer.31
Ethylenediaminetetraacetic acid (EDTA) is indeed a widely used chelating agent due to its versatile chemical properties and its ability to form stable complexes with metal ions.32 In recent years, EDTA intercalated in LDH has been increasingly utilized as a supplementary substance for treating wastewater with heavy metal content and as a cleansing solution for remediating soil contaminated by heavy metals, owing to its wealth of functional groups, notably carboxyl and amino.33–35 However, the unexplored potential of using EDTA as a post-treatment inside LDH galleries for corrosion inhibition and photocatalysis remains a fascinating path for future studies. This study aims to investigate a novel technique wherein EDTA anions are intercalated into MgAl LDH layers on a PEO-coated AZ31 Mg alloy, aiming to create a multifunctional material that facilitates both corrosion protection and photocatalytic degradation of organic dyes. Herein, we conducted a comparative analysis of surface modification techniques for MgAl LDH, specifically using EDTA and the Zn–EDTA complex to determine which method offers the most effective corrosion protection and photocatalytic degradation when subjected to a hydrothermal approach. The formation of the defective layer on the AZ31 magnesium alloy is crucial in the production of the LDH–EDTA complex, leading to the creation of hybrid organic–inorganic composites with improved electrochemical properties. The study's findings show that the incorporation of [EDTA]4− anions into LDH galleries, favored at higher pH levels, leads to the formation of smaller and denser hexagonal flakes. These flakes uniformly cover the entire surface of the porous layer, providing effective corrosion protection. On the other hand, at lower pH values, the introduction of [Zn(EDTA)]2− anion complexes into the LDH layers leads to the dissolution of metals originally present within the LDH layers, resulting in the formation of defects. Interestingly, these defects can be advantageous for photocatalytic activity as the increased porosity resulting from these defects offers a larger surface area, enhancing the potential for photocatalytic activation and accelerating the degradation rate of substances. As a result, LDH–EDTA–Zn specimens exhibit a modest reduction in their corrosion prevention capabilities compared to LDH–EDTA. However, they demonstrate a significant improvement in their capacity for photocatalysis. Our research suggests an effective strategy for achieving a harmonious balance between structural and functional characteristics, involving moderate corrosion inhibition alongside superior photocatalytic performance through the surface modification of LDH. This makes it a suitable choice for widespread use in industrial applications. An in-depth electrochemical study is presented, investigating the corrosion protection and photocatalytic degradation mechanisms for the prepared samples, complemented by a detailed explanation of the interaction between the EDTA–Zn complex and the LDH surface through DFT calculations.
2. Experimental methods
2.1 Fabrication of the inorganic defective layer
An AZ31 magnesium alloy with the following chemical composition served as the substrate: 3.2% Al, 0.8 wt% Zn, 0.4 wt% Mn, 0.1 wt% Sn, 0.01 wt% Si, 0.003% Fe, 0.002% Ni, 0.0001 wt% Cu, and balanced Mg. The Mg alloy is cut into 20 × 20 × 3 mm3 pieces, and the pieces are then polished with silicon abrasive sheets of up to 2000 grit. The alloy is polished and then dried after being cleaned with ethanol. A current density of 100 mA cm−2 is delivered to the PEO coatings for 5 minutes at a frequency of 60 Hz. A water bath maintained the room temperature while the electrolyte used comprised 7 g per L KOH and 4 g per L NaAlO2, consistently blended. The result is designated as the PEO product.
2.2 Synthesis of Mg–Al LDH film
Using a conventional hydrothermal method, a PEO specimen is coated with Mg–Al LDH. In 300 mL deionized water, 0.06 M Mg(NO3)·6H2O and 0.03 M Al(NO3)·9H2O are dissolved and agitated for 30 minutes. To keep the pH at 10, 1 M NaOH was gradually added. The PEO specimen is placed vertically in a Teflon-lined hydrothermal container, and the solution is heated for 12 hours at 125 °C. The LDH-coated specimen is washed with deionized water and air-dried after cooling, yielding the coated substance known as LDH.
2.3 Synthesis of LDH–EDTA and LDH–EDTA–Zn films
The LDH samples are put in a 100 mL aqueous solution containing 0.1 M ethylenediaminetetraacetic acid (EDTA). The pH of the solution is adjusted to 9 by adding 0.1 M NaOH. The resulting solution is then put in a reactor and kept at a temperature of 65 °C for 12 hours. Following a certain interval, the acquired sample is thoroughly cleaned with ethanol and allowed to air-dry at room temperature. The resultant substance is identified as LDH–EDTA. The LDH–EDTA–Zn film is created by dissolving 0.1 M EDTA in 100 mL of an aqueous solution, referred to as solution A. Simultaneously, another 100 mL of an aqueous solution is employed to dissolve 0.1 M Zn(NO3)2·2H2O, denoted as solution B. Following this, with the assistance of a magnetic stirrer, solution B is slowly added drop by drop to solution A. Initially, the pH of the solution is set to 9 using NaOH, but this does not lead to deposition on the LDH surface. To achieve a uniform deposition on the LDH surface, the final pH is carefully fine-tuned to 4. Following that, the produced solution is placed in an autoclave and exposed to a temperature of 65 °C for 12 hours. Thereafter, the resulting sample is carefully washed with ethanol and allowed to dry naturally at room temperature.
2.4 Microstructural and compositional analysis
A scanning electron microscope (SEM-HITACHI, PT-S8200) equipped with a dispersive X-ray spectroscopy (EDS) instrument for elemental analysis is employed to evaluate the surface and cross-sectional characteristics. X-ray diffraction (XRD) patterns are obtained using a RIGAKU D-MAX 2500 apparatus to evaluate chemical composition. The XRD scans had a step size of 0.01° and a scanned cover range of 5 to 80°. The chemical composition of the surfaces is thoroughly examined using X-ray photoelectron spectroscopy (XPS) on a K-alpha+ ESCALAB 220i instrument. Furthermore, the samples' infrared spectra are obtained via FTIR-ATR spectroscopy on PerkinElmer Spectrum 100 equipment, with wavenumbers ranging from 500 to 4000 cm−1.
2.5 Electrochemical evaluation
The electrochemical performance of the materials was evaluated through a series of electrochemical assessments, including potentiodynamic polarization (PDP) and electrochemical impedance spectroscopy (EIS). A platinum (Pt) plate was employed as the counter electrode, the sample with a 1 cm2 surface area served as the working electrode, and a silver/silver chloride (Ag/AgCl) electrode acted as the reference electrode. To stabilize the open circuit potential, the samples were immersed in a 3.5% NaCl solution for 5 hours, followed by PDP testing in which a voltage range of −250 mV to +400 mV was applied in comparison to the open circuit potential (OCP) at a rate of 1 mV s−1. In parallel, EIS measurements are taken across frequencies ranging from 106 Hz to 0.1 Hz, using a sinusoidal voltage of 10 mV rms. For data reliability, each test is conducted a minimum of three times.
2.6 Photocatalytic evaluations
The photocatalytic properties of PEO, LDH, LDH–EDTA, and LDH–EDTA–Zn samples are examined at room temperature by their ability to degrade rhodamine B (RhB) molecules. The samples (with a surface area of 400 mm2) are immersed in a 40 mL solution containing 20 mg L−1 RhB during the experiment. The mechanism of breakdown is initiated with the addition of hydrogen peroxide (H2O2). Before exposing the mixture to light, it is kept in the dark and stirred for 30 minutes. This allowed the RhB molecules to establish an equilibrium between being adsorbed onto and desorbed from the catalyst surface. For photocatalytic degradation, a UV-light source (Ultra Vitalux OSRAM lamp, 300 W) is utilized to illuminate the dye within the glass beaker for 50 minutes, along with 0.2 mL of H2O2. The absorbance peak intensities of the RhB solution are assessed using a UV-vis spectrometer (Agilent Cary 5000) and a 2 mL RHB solution is collected every 10 minutes. The dye degradation is estimated by reducing the absorbance peak intensities of the individual dye solution. Each measurement is reproduced at least 3 times under similar experimental conditions to ensure reliability. The UV-vis absorbance is measured across a wavelength range of 300 nm to 800 nm. The band gap of the material is calculated using Tauc's plot equation: αhν = A(h − Eg)n. In this equation, α represents the absorption coefficient, h is Planck's constant, ν stands for the light source frequency, A is the optical constant, and Eg represents the optical band gap. For materials and spectra with direct transmission mode, the value of n is set to 1/2.36
Furthermore, to investigate the initial reactive species involved in the degradation of rhodamine by the advanced catalyst LDH–EDTA–Zn, scavengers such as 10 mM isopropanol (IPA) and 10 mM benzoquinone (BQ) are employed to capture hydroxyl radicals (OH˙) and superoxide radicals (O2˙) in rhodamine solution. Moreover, the stability and recyclability of the premium photocatalyst for RhB degradation are assessed through recycling investigations using the LDH–EDTA–Zn catalyst with a consistent catalyst-to-dye ratio. The LDH–EDTA–Zn specimens are subjected to five cycles throughout the study. To ensure the avoidance of contamination, each specimen undergoes thorough cleaning after each degradation phase, involving the use of ethanol and deionized water, followed by a drying stage.
2.7 Theoretical calculations
We conducted a comprehensive analysis of the EDTA–Zn complex's structure using Gaussian 09 W, which involved geometry optimization and computational investigations. For these calculations, we utilized density functional theory (DFT) and the widely used B3LYP functional developed by Becke, incorporating three parameters and Lee–Yang–Parr for accuracy.37 Furthermore, in order to acquire a comprehensive understanding of the electronic attributes and bonding characteristics inherent to the EDTA–Zn complex structure, a series of theoretical calculations are undertaken. These calculations encompassed various key parameters such as the total density of states (TDOS), projected densities of states (PDOS), molecular electrostatic potential (MEP), and electron localization function (ELF), as well as the reduced density gradient (RDG) and its corresponding scatter plot. These computations are conducted employing specialized software tools, namely multiwfn and visual molecular dynamics (VMD).38 Materials Studio 2020 is used to study the charge transfer behavior and adsorption configuration of Zn–EDTA on Mg–Al LDH layers, employing the DMOL3 classical simulation engine. The LDH model is constructed with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Mg:Al ratio, and Grimme's custom method for dispersion-corrected density functional theory (DFT-D) was applied. The simulation used the Perdew–Burke–Ernzerhof (PBE) functional under the generalized gradient approximation (GGA) with fine quality, based on the following points: (1) an energy tolerance of 10−5 eV, (2) a displacement of 0.005 Å, and (3) a force of 0.05 eV Å−1. The cut-off energy is determined to be 340 eV, along with 3 × 3 × 1 k-points geometric optimization. The Gaussian smearing width is set to be 0.2 eV.
:1 Mg:Al ratio, and Grimme's custom method for dispersion-corrected density functional theory (DFT-D) was applied. The simulation used the Perdew–Burke–Ernzerhof (PBE) functional under the generalized gradient approximation (GGA) with fine quality, based on the following points: (1) an energy tolerance of 10−5 eV, (2) a displacement of 0.005 Å, and (3) a force of 0.05 eV Å−1. The cut-off energy is determined to be 340 eV, along with 3 × 3 × 1 k-points geometric optimization. The Gaussian smearing width is set to be 0.2 eV.
3. Results and discussion
3.1 Morphological analysis of the coating
Fig. 1 depicts the surface morphologies of the PEO, LDH, LDH–EDTA, and LDH–EDTA–Zn samples, along with magnified images and EDS analyses. Fig. 1(a) display a range of defects and micro-pores on the surface of the PEO sample. A closer look at the magnified image in Fig. 1(b) emphasizes these characteristics even more. The imperfections and pores are an immediate result of the discharge process's gas development, combined with the strong intensity of the plasma during the coating process.4Fig. 1(c) exhibit the EDS spectrum that identifies magnesium (Mg), aluminum (Al), and oxygen (O) as the PEO coating's main components. Additionally, small amounts of potassium (K) and sodium (Na) have been detected in the coating. The comparatively weak signals in the spectrum assigned to the elements Na and K indicate that these substances are not major components of the PEO coating. A PEO coating's surface includes numerous holes that allow corrosive ions to slowly deteriorate the underlying substrate by making their way through the coating and into the substrate. In response to this concern, an LDH film is created using hydrothermal synthesis techniques, as shown in Fig. 1(d) and with a more detailed view in Fig. 1(e). This film, which is made up of linked hexagonal nanoflakes, provides a strong and effective protective layer over the PEO-coated surface, serving as a reliable barrier against potential damage. The EDX analysis demonstrated in Fig. 1(f) that the LDH coating is made up of components including Mg, Al, O, and N. These components are remarkably similar to those found in the PEO coating. Particularly, there is a higher presence of O, while the amounts of Mg and Al are reduced. This could potentially be attributed to the dissolution of the PEO coating at elevated temperatures. Additionally, a small quantity of nitrogen is detected, indicating the inclusion of nitrate within the LDH layers. An obvious change in the structure of the LDH–EDTA film has been seen after immersing the LDH film in an EDTA solution, as can be seen in Fig. 1(g). Further inspection reveals that the LDH–EDTA film is more closely packed and has thinner flakes than the LDH film, as seen in the enlarged image in Fig. 1(f). This change in flake size is attributed to fragmentation, a phenomenon occurring during the initial rapid anion exchange stages.39 The presence of carbon relative to the main components (Mg, Al, and O) in the EDS analysis of the LDH–EDTA film shown in Fig. 1(i) demonstrates that the anion-exchange method used to successfully intercalate EDTA anions into the interlayer galleries of the LDH structure was effective. On the other hand, utilizing the Zn–EDTA complex to alter LDH (creating the LDH–EDTA–Zn film) has the dual effect of lowering the micro-pore dimensions inside the LDH film as well as compacting the surface, which in consequence reduced flake size (Fig. 1(j)). The magnified image (Fig. 1(k)) revealed more compact surfaces of LDH flakes, effectively filling the gaps between them. The strong and intricate bond formed by [Zn(EDTA)]2−, a complex of EDTA and Zn2+, played a vital role. This bond is established prior to the intercalation process. Consequently, the nitrate anions present in the initial LDH interlayer were exchanged with [Zn(EDTA)]2− complexes from the solution.35 The LDH–EDTA–Zn EDS analysis shown in Fig. 1(l) indicated the identification of Zn and C elements in addition to the major components. These results explained the successful inclusion of a complex made of EDTA and Zn2+ into the layers of LDH, which caused a decrease in the amounts of other major components.
 |
| | Fig. 1 Surface morphologies along with corresponding magnified images and EDS analysis for (a–c) PEO, (d–f) LDH, (g–i) LDH–EDTA, and (j–l) LDH–EDTA–Zn, respectively. The images exhibit distinctive micro-pores and flake-like structures, highlighting the transformation of the surface after successive LDH-based treatments. EDS analysis provides insight into elemental composition changes. | |
The elemental mappings depicted in Fig. 2 illustrate the distribution of elements on the surface of PEO, LDH, LDH–EDTA, and LDH–EDTA–Zn respectively. The findings indicate that both the underlying substrate and the electrolyte played active roles in the plasma chemical oxidation events, enabling the formation of these coatings. Fig. 2(a), which shows a balanced distribution of all the different elements, clearly displays the porous nature of the PEO film. After the deposition and subsequent application of LDH, there is a substantial increase in the concentrations of Mg, Al, and O, with only minimal traces of nitrogen detected. However, as clearly shown in Fig. 2(b), this LDH material efficiently covers up any pre-existing flaws or imperfections in the first PEO coating. Upon conducting surface treatment on LDH, the noticeable increase in oxygen content was accompanied by the emergence of additional C and Zn content, as depicted in Fig. 2(c) and (d). These alterations played a highly effective role in sealing the defects present in LDH flakes.
 |
| | Fig. 2 Layered images and elemental mapping on the surface of (a) PEO, (b) LDH, (c) LDH–Zn and (d) LDH–EDTA–Zn, respectively. Elements such as O, Mg, Al, C, N, Na, K, and Zn are identified throughout the entire surface. The scale bar for the layered image of all samples is 5 μm. | |
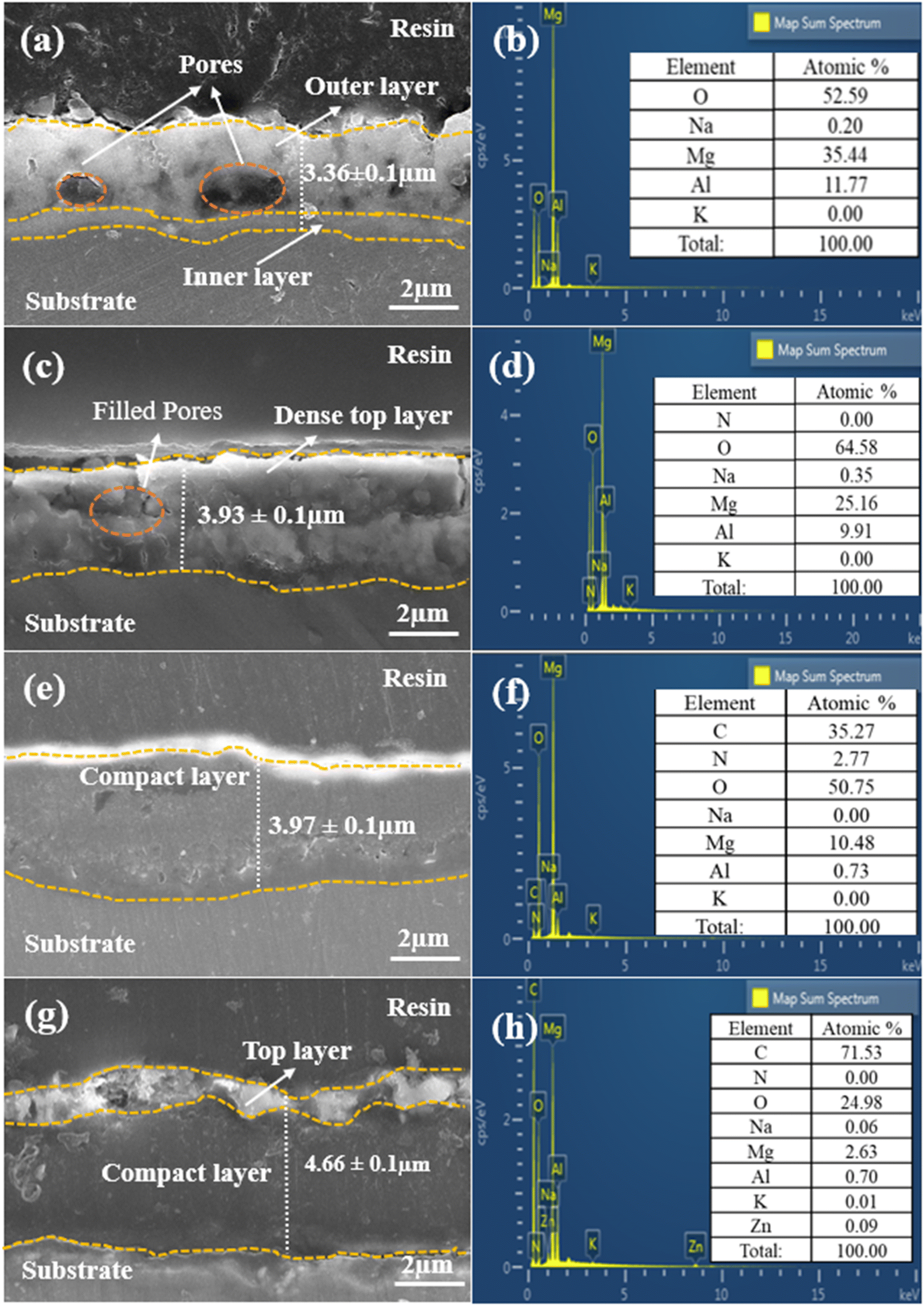
Fig. 3 presents a comprehensive view of the cross-sectional images of all prepared samples, utilizing EDS layered images and elemental analysis to provide a detailed understanding of their microstructures and morphological characteristics. In Fig. 3(a), we can observe the outstanding adhesion strength between the PEO film and the AZ31 Mg alloy substrate, with distinct inner compact and outer porous layers clearly evident. The average thickness of the PEO film, measured at four different points, is 3.36 ± 0.1 μm. The porous structure within the PEO film, represented by the circles in the image, is the result of a series of plasma discharges that create an interconnected network of imperfections within the film. The elemental analysis results in Fig. 3(b) reveal the presence of elements such as O, Mg, and Al in the PEO film. However, potassium (K) and sodium (Na) are detected in the film in trace amounts, indicating their minimal concentration. The cross-sectional analysis of LDH, as depicted in Fig. 3(c), reveals that subjecting the PEO film to hydrothermal conditions led to the growth of LDH flakes on the surface of the film. These flakes created a dense top layer, filling the gaps in the outer layer and raising the overall average coating thickness to 3.93 ± 0.1 μm. The EDS analysis in Fig. 3(d) confirmed the presence of elements consistent with those in the PEO context but revealed a significant decrease in Mg and Al levels, due to PEO film dissolution, accompanied by an increase in O concentration possibly linked to the electrolyte composition.40Fig. 3(e) provides a cross-sectional view of the LDH–EDTA coating, and it is worth noting that post-treatment has resulted in an increase in both thickness and density, measuring around 3.97 ± 0.1 μm. This indicates that the upper layer has experienced a transformation towards increased density and compactness due to the substitution of NO3− anions with EDTA4− within the LDH galleries. This observation corresponds with the results obtained from the analysis of surface morphology, as depicted in Fig. 1(g). The EDS analysis performed on LDH–EDTA, as illustrated in Fig. 3(f), revealed a significant reduction in the levels of Mg, Al, and O elements. This reduction is a result of the hydrothermal process and the dissolution of the coating. In contrast, the carbon content is found to be the highest among all the elements. This observation suggests that EDTA has been successfully integrated into the LDH layers, leading to a denser and more compact surface. This layer not only improves the stability of the coating but also enhances its corrosion resistance. The EDS analysis depicted in Fig. 3(h) for LDH–EDTA–Zn indicates a notably higher carbon content when compared to the LDH–EDTA samples. In the comparison between LDH–EDTA and LDH–EDTA–Zn, it becomes evident that the cross-sectional structure of LDH–EDTA–Zn, as shown in Fig. 3(g), exhibits a denser configuration. This denseness is characterized by a higher concentration of [Zn(EDTA)]2− ions intercalated within the LDH galleries, as supported by the elevated carbon content identified in the EDS analysis. Furthermore, there is an observable thin and irregular topmost layer of EDTA–Zn, contributing to an overall augmentation in the coating thickness to approximately 4.66 ± 0.1 μm. This difference is attributable to the strong chelation ability of Zn2+ ions with EDTA molecules, leading to the creation of a more stable and long-lasting coating.41,42 Consequently, this coating may effectively impede the infiltration of corrosive ions, enhancing its corrosion resistance properties.
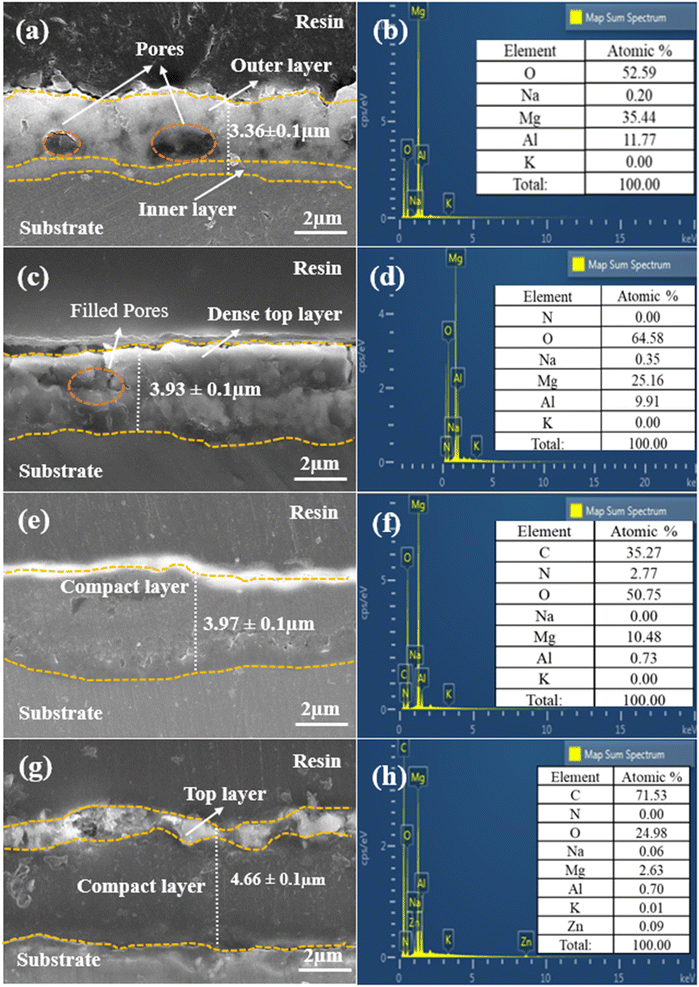 |
| | Fig. 3 Cross-sectional morphologies and corresponding EDS analysis of (a and b) the PEO sample, (c and d) LDH sample, (e and f) LDH–EDTA sample, and (g and h) LDH–EDTA–Zn sample, respectively. (a) The circles highlighted indicate the diverse range of pores observed in the PEO coating. | |
3.2 Compositional analysis of the coating
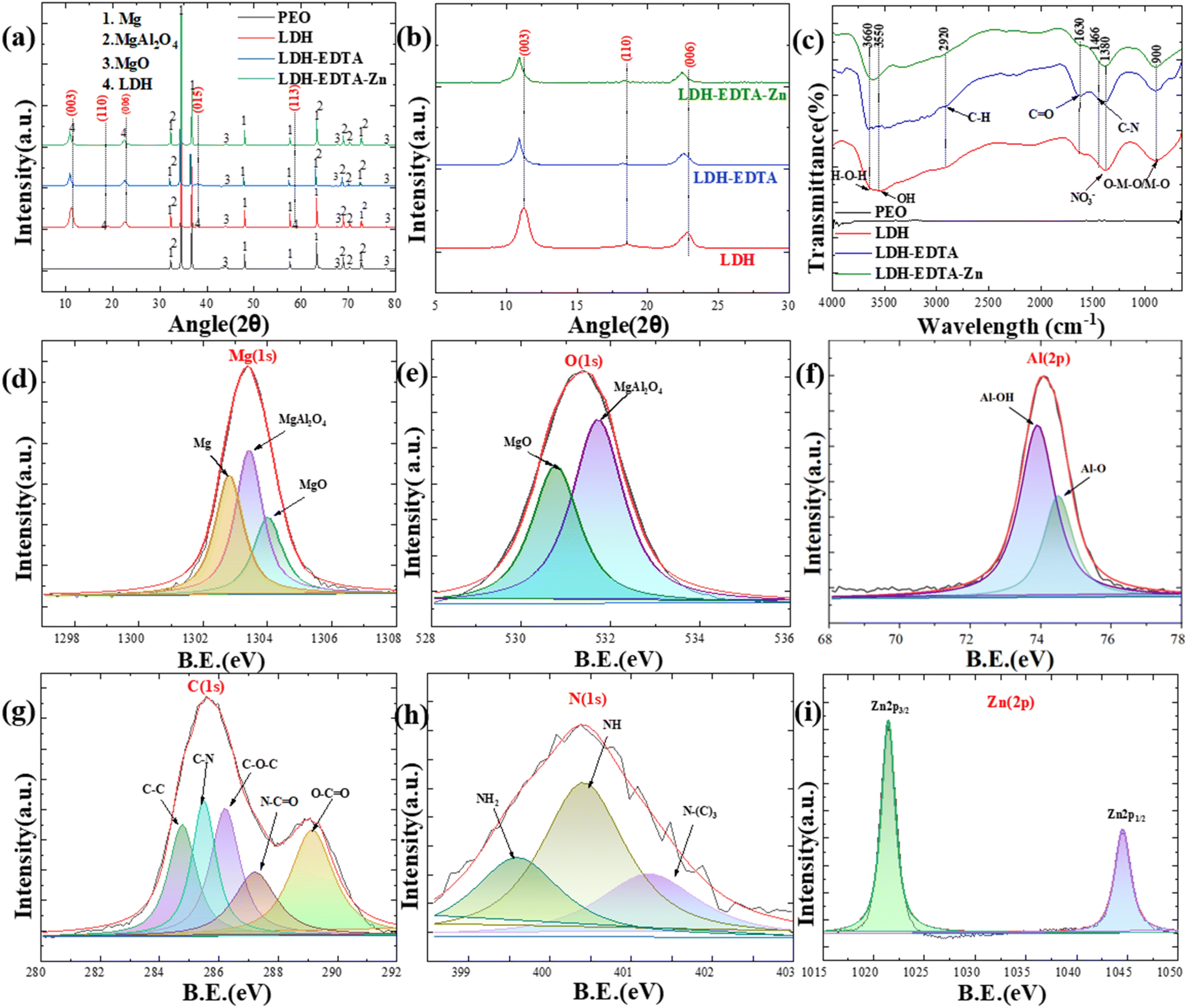
Fig. 4(a) illustrates the XRD patterns of PEO, LDH, LDH–EDTA, and LDH–EDTA–Zn across an angle range of 5–80° (2θ), which are used to analyze and determine the structural compositions of each compound. The execution of a PEO film growth technique on an AZ31 Mg alloy substrate, facilitated by an aluminate electrolyte solution, yielded multiple peaks indicative of various magnesium compounds, including magnesium (Mg) (JCPDS: 00-035-0821), magnesium oxide (MgO) (JCPDS: 00-045-0946), and magnesium aluminate (MgAl2O4) (JCPDS: 00-033-0853). The recognition of Mg peaks correlates with X-rays penetrating into the porous layer and exposing the underlying AZ31 Mg alloy substrate. The formation of MgO is caused by the outward migration of Mg2+ ions from the substrate into micro-discharge channels, which is accompanied by the simultaneous inward migration of O2− from the electrolyte into these finely structured micro-discharge channels (eqn (1)):
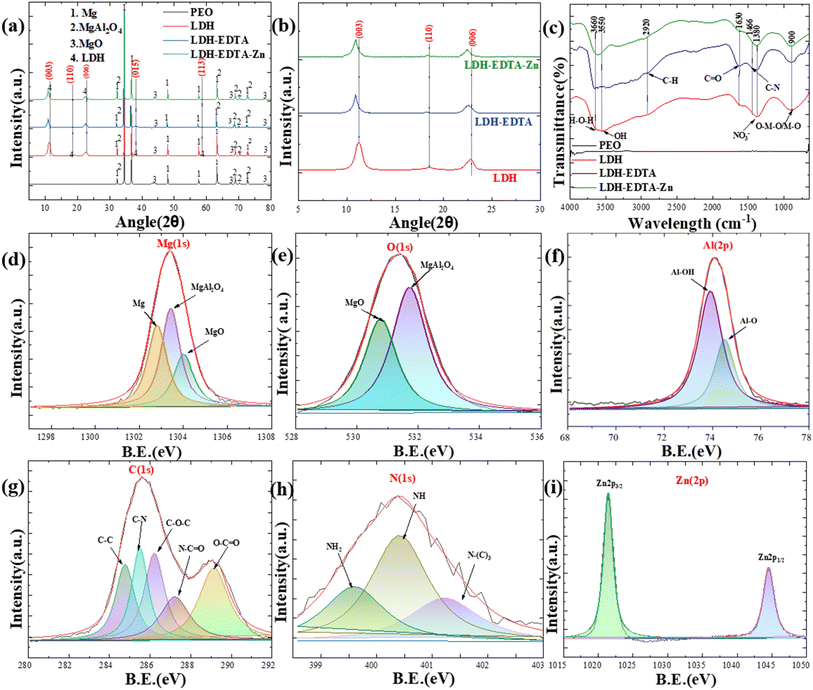 |
| | Fig. 4 Compositional analysis of PEO, LDH, LDH–EDTA, and LDH–EDTA–Zn. (a) XRD patterns for all the samples are obtained by scanning in the range of 5 to 80° using a Cu Kα radiation source. (b) XRD patterns of three different samples, namely LDH, LDH–EDTA, and LDH–EDTA–Zn, are obtained by scanning in the range of 5 to 30° respectively. (c) FT-IR spectra covering the wavelength range of 4000–650 cm−1 for all samples. XPS spectra of (d and e) the PEO film for Mg(1s) and O(1s), (f) LDH film for Al(2p), (g and h) LDH–EDTA film for C(1s) and N(1s), and (i) LDH–EDTA–Zn film for Zn(2p). | |
Similarly, the negatively charged AlO2− ions formed by the hydrolysis of NaAlO2 are attracted to the anode and interact with surrounding positively charged Mg2+ ions to create magnesium aluminate spinel (eqn (2)):
| | | Mg2+ + 2AlO2− → MgAl2O4 | (2) |
The precise identification of distinct and well-defined reflections aligned precisely with the (003), (110), (006), (015), and (113) crystallographic planes, as defined in the JCPDS 00-048-0601 and 00-035-0964 databases, serves as strong support for the successful intercalation of LDH within the PEO coatings. The observed interlayer spacings of 0.787 nm, 0.486 nm, 0.401 nm, 0.249 nm, and 0.180 nm (using Bragg's law (2dsinθ = mλ)) at angles of about 11.3°, 18.5°, 22.6°, 38.2°, and 58.6° offer convincing evidence for the effective development of LDH and are consistent with observations in the literature.43,44 In Fig. 4(b), a focused scan range of 5° to 30° is employed to enhance the resolution of XRD patterns for crystal planes (003), (006), and (110). Within this limited range, three distinct peaks are clearly visible at approximately 11.3°, 18.5°, and 22.6°, corresponding to the indicated LDH crystal planes. The XRD patterns for (003), (006), and (110) in the LDH–EDTA film displayed a considerable shift towards lower angles, about 10.8°, 18.2°, and 22.4° (2θ), respectively. These changes revealed an increase in interlayer distances, with approximate values of 0.82, 0.49, and 0.404 nm. This growth is caused by the addition of EDTA anions, which have greater dimensions than the NO3− found in the LDH structure. These findings align with prior research reported in the existing literature.45–47 The XRD patterns for the (003), (006), and (110) peaks for the LDH–EDTA–Zn showed similar shifts, suggesting a reduction in their angles of around 10.8°, 18.2°, and 22.4° (2θ), respectively. The observed modifications in XRD patterns are attributed to the crystal growth shift caused by the addition of [Zn(EDTA)]2− anions, which have larger dimensions than the NO3− ions present in the LDH structure.
In Fig. 4(c), the FT-IR analysis provides a comprehensive insight into the chemical bonds and functional groups present in the prepared samples, allowing for a detailed understanding of their composition. The investigation revealed a large absorption band in the Mg–Al LDH film at 3550 cm−1 and 3660 cm−1, which is ascribed to O–H and H–O–H bond stretching vibrations. The presence of water molecules making hydrogen bonds with nitrate ions in the LDH interlayer is revealed by this band's broad and extended structure.48 The band at approximately 1380 cm−1 is associated with the stretching vibration of NO3−.49 The lower-frequency bands noticed at around 900 cm−1 are caused by lattice vibrations and can be ascribed to the (M−O) and (M–O–M) modes, respectively.45 The FT-IR spectra of LDH–EDTA suggest strong peaks at 1630 cm−1, which correspond to the asymmetric vibrations of carboxylate (COO−) groups. Furthermore, the less pronounced peaks at 2920 cm−1 and 1466 cm−1 can be ascribed to the existence of C–H and C–N groups, respectively.50,51 The LDH–EDTA–Zn spectra display distinct bands similar to those observed in LDH–EDTA, confirming the presence of [Zn(EDTA)]2− anions inside the LDH layer.
Furthermore, XPS analysis is used to determine the chemical states of the prepared specimens, as indicated in Fig. 4(d)–(i). We observed three distinct peaks with binding energies of ∼1303.18 eV, ∼1303.98 eV, and ∼1304.08 eV, when we deconvoluted the Mg(1s) spectra for the PEO sample in Fig. 4(d). These peaks are attributed to the presence of Mg, MgAl2O4, and MgO inside the sample.52Fig. 4(e) displays a prominent peak in the PEO sample's O(1s) spectra at ∼531.28 eV, primarily attributed to the presence of oxygen in the MgAl2O4 form.53 Additionally, there is another evident peak with a binding energy at ∼530.38 eV, indicative of the presence of MgO.54Fig. 4(f) shows two strong peaks in the Al(2p) spectra at about 73.88 and 74.48 eV, indicating the presence of Al–O and Al–OH in the LDH film.55 The C(1s) binding energy peaks in the LDH–EDTA spectrum demonstrated in Fig. 4(g) are consistent with specific chemical bonds and functional groups, including C–C at ∼284.6 eV, C–N at ∼285.5 eV, C–O–C at ∼286.2 eV, N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at ∼287.0 eV, and O–CO at ∼288.9 eV respectively.56,57 Moreover, Fig. 4(h) reveals the presence of nitrogen-binding energy peaks, particularly those associated with primary, secondary, and tertiary amine groups, with values at ∼399.2, ∼400.3, and ∼401.7 eV, respectively which supports the effective functionalization of EDTA onto LDH.58 The strong spin–orbit coupling is evident in Fig. 4(i), where the Zn(2p) signal splits into two symmetrical peaks, Zn(2p3/2) and Zn(2p1/2), at ∼1021 eV and ∼1044 eV, respectively, with a spin–orbit splitting value of 23 eV, closely matching the findings for Zn2+ bound within the EDTA matrix.59 The other peaks for PEO and LDH samples are shown in Fig. S1.†
O at ∼287.0 eV, and O–CO at ∼288.9 eV respectively.56,57 Moreover, Fig. 4(h) reveals the presence of nitrogen-binding energy peaks, particularly those associated with primary, secondary, and tertiary amine groups, with values at ∼399.2, ∼400.3, and ∼401.7 eV, respectively which supports the effective functionalization of EDTA onto LDH.58 The strong spin–orbit coupling is evident in Fig. 4(i), where the Zn(2p) signal splits into two symmetrical peaks, Zn(2p3/2) and Zn(2p1/2), at ∼1021 eV and ∼1044 eV, respectively, with a spin–orbit splitting value of 23 eV, closely matching the findings for Zn2+ bound within the EDTA matrix.59 The other peaks for PEO and LDH samples are shown in Fig. S1.†
3.3 Electrochemical performance
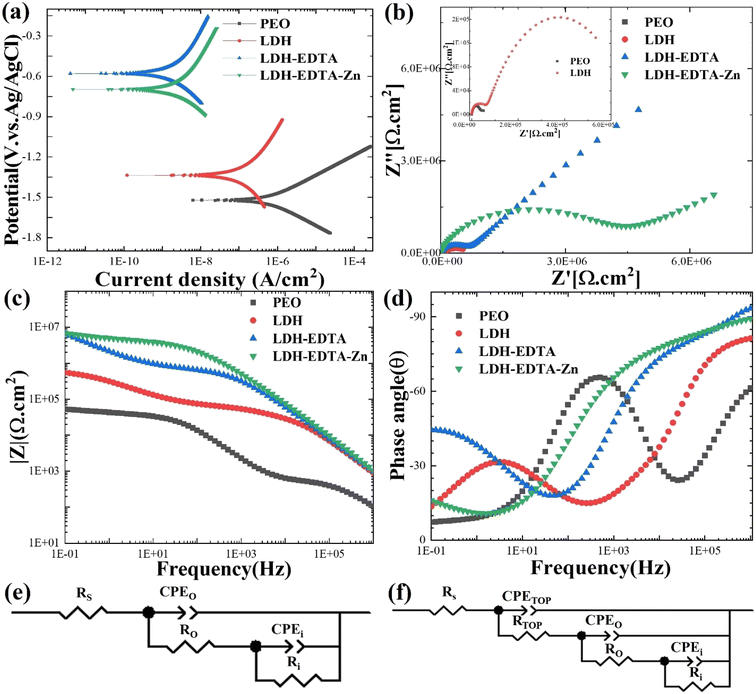
Fig. 5(a) presents the results of PDP assessments performed on a range of samples, including PEO, LDH, LDH–EDTA, and LDH–EDTA–Zn, all of which were exposed to a 3.5 wt% NaCl solution. The PDP curve for the AZ31 Mg alloy substrate is presented in Fig. S2.† The polarization resistance of all prepared samples is calculated using the Stern–Geary formula (eqn (3)):w| | 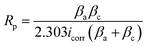 | (3) |
here βa represents the anodic Tafel constant, while βc represents the cathodic Tafel constant. The corrosion potential (Ecorr) and corrosion current density (Icorr) are calculated from the PDP curves using the Tafel method, and these results are shown in Table 1. It is critical to note that a high Ecorr value and a low Icorr value in the tested sample reflect the coating's extraordinary ability to resist corrosion.60 Based on prior studies, PEO samples have the greatest Icorr and the lowest Ecorr, suggesting a greater susceptibility to corrosion. However, after applying post-treatments with LDH, LDH–EDTA, and LDH–EDTA–Zn, we found a considerable decrease in Icorr. Among all devised samples, LDH–EDTA and LDH–EDTA–Zn have the lowest Icorr values and the highest Ecorr values, suggesting the most efficient corrosion prevention. It's important to highlight that the Icorr values for LDH–EDTA–Zn and LDH–EDTA are quite similar, indicating a high degree of corrosion resistance in both cases. However, there is a slight decrease in the Ecorr value observed for LDH–EDTA–Zn. This decline in Ecorr could be attributed to the presence of the [Zn(EDTA)]2− anion complex, which has the potential to dissolve some of the existing divalent metal ions within the LDH structure. Consequently, this may result in a slightly reduced stability when compared to LDH–EDTA.50 The improved corrosion resistance found in the LDH–EDTA and LDH–EDTA–Zn samples is due to the efficient sealing of micro-pores and the formation of metal complexes that function as corrosion inhibitors. The poor performance of the PEO sample, on the other hand, is owing to its larger porosity, weaker stability, and less durability.
 |
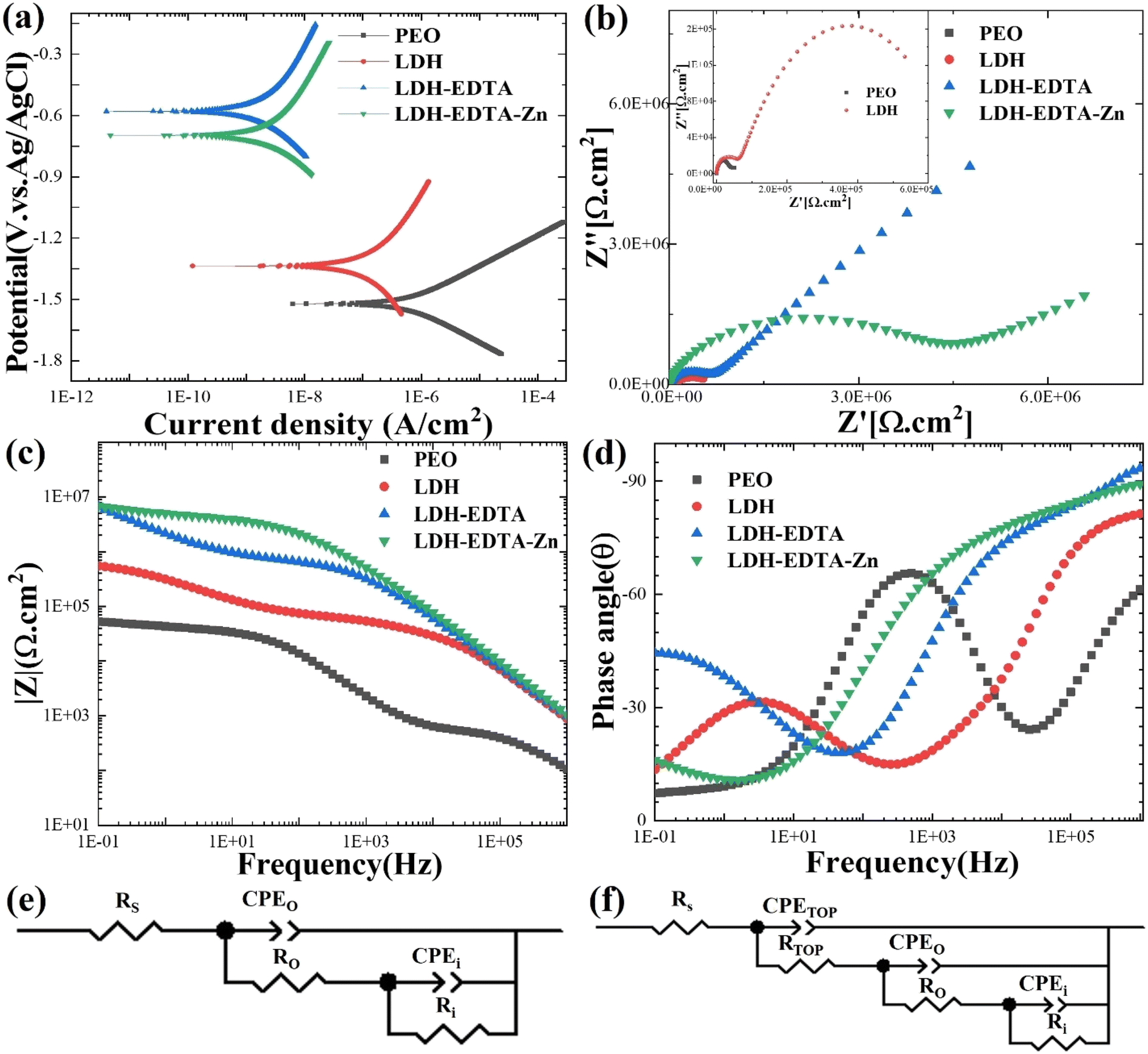
| | Fig. 5 (a–d) PDP and EIS results for PEO, LDH, LDH–EDTA, and LDH–EDTA–Zn samples immersed in 3.5% NaCl solution for 5 hours: (a) PDP curves, (b) Nyquist plots with inset magnified plots for PEO and LDH samples, (c) impedance plots, and (d) phase plots, respectively. The proposed fitted circuit model (e) for PEO and (f) for LDH, LDH–EDTA, and LDH–EDTA–Zn samples, respectively. | |
Table 1 The potentiodynamic polarization test results for PEO, LDH, LDH–EDTA, and LDH–EDTA–Zn films formed on AZ31 in 3.5% NaCl solution
| Film |
E
corr (V) |
I
corr (A cm−2) |
β
a (V dec−1) |
−βc (V dec−1) |
R
p (Ω cm2) |
| PEO |
−1.52 |
7.3 × 10−7 |
150 × 10−3 |
153.1 × 10−3 |
4.5 × 104 |
| LDH |
−1.32 |
1.35 × 10−7 |
540.8 × 10−3 |
663.8 × 10−3 |
9.5 × 106 |
| LDH–EDTA |
−0.70 |
2.64 × 10−9 |
589.8 × 10−3 |
363.5 × 10−3 |
6.76 × 107 |
| LDH–EDTA–Zn |
−0.56 |
2.12 × 10−9 |
549.6 × 10−3 |
338.2 × 10−3 |
2.73 × 107 |
To achieve a comprehensive grasp of the electrochemical characteristics displayed by the prepared samples, we performed electrochemical impedance spectroscopy (EIS) experiments utilizing a solution comprising 3.5 wt% of NaCl. Fig. 5(b) depicts Nyquist plots of the PEO, LDH, LDH–EDTA, and LDH–EDTA–Zn samples. The EIS curves for the PEO and LDH samples are too small to be visible, so we've included inset images of both samples to enhance their visibility. In this analysis, the diameter of the capacitive loop is more significant, as it is directly tied to the magnitude of the material's capacitive reactance. A larger capacitive loop diameter indicates higher capacitance and a lower corrosion rate, while a smaller diameter suggests lower capacitance and a higher corrosion rate.61 The Nyquist plots and polarization curves coincide, confirming that the LDH–EDTA and LDH–EDTA–Zn samples have the greatest impedance values. This is evident from the greater semicircle diameter and the greatest surface area under the curve, both of which imply superior corrosion resistance. In contrast, the PEO sample has the lowest corrosion resistance, as shown by its smaller semicircle diameter and lower impedance value. These findings underscore the effectiveness of LDH–EDTA and LDH–EDTA–Zn in effectively inhibiting the penetration of corrosive agents, making them excellent choices for corrosion prevention.
The corresponding Bode plots for PEO, LDH, LDH–EDTA, and LDH–EDTA–Zn are provided in Fig. 5(c) and (d), where the absolute impedance |Z| values and their phase angles (θ) are shown as functions of frequency (f), respectively. The magnitude of low-frequency impedance (|Z|) is frequently employed to represent the total resistance of the protective coatings.62,63 The impedance value of the post treated films showed a significant increase after applying the sealing treatment to the PEO layer. In particular, the |Z|0.1Hz values for the LDH–EDTA and LDH–EDTA–Zn samples are around two orders of magnitude greater than that of the PEO sample. Additionally, it should be noted that the PEO sample had an impedance value that is around one order of magnitude lower than that of the LDH sample. The high-frequency range is indicative of the dielectric properties exhibited by the outer coating layer, whereas the lower-frequency range is associated with the attributes of the inner layer.64 The phase angle Bode plot indicates two distinct time constants for outer and inner layers. Notably, the LDH–EDTA and LDH–EDTA–Zn samples exhibit a peak phase angle at 106 Hz, higher than that of LDH and PEO samples. This suggests improved corrosion resistance due to challenges in ion migration during corrosion tests.
To evaluate and compare the electrochemical characteristics of the specimens systematically and thoroughly, the impedance data obtained from EIS are analyzed by fitting it to a specialized equivalent circuit model designed for both the PEO-coated and post-treated samples, as illustrated in Fig. 5(e) and (f). The circuit model depicted in Fig. 5(e) is employed for fitting PEO, whereas the circuit model shown in Fig. 5(f) is utilized to simulate the EIS results for LDH, LDH–EDTA, and LDH–EDTA–Zn samples, respectively. In both circuit models, the variables Rs, Ro, and Ri are employed in both circuit configurations to represent the resistance levels of the solution, outer layer, and inner layer, respectively, while CPEo and CPEi denote the corresponding constant phase elements for the outer and inner layers. For the LDH, LDH–EDTA, and LDH–EDTA–Zn samples, an additional set of parameters, namely Rtop and CPEtop, are introduced into the circuit model. These parameters are introduced to better describe the effectiveness of the sealing treatment and to optimize the fitting of the data. The absence of significant influence from the diffusion of Cl− ions in the post-treated layer allowed the model presented in Fig. 5(f) to achieve the best fit with a low chi-squared value, without requiring a Warburg element (W) in series with Ri. Detailed results of this fitting process are provided in Table 2. When the three time constants of the equivalent circuit significantly overlap, it becomes challenging to distinguish them in the impedance spectra.65 It's worth noting that within PEO coatings, the Ri values exceeded their corresponding Ro values, which is attributed to the dense and flawless morphology of the inner layer, in contrast to the more porous structure of the outer layer. The total resistance (Rt) within a complex coating is determined by the summation of individual resistances, including Rtop, Ro, and Rin. Upon examining the total resistance across all the prepared samples, it becomes evident that LDH–EDTA exhibits a marginally higher value at 6.59 × 107 Ω cm2, while LDH–EDTA–Zn registers a slightly lower value at 4.95 × 107 Ω cm2. This implies that the [Zn(EDTA)]2− anion complex might dissolve some of the divalent metals present in the LDH galleries.66 It is also worth noting that the aforementioned resistance values are greater than those of PEO and LDH. This clearly demonstrates the sealing materials' superior chemical stability and durability.
Table 2 Results of EIS fitting of PEO, LDH, LDH–EDTA, and LDH–EDTA–Zn samples at an immersion time of 5 h in 3.5% NaCl solution
| Sample |
CPEtop |
R
top (Ω cm2) |
CPEo |
R
o (Ω cm2) |
CPEi |
R
i (Ω cm2) |
χ
2 (×10−4) |
|
Y
top (S sn cm−2) |
n
top
|
Y
o (S sn cm−2) |
n
o
|
Y
o (S sn cm−2) |
n
i
|
| PEO |
— |
— |
— |
1.88 × 10−7 |
0.88 |
3.04 × 104 |
2.95 × 10−5 |
0.38 |
4.69 × 105 |
2.79 |
| LDH |
6.08 × 10−10 |
0.92 |
3.10 ×104 |
3.25 × 10−8 |
0.71 |
3.49 × 104 |
8.50 × 10−7 |
0.61 |
1.61 × 106 |
3.80 |
| LDH–EDTA |
1.92 × 10−10 |
0.97 |
5.80 ×104 |
3.14 × 10−9 |
0.69 |
6.83 × 105 |
1.96 × 10−7 |
0.58 |
6.52 × 107 |
3.51 |
| LDH–EDTA–Zn |
1.91 × 10−10 |
0.99 |
4.96 ×104 |
4.02 × 10−9 |
0.64 |
4.24 × 106 |
3.80 × 10−7 |
0.43 |
4.53 × 107 |
3.29 |
3.4 Photocatalytic performance
Rhodamine B (RhB), a water-soluble cationic dye with potential toxicity and carcinogenicity, remains relatively stable in wastewater, accumulates in organisms, and can cause irritation to the respiratory tract, eyes, and human skin.67,68 Hence, in addition to improving the corrosion resistance of coatings used on industrial equipment, there is also significant interest in understanding the photocatalytic characteristics of these coatings. This interest arises because the coated products will be deployed in industries where organic dyes are employed. The photocatalytic performance of PEO, LDH, LDH–EDTA, and LDH–EDTA–Zn samples has been evaluated using the degradation of RhB dye as a model. In Fig. 6(a)–(d), a clear peak associated with RhB, usually found at around 550 nm, is observed, exhibiting a gradual decline in intensity with prolonged exposure time.69 This diminishing peak intensity signifies the ongoing degradation of the RhB dye caused by the photocatalytic materials undergoing evaluation. The peak at 0 minutes is a baseline test for evaluating the UV-visible absorbance of the produced RhB solution prior to photocatalytic treatment. The RhB solution is subjected to treatment at different time intervals of up to 50 minutes. Since each of the prepared samples exhibited photocatalytic behavior, they facilitated the decolorization of the RhB solution within varying periods. Specifically, PEO has taken 50 minutes for decolorization, LDH achieved it in 35 minutes, LDH–EDTA reduced the time to 30 minutes, and LDH–EDTA–Zn demonstrated the fastest decolorization, accomplishing it in just 25 minutes. The degradation efficiency (η) of RhB is calculated as (eqn (4)):| | 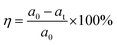 | (4) |
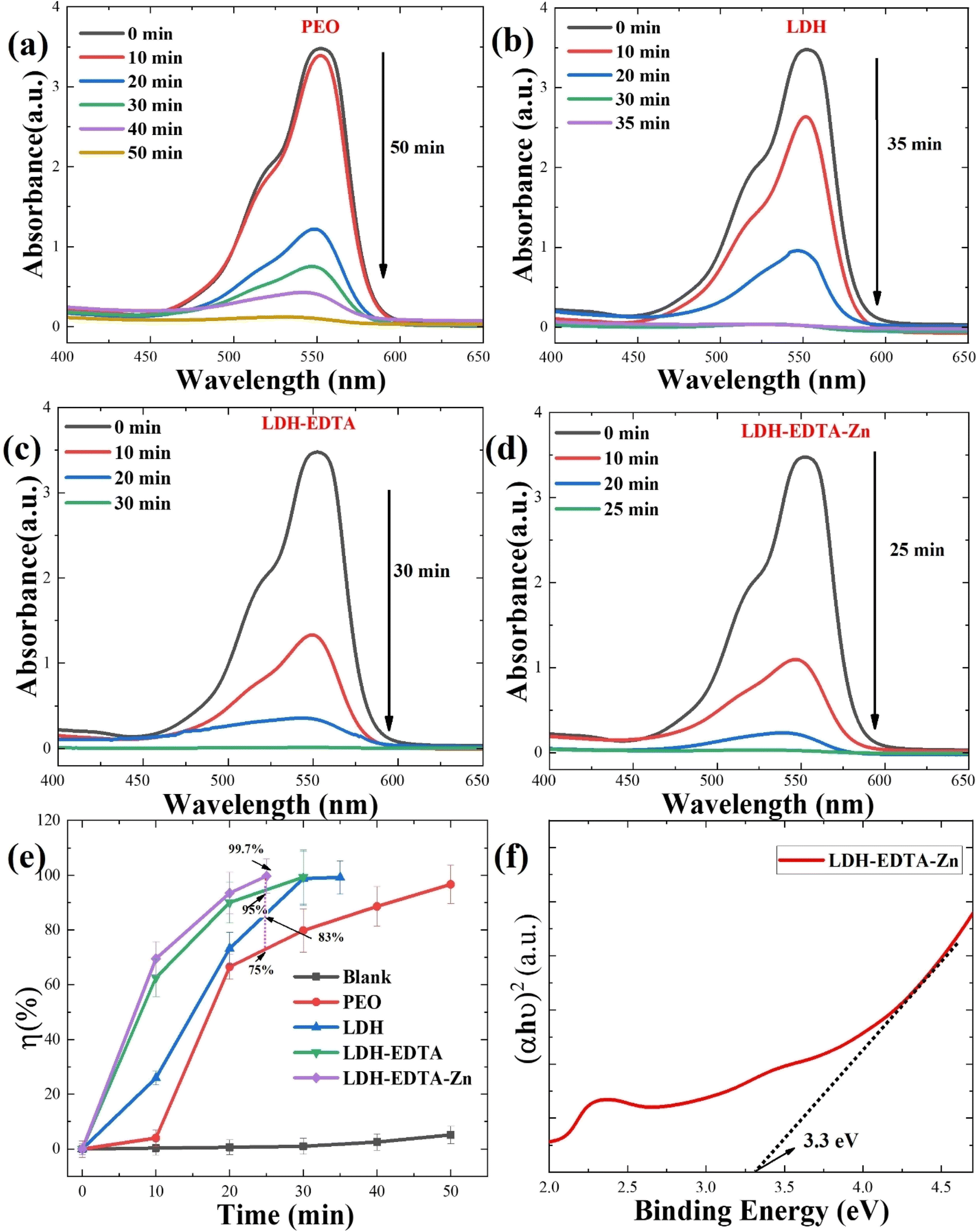
where a0 is the initial intensity of RhB and at is the intensity at a certain time during the photocatalytic process. Fig. 6(e) depicts the variation in photocatalytic degradation efficiency between the four samples. Notably, there is a significant difference in photocatalytic efficiency before and after post-treatment. It is noteworthy that the LDH–EDTA–Zn photocatalyst demonstrated remarkable efficiency in rapidly degrading the dye, achieving an impressive 99.7% removal within just 25 minutes. In comparison, LDH–EDTA exhibited a substantial degradation rate of 99.5% within 30 minutes, while LDH alone reached 99.1% within 35 minutes. PEO, although effective, showed a slightly lower degradation rate of 96.6% within 50 minutes. Conversely, the solution without any catalyst (blank) displayed minimal degradation, with only a 5% removal after 50 minutes, highlighting the challenging degradation of rhodamine B dye in the absence of a catalyst. The corresponding UV-vis absorption spectra for the blank solution (without catalyst) are shown in Fig. S3.† In a direct comparison of all samples within a 25 minutes duration, LDH–EDTA–Zn demonstrated the highest degradation efficiency, boasting an exceptional 99.7%. In contrast, EDTA achieved a degradation rate of 95.4%, LDH reached 83%, and PEO lagged behind at 75%. The evaluation of optical characteristics, particularly bandgap measurements, is an essential method for establishing the exact light wavelengths necessary for the photocatalytic degradation of dye molecules. Fig. 6(f) exhibit the bandgap plot for the LDH–EDTA–Zn catalyst. The remarkably narrow bandgap value of 3.3 eV, as well as high absorption edge values around 550 nm and the presence of compact, densely packed LDH flakes with reduced sizes, completely demonstrate the superior photocatalytic activity of LDH–EDTA–Zn when compared to the other prepared catalysts.
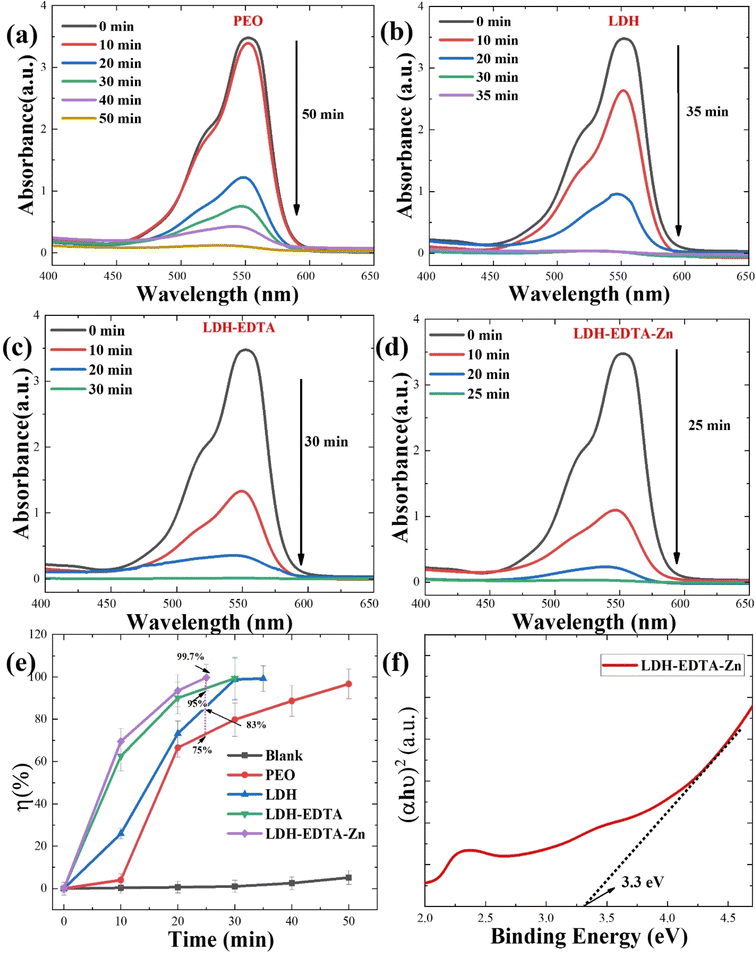 |
| | Fig. 6 UV-vis absorbance spectra displaying the photocatalytic activity of (a) PEO, (b) LDH, (c) LDH–EDTA, and (d) LDH–EDTA–Zn, at different time periods up to 50 minutes for degradation of organic dye rhodamine B. (e) Photocatalytic degradation efficiencies of RhB dyes with all prepared samples. (f) Determination of the energy band for the best catalyst (LDH–EDTA–Zn) which displayed a narrow energy gap of around 3.3 eV. | |
To assess the adsorption capabilities of our premier catalyst LDH–EDTA–Zn for dye molecules under varying conditions, we conducted UV-vis absorbance measurements in both dark and visible light conditions. The absorbance spectra under visible light conditions are depicted in Fig. 6(d), while the absorbance spectra for dark conditions can be found in Fig. S4(a).† Notably, the photocatalytic degradation under visible light is substantial, reaching 99.7% within the first 25 minutes. In contrast, under dark conditions, the degradation is notably limited, with only a 4% reduction observed as shown in Fig. S4(b).† This observation underscores the challenging nature of degrading Rhodamine B dye in the absence of light.
Furthermore, to examine the role of H2O2 in the photocatalytic degradation process, we conducted a comparative analysis of the photocatalytic degradation efficiencies using the best catalyst LDH–EDTA–Zn with and without the addition of H2O2, as detailed in Fig. S5.† The UV-vis absorbance spectra in the absence of H2O2 for LDH–EDTA–Zn are illustrated in Fig. S5(a).† These spectra indicate that, during the first 10 minutes, the degradation rate is relatively low at around 23%. However, in the presence of H2O2, the degradation efficiency after 10 minutes is notably higher at 61%. Similarly, the photocatalytic degradation efficiencies, as depicted in Fig. S5(b),† indicate that after 25 minutes, the LDH–EDTA–Zn sample without H2O2 exhibited a degradation efficiency of 99.3%, while in the presence of H2O2, it reached 99.7%. This suggests that H2O2 does not significantly affect the overall degradation efficiencies; however, it does accelerate the initial stages of the photocatalytic degradation process.
3.5 Scavenger studies and recyclability of photocatalysts
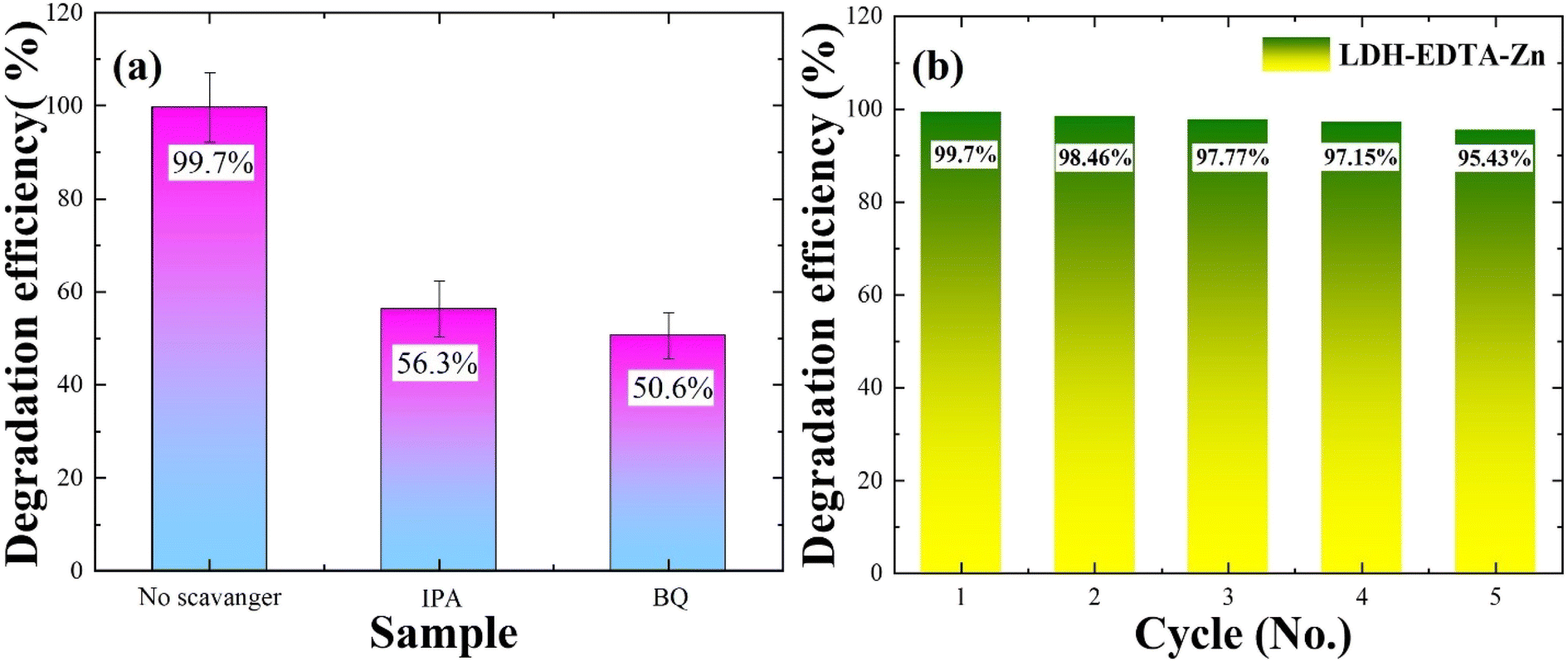
To enhance our comprehension of the role played by oxidizing species in the photocatalytic degradation process, we conducted scavenger studies aimed at capturing oxidants like OH˙ radicals and O2˙ radicals. Benzoquinone (BQ) was employed as a scavenger to trap O2˙ radicals, while isopropanol (IPA) was utilized to capture OH˙ radicals, respectively.70,71 The UV-vis absorption spectra for rhodamine degradation in the presence of IPA and BQ are illustrated in Fig. S6(a) and (b),† respectively. In Fig. 7(a), the photocatalytic degradation efficiency of the rhodamine B solution reached 99.7% in the absence of a scavenger. However, when IPA is introduced, the efficiency drops to 56.3%, and with the addition of BQ, it further diminishes to 50.6%. The decline from 99.7% to 56.3% with IPA suggests its effective trapping of OH˙ radicals, while the reduction to 50% with BQ points to the substantial trapping of O2˙ radicals. According to the results mentioned above, it can be inferred that all active species played a role in the photocatalytic decomposition of RhB, with O2˙ radicals exerting a greater influence compared to OH˙ radicals. According to Wu et al.,69 their report highlights a reduction in the degradation efficiency of rhodamine, decreasing from 98% to 76% upon the addition of isopropanol (IPA). This observation underscores the significant involvement of OH• radicals as the most active species in the photocatalytic process. Similarly, in another report,72 the degradation rate of RhB witnessed a substantial reduction from 99.8% to 41.5% and 43.8% upon the addition of BQ. This suggests that O2˙ radicals played crucial and noteworthy roles in the photodegradation process of rhodamine. These reports align closely with our findings, indicating that the primary agents responsible for the degradation of rhodamine are OH˙ and O2˙ radicals.
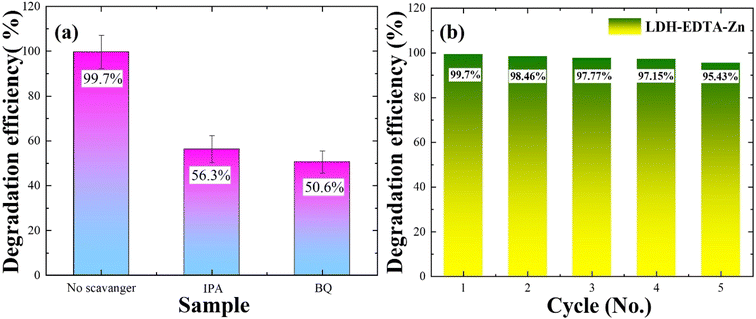 |
| | Fig. 7 (a) Radical trapping experiment for the photocatalytic degradation of Rhodamine B. (b) RhB degradation efficiency by the LDH–EDTA–Zn catalyst in five cycles. After the fifth cycle, even the degradation efficiency is more than 95% which displays the exceptional stability and recyclability of LDH–EDTA–Zn. | |
The stability and recyclability of a catalyst are important factors when considering a photocatalyst for RhB degradation. In this context, recycling investigations are conducted with the LDH–EDTA–Zn catalyst, maintaining a consistent catalyst-to-dye ratio. Throughout this study, the LDH–EDTA–Zn specimens are employed for five cycles. To avoid contamination, each specimen is carefully cleaned after each degrading phase using ethanol and deionized water, followed by a drying stage. Fig. S7(a)–(e)† demonstrate the photocatalytic activity of the LDH–EDTA–Zn catalyst during all five cycles of RhB dye degradation in the presence of visible light. As shown in the figures, there is a notable and quick reduction in the RhB absorption spectra under visible light irradiation within 25 minutes for each cycle. When compared to other systems, this clearly displays the catalyst's remarkable photocatalytic efficiency. In Fig. 7(b), the chart depicts the degradation efficiencies across five consecutive cycles. Initially, in the first cycle, the photocatalytic efficiency was an impressive 99.7%. Subsequently, in the second cycle, it exhibited a minor decline to 98.46%. This decrease continued, with values reaching 97.7% in the third cycle, 97.15% in the fourth cycle, and stabilizing at 95.4% after the fifth cycle. Notably, even after five cycles, this efficiency remained superior to the degradation achieved by LDH and PEO methods. These findings highlight the LDH–EDTA–Zn composite's extraordinary potential as a catalyst, which is characterized by high photocatalytic activity, stability, and reusability.
To assess the stability of the coating following photocatalytic degradation, the surface morphologies of all prepared samples are depicted in Fig. 8. The extensive presence of micropores and defects across a range of scales on the surface of PEO led to its pronounced degradation post-photocatalysis, as depicted in Fig. 8(a). The magnified image (Fig. 8(b)) corresponding to the high degradation of PEO demonstrates extensive surface destruction, resulting in the formation of bulges and indicating diminished stability and durability of the inorganic coating. While the surface morphology of LDH in Fig. 8(c) suggests good stability with minimal degradation as compared to PEO, a closer look at the magnified image in Fig. 8(d) reveals the dissolution and merging of LDH flakes, indicating potential structural alterations. Concerning the LDH–EDTA sample depicted in Fig. 8(e), it demonstrates minimal degradation, displaying the highest surface stability. Moreover, the magnified image (Fig. 8(f)) reveals that LDH flakes are not effectively dissolved, indicating a high level of stability, consistent with the results obtained from EIS as mentioned above. Following five cycles, the surface morphology of our premium catalyst, LDH–EDTA–Zn, displayed in Fig. 8(g), reveals increased degradation compared to LDH–EDTA. Upon closer inspection of the magnified image (Fig. 8(h)), the dissolution of LDH flakes is evident, contributing to a rise in surface porosity. The findings emphasize the impressive stability of LDH–EDTA, highlighting its potential for practical applications as a highly effective anticorrosive material. The observed degradation in LDH–EDTA–Zn not only highlights its effectiveness in photocatalytic degradation but also suggests superior photocatalytic performance, particularly evidenced by the highest degradation observed after five cycles.
 |
| | Fig. 8 Surface morphologies observed after photocatalysis for different samples: (a) PEO, (c) LDH, (e) LDH–EDTA, and (g) LDH–EDTA–Zn. Corresponding magnified images for each sample are presented in figures (b), (d), (f), and (h), respectively. | |
As the integration of CO32− into LDH galleries post-photocatalysis is feasible, we performed FTIR analyses on the LDH, LDH–EDTA, and LDH–EDTA–Zn samples, as illustrated in Fig. S8.† The results revealed a distinct absorption band at ∼3550 cm−1, which is attributed to the stretching vibrations of O–H bonds related to LDH. Additionally, evident peaks at ∼1630 cm−1 and ∼1496 cm−1 are identified, indicating the asymmetric vibrations of carboxylate (COO−) groups and the symmetric bending of NH3+. These findings suggest the deposition of rhodamine on the LDH surface.73 Importantly, the absence of a discernible peak associated with CO32−, typically found around ∼1380 cm−1, is notable in the spectrum, indicating that carbonate anions are not intercalated after photocatalysis.
3.6 Theoretical calculations
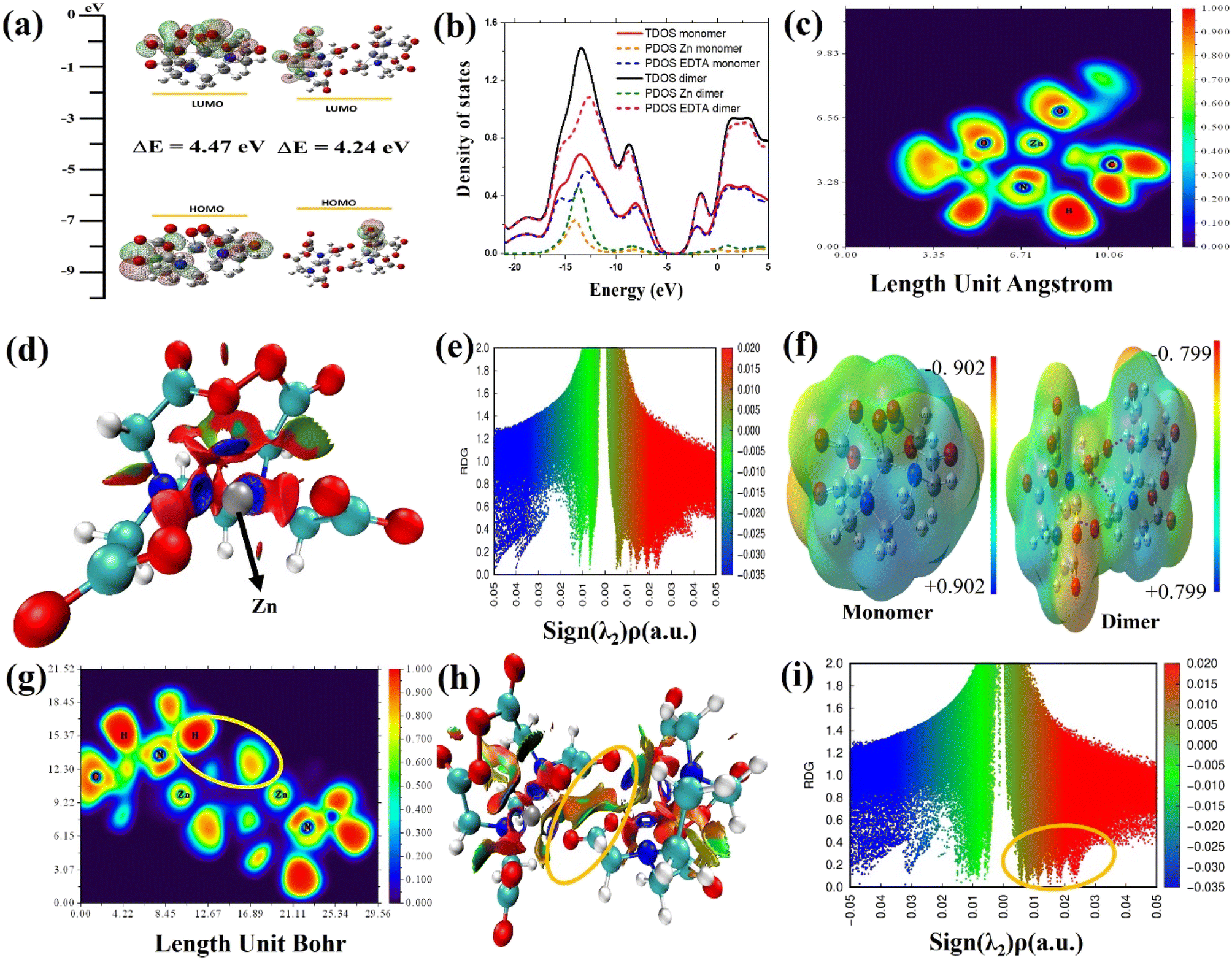
The DFT method is used to study how EDTA–Zn molecules interact with the Mg–Al LDH surface. It also examined charge transfer behavior and various properties of the molecular network formed by EDTA–Zn monomers and dimers as shown in Fig. 9. This analysis includes visualizing HOMO and LUMO states, assessing DOS, PDOS, ELF, MEP, and RDG for both the individual EDTA–Zn monomer and dimer structures. In Fig. 9(a), both the monomer and dimer forms of Zn–EDTA exhibit a significant HOMO–LUMO region centered around the EDTA molecules. Theoretical calculations reveal a smaller energy gap in the dimer (4.24 eV), indicating a higher degree of EDTA–Zn incorporation and consequently more reactive molecules available for interaction with the LDH surface.74 These findings are further explained by the TDOS visualization in Fig. 9(b), where the region of zero DOS (approximately −5.0 eV) is noticeably reduced in the dimer (depicted by the black line) in comparison to the monomer (represented by the red line). The presence of this region implies a lack of available electronic states for electrons to occupy. Therefore, a smaller gap signifies a greater reactivity of the molecules. Furthermore, the PDOS (partial density of states) for each EDTA–Zn monomer (Zn in yellow, EDTA in blue) and dimer (Zn in green, EDTA in purple) is visualized. Notably, there is a discernible shift towards positive energy values in the PDOS of Zn upon transformation from the monomer to the dimer. The ELF map in Fig. 9(c) visually represents the electron density distribution in the molecule, with colors in the map representing ELF values from 0.0 to 1.0. High ELF values (1.0) indicate localized bonding and non-bonding electron regions, while lower values (<0.5) represent delocalized electron regions. High ELF regions around hydrogen atoms show localized bonding and non-bonding electrons, while blue regions around carbon, nitrogen and oxygen atoms indicate delocalized electron clouds. The RDG method is a valuable tool for understanding intermolecular interactions, especially the bonding mechanism between Zn–EDTA molecules in Fig. 9(d). This method employs a color-coded scheme on its isosurface, where green indicates weak van der Waals attractive interactions, blue represents strong attractive interactions, and red corresponds to steric repulsion. In our particular case, after the incorporation of Zn into the EDTA molecule, it becomes apparent that Zn forms strong attractive interactions with nearby nitrogen and oxygen atoms, evident from the blue regions surrounding Zn in the EDTA–Zn monomer. Simultaneously, a noticeable increase in steric effects is observed, primarily originating from the deformation of the benzene rings when Zn is integrated into the EDTA molecule. In Fig. 9(e), the RDG scatter plot shows RDG S plotted against the sign of λ2(ρ) for the EDTA–Zn monomer. Sign λ2(ρ) corresponds to the second eigenvalue of the electron density Hessian matrix. The purpose of incorporating sign λ2(ρ) is to distinguish interactions as either attractive or repulsive. To be specific, a sign λ2(ρ) value less than 0 denotes an attractive interaction, such as a hydrogen bond, while a sign λ2(ρ) value greater than 0 signifies a repulsive interaction, like a steric effect. The EDTA–Zn monomer and dimer are analyzed for their charge distribution and identification of potential electrophilic and nucleophilic sites using MEP, as depicted in Fig. 9(e). The MEP maps highlight the negative charge regions of the molecule in red and yellow, indicating electrophilic reactivity, while blue regions represent positive charge, suggesting nucleophilic reactivity. The MEP color scale ranges from −0.902 to 0.902 for the monomer and −0.799 to 0.799 for the dimer, with blue indicating the strongest attractive forces and red indicating the strongest repulsive forces. In the context of an EDTA–Zn monomer, the oxygen atoms within the carboxylic group are characterized by a pronounced repulsive effect. Conversely, on the other hand, the hydrogen atoms connected to the carbon atom exhibit a substantial attractive force. The strong interaction between hydrogen atoms at the nucleophilic sites of one monomer and oxygen atoms at the electrophilic sites of the other monomer drives the formation of the EDTA–Zn complex. In Fig. 9(g), the ELF image of the EDTA–Zn dimer illustrates the existence of localized bonding and non-bonding electrons around the hydrogen atoms, indicated by the red regions. Furthermore, the ELF image reveals the presence of delocalized electron clouds surrounding the carbon and oxygen atoms, resembling the patterns observed in the ELF image of the EDTA–Zn monomer. Additionally, the image highlights a yellow circle, signifying a strong interaction between the hydrogen atoms of one monomer and the oxygen atoms of the other monomer. RDG analysis was performed on the EDTA–Zn dimer, and the results are visually represented in Fig. 9(h). Importantly, our investigation revealed that the oxygen atoms at electrophilic sites within one monomer formed more favorable primary bonds with the hydrogen atoms situated at nucleophilic sites within other monomers in the self-assembled system. This crucial interaction is prominently highlighted within a yellow circle, facilitating the establishment of appropriate hydrogen bond interactions. Furthermore, the RDG scatter plot for the dimer, particularly in the vicinity of positive peak values of λ2(ρ) (0.02 a.u), exhibited a significant spike, marked by a yellow circle in Fig. 9(i), which strongly indicates the presence of hydrogen interactions. In brief, the predominant factor responsible for the formation of the self-assembled network in the EDTA–Zn system is primarily driven by intermolecular interactions, with a significant contribution from hydrogen bonds facilitated by the oxygen atoms at electrophilic sites on one monomer and the hydrogen atoms at nucleophilic sites on another monomer.
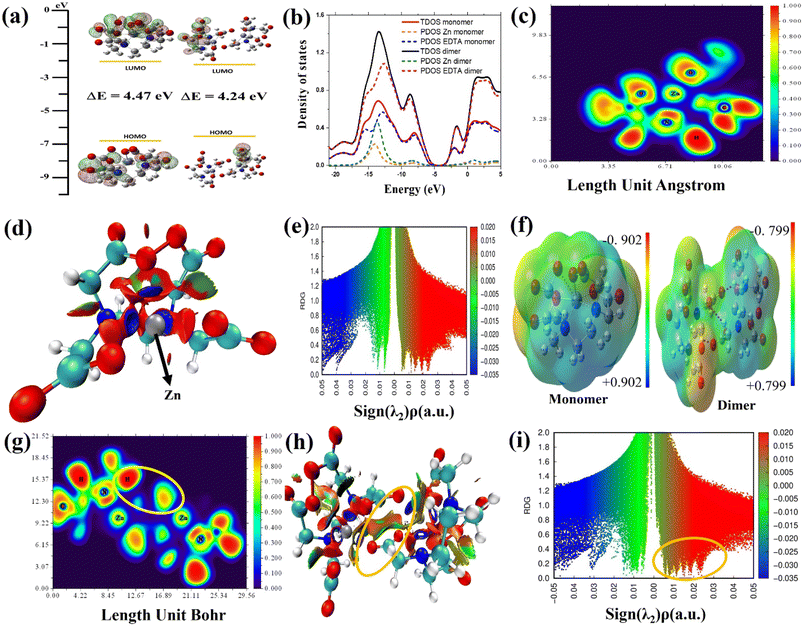 |
| | Fig. 9 Electronic structure analysis of the EDTA–Zn monomer and dimer. (a) The HOMO and LUMO visualization. (b) Density of states (DOS) distribution, (c) ELF visualization, and (d and e) RDG visualization and scatter plot for the EDTA–Zn monomer, respectively. (f) MEP visualization for both the monomer and dimer. (g–i) The ELF visualization, RDG visualization, and scatter plot for the EDTA–Zn dimer, respectively. | |
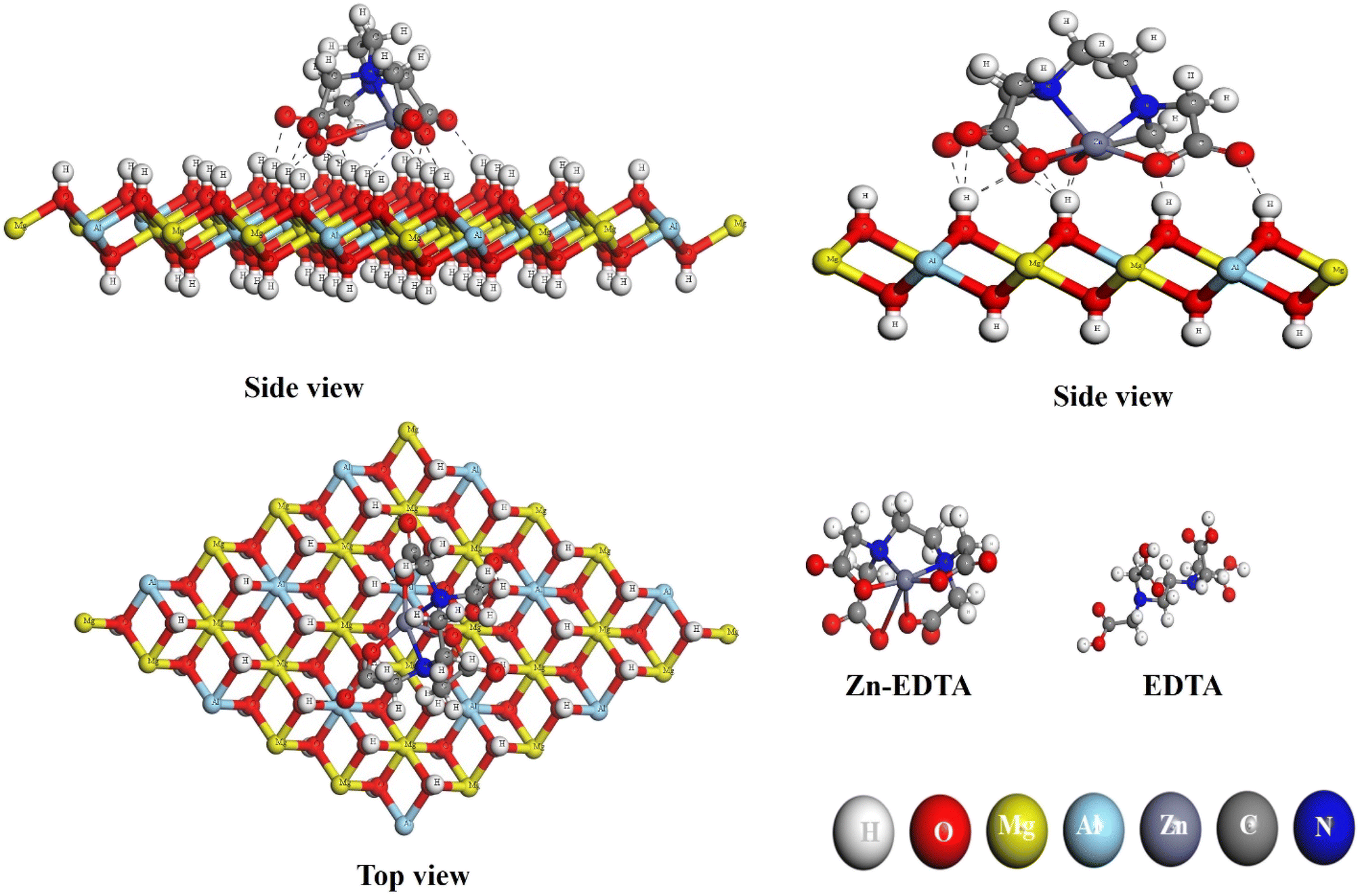
Fig. 10 offers a comprehensive visual representation of the final chemical interaction between the EDTA–Zn monomer and the Mg–Al LDH surface. It effectively captures this interaction from various angles, highlighting the conclusive state of their bonding. When observed from the side, the interaction becomes prominently visible, driven by the electrophilic sites on the oxygen atoms. These electrophilic sites facilitate the formation of hydrogen bonds with varying bond lengths (within 2.5 Å), connecting them to the hydrogen atoms located on the LDH layer's surface. Notably, this bond formation results in the intriguing formation of a self-assembled network. Oxygen atoms from one monomer's electrophilic site establish hydrogen bonds with the nucleophilic site of another monomer, spontaneously forming this network. Consequently, the oxygen atoms within the EDTA–Zn self-assembled network interact randomly with the hydrogen atoms on the surface of Mg–Al LDH.
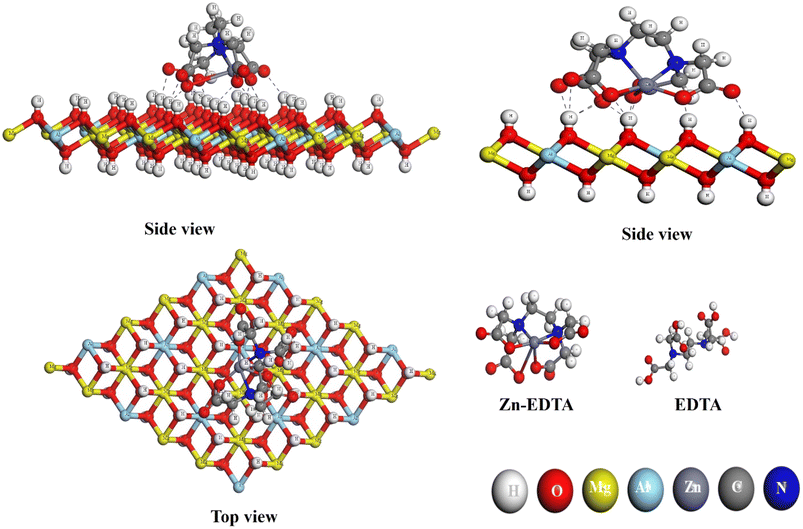 |
| | Fig. 10 EDTA–Zn monomer configuration on the layers of Mg–Al LDH from both side and top perspectives, respectively. | |
3.7 Formation and inhibition mechanisms
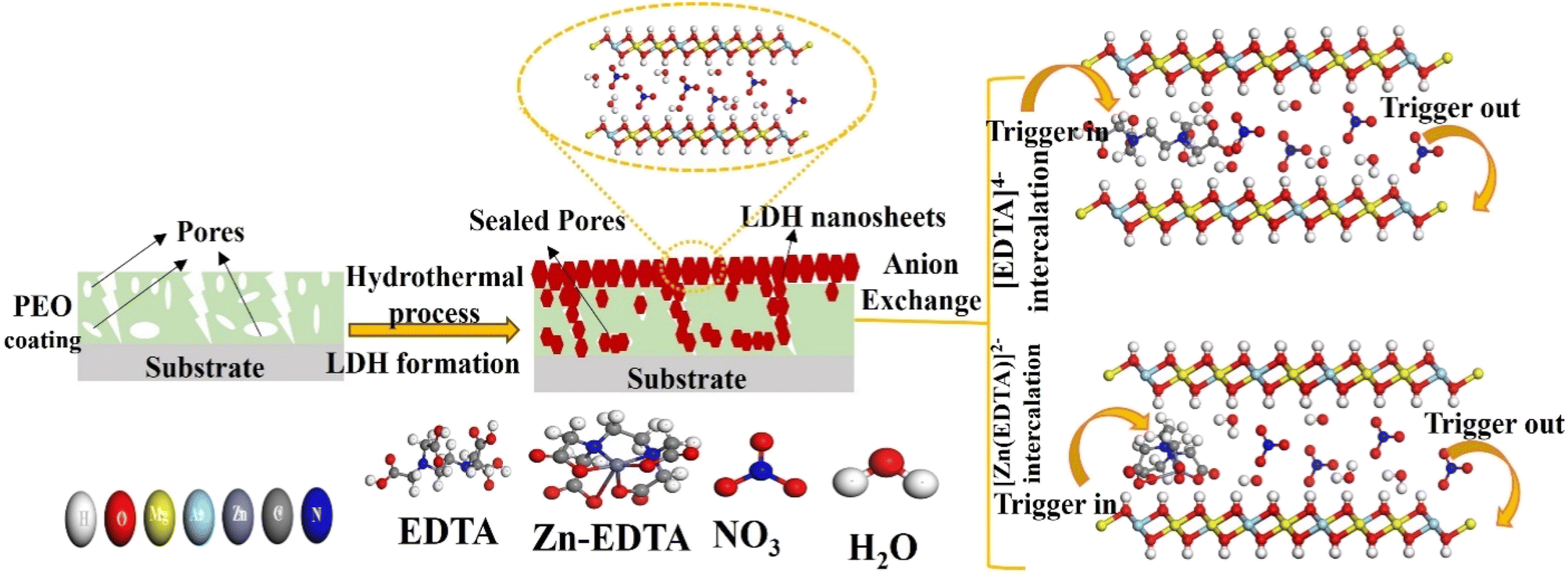
Fig. 11 illustrates the formation mechanism of PEO, LDH, LDH–EDTA and LDH–EDTA–Zn on the AZ31 alloy. The PEO-coated film formed by utilizing aluminate electrolyte exhibits a distinct pore structure, with a compact inner layer and a porous outer layer, containing MgAl2O4 and MgO as main compounds. The rapid and substantial dissolution of the porous inorganic PEO coating occurred when the specimen was submerged in a NaCl solution due to the infiltration of highly reactive chloride ions into the coating's pores and defects. To enhance the stability of the coating, a layer of LDH flakes are grown on the PEO surface. These LDH flakes completely covered the entire surface of the coating, effectively sealing all the pores. The growth of LDH nanoflakes on the PEO layer is achieved using a hydrothermal approach, which relies on the dissolution of the substrate and the hydration of the PEO film. Under hydrothermal conditions, MgO undergoes hydrolysis, resulting in the creation of Mg(OH)2. Additionally, the dissolution of MgAl2O4 results in the formation of both MgO and Al2O3. Finally, Al2O3 interacts with Mg(OH)2, NO3− ions, and the water present in the solution, leading to the production of LDH.75,76 The chemical reactions involved in the formation of LDH are given in the following equations77,78 (eqn (5)–(8)):| | | Al2O3 + 3H2O + 2OH− → 2Al(OH)4− | (7) |
| | | Mg(OH)2 + Al(OH)4− + H2O + NO3− → MgAl–NO3–LDH | (8) |
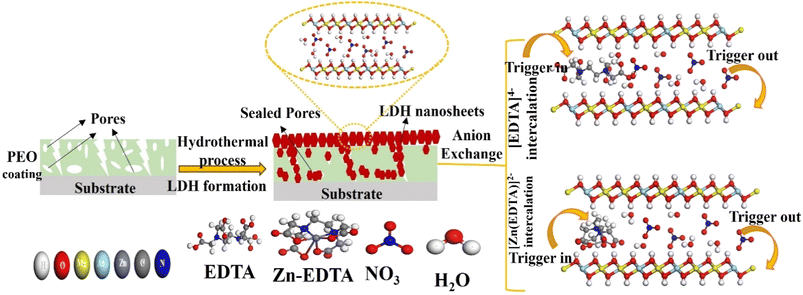 |
| | Fig. 11 Schematic representation of the formation mechanism for a PEO/LDH film and the anion exchange of [EDTA]4− and [Zn(EDTA)]2− within the LDH galleries with the NO3− anion. | |
Hydrothermal growth leads to a compact structure with random shapes and suitable flake sizes, improving corrosion resistance by effectively sealing pores, especially compared to PEO films. Additionally, LDHs with NO3− interlayer anions act as effective nano-traps for Cl− ions, creating a concentration gradient barrier against them, further enhancing their corrosion resistance.1 The structural defects within the LDH flakes are addressed through anion exchange using EDTA within the LDH galleries, carried out under hydrothermal conditions. This process leads to the replacement of NO3− anions by EDTA4−, resulting in LDH flakes that are not only denser but also smaller in size. The size disparity in LDH flakes can be attributed to the differential rates of LDH crystal formation. A rapid nucleation rate results in a greater quantity of LDH flakes, which tend to have smaller average dimensions. This outcome is primarily influenced by the strong chelation ability of EDTA, which readily forms stable complexes with existing metal ions.79 Moreover, in an alkaline solution, EDTA exists as the tetravalent anion [EDTA]4−, which forms highly stable complexes with Mg2+ and Al3+ ions within LDH galleries. This interaction facilitates the recrystallization of LDH flakes, resulting in a denser and more stable LDH film structure. This compact LDH film serves as an effective barrier in aggressive NaCl solutions, preventing corrosion and film damage.48 The chemical reaction involved in anion exchange is represented in the following equation (eqn (9)):
| | | MgAl–NO3–LDH + H2O + EDTA → MgAl–EDTA–LDH + NO3− | (9) |
On the other hand, when the Zn–EDTA complex is introduced into LDH under acidic conditions (pH = 4), a reaction occurs where EDTA4− ions interact with Zn2+ ions, resulting in the formation of [Zn(EDTA)]2− anions. These anions replace the NO3− anions within the LDH layers through anion exchange. The Zn–EDTA complex exhibits strong chelating abilities, leading to the formation of an uneven surface layer on the LDH which is consistent with results of cross sectional morphology as discussed earlier. However, it is worth noting that the Zn–EDTA complex also dissolves the existing metals present in the LDH under the acidic environment, introducing structural defects.80,81 As a consequence, the electrochemical stability of LDH–EDTA–Zn is slightly lower compared to that of LDH–EDTA. Nevertheless, this property enhances porosity within the LDH layers, which proves advantageous for its photocatalytic properties. LDH–EDTA–Zn offers corrosion protection that is as effective as LDH–EDTA, but it excels in photocatalytic degradation properties, making it the best sample for dual applications.
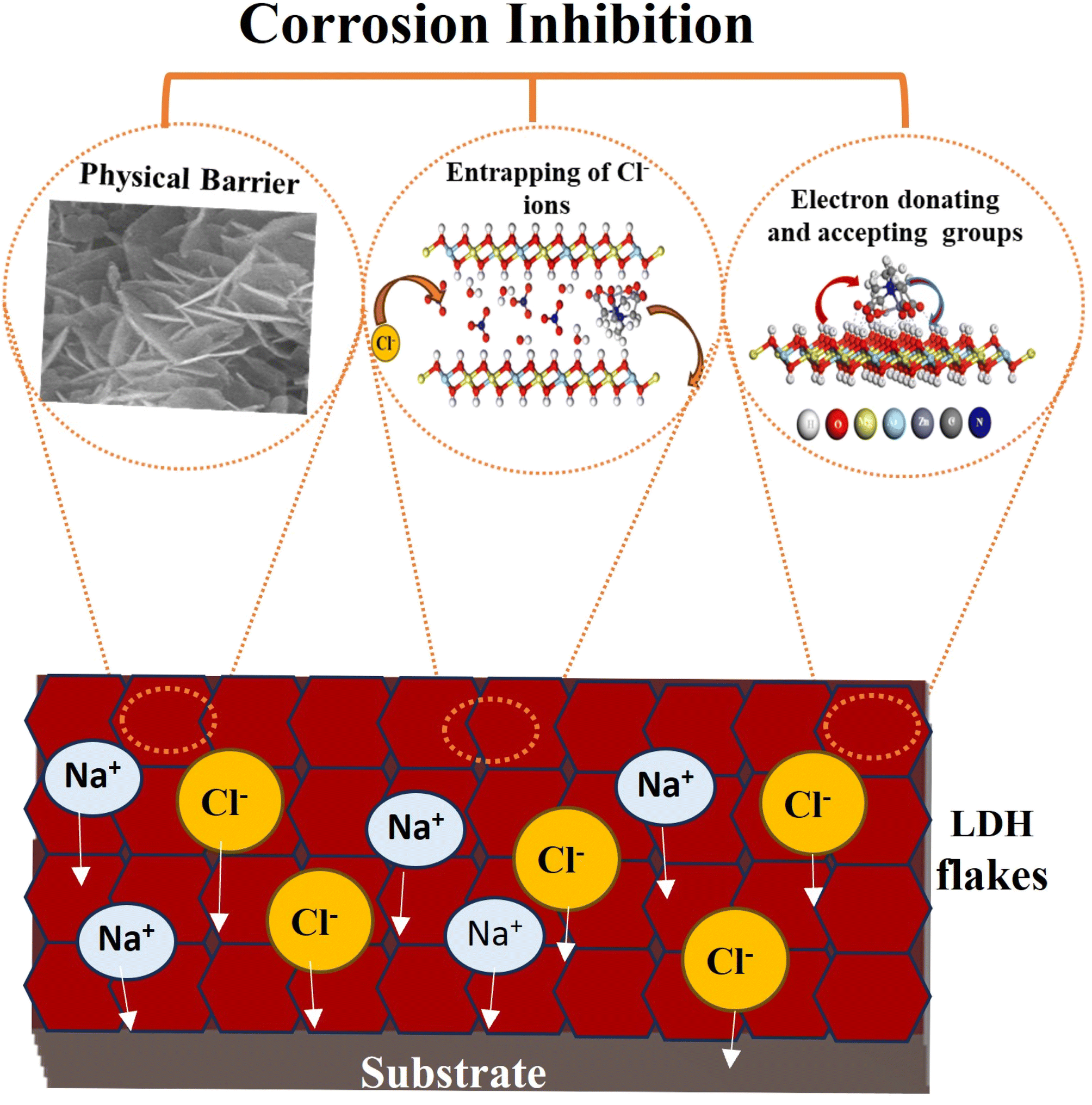
Fig. 12 illustrates the detailed corrosion inhibition mechanism revealed by the LDH–EDTA and LDH–EDTA–Zn samples. The corrosion protection offered by LDH films is the result of a multi-dimensional influence, encompassing not just the typical physical barrier but also the chemical inhibition of electron-donating functional groups. Additionally, these films possess ion-exchange capabilities that effectively engage with corrosive chloride ions. (i) The existence of self-arranged EDTA–Zn structures results in strong interaction with LDH, resulting in a significant enhancement in the electrochemical stability of the compound coatings. This layer offers extensive protection across the LDH exterior, proficiently sealing any imperfections and effectively blocking the infiltration of destructive chloride ions into the Mg substrate. (ii) The remarkable anion-exchange capacity of LDHs enables the release of anionic species from the LDH interlayers into corrosive environments by replacing them with corrosive chloride anions. All of this isolates aggressive chloride ions within the interlayers, leading to a subsequent reduction in their concentrations and, consequently, an improved corrosion protection effect. (iii) Electron-donating functional groups enhance the inhibition of corrosive ions, whereas electron-removing functional groups act as a hindrance.82 In the case of adsorbed EDTA–Zn species, there are two electron-donating functional groups, namely C–N and –OH, and one electron-withdrawing group (CO). Consequently, due to the majority of electron-donating functional groups, they exert a dominant influence, effectively creating an effective protective barrier around the LDH and PEO layers, thus hindering the penetration of corrosive ions. Sometimes if chloride ions, known for their corrosive properties, unintentionally infiltrate the LDH flakes and dissolve the PEO coating to access the substrate, they will engage with the metal ions, resulting in the creation of complexes with EDTA because of its potent chelating abilities. As a result, this procedure will ensure the protection of the metal surfaces beneath. In summary, the EDTA–Zn network formation plays a pivotal role in creating robust connections with LDH, leading to enhanced stability. Furthermore, the increased stability can be attributed to the greater number of electron-donating functional groups in both LDH–EDTA and LDH–EDTA–Zn samples, as well as the effective capture of chloride ions through ion exchange. These findings are consistent with the results obtained from the EIS and polarization curves.
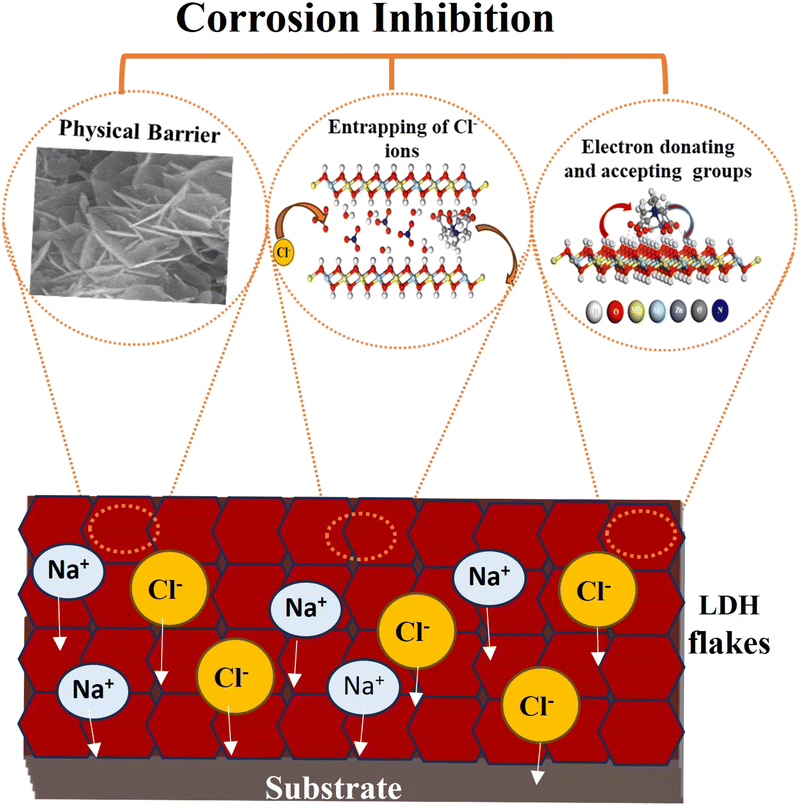 |
| | Fig. 12 The corrosion inhibition mechanism of LDH–EDTA and LDH–EDTA–Zn relies on a multifaceted approach that includes the creation of a physical barrier, the entrapment of chlorides through ion exchange, and the involvement of electron-donating and electron-accepting functional groups. | |
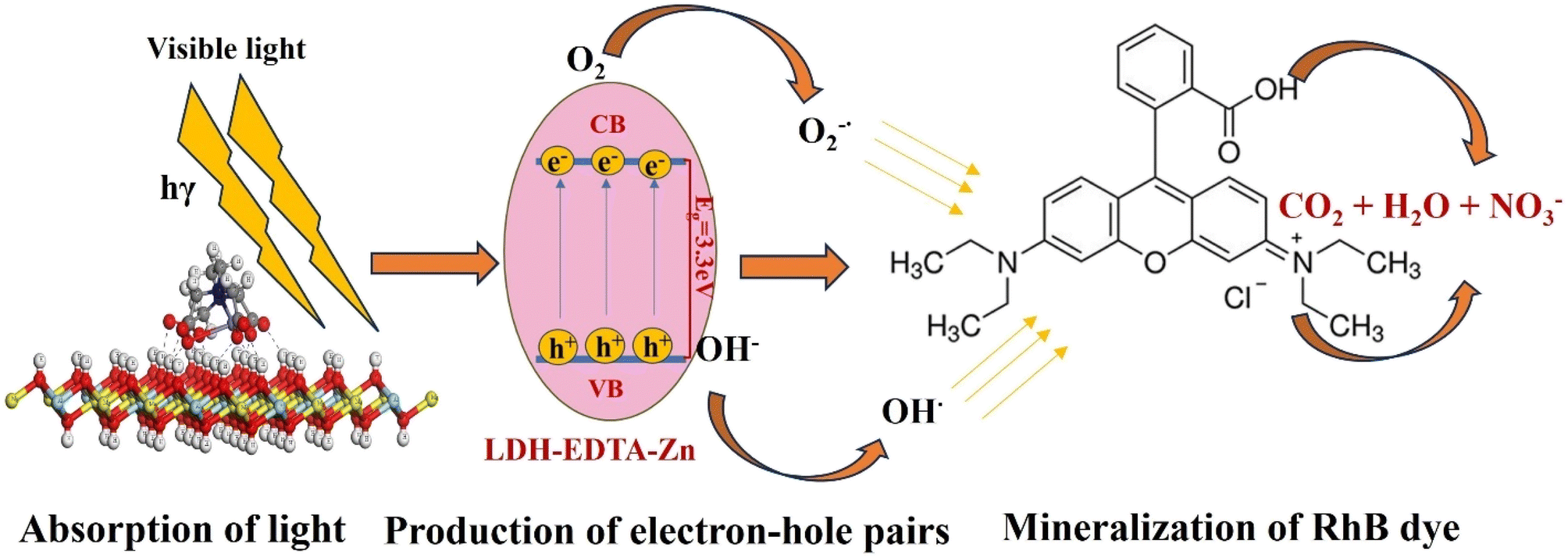
3.8 Photocatalytic degradation mechanism
The efficiency of photocatalytic activity is determined by how materials absorb light and the mobility of electron–hole pairs formed as a result of this absorption. This relates directly to the electronic structure of the material, and improved absorption and mobility enhance the probability of electrons and holes acquiring active reaction sites on the photocatalyst's surface.83 Based on our experimental data, the potential mechanism for LDH–EDTA–Zn is proposed and discussed below.
Initially, when LDH–EDTA–Zn absorbs a photon with energy equivalent to or larger than its band gap, it excites electrons, shifting them from the valence to the conduction band and generating electron–hole pairs (eqn (10)):
| | | LDH–EDTA–Zn + hγ → (h+)VB + (e−)CB | (10) |
Secondly, the aggressive interactions between the electron–hole pairs and the initiator molecules (H2O2) led to the production of highly reactive hydroxyl radicals (eqn (11) and (12)):
| | 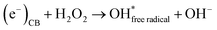 | (11) |
| | 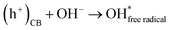 | (12) |
Moreover, electron–hole pairs can react with water and dissolved oxygen, leading to increased generation of hydroxyl and hydroperoxyl radicals (eqn (13)–(15)):
| | | (e−)CB + O2(dissolved) → O2− | (13) |
| | 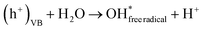 | (14) |
Finally, these hydroxyl radicals and superoxide radicals played a pivotal role in oxidizing RhB molecules into intermediate compounds, resulting in harmless end products such as CO2, NO3−, and H2O84 (eqn (16)):
| | | O2− + OH* + RhB → intermediate compounds → CO2 + H2O + NO3− | (16) |
Furthermore, the hydroxide groups on the LDH–EDTA–Zn surface successfully captured photoinduced holes (h+), avoiding electron recombination and significantly boosting photocatalytic activity.83 The LDH–EDTA–Zn complex, characterized by a low band gap of around 3.3 eV, effectively extends the lifespan of photoinduced electron–hole pairs. This results in a remarkable enhancement of up to 99.7% in the degradation rate of RhB. The accelerated photocatalytic degradation of RhB into non-hazardous compounds is primarily attributed to the immediate reactivity of radical species, specifically hydroxide and superoxide radicals. Experimental evidence, such as scavenger tests, has demonstrated that superoxides play a more significant role compared to hydroxides in the overall degradation process. The schematic illustration of rhodamine degradation by LDH–EDTA–Zn is depicted in Fig. 13. While numerous strategies have been explored in previous literature for the utilization of LDH-based photocatalysts in the degradation of RhB dye in aqueous solutions, our study introduced a novel LDH–EDTA–Zn catalyst. Furthermore, it is noteworthy to observe that within the LDH–EDTA–Zn sample, EDTA not only forms bonds with zinc but also dissolves any existing metals present in the LDH when subjected to an acidic solution. This phenomenon introduces structural irregularities. As a result, it enhances the porosity within the LDH layers, a quality that proves advantageous for its photocatalytic performance.81 Moreover, to highlight the advancements in this study, a comparative table has been constructed, featuring previously reported LDH, MOF, and general catalysts. This newly developed catalyst stands out by showing markedly enhanced catalytic activity within a brief period. The promising features, such as its superior catalytic efficiency within a short period, are summarized in Table 3.
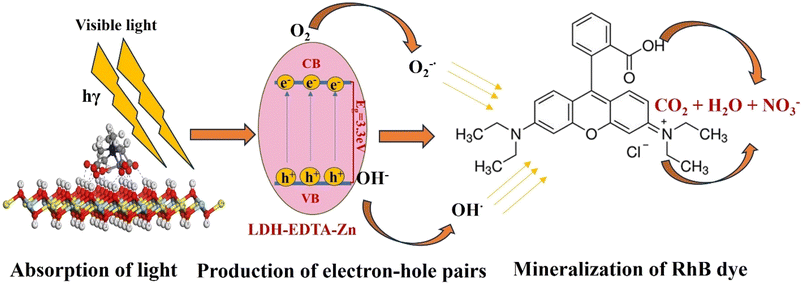 |
| | Fig. 13 A schematic representation of the photocatalytic degradation mechanism employed for RhB solution using the LDH–EDTA–Zn sample in the presence of an H2O2 initiator. | |
Table 3 Comparison of the photocatalytic behavior of LDH–EDTA–Zn with other LDH-based catalysts for RhB degradation
| Photocatalyst |
RhB (mg L−1) |
Light source |
Efficiency (%) |
Time (min) |
Ref. |
| BiOCl–NiFe–LDH |
20 |
Visible light |
93.3 |
120 |
85
|
| Zn–Al-LDH |
2 |
UV light |
98.2 |
100 |
69
|
| Zn/M–NO3-LDH (M = Al, Fe, Ti, and Fe/Ti) |
3 |
Visible light |
72 |
120 |
83
|
| Bi2O3–CoAl-LDHs |
50 |
Visible light |
90.36 |
180 |
84
|
| Ag2WO4/Zn–Cr LDH |
10 |
Visible light |
99.5 |
100 |
86
|
| CeO2–CoAl-LDH |
10 |
UV light |
96.9 |
30 |
87
|
| NiFe-LDH/N-rGO/g-C3N4 |
20 |
Visible light |
95 |
120 |
88
|
| MoS2/NiFe-LDH |
20 |
Visible light |
90 |
120 |
89
|
| Zn-MOF |
5 |
UV light |
94.1 |
150 |
90
|
| N–BiVO4 |
0.6 |
Visible light |
93.1 |
180 |
91
|
| BiOCl/Ni-MOF-74 |
25 |
UV light |
97 |
100 |
92
|
| Bi2WO6/Bi-MOF |
10 |
UV light |
96 |
60 |
93
|
| ZnO/CuO |
50 |
Visible light |
75.4 |
300 |
94
|
| MgAl-LDH |
20 |
Visible light |
99 |
35 |
This work |
| LDH–EDTA |
20 |
Visible light |
99.4 |
30 |
This work |
| LDH–EDTA–Zn |
20 |
Visible light |
99.7 |
25 |
This work |
4. Conclusions
In this study, a dual-functional material was successfully fabricated on an AZ31 alloy substrate through PEO, where EDTA–Zn complex molecules are intercalated into MgAl LDH galleries under acidic conditions, and it exhibited both high chemical stability and photocatalytic activity. The research results revealed that the PEO coating exhibited a diverse array of micropores, which are completely surrounded by flake-like structures. This envelopment effectively sealed the pores, consequently enhancing their overall stability. The findings also highlighted that the intercalation of [EDTA]4− and [Zn(EDTA)]2− anions in the LDH film had a pronounced impact on the formation of the LDH film, resulting in a more compact arrangement of smaller LDH nanosheets. The electrochemical analysis results reveal that LDH–EDTA–Zn, while offering slightly lower corrosion protection than LDH–EDTA, stands out due to its exceptional performance in the photocatalytic degradation of Rhodamine dye, making it the preferred choice among the samples. The improved photocatalytic performance of LDH–EDTA–Zn is attributed to smaller, denser flakes with a reduced energy band gap, while enhanced electrochemical stability is achieved through self-assembled EDTA–Zn complexes that seal pores and trap chloride ions via ion exchange in LDH galleries, and the presence of numerous electron-donating functional groups. Moreover, theoretical calculations revealed that EDTA–Zn monomers possessed both electrophilic and nucleophilic characteristics, enabling the formation of a self-assembled EDTA–Zn complex network. This network effectively sealed surface imperfections and defects on the LDH substrate, significantly enhancing the coating's compactness, and notably, these theoretical findings closely matched the experimental results, further validating the study's significance and reliability.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) funded by the Korean government (MSIT) (No. 2022R1A2C1006743).
References
- G. Zhang, L. Wu, A. Tang, Y. Ma, G.-L. Song, D. Zheng, B. Jiang, A. Atrens and F. Pan, Corros. Sci., 2018, 139, 370–382 CrossRef CAS.
- M. Kaseem, A. Repycha Safira and A. Fattah-alhosseini, J. Magnesium Alloys, 2023 DOI:10.1016/j.jma.2023.11.004.
- G. Zhang, E. Jiang, L. Wu, A. Tang, A. Atrens and F. Pan, J. Magnesium Alloys, 2022, 10, 1351–1357 CrossRef CAS.
- M. Kaseem, T. Hussain, U. R. Zeeshan, H. W. Yang, B. Dikici and Y. G. Ko, Chem. Eng. J., 2021, 420, 129737 CrossRef CAS.
- M. A. Khan, A. Fattah-alhosseini and M. Kaseem, Inorg. Chem. Commun., 2023, 153, 110739 CrossRef CAS.
- A. Ali Khan, M. Tahir and N. Khan, J. Energy Chem., 2023, 84, 242–276 CrossRef CAS.
- Y. Li, X. Lu, M. Serdechnova, C. Blawert, M. L. Zheludkevich, K. Qian, T. Zhang and F. Wang, J. Magnesium Alloys, 2023, 11, 1236–1246 CrossRef CAS.
- S. Jung Kim, H. Choi, J. Ho Ryu, K. Min Kim, S. Mhin, A. Kumar Nayak, J. Bang, M. Je, G. Ali, K. Yoon Chung, K.-H. Na, W.-Y. Choi, S. Yeo, J. Uk Jang and H. Han, J. Energy Chem., 2023, 81, 82–92 CrossRef CAS.
- K. Gu, X. Zhu, D. Wang, N. Zhang, G. Huang, W. Li, P. Long, J. Tian, Y. Zou, Y. Wang, R. Chen and S. Wang, J. Energy Chem., 2021, 60, 121–126 CrossRef CAS.
- Y. Zhao, X. Jia, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung, D. O'Hare and T. Zhang, Adv. Energy Mater., 2016, 6, 1501974 CrossRef.
- X. Xiong, Y. Zhao, R. Shi, W. Yin, Y. Zhao, G. I. N. Waterhouse and T. Zhang, Sci. Bull., 2020, 65, 987–994 CrossRef CAS PubMed.
- Y. Zhao, G. Chen, T. Bian, C. Zhou, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung, L. J. Smith, D. O'Hare and T. Zhang, Adv. Mater., 2015, 27, 7824–7831 CrossRef CAS PubMed.
- E. R. soliman, Y. H. Kotp, E. R. Souaya, K. A. Guindy and R. G. M. Ibrahim, Composites, Part B, 2019, 175, 107131 CrossRef CAS.
- J. Tedim, M. L. Zheludkevich, A. C. Bastos, A. N. Salak, A. D. Lisenkov and M. G. S. Ferreira, Electrochim. Acta, 2014, 117, 164–171 CrossRef CAS.
- M. Serdechnova, A. N. Salak, F. S. Barbosa, D. E. L. Vieira, J. Tedim, M. L. Zheludkevich and M. G. S. Ferreira, J. Solid State Chem., 2016, 233, 158–165 CrossRef CAS.
- Y. Chen, L. Wu, W. Yao, Y. Chen, Z. Zhong, W. Ci, J. Wu, Z. Xie, Y. Yuan and F. Pan, Corros. Sci., 2022, 194, 109941 CrossRef CAS.
- C. M. Terzi, E. H. dos Santos, C. Carvalho, V. Prevot, F. Wypych, C. Forano and S. Nakagaki, Catal. Today, 2023, 422, 114221 CrossRef CAS.
- D. Jiang, X. Xia, J. Hou, G. Cai, X. Zhang and Z. Dong, Chem. Eng. J., 2019, 373, 285–297 CrossRef CAS.
- B. Kuznetsov, M. Serdechnova, J. Tedim, M. Starykevich, S. Kallip, M. P. Oliveira, T. Hack, S. Nixon, M. G. S. Ferreira and M. L. Zheludkevich, RSC Adv., 2016, 6, 13942–13952 RSC.
- J.-l. Chen, L. Fang, F. Wu, X.-g. Zeng, J. Hu, S.-f. Zhang, B. Jiang and H.-j. Luo, Trans. Nonferrous Met. Soc. China, 2020, 30, 2424–2434 CrossRef CAS.
- P. Benito, F. M. Labajos, L. Mafra, J. Rocha and V. Rives, J. Solid State Chem., 2009, 182, 18–26 CrossRef CAS.
- R.-C. Zeng, Z.-G. Liu, F. Zhang, S.-Q. Li, H.-Z. Cui and E.-H. Han, J. Mater. Chem. A, 2014, 2, 13049–13057 RSC.
- M. Kaseem, T. Hussain, Z. U. Rehman, M. J. Banisalman and Y. G. Ko, Surf. Coat. Technol., 2020, 399, 126200 CrossRef CAS.
- A. López-Ortega, J. L. Arana, E. Rodríguez and R. Bayón, Corros. Sci., 2018, 143, 258–280 CrossRef.
- S. Stojadinović, N. Tadić, N. Radić, B. Grbić and R. Vasilić, Surf. Coat. Technol., 2018, 337, 279–289 CrossRef.
- G. I. Marinina, M. S. Vasilyeva, A. S. Lapina, A. Y. Ustinov and V. S. Rudnev, J. Electroanal. Chem., 2013, 689, 262–268 CrossRef CAS.
- T. S. N. Sankara Narayanan, I. S. Park and M. H. Lee, Prog. Mater. Sci., 2014, 60, 1–71 CrossRef CAS.
- M. Kaseem, T. Hussain and Y. G. Ko, J. Alloys Compd., 2020, 822, 153566 CrossRef CAS.
- F. Chen, P. Yu and Y. Zhang, J. Alloys Compd., 2017, 711, 342–348 CrossRef CAS.
- M. Mohedano, M. Serdechnova, M. Starykevich, S. Karpushenkov, A. C. Bouali, M. G. S. Ferreira and M. L. Zheludkevich, Mater. Des., 2017, 120, 36–46 CrossRef CAS.
- Y. Liu, T. Yu, R. Cai, Y. Li, W. Yang and J. Caro, RSC Adv., 2015, 5, 29552–29557 RSC.
- P. Wu, J. Zhou, X. Wang, Y. Dai, Z. Dang, N. Zhu, P. Li and J. Wu, Desalination, 2011, 277, 288–295 CrossRef CAS.
- L. Wang, L. Yang, Y. Li, Y. Zhang, X. Ma and Z. Ye, Chem. Eng. J., 2010, 163, 364–372 CrossRef CAS.
- S. V. Prasanna, R. A. P. Rao and P. V. Kamath, J. Colloid Interface Sci., 2006, 304, 292–299 CrossRef CAS PubMed.
- R. Rojas, M. R. Perez, E. M. Erro, P. I. Ortiz, M. A. Ulibarri and C. E. Giacomelli, J. Colloid Interface Sci., 2009, 331, 425–431 CrossRef CAS PubMed.
- R. Rusdi, A. A. Rahman, N. S. Mohamed, N. Kamarudin and N. Kamarulzaman, Powder Technol., 2011, 210, 18–22 CrossRef CAS.
- M. Štekláč and M. Breza, Acta Chim. Slovaca, 2018, 11, 6–10 CrossRef.
- M. Chafiq, A. Chaouiki, T. Suhartono, F. Hazmatulhaq and Y. G. Ko, Chem. Eng. J., 2023, 462, 142239 CrossRef CAS.
- M. Kaseem and Y. G. Ko, Ultrason. Sonochem., 2018, 49, 316–324 CrossRef CAS PubMed.
- C.-Y. Li, L. Gao, X.-L. Fan, R.-C. Zeng, D.-C. Chen and K.-Q. Zhi, Bioact. Mater., 2020, 5, 364–376 Search PubMed.
- S. Kong, L. Wang, M. Xie, Y. Ma and L. Su, J. Electroanal. Chem., 2022, 925, 116927 CrossRef CAS.
- S. S. Montiel Guerrero, Y. E. Durmus, K. Dzieciol, S. Basak, H. Tempel, S. van Waasen, H. Kungl and R.-A. Eichel, Batteries Supercaps, 2021, 4, 1830–1842 CrossRef CAS.
- F. Shi, J. Zhao, M. Tabish, J. Wang, P. Liu and J. Chang, J. Magnesium Alloys, 2023, 11(7), 2541–2557 CrossRef CAS.
- G. A. Borges, G. M. D. Ferreira, K. P. F. Siqueira, A. Dias, K. O. N. Navarro, S. J. B. E. Silva, G. D. Rodrigues and A. B. Mageste, J. Colloid Interface Sci., 2020, 575, 194–205 CrossRef CAS PubMed.
- H. Chen, J. Lin, N. Zhang, L. Chen, S. Zhong, Y. Wang, W. Zhang and Q. Ling, J. Hazard. Mater., 2018, 345, 1–9 CrossRef CAS PubMed.
- M. Kaseem and Y. G. Ko, J. Alloys Compd., 2019, 787, 772–778 CrossRef CAS.
- G. A. Gebreslase, M. V. Martínez-Huerta and M. J. Lázaro, J. Energy Chem., 2022, 67, 101–137 CrossRef CAS.
- Z. Dou, Y. Zhang, T. Shulha, R. Cui, M. Serdechnova, H. Tian, T. Yan, C. Blawert, L. Li, M. L. Zheludkevich and F. Chen, Surf. Coat. Technol., 2022, 439, 128414 CrossRef CAS.
- L. Wu, D. Yang, G. Zhang, Z. Zhang, S. Zhang, A. Tang and F. Pan, Appl. Surf. Sci., 2018, 431, 177–186 CrossRef CAS.
- T. Stimpfling, F. Leroux and H. Hintze-Bruening, Appl. Clay Sci., 2013, 83–84, 32–41 CrossRef CAS.
- M. R. Pérez, I. Pavlovic, C. Barriga, J. Cornejo, M. C. Hermosín and M. A. Ulibarri, Appl. Clay Sci., 2006, 32, 245–251 CrossRef.
- M. Kaseem and Y. G. Ko, Composites, Part B, 2019, 176, 107225 CrossRef CAS.
- R. Blanga, L. Burstein, M. Berman, S. G. Greenbaum and D. Golodnitsky, J. Electrochem. Soc., 2015, 162, D3084 CrossRef CAS.
- S. K. Alla, A. D. Verma, V. Kumar, R. K. Mandal, I. Sinha and N. K. Prasad, RSC Adv., 2016, 6, 61927–61933 RSC.
- G. Zhang, L. Wu, A. Tang, B. Weng, A. Atrens, S. Ma, L. Liu and F. Pan, RSC Adv., 2018, 8, 2248–2259 RSC.
- Z. Chen, S. Pronkin, T.-P. Fellinger, K. Kailasam, G. Vilé, D. Albani, F. Krumeich, R. Leary, J. Barnard, J. M. Thomas, J. Pérez-Ramírez, M. Antonietti and D. Dontsova, ACS Nano, 2016, 10, 3166–3175 CrossRef CAS PubMed.
- S. Ravi, S. Zhang, Y.-R. Lee, K.-K. Kang, J.-M. Kim, J.-W. Ahn and W.-S. Ahn, J. Ind. Eng. Chem., 2018, 67, 210–218 CrossRef CAS.
- S. Ravi, P. Puthiaraj and W.-S. Ahn, J. CO2 Util., 2017, 21, 450–458 CrossRef CAS.
- A. S. Haja Hameed, C. Karthikeyan, S. Sasikumar, V. Senthil Kumar, S. Kumaresan and G. Ravi, J. Mater. Chem. B, 2013, 1, 5950–5962 RSC.
- H. A. Jaffery, M. F. M. Sabri, S. M. Said, S. W. Hasan, I. H. Sajid, N. I. M. Nordin, M. M. I. Megat Hasnan, D. A. Shnawah and C. V. Moorthy, J. Alloys Compd., 2019, 810, 151925 CrossRef CAS.
- T. Zehra, B. Dikici, A. Dafali and M. Kaseem, Prog. Org. Coat., 2023, 182, 107677 CrossRef CAS.
- J. Tedim, M. L. Zheludkevich, A. N. Salak, A. Lisenkov and M. G. S. Ferreira, J. Mater. Chem., 2011, 21, 15464–15470 RSC.
- M. Kaseem, B. Dikici, A. Dafali and A. Fattah-alhosseini, J. Magnesium Alloys, 2023, 11, 1618–1628 CrossRef CAS.
- M. Kaseem, S. Fatimah, N. Nashrah and Y. G. Ko, Prog. Mater. Sci., 2021, 117, 100735 CrossRef CAS.
- M. Mohedano, C. Blawert and M. L. Zheludkevich, Mater. Des., 2015, 86, 735–744 CrossRef CAS.
- H. Tamura, N. Ito, M. Kitano and S. Takasaki, Corros. Sci., 2001, 43, 1675–1691 CrossRef CAS.
- N. S. Topare, D. S. Bhutada and P. G. Bansod, Mater. Today: Proc., 2022, 51, 125–128 CAS.
- X. Peng, S. Xie, S. Xiong, R. Li, P. Wang, X. Zhang, Z. Liu, L. Hu, B. Gao, P. Kelly and P. K. Chu, J. Energy Chem., 2023, 81, 574–582 CrossRef CAS.
- S. Wu, H. Liang, Z. Zhang, Q. Zhang, Q. Han, J. Wang, M. Gao, H. Fan, J. Yang and J. Lang, Opt. Mater., 2022, 131, 112636 CrossRef CAS.
- N. Li, M. He, X. Lu, L. Liang, R. Li, B. Yan and G. Chen, Sci. Total Environ., 2021, 761, 143268 CrossRef CAS PubMed.
- Q. Chen, L. Wu, X. Zhao and X.-J. Yang, J. Mol. Liq., 2022, 353, 118794 CrossRef CAS.
- H. He, L. Zhao, L. Wei, J. Wang, W. Mao, S. Liu, Y. Deng, Z. Zhan, H. Bai and B. Liang, Mater. Lett., 2023, 343, 134405 CrossRef CAS.
- M. Mercina, J. Reena Priya, D. Jayaraman and V. Joseph, Mater. Today: Proc., 2022, 49, 2607–2610 CAS.
- J. Priscilla, D. Arul Dhas, I. Hubert Joe and S. Balachandran, J. Mol. Struct., 2023, 1273, 134261 CrossRef CAS.
- G. Zhang, L. Wu, A. Tang, H. Pan, Y. Ma, Q. Zhan, Q. Tan, F. Pan and A. Atrens, J. Electrochem. Soc., 2018, 165, C317 CrossRef CAS.
- B.-s. Liu, Y.-f. Kuang, Y.-s. Chai, D.-q. Fang, M.-g. Zhang and Y.-h. Wei, J. Magnesium Alloys, 2016, 4, 220–229 CrossRef CAS.
- A. Atrens, G.-L. Song, F. Cao, Z. Shi and P. K. Bowen, J. Magnesium Alloys, 2013, 1, 177–200 CrossRef CAS.
- Z. P. Xu and G. Q. Lu, Chem. Mater., 2005, 17, 1055–1062 CrossRef CAS.
- E. Petrova, M. Serdechnova, T. Shulha, S. V. Lamaka, D. C. F. Wieland, P. Karlova, C. Blawert, M. Starykevich and M. L. Zheludkevich, Sci. Rep., 2020, 10, 8645 CrossRef CAS PubMed.
- Y. Wu, X. Wang, X. Zhang, Y. Lu, M. Chen, Y. Sun and P. Ye, Chemosphere, 2022, 298, 134332 CrossRef CAS PubMed.
- M. Wei and J. Chen, Environ. Sci. Pollut. Res., 2016, 23, 23123–23133 CrossRef CAS PubMed.
- H. Assad and A. Kumar, J. Mol. Liq., 2021, 344, 117755 CrossRef CAS.
- S.-J. Xia, F.-X. Liu, Z.-M. Ni, J.-L. Xue and P.-P. Qian, J. Colloid Interface Sci., 2013, 405, 195–200 CrossRef CAS PubMed.
- L. Zhang, Y. Meng, H. Shen, J. Li, C. Yang, B. Xie and S. Xia, Appl. Surf. Sci., 2021, 567, 150760 CrossRef CAS.
- J. Ma, J. Ding, L. Yu, L. Li, Y. Kong and S. Komarneni, Appl. Clay Sci., 2015, 109–110, 76–82 CrossRef CAS.
- J. Zhu, H. Fan, J. Sun and S. Ai, Sep. Purif. Technol., 2013, 120, 134–140 CrossRef CAS.
- C. Yang, G. Zhang, Y. Meng, G. Pan, Z. Ni and S. Xia, J. Hazard. Mater., 2021, 408, 124908 CrossRef CAS PubMed.
- S. Nayak and K. M. Parida, Sci. Rep., 2019, 9, 2458 CrossRef PubMed.
- S. Nayak, G. Swain and K. Parida, ACS Appl. Mater. Interfaces, 2019, 11, 20923–20942 CrossRef CAS PubMed.
- Y. N. Hendri, Y. Rati, A. K. E. Auni, R. Marlina, M. M. Munir, A. Patah and Y. Darma, Mater. Today Commun., 2023, 37, 107011 CrossRef CAS.
- Y. Song, J. Lu, M. Li, Y. Yan, N. Liu, H. Kang, Y. Wang and R. Wang, J. Photochem. Photobiol., A, 2023, 445, 115046 CrossRef CAS.
- M. Xu, H. Hu, J. Jin, Y. Yang, W. Jin, F. Dong, Z. Zhang, M. Shao and Y. Wan, Mater. Sci. Semicond. Process., 2023, 168, 107832 CrossRef CAS.
- Y. Yang, M. Xu, L. Ai, N. Guo, C. Leng, C. Tan, M. Lu, L. Wang, L. Huang and D. Jia, J. Environ. Chem. Eng., 2023, 11, 109873 CrossRef CAS.
- G. Cabello-Guzmán, M. Seguel, L. Fernández, C. Caro, C. Suarez, M. Matus, C. Cifuentes, F. Bustos and K. Ariz, Inorg. Chem. Commun., 2023, 152, 110695 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 and
Mosab
Kaseem
and
Mosab
Kaseem