DOI:
10.1039/D3TA07951A
(Paper)
J. Mater. Chem. A, 2024,
12, 5711-5718
Coordination environment dominated catalytic selectivity of photocatalytic hydrogen and oxygen reduction over switchable gallium and nitrogen active sites†
Received
22nd December 2023
, Accepted 29th January 2024
First published on 29th January 2024
Abstract
Catalytic properties of single-atom catalysts are very sensitive to the geometric interaction between metal sites and their supports. Their catalytic behavior is closely related to the local coordination environment of metal sites. Herein, Ga–N4, Ga–N5 and Ga–N6 coordinated structures were obtained by tuning the coordination environment of a gallium (Ga) single atom. The Ga–N4 structure preferred photocatalytic hydrogen reduction for hydrogen with ∼96.4% selectivity. In contrast, the Ga–N6 structure favored photocatalytic oxygen reduction for hydrogen peroxide (H2O2) with 100% selectivity. In situ infrared (IR) spectra and density functional theory (DFT) calculations verified that the Ga metal site was the active center in the Ga–N4 structure while the N site was the active center in the Ga–N6 structure. This study demonstrated the coordination number dominated catalytic selectivity over Ga–Nx switchable active sites, providing a new insight for the design of single-atom catalysts.
1. Introduction
Single-atom catalysts have received considerable attention due to their tunable electronic structures and maximum utilization of isolated metal atoms.1–4 Accordingly, they have high activity, excellent selectivity and superior stability.5 To date, they have been extensively studied in catalytic reactions.6–11 For example, platinum (Pt) single atoms on FeOx had a high activity for carbon monoxide (CO) oxidation.12 Iridium (Ir) single atoms in Ni–Fe–S nanosheets could maximize the water oxidation activity.13 Some interesting studies also show the high selectivity of single-atom catalysts. For example, Pt single atoms on graphene with double vacancies had a high selectivity for carbon dioxide (CO2) electroreduction to methanol production.14 Ferrum (Fe) single atoms on carbon nitrides had 100% selectivity for 1O2 generation.15 In addition, single-atom catalysts with a high density were also reported.16–18 The corresponding metal content could reach up to 23 wt% or even up to 40 wt%. In these reported studies, metal atoms, connected to the local environment of supports, are nearly completely considered as active centers in these single-atom catalysts. There is no doubt that these dispersed metals bridge homogeneous and heterogeneous catalysis, offering new perspectives for controlling the activity and selectivity.19–22 However, the great challenge and urgent task is to clarify the catalytic behavior of metal sites with their local coordination environments.
Here we disclosed for the first time that the active center and selectivity of gallium-based single-atom catalysts could be dramatically tuned by using the doping level of Ga metal. From low to high Ga metal doping, Ga–N4, Ga–N5, and Ga–N6 coordinated structures were obtained. Ga atoms were the active sites in Ga–N4 structures, while N atoms were the active sites in Ga–N6 structures. The switchable active centers were responsible for the excellent selectivity of photocatalytic hydrogen reduction reactions in Ga–N4 structures and the photocatalytic oxygen reduction reactions in Ga–N6 structures. Such compelling results elucidated the dependence among catalytic activity, reaction selection and metal loading, demonstrating the coordination number dominated catalytic selectivity over switchable Ga and N active sites.
2. Experimental
2.1. Preparation of pure g-C3N4 nanosheets
Pure g-C3N4 nanosheets were prepared according to a previous report.5 Typically, 5 g of urea was placed in a porcelain boat, which was heated to 550 °C in an electric furnace with a heating rate of 5 °C min−1 under a high-purity Argon atmosphere. After the annealing process at 550 °C for 2 h, the furnace was gradually cooled down to room temperature. The g-C3N4 nanosheets in light yellow color were acquired.
2.2. Synthesis of Ga-doped g-C3N4 nanosheets
The preparation of Ga-doped g-C3N4 nanosheets followed the same procedure as that for pure g-C3N4 nanosheets. GaCl3 powders were dispersed in 30 mL of deionized water with strong ultrasonic treatment to form a transparent solution. Then 5 g of urea was dissolved in the transparent solution to obtain the initial mixed solution. The mixed solution was rapidly frozen using liquid nitrogen and then dried by using a freeze drier to obtain the precursors of uniformly mixed GaCl3 and urea. The precursors were then transferred into an electronic furnace for the same procedure as that for the preparation of g-C3N4 nanosheets. By changing the incorporated content of GaCl3, Ga-doped g-C3N4 nanosheets were obtained, which were denoted as sample #1, sample #2, and sample #3.
2.3. Characterization
TEM bright field images were characterized by using an FEI Talos. Atomic-scale Cs-corrected HAADF-STEM images were obtained by using an FEI Titan Cubed Themis G2 operated at 300 kV. Energy dispersive X-ray spectroscopy (EDS) elemental maps were performed by using an FEI Talos. The microstructures of the prepared samples were identified by X-ray diffraction (XRD, Bruker D8) with Cu Kα radiation (λ = 1.5418 Å). X-ray photoelectron spectroscopy (XPS) measurements were performed by using a Thermo Scientific spectrometer (ESCA Lab 250). UV-vis diffuse reflectance spectra were analyzed by using a spectrophotometer (Shimadzu, UV-2600). Photoluminescence (PL) spectra were recorded by using a Hitachi F-700. Time-resolved fluorescence spectra were collected by using an Edinburgh FLS980 fluorescence spectrometer. The accurate content of the Ga element was analyzed by inductively coupled plasma-atomic emission spectrometry (ICP-AES, Agilent 730).
2.4.
Ex situ and in situ TPV experiments
A third-harmonic Nd:YAG laser (λ = 355 nm) was used for ex situ TPV measurements. The working electrode was prepared by loading photocatalysts onto a Pt net. Pt wire was used as the counter electrode. An oscilloscope was used to collect TPV signals. For in situ TPV measurements, the working electrode was prepared by coating the slurry containing photocatalysts onto ITO glass. N2, O2 and tiny amounts of water were coupled with anhydrous acetonitrile during in situ TPV tests.
2.5. XAFS measurements and analysis
X-ray absorption spectra (Ga K-edge) were recorded at the 1W1B station in the Beijing Synchrotron Radiation Facility (BSRF). The storage rings were operated at 2.5 GeV with an average current of 0.25 A. A Si (111) double-crystal monochromator was used for data collection in transmission/fluorescence mode based on an ionization chamber. All spectra were obtained under ambient conditions.
EXAFS data were analyzed by the standard procedure based on the ATHENA module as implemented in the IFEFFIT software package. The k3-weighted EXAFS spectra were acquired by deducting the post-edge background from the total absorption, and normalizing them according to the edge-jump step. Then k3-weighted χ(k) data of the Ga K-edge were converted to real (R) space using a Hanning window (dk = 1.0 Å−1). The ARTEMIS module based on IFEFFIT software was employed for least-squares curve parameter fitting to obtain the structural information around central atoms. The k- and R-range for k3 weighting for the fitting is 2.4–12.2 Å−1 and 1.0–2.0 Å, respectively. The coordination number (CN), bond length (R), Debye–Waller factor (σ2) and E0 shift (ΔE0) were obtained without any additional conditions. The WT data were analyzed by MatLab R2021a.
2.6. Photocatalytic H2 tests
The photocatalytic H2 tests were performed by using a Perfectlight Labsolar 6A system (Beijing Perfectlight Technology Co., Ltd). A 300 W Xe lamp was used as the light source. During the H2 tests, 10 mg of the prepared photocatalysts was mixed with 15 mL of ultra-pure water and 5 mL of isopropanol (IPA) as the sacrificial agent with ultrasonic treatment to obtain a well-dispersed solution. Then high-purity N2 was introduced into the solution and aerated for 30 min to remove the dissolved O2. During the photocatalytic process, the system was continuously evacuated by using a vacuum pump. The H2 yield was evaluated by using a gas chromatograph equipped with a thermal conductivity detector (TCD). Meanwhile, a certain amount of reaction solution was extracted via the gasket port in the reactor. The H2O2 yield was examined using 0.01 M of Ce2SO4 solution with O-diphenylene iron as the tracer agent. For the test with O2, high-purity O2 was injected into the closed system from the sampling port.
2.7. Photocatalytic H2O2 tests
The photocatalytic H2O2 experiments were performed on a Teflon-linked reactor (Shanghai Yanzheng Experimental Instrument Co., Ltd). 5 mg of photocatalysts was added to 40 mL of ultrapure water with 25% IPA as the sacrificial agent inside. Then 10 μL of HCl solution was dropped into the above solution to prevent the decomposition of the generated H2O2. The solution was subjected to ultrasonic treatment for 1 h with an ice bath. Afterwards, O2 was introduced into the solution and bubbled for 30 min to achieve the absorption–desorption equilibrium under dark conditions. A 300 W Xe lamp (Beijing Perfectlight Technology Co., Ltd.) was employed for light irradiation with an AM 1.5G filter. 3 mL of solution was extracted at specific time intervals for centrifugation to obtain the supernatant after removing the photocatalysts inside. The H2O2 yield was obtained by using 0.01 M of Ce2SO4 solution with O-diphenylene iron as the tracer agent. The benchmark test on reaction solution before light irradiation was performed to remove the background effect. Meanwhile, a certain amount of reaction gas was extracted via the gasket port in the reactor and kept in an air bag. Then, the gas was injected into a gas chromatograph (Shimadzu, 2014C) to obtain the H2 yield.
2.8. Electrochemical measurements
A Biologic VSP 300 was employed to perform electrochemical measurements. 0.1 M of Na2SO4 solution was chosen as the electrolyte. A graphite rod was used as the counter electrode. A saturated calomel electrode was selected as the reference electrode. The working electrode was prepared as follows. 10 mg of the photocatalyst was added to 2 mL of ethanol with 90 μL of naphthol. Then the solution was subjected to ultrasonic treatment for 30 min to form a slurry. The slurry was coated on a FTO glass and dried at 80 °C in a vacuum oven. The electrochemical impedance spectra (EIS) were measured within the frequency range of 1 mHz to 1 MHz at room temperature. Transient photocurrent density was obtained by using an electronic shutter with a switching frequency of 10 s under light irradiation.
2.9.
In situ DRIFTS measurements
A Nicolet IS50 device was employed to perform in situ DRIFTS measurements. Before the tests, the reaction chamber was cleaned by heating the reactor at 120°C for 30 min to remove the moisture inside. High-purity Ar gas was continuously purged into the reactor for 30 min to remove the residual air. Photocatalysts were pre-treated under vacuum conditions for 30 min under the protection of an Ar atmosphere. Afterwards, an infrared spectrum was collected as background. Then a series of IR data were collected under different experimental conditions (N2 or O2) at different time intervals under light irradiation.
2.10. Theoretical methods
DFT calculations were performed by using the VASP 6.3.0 code.23,24 The Perdew–Burke–Ernzerhof (PBE) method with generalized gradient approximation (GGA) was used to treat the exchange and correlation interaction.25 The empirical DFT-D3 (BJ) correction was employed to handle the van der Waals interaction due to the presence of C, N, O and H elements.26,27 The spin-polarization was included during the calculation of singlet state and triplet state oxygen. The cutoff energy was set to 400 eV. During the optimization, a 2 × 2 × 1 k-mesh was utilized. The energy convergence and the force convergence were set to 10−4 eV and 0.02 eV Å−1, respectively. The numerical accuracy was carefully tested. The valence electrons of H (1s1), C (2s2 and 2p2), N (2s2 and 2p3), O (2s2 and 2p4) and Ga (4s2 and 4p1) were used during self-consistent field calculations.
2.11. Structural models
A two-dimensional C3N4 monolayer (14.27 Å × 12.35 Å × 20 Å, γ = 90°) was constructed according to the structural parameters of bulk C3N4 (7.135 Å × 12.353 Å × 7.138 Å, γ = 90°). An adequate vacuum layer of 20 Å was set to prevent the periodic effect. The structural models with Ga–N4 and Ga–N6 coordinates were constructed. Due to the surface undulation of C3N4, the bond length of Ga–N was estimated to be in the reasonable range of 2.4–3.0 Å. The adsorption energy of Ga atom was defined as:| | | Eads = EGa–C3N4 − EC3N4 − EGa, | (1) |
where EGa–C3N4 and EC3N4 represent the energy of Ga-adsorbed C3N4 and pristine C3N4. EGa is the energy of the Ga atom in its most stable bulk phase.
2.12. ORR calculations
The oxygen reduction reaction (ORR) can be described as the following processes:28| | | O2 + e− + H+ + * → *OOH | (2) |
| | | *OOH + e− + H+ → * + H2O2 | (3) |
According to the above equations, the catalytic performance was evaluated by using free energy ΔG, which is defined as:
| | | ΔG = Eads + ΔEZPE − TΔS | (4) |
| | | ΔGO2(U) = 4 × 1.23 − 4U | (5) |
| | | ΔG*OOH(U) = G*OOH(U) + 1.5G(H2) − 2G(H2O) − G(*) − 3U | (6) |
| | | ΔG*H2O2(U) = G*H2O2(U) + G(H2) − 2G(H2O) − G(*) − 2U | (7) |
where Δ
EZPE represents the vibrations of all degrees of freedom of adsorbates under the harmonic approximation and
T and Δ
S are temperature and entropy change.
3. Results and discussion
3.1. Different Ga-coordinated structures
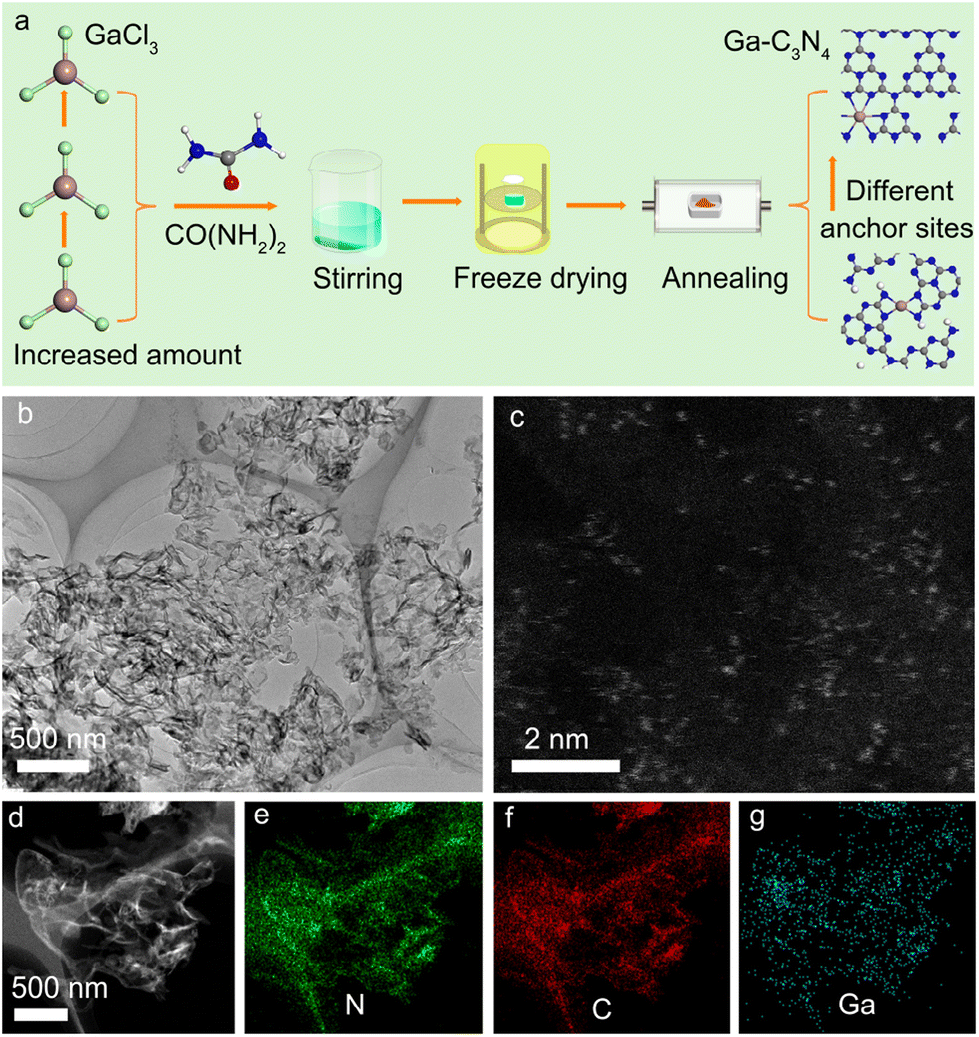
Fig. 1a shows the preparation process of Ga-coordinated C3N4 nanosheets. Gallium trichloride (GaCl3), used as the Ga source, was dissolved in de-ionized water with urea (CO(NH2)2) to form a transparent solution under ultrasonic treatment. Then, the solution was rapidly frozen by using liquid nitrogen and dried in a freeze dryer. Ga-coordinated C3N4 nanosheets were obtained by annealing the freeze-dried precursors in an electric furnace. By changing the GaCl3 additive amount, a series of Ga-coordinated C3N4 samples were acquired. Fig. 1b shows the representative TEM image of the Ga-coordinated C3N4 sample, in which the nanosheet structure was observed. The atomic-scale high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image in Fig. 1c demonstrated that Ga atoms (bright spots) were monodispersed in C3N4 nanosheets. The energy dispersive X-ray spectra (EDS) elemental maps in Fig. 1d–g verified the presence of N, C, and Ga. The Ga high-resolution X-ray photoelectron spectroscopy (XPS) image in Fig. S1† further confirmed the Ga incorporation.29 X-ray diffraction (XRD) data (Fig. S2†) showed the microstructure of Ga-coordinated C3N4 nanosheets.5 Ga incorporation did not change the micro-structure of g-C3N4 nanosheets. Fourier transform infrared (FTIR) spectra in Fig. S3† showed a typical absorption peak at ∼740 cm−1, verifying that Ga atoms were bonded to N atoms.30
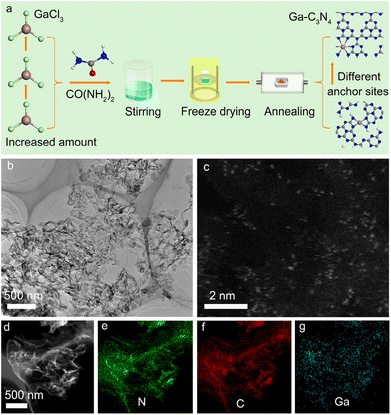 |
| | Fig. 1 (a) Schematic diagram of the preparation process of different Ga anchor sites in g-C3N4 by changing Ga doping concentrations. (b) Low magnification TEM image of a representative Ga-doped g-C3N4 sample. (c) Atomic-scale aberration-corrected HAADF-STEM image of Ga single atoms distributed in g-C3N4. (d) HAADF-STEM image of a representative Ga-doped g-C3N4 sample. (e–g) The EDS maps of N, C, and Ga elements, respectively. | |
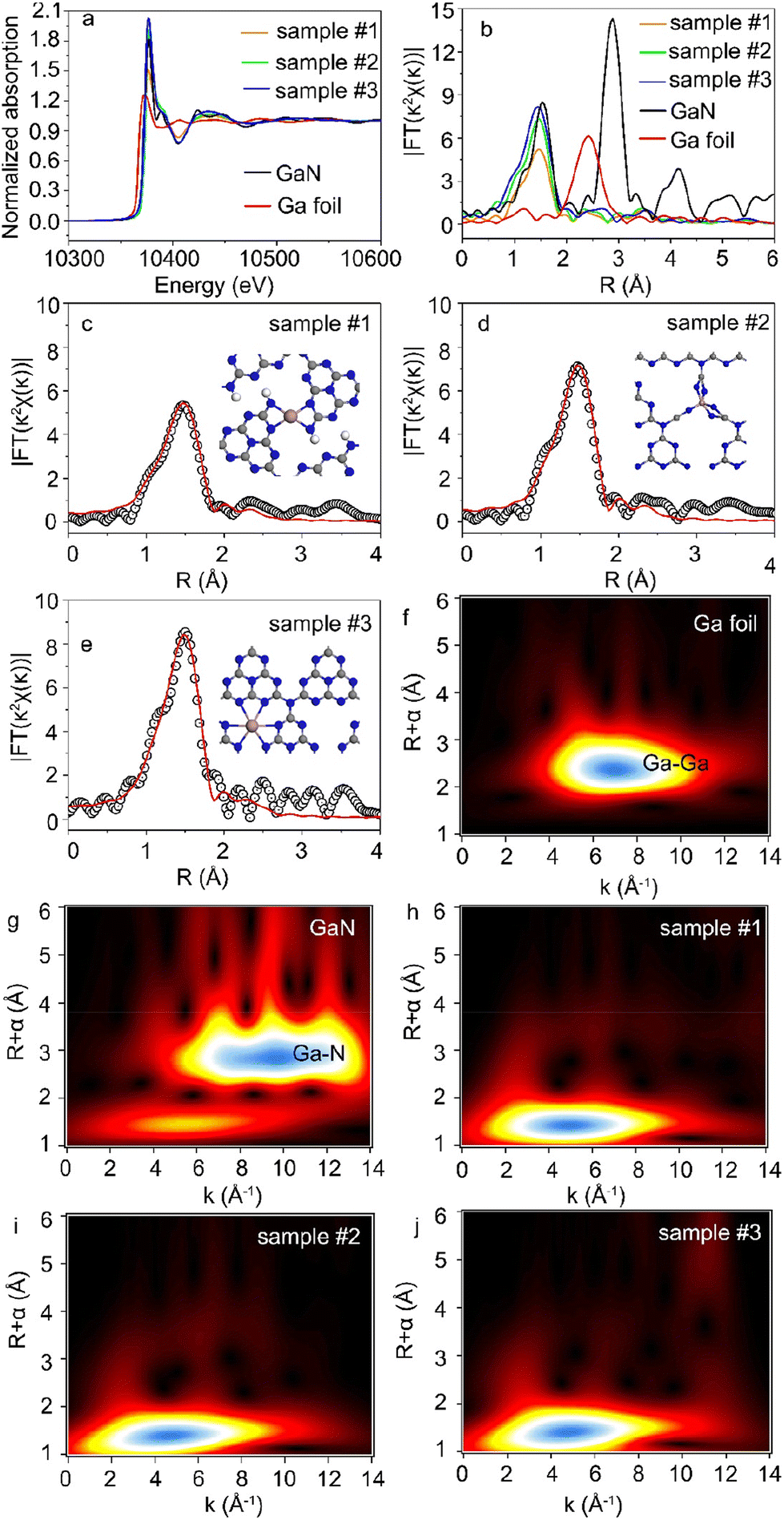
X-ray absorption near-edge structure (XANES) and Fourier-transformed extended X-ray absorption fine structure (FT-EXAFS) were used to study the chemical state and local coordination environment of Ga single atoms.31,32Fig. 2a shows the Ga K-edge XANES spectra of Ga-C3N4 samples. The data of Ga foil and gallium nitride (GaN) samples were also shown for comparison. The absorption edge of Ga-C3N4 samples was higher than that of Ga foil and GaN references, indicating that the Ga average valence state in Ga-C3N4 samples was higher than that in the GaN standard sample.33 Meanwhile, this also indicated that Ga atoms in Ga-C3N4 samples were not in the metallic state. In addition, a high intensity of the white line peak corresponds to a high proportion of the single-atom structures and high coordination numbers.34 The intensity of the white line peak in sample #3 was higher than that in samples #1 and 2, indicating that the Ga single atom in sample #3 had a higher coordination number. Accordingly, the Ga coordination structure could be tuned by using different Ga doping levels in C3N4 samples.
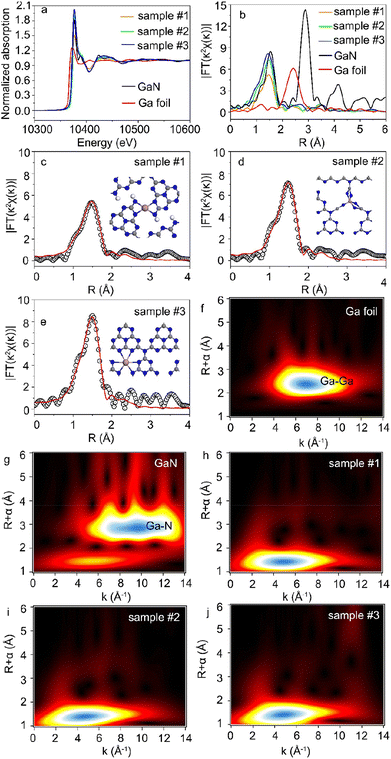 |
| | Fig. 2 (a) XANES spectra of the Ga K-edge for Ga foil, GaN, and Ga-C3N4. (b) Fourier-transform EXAFS spectra of Ga foil, GaN, and Ga-C3N4. (c–e) Fitted FT-EXAFS curve in R space for Ga-C3N4. (f and g). WT-EXAFS contour plots of Ga foil, GaN, and Ga-C3N4, respectively. (h–j) WT-EXAFS contour plots of sample #1, sample #2 and sample #3, respectively. | |
As shown in Fig. 2b, the dominant peak of Ga–Ga bonds in Ga foil was located at ∼2.39 Å in the R space of FT-EXAFS. The notable peaks at 1.44 Å and 2.73 Å correspond to the Ga–N and Ga–Ga bonds in the GaN sample, respectively. There was no Ga–Ga peak in the FT-EXAFS spectra of the Ga-C3N4 sample, indicating that the Ga atoms were monodispersed in g-C3N4 nanosheets, consistent with the HAADF-STEM image in Fig. 1c. The Ga–N first coordination shell at 1.47 Å in R space was observed in the Ga-C3N4 sample, indicating that Ga atoms were surrounded by N atoms in the local coordination environment of the Ga-C3N4 sample. The least-squares FT-EXAFS fittings are shown in Fig. 2c–e, S4 and S5†, and the detailed fitting data were shown in Table S1.† For a low Ga doping level, Ga atoms in g-C3N4 nanosheets (sample #1) preferred to form a four coordinated structure with the surrounding N atoms (Ga–N4). The Ga–N5 coordinated structure was observed in sample #2. With the increase in Ga loading concentration, the Ga–N6 coordinated structure was obtained (sample #3), indicating that the Ga doping level could significantly change the local coordination environment of Ga atoms in g-C3N4 nanosheets. Accordingly, different single-atom coordination environments could be realized by changing the atomic doping level. Fig. 2f–j show the wavelet transform (WT) contour plots of Ga foil, GaN and Ga-C3N4 samples. The intensity maximum in the contour plot of the Ga-C3N4 sample corresponds to a k value of ∼5 Å−1 for the Ga–N path, clearly different from that of the Ga–Ga path (∼7 Å−1) in the Ga foil, further verifying the monodispersed Ga atoms in g-C3N4 nanosheets.
3.2. From photocatalytic hydrogen reduction to oxygen reduction
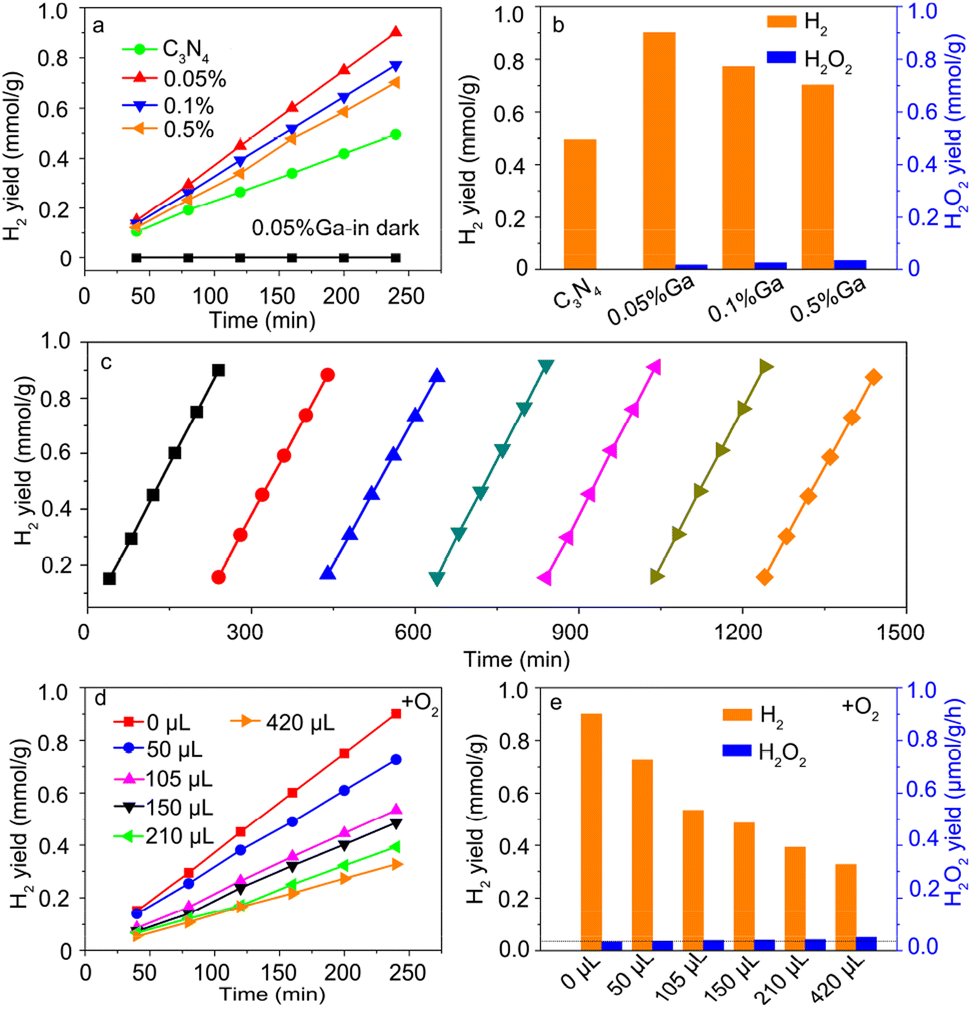
Fig. 3a shows the photocatalytic hydrogen (H2) evolution as a function of the irradiation time. The H2 yield at 4 h is summarized in Fig. 3b. For the pure g-C3N4 sample, the H2 yield was ∼0.49 mmol g−1, comparable to the reported data.5 The H2 yield of the Ga-doped sample was enhanced compared with that of pure g-C3N4, indicating that Ga incorporation contributed to photocatalytic H2 evolution. However, the H2 yield decreased for a high Ga doping concentration. The Ga–N4 coordinated sample had the highest H2 yield, consistent with the reported data.32 The H2O2 yield at 4 h was also examined. As shown in Fig. 3b, the H2O2 yield was remarkably lower than the H2 yield in the Ga–N4 coordinated sample. The H2 yield at 4 h for the Ga–N4 coordinated sample was ∼0.9 mmol g−1, significantly higher than the H2O2 yield (∼0.03 mmol g−1), showing a high selectivity of 96.4% for hydrogen reduction. Fig. 3c shows the cycling stability of the Ga–N4 coordinated sample. After seven-round cycling tests, the photocatalytic H2 yield showed nearly no degradation, indicating the superior photocatalytic stability in the Ga–N4 coordinated sample.
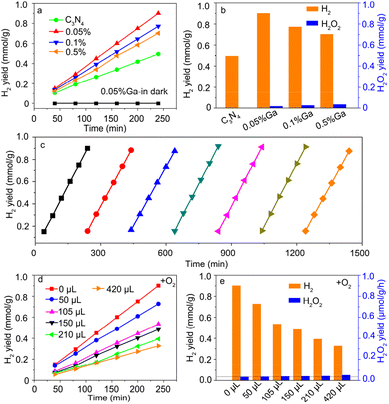 |
| | Fig. 3 (a) Photocatalytic H2 yield as a function of reaction time. (b) The H2 yield of the prepared samples at 4 h. The corresponding H2O2 yield in reaction solution was also measured. (c) Cycling stability of photocatalytic H2 yield. (d) Adding different O2 amounts into the reactor during the photocatalytic H2 evolution process based on the Ga–N4 coordinated sample. (e) The photocatalytic H2 yield of the optimized sample with the addition of different O2 amounts at 4 h. The corresponding H2O2 yield in reaction solution was also measured. | |
To check the competition of H2 and H2O2 photoproduction, high-purity oxygen (O2) was injected into the reactor during photocatalytic H2 reduction. Fig. 3d shows the H2 evolution after injecting O2, and Fig. 3e summarizes the H2 yield at 4 h. Without O2 addition, the H2 yield was ∼0.9 mmol g−1. With O2 addition, the H2 yield obviously decreased. The corresponding H2O2 yield also increased, indicating the competition of photocatalytic products. Nonetheless, H2 production still dominated the photocatalytic activity. To sum up, the Ga–N4 coordinated structure has the optimal H2 yield, indicating that Ga with a low coordination environment is beneficial for H2 evolution.
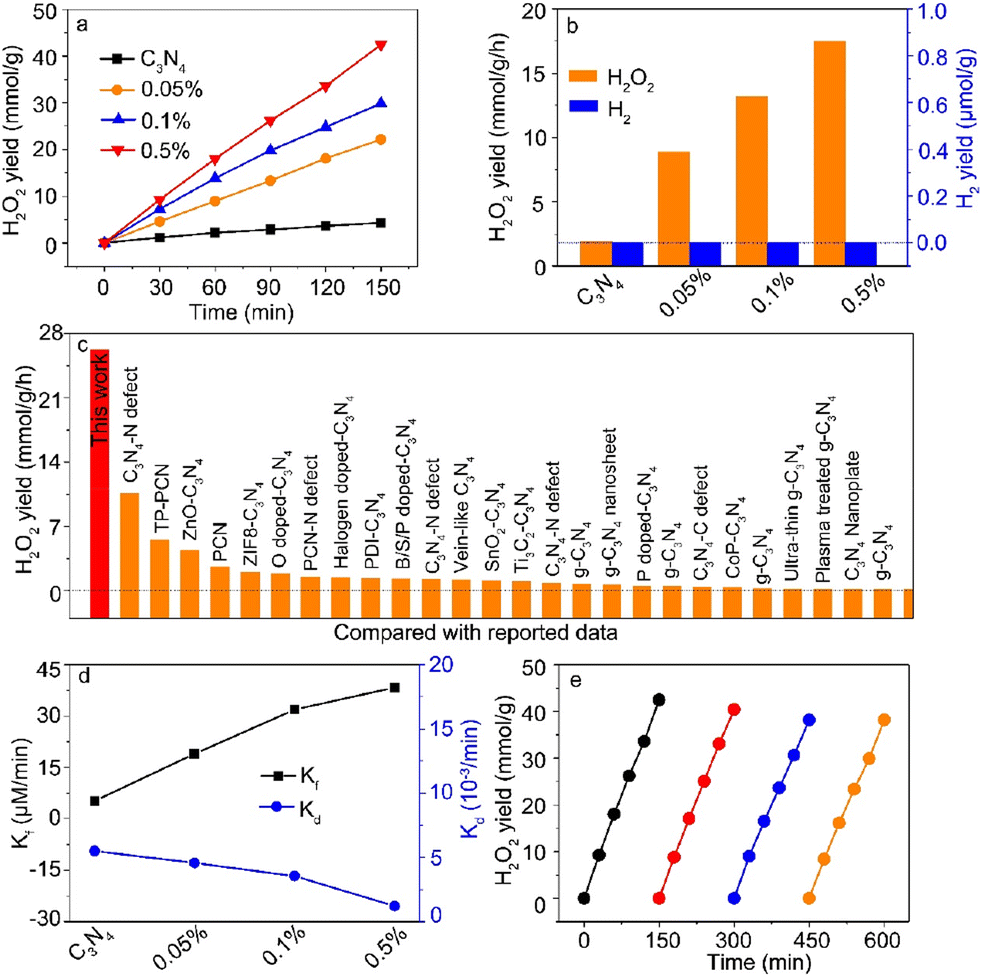
To further study the competition between hydrogen reduction and oxygen reduction, high-purity O2 with a pressure of 1.8 MPa was maintained in the reactor during light irradiation. Meanwhile, we need to keep in mind that the increased Ga contents in C3N4 nanosheets led to the change of the Ga coordination number from Ga–N4 to Ga–N6. Fig. 4a shows the photocatalytic H2O2 yield as a function of irradiation time, and the corresponding H2O2 yield is shown in Fig. 4b. In sharp contrast, the Ga–N4 coordinated structure had the highest H2 yield for hydrogen reduction, while the Ga–N6 coordinated sample had the highest H2O2 yield of ∼17.5 mmol g−1 h−1. This indicated that the Ga–N6 coordinated structure preferred oxygen reduction compared with the Ga–N4 coordinated structure. Moreover, there was nearly no H2 yield as shown in Fig. 4b, indicating that the Ga-doped g-C3N4 samples had nearly 100% selectivity for the H2O2 product (Fig. S6†). Fig. 4c shows the comparison of photocatalytic H2O2 yield with reported data. The H2O2 yield of the Ga–N6 coordinated structure was obviously higher than that in all reported data. As shown in Fig. 4d, the highest formation rate and lowest decomposition rate demonstrated the robust photocatalytic H2O2 ability of the Ga–N6 coordinated structure. The H2O2 yield after four-round cycling tests (Fig. 4e) was ∼38.2 mmol g−1, which is ∼93% of the yield of the first-round test (42.4 mmol g−1). This fully demonstrated the durable H2O2 cycling stability of the Ga–N6 coordinated structure.
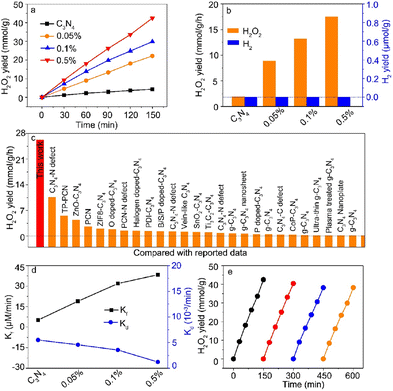 |
| | Fig. 4 (a) Photocatalytic H2O2 yield as a function of reaction time. (b) The H2O2 yield of the prepared samples at 1 h. The corresponding H2 yield was also examined. (c) Comparison of the H2O2 yield between our prepared sample and reported data. (d) Formation and decomposition rates of H2O2. (e) Cycling stability of the Ga–N6 coordinated sample. | |
3.3. Variable active centers
To identify the important role of Ga metal loading in catalytic properties, photo-generated carrier dynamics were studied. As seen from the photoluminescence (PL) spectra in Fig. S7,† efficient charge separation in C3N4 nanosheets was realized on Ga metal loading, which was further confirmed by using the transient photocurrent (Fig. S9†). The electrochemical impedance spectra (EIS) shown in Fig. S8† indicated the relatively lower interfacial resistance in Ga-coordinated C3N4 samples compared with the pure C3N4 sample. The transient photovoltage (TPV) data (Fig. S10†) and time-resolved PL spectra (Fig. S11†) indicated the longer photo-generated carrier lifetime, indirectly verifying the highly improved charge separation on Ga metal loading.
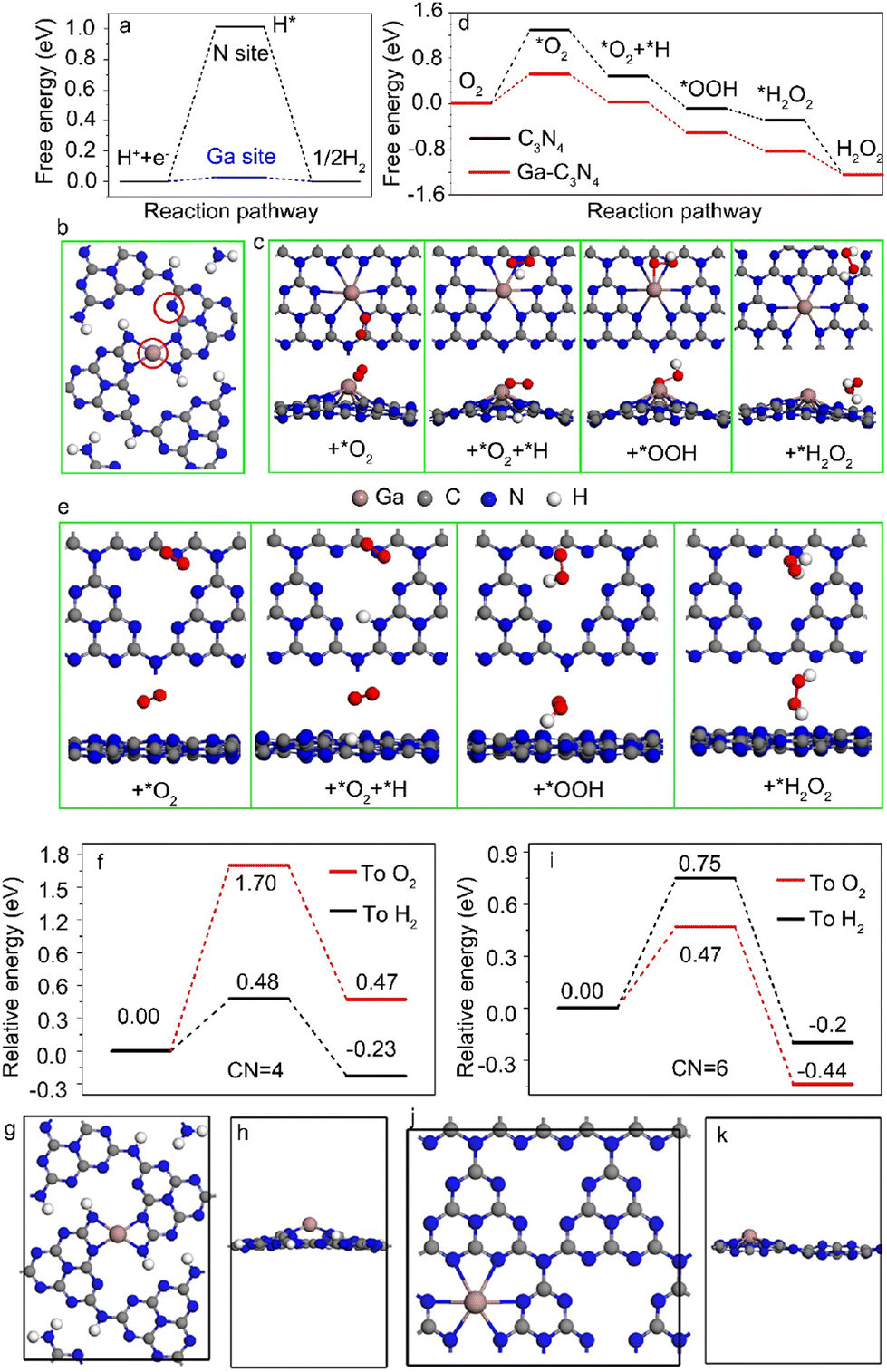
DFT calculations were performed to reveal the photocatalytic selectivity of Ga–N4 and Ga–N6 coordinated samples. The free energy profile of the Ga–N4 coordinated sample is shown in Fig. 5a. Both the Ga site and N site indicated by red circles in Fig. 5b were selected as potential active centers. The free energy for the H* intermediate on the Ga site was closer to zero than that on the N site, demonstrating that the Ga site was the active center for hydrogen reduction. In contrast, the *O2 intermediate preferred to adsorb on the N site for photocatalytic H2O2 in Ga–N6 coordinated samples. The adsorption of the *O2 intermediate on Ga and C sites corresponded to higher barriers compared with the N site as shown in Fig. S12,† verifying that the N site was the active center for the Ga–N6 coordinated sample. Furthermore, the optimized adsorbed sites for the intermediates also demonstrated that intermediates were more stable on the N site than the Ga and C sites as shown in Fig. 5c. The potential reaction pathway was estimated using the calculated free energies as shown in Fig. 5d. The free energy profile of pure C3N4 was also shown for comparison. The corresponding stable adsorption sites are shown in Fig. 5e. The free energy calculation based on the Ga–N6 coordinated sample was better than that of pure C3N4, verifying that Ga–N6 coordinated samples preferred oxygen reduction.
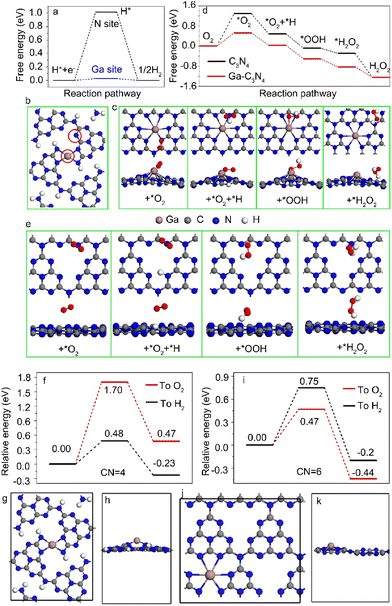 |
| | Fig. 5 (a) Free energy diagram for hydrogen adsorption on N and Ga sites in Ga–N4 coordinated samples, respectively. (b) The corresponding structural models of Ga–N4 coordinated samples for hydrogen adsorption on N and Ga sites as indicated by red circles. (c) Structural models of the adsorption of intermediates in Ga–N6 coordinated samples. (d) Free energy diagram for oxygen adsorption on C3N4 and Ga–N6 coordinated samples. (e) Structural models of the adsorption of intermediates in pure g-C3N4 samples. (f) DFT calculations of the energy barrier for the proton transfer to H2 and O2 in Ga doped C3N4 with four coordination number (CN = 4). (g and h) The optimized structural model of Ga-C3N4 with four coordination number (CN = 4) from the top and side view, respectively. (i) DFT calculations of the energy barrier for the proton transfer to H2 and O2 in Ga doped C3N4 with six coordination number (CN = 6). (j and k) The optimized structural model of Ga–C3N4 with six coordination number (CN = 6) from the top and side view, respectively. | |
In situ diffuse reflectance infrared transform spectroscopy (DRIFTS) was utilized to further confirm the N active sites for oxygen reduction in the Ga–N6 coordinated sample. The DRIFTS spectrum had no apparent variation at 0, 10 and 20 min under a nitrogen (N2) atmosphere, as shown in Fig. S13.† No obvious IR peak could be observed for Ga–O,35 at ∼685 cm−1 and for N–O,5 at ∼930 cm−1 and ∼1357 cm−1. However, the IR peaks at ∼930 cm−1 and ∼1357 cm−1 were clearly observed under an O2 atmosphere. The peak intensities at 1, 5, 10, 20, 30 and 40 min indicated the continuous adsorption of O2 molecules on the N sites. There was no clear variation at ∼685 cm−1, verifying that the N site in the Ga–N6 coordinated sample was the active center of oxygen reduction. Therefore, the Ga metal loading concentration regulated the Ga coordination environment in g-C3N4 nanosheets, and further determined the variable active center for different photocatalytic selectivities.
3.4. Different proton transfer paths
The proton transfer path was studied by DFT calculations to demonstrate the catalytic selectivity of hydrogen reduction and oxygen reduction in Ga–N4 and Ga–N6 coordinated samples, respectively. The energy barrier of proton transfer in the Ga–N4 coordinated sample is shown in Fig. 5f. The corresponding structural model is shown in Fig. 5g and h. As shown in Fig. 5f, the energy barrier of the proton transfer to H2 was lower than that of the proton transfer to O2, indicating that hydrogen reduction was easier than oxygen reduction in the Ga–N4 coordinated sample. In sharp contrast, as shown in Fig. 5i and k, the energy barrier of proton transfer to O2 was 0.47 eV, 0.28 eV lower than that for H2, demonstrating that oxygen reduction was easier than hydrogen reduction in the Ga–N6 coordinated sample. DFT results clearly explained our experimental data at the atomic level, i.e., Ga–N4 and Ga–N6 coordinated samples, respectively, preferred the hydrogen and oxygen reduction due to their different proton transfer paths.
3.5. Photocatalytic H2O2 mechanism
The photocatalytic H2O2 process includes several possible pathways for the oxygen reduction reaction (ORR). The 2e− process is described as:| | | O2 + 2H+ + 2e− → H2O2. | (8) |
The 4e− pathway is defined as:
| | | O2 + 4H+ + 4e− → 2H2O2. | (9) |
The two-step one electron pathway is described as:
| | | ˙O2− + 2H + e− + H2O2. | (11) |
The other half-reaction for the water oxidation reaction (WOR) is described by using the following equations.
The 2e− process:
| | | 2H2O + 2h+ → H2O2 + 2H+ | (12) |
The 4e− process:
| | | 2H2O + 4h+ → O2 + 4H+ | (13) |
The two-step one electron process:
To further check the photocatalytic H2O2 mechanism, we performed the control experiments. The oxygen gas was replaced by high-purity Ar gas. After photocatalytic tests, the gas in the reactor was collected and examined by on-line gas chromatography with thermal conductivity detection (GC-TCD). As shown in Fig. S14,† no H2 signal (∼1.4 min) was detected, indicating that the photocatalytic process did not produce H2 gas. This further demonstrated the good selectivity of photocatalytic H2O2 production. There was no O2 signal as well, indicating that there was no WOR process. This is because the IPA sacrificial agent was added to react with holes. Accordingly, the whole WOR process was absent. The corresponding H2O2 yield was estimated to be neglectable, indicating that the ORR process dominated the photocatalytic H2O2 yield. To examine the intermediates, electron spin resonance (ESR) spectra were obtained with 2,2,6,6-tetramethylpiperidine-1-oxyl (TMPO) as the radical trapping agent. As shown in Fig. S15,† there were no superoxide radicals (˙O2−) under dark conditions. In contrast, the ˙O2− signal was clearly observed under light irradiation. Similarly, the ˙OOH radicals were also detected under light irradiation as shown in Fig. S16.† This indicates that the O2 molecules received photo-generated electrons to generate the corresponding ˙O2− and ˙OOH radicals, confirming the DFT calculations. Meanwhile, these experiments also demonstrated that the photocatalytic H2O2 evolution proceeded according to the two-step one electron pathway.
4. Conclusions
In summary, the catalytic activity and selectivity of Ga-coordinated catalysts highly depended on the Ga coordination environment in g-C3N4 nanosheets. Ga–N4, Ga–N5 and Ga–N6 coordinated structures could be obtained by gradually tuning the Ga doping concentration. Furthermore, the change in the Ga coordination environments resulted in the variation of the active center from the Ga site in the Ga–N4 coordinated structure to the N site in the Ga–N6 coordinated structure. Interestingly, the Ga–N4 coordinated structure exhibited optimal hydrogen reduction with ∼96.4% catalytic selectivity. In sharp contrast, the Ga–N6 coordinated structure exhibited superior oxygen reduction with 100% selectivity. The different proton transfer paths in the Ga–N4 and Ga–N6 coordinated structures were confirmed by DFT calculations and were responsible for the high selectivity from hydrogen reduction to oxygen reduction, respectively. The Ga–N6 coordinated structure with 0.5% Ga metal loading had a ∼42.4 mmol g−1 superhigh H2O2 yield, higher than that of all explored and referenced materials. Our results here indicated that the metal loading concentration could obviously change the catalytic active center and selectivity, which should be carefully considered for single-atom catalysts.
Author contributions
Chunqiang Zhuang: conceptualization, supervision, writing – original draft, writing – review & editing, and funding acquisition; Weiming Li: data curation and formal analysis; Yuan Chang: data curation and formal analysis; Shijie Li: data curation and formal analysis; Yihong Zhang: data curation and formal analysis; Yuanli Li: data curation and formal analysis; Ge Chen: conceptualization; Junfeng Gao: conceptualization; Zhenhui Kang: conceptualization and writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (12274011 and 12074053), Beijing Outstanding Young Scientists Projects (BJJWZYJH01201910005018), and Basic Science Center Program for Multiphase Evolution in Hypergravity of the National Natural Science Foundation of China (No. 51988101).
Notes and references
- Y. Ren, Y. Tang, L. Zhang, X. Liu, L. Li, S. Miao, D. Sheng Su, A. Wang, J. Li and T. Zhang, Nat. Commun., 2019, 10, 4500 CrossRef PubMed.
- X. Li, H. Rong, J. Zhang, D. Wang and Y. Li, Nano Res., 2020, 13, 1842–1855 CrossRef CAS.
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef.
- J. Shan, C. Ye, Y. Jiang, M. Jaroniec, Y. Zheng and S.-Z. Qiao, Sci. Adv., 2022, 8, eabo0762 CrossRef CAS PubMed.
- C. Zhuang, W. Li, T. Zhang, J. Li, Y. Zhang, G. Chen, H. Li, Z. Kang, J. Zou and X. Han, Nano Energy, 2023, 108, 108225 CrossRef CAS.
- Z.-H. Xue, D. Luan, H. Zhang and X. W. Lou, Joule, 2022, 6, 92–133 CrossRef CAS.
- X. Wu, H. Zhang, J. Dong, M. Qiu, J. Kong, Y. Zhang, Y. Li, G. Xu, J. Zhang and J. Ye, Nano Energy, 2018, 45, 109–117 CrossRef CAS.
- X. Li, W. Bi, L. Zhang, S. Tao, W. Chu, Q. Zhang, Y. Luo, C. Wu and Y. Xie, Adv. Mater., 2016, 28, 2427–2431 CrossRef CAS PubMed.
- J.-X. Peng, W. Yang, Z. Jia, L. Jiao and H.-L. Jiang, Nano Res., 2022, 15, 10063–10069 CrossRef CAS.
- T. Cao, R. Lin, S. Liu, W.-C. M. Cheong, Z. Li, K. Wu, Y. Zhu, X. Wang, J. Zhang, Q. Li, X. Liang, N. Fu, C. Chen, D. Wang, Q. Peng and Y. Li, Nano Res., 2022, 15, 3959–3963 CrossRef CAS.
- X. Zheng, B. Li, Q. Wang, D. Wang and Y. Li, Nano Res., 2022, 15, 7806–7839 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- Z. Lei, W. Cai, Y. Rao, K. Wang, Y. Jiang, Y. Liu, X. Jin, J. Li, Z. Lv, S. Jiao, W. Zhang, P. Yan, S. Zhang and R. Cao, Nat. Commun., 2022, 13, 24 CrossRef CAS PubMed.
- S. Back, J. Lim, N.-Y. Kim, Y.-H. Kim and Y. Jung, Chem. Sci., 2017, 8, 1090–1096 RSC.
- L.-S. Zhang, X.-H. Jiang, Z.-A. Zhong, L. Tian, Q. Sun, Y.-T. Cui, X. Lu, J.-P. Zou and S.-L. Luo, Angew. Chem., Int. Ed., 2021, 60, 21751 CrossRef CAS PubMed.
- L. Han, H. Cheng, W. Liu, H. Li, P. Ou, R. Lin, H.-T. Wang, C.-W. Pao, A. R. Head, C.-H. Wang, X. Tong, C.-J. Sun, W.-F. Pong, J. Luo, J.-C. Zheng and H. L. Xin, Nat. Mater., 2022, 21, 681–688 CrossRef CAS PubMed.
- X. Hai, S. Xi, S. Mitchell, K. Harrath, H. Xu, D. F. Akl, D. Kong, J. Li, Z. Li, T. Sun, H. Yang, Y. Cui, C. Su, X. Zhao, J. Li, J. Pérez-Ramírez and J. Lu, Nat. Nanotechnol., 2022, 17, 174–181 CrossRef CAS PubMed.
- H. Zhang and X. W. Lou, Nat. Synth., 2023, 2, 81–82 CrossRef.
- G. Zhao, H. Liu and J. Ye, Nano Today, 2018, 19, 108–125 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- C. Gao, J. Wang, H. Xu and Y. Xiong, Chem. Soc. Rev., 2017, 46, 2799–2823 RSC.
- R. Qin, K. Liu, Q. Wu and N. Zheng, Chem. Rev., 2020, 120, 11810–11899 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- Y. Imazeki, M. Sato, T. Takeda, M. Kobayashi, S. Yamamoto, I. Matsuda, J. Yoshinobu, M. Sugiyama and Y. Nakano, J. Phys. Chem. C, 2021, 125, 9011–9019 CrossRef CAS.
- S. Ito, H. Kobayashi, K. Araki, K. Suzuki, N. Sawaki, K. Yamashita, Y. Honda and H. Amano, J. Cryst. Growth, 2015, 414, 56–61 CrossRef CAS.
- H. Tan, P. Zhou, M. Liu, Q. Zhang, F. Liu, H. Guo, Y. Zhou, Y. Chen, L. Zeng, L. Gu, Z. Zheng, M. Tong and S. Guo, Nat. Synth., 2023, 2, 557–563 CrossRef.
- W. Jiang, Y. Zhao, X. Zong, H. Nie, L. Niu, L. An, D. Qu, X. Wang, Z. Kang and Z. Sun, Angew. Chem., Int. Ed., 2021, 60, 6124–6129 CrossRef CAS PubMed.
- X. Liu, L. Zheng, C. Han, H. Zong, G. Yang, S. Lin, A. Kumar, A. R. Jadhav, N. Q. Tran, Y. Hwang, J. Lee, S. Vasimalla, Z. Chen, S.-G. Kim and H. Lee, Adv. Funct. Mater., 2021, 31, 2100547 CrossRef CAS.
- D. Yu, Y. Ma, F. Hu, C.-C. Lin, L. Li, H.-Y. Chen, X. Han and S. Peng, Adv. Energy Mater., 2021, 11, 2101242 CrossRef CAS.
- J. Yang, Y. Zhao and R. L. Frost, Spectrochim. Acta, Part A, 2009, 74, 398–403 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Weiming
Li
a,
Weiming
Li