
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-functional perovskite oxide Pr0.6Sr0.4Mn0.2Fe0.7Ni0.1O3−δ as an efficient quasi-symmetric electrode for solid oxide fuel/electrolysis cells†
Junil
Choi
 a,
Daehee
Jang
a,
Minho
Kim
a,
Jungseub
Ha
a,
Hwichan
Ahn
a and
Won Bae
Kim
*ab
a,
Daehee
Jang
a,
Minho
Kim
a,
Jungseub
Ha
a,
Hwichan
Ahn
a and
Won Bae
Kim
*ab
aDepartment of Chemical Engineering, Pohang University of Science and Technology (POSTECH), 77 Cheongam-ro, Nam-gu, Pohang, Gyeongbuk 37673, Republic of Korea. E-mail: kimwb@postech.ac.kr
bGraduate Institute of Ferrous & Eco Materials Technology, Pohang University of Science and Technology (POSTECH), 77 Cheongam-ro, Nam-gu, Pohang, Gyeongbuk 37673, Republic of Korea
First published on 30th April 2024
Abstract
Solid oxide cells (SOCs) have recently gained attention as an efficient energy conversion system for a smart-grid that can resolve the intermittency of renewable energy. In particular, a symmetric configuration of the electrode has been developed to minimize fabrication cost and relieve compatibility issues. In this work, PSMFN (Pr0.6Sr0.4Mn0.2Fe0.7Ni0.1O3−δ) was developed and evaluated as a quasi-symmetric electrode for a solid oxide cell (Q-SSOC). A solid oxide cell fashioned with PSMFN–GDC (Ce0.9Gd0.1O2) exhibited an electrochemical performance of 0.702 W cm−2 for H2-SOFC and 1.02 A cm−2 at a voltage of 1.5 V for CO2-SOEC at 800 °C. The physiochemical and electrochemical properties of PSMFN were investigated to elaborate this notable electrochemical performance of PSMFN–GDC. X-ray diffraction (XRD), microscopic analyses, and X-ray absorption near edge structure (XANES) indicated the occurrence of an exsolution of Fe–Ni alloy nanoparticles (NPs) along with a phase transition from the perovskite to the Ruddlesden–Popper structure. In addition, X-ray photoelectron spectroscopy (XPS) was utilized to elucidate the change in surface oxygen vacancies of the reduced PSMFN form. Furthermore, electrical conductivities and area specific resistances under an air atmosphere were measured to investigate the activities of PSMFN towards oxygen electrode reactions. From these analyses, PSMFN can provide numerous active sites both for electrode reactions with oxygen as well as fuel, which make it a promising quasi-symmetric electrode for an SOC.
Introduction
Solid oxide cells (SOCs), which can be operated under both modes of a galvanic cell (e.g., solid oxide fuel cells, SOFCs) and an electrolytic cell (e.g., solid oxide electrolysis cells, SOECs), can play a key technological role in resolving the intermittency issue of renewable energies and relieving global warming by the greenhouse gas effect.1–6 Moreover, various types of fuel, even carbon-containing gas, can be converted into electricity with lower emission of greenhouse gas,7–9 while superfluous energy produced during the night-time from renewable energy sources can be stored in forms of chemical energy; for instance, converting carbon dioxide (CO2) into carbon monoxide (CO).10,11 The high-temperature operation, where heat demand is largely diminished, gives a high theoretical thermodynamic efficiency and fast electrode kinetics compared to other electrochemical systems.In an SOC configuration, materials for the fuel electrode and air electrode, which are separated by a gastight solid electrolyte, should have sufficient electrical conductivity over a wide range of pO2 and catalytic activity towards several reactions, including the hydrogen oxidation reaction (HOR), CO2 reduction reaction (CO2RR) for a fuel electrode and the oxygen reduction reaction (ORR), and the oxygen evolution reaction (OER) for an air electrode. Nowadays, symmetric SOCs (SSOCs), which adopt an identical electrode material for anode and cathode, have been developed with advantages over conventional SOCs, such as simplified preparation procedure, low cost in fabrication, and well-matched thermo-chemical compatibility.12,13 Irvine's group has suggested the concept of a symmetric configuration in an SOFC, and demonstrated the versatility of a redox-stable (La0.75Sr0.25)Cr0.5Mn0.5O3−δ electrode for the operation of a hydrogen/hydrocarbon fuel cell (FC) and a water electrolysis cell (EC).14 Indeed, the electrode material of an SSOC should be electrochemically active under both a reducing and an oxidizing gas atmosphere. Recently, a quasi-symmetric configuration has been studied for solid oxide cells (Q-SSOCs), where two similar electrode materials are optimally combined to further enhance the electrochemical performance of SSOCs.15,16 This configuration can also be obtained via a follow-up process after electrode fabrication, such as annealing in a reducing atmosphere.17,18
A perovskite oxide (ABO3) can be employed as the effective electrode material for an SOC since it has mixed ionic and electronic conducting (MIEC) and redox-stable properties depending on its versatile combinations of A/B cations. For most perovskite oxides, the component of the B-site cations is closely correlated with the electronic and catalytic properties of the parent oxides. For example, Fe-based perovskite oxide, where the redox pair of Fe3+/Fe4+ plays a critical role in its electrocatalytic activity, has been studied as an electrode material for SOCs.19–21 However, use of this perovskite oxide could lead to inferior electrochemical performance compared with a conventional Ni-based electrode. Recently, the exsolution method has been applied to enhance the electrochemical performance of perovskite oxide materials because evenly distributed metal or alloy nanoparticles (NPs) could be formed in situ on the surface during the reduction process with easy control over the size of the NPs.22–24 Nevertheless, the exsolution method has a limitation on the amounts of NPs, since excessive amounts of exsolved NPs on the surface leave a large amount of B-site vacancies inside the lattice, leading to stability issues for the crystal structure. To overcome this limitation, tuning the A-site deficiency of the host oxide has been demonstrated as an effective way to increase the amount of exsolved NPs and to facilitate the exsolution of less reducible cations.25 On the other hand, several perovskite oxides have been reported to be transformed into a layered perovskite oxide such as a Ruddlesden–Popper (RP) oxide and this transition can induce a large number of NPs to be exsolved according to its stoichiometry.26–28 A phase transformation concurrent with the exsolution process can occur due to different vacancy formation behaviors on the AO layer and the BO2 layer. H. Kim et al. demonstrated a DFT calculation to estimate the Gvf-O (Gibbs free energy of oxygen vacancy formation) on the AO and BO2 layers of Pr0.5Sr0.5TO3−δ (T: Mn, Fe, Co, Ni) and revealed that positive oxygen vacancy formation on the AO layer and negative formation on the BO2 layer can lead to a complete phase transformation from single perovskite oxide to Ruddlesden–Popper oxide, where Mn and Fe are used as B-site components.29
Herein, we designed a novel perovskite oxide, Pr0.6Sr0.4Mn0.2Fe0.7Ni0.1O3−δ (PSMFN), as an efficient electrode material for a quasi-symmetric SOC and evaluated it as an efficient electrode material for HOR, CO2RR, ORR, and OER in an SOC. Especially under fuel conditions, in situ reconstructed PSMFN, which is composed of a Ruddlesden–Popper oxide (R.P. PSMFN) with Ni–Fe alloy NPs decorated on the surface has been applied to the fuel electrode of a Q-SSOC. Exsolved metal NPs have shown high catalytic activity towards both HOR in SOFC mode and CO2RR in SOEC mode.20,30,31 In addition, Ni doping on ferrite oxide can induce the formation of an Ni–Fe alloy, which can drastically enhance the electrochemical performance of the oxide. Furthermore, several studies have demonstrated that ferrite-based perovskite oxides have shown catalytic activity against OER/ORR as good as that of cobalt-based perovskite oxide.32–34 X-ray diffraction (XRD) and microscopic analyses were conducted to investigate the occurrence of exsolution. Electrochemical analyses, such as X-ray absorption fine structure (XAFS), X-ray photoelectron spectroscopy (XPS), and electrochemical impedance spectroscopy (EIS) were performed to elucidate the electrochemical performance of PSMFN for Q-SSOC.
Experimental section
Catalyst preparation
PSMFN was synthesized by a modified a sol–gel method, as described below: stoichiometric amounts of metal nitrate precursors, Pr(NO3)3·6H2O (Sigma-Aldrich, 99.9%), Sr(NO3)2 (Sigma-Aldrich, 99.0%), Mn(NO3)2·4H2O (Alfa Aesar, 98%), Fe(NO3)3·9H2O (Sigma-Aldrich, 98%), Ni(NO3)2·6H2O (Samchun, 98%), were prepared and dissolved in DI water. Chelating agents, citric acid (Daejung, anhydrous) and ethylenediaminetetraacetic acid (Sigma-Aldrich), were also dissolved in the above solution, followed by adjusting the pH of the solution to 8. After that, the precursor solution was heated on a hot plate at 90 °C with vigorous stirring until the solution became a brown gel. The gelled solution was then pre-heated at 200 °C for 1 h and pulverized to get a fully dehydrated and fine powder. The final product was obtained by calcination at 1000 °C for 5 h.Cell fabrication
An electrolyte-supported symmetric solid oxide cell was prepared for an electrochemical performance test. A button-shaped LSGM (La0.8Sr0.2Ga0.8Mg0.2O3−δ, fuel cell material) electrolyte pellet was fabricated by a dry-pressing method. The pressed pellet was sintered twice in an air atmosphere at 1200 °C for 3 h and 1450 °C for 15 h. The sintered electrolyte pellet has a diameter of 17 mm with a thickness of 230 μm without any secondary phase observed in the XRD pattern (Fig. S1†). The as-prepared PSMFN was ball milled by a planetary mixer with an ion conducting material, GDC (Ce0.9Gd0.1O2, UHSA Anan Kasei), in a weight ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 at 400 rpm for 1 h. The obtained powder was mixed with an organic binder (6% ethyl cellulose in α-terpineol solution) in a weight ratio of 1:1.1 and pasted on both sides of the electrolyte pellet by a screen-printing method, followed by calcination at 1000 °C for 1 h. The fabricated porous electrode has an area of 0.283 cm2 with a thickness of 20 nm, and is strongly attached onto the surface of the electrolyte (Fig. S2†). A bar-shaped pellet with dimensions of 3.5 × 1.6 × 19 mm was dry-pressed and sintered at 1350 °C for 10 h for the measurement of electrical conductivity.
:4 at 400 rpm for 1 h. The obtained powder was mixed with an organic binder (6% ethyl cellulose in α-terpineol solution) in a weight ratio of 1:1.1 and pasted on both sides of the electrolyte pellet by a screen-printing method, followed by calcination at 1000 °C for 1 h. The fabricated porous electrode has an area of 0.283 cm2 with a thickness of 20 nm, and is strongly attached onto the surface of the electrolyte (Fig. S2†). A bar-shaped pellet with dimensions of 3.5 × 1.6 × 19 mm was dry-pressed and sintered at 1350 °C for 10 h for the measurement of electrical conductivity.
Physicochemical characterization
X-ray diffraction (XRD) analysis was conducted to investigate the crystal structure of the prepared PSMFN, reduced PSMFN and PSMFN–GDC composite using a Rigaku Ultima IV with 40 kV, 30 mA conditions within the 2θ range of 20–80°. Field-emission scanning electron microscopy (FE-SEM, S-4800, HITACHI) and mini-SEM (SNE-4000M, SEC) were employed to examine the surface morphology of the electrode catalyst and cross-sectional image of the fabricated solid oxide cell. Transmission electron microscopy (TEM, JEM-2200FS, JEOL) and electron energy loss spectroscopy (EELS) were also utilized to investigate the microstructure and elemental distribution of the exsolved metal NPs at 200 kV. X-ray absorption fine structure (XAFS) was conducted to inspect the electronic structure and estimated oxidation states of the transition metal cations on the 7D beamline of Pohang Accelerator Laboratory (PLS-II). X-ray absorption near edge structure (XANES) was analyzed using Athena software. X-ray photoelectron spectroscopy (XPS) was adopted to examine the surface oxygen species of the electrode catalysts using high-performance XPS (K-Alpha XPS system, Thermo Scientific). H2-temperature programmed reduction (H2-TPR) was carried out to investigate the reduction behavior of the prepared electrode catalyst. 40 mg of PSMFN was analyzed by a flow-type reactor (AutoChem II Chemisorption Analyzer, Micromeritics) under a 4% H2/Ar atmosphere at a flow rate of 50 mL min−1 in a temperature range from 100 °C to 800 °C.Electrochemical measurements
The electrical conductivities of PSMFN were measured using a four-probe method under air, 20% H2/N2, and 30% CO/CO2 at a total flow rate of 30 mL min−1 in the temperature range from 550 to 800 °C. Pt wire and Ag paste were used to connect the four probes. The electrochemical performance of a PSMFN–GDC symmetric cell was evaluated under both FC and EC operation. 100% H2 and air were fed into each fuel and oxygen electrode side, respectively, and 30% CO/CO2 was flowed into the fuel side, while the oxygen electrode was exposed to ambient air conditions. The total flow rate of the feed gas was 50 mL min−1. The electrolyte-supported button cell was mounted on an alumina tube using ceramic adhesive (552-VFG, AREMCO) as a sealant and Ag paste and wire was used as a current collector. Before the cell test, the fuel electrode had been previously annealed in a 100% H2 atmosphere for 0.5 h to induce the exsolution process of PSMFN. Electrochemical impedance spectroscopy (EIS, MP2, ZiveLab) was utilized to measure the ohmic and polarization resistance of the symmetric cell under OCV and applied voltage conditions with a voltage amplitude of 28.3 mV in the frequency range of 100 MHz to 10 mHz. Further distribution of relaxation times (DRT) analysis was performed with the MATLAB DRTtools developed by Ciucci et al.35 A half-cell test of the symmetric cell was conducted to estimate the HOR and ORR activities of the PSMFN–GDC electrode in air and a 100% H2 atmosphere. Both sides of the electrodes were exposed to the same gas atmosphere and EIS profiles were obtained under OCV conditions. Product analysis of CO2 electrolysis was carried out to calculate the CO production rate and the Faraday efficiency was determined by gas chromatography (Agilent, 7890B).Results and discussion
Structural and microscopic analyses
Fig. 1(a) depicts the reduction process of PSMFN, which has been developed and utilized as a quasi-symmetric electrode in this study. A single perovskite oxide, PSMFN, which can be denoted by the SrFeO2.97 (PDF #40-0905) phase in XRD analysis (Fig. 1(b)), is transformed into the R.P. phase of SrPrFeO4 (PDF #32-1237), which will now be denoted as R.P. PSMFN, during an annealing process under a 20% H2/N2 atmosphere at 800 °C for 10 h. Simultaneously, newly formed signals at 2θ values of 43.6°, 57.8°, and 74.7° are detected in the XRD patterns and identified as an alloy phase of Fe0.64Ni0.36 (PDF #47-1405). The above reduction behavior can be expressed with the equation: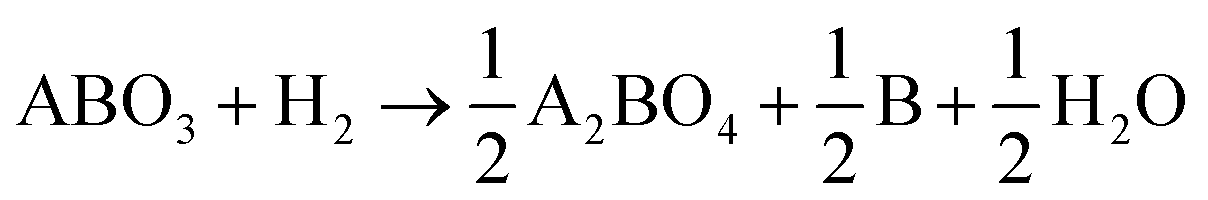 | (1) |
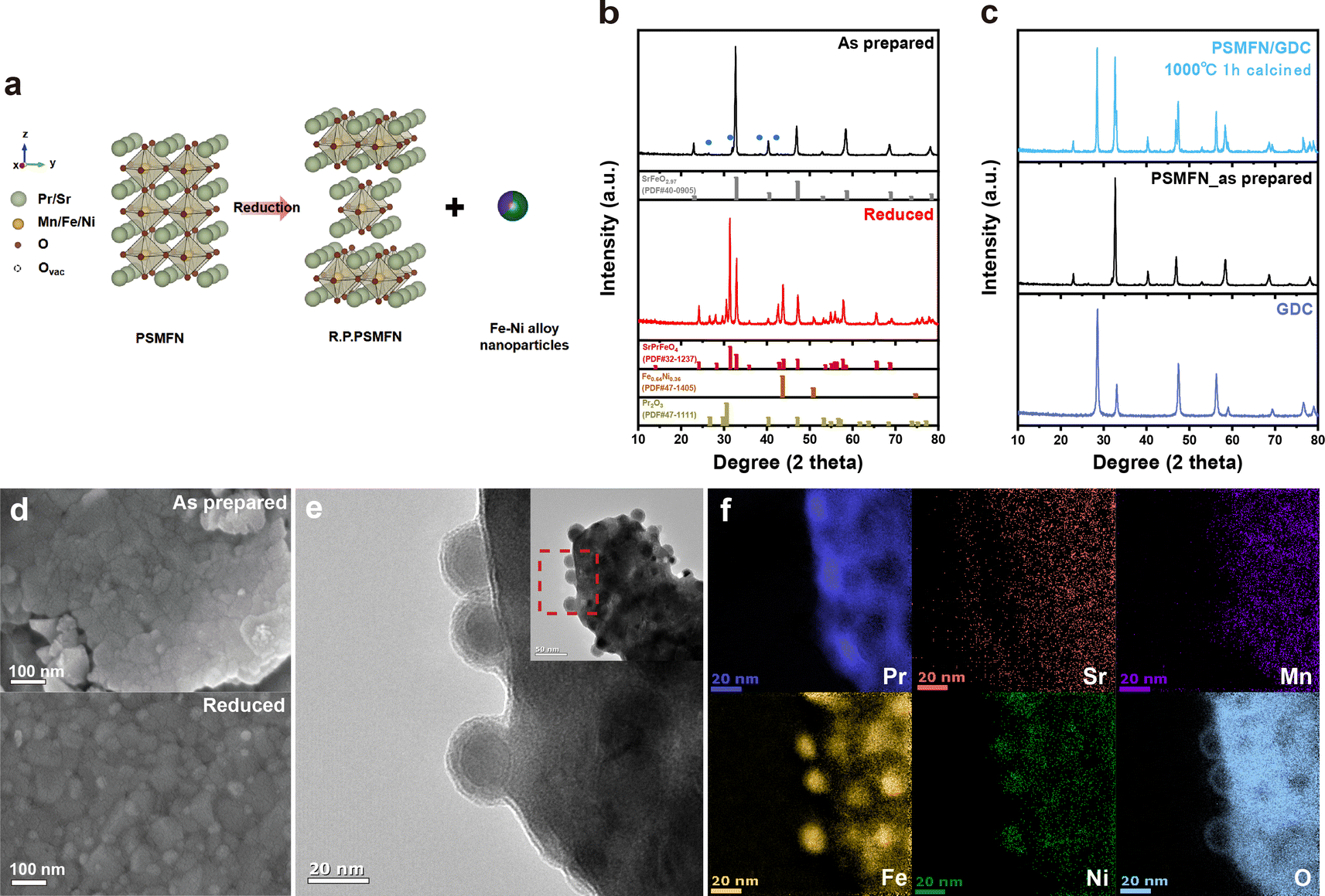 | ||
| Fig. 1 Structural and microscopic analyses of PSMFN. (a) Schematic diagram of the exsolution process with concurrent phase transformation from perovskite to Ruddlesden–Popper oxide. (b) XRD patterns of the as-prepared and reduced PSMFN with corresponding PDF standard patterns and of (c) the PSMFN–GDC mixture annealed at 1000 °C for 1 h. (d) SEM images of as-prepared and reduced PSMFN. (e) Magnified TEM image of reduced PSMFN (red box with dashed line) and (f) corresponding EELS mappings at the Pr L-edge, Sr L-edge, Mn K-edge, Fe K-edge, Ni K-edge and O K-edge, respectively. | ||
According to the above equation, a large amount of B-site metal cations can be exsolved to a separate metal phase due to the difference in stoichiometry between the two phases along with the phase transition. The stray peaks, mainly located at around 32° and marked by blue circles in Fig. 1b, could be assigned to the Pr0.96Mn0.982O3 (PDF #85-2203) phase, which is known to be decomposed into Pr2O3 and MnO under a reduction atmosphere. On the other hand, an ionic conducting material, GDC, was mixed with PSMFN to enlarge the triple phase boundaries (TPBs) for the electrode reaction. Fig. 1(c) suggests that PSMFN and GDC represented good material compatibility under electrode sintering conditions, which were calcination at 1000 °C for 1 h in ambient air, by revealing no additional phase formation between those two materials after sintering.
Microscopic analyses of reduced PSMFN were conducted to further verify the formation of an alloy phase. The TEM image and corresponding EELS mapping of reduced PSMFN (Fig. 1(d) and (e)) show that the newly formed Fe–Ni alloy NPs are present with spherical shape with a diameter of ∼30 nm, and are evenly distributed and deeply socketed onto the surface of the oxide. Meanwhile, polycrystallinity of the support oxide was observed, which might be due to the widespread phase reconstruction during the in situ exsolution process, as shown in the HR-TEM image (Fig. S3†). Small peaks from Pr2O3 were found in the XRD analysis of reduced PSMFN and the Pr2O3 seemed to be delocalized in the vicinity of Fe–Ni alloy particles, as can be seen in the Pr mapping in Fig. 1f. Similar behavior for the segregation of A-site cations during the exsolution process has been reported; for example, BaO islands appeared near the exsolved Ni nanoparticles on a BCZYYb support oxide.36 In this work, Pr2O3 segregation might take place due to the intrinsic high-valence state of praseodymium (Pr3+/Pr4+). From the XRD and microscopic analyses, it was clearly observed that the as-prepared PSMFN undergoes a phase transition from single perovskite oxide to R.P. oxide with newly exsolved Fe–Ni ally nanoparticles on the surface of the oxide.
XANES analysis
In a perovskite-type oxide, the oxidation state of the B-site cation plays an important role in charge transfer and catalytic activity against an electrochemical reaction, where Bn+–O–B(n−1)+ is usually known as the charge transfer pathway.37 The relatively low catalytic activity of perovskite oxides can be ascribed to the AO termination of the perovskite oxide surface, due to its larger ionic radius compared to the B-site cation.38,39 In this context, exsolution can be another strategy to expose the B-site cations to the surface, further reducing these cations to the metal or alloy NPs. Therefore, the oxidation states of B-site cations should be investigated to assess the electrochemical properties of their prepared catalysts. For a 3d or 4d transition metal, the absorption edge shape and position in XANES spectra are highly dependent on the electronic structure of the species, where electron excitation from the s orbital to the p orbital occurs by X-ray absorption.40 In other words, the main absorption edge position of the transition metal is highly sensitive to the oxidation states of the metal species. To estimate the change in oxidation states of the B-site cations, X-ray absorption near edge spectra (XANES) of PSMFN and reduced PSMFN at the Fe K-edge and Ni K-edge were obtained and analysed (Fig. 2(a) and (c)). Based on the obtained 1st-derivative curves of the normalized XANES spectra (Fig. S4†), the energies of the edge positions and oxidation states of the reference oxide are interpolated to estimate the oxidation states of Fe and Ni species for the as-prepared and reduced PSMFN. In the as-prepared PSMFN, the average oxidation state of Fe cations is calculated to be 3.75+, which represents mixed states of Fe3+ and Fe4+ in the perovskite lattice (Fig. 2(b)). After reduction in an H2 atmosphere, two edge positions of the XANES spectra at the Fe K-edge were simultaneously observed at 7112 and 7127 eV, which can be assigned to the edge energy of the Fe0 phase and Fe3.56+ in the lattice, respectively. These results imply that the exsolution of Fe species took place successfully in agreement with the microstructural analyses, and the Fe species are also present in a mixed state of Fe3+/Fe4+ in the R.P. phase. Similar behavior is also observed for Ni species; the oxidation state of Ni species in the as-prepared PSMFN, 2.47+, is reduced with an emerging state of Ni0 in the reduced PSMFN sample. Notably, Ni species seems to remain in the lattice structure of R.P. PSMFN with an observed oxidation state of 1.91+ in its XANES spectra. In a low oxygen partial pressure (pO2), all the oxygen atoms of the perovskite oxide can be released and neutral oxygen vacancies, which are composed of positively charged oxygen vacancies and two electrons, can be produced. This neutral oxygen vacancy reduces the neighboring cation, especially a transition metal cation in the B site of perovskite oxide, as expressed by eqn (2) below using Kröger–Vink notation:41,42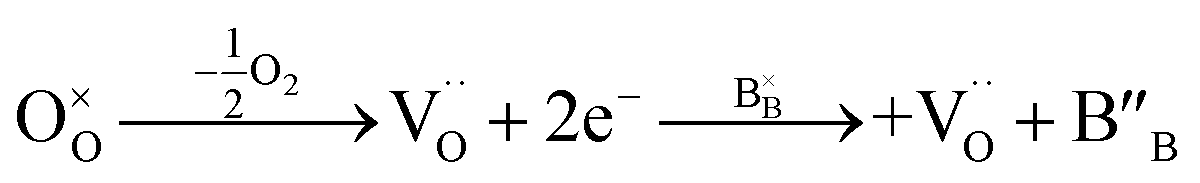 | (2) |
 | ||
| Fig. 2 Edge energy analyses for the oxidation states of transition metal ions in PSMFN. Normalized X-ray absorption near edge spectra of PSMFN, reduced PSMFN and reference oxide at (a) the Fe K-edge, (c) the Ni K-edge. Corresponding linear interpolations of edge positions for PSMFN, reduced PSMFN and reference oxides at (b) the Fe K-edge, (d) the Ni K-edge. | ||
For the material characteristics of PSMFN, oxygen vacancy concentration increases up to a critical point, where the perovskite structure cannot tolerate the large amounts of defects, and phase reconstruction and exsolution of Fe and Ni cations occur simultaneously. As a consequence, metallic Fe0 and Ni0 phases were observed with a reduced lattice oxidation state of B-site cations in the R.P. PSMFN.
Redox behavior and electrical properties
The redox behavior of PSMFN was further investigated with the H2-TPR profiles obtained in a 4% H2/Ar atmosphere. As featured in Fig. 3(a), the TPR profile of PSMFN have three significant peaks, one is located at ca. 250 °C, another is at ca. 350 °C and the third is at ca. 700 °C. As mentioned in the above section, a reducing atmosphere under the condition of a low oxygen partial pressure can create oxygen vacancies with the reduction of neighboring transition metal cations in B sites. The first reduction peak near 250 °C can be attributed to the reduction processes of Mn4+ and Fe4+ to Mn3+ and Fe3+, whereas the other peak positioned at about 350 °C could be assigned to the reduction processes of Mn3+, Fe3+, and Ni3+ to Mn2+, Fe2+, and Ni2+.43–45 When the temperature is raised to over 600 °C, further reduction causes the formation of a large amount of oxygen vacancies, which can destabilize the perovskite lattice and thus induce exsolution of the Fe–Ni alloy with a concurrent phase transition to a Ruddlesden–Popper structure, resulting in a large H2O peak in the TPR profile.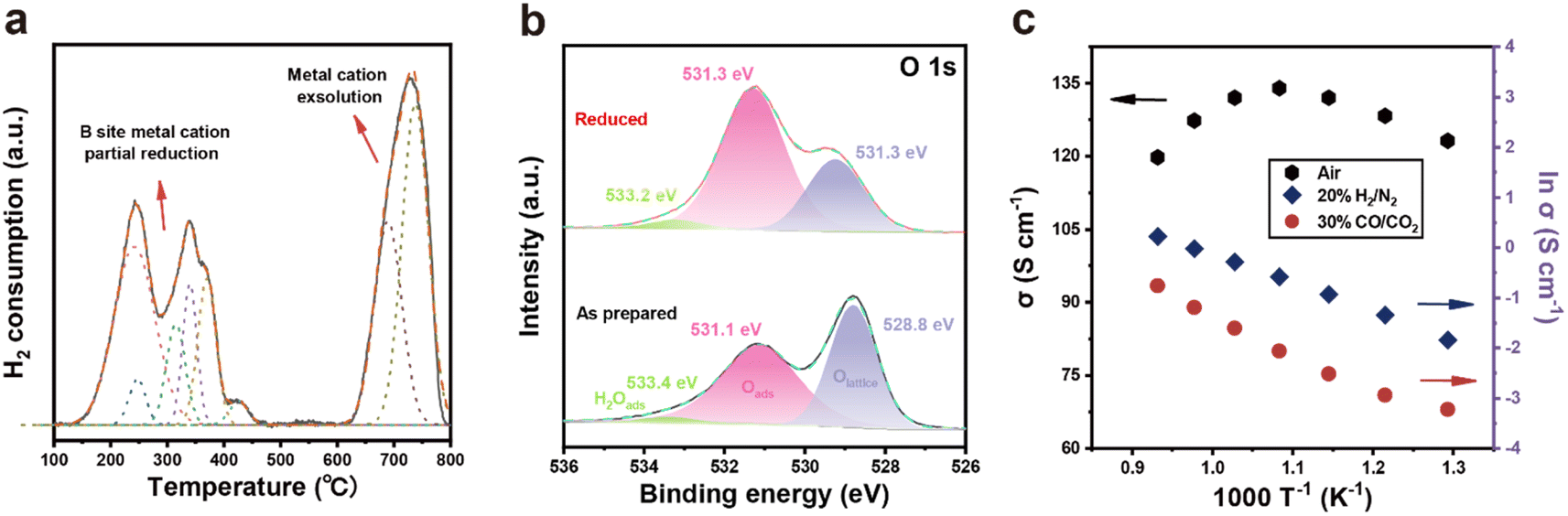 | ||
| Fig. 3 Redox behavior and electrical properties of PSMFN. (a) H2-TPR profile of PSMFN under 4% H2/Ar atmosphere at temperatures from 100 °C to 800 °C with a ramping rate of 5 °C min−1. (b) O 1s XPS spectra of PSMFN and reduced PSMFN. (c) Electrical properties of PSMFN under air (left axis), 20% H2/N2, and 30% CO/CO2 atmosphere (right axis) in a temperature range from 550 °C to 800 °C. | ||
Even if phase reconstruction occurred to rebalance the unstable perovskite structure with high oxygen vacancy concentration, large amounts of oxygen vacancies can still remain on the lattice after the exsolution process is complete. Fig. 3(b) represents the electronic structure of O 1s in the XPS spectra of the as-prepared and reduced PSMFN samples. The O 1s spectrum of a typical perovskite oxide usually shows three characteristic peaks; one located near a binding energy of 529 eV is related to lattice oxygen species, another positioned at ∼531 eV is attributed to surface-adsorbed species such as –OH groups and O2, and the other one at the highest binding energy is correlated with adsorbed H2O species.46,47 For lattice oxygen species, a slight increase in binding energy in the reduced PSMFN could be associated with varied electronic structures within the lattice oxygen (O2−). More importantly, the ratio of surface-adsorbed species to lattice oxygen greatly increased in the reduced sample and the amount of surface-adsorbed species is directly connected with the adsorption sites of oxygen-containing gas molecules, thus indicating oxygen vacancies on the surface of the oxides. Therefore, the XPS spectra of O 1s exhibited an increased number of oxygen vacancy species over the reduced PSMFN even after the phase reconstruction had occurred. This large number of created oxygen vacancies could also contribute to the superior fuel electrode kinetics in both operations of the SOC.
The electrical conductivities of PSMFN were also measured by the 4-probe method under the operating conditions of a quasi-symmetric electrode in an SOC in the temperature range from 550 °C to 800 °C with air, H2, and CO2 atmospheres, respectively (Fig. 3(c)). In the air atmosphere, the electrical conductivity of PSMFN increased with temperature until it reached a maximum value at 650 °C, then decreased at temperatures higher than 800 °C, indicating that the electrical conducting behavior of the PSMFN has transformed from semi-conducting to metallic behavior. Such a transition of conduction behavior is typically observed for a perovskite-related mixed conductor governed by a small polaron hopping mechanism.48,49 Ferrite perovskite oxide, represented by LaFeO3, shows p-type conduction behavior where small polaron hopping is thermally activated below ∼600 °C via the Fe3+–O–Fe4+ bond pathway. As the temperature rises further, however, a substantially increased number of oxygen vacancies disturbs the three-dimensional Fe–O–Fe network, resulting in decreased electrical conductivity.30,50 In other words, the creation of electrons induced by oxygen vacancy formation (eqn (2)), diminishes the main charge carriers, i.e., holes, of ferrite oxide by a recombination process and reduces the redox pairs by facilitating the conversion of Fe4+ to Fe3+. As shown in Fig. 3(c), the electrical conductivity of PSMFN is 120 S cm−1 at 800 °C, which is still a little bit larger than the threshold for application to an air electrode of an SOC (100 S cm−1).51 Before measuring the electrical conductivity of PSMFN under 20% H2/N2 and 30% CO/CO2 atmospheres, an annealing process under 20% H2/N2 for 0.5 h was carried out in order to induce exsolution and phase transition. Under both atmospheric conditions, the electrical conductivities are significantly lower than that under the air atmosphere. These lower conductivities are also associated with p-type conduction; a low oxygen partial pressure leads to a substantial loss of lattice oxygen species through liberation, decreasing the charge carriers and electrical conductivities of PSMFN. Specifically, under the H2/N2 atmosphere, the reduced PSMFN has a conductivity value of 1.66 S cm−1, which is comparable with those of reported symmetrical electrode catalysts, such as (La0.75Sr0.25)0.9Cr0.5Mn0.45Ru0.05O3−δ (1.6 S cm−1 at 800 °C),50 and La0.5Sr0.5Fe0.9Mo0.1O3−δ (1.1 S cm−1 at 850 °C).52
Performance evaluation on SOFC mode
Due to its high electrical conductivity under an air atmosphere and large amounts of TPBs attained by in situ exsolution of Fe–Ni alloy NPs with phase reconstruction, it is demonstrated that PSMFN can be simultaneously employed as both fuel and air electrodes. Therefore, an LSGM-based PSMFN–GDC quasi-symmetric cell was evaluated under fuel cell operating conditions, as shown in Fig. 4. Symmetric half-cell tests under air and H2 atmospheres were conducted to measure the ORR/OER and HOR activities of PSMFN–GDC. For HOR activity, both sides of the symmetric cell were treated under 100% H2 for 0.5 h to induce the exsolution and phase transition of PSMFN–GDC. Fig. 4(a) shows the temperature dependency of area specific resistance (ASR) and the calculated activation energy of the PSMFN–GDC electrode. In the Arrhenius plot, the ASR of PSMFN–GDC shows nearly linear correlation with temperature, which indicates thermally activated electrode kinetics in fuel cell conditions. In particular, the ASR value of PSMFN–GDC at 800 °C is 0.0371 Ω cm2, which is significantly lower than that of Sr2Fe1.5Mo0.5O6−Ω (0.24 Ω cm2)53 and comparable with a typical double perovskite (Pr,Ba)0.95(Fe0.95W0.05)2O5+δ 0.0307 Ω cm2,54 demonstrating the validity and superiority of ferrite oxide as an oxygen electrode. For HOR activity, the PSMFN–GDC exhibited an ASR of 0.290 Ω cm2 at 800 °C with an activation energy of 0.80 eV. These electrodes show a lower ASR value and activation energy against HOR compared to similar ferrite-oxide-based electrodes, such as Pr0.4Sr0.6Fe0.875Mo0.125O3−δ (1.6 Ω cm2, 0.95 eV),55 which might be attributed to the fast ion transport kinetics of the transformed Ruddlesden–Popper structure, the large number of oxygen vacancies created and exsolved alloy NPs. The performance of the PSMFN–GDC symmetric cell was evaluated under an FC mode with dry H2 flow in a fuel electrode and air flow in an oxygen electrode in Fig. 4(b) with the corresponding EIS profiles given in Fig. 4(c). Prior reduction before the performance test was also conducted in the same way as for the half-cell test. These electrodes exhibited peak power densities (PPD) of 0.702, 0.428, and 0.250 W cm−2 with corresponding polarization resistances of 0.370, 0.542, and 0.876 Ω cm2 under OCV at 800, 750, and 700 °C, respectively. The impressive PPD at 800 °C is comparable with other reported electrodes adopted in symmetric SOFC operation, as shown in Fig. 4(d). To further investigate the elemental electrode process under the FC mode, EIS data under applied voltage were analyzed using distribution of relaxation time (DRT), as shown in Fig. 4(e). In the DRT plots, the area of each peak directly represents the polarization resistance of the electrode sub-process occurring in the characteristic frequency region. Under all applied voltage conditions, the DRT plots showed four peaks, which are labelled P1F (103 to 104 Hz), P2F (102 to 103 Hz), P3F (100 to 102 Hz), and P4F (10−1 to 100 Hz). According to previous study, the P1F, P2F, and P3F peaks represent fuel electrode kinetics, where the P1F is attributed to charge transport and the others reflect oxygen-transport-related processes in the fuel electrode.56 On the other hand, the P4F process is related mainly to the cathodic reaction, i.e., ORR. Under OCV conditions, the cathodic reaction (P4F) makes a larger contribution to overall polarization compared with P1F, P2F, and P3F, indicating sluggish ORR in FC operation. In addition, while oxygen-transport-related processes, P1F and P3F, show negligible change in voltage, charge transfer kinetics, represented by P2F and P4F, could exhibit great dependency on voltage. In particular, cathodic polarization is greatly reduced with an applied voltage compared to polarization of the fuel electrode. A relatively low contribution from fuel electrode kinetics to overall polarization could be ascribed to enhanced fuel electrode kinetics by in situ exsolution and sluggish ORR kinetics in SOFC. A long-term stability test was conducted under a constant current density of 280 mA cm−2 at 750 °C for 100 h, where no significant voltage decay was observed, indicating the robustness of PSMFN–GDC as a symmetric electrode for SOFC (Fig. 4(f)).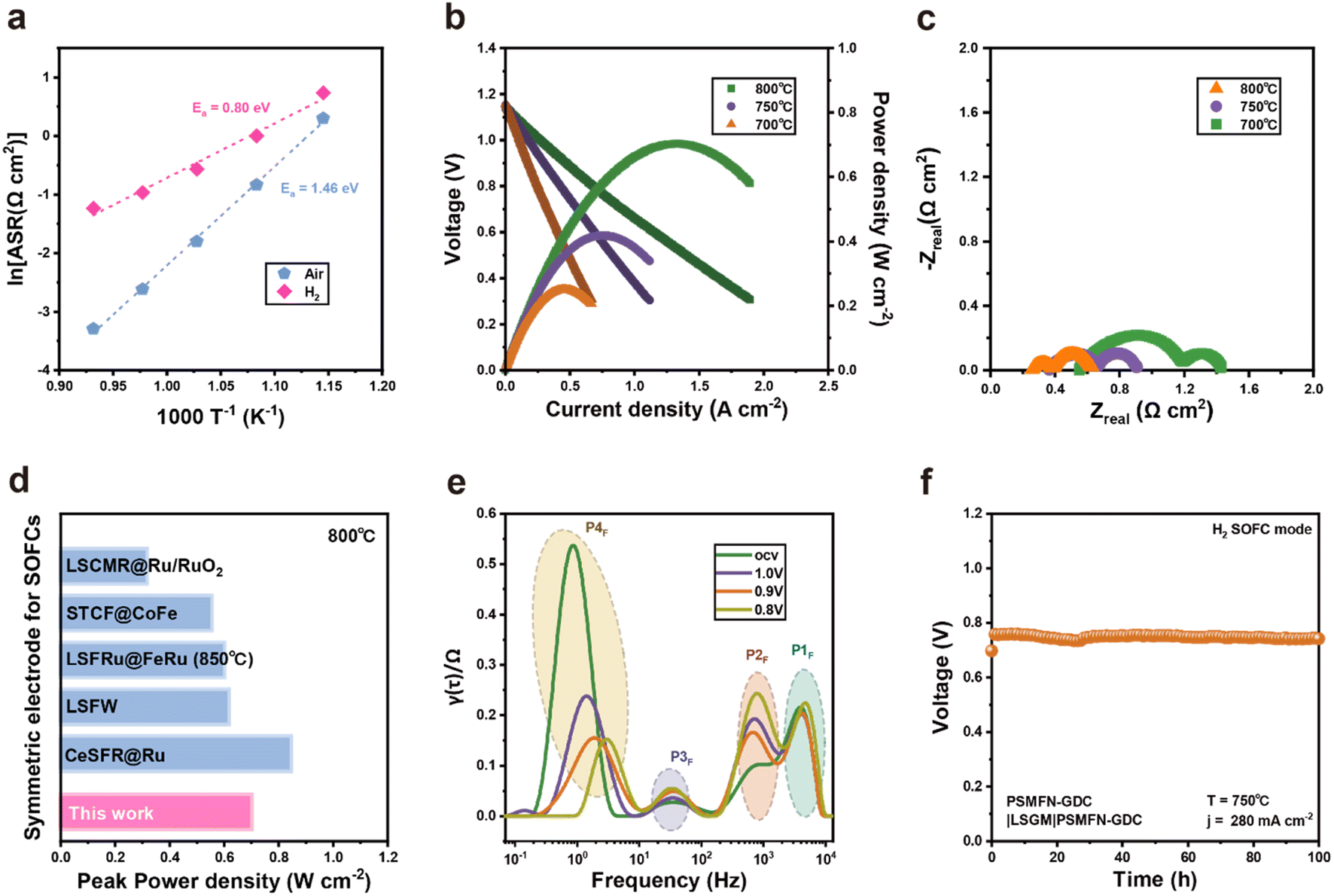 | ||
| Fig. 4 Electrochemical performances of PSMFN in quasi-symmetric SOFC mode with a configuration of PSMFN–GDC|LSGM|PSMFN–GDC. (a) Arrhenius plots for the area specific resistances of a PSMFN–GDC electrode in 100% H2 and air atmospheres. (b) I–V–P plots of a PSMFN–GDC quasi-symmetric cell under 100% H2 fuel conditions and corresponding (c) Nyquist plots under OCV conditions at temperatures of 800 °C, 750 °C, and 700 °C. (d) Peak power density comparison with other symmetric SOFC electrodes in 100% H2. (e) DRT plots of the PSMFN–GDC cell at OCV and applied voltage conditions for SOFC operation at 800 °C. (f) Long-term test of the PSMFN–GDC cell under constant current operation of 280 mA cm−2 at 750 °C. | ||
Performance evaluation on SOEC mode
The electrochemical performance of the PSMFN–GDC electrode was also evaluated under an EC mode for CO2 electrolysis (Fig. 5). The symmetric cell demonstrated notable current densities of −1.02, −0.508, and −0.240 A cm−2 at a voltage of 1.5 V at 800, 750 and 700 °C, respectively (Fig. 5(a)). The EIS profiles in Fig. 5(b) also exhibit corresponding polarization resistances under OCV conditions, which are 0.752, 1.616, and 3.71 Ω cm2 at each temperature. The current density values of the PSMFN–GDC symmetric cell against CO2 electrolysis at 800 °C is much higher than for previously studied symmetric electrode catalysts, as summarized and listed in Fig. 5(c). DRT analysis of the EIS plots was also conducted to elucidate the electrode process of CO2 electrolysis in SOEC. As shown in Fig. 5(d), the DRT plots present four elemental processes of oxygen ion transportation between electrolyte and electrode (P1E), OER (P2E), CO2 adsorption (P3E), and dissociation of carbonate intermediates on the cathode surface (P4E).10,57 In particular, the P3E process made the majority of the contribution to total polarization resistance, indicating that CO2 adsorption could be the rate-limiting step for CO2 electrolysis in the SOEC system, in agreement with previous studies. Also, apart from the P1E process, three elemental processes were accelerated by applying a voltage, especially for the P3E process. Notably, the shoulder peak beside P3E (P3aE) emerged under a high imposed voltage of 1.2 V, where this peak can be caused by a finite Warburg effect that is indicative of gas diffusion and surface processes.58 Outlet gas analysis of CO2 electrolysis leads to a CO production rate of 158 μmol cm−2 min−1 with a high Faraday efficiency of 91% at 0.5 V vs. OCV, as demonstrated in Fig. 5(e). A long-term test under a constant current density of 0.39 A cm−2 also demonstrated the voltage profile with a decay rate of 0.621 mV h−1, indicating the stable behavior of the PSMFN–GDC symmetric electrode for CO2 electrolysis under the EC mode.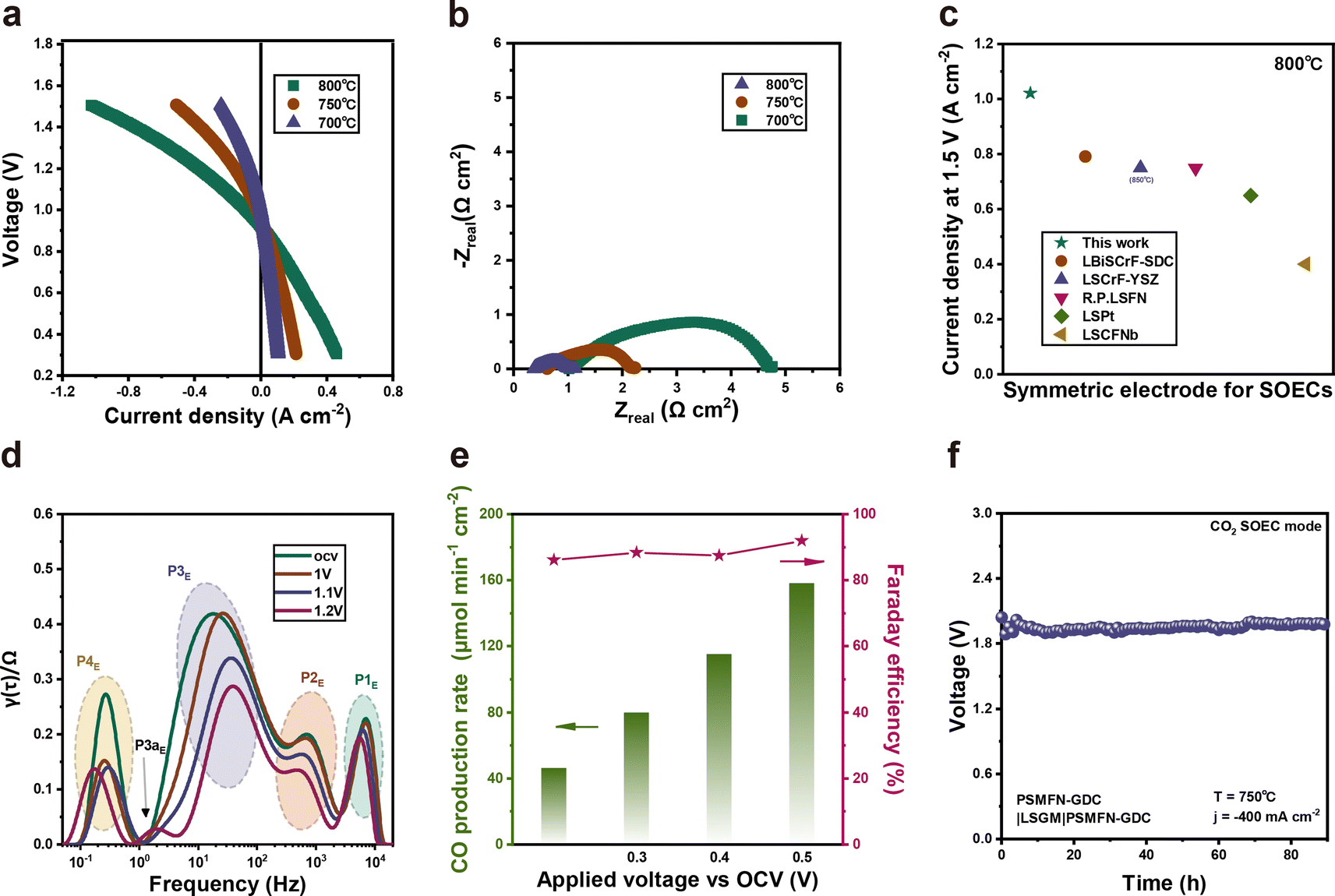 | ||
| Fig. 5 PSMNF–GDC quasi-symmetric cell for CO2 electrolysis performance evaluation. (a) I–V plots of PSMFN–GDC cell for CO2 electrolysis under 30% CO/CO2 and (b) Nyquist plots under OCV at 800 °C to 700 °C. (c) CO2 electrolysis performance comparison with other symmetric SOEC electrodes. (d) DRT analyses of PSMFN–GDC cell under applied voltage conditions for CO2 electrolysis at 800 °C. (e) Production rates of CO and corresponding Faraday efficiencies under applied voltage of 0.2–0.5 V vs. OCV at 800 °C. (f) Long-term stability test under constant current density of 280 mA cm−2 at 750 °C. | ||
Conclusions
In this work, PSMFN was prepared and evaluated as an efficient electrode material for Q-SSOCs. A fabricated PSMFN–GDC quasi-symmetric cell exhibited a peak power density of 0.702 W cm−2 under an H2-fed FC mode and current density of 1.02 A cm−2 at a voltage of 1.5 V under CO2 electrolysis EC mode at 800 °C. The physiochemical and electrochemical properties of PSMFN were investigated to support the electrochemical performance of a PSMFN–GDC electrode in symmetric application. Under a fuel atmosphere, PSMFN was successfully phase-transformed from single perovskite to an R.P. structure along with a simultaneous exsolution process to produce large amounts of Fe–Ni NPs. These in situ exsolved Fe–Ni alloy NPs were thoroughly characterized by microscopic analyses and the great number of surface oxygen vacancies produced were examined by O 1s XPS. Furthermore, a suitable electrical conductivity of 120 S cm−1 and significantly low area specific resistance of 0.0371 Ω cm2 in an air atmosphere can ensure sufficient activity towards ORR and OER. Therefore, the quasi-symmetric application of a PSMFN electrode on SOC could be a promising strategy for an efficient energy conversion system.Author contributions
Choi J.: investigation, synthesis, formal analysis, writing – original draft, visualization. Jang D., Kim M., Ha J.: data curation. Ahn H.: writing, visualization – review & editing. Kim W. B.: project administration, supervision, conceptualization, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Korea Government (MOTIE) (No. 20212010100040), and by “Human Resources Program in Energy Technology” of the Korea Institute of Energy Technology Evaluation and Planning (KETEP), granted financial resource from the Ministry of Trade, Industry & Energy, Republic of Korea (No. 20204010600100), and by the National Research Foundation of Korea (NRF) grant funded by the Korea Government (MSIT) (No. 2021R1A5A1084921).References
- R. Murphy, Y. Zhou, L. Zhang, L. Soule, W. Zhang, Y. Chen and M. Liu, Adv. Funct. Mater., 2020, 30, 2002265 CrossRef CAS.
- W. Zhang, Y. Zhou, E. Liu, Y. Ding, Z. Luo, T. Li, N. Kane, B. Zhao, Y. Niu, Y. Liu and M. Liu, Appl. Catal., B, 2021, 299, 120631 CrossRef CAS.
- M. A. Laguna-Bercero, J. Power Sources, 2012, 203, 4–16 CrossRef CAS.
- J. C. Ruiz-Morales, D. Marrero-López, J. Canales-Vázquez and J. T. S. Irvine, RSC Adv., 2011, 1, 1403–1414 RSC.
- A. Brisse, J. Schefold and M. Zahid, Int. J. Hydrogen Energy, 2008, 33, 5375–5382 CrossRef CAS.
- O. A. Marina, L. R. Pederson, M. C. Williams, G. W. Coffey, K. D. Meinhardt, C. D. Nguyen and E. C. Thomsen, J. Electrochem. Soc., 2007, 154, B452 CrossRef CAS.
- Y. Chen, B. deGlee, Y. Tang, Z. Wang, B. Zhao, Y. Wei, L. Zhang, S. Yoo, K. Pei, J. H. Kim, Y. Ding, P. Hu, F. F. Tao and M. Liu, Nat. Energy, 2018, 3, 1042–1050 CrossRef CAS.
- W. Zhang, X. Hu, Y. Zhou, Z. Luo, G. Nam, Y. Ding, T. Li, Z. Liu, Y. Ahn, N. Kane, W. Wang, J. Hou, D. Spradling and M. Liu, Adv. Energy Mater., 2022, 12, 2202928 CrossRef CAS.
- S. Park, J. M. Vohs and R. J. Gorte, Nature, 2000, 404, 265–267 CrossRef CAS.
- S. Lee, M. Kim, K. T. Lee, J. T. S. Irvine and T. H. Shin, Adv. Energy Mater., 2021, 11, 2202928 Search PubMed.
- K. T. Bae, I. Jeong, A. Akromjon, H. N. Im and K. T. Lee, Chem. Eng. J., 2023, 472, 145015 CrossRef CAS.
- B. Niu, C. Lu, W. Yi, S. Luo, X. Li, X. Zhong, X. Zhao and B. Xu, Appl. Catal., B, 2020, 270, 118842 CrossRef CAS.
- Q. Liu, X. Dong, G. Xiao, F. Zhao and F. Chen, Adv. Mater., 2010, 22, 5478–5482 CrossRef CAS PubMed.
- D. M. Bastidas, S. Tao and J. T. S. Irvine, J. Mater. Chem., 2006, 16, 1603–1605 RSC.
- K. Zheng, J. Lach, P. Czaja, M. Gogacz, P. Czach, A. Brzoza-Kos, P. Winiarz and J. Luo, J. Power Sources, 2023, 587, 233707 CrossRef CAS.
- Y. Zhang, X. Zhang, J. Ni and C. Ni, ACS Appl. Mater. Interfaces, 2023, 15, 3974–3984 CrossRef CAS PubMed.
- P. Li, Y. Xuan, B. Jiang, S. Zhang and C. Xia, Electrochem. Commun., 2022, 134, 107188 CrossRef CAS.
- Y. Chen, Z. Cheng, Y. Yang, Q. Gu, D. Tian, X. Lu, W. Yu and B. Lin, J. Power Sources, 2016, 310, 109–117 CrossRef CAS.
- B. Li, S. He, J. Li, X. Yue, J. T. S. Irvine, D. Xie, J. Ni and C. Ni, ACS Catal., 2020, 10, 14398–14409 CrossRef CAS.
- A. P. Panunzi, L. Duranti, I. Luisetto, N. Lisi, M. Marelli and E. Di Bartolomeo, Chem. Eng. J., 2023, 471, 144448 CrossRef CAS.
- B. Zhang, Y. Wan, Z. Hua, K. Tang and C. Xia, ACS Appl. Energy Mater., 2021, 4, 8401–8409 CrossRef CAS.
- O. Kwon, S. Sengodan, K. Kim, G. Kim, H. Y. Jeong, J. Shin, Y. W. Ju, J. W. Han and G. Kim, Nat. Commun., 2017, 8, 15967 CrossRef CAS PubMed.
- Y. Gao, D. Chen, M. Saccoccio, Z. Lu and F. Ciucci, Nano Energy, 2016, 27, 499–508 CrossRef CAS.
- D. Neagu, T. S. Oh, D. N. Miller, H. Ménard, S. M. Bukhari, S. R. Gamble, R. J. Gorte, J. M. Vohs and J. T. S. Irvine, Nat. Commun., 2015, 6, 8120 CrossRef PubMed.
- D. Neagu, G. Tsekouras, D. N. Miller, H. Ménard and J. T. S. Irvine, Nat. Chem., 2013, 5, 916–923 CrossRef CAS PubMed.
- M. Qin, Y. Xiao, H. Yang, T. Tan, Z. Wang, X. Fan and C. Yang, Appl. Catal., B, 2021, 299, 120613 CrossRef CAS.
- J. Choi, M. Kim, S. K. Kang, J. Kim, J. Ha, H. H. Shin, T. Park and W. B. Kim, ACS Sustain. Chem. Eng., 2022, 10, 7595–7602 CrossRef CAS.
- J. Choi, S. Park, H. Han, M. Kim, M. Park, J. Han and W. B. Kim, J. Mater. Chem. A, 2021, 9, 8740–8748 RSC.
- H. Kim, C. Lim, O. Kwon, J. Oh, M. T. Curnan, H. Y. Jeong, S. Choi, J. W. Han and G. Kim, Nat. Commun., 2021, 12, 6814 CrossRef CAS PubMed.
- M. Marasi, L. Duranti, I. Luisetto, E. Fabbri, S. Licoccia and E. Di Bartolomeo, J. Power Sources, 2023, 555, 232399 CrossRef CAS.
- Y. F. Sun, Y. Q. Zhang, J. Chen, J. H. Li, Y. T. Zhu, Y. M. Zeng, B. S. Amirkhiz, J. Li, B. Hua and J. L. Luo, Nano Lett., 2016, 16, 5303–5309 CrossRef CAS.
- M. Chen, S. Paulson, W. H. Kan, V. Thangadurai and V. Birss, J. Mater. Chem. A, 2015, 3, 22614–22626 RSC.
- N. A. Baharuddin, N. A. M. N. Aman, A. Muchtar, M. R. Somalu, A. A. Samat and M. I. Aznam, Ceram. Int., 2019, 45, 12903–12909 CrossRef CAS.
- H. Choi, A. Fuller, J. Davis, C. Wielgus and U. S. Ozkan, Appl. Catal., B, 2012, 127, 336–341 CrossRef CAS.
- T. H. Wan, M. Saccoccio, C. Chen and F. Ciucci, Electrochim. Acta, 2015, 184, 483–499 CrossRef CAS.
- Y. Song, W. Wang, L. Ge, X. Xu, Z. Zhang, P. S. B. Julião, W. Zhou and Z. Shao, Adv. Sci., 2017, 4, 1700337 CrossRef PubMed.
- J. Mizusaki, Solid State Ionics, 1992, 52, 79–91 CrossRef CAS.
- X. Xi, X. W. Wang, Y. Fan, Q. Wang, Y. Lu, J. Li, L. Shao, J. L. Luo and X. Z. Fu, J. Power Sources, 2021, 482, 228981 CrossRef CAS.
- J. Druce, H. Téllez, M. Burriel, M. D. Sharp, L. J. Fawcett, S. N. Cook, D. S. McPhail, T. Ishihara, H. H. Brongersma and J. A. Kilner, Energy Environ. Sci., 2014, 7, 3593–3599 RSC.
- H. Sun and W. Zhou, Energy Fuels, 2021, 35, 5716–5737 CrossRef CAS.
- A. B. Muñoz-García, A. M. Ritzmann, M. Pavone, J. A. Keith and E. A. Carter, Acc. Chem. Res., 2014, 47, 3340–3348 CrossRef PubMed.
- R. B. Wexler, G. S. Gautam, E. B. Stechel and E. A. Carter, J. Am. Chem. Soc., 2021, 143, 13212–13227 CrossRef CAS PubMed.
- K. Zhao, A. Zheng, H. Li, F. He, Z. Huang, G. Wei, Y. Shen and Z. Zhao, Appl. Catal., B, 2017, 219, 672–682 CrossRef CAS.
- S. Shah, M. Xu, X. Pan and K. L. Gilliard-Abdulaziz, ACS Appl. Nano Mater., 2021, 4, 8626–8636 CrossRef CAS.
- F. He, J. Chen, S. Liu, Z. Huang, G. Wei, G. Wang, Y. Cao and K. Zhao, Int. J. Hydrogen Energy, 2019, 44, 10265–10276 CrossRef CAS.
- B. Yang, J. Bian, L. Wang, J. Wang, Y. Du, Z. Wang, C. Wu and Y. Yang, Phys. Chem. Chem. Phys., 2019, 21, 11697–11704 RSC.
- T. J. Frankcombe and Y. Liu, Chem. Mater., 2023, 35, 5468–5474 CrossRef CAS.
- M. Chen, S. Paulson, V. Thangadurai and V. Birss, ECS Trans., 2012, 45, 343–348 CrossRef CAS.
- J. Mizusaki, J. Tabuchi, T. Matsuura, S. Yamauchi and K. Fueki, J. Electrochem. Soc., 1989, 136, 2082 CrossRef CAS.
- L. Bian, C. Liu, S. Li, J. Peng, X. Li, L. Guan, Y. Liu, J. H. Peng, S. An and X. Song, Int. J. Hydrogen Energy, 2020, 45, 19813–19822 CrossRef CAS.
- A. P. Panunzi, L. Duranti, I. Luisetto, N. Lisi, M. Marelli and E. Di Bartolomeo, Chem. Eng. J., 2023, 471, 144448 CrossRef CAS.
- J. Wang, J. Zhou, J. Yang, D. Neagu, L. Fu, Z. Lian, T. H. Shin and K. Wu, Adv. Mater. Interfaces, 2020, 7, 2000828 CrossRef CAS.
- Q. Liu, X. Dong, G. Xiao, F. Zhao and F. Chen, Adv. Mater., 2010, 22, 5478–5482 CrossRef CAS PubMed.
- B. Zhang, Y. Wan, Z. Hua, K. Tang and C. Xia, ACS Appl. Energy Mater., 2021, 4, 8401–8409 CrossRef CAS.
- D. Zhang, K. Zhang, T. He, N. Yu, Y. Zhao, Y. Wang and T. Liu, Int. J. Hydrogen Energy, 2020, 45, 21825–21835 CrossRef CAS.
- S. M. Choi, H. An, K. J. Yoon, B. K. Kim, H. W. Lee, J. W. Son, H. Kim, D. Shin, H. I. Ji and J. H. Lee, Appl. Energy, 2019, 233–234, 29–36 CrossRef CAS.
- X. Zhang, Y. Song, F. Guan, Y. Zhou, H. Lv, G. Wang and X. Bao, J. Catal., 2018, 359, 8–16 CrossRef CAS.
- I. D. Unachukwu, V. Vibhu, J. Uecker, I. C. Vinke, R. A. Eichel and L. G. J. B. de Haart, J. CO2 Util., 2023, 69, 102423 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Cross-sectional SEM images, HR-TEM image, 1st derivative curves of XANES spectra, XPS survey, additional electrochemical properties, and EIS profiles. See DOI: https://doi.org/10.1039/d3ta08110a |
| This journal is © The Royal Society of Chemistry 2024 |