
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImine-linked 2D covalent organic frameworks based on benzotrithiophene for visible-light-driven selective aerobic sulfoxidation†
Fengwei
Huang
,
Yuexin
Wang
,
Xiaoyun
Dong
and
Xianjun
Lang
 *
*
Hubei Key Lab on Organic and Polymeric Optoelectronic Materials, College of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, China. E-mail: xianjunlang@whu.edu.cn
First published on 6th February 2024
Abstract
Recently, emerging visible-light photocatalysts such as covalent organic frameworks (COFs) have undergone rapid development due to the molecular tunability and structural diversity. Imine takes an overriding role in reversible covalent bonds to form COFs, which usually lack high activity because the polarity of imine hampers light-driven electron transfer. In principle, this barrier can be overcome by exploring the electron push–pull effect of the molecular building units. In this work, with benzotrithiophene as the electron donor, two imine-linked 2D (two-dimensional) COFs were successfully synthesized by the condensation of BTT (benzo[1,2-b:3,4-b′:5,6-b′′]trithiophene-2,5,8-tricarbaldehyde) with TAPT (2,4,6-tris(4-aminophenyl)-1,3,5-triazine) and TAPB (1,3,5-tris(4-aminophenyl)benzene), giving BTT-TAPT-COF and BTT-TAPB-COF, respectively. The density functional theory calculation predicted that BTT-TAPT-COF with an electron-withdrawing triazine core can boost charge separation, and the superiority was further verified by optoelectronic properties. Both imine-linked 2D COFs based on benzotrithiophene can activate dioxygen, in which BTT-TAPT-COF exhibits better activity based on the prominent properties. BTT-TAPT-COF was found to be a photocatalyst for blue-light-driven selective aerobic sulfoxidation with outstanding versatility and reusability. This work paints a clear picture of how the electron push–pull effect of the building units of COFs regulates the ensuing activity in visible-light photocatalysis.
1. Introduction
Covalent organic frameworks (COFs) are crystalline porous materials organized by molecular building units of light elements via strong covalent bonds.1–3 The abundant building units and various synthesis methods greatly enrich the diversity of COFs.4–8 As potential materials, numerous applications including energy storage,9–11 sensing,12–14 adsorption,15–18 and catalysis19–23 have been developed based on COFs. Equipped with adjusted band positions and functionalized skeletons, COFs with inherent porosity are outstanding materials for efficient photocatalysis.24–26 Notably, the periodic crystalline structure and the large π conjugation favor the light-driven delocalization and transfer of electrons. Therefore, COF photocatalysis has triggered keen interest in selective organic conversions.27–33Imine-linked COFs with reversible covalent bonds have been widely used in various realms; however, high photocatalytic activity of the COFs is hindered due to the polarity of imine which hampers light-driven electron transfer. In principle, this barrier can be overcome by exploring the electron push–pull effect of the molecular building units.34 The photocatalytic activity of COFs can be boosted through ingenious molecular design of the building units. Inspired by the rapid development of graphitic carbon nitride,35–40 triazine units provide great inspiration in photocatalysis for further investigation. Moreover, covalent triazine frameworks, with heteroatom effects and structural durability, have provided fascinating platforms in a number of redox reactions.41–44 As an excellent light-harvesting unit, the triazine core has possessed outstanding flexibility in fashioning photoactive COFs for visible-light photocatalysis.45–48 The apparent electron-withdrawing effect and the rigid plane of the triazine ring underpin a great deal of emerging applications of triazine-cored COFs due to the highly tunable structures.49–53 Several functionalized N-atoms endow triazine-cored COFs with adequate electron-withdrawing effect. Therefore, multiple reports have verified triazine-cored COFs with outstanding ability in various fields of photocatalysis.54–56
In this work, benzotrithiophene, a significant electron-donating unit for COFs,57–62 is adopted to work with an electron-withdrawing triazine unit for the synthesis of photoactive COFs. The good rigidity of both building units ensures the planarity, and the obvious complementarity of electron push–pull effect is conducive to the donor–acceptor (D–A) pair. Therefore, TAPT (2,4,6-tris(4-aminophenyl)-1,3,5-triazine) and TAPB (1,3,5-tris(4-aminophenyl)benzene) are separately adopted to frame imine-linked 2D (two-dimensional) COFs with BTT (benzo[1,2-b:3,4-b′:5,6-b′′]trithiophene-2,5,8-tricarbaldehyde), giving BTT-TAPT-COF and BTT-TAPB-COF, respectively. The visible-light-driven selective aerobic sulfoxidation is used as a model reaction to evaluate the photocatalytic activity of BTT-TAPT-COF and BTT-TAPB-COF. BTT-TAPT-COF better facilitates the transfer of the photogenerated charge carriers compared with BTT-TAPB-COF owing to the superior D–A pair. As such, BTT-TAPT-COF is found to be a superior photocatalyst for blue-light-driven selective aerobic sulfoxidation with outstanding versatility and reusability. This work highlights the essence of molecular design based on electronic effect of COFs to realize efficient visible-light photocatalysis for selective organic conversion.
2. Results and discussion
Two imine-linked 2D COFs based on benzotrithiophene were synthesized by the condensation of BTT with TAPT and TAPB57,58 using acetic acid as a catalyst (Fig. 1a), giving BTT-TAPT-COF and BTT-TAPB-COF, respectively. More detailed synthesis steps are provided in the ESI.† Density functional theory (DFT) calculation was first performed to investigate the orbital electron distribution of BTT-TAPT-COF and BTT-TAPB-COF. As illustrated in Fig. 1b and c, the electrostatic potential (ESP) map of BTT-TAPT-COF suggested that the negative charge maxima (red area) were distributed in the triazine unit and N atoms in C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N. As for BTT-TAPB-COF, the negative charge maxima were distributed in N atoms in CN (Fig. 1c). Moreover, the dipole moment of BTT-TAPT-COF (2.145 Debye) is larger than that of BTT-TAPB-COF (1.423 Debye) due to more apparent electron push–pull effect.
N. As for BTT-TAPB-COF, the negative charge maxima were distributed in N atoms in CN (Fig. 1c). Moreover, the dipole moment of BTT-TAPT-COF (2.145 Debye) is larger than that of BTT-TAPB-COF (1.423 Debye) due to more apparent electron push–pull effect.
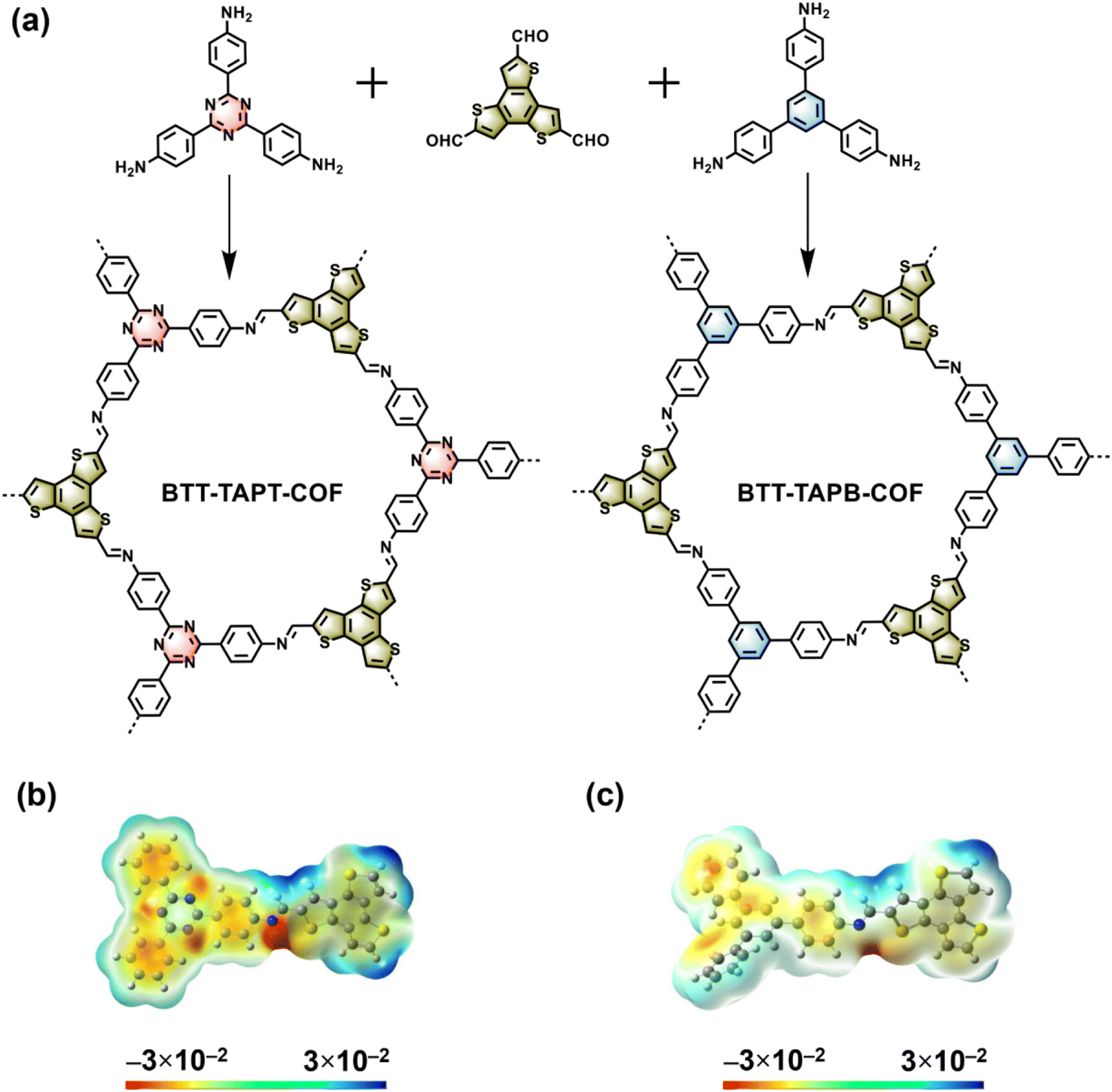 | ||
| Fig. 1 (a) Syntheses of BTT-TAPT-COF and BTT-TAPB-COF. ESP maps of BTT-TAPT-COF (b) and BTT-TAPB-COF (c). | ||
The localizations of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) for the two imine-linked 2D COFs based on benzotrithiophene are displayed. For BTT-TAPT-COF, the HOMO was distributed at the BTT unit, and the LUMO was generally distributed at the triazine, indicating it is a good D–A pair for efficient charge separation (Fig. 2a). The same simulation on BTT-TAPB-COF indicated that it is structurally similar to BTT-TAPT-COF. In contrast, with no triazine unit in the framework, the HOMO and LUMO were localized on the BTT and phenyl units (Fig. 2b). Upon comparison, BTT-TAPT-COF equipped with triazine cores demonstrated more apparent electron push–pull effect than BTT-TAPB-COF. Based on the HOMO and LUMO locations, the calculated bandgap of BTT-TAPT-COF is narrower than that of BTT-TAPB-COF.
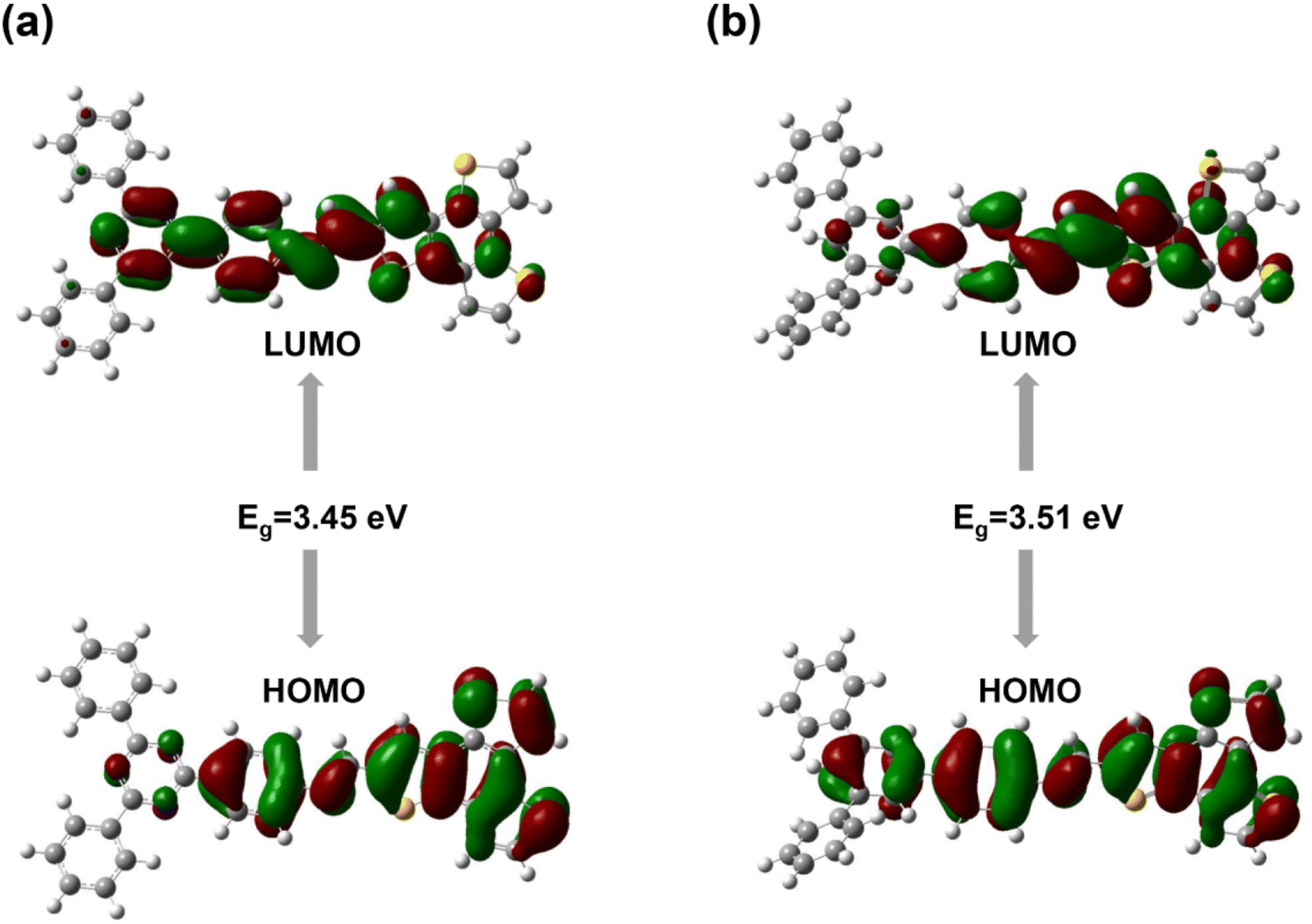 | ||
| Fig. 2 Calculated orbital distributions of BTT-TAPT-COF (a) and BTT-TAPB-COF (b). | ||
The crystal structures of BTT-TAPT-COF and BTT-TAPB-COF were determined by powder X-ray diffraction (PXRD). The high-intensity peak at 4.7° of BTT-TAPT-COF is associated with the periodicities in (100) plane, and other obvious peaks at 8.1°, 9.4° and 12.4° are separately attributed to the reflections from (200), (210), and (220) planes, indicative of a well-defined ordered columnar array (Fig. 3a). Upon switching triazine unit to phenyl unit, BTT-TAPB-COF revealed peak positions in the PXRD pattern similar to those in BTT-TAPT-COF (Fig. 3b). Pawley refinements were used to simulate the theoretical PXRD pattern in order to analyze the geometrical structure. The experimental PXRD patterns of BTT-TAPT-COF and BTT-TAPB-COF are well consistent with the simulated data, including peak position and relative ratio, which implies the accuracy of the obtained two imine-linked 2D COFs based on benzotrithiophene. The Pawley refinement revealed the correctness of these peak assignments (Fig. 3a and b), in which the profile and weighted profile values were 4.99% and 6.94% for BTT-TAPT-COF, and 5.39% and 7.23% for BTT-TAPB-COF, respectively. Eclipsed stacking structures were simulated for BTT-TAPT-COF and BTT-TAPB-COF, which are in good agreement with the experimental results. Top views and side views of the two imine-linked 2D COFs based on benzotrithiophene displayed AA stacking (Fig. 3c–f). BTT-TAPT-COF showed refined cell parameters of α = β = 90° and γ = 120°, a = 21.98 Å, b = 21.98 Å, c = 3.48 Å. BTT-TAPB-COF showed refined cell parameters of α = β = 90° and γ = 120°, a = 22.1 Å, b = 21.95 Å, c = 3.57 Å. The optimized structures of BTT-TAPT-COF and BTT-TAPB-COF are also displayed in Fig. S1 and S2,† respectively.
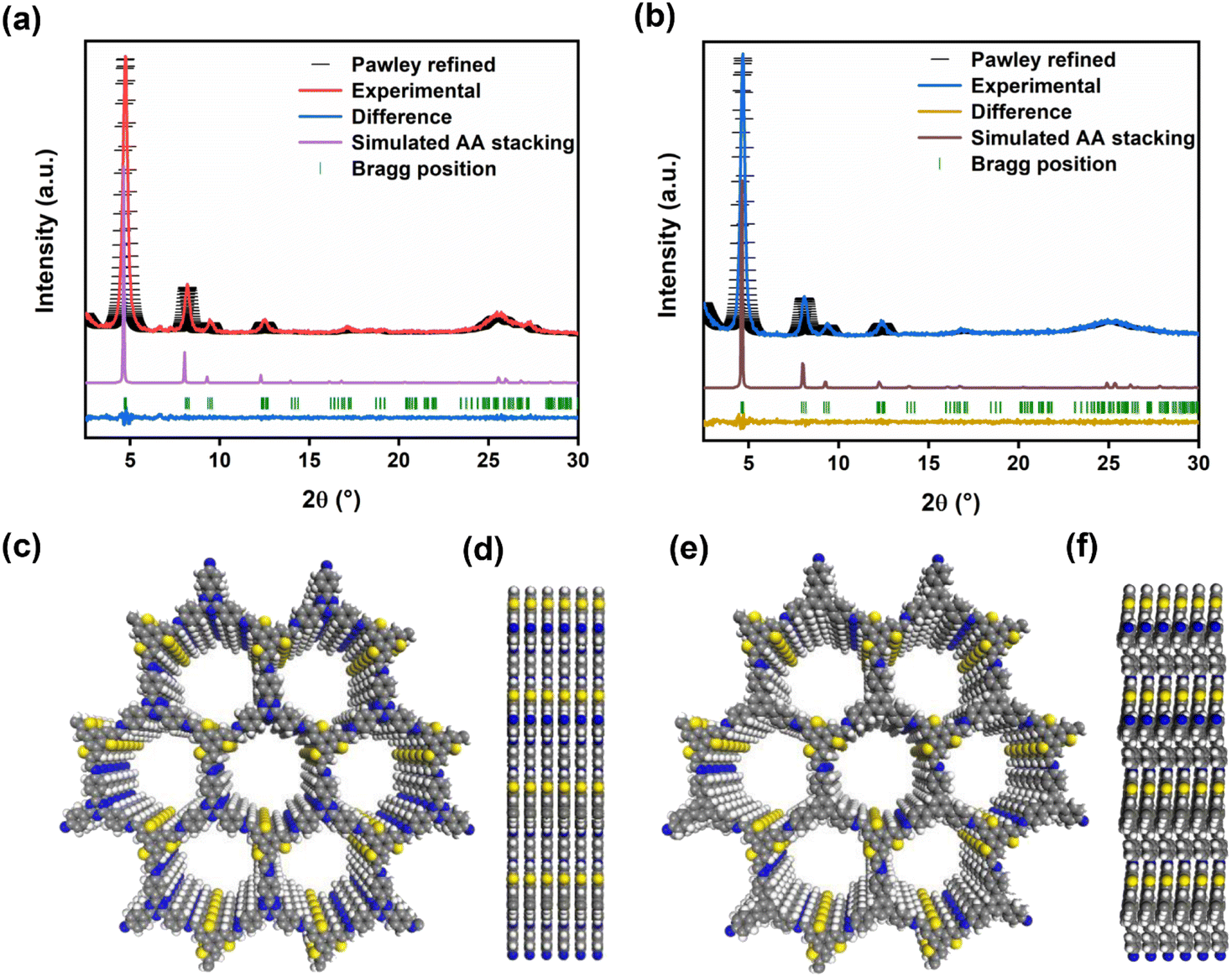 | ||
| Fig. 3 PXRD pattern analysis of BTT-TAPT-COF (a) and BTT-TAPB-COF (b) through Pawley refinement. Top views (c) and side views (d) of BTT-TAPT-COF. Top views (e) and side views (f) of BTT-TAPB-COF. | ||
BTT-TAPT-COF and BTT-TAPB-COF showed specific surface areas of 988 and 1184 m2 g−1, respectively, based on Brunauer–Emmett–Teller (BET) method according to the N2 sorption isotherms (Fig. 4a and b). The pore volume of BTT-TAPT-COF and BTT-TAPB-COF are 0.344 and 0.178 cm3 g−1, respectively. Due to the similar structure, pore diameters of BTT-TAPT-COF and BTT-TAPB-COF are both centered at about 1.5 nm, using non-local DFT calculation (Fig. S3 and S4†). The solid-state nuclear magnetic resonance (NMR) analysis of BTT-TAPT-COF and BTT-TAPB-COF was performed by using classical 13C cross-polarization magic angle spinning (CP-MAS) spectra. As displayed in Fig. 4c, the chemical shift at approximately 150 ppm is attributed to CN, and the chemical shift at about 167 ppm corresponds to the C-atoms on the triazine unit. The signals of CN at approximately 1620 and 1618 cm−1 in the Fourier transform infrared (FTIR) spectra of BTT-TAPT-COF and BTT-TAPB-COF are displayed (Fig. 4d), further confirming the triumphal establishment of imine. From the scanning electron microscope (SEM) images, one can observe that BTT-TAPT-COF and BTT-TAPB-COF are composed of stacked rods (Fig. S5a and b†). Transmission electron microscopy (TEM) images of BTT-TAPT-COF and BTT-TAPB-COF showed long-ordered channels, suggesting the overall porous morphology (Fig. S5c and d†).
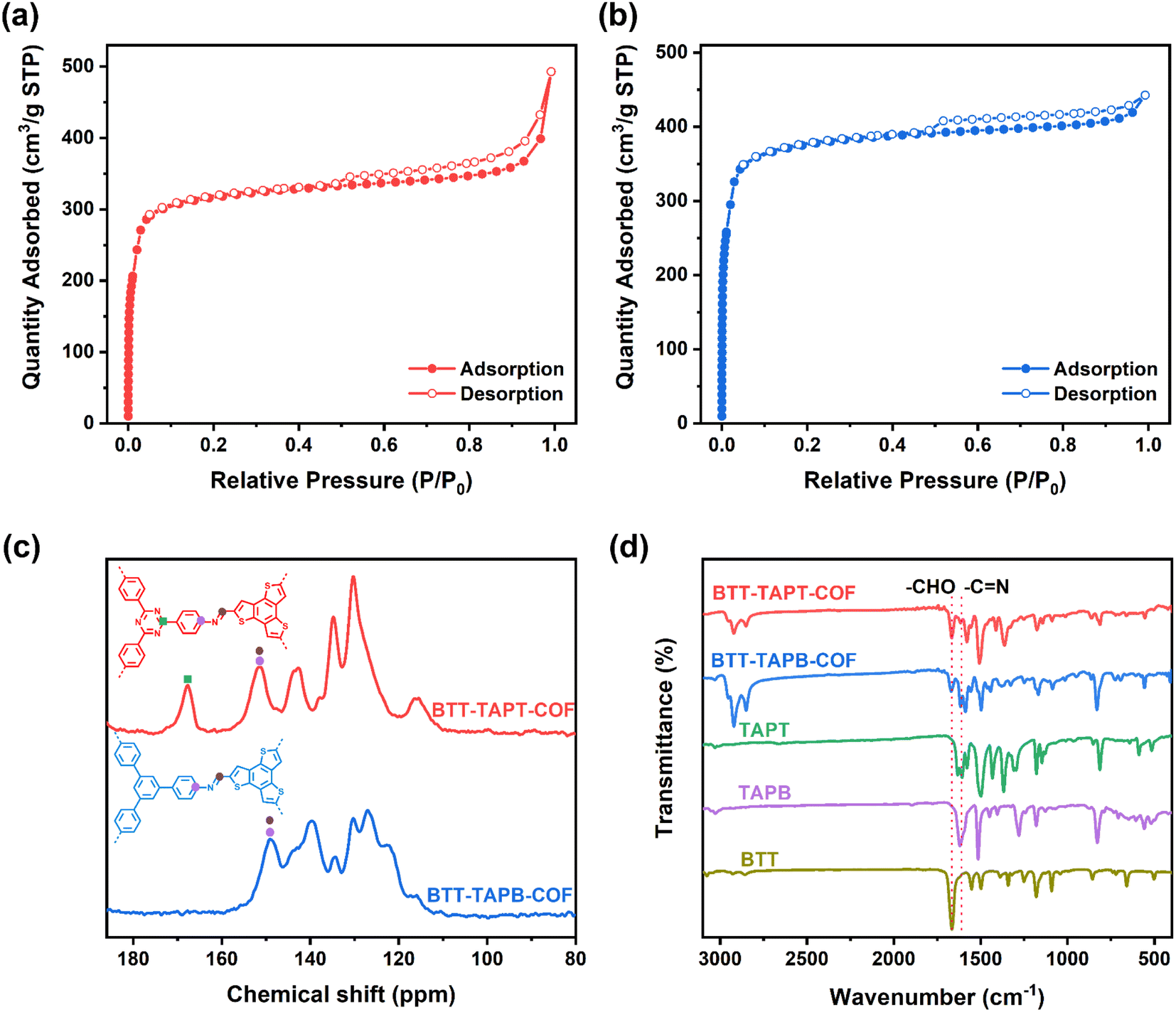 | ||
| Fig. 4 N2 sorption isotherms of BTT-TAPT-COF (a) and BTT-TAPB-COF (b). (c) Solid-state 13C NMR spectra of BTT-TAPT-COF and BTT-TAPB-COF. (d) FTIR spectra of BTT-TAPT-COF, BTT-TAPB-COF and BTT, TAPT, and TAPB. | ||
Solid-state ultraviolet (UV)-visible spectroscopy was applied to evaluate the light-harvesting properties of the two imine-linked 2D COFs; the spectra showed broad absorbance mainly in the range from 250 and 600 nm for BTT-TAPT-COF and BTT-TAPB-COF (Fig. 5a). The bandgaps for BTT-TAPT-COF and BTT-TAPB-COF were calculated to be 2.39 and 2.42 eV, respectively, according to the Tauc plots (Fig. 5b). Benefitting from the strong electron-withdrawing ability of the triazine unit, BTT-TAPT-COF has more apparent electron push–pull effect than BTT-TAPB-COF, which enables it a better photogenerated charge separation ability. This tendency is also reflected in Fig. 5c with a higher photocurrent intensity of BTT-TAPT-COF than that of BTT-TAPB-COF. In addition, from the Nyquist plots of the electrochemical impedance spectroscopy (EIS), it can be concluded that BTT-TAPT-COF has less charge transfer resistance compared to BTT-TAPB-COF (Fig. 5d), which is favorable for charge carrier transfer. Mott–Schottky analysis was carried out under three different frequencies to evaluate the flat band position (Vfb) of BTT-TAPT-COF and BTT-TAPB-COF. Both BTT-TAPT-COF and BTT-TAPB-COF showed characteristics of n-type semiconductors with positive slopes. Based on the Mott–Schottky plots, the Vfb of BTT-TAPT-COF and BTT-TAPB-COF were calculated as −0.83 and −0.64 V versus Ag/AgCl, respectively (Fig. 5e and f). This potential is sufficient for the activation of O2 to superoxide (O2˙−); therefore, these two imine-linked 2D COFs are expected to carry out selective organic conversion by visible-light photocatalysis. Based on the above discussion, the band positions of the two imine-linked 2D COFs were supplied (Fig. S6†). Additionally, thermogravimetry analysis curves for BTT-TAPT-COF and BTT-TAPB-COF demonstrated that both COFs were equipped with good thermal stability below 500 °C (Fig. S7†). Moreover, BTT-TAPT-COF and BTT-TAPB-COF both maintained good crystallinity in acid and base solutions (Fig. S8†), further confirming their robustness.
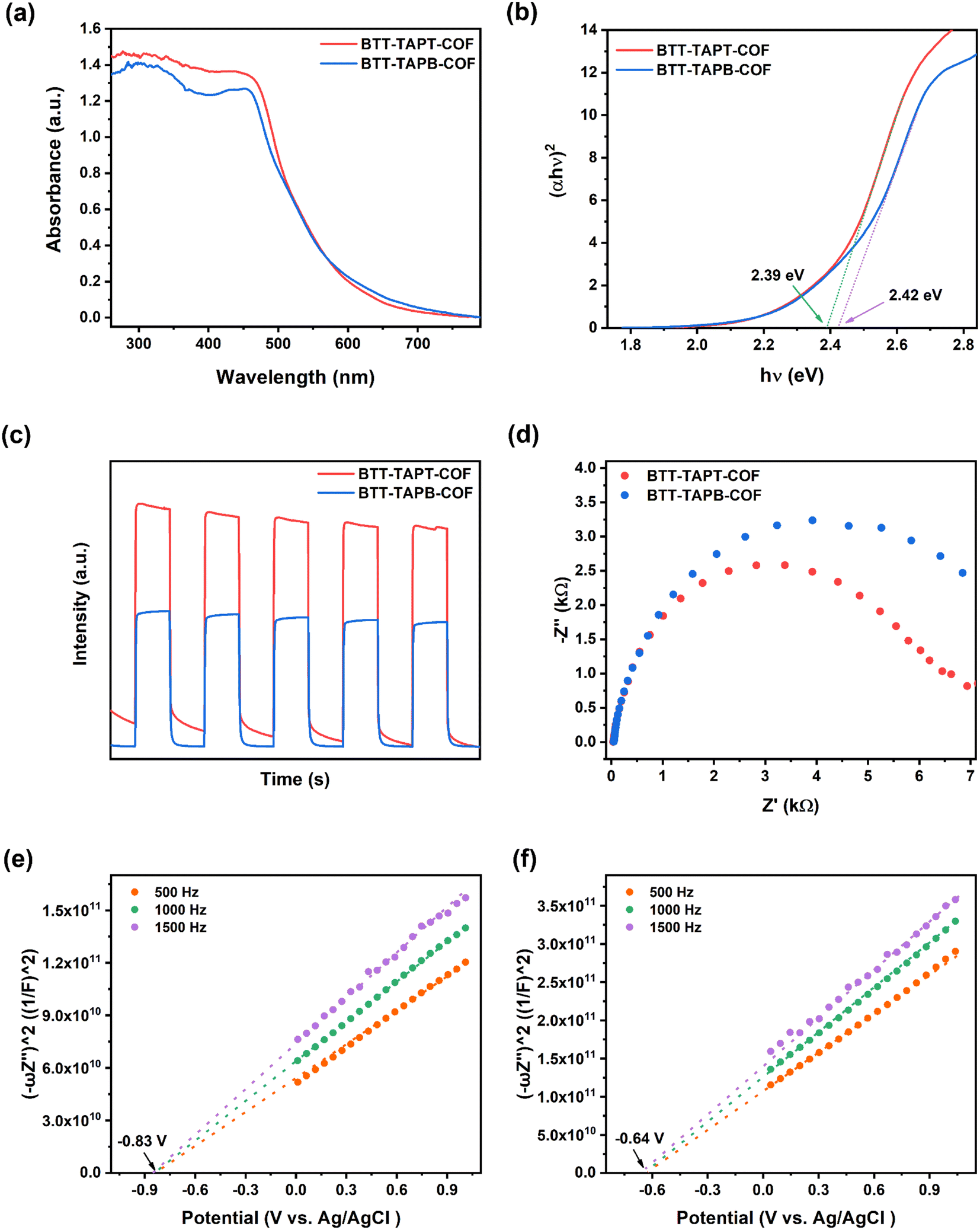 | ||
| Fig. 5 Solid-state UV-visible spectra (a), Tauc plots (b), transient photocurrent responses (c), and Nyquist plots of EIS (d) of BTT-TAPT-COF and BTT-TAPB-COF. Mott–Schottky plots of (e) BTT-TAPT-COF and (f) BTT-TAPB-COF. | ||
Next, visible-light-driven selective aerobic sulfoxidation was adopted as the model reaction to assess the photocatalytic activity of both imine-linked 2D COFs based on benzotrithiophene. Selective sulfoxidation was used to evaluate the difference in photocatalytic activity between the two structurally distinct COFs. Sulfoxides play a significant role as intermediates in organic synthesis, and therefore convenient and practical methods for forming organic sulfoxides have been extensively exploited.63–68 In addition to being useful, using it as a model reaction has important implications for studying the structure–activity relationship of COFs. More significantly, N-atoms in triazine are often evenly distributed, which can aid in charge carrier transfer and improve thermal stability. With similar light absorption, the yield of organic sulfoxide by BTT-TAPT-COF photocatalysis under the irradiation of blue light-emitting diodes (LEDs) far exceeded that by BTT-TAPB-COF, which benefits the strong electron push–pull effect caused by the triazine units. When methyl phenyl sulfide or its derivatives were selected for comparison, BTT-TAPT-COF was found to be better than BTT-TAPB-COF for blue-light-driven selective aerobic sulfoxidation in terms of yields for organic sulfoxides (Fig. 6a). For comparison, the kinetic curves of blue-light-driven selective aerobic sulfoxidation of methyl phenyl sulfide were investigated by BTT-TAPT-COF and BTT-TAPT-COF photocatalysis (Fig. 6b). Delightly, sulfoxidation cannot only smoothly occur, but was equipped with outstanding reusability (Fig. S9†), implying its robustness. Additionally, the PXRD pattern and FTIR spectra of the recycled BTT-TAPT-COF were identical to the fresh sample (Fig. S10 and S11†). The absorption range of BTT-TAPT-COF after irradiation showed a certain redshift, which is consistent with the increased activity of recycling experiments (Fig. S12†). In addition to blue LEDs, green and white LEDs can also drive selective aerobic sulfoxidation, yet with inferior conversions of methyl phenyl sulfide (Fig. S13†). It is reported that TEMPO (2,2,6,6-tetramethylpiperidine-N-oxyl) can be adopted as an outstanding redox mediator to accelerate the visible-light-driven aerobic sulfoxidation, and cooperative photocatalysis of COFs with TEMPO has displayed certain generality. However, by adding a catalytic amount of TEMPO, no obvious enhancement of the conversion of methyl phenyl sulfide was observed. Thus, the concept of cooperative photocatalysis with TEMPO is not applicable here.
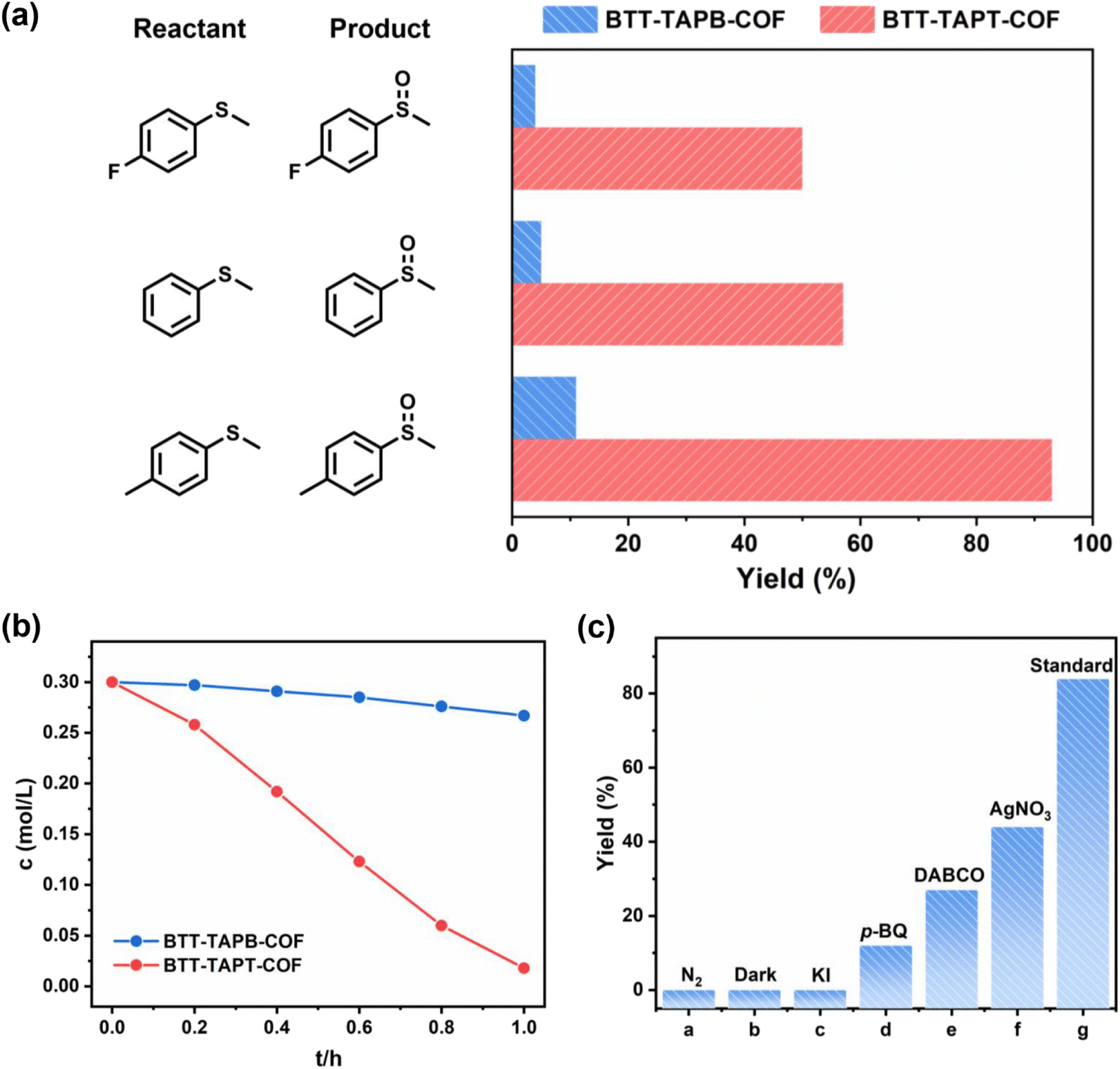 | ||
| Fig. 6 (a) The comparison for the blue-light-driven selective aerobic sulfoxidation of three sulfides by BTT-TAPT-COF and BTT-TAPB-COF photocatalysis. (b) Kinetic curves for the blue-light-driven selective aerobic sulfoxidation by BTT-TAPT-COF and BTT-TAPB-COF photocatalysis. (c) Quenching experiments for the blue-light-driven selective aerobic sulfoxidation. Standard reaction conditions: methyl phenyl sulfide (0.3 mmol), BTT-TAPT-COF (5 mg), blue LEDs (3 W × 4), CH3OH (1 mL), and O2 (0.1 MPa), reaction time: [a] 0.6 h and [c] 0.9 h. Determined by GC-FID, yield of methyl phenyl sulfoxide. | ||
Notably, the sulfoxidation of methyl phenyl sulfoxide showed an extremely poor result under an inert N2 atmosphere (Fig. 6c(a)). Trace conversion was monitored without light irradiation, revealing the blue-light-driven nature of photocatalysis (Fig. 6c(b)). Therefore, the effect of the light intensity was evaluated, implying that the reaction is light intensity dependent (Fig. S14†). The spectrum of the blue LED is displayed in Fig. S15.† To investigate the mechanism of this blue-light-driven selective aerobic sulfoxidation by BTT-TAPT-COF photocatalysis, different quenchers were added to determine the possible reactive oxygen species (ROS). When KI was used as a hole (h+) scavenger, no sulfoxide product was detected (Fig. 6c(c)). When p-benzoquinone (p-BQ) and 1,4-diazabicyclo[2.2.2]octane (DABCO) were added into the system as free radical trapping agents for O2˙− and singlet oxygen (1O2), respectively, sulfoxidation was hampered, which confirmed that both free radicals contributed to the formation of sulfoxide product (Fig. 6c(d and e)). By using AgNO3 as an electron (e−) scavenger, a conversion of 46% was observed, indicating the major role of e− during sulfoxidation (Fig. 6c(f)). It is speculated that the participation of e− should be present during O2 activation to generate O2˙−. To demonstrate the involvement of 1O2, kinetic studies were carried out in CH3OH and CD3OD, showing that the rate of sulfoxidation was increased slightly in CD3OD because CD3OD could extend the lifetime of 1O2 (Fig. 7a). This also confirmed the minor role of 1O2.
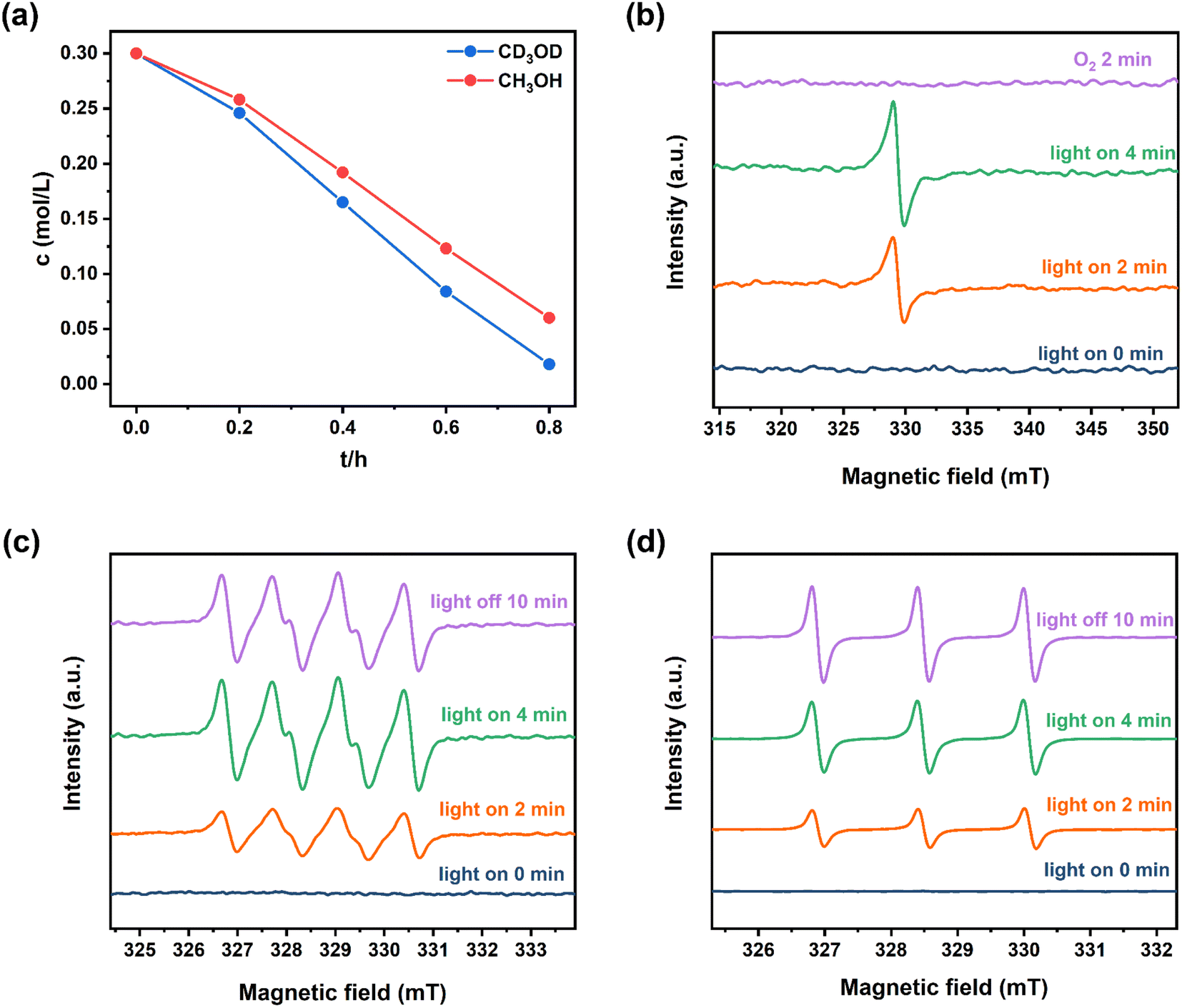 | ||
| Fig. 7 (a) Kinetic curves for the blue-light-driven selective aerobic sulfoxidation in CH3OH and CD3OD by BTT-TAPT-COF photocatalysis. Methyl phenyl sulfide (0.3 mmol), BTT-TAPT-COF (5 mg), blue LEDs (3 W × 4), CH3OH (1 mL), and O2 (0.1 MPa). EPR detection of the evolution of e− (b), O2˙− probed by DMPO (c) and 1O2 probed by TMPD (d) by BTT-TAPT-COF photocatalysis under visible-light irradiation. | ||
The evolution of e− and ROS during selective aerobic sulfoxidation was explored by electron paramagnetic resonance (EPR) spectroscopy. The signal of e− exhibited a significant improvement under visible light irradiation and faded by the consumption with O2 (Fig. 7b). Furthermore, detectable by EPR (Fig. 7c), the O2˙− signal intensified with the expansion of the light irradiation time after being captured by 5,5-dimethyl-1-pyrroline N-oxide (DMPO). The signal intensity of 2,2,6,6-tetramethyl-4-piperidone (TMPD)-captured 1O2 showed a similar trend to that of O2˙− (Fig. 7d). In summary, the quenching experiments and EPR results showed the generation of O2˙− and 1O2 to carry out the blue-light-driven selective aerobic sulfoxidation, and a possible mechanism of BTT-TAPT-COF photocatalysis is suggested (Fig. 8). First, BTT-TAPT-COF, as a photocatalyst, is driven by blue light to produce photogenerated e− and h+. Photogenerated e− and h+ are then used to activate O2 to form O2˙− and to induce substrates to form radical cations of methyl phenyl sulfide. For further sulfoxidation, O2˙− attacks the S-centered radical cation by nucleophilic characteristics to obtain peroxy sulfoxide intermediates, which are converted to the product, methyl phenyl sulfoxide by CH3OH. Furthermore, the pathway for the involvement of 1O2 is also depicted. Peroxy sulfoxide intermediates can be directly generated from the substrate methyl phenyl sulfide with 1O2via energy transfer for the subsequent conversion to methyl phenyl sulfoxide by CH3OH.
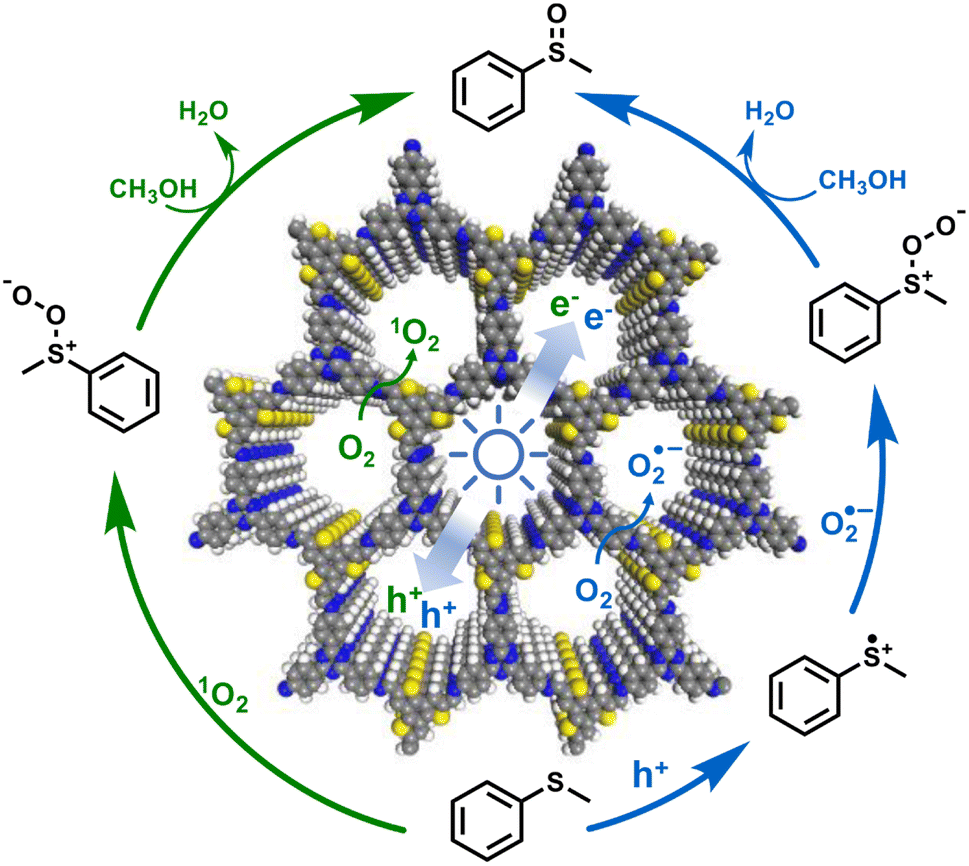 | ||
| Fig. 8 A possible mechanism for the blue-light-driven selective aerobic sulfoxidation by BTT-TAPT-COF photocatalysis. | ||
As summarized in Table 1, several organic sulfides were screened as the substrate in the blue-light-driven aerobic sulfoxidation by BTT-TAPT-COF photocatalysis with outstanding activity. The effect of substitutions on the aromatic ring of methyl phenyl sulfide was then investigated. The variation of activities among the para-substituted methyl phenyl sulfide resulted from the electronic effect, in which the electron-donating substituted groups played a promoting role and the electron-withdrawing substituted groups played an inhibiting role (Table 1, entries 1–7). Compared with para-substitution, the sulfoxidation for both ortho- and meta-substituted methyl phenyl sulfides of the same group exhibited a slower rate due to the steric effect (Table 1, entries 8–11). As for aliphatic sulfides, the sulfoxidation for tetrahydrothiophene and dibutylsulfane also exhibited outstanding activity towards forming corresponding sulfoxides (Table 1, entries 12 and 13).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Product | t (h) | Conv.b (%) | Sel.b (%) |
| a Standard reaction conditions: organic sulfide (0.3 mmol), BTT-TAPT-COF (5 mg), blue LEDs (3 W × 4), CH3OH (1 mL), and O2 (0.1 MPa). b Determined by GC-FID, conversion for sulfide, and selectivity for sulfoxide. | |||||
| 1 |
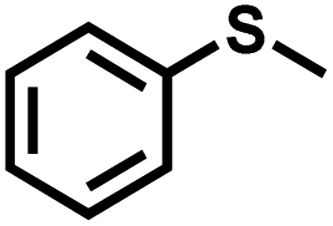
|
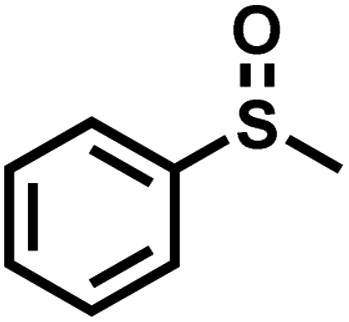
|
1.0 | 90 | 99 |
| 2 |
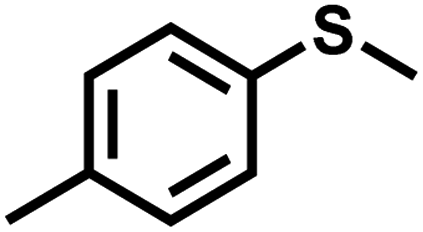
|
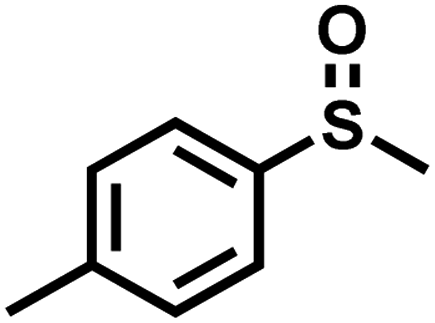
|
0.6 | 93 | 99 |
| 3 |
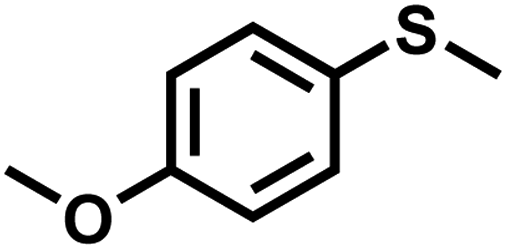
|
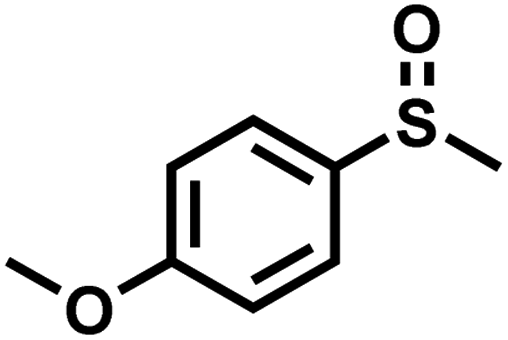
|
0.6 | 97 | 99 |
| 4 |
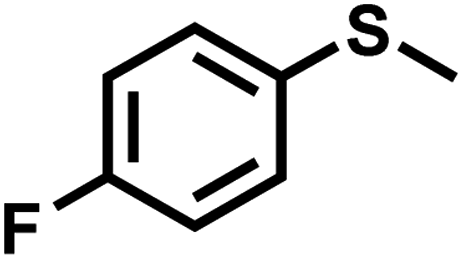
|
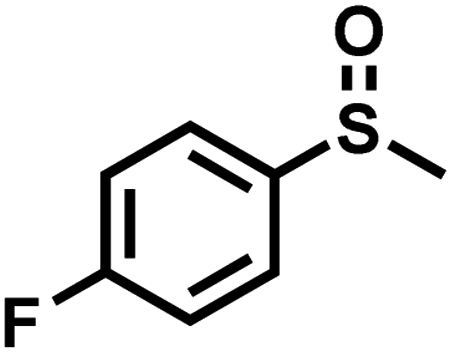
|
1.2 | 92 | 99 |
| 5 |
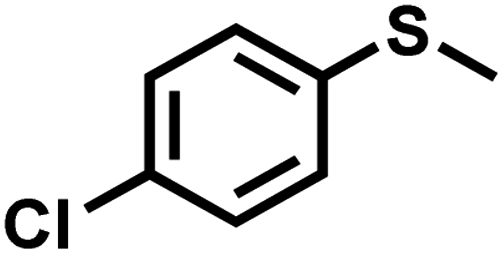
|
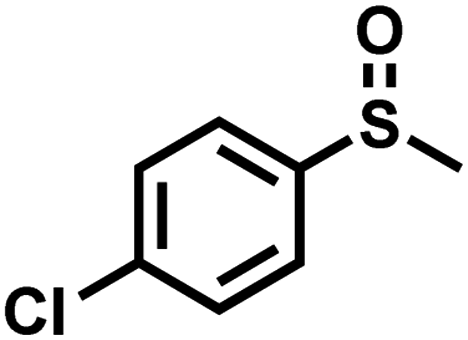
|
1.5 | 93 | 99 |
| 6 |
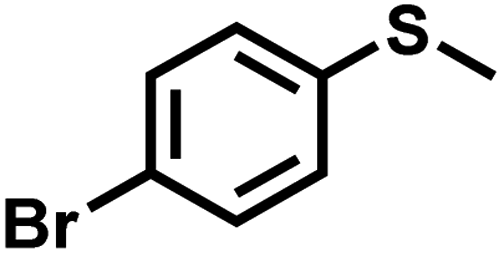
|
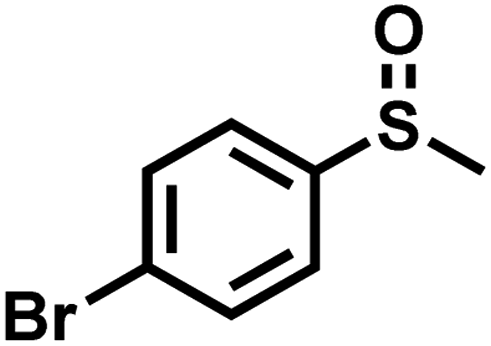
|
1.4 | 90 | 98 |
| 7 |
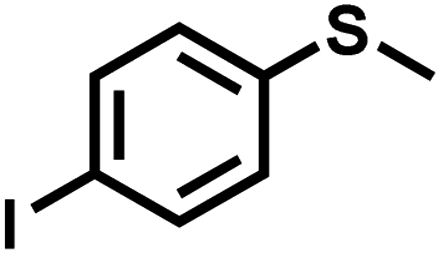
|
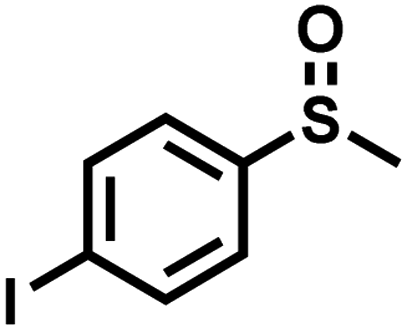
|
1.2 | 90 | 97 |
| 8 |
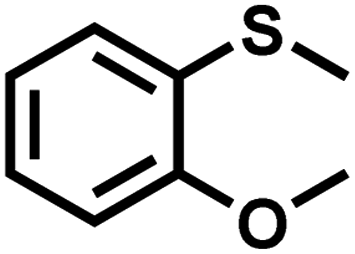
|
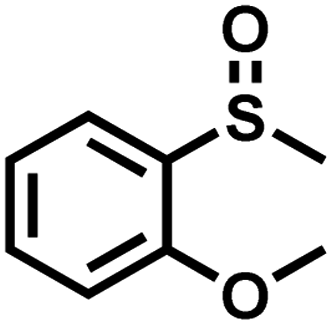
|
1.0 | 95 | 99 |
| 9 |
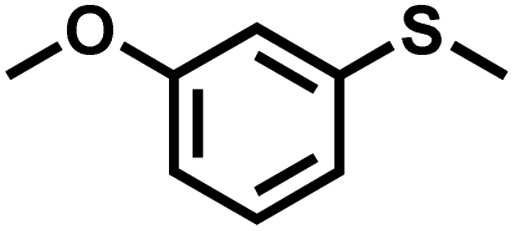
|
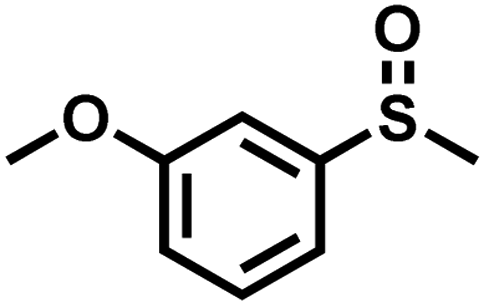
|
0.6 | 90 | 99 |
| 10 |
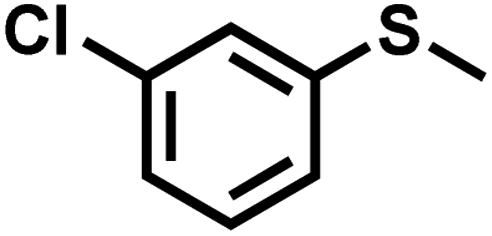
|
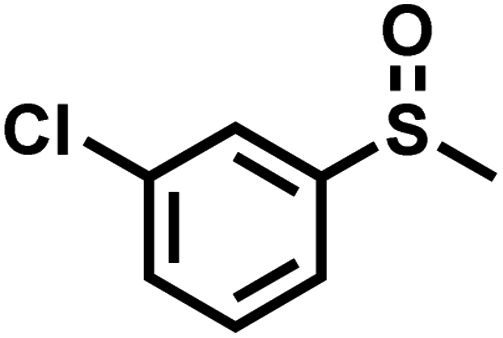
|
2.1 | 90 | 99 |
| 11 |
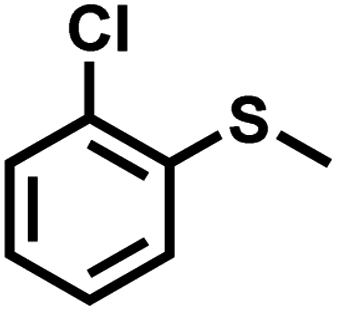
|
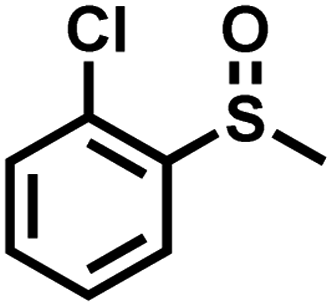
|
3.0 | 90 | 99 |
| 12 |
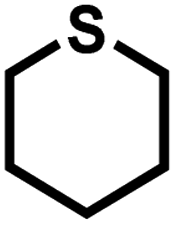
|
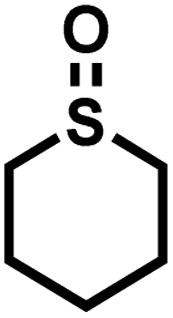
|
0.5 | 98 | 97 |
| 13 |
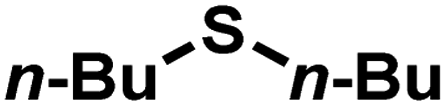
|
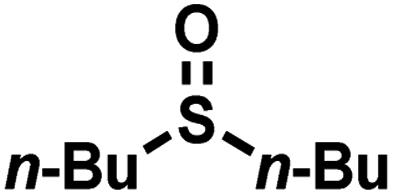
|
0.6 | 96 | 99 |

3. Conclusions
In summary, two imine-linked 2D COFs based on benzotrithiophene, BTT-TAPT-COF and BTT-TAPB-COF, were adapted for visible-light-driven photocatalytic selective aerobic sulfoxidation. Using the triazine unit as the electron acceptor, BTT-TAPT-COF was equipped with more apparent electron push–pull effect than BTT-TAPB-COF, leading to enhanced electron separation efficiency and carrier transfer capacity. Therefore, the photocatalytic activity of BTT-TAPT-COF was better than that of BTT-TAPB-COF for blue-light-driven selective aerobic sulfoxidation. Through DFT calculation, the superiority of BTT-TAPT-COF to BTT-TAPB-COF was further proved. This was also a successful example of accurately regulating the photocatalytic activity of COFs by regulating the molecular building units. BTT-TAPT-COF, with the promising insertion of the triazine unit into a customized COF, is a photocatalyst for blue-light-driven selective selective aerobic sulfoxidation with outstanding versatility and stability. This work provides new insights into the essence of electron push–pull effect of the building units of COFs for visible-light-driven selective organic conversion.Author contributions
Fengwei Huang: investigation; writing – original draft. Yuexin Wang: investigation, formal analysis. Xiaoyun Dong: formal analysis. Xianjun Lang: conceptualization, supervision, writing – review and editing and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants 22072108 and 22372124). The numerical calculations were done on the supercomputing system in the Supercomputing Center of Wuhan University. We also acknowledge the Core Facility of Wuhan University and the Center for Electron Microscopy at Wuhan University for support to materials characterizations.References
- K. T. Tan, S. Ghosh, Z. Y. Wang, F. X. Wen, D. Rodríguez-San-Miguel, J. Feng, N. Huang, W. Wang, F. Zamora, X. L. Feng, A. Thomas and D. L. Jiang, Nat. Rev. Methods Primers, 2023, 3, 1 CrossRef CAS.
- Y. Q. Xia, W. F. Zhang, S. Yang, L. P. Wang and G. Yu, Adv. Mater., 2023, 202301190 Search PubMed.
- Y. C. Wang, Y. J. Zhao and Z. B. Li, Macromol. Rapid Commun., 2022, 43, 2200108 CrossRef CAS PubMed.
- Q. Guan, L. L. Zhou and Y. B. Dong, J. Am. Chem. Soc., 2023, 145, 1475–1496 CrossRef CAS PubMed.
- J. Y. Hu, Z. Y. Huang and Y. Liu, Angew. Chem., Int. Ed., 2023, 62, e202306999 CrossRef CAS PubMed.
- Y. S. Li, W. B. Chen, G. L. Xing, D. L. Jiang and L. Chen, Chem. Soc. Rev., 2020, 49, 2852–2868 RSC.
- Y. Q. Zhang, J. Guo, G. Han, Y. P. Bai, Q. C. Ge, J. Ma, C. H. Lau and L. Shao, Sci. Adv., 2021, 7, eabe8706 CrossRef CAS PubMed.
- S. D. Huang, B. Y. Zhang, D. S. Wu, Y. F. Xu, H. Y. Hu, F. Duan, H. Zhu, M. L. Du and S. L. Lu, Appl. Catal., B, 2024, 340, 123216 CrossRef CAS.
- C. Li, Q. Li, Y. V. Kaneti, D. Hou, Y. Yamauchi and Y. Y. Mai, Chem. Soc. Rev., 2020, 49, 4681–4736 RSC.
- Y. F. Zhi, Z. R. Wang, H. L. Zhang and Q. C. Zhang, Small, 2020, 16, 2001070 CrossRef CAS PubMed.
- J. Li, X. C. Jing, Q. Q. Li, S. W. Li, X. Gao, X. Feng and B. Wang, Chem. Soc. Rev., 2020, 49, 3565–3604 RSC.
- S. Jhulki, A. M. Evans, X. L. Hao, M. W. Cooper, C. H. Feriante, J. Leisen, H. Li, D. Lam, M. C. Hersam, S. Barlow, J. L. Brédas, W. R. Dichtel and S. R. Marder, J. Am. Chem. Soc., 2020, 142, 783–791 CrossRef CAS PubMed.
- T. Skorjanc, D. Shetty and M. Valant, ACS Sens., 2021, 6, 1461–1481 CrossRef CAS PubMed.
- L. L. Guo, L. Yang, M. Y. Li, L. J. Kuang, Y. H. Song and L. Wang, Coord. Chem. Rev., 2021, 440, 213957 CrossRef CAS.
- W. R. Cui, C. R. Zhang, W. Jiang, F. F. Li, R. P. Liang, J. W. Liu and J. D. Qiu, Nat. Commun., 2020, 11, 436 CrossRef CAS PubMed.
- Y. Q. Xie, T. T. Pan, Q. Lei, C. L. Chen, X. L. Dong, Y. Y. Yuan, J. Shen, Y. C. Cai, C. H. Zhou, I. Pinnau and Y. Han, Angew. Chem., Int. Ed., 2021, 60, 22432–22440 CrossRef CAS PubMed.
- W. R. Cui, F. F. Li, R. H. Xu, C. R. Zhang, X. R. Chen, R. H. Yan, R. P. Liang and J. D. Qiu, Angew. Chem., Int. Ed., 2020, 59, 17684–17690 CrossRef CAS PubMed.
- X. H. Guo, Y. Li, M. C. Zhang, K. C. Cao, Y. Tian, Y. Qi, S. J. Li, K. Li, X. Q. Yu and L. J. Ma, Angew. Chem., Int. Ed., 2020, 59, 22697–22705 CrossRef CAS PubMed.
- Z. J. Yong and T. Y. Ma, Angew. Chem., Int. Ed., 2023, 62, e202308980 CrossRef CAS PubMed.
- T. He and Y. L. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202303086 CrossRef CAS PubMed.
- A. Wagner, C. D. Sahm and E. Reisner, Nat. Catal., 2020, 3, 775–786 CrossRef CAS.
- T. Li, P. L. Zhang, L. Z. Dong and Y. Q. Lan, Angew. Chem., Int. Ed., 2024, 63, e202318180 Search PubMed.
- H. Y. Hu, R. Y. Miao, F. L. Yang, F. Duan, H. Zhu, Y. M. Hu, M. L. Du and S. L. Lu, Adv. Energy Mater., 2024, 14, 202302608 Search PubMed.
- E. B. Zhou, X. Zhang, L. Zhu, E. C. Chai, J. S. Chen, J. Li, D. Q. Yuan, L. T. Kang, Q. F. Sun and Y. B. Wang, Sci. Adv., 2024, 10, eadk8564 CrossRef CAS PubMed.
- N. Y. Huang, Y. T. Zheng, D. Chen, Z. Y. Chen, C. Z. Huang and Q. Xu, Chem. Soc. Rev., 2023, 52, 7949–8004 RSC.
- Z. Z. Liang, R. C. Shen, Y. H. Ng, Y. Fu, T. Y. Ma, P. Zhang, Y. J. Li and X. Li, Chem Catal., 2022, 2, 2157–2228 CrossRef CAS.
- A. López-Magano, S. Daliran, A. R. Oveisi, R. Mas-Ballesté, A. Dhakshinamoorthy, J. Alemán, H. Garcia and R. Luque, Adv. Mater., 2023, 35, 2209475 CrossRef PubMed.
- Q. Yang, M. L. Luo, K. W. Liu, H. M. Cao and H. J. Yan, Appl. Catal., B, 2020, 276, 119174 CrossRef CAS.
- H. R. Liu, C. Z. Li, H. Li, Y. Q. Ren, J. Chen, J. T. Tang and Q. H. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 20354–20365 CrossRef CAS PubMed.
- S. Li, L. Li, Y. J. Li, L. Dai, C. X. Liu, Y. Z. Liu, J. N. Li, J. N. Lv, P. F. Li and B. Wang, ACS Catal., 2020, 10, 8717–8726 CrossRef CAS.
- T. Banerjee, F. Podjaski, J. Kröger, B. P. Biswal and B. V. Lotsch, Nat. Rev. Mater., 2021, 6, 168–190 CrossRef CAS.
- G. B. Wang, K. H. Xie, H. P. Xu, Y. J. Wang, F. Zhao, Y. Geng and Y. B. Dong, Coord. Chem. Rev., 2022, 472, 214774 CrossRef CAS.
- Z. W. Zhang, J. Jia, Y. F. Zhi, S. Ma and X. M. Liu, Chem. Soc. Rev., 2022, 51, 2444–2490 RSC.
- W. K. Qin, C. H. Tung and L. Z. Wu, J. Mater. Chem. A, 2023, 11, 12521–12538 RSC.
- M. L. Luo, Q. Yang, W. B. Yang, J. H. Wang, F. F. He, K. W. Liu, H. M. Cao and H. J. Yan, Small, 2020, 16, 2001100 CrossRef CAS PubMed.
- J. L. Wang and S. Z. Wang, Coord. Chem. Rev., 2022, 453, 214338 CrossRef CAS.
- Q. H. Zhu, Z. H. Xu, B. C. Qiu, M. Y. Xing and J. L. Zhang, Small, 2021, 17, 2101070 CrossRef CAS PubMed.
- G. Z. S. Ling, S. F. Ng and W. J. Ong, Adv. Funct. Mater., 2022, 32, 2111875 CrossRef CAS.
- T. Sun, C. X. Wang and Y. X. Xu, Chem. Res. Chin. Univ., 2020, 36, 640–647 CrossRef CAS.
- Y. H. Wang, L. Z. Liu, T. Y. Ma, Y. H. Zhang and H. W. Huang, Adv. Funct. Mater., 2021, 31, 2102540 CrossRef CAS.
- C. B. Wu, Z. Y. Teng, C. Yang, F. S. Chen, H. B. Yang, L. Wang, H. X. Xu, B. Liu, G. F. Zheng and Q. Han, Adv. Mater., 2022, 34, 2110266 CrossRef CAS PubMed.
- X. L. Hu, Z. Zhan, J. Q. Zhang, I. Hussain and B. E. Tan, Nat. Commun., 2021, 12, 6596 CrossRef CAS PubMed.
- W. Huang, N. Huber, S. Jiang, K. Landfester and K. A. I. Zhang, Angew. Chem., Int. Ed., 2020, 59, 18368–18373 CrossRef CAS PubMed.
- Z. Q. Wang, S. Gu, L. J. Cao, L. Kong, Z. Y. Wang, N. Qin, M. Q. Li, W. Luo, J. J. Chen, S. S. Wu, G. Y. Liu, H. M. Yuan, Y. F. Bai, K. L. Zhang and Z. G. Lu, ACS Appl. Mater. Interfaces, 2021, 13, 514–521 CrossRef CAS PubMed.
- Y. L. Yang, H. Y. Niu, L. Xu, H. Zhang and Y. Q. Cai, Appl. Catal., B, 2020, 269, 118799 CrossRef CAS.
- H. Z. Wang, C. Yang, F. S. Chen, G. F. Zheng and Q. Han, Angew. Chem., Int. Ed., 2022, 61, e202202328 CrossRef CAS PubMed.
- H. M. Hao, F. L. Zhang, X. Y. Dong and X. J. Lang, Appl. Catal., B, 2021, 299, 120691 CrossRef CAS.
- F. L. Zhang, X. Y. Dong, Y. X. Wang and X. J. Lang, Small, 2023, 19, 202302456 Search PubMed.
- Z. P. Lei, L. J. Wayment, J. R. Cahn, H. X. Chen, S. F. Huang, X. B. Wang, Y. H. Jin, S. Sharma and W. Zhang, J. Am. Chem. Soc., 2022, 144, 17737–17742 CrossRef CAS PubMed.
- Y. Q. Cai, Z. T. Gong, Q. Rong, J. M. Liu, L. F. Yao, F. X. Cheng, J. J. Liu, S. B. Xia and H. Guo, Appl. Surf. Sci., 2022, 594, 153481 CrossRef CAS.
- R. X. Sun, X. Y. Wang, X. P. Wang and B. E. Tan, Angew. Chem., Int. Ed., 2022, 61, e202117668 CrossRef CAS PubMed.
- M. J. Liu, J. N. Liu, K. Zhou, J. W. Chen, Q. Sun, Z. B. Bao, Q. W. Yang, Y. W. Yang, Q. L. Ren and Z. G. Zhang, Adv. Sci., 2021, 8, 2100631 CrossRef CAS PubMed.
- G. X. Jiang, W. W. Zou, Z. Y. Ou, L. H. Zhang, W. F. Zhang, X. J. Wang, H. Y. Song, Z. M. Cui, Z. X. Liang and L. Du, Angew. Chem., Int. Ed., 2022, 61, e202208086 CrossRef CAS PubMed.
- Z. J. Gu, J. J. Wang, Z. Shan, M. M. Wu, T. T. Liu, L. Song, G. X. Wang, X. H. Ju, J. Su and G. Zhang, J. Mater. Chem. A, 2022, 10, 17624–17632 RSC.
- W. Zhou, X. Wang, W. L. Zhao, N. J. Lu, D. Cong, Z. Li, P. G. Han, G. Q. Ren, L. Sun, C. C. Liu and W. Q. Deng, Nat. Commun., 2023, 14, 6971 CrossRef CAS PubMed.
- Z. P. Li, J. A. Wang, S. Ma, Z. W. Zhang, Y. F. Zhi, F. C. Zhang, H. Xia, G. Henkelman and X. M. Liu, Appl. Catal., B, 2022, 310, 121335 CrossRef CAS.
- S. K. Chang, C. Y. Li, H. Li, L. K. Zhu and Q. R. Fang, Chem. Res. Chin. Univ., 2022, 38, 396–401 CrossRef CAS.
- Z. P. Li, Z. W. Zhang, R. M. Xie, C. Z. Li, Q. K. Sun, W. Shi, W. C. Chu, Y. Y. Long, H. Li and X. M. Liu, Adv. Funct. Mater., 2022, 32, 2112553 CrossRef CAS.
- F. W. Huang, Y. X. Wang, X. Y. Dong and X. J. Lang, Sci. China: Chem., 2023, 66, 3290–3296 CrossRef CAS.
- S. Li, L. Dai, L. Li, A. W. Dong, J. N. Li, X. J. Meng, B. Wang and P. F. Li, J. Mater. Chem. A, 2022, 10, 13325–13332 RSC.
- Z. P. Li, T. Q. Deng, S. Ma, Z. W. Zhang, G. Wu, J. A. Wang, Q. Z. Li, H. Xia, S. W. Yang and X. M. Liu, J. Am. Chem. Soc., 2023, 145, 8364–8374 CAS.
- H. T. Wei, J. Ning, X. D. Cao, X. H. Li and L. Hao, J. Am. Chem. Soc., 2018, 140, 11618–11622 CrossRef CAS PubMed.
- A. López-Magano, S. Daliran, A. R. Oveisi, R. Mas-Ballesté, A. Dhakshinamoorthy, J. Alemán, H. Garcia and R. Luque, Adv. Mater., 2023, 35, 202209475 CrossRef PubMed.
- J. F. Li, Z. Y. An, J. Y. Sun, C. Y. Tan, D. Gao, Y. Tan and Y. Y. Jiang, ACS Appl. Mater. Interfaces, 2020, 12, 35475–35481 CrossRef CAS PubMed.
- H. C. Shan, D. Cai, X. X. Zhang, Q. Zhu, P. Y. Qin and J. Baeyens, Chem. Eng. J., 2022, 432, 134288 CrossRef CAS.
- S. Suleman, Y. Zhang, Y. Y. Qian, J. W. Zhang, Z. Y. Lin, Ö. Metin, Z. Meng and H. L. Jiang, Angew. Chem., Int. Ed., 2024, 63, e202314988 CrossRef CAS PubMed.
- C. J. Wu, X. Y. Li, T. R. Li, M. Z. Shao, L. J. Niu, X. F. Lu, J. L. Kan, Y. Geng and Y. B. Dong, J. Am. Chem. Soc., 2022, 144, 18750–18755 CrossRef CAS PubMed.
- D. Chen, W. B. Chen, G. Zhang, S. Li, W. H. Chen, G. L. Xing and L. Chen, ACS Catal., 2022, 12, 616–623 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta00087k |
| This journal is © The Royal Society of Chemistry 2024 |