
Constructing oxygen vacancies by doping Mo into spinel Co3O4 to trigger a fast oxide path mechanism for acidic oxygen evolution reaction†
Lang
Sun
,
Min
Feng
,
Yang
Peng
,
Xu
Zhao
,
Yiqun
Shao
,
Xin
Yue
 * and
Shaoming
Huang
*
* and
Shaoming
Huang
*
Guangzhou Key Laboratory of Low-Dimensional Materials and Energy Storage Devices, Collaborative Innovation Center of Advanced Energy Materials, School of Materials and Energy, Guangdong University of Technology, Guangzhou, 510006, P. R. China. E-mail: xinyue@gdut.edu.cn; smhuang@gdut.edu.cn
First published on 5th March 2024
Abstract
The development of non-precious metal electrocatalysts for acidic oxygen evolution reaction (OER) that are highly durable, cost-effective, and efficient is crucial to advancing the use of proton exchange membrane water electrolyzers (PEMWEs). Because of its reasonable activity and stability, spinel Co3O4 is regarded as a potential candidate for acidic OER. However, it is still far from industrial requirements owing to the sluggish adsorbate evolution mechanism (AEM) for the OER in acids. Herein, we report the enhancement of the activity and stability of acidic OER on Co3O4 by doping a small amount of Mo (∼0.5 wt%). The oxygen vacancies are created by doping Mo to activate the lattice oxygen followed by etching away from the lattice, effectively promoting the fast oxide path mechanism (OPM) for the OER. Meanwhile, its stability is boosted by introducing Mo with variable valence states. As a result, as-fabricated Mo-doped oxygen vacancy enriched Co3O4 (VO-MoxCo3−xO4) exhibits efficient activity with a current density of 100 mA cm−2 at an overpotential of 490 mV and Tafel slope of 102.5 mV dec−1 towards acidic OER. Meanwhile, VO-MoxCo3−xO4 exhibits high stability and can maintain a current density of 10 mA cm−2 for 30 h.
Introduction
The production of hydrogen (H2) through electrochemical water splitting is considered to offer a means of storing renewable electricity in the form of H2 molecules.1 The proton exchange membrane water electrolyzer (PEMWE) is recognized as a promising alternative to the alkaline water electrolyzer due to its superior gas purity and conversion efficiency, lower ohmic loss, higher current density, stable operational performance, and so on.2 However, the oxygen evolution reaction (OER) in an acidic medium involves a four-electron–proton transfer process and the formation of O–O bonds, which necessitates a high overpotential to overcome the energy barrier, thereby resulting in sluggish kinetics.2,3 The rapid decline in catalytic activity poses a more severe challenge for electrocatalysts, because they work in harsh environments, including at low pH values (∼1), high potential windows, and saturated oxygen concentrations.3,4 Precious metal-based electrocatalysts, such as IrO2 and RuO2, exhibit satisfactory activity for acidic OER. However, their high cost, limited resources, and poor stability greatly hinder the practical implementation.5 Consequently, it is crucial to promote the widespread use of PEMWEs by developing non-precious metal electrocatalysts with high efficiency, stability and low cost.By virtue of its relatively high activity and stability, spinel Co3O4 is considered to have potential to replace precious metal-based electrocatalysts for acidic OER.6 However, owing to the slower adsorbate evolution mechanism (AEM) on the surface of Co3O4 for the OER, it is still far from realistic requirements. Three oxygen reaction intermediates of *OH, *O, and *OOH are generated and transformed during the AEM pathway (* denotes an active site). The binding energies of these intermediates adsorbed on the electrocatalysts are linearly correlated and therefore follow a scaling relationship.7 This restriction makes a large theoretical overpotential of approximately 370 mV necessary to drive the OER via the AEM pathway.8 Some electrocatalysts have reportedly shown increased activity surpassing the theoretical limit because they catalyze the OER through a kinetically favorable lattice-oxygen-mediated mechanism (LOM). Lattice oxygen and O-radicals combine to form oxygen gas, preventing the production of *OOH.9 Nevertheless, the formation of oxygen vacancies during the LOM process leads to the etching away of metal species from surfaces, which causes the electrocatalysts to degrade quickly.10 The recently reported oxide path mechanism (OPM), which allows direct oxygen radical self-coupling to be the oxygen bridge (O–O) without generating oxygen vacancies and additional intermediates like *OOH, is deemed to be the fastest and most stable pathway for the OER in an acidic environment.11 The design of non-precious metal electrocatalysts for the OER in an acidic solution to undergo the OPM pathway, thus, calls for a feasible strategy.
To the best of our knowledge, the OPM pathway for the OER in acid has been found only on limited electrocatalysts, primarily Ru-based heterogeneous catalysts, such as Ru-MOF-400 and 12Ru/MnO2.11–13 Building oxygen vacancies has been reportedly presented to effectively enhance the activity of the OER in alkaline media by triggering the OPM pathway on Co3O4.14,15 However, when applying this strategy in an acidic medium, Co3O4 would be rapidly degraded in acidic electrolyte owing to the oxygen vacancy enriched surface, which eventually causes the activity to drop.16 Fortunately, it has been proposed that modulating the d-band structure of Co by doping or substituting metal-cations such as Mn, Ti, Mo, and so on with variable valence states into Co3O4 can efficiently improve the stability of acidic OER.17–19 In recent years, introducing cations with variable valence states, for example, Mn, into the lattice of spinel Co3O4 has been proven to improve the stability of acidic OER on the as-synthesized electrocatalysts.17,18 This is because the introduced cations would be oxidized to higher valence states during the OER process. This oxidative conversion would transfer the electrons to the closest Co sites, which reduces the valence states of Co3+ species to the more stable Co2+ species.17 Meanwhile, the O 2p band center is typically upshifted by introducing Mo into metal oxide, which weakens the metal–oxygen bonds and ultimately builds oxygen vacancies inside metal oxide.20
Therefore, in this report, an approach of doping a small amount of Mo (∼0.5 wt%) into Co3O4 is reported to enhance the activity and stability of the OER in acidic media since it can activate the lattice oxygen to be etched away from the lattice for creating oxygen vacancies. The generated oxygen vacancies can effectively promote the fast OPM pathway, and simultaneously the introduction of Mo with variable valence states will improve the stability. We successfully prepare a very small amount of Mo-doped oxygen vacancy enriched Co3O4 (VO-MoxCo3−xO4) through hydrothermal treatment followed by annealing under an air atmosphere. As a result, Co3O4 requires an overpotential of 490 mV to attain a current density of 10 mA cm−2. Owing to the fast OPM pathway triggered by creating oxygen vacancies in its lattice, as-fabricated VO-MoxCo3−xO4 exhibits an enhanced activity, which reaches 10 and 100 mA cm−2 at overpotentials of 420 and 490 mV, respectively, being superior to Ir/C. Meanwhile, VO-MoxCo3−xO4 possesses the fastest kinetics (with a Tafel slope of only 102.5 mV dec−1) towards the OER in acid when compared with those of pure Co3O4 and Ir/C electrocatalysts. VO-MoxCo3−xO4 exhibits high stability towards acidic OER and can maintain a current density of 10 mA cm−2 for 30 h. The mechanism for enhancing the OPM on VO-MoxCo3−xO4 has been studied by detailed characterization together with theoretical calculations.
Results and discussion
The synthetic route for VO-MoxCo3−xO4 is schematically illustrated in Fig. 1a, involving hydrothermal treatment followed by the annealing process under an air atmosphere. Cobalt nitrate hexahydrate (Co(NO3)2·6H2O) and ammonium molybdate tetrahydrate ((NH4)6Mo7O24·4H2O) were used as the precursors of Co and Mo elements, respectively. Mo doped Co(OH)2 (Co1−xMox(OH)2) was generated after hydrothermal treatment as the intermediate (Fig. S1 in the ESI†). Lattice oxygen was then activated to be etched away from the as-prepared MoxCo3−xO4 when it was annealed under an air atmosphere. Inductively coupled plasma atomic emission spectroscopy (ICP-AES) was used to determine the atomic ratio of Mo to Co in VO-MoxCo3−xO4, which is about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :230. Thus, the amount of Mo in VO-MoxCo3−xO4 can be obtained to be ∼0.5 wt%. For comparison, pure Co3O4 was also prepared through a similar method (for details see the Experimental section, ESI†). X-ray diffraction (XRD) was used to investigate the crystal structures of VO-MoxCo3−xO4 and Co3O4, as shown in Fig. 1b. In the XRD patterns of VO-MoxCo3−xO4 and Co3O4, the diffraction peaks at 19.0°, 31.3°, 36.8°, 38.5°, 44.8°, 55.7°, 59.4°, and 65.2° correspond to the (111), (220), (311), (222), (400), (422), (511) and (440) facets of cubic spinel Co3O4 (PDF# 42-1467).17
:230. Thus, the amount of Mo in VO-MoxCo3−xO4 can be obtained to be ∼0.5 wt%. For comparison, pure Co3O4 was also prepared through a similar method (for details see the Experimental section, ESI†). X-ray diffraction (XRD) was used to investigate the crystal structures of VO-MoxCo3−xO4 and Co3O4, as shown in Fig. 1b. In the XRD patterns of VO-MoxCo3−xO4 and Co3O4, the diffraction peaks at 19.0°, 31.3°, 36.8°, 38.5°, 44.8°, 55.7°, 59.4°, and 65.2° correspond to the (111), (220), (311), (222), (400), (422), (511) and (440) facets of cubic spinel Co3O4 (PDF# 42-1467).17
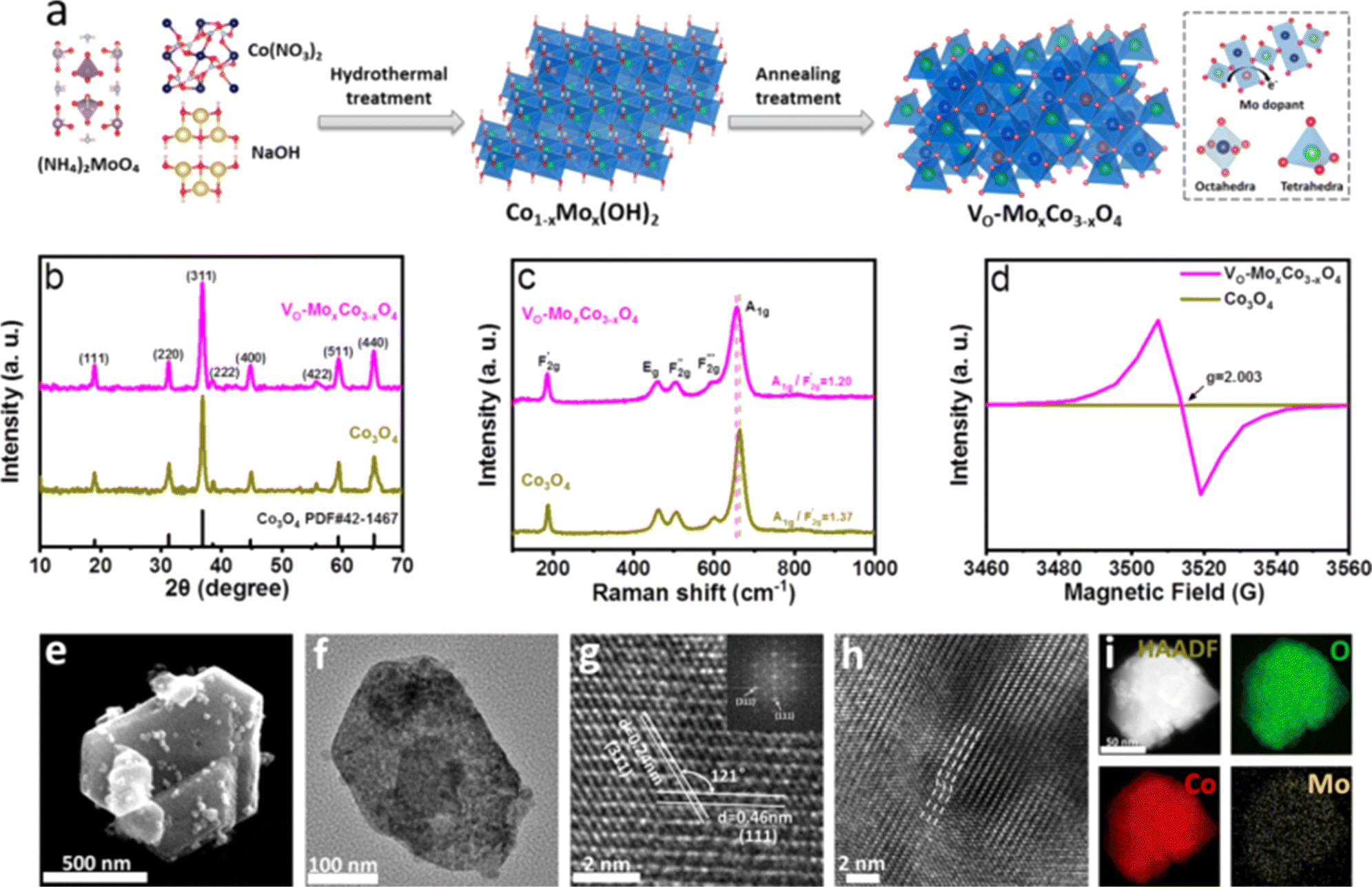 | ||
| Fig. 1 (a) Scheme of the synthetic process for VO-MoxCo3−xO4. (b) XRD patterns of VO-MoxCo3−xO4 and Co3O4. (c) Raman spectra of VO-MoxCo3−xO4 and Co3O4. (d) EPR spectra of VO-MoxCo3−xO4 and Co3O4. (e) SEM image of VO-MoxCo3−xO4. (f–h) TEM images of VO-MoxCo3−xO4. (i) HAADF-STEM and corresponding elemental mapping images of VO-MoxCo3−xO4. | ||
The lattice dynamics of VO-MoxCo3−xO4 and Co3O4 were confirmed by Raman and Fourier transform infrared (FT-IR) spectroscopy (Fig. 1c, S2 and S3, ESI†). A1g, Eg, and three F2g modes are the five typical Raman active modes of cubic spinel oxides with an Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group,21 as shown in Fig. 1c. The A1g mode is related to the Co3+–O stretching in the octahedral CoO6 unit, and the
m space group,21 as shown in Fig. 1c. The A1g mode is related to the Co3+–O stretching in the octahedral CoO6 unit, and the 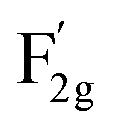 mode corresponds to the Co2+–O stretching in the tetrahedral CoO4 geometry.22,23 The A1g mode in the Raman spectrum of VO-MoxCo3−xO4 at 655.4 cm−1, has been found to red-shift by 7.9 cm−1 compared to that of Co3O4 (Fig. S2, ESI†). This is ascribed to the lattice disorder increased by oxygen vacancies.24 The intensity ratio of the A1g to
mode corresponds to the Co2+–O stretching in the tetrahedral CoO4 geometry.22,23 The A1g mode in the Raman spectrum of VO-MoxCo3−xO4 at 655.4 cm−1, has been found to red-shift by 7.9 cm−1 compared to that of Co3O4 (Fig. S2, ESI†). This is ascribed to the lattice disorder increased by oxygen vacancies.24 The intensity ratio of the A1g to 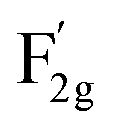 band
band 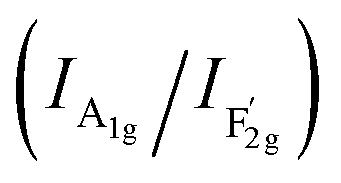 shows the occupied degree of octahedral sites.25
shows the occupied degree of octahedral sites.25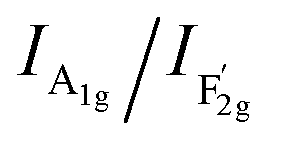 of VO-MoxCo3−xO4 is 1.20, which is lower than that of Co3O4 (1.37). This is related to the presence of Co vacancies caused by the oxidation of the closest Mo cations.26,27 The FT-IR results can also be used to identify similar situations involving the variation in octahedral sites (Fig. S3, ESI†). Electron paramagnetic resonance (EPR) spectroscopy was employed to examine the structure of defects in VO-MoxCo3−xO4 since it is sensitive to unpaired electrons, as illustrated in Fig. 1d. In the EPR spectrum of Co3O4, there is no discernible signal, indicating a pure crystal without defects. The EPR spectrum of VO-MoxCo3−xO4 shows a sharp increase in the intensity of the g-factor at 2.003, suggesting the successful construction of oxygen vacancies by doping Mo into Co3O4.28
of VO-MoxCo3−xO4 is 1.20, which is lower than that of Co3O4 (1.37). This is related to the presence of Co vacancies caused by the oxidation of the closest Mo cations.26,27 The FT-IR results can also be used to identify similar situations involving the variation in octahedral sites (Fig. S3, ESI†). Electron paramagnetic resonance (EPR) spectroscopy was employed to examine the structure of defects in VO-MoxCo3−xO4 since it is sensitive to unpaired electrons, as illustrated in Fig. 1d. In the EPR spectrum of Co3O4, there is no discernible signal, indicating a pure crystal without defects. The EPR spectrum of VO-MoxCo3−xO4 shows a sharp increase in the intensity of the g-factor at 2.003, suggesting the successful construction of oxygen vacancies by doping Mo into Co3O4.28
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) techniques were used to analyze the morphologies of VO-MoxCo3−xO4 and Co3O4 (Fig. 1e–i and S4, ESI†). VO-MoxCo3−xO4 consists of nanoparticles with sizes ranging from 50 to 500 nm (Fig. 1e). A similar morphology of Co3O4 can be observed in the SEM image as well (Fig. S4a†). A typical TEM image of VO-MoxCo3−xO4 with a particle size of 200 nm is depicted in Fig. 1f. As observed in the high-resolution TEM (HR-TEM) image of VO-MoxCo3−xO4 in Fig. 1g, the interplanar spacings of 0.24 and 0.46 nm can be assigned to the (311) and (111) facets of VO-MoxCo3−xO4, respectively. Additionally, the HR-TEM image of Co3O4 shows the crystal indices of 0.24 and 0.28 nm, respectively, which is corresponding to the (311) and (220) planes of Co3O4 (Fig. S4c†). Moreover, lattice distortion can be observed in the TEM image of VO-MoxCo3−xO4 presented in Fig. 1h, which originates from the lattice strain caused by doping Mo with a larger radius and creating oxygen vacancies.29 The uniform distribution of Co, Mo, and O elements can be found across VO-MoxCo3−xO4 according to the high angle annular dark field-scanning transmission electron microscope (HAADF-STEM) and corresponding elemental mapping images (Fig. 1i). Similar situations regarding the distribution of elements can be also observed for Co3O4 (Fig. S4d–f, ESI†).
X-ray photoelectron spectroscopy (XPS) was used to study the surface chemical state of VO-MoxCo3−xO4 and Co3O4 (Fig. S5–S8, ESI†), which suggests lower valence states of Co along with a higher concentration of oxygen vacancies in VO-MoxCo3−xO4. According to the results of XPS spectroscopy, the atomic ratio of Mo to Co in the near-surface of VO-MoxCo3−xO4 is obtained to be 3:247 (Table S1, ESI†). Based on the result of ICP, the total atomic ratio of Mo to Co in VO-MoxCo3−xO4 is 1:230. Therefore, Mo cations should mainly be distributed in the near-surface of VO-MoxCo3−xO4. X-ray absorption fine structure spectroscopy (XAFS) was performed to investigate the electronic structure and coordination environment of VO-MoxCo3−xO4 (Fig. 2). The X-ray absorption near edge structure (XANES) spectra of Co K edges for VO-MoxCo3−xO4, Co3O4 and other compounds are displayed in Fig. 2a. As observed in the XANES spectra, CoO and Co2O3 display white line intensities at 7725.1 and 7730.3 eV, corresponding to Co2+ and Co3+, respectively. The XANES spectrum of Co3O4 presents a white line intensity at 7729.2 eV, suggesting the coexistence of Co2+ and Co3+ in the lattice,30 while VO-MoxCo3−xO4 exhibits a white line intensity at 7729.0 eV, representing a decline of valence states of Co compared with that of Co3O4. Additionally, by examining the absorption energy location of the highest peak of the first derivative for the Co K-edge XANES spectra, the valence states of Co cations in various materials can be calculated31 (Fig. 2b and S9, ESI†). The average valence state of Co in VO-MoxCo3−xO4 is obtained to be Co2.72+, which is slightly lower than that of Co3O4 (Co2.76+) (Fig. 2b). This should be attributed to the creation of oxygen vacancies to lead to a lower coordination number with oxygen, and the doping of Mo cations with lower electronegativity.32 In general, a lower valence state is in favour of enhancing the stability towards the OER because it will prevent the dissolution of Co into acidic solutions during the OER process.33,34
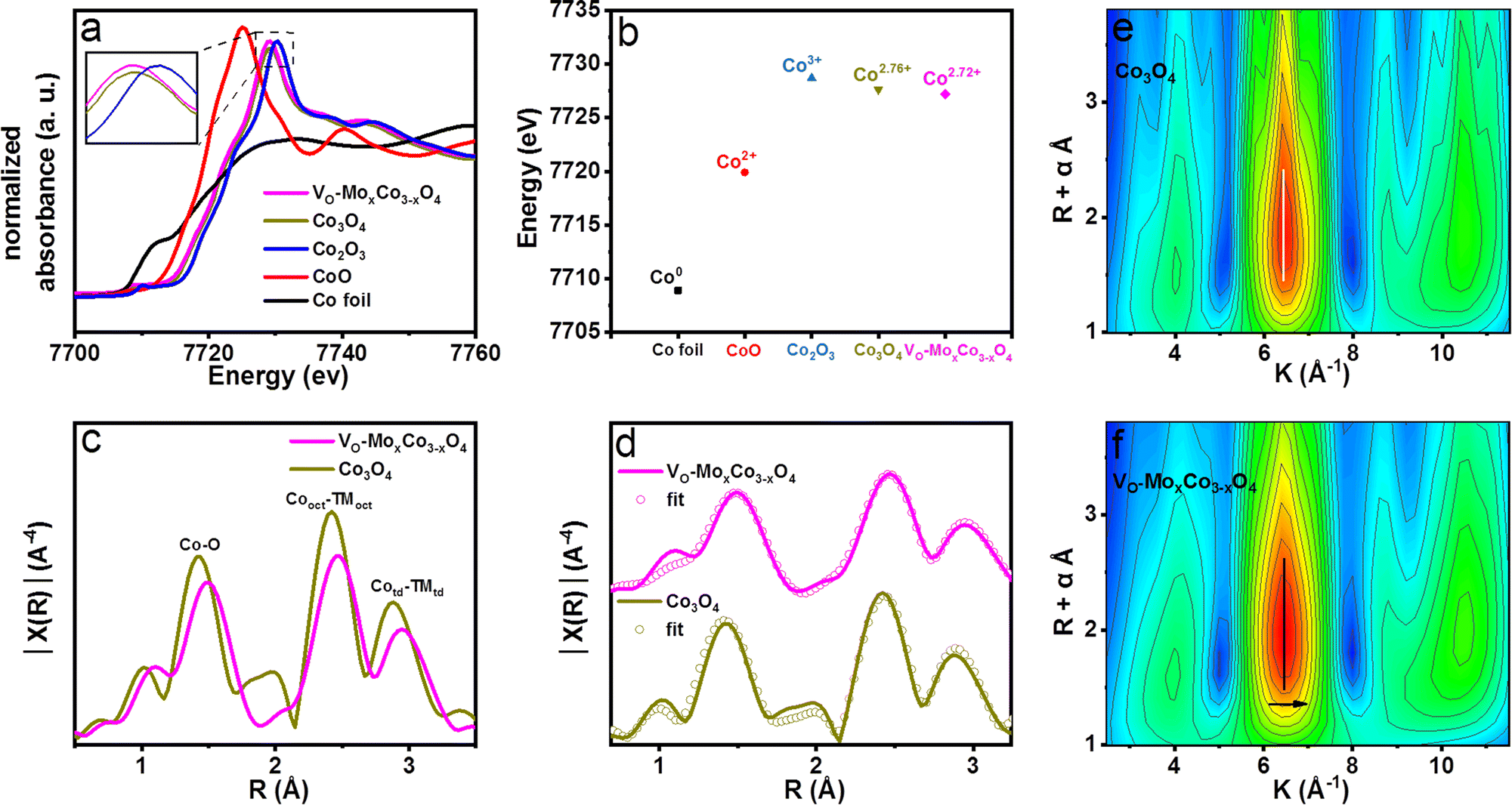 | ||
| Fig. 2 (a) Normalized XANES spectra at the Co K-edge of VO-MoxCo3−xO4, Co3O4 and comparisons. (b) Relationships between valence states and positions of the highest peak in the first derivative of XANES spectra at the Co K-edge of VO-MoxCo3−xO4, Co3O4 and reference materials. (c) Resulting Fourier-transform (FT) k3 weighted χ(k)-function of Co K-edge EXAFS spectra for VO-MoxCo3−xO4 and Co3O4. (d) Experimental data and fits of the Co K-edge EXAFS spectra for VO-MoxCo3−xO4 and Co3O4. Wavelet transforms (WT) for the k3 weighted EXAFS contour plots of (e) Co3O4 and (f) VO-MoxCo3−xO4. | ||
The Fourier transform (FT) Co K-edge extended X-ray absorption fine structure (EXAFS) spectra used to further analyze the coordination environments of VO-MoxCo3−xO4 and Co3O4 are depicted in Fig. 2c. The peak at around 1.5 Å is typically attributed to the TM–O bonds.35 The peak at around 2.5 Å is assigned to the bonds between the nearest octahedral cations (TMoct–TMoct).36 The peak at ∼3.0 Å is a characteristic peak for tetrahedral cations (TMtd), representing the bonds between tetrahedral cations with the closest octahedral (TMoct–TMtd) and tetrahedral (TMtd–TMtd) cations.37 In addition, the experimental EXAFS spectra of VO-MoxCo3−xO4 and Co3O4 were well-fitted to the cubic structure of Co3O4 (Fdm) (Fig. 2d and Table S2, ESI†). As a result, the amplitude of the Co–O characteristic peak in the EXAFS spectrum of VO-MoxCo3−xO4 decreases when compared with that of Co3O4, suggesting the successful formation of oxygen vacancies in octahedral geometry.38 Meanwhile, the bond length of Co–O in VO-MoxCo3−xO4 is found to be larger than that of Co3O4, which is fitted to be a Cooct–O bond with a coordination number (CN) of 5.375 and a bond length of 1.994 Å (Table S2, ESI†). While the Cooct–O bond in Co3O4 is fitted to be a CN of 6.160 and a bond length of 1.953 Å. This may be related to the modulation of the electronic structure of Co cations by introducing oxygen vacancies and doping Mo with variable valence states into the lattice, which results in a decrease in the average valence states of Co, thereby regulating the Co–O bonds in TMO6 units.39 Furthermore, the amplitudes of the octahedral and tetrahedral characteristic peaks in the spectrum of VO-MoxCo3−xO4 are observed to decrease, corresponding to the creation of Cooct defects by inducing the oxidation of the nearest Mo cations.40 The fitted bond length of Cooct–Cooct in VO-MoxCo3−xO4 is 2.869 Å, which is slightly longer than that of Co3O4 (2.861 Å), resulting in a right-shift of the Cooct characteristic peak in the EXAFS spectrum. Generally, it is considered to match well with the bond length of the O–O radical for enhancing the OPM pathway when the distance of the neighboring active sites is regulated to be 2.8–2.9 Å.11–13 Cooct is believed to be the real active species of spinel oxides for the OER because it is preferentially exposed on the surface.38,39 Therefore, owing to the appropriate distance of Cooct–Cooct, VO-MoxCo3−xO4 and Co3O4 have been deemed to have the conditions to achieve the OPM pathway for acidic OER.13 The wavelet transform (WT) of the k3-weighted EXAFS spectra at the VO-MoxCo3−xO4 and Co3O4 Co K edges are shown in Fig. 2e and f. When compared to Co3O4, the Cooct–Cooct coordination characteristics of VO-MoxCo3−xO4 exhibit a slight right-shift. This is connected to doping of the heavier element of Mo into VO-MoxCo3−xO4.41
The effects on the performance of acidic OER by creating oxygen vacancies in Co3O4 were examined by obtaining the linear sweep voltammogram (LSV) curve of VO-MoxCo3−xO4 in 0.1 M HClO4 solution (Fig. 3a). Polarization curves of the OER were obtained on a Co3O4, Ir/C carbon paper electrode for comparison as well. All polarization curves were iR-corrected. It is worth noting that the synthetic condition for VO-MoxCo3−xO4 was optimized (for details see Experimental section and Fig. S10, ESI†). As displayed in Fig. 3a, the carbon paper used as a working electrode only exhibits ignorable activity towards OER in acidic media. Ir/C has been proven to be oxidized to IrO2/C during the catalytic process.42 At an overpotential of 460 mV, the Ir/C electrocatalyst can reach a current density of 10 mA cm−2. When compared to Ir/C, Co3O4 shows inferior activity for OER, which can achieve a current density of 10 mA cm−2 at an overpotential of 490 mV. Interestingly, VO-MoxCo3−xO4 displays an overpotential of 420 mV to attain 10 mA cm−2. Additionally, it only requires an overpotential of 490 mV to achieve a current density of 100 mA cm−2. It suggests that the activity of Co3O4 can be significantly enhanced by constructing oxygen vacancies via doping Mo into the lattice. As shown in Fig. 3b, VO-MoxCo3−xO4 exhibits the lowest Tafel slope of only 102.5 mV dec−1 compared to those of Co3O4 (121.8 mV dec−1), and Ir/C (164.7 mV dec−1), indicating fastest kinetics of OER process.43 As shown in Fig. 3c, Vo-MoxCo3−xO4 is at a similar level in terms of activity (overpotential to achieve 10 mA cm−2) and kinetics (Tafel slope) when compared with recently reported state-of-the-art non-precious metal electrocatalysts for acidic OER (Table S3, ESI†). Furthermore, compared to Co3O4 and Ir/C, as-synthesized VO-MoxCo3−xO4 possesses larger electrochemical surface area (ECSA)-normalized current density, turnover frequency (TOF), and mass-activity, and a smaller reaction resistance for acidic OER (Fig. S11–S13 and Table S4, ESI†). This implies that VO-MoxCo3−xO4 exhibits a higher intrinsic activity towards acidic OER.44 To assess the stability of the OER in acids, the chronopotentiometric curves of VO-MoxCo3−xO4 and Co3O4 were obtained at 10 mA cm−2 (Fig. 3d). The initial potential of 1.65 V vs. RHE for VO-MoxCo3−xO4 is observed to only increase to 1.72 V vs. RHE after 30 h. Moreover, it is noticed that the morphology, valence states, and crystal structure of VO-MoxCo3−xO4 were unchanged before and after the stability test, suggesting high structural stability of the catalyst (Fig. S14–17, ESI†). However, the initial potential of Co3O4 is 1.70 V vs. RHE and the potential rapidly increases to 2.4 V vs. RHE after 6.1 h, completely losing its catalytic capability. Consequently, the as-prepared VO-MoxCo3−xO4 is at a similar level compared with the current reported state-of-the-art Co-based electrocatalysts in stability for acidic OER (Table S5, ESI†). Defects in the electrocatalysts commonly harm the long-term stability of the OER in acids.10 However, doping a small amount of Mo can supply electrons to the Co sites during the catalytic process to hinder the dissolution of Co in an acidic solution.19
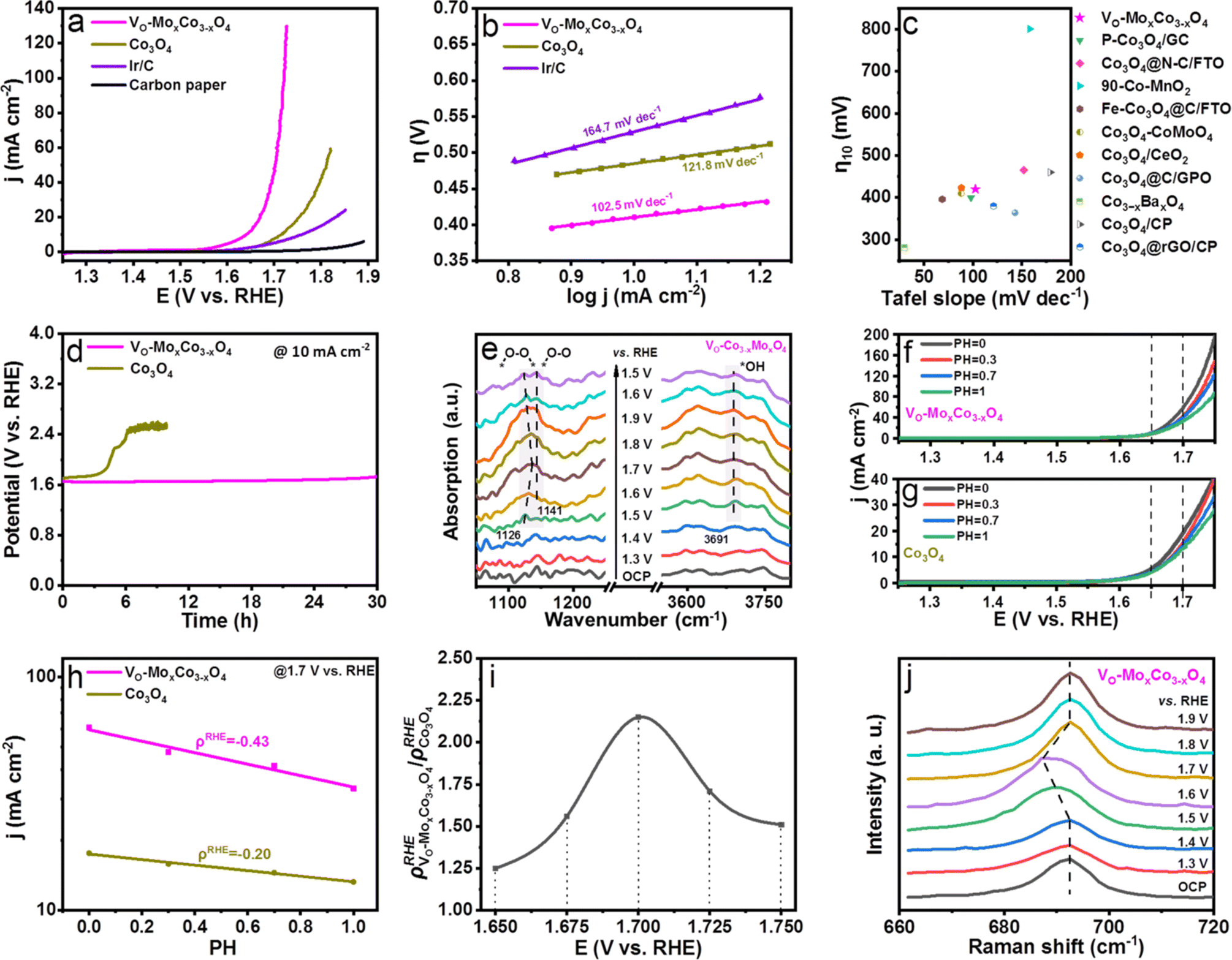 | ||
| Fig. 3 (a) LSV curves of the OER on VO-MoxCo3−xO4, Co3O4, Ir/C and carbon paper electrocatalysts in 0.1 M HClO4 at 25 °C with scan rates of 5 mV s−1. (b) Tafel plots of VO-MoxCo3−xO4, Co3O4, Ir/C and carbon paper electrocatalysts. (c) Comparisons of Vo-MoxCo3−xO4 in terms of activity and kinetics with currently reported state-of-the-art non-precious metal electrocatalysts for acidic OER. (d) The chronopotentiometric curves of the OER on VO-MoxCo3−xO4 and Co3O4 electrocatalysts at 10 mA cm−2 in 0.1 M HClO4 (25 °C). (e) In situ FT-IR spectra recorded at various applied potentials (vs. RHE) in 0.1 M HClO4 electrolyte on (a) VO-MoxCo3−xO4. LSV curves of (f) VO-MoxCo3−xO4 and (g) Co3O4 in acid electrolytes with different pH values. (h) OER specific activities of VO-MoxCo3−xO4 and Co3O4 at 1.7 V vs. RHE against the pH values. (i) Relationship between the ratios of (ρRHE VO-MoxCo3−xO4/ρRHE Co3O4) at different potentials. (j) In situ Raman spectra of VO-MoxCo3−xO4 obtained on stepping the potential from 1.3 to 1.9 V vs. RHE in 0.1 M HClO4 electrolyte. | ||
The variations in surface reaction intermediates were examined by in situ electrochemical Fourier transform infrared (FT-IR) spectroscopy to investigate the mechanism of the OER (Fig. 3e, S18 and S19, ESI†). The catalysts were uniformly dispersed onto a carbon paper electrode before being assembled in an infrared reflection setup (for details see Electrochemical measurements†). As shown in Fig. 3e, when the potentials increase to approach the OER region (≥1.5 V vs. RHE), distinctive adsorption bands at 1126 and 1141 cm−1 can be detected, in the in situ FT-IR spectra of VO-MoxCo3−xO4, which are observed to be potential-dependent. The intensities of these bands increase dramatically as the potential increases. This shows that the bands are formed as a function of the applied potentials. The peak at 1126 cm−1 is related to the generation of an O–O bond, which frequently represents the oxygen bridges between neighboring metal sites (*–O–O–*) formed in the OPM-type OER process.11,13 Below 1.70 V vs. RHE, this adsorption band blue-shifts to a higher frequency as potential increases, indicating strengthened bonding of metal sites with O–O radicals,45 while above 1.70 V vs. RHE, the band gradually red-shifts to a lower frequency, suggesting a weakened bonding.46 The peak at 1141 cm−1 is due to linearly bound superoxide species (*–O–O), which is the intermediate before O2 release.13 The potential-dependent adsorption bands detected at 3691 cm−1 are assigned to *OH, a typical intermediate of the OPM pathway for the OER.11 However, these characteristic peaks cannot be observed in the in situ FT-IR spectra of Co3O4, because it is extensively believed that acidic OER is catalyzed through the AEM pathway on Co3O4 (ref. 13) (Fig. S18, ESI†). Doping Mo has been proven to be unable to promote the OER process on Co3O4 through the OPM pathway since the resulting larger Co–Co distance inhibits direct O–O radical coupling, as shown by the results of EXAFS.13,19 Thus, creating oxygen vacancies in the lattice is considered to be the key to inducing the OPM.
To investigate the proton–electron transfer kinetics, the acidic OER activities of VO-MoxCo3−xO4 and Co3O4 were determined at pH values of 0, 0.3, 0.7, and 1.0 (Fig. 3f,g, and S20, ESI†).47–51 VO-MoxCo3−xO4 exhibits a stronger pH-dependent feature compared to Co3O4. Generally, it is an important parameter to evaluate the degree of proton reaction on the RHE scale which is used to calculate the current densities on the log scale as a function of pH (ρRHE = ∂(logj)/∂pH) (Fig. 3h and S20, ESI†).47Fig. 3i presents the relationship between the ratios of ρRHE (ρRHE VO-MoxCo3−xO4/ρRHE Co3O4) obtained on VO-MoxCo3−xO4 and Co3O4 at 1.650, 1.675, 1.700, 1.725, and 1.750 V vs. RHE, respectively. As a result, Co3O4 presents a lower ρRHE at the OER ranging from 1.650 to 1.750 V vs. RHE as compared with those of VO-MoxCo3−xO4, implying a lower degree of decoupled proton–electron transfer during the rate determining step (RDS) of the AEM, i.e., the deprotonation of *OOH. In contrast, higher values of ρRHE suggest a unrelated proton–electron transfer process on VO-MoxCo3−xO4, which means a strong involvement of the OPM pathway during the OER. Furthermore, the ρRHE VO-MoxCo3−xO4/ρRHE Co3O4 begins to increase gradually as the potential increases and reaches its maximum value of 2.15 at 1.700 V vs. RHE before descending. This indicates that at 1.70 V vs. RHE, participation of the OPM pathway in acidic OER on VO-MoxCo3−xO4 is at its maximum.
The mechanism of the OER was clarified by electrochemical in situ Raman spectroscopy as well (Fig. 3j and S21, ESI†). The A1g modes on the in situ Raman spectra of VO-MoxCo3−xO4 have been found to redshift compared with those of Co3O4 at each applied potential. This means that oxygen vacancies are still present in the as-prepared VO-MoxCo3−xO4 during the OER process.24 When compared to the open-circuit potential (OCP), the in situ Raman spectra of Co3O4 at potentials from 1.3 to 1.9 V vs. RHE remain unchanged, exhibiting characteristic bands of spinel oxides.16 However, the A1g band in the in situ Raman spectrum of VO-MoxCo3−xO4, the characteristic peak of octahedral sites, is found to gradually redshift from 692 cm−1 at 1.3 V vs. RHE to 687 cm−1 at 1.6 V vs. RHE. This usually represents the formation of highly active species of Co4+ in octahedral interstices during the OER process.52 Generally, the tetravalent species are in favour of the fast deprotonation of hydroxyl by facilitating the hybridization of their t2g orbitals with the O 2p band. This ultimately boosts the direct O–O radical coupling.53,54 This is the reason why the most strengthened bonding of *–O–O–* can be detected and the strongest participation of the OPM pathway has been found at approximately 1.7 V vs. RHE.
To clarify the origin of enhanced activity on VO-MoxCo3−xO4, density functional theory (DFT) simulations were performed to investigate the reaction thermodynamics and equilibrium variation during the OER process (Fig. 4). Two types of computational models were established, including Mo-doped oxygen vacancy enriched Co3O4 and pure Co3O4, representing VO-MoxCo3−xO4 and Co3O4, respectively (Fig. S22 and S23, ESI†). Commonly, the OER process on the surface of electrocatalysts via the AEM pathway involves the adsorption and transformation of three key reaction intermediates of *OH, *O, and *OOH, as depicted in Fig. 4a.7 As shown in Fig. 4b, the Gibbs free energy diagrams of the OER through the AEM pathway on VO-MoxCo3−xO4 and Co3O4 are constructed based on the optimized structures and the energies of the adsorbed intermediates on the surfaces (Fig. S22 and S23, ESI†). It can be concluded that VO-MoxCo3−xO4 possesses a lower free energy increase of 2.22 eV than Co3O4 (3.05 eV), indicating a better activity via the AEM (Fig. 4c). It should be noted that the process of transforming *O into *OOH is frequently deemed to be the potential-determining step (PDS), according to the scaling relationship.7 On the other hand, the key catalytic process during the OPM pathway is generally believed to be a synergistic proton–electron-transfer reaction, involving the adsorptions of hydroxyl species (OH*–* and OH*–*OH), sequential deprotonations (O*–*OH and O*–*O), and ultimately directly coupling to O–O radicals for the generation of O2, as presented in Fig. 4a.11 The Gibbs free energy diagrams of the OPM pathway for the OER on VO-MoxCo3−xO4 and Co3O4 are displayed in Fig. 4d, which are obtained based on the optimized structures and energies (Fig. S22 and S23, ESI†). The process of O–O radical coupling to generate O2 is proven to be the PDS.11 VO-MoxCo3−xO4 shows a minimal free energy increase of 1.85 eV, which is lower than that of Co3O4 (2.76 eV) (Fig. 4e). Meanwhile, the superiority of the OPM over the AEM has been demonstrated, owing to the lower free energy increase.
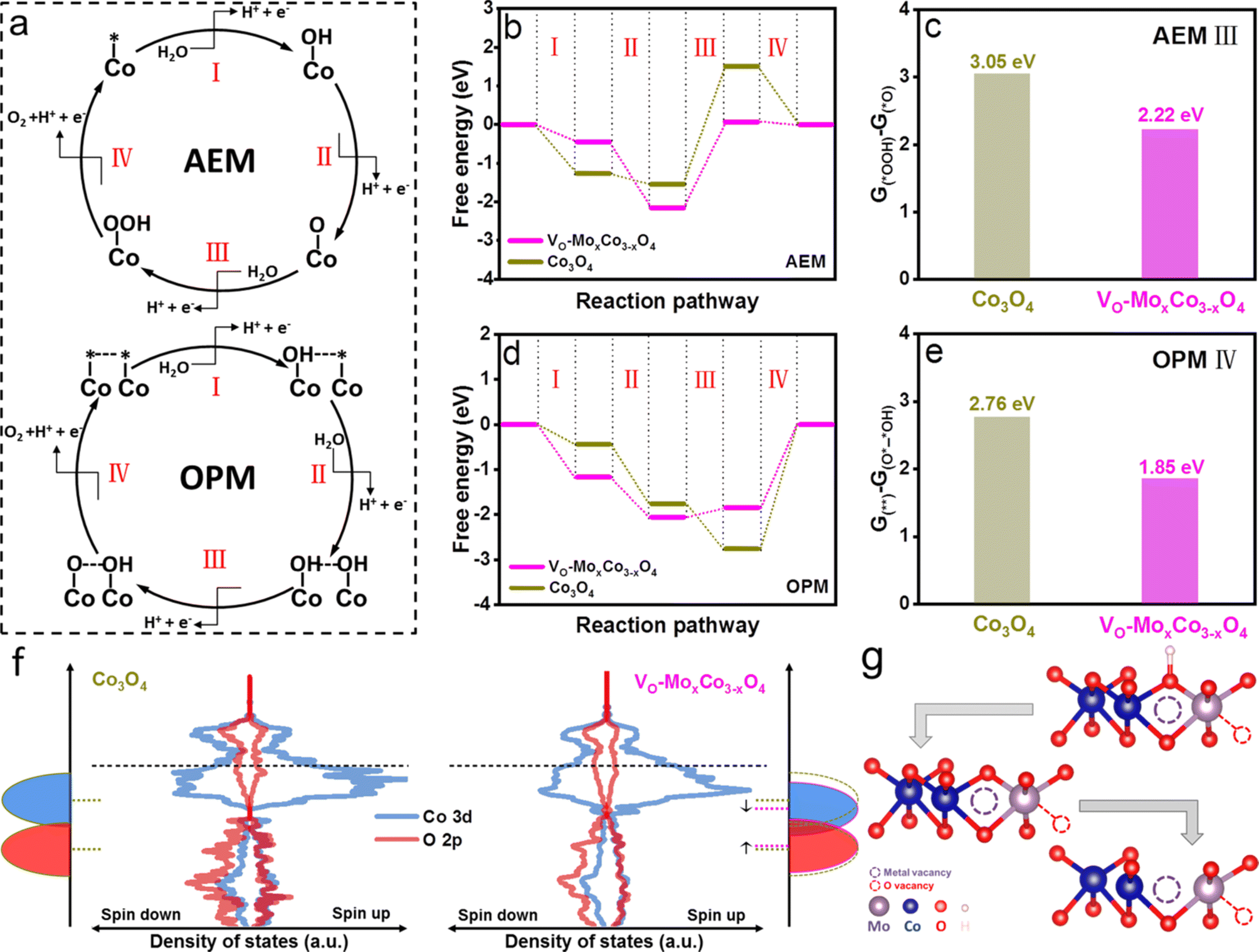 | ||
| Fig. 4 (a) Schematic illustration of mechanisms for the OER through AEM and OPM pathways. (b) Gibbs free energy diagrams of the AEM pathway on VO-MoxCo3−xO4 and Co3O4. (c) Histogram of free energy increases of Co3O4 and VO-MoxCo3−xO4 during the PDS of the AEM pathway (AEM III) at U = 1.23 V. (d) Gibbs free energy diagrams of the OPM pathway on VO-MoxCo3−xO4 and Co3O4. (e) Histogram of free energy increases of Co3O4 and VO-MoxCo3−xO4 during the PDS of the OPM pathway (OPM IV) at U = 1.23 V. (f) The curves of density of states and schematic diagrams of Co 3d and O 2p orbitals in VO-MoxCo3−xO4 and Co3O4. (g) The optimized structures of key intermediates adsorbed on the surface of VO-MoxCo3−xO4 during the OPM pathway. | ||
The density of states (DOS) and band centers of Co 3d and O 2p orbitals were calculated to provide insight into the difference in the electronic structures of VO-MoxCo3−xO4 and Co3O4 (Fig. 4f and S24, ESI†). Below the Fermi level, a downshifting Co 3d band and an upshifting O 2p band can be found when compared with Co3O4, indicating a more significant overlap of Co 3d and O 2p orbitals.9 Generally, this will strengthen the metal–oxygen bonds in the lattice, thereby reducing the possibility of the LOM occurring on the surface of VO-MoxCo3−xO4.20 At the same time, the downshifting Co 3d band facilitates the hybridization of its t2g orbitals with unbounded O 2p orbitals, which is beneficial for the adsorption of hydroxyl as well as deprotonation to form *O.54 Due to the appropriate distance between the closest Cooct in VO-MoxCo3−xO4, neighboring adsorbed oxygen species will tend to directly couple for the formation of O–O radicals,13 as exhibited in Fig. 4g. Thus, constructing oxygen vacancies by doping Mo tends to push the thermodynamic equilibrium of the OER positively and decreases the free energy increase of the PDS, ultimately lowering the overpotential of the OER process.
Conclusions
In summary, an approach of doping a small amount of Mo heteroatoms (∼0.5 wt%) into Co3O4 was reported to enhance the activity of the OER in acids. It has been experimentally proved that the doping can activate lattice oxygen which can be etched away from the lattice afterward for building oxygen vacancies. The achieved oxygen vacancies boost the faster OPM pathway, and simultaneously Mo with variable valence states can promote the stability. EPR results demonstrated the successful creation of oxygen vacancies in doped Co3O4. Synchrotron radiation characterization confirmed the variations of the valence states and coordination environments as well as bond lengths. As a result, as-synthesized Mo-doped oxygen vacancy enriched Co3O4 (VO-MoxCo3−xO4) exhibited the most efficient catalytic activity (requires overpotentials of 420 and 490 mV to reach 10 and 100 mA cm−2, respectively) and fastest kinetics (a Tafel slope of only 102.5 mV dec−1) for the OER in an acidic medium, as compared with those of Co3O4 and Ir/C. VO-MoxCo3−xO4 towards acidic OER exhibited high stability and can maintain a current density of 10 mA cm−2 for 30 h. Detailed electrochemical in situ investigations revealed that the OPM pathway for the OER was enhanced by building oxygen vacancies, due to directly observing the key reaction intermediates of *–O–O–*, *–O–O, and *OH. Meanwhile, owing to the generation of tetravalent species, which was beneficial for the adsorption as well as deprotonation of hydroxyl, the strongest participation of the OPM pathway has been found at approximately 1.7 V vs. RHE. Further DFT calculations clarified that the tuned Co 3d and O 2p orbitals suppressed the LOM pathway and enhanced the adsorption and rapid deprotonation of hydroxyl (the key intermediate), facilitating direct O–O radical coupling and ultimately, the promotion of the OPM pathway. This study is of significance for designing and developing highly efficient and stable non-precious metal electrocatalysts for the OER in an acidic medium, thus promoting the large-scale applications of PEMWEs.Author contributions
X. Y. and S. H. proposed the concept and supervised the work. L. S. synthesized the materials and performed the experiments with the help of M. F., Y. P., X. Z. and Y. S.; X. Y. and S. H. analyzed the data and co-wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51920105004), the Natural Science Foundation of Guangdong Province (2023A1515030188), the Science and Technology Program of Guangzhou (2023A04J1373), and the Special Fund for the Cultivation of Guangdong College Students' Scientific and Technological Innovation (“Climbing Program” Special Funds, pdjh2023a0160).Notes and references
- Y. Chen, H. Li, J. Wang, Y. Du, S. Xi, Y. Sun, M. Sherburne, J. W. Ager, A. C. Fisher and Z. J. Xu, Nat. Commun., 2019, 10, 572 CrossRef CAS PubMed.
- J. Wang, L. Han, B. Huang, Q. Shao, H. L. Xin and X. Huang, Nat. Commun., 2019, 10, 5692 CrossRef CAS.
- H. N. Nong, T. Reier, H.-S. Oh, M. Gliech, P. Paciok, T. H. T. Vu, D. Teschner, M. Heggen, V. Petkov, R. Schlögl, T. Jones and P. Strasser, Nat. Catal., 2018, 1, 841–851 CrossRef CAS.
- T. Reier, H. N. Nong, D. Teschner, R. Schlögl and P. Strasser, Adv. Energy Mater., 2016, 7, 1601275 CrossRef.
- J. Feng, F. Lv, W. Zhang, P. Li, K. Wang, C. Yang, B. Wang, Y. Yang, J. Zhou, F. Lin, G. C. Wang and S. Guo, Adv. Mater., 2017, 29, 1703798 CrossRef.
- X. Yang, H. Li, A.-Y. Lu, S. Min, Z. Idriss, M. N. Hedhili, K.-W. Huang, H. Idriss and L.-J. Li, Nano Energy, 2016, 25, 42–50 CrossRef CAS.
- J. Yu, Q. He, G. Yang, W. Zhou, Z. Shao and M. Ni, ACS Catal., 2019, 9, 9973–10011 CrossRef CAS.
- J. Song, C. Wei, Z.-F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- K. Xiao, Y. Wang, P. Wu, L. Hou and Z. Q. Liu, Angew. Chem., Int. Ed., 2023, 62, e202301408 CrossRef CAS PubMed.
- C. Roy, R. R. Rao, K. A. Stoerzinger, J. Hwang, J. Rossmeisl, I. Chorkendorff, Y. Shao-Horn and I. E. L. Stephens, ACS Energy Lett., 2018, 3, 2045–2051 CrossRef CAS.
- C. Lin, J.-L. Li, X. Li, S. Yang, W. Luo, Y. Zhang, S.-H. Kim, D.-H. Kim, S. S. Shinde, Y.-F. Li, Z.-P. Liu, Z. Jiang and J.-H. Lee, Nat. Catal., 2021, 4, 1012–1023 CrossRef CAS.
- J. Liang, X. Gao, K. Xu, J. Lu, D. Liu, Z. Zhao, E. C. M. Tse, Z. Peng, W. Zhang and J. Liu, Small, 2023, 2304889 CrossRef CAS PubMed.
- N. Wang, P. Ou, R. K. Miao, Y. Chang, Z. Wang, S.-F. Hung, J. Abed, A. Ozden, H.-Y. Chen, H.-L. Wu, J. E. Huang, D. Zhou, W. Ni, L. Fan, Y. Yan, T. Peng, D. Sinton, Y. Liu, H. Liang and E. H. Sargent, J. Am. Chem. Soc., 2023, 145, 7829–7836 CrossRef CAS PubMed.
- M. Okamura, M. Kondo, R. Kuga, Y. Kurashige, T. Yanai, S. Hayami, V. K. K. Praneeth, M. Yoshida, K. Yoneda, S. Kawata and S. Masaoka, Nature, 2016, 530, 465–468 CrossRef CAS PubMed.
- F. Song, M. M. Busch, B. Lassalle-Kaiser, C.-S. Hsu, E. Petkucheva, M. Bensimon, H. M. Chen, C. Corminboeuf and X. Hu, ACS Cent. Sci., 2019, 5, 558–568 CrossRef CAS PubMed.
- K. Natarajan, E. Munirathinam and T. C. K. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 27140–27148 CrossRef CAS PubMed.
- A. Li, S. Kong, C. Guo, H. Ooka, K. Adachi, D. Hashizume, Q. Jiang, H. Han, J. Xiao and R. Nakamura, Nat. Catal., 2022, 5, 109–118 CrossRef CAS.
- S. Anantharaj, K. Karthick and S. Kundu, Inorg. Chem., 2019, 58, 8570–8576 CrossRef CAS PubMed.
- W. Han, Y. Qian, F. Zhang, Y. He, P. Li and X. Zhang, Chem. Eng. J., 2023, 473, 145353 CrossRef CAS.
- Z. He, J. Zhang, Z. Gong, H. Lei, D. Zhou, N. Zhang, W. Mai, S. Zhao and Y. Chen, Nat. Commun., 2022, 13, 2191 CrossRef CAS PubMed.
- Y. Qi, X. Xiao, Y. Mei, L. Xiong, L. Chen, X. Lin, Z. Lin, S. Sun, B. Han, D. Yang, Y. Qin and X. Qiu, Adv. Funct. Mater., 2022, 32, 2111615 CrossRef CAS.
- S. K. Tripathy, M. Christy, N.-H. Park, E.-K. Suh, S. Anand and Y.-T. Yu, Mater. Lett., 2008, 62, 1006–1009 CrossRef CAS.
- J. Shan, C. Ye, S. Chen, T. Sun, Y. Jiao, L. Liu, C. Zhu, L. Song, Y. Han, M. Jaroniec, Y. Zhu, Y. Zheng and S.-Z. Qiao, J. Am. Chem. Soc., 2021, 143, 5201–5211 CrossRef CAS PubMed.
- J. Qian, X. Liu, C. Zhong, G. Xu, H. Li, W. Zhou, B. You, F. Wang, D. Gao and D. Chao, Adv. Funct. Mater., 2023, 33, 2212021 CrossRef CAS.
- W. Zhu, F. Yao, K. Cheng, M. Zhao, C.-J. Yang, C.-L. Dong, Q. Hong, Q. Jiang, Z. Wang and H. Liang, J. Am. Chem. Soc., 2023, 145, 17995–18006 CrossRef CAS PubMed.
- Z. Xiong, B. Tang, Z. Fang, C. Yang and S. Zhang, J. Alloys Compd., 2017, 723, 580–588 CrossRef CAS.
- X. Wu, W. Wang, N. Song, X. Yang, S. Khaimanov and N. Tsidaeva, Chem. Eng. J., 2016, 306, 382–392 CrossRef CAS.
- M.-M. Wang, L.-J. Liu, J.-R. Xi, Y. Ding, P.-X. Liu, L. Mao, B.-J. Ni, W.-K. Wang and J. Xu, Chem. Eng. J., 2023, 451, 138605 CrossRef CAS.
- Z. Hou, C. Cui, Y. Li, Y. Gao, D. Zhu, Y. Gu, G. Pan, Y. Zhu and T. Zhang, Adv. Mater., 2023, 35, 2209876 CrossRef CAS.
- L. Chong, J. Wen, E. Song, Z. Yang, I. D. Bloom and W. Ding, Adv. Energy Mater., 2023, 13, 2302306 CrossRef CAS.
- Z. Li, Y. Zhang, Y. Feng, C. Q. Cheng, K. W. Qiu, C. K. Dong, H. Liu and X. W. Du, Adv. Funct. Mater., 2019, 29, 1903444 CrossRef.
- C. Hao, X. Li, H. Huang, L. Ge, Z. Fu, Y. Lu, Y. Wang, S. Zhang and Z. Cheng, ACS Energy Lett., 2023, 8, 4506–4513 CrossRef CAS.
- L. Li, X. Cao, J. Huo, J. Qu, W. Chen, C. Liu, Y. Zhao, H. Liu and G. Wang, J. Energy Chem., 2023, 76, 195–213 CrossRef CAS.
- J. Wang, J. Liu, B. Zhang, F. Cheng, Y. Ruan, X. Ji, K. Xu, C. Chen, L. Miao and J. Jiang, Nano Energy, 2018, 53, 144–151 CrossRef CAS.
- Y. Zhang, Z. Zhang, G. Jiang, A. H. Mamaghani, S. Sy, R. Gao, Y. Jiang, Y. Deng, Z. Bai, L. Yang, A. Yu and Z. Chen, Nano Energy, 2022, 100, 107425 CrossRef CAS.
- X. Li, L. Ge, Y. Du, H. Huang, Y. Ha, Z. Fu, Y. Lu, W. Yang, X. Wang and Z. Cheng, ACS Nano, 2023, 17, 6811–6821 CrossRef CAS PubMed.
- J. Zhang, J. Qian, J. Ran, P. Xi, L. Yang and D. Gao, ACS Catal., 2020, 10, 12376–12384 CrossRef CAS.
- Z.-F. Huang, J. Song, Y. Du, S. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. J. Xu and X. Wang, Nat. Energy, 2019, 4, 329–338 CrossRef CAS.
- T. Wu, S. Sun, J. Song, S. Xi, Y. Du, B. Chen, W. A. Sasangka, H. Liao, C. L. Gan, G. G. Scherer, L. Zeng, H. Wang, H. Li, A. Grimaud and Z. J. Xu, Nat. Catal., 2019, 2, 763–772 CrossRef CAS.
- L. An, H. Zhang, J. Zhu, S. Xi, B. Huang, M. Sun, Y. Peng, P. Xi and C. H. Yan, Angew. Chem., Int. Ed., 2023, 62, e202214600 CrossRef CAS PubMed.
- M. Feng, J. Huang, Y. Peng, C. Huang, X. Yue and S. Huang, ACS Nano, 2022, 16, 13834–13844 CrossRef CAS PubMed.
- C. Spöri, P. Briois, H. N. Nong, T. Reier, A. Billard, S. Kühl, D. Teschner and P. Strasser, ACS Catal., 2019, 9, 6653–6663 CrossRef.
- Y. Peng, C. Huang, J. Huang, M. Feng, X. Qiu, X. Yue and S. Huang, Adv. Funct. Mater., 2022, 32, 2201011 CrossRef CAS.
- X. Yue, X. Qin, Y. Chen, Y. Peng, C. Liang, M. Feng, X. Qiu, M. Shao and S. Huang, Adv. Sci., 2021, 8, 2101653 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS.
- M. Zhang, M. de Respinis and H. Frei, Nat. Chem., 2014, 6, 362–367 CrossRef CAS PubMed.
- L. Giordano, B. Han, M. Risch, W. T. Hong, R. R. Rao, K. A. Stoerzinger and Y. Shao-Horn, Catal. Today, 2016, 262, 2–10 CrossRef CAS.
- A. K. Tomar, U. N. Pan, N. H. Kim and J. H. Lee, ACS Energy Lett., 2023, 8, 565–573 CrossRef CAS.
- R. Chen, Z. Wang, S. Chen, W. Wu, Y. Zhu, J. Zhong and N. Cheng, ACS Energy Lett., 2023, 8, 3504–3511 CrossRef CAS.
- Z.-M. Wang, Q.-L. Hong, X.-H. Wang, H. Huang, Y. Chen and S.-N. Li, Acta Phys.-Chim. Sin., 2023, 39, 2303028 Search PubMed.
- Q. Xue, Z. Wang, Y. Ding, F. Li and Y. Chen, Chin. J. Catal., 2023, 45, 6 CrossRef CAS.
- J. Huang, H. Sheng, R. D. Ross, J. Han, X. Wang, B. Song and S. Jin, Nat. Commun., 2021, 12, 3036 CrossRef CAS.
- X. L. Liu, Y. C. Jiang, J. T. Huang, W. Zhong, B. He, P. J. Jin and Y. Chen, Carbon Energy, 2023, 5, e367 CrossRef CAS.
- N. Zhang, X. Feng, D. Rao, X. Deng, L. Cai, B. Qiu, R. Long, Y. Xiong, Y. Lu and Y. Chai, Nat. Commun., 2020, 11, 4066 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta00655k |
| This journal is © The Royal Society of Chemistry 2024 |