
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Band engineering in iron and silver co-doped double perovskite nanocrystals for selective photocatalytic CO2 reduction†
Razi
Ahmad
*a,
Yu
Zhang
a,
Jan
Navrátil
 ab,
Piotr
Błoński
ae,
Lukáš
Zdražil
af,
Sergii
Kalytchuk
a,
Alberto
Naldoni
*ac,
Andrey L.
Rogach
de,
Michal
Otyepka
ae,
Radek
Zboril
*af and
Štěpán
Kment
*af
ab,
Piotr
Błoński
ae,
Lukáš
Zdražil
af,
Sergii
Kalytchuk
a,
Alberto
Naldoni
*ac,
Andrey L.
Rogach
de,
Michal
Otyepka
ae,
Radek
Zboril
*af and
Štěpán
Kment
*af
aRegional Centre of Advanced Technologies and Materials, Czech Advanced Technology and Research Institute, Palacký University Olomouc, Šlechtitelů 27, Olomouc, 783 71, Czech Republic. E-mail: razi.ahmad@upol.cz; radek.zboril@upol.cz; stepan.kment@upol.cz
bDepartment of Physical Chemistry, Faculty of Science, Palacký University Olomouc, 17. listopadu 1192/12, 779 00 Olomouc, Czech Republic
cDepartment of Chemistry, NIS Centre, University of Turin, Turin 10125, Italy. E-mail: alberto.naldoni@unito.it
dDepartment of Materials Science and Engineering, Center for Functional Photonics (CFP), City University of Hong Kong, 83 Tat Chee Avenue, Hong Kong SAR, China
eIT4Innovations, VŠB – Technical University of Ostrava, 17. listopadu 2172/15, 708 00 Ostrava-Poruba, Czech Republic
fNanotechnology Centre, Centre for Energy and Environmental Technologies, VŠB–Technical University of Ostrava, 17. listopadu 2172/15, 708 00 Ostrava-Poruba, Czech Republic
First published on 6th August 2024
Abstract
Double metal cation halide perovskites are promising alternatives to lead halide perovskites due to their exceptional compositional flexibility and stability. However, their utilization in solar-light harvesting applications has been hindered by their large band gap and the complexity of producing doped or alloyed materials with desirable optoelectronic properties. In this study, we report the colloidal synthesis of iron-doped Cs2NaInCl6 double perovskite nanocrystals (NCs), leading to a significant extension of the absorption edge from 330 nm to 505 nm. We also demonstrate that simultaneous doping with Fe3+ and Ag+ ions allows significant reduction of the optical band gap and precise tuning of electronic band structures of the resulting NCs. The enhanced absorption in the visible region is attributed to the substitution of In-5s by the Fe-3d state, while the introduction of the Ag 4d state upshifts the valence band maximum, inducing a transformative change in the band structure, as confirmed by density functional theory (DFT) calculations. Remarkably, by precisely controlling the band positions of the Fe3+-doped Cs2Ag0.5Na0.5InCl6 NCs, we accomplished the selective photocatalytic reduction of CO2 into CH4, making them readily available for solar-energy conversion technologies.
Introduction
Solution-processable hybrid lead halide perovskites have garnered unprecedented attention in the realm of low-cost optoelectronic devices, thanks to their remarkable optical and electronic properties.1 These materials exhibit a large absorption coefficient, high charge carrier mobility, long diffusion length, and useful defect tolerance, leading to significant advancements in optoelectronic devices like solar cells and light-emitting diodes.2,3 However, the practical utilization of lead-based perovskite materials faces two major obstacles: poor long-term stability and toxicity concerns.4 Consequently, significant efforts have been dedicated to the exploration of stable and environmentally benign materials, aiming to achieve optical and electronic properties comparable to those of lead halide perovskites.5–10 Among these, lead-free halide double perovskites (DPs) with a formula A2MIMIIIX6 (where A = Cs+, and X = Cl−, Br−, I) can be formed by heterovalent substitution of Pb2+ cations with a pair of monovalent (MI = Na+, K+, Ag+) and trivalent (MIII = Bi3+, Sb3+, In3+) cations. They have attracted considerable attention due to several desirable features, including superior material stability, less toxic elements, compositional flexibility, and attractive optoelectronic properties.9–11 Specifically, Cs2AgBiX6 (X = Br, Cl), Cs2AgInCl6, and Cs2NaInCl6 have been recognized as promising candidates across a diverse spectrum of applications. These encompass solar cells, photocatalysis, light-emitting diodes, luminescent solar concentrators, and scintillators.12–16 In particular, Cs2AgBiBr6 is the only example of DP that has been extensively investigated as a lead-free light absorber layer in perovskite solar cells and as a photocatalyst for CO2 reduction, H2 production and organic synthesis.13,17–22 However, Cs2AgBiBr6 DP has an indirect band gap, which is not ideal for optoelectronic and photocatalytic applications as both photons and phonons are involved in the optical absorption and recombination processes, which results in relatively weak oscillator strength for optical absorption and low photoluminescence (PL) quantum yields (QY), respectively. Thus, despite the commendable material stability and direct band gap characteristics exhibited by Cs2AgInCl6 and Cs2NaInCl6, these DP materials are not suitable for solar cells and photocatalysis application due to a large (∼4 eV) and parity-forbidden band gap.Previous studies have demonstrated that doping and/or alloying are promising strategies to significantly alter the optical and electronic properties of DPs.14,23,24 For example, Sb3+, Sn2+, Cu+ and Fe3+ doping of Cs2AgBiBr6 DP materials has been used to enhance their light absorption capability, resulting in a broadband absorption range from the visible to the NIR region.25–28 Similarly, Fe3+ has been incorporated into Cs2AgInCl6 DPs, demonstrating extended absorption near ≈800 nm.29,30 Nevertheless, the majority of doping strategies aimed at enhancing light absorption have been predominantly focused on single crystals or microcrystals of DPs. Consequently, the optoelectronic and photocatalytic applications of doped/alloyed DPs have been hindered due to their limited solubility in common organic solvents, which is highly desirable for thin-film processing via the spin/dip coating method and to obtain stable colloidal dispersions for photocatalysis. In this regard, the colloidal synthesis of doped/alloyed DP nanocrystals (NCs) is an attractive strategy to overcome these limitations and make them available for solar-energy conversion technologies.13,19,31
In the case of DP NCs, however, there remains a necessity to implement doping strategies for the purpose of fine-tuning their electronic and optical properties. For instance, diverse transition metals and lanthanide ions have been introduced into Cs2MInCl6 (M = Ag, Na) chloride-based DP NCs to improve their PLQYs, with the aim of utilizing them in light emitting diodes.31–38 Another challenging aspect of compositional engineering for DP NCs involves the precise control over the doping concentration. Ning et al. used a Bi![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe molar ratio of 3:2 to achieve only 1.4% Fe doping in Cs2AgBiBr6 single crystals.39 In another report by Liu et al., approximately 1% of Fe ions w.r.t. Bi ions were successfully incorporated into the Cs2AgBiBr6 lattice, when equimolar ratio of Fe and Bi precursors were used during synthesis.27 Han et al. emphasized the formidable challenge associated with synthesizing Fe-based DP NCs using a colloidal route, an attempt that ultimately yielded unsuccessful results.40 The formation of the desired composition and phase of DP NCs requires precise control on growth kinetics. In other words, the fast reaction kinetics along with balanced reactivity of precursors during nucleation and growth is critical to achieve the desired composition of DP NCs.36,41 In this context, the utilization of a highly reactive halide source could be advantageous to initiate the prompt nucleation and growth processes that may result in the desired chemical composition and optoelectronic properties.
:Fe molar ratio of 3:2 to achieve only 1.4% Fe doping in Cs2AgBiBr6 single crystals.39 In another report by Liu et al., approximately 1% of Fe ions w.r.t. Bi ions were successfully incorporated into the Cs2AgBiBr6 lattice, when equimolar ratio of Fe and Bi precursors were used during synthesis.27 Han et al. emphasized the formidable challenge associated with synthesizing Fe-based DP NCs using a colloidal route, an attempt that ultimately yielded unsuccessful results.40 The formation of the desired composition and phase of DP NCs requires precise control on growth kinetics. In other words, the fast reaction kinetics along with balanced reactivity of precursors during nucleation and growth is critical to achieve the desired composition of DP NCs.36,41 In this context, the utilization of a highly reactive halide source could be advantageous to initiate the prompt nucleation and growth processes that may result in the desired chemical composition and optoelectronic properties.
Herein, we introduce the colloidal synthesis of Fe3+-doped Cs2AgxNa1−xInCl6 (x = 0, 0.5 and 1) DP NCs, and demonstrate their application in photocatalytic CO2 reduction. Iron doping significantly extends the absorption edge of CsNaInCl6 NCs from 330 nm to 505 nm. Moreover, simultaneous doping by Fe3+ and Ag+ allows for significant reduction in the optical band gap and electronic band structure tuning of CsNaInCl6 NCs. Electron paramagnetic resonance (EPR) measurements reveal that iron is incorporated in the Fe3+ state, which apparently replaces In3+ sites in the host DP NCs. Importantly, the Fe3+-doped DP NCs tolerated polar solvents during the purification process, which is highly desirable for their application in thin-film devices and photocatalysis. The purified NCs exhibited high colloidal and structural stability under ambient conditions due to efficient surface passivation with dual cationic ligands. The Fe- and Ag- co-doped Cs2NaInCl6 DP NCs are explored for photocatalytic CO2 reduction and demonstrate selective reduction of CO2 into CH4 due to favourable energetic band edge positions. We employed density functional theory (DFT) calculations to substantiate that by co-doping Cs2NaInCl6 DPs with iron and silver, we can significantly enhance their light absorption, enhance charge separation efficiency, and adjust the reduction potentials. Furthermore, this compositional modification boosts CO2 adsorption and activation, so these synergistic improvements are crucial for the selective reduction of CO2 to methane (CH4). Our findings introduce a new approach for the design and synthesis of highly stable lead-free DP NCs that can be processed and employed in a variety of optoelectronic applications, including photocatalysis.
Experimental section
Materials
Cesium carbonate (99.99%), silver nitrate (99.9%), indium(III) acetate (99.99%), iron(III) acetylacetonate (99.99%), 1-octadecene (ODE, 90%), oleic acid (OA, 90%), oleylamine (OLA, 90%), trioctylphosphine (TOP, 90%), benzoyl chloride (98%), and methyl acetate (99.5%) were purchased from Sigma Aldrich; sodium acetate (99%) was purchased from Alfa Aesar. All chemicals were used without any further purification.Synthesis of Cs2AgxNa1−xInCl6 (x = 0, 0.5 and 1) NCs
In a typical synthesis of Cs2NaInCl6 NCs, cesium carbonate (0.3 mmol), sodium acetate (0.3 mmol), indium(III) acetate (0.3 mmol), ODE (10 mL), OA (1 mL), and OLA (0.5 mL) were loaded in a four-neck flask under a N2 atmosphere. The reaction mixture was heated to 110 °C under continuous stirring for 20 min. At this stage all precursors were fully dissolved and the solution became optically clear, then 0.3 mL TOP was injected into the hot reaction mixture and the solution was kept for another 10 min under N2 protection. Next, the temperature of the solution was increased to 140 °C and 300 μL benzoyl chloride was swiftly injected into the hot reaction mixture which initiated a prompt nucleation and growth process, indicating the formation of Cs2NaInCl6 NCs. After 5 s, the reaction mixture was rapidly cooled down in an ice-water bath.Cs2Ag0.5Na0.5InCl6 alloyed NCs were synthesized by a similar procedure as mentioned above, with slight modifications. In brief, 0.3 mmol of cesium carbonate, 0.3 mmol indium(III) acetate, 0.15 mmol sodium acetate, and 10 mL of ODE together with 1 mL OA and 0.5 mL OLA were heated to 110 °C under vigorous stirring in a N2 atmosphere. Once the reaction solution became clear and transparent, 0.3 mL TOP followed by premixed Ag–TOP solution (0.15 mmol AgNO3 in 0.15 mL TOP) was sequentially injected into the hot reaction mixture and the solution was kept for another 10 min under N2 protection. For the synthesis of Cs2AgInCl6 NCs, sodium acetate was completely replaced by Ag–TOP solution (0.3 mmol AgNO3 in 0.3 mL TOP). Finally, the reaction flask was heated to 140 °C and 300 μL of neat benzoyl chloride was swiftly injected into the hot reaction mixture which resulted in prompt nucleation and growth of NCs. After 5 s, the reaction flask was rapidly cooled down in an ice-water bath.
Synthesis of Fe3+-doped Cs2AgxNa1−xInCl6 (x = 0, 0.5 and 1) NCs
Fe3+-doped Cs2AgxNa1−xInCl6 (x = 0, 0.5 and 1) NCs were synthesized by the same procedure mentioned above by the substitution of indium(III) acetate with iron(III) acetylacetonate, while amounts of the other precursors and synthesis conditions remained the same. In the precursor solution, 25% and 50% iron(III) acetylacetonate substitute an equimolar amount of indium(III) acetate, while the total amount of iron(III) acetylacetonate and indium(III) acetate was fixed to 0.3 mmol. Upon benzoyl chloride injection, the color of reaction mixture was rapidly changed from dark red to yellow or yellow-orange or orange-red color depending upon the Na/Ag composition, indicating the formation of Fe3+-doped Cs2AgxNa1−xInCl6 (x = 0, 0.5 and 1) NCs.Photocatalytic CO2 reduction experiments
The CO2 reduction experiments were performed in a sealed Pyrex bottle loaded with an ethyl acetate/isopropanol mixture. In a typical reaction, 3 mg of extensively washed and dried NC powder were dispersed in a solvent mixture of 24 mL ethyl acetate and 1 mL of isopropanol. The reactor was purged with CO2 for about 30 min in order to eliminate air and to form a saturated CO2 atmosphere. The photocatalytic reaction was initiated by illuminating the reactor with a light source of intensity 150 mW cm−2 using a solar simulator equipped with an AM 1.5G filter. The photocatalytic reaction was performed for 6 h at room temperature. The photocatalytic reaction products were quantified through headspace gas analysis by gas chromatography with a flame ionization detector.Results and discussion
Synthesis and purification of iron and silver co-doped double perovskite nanocrystals
Fe3+-doped Cs2AgxNa1−xInCl6 (x = 0, 0.5 and 1) DP NCs were synthesized based on a previously developed colloidal hot injection approach with slight modifications (see the schematic illustration of the synthesis procedure in Scheme 1).35,36 This approach relies on the use of strongly coordinated silver–trioctylphosphine (Ag–TOP) and benzoyl chloride as the alternative silver and chloride source, respectively, along with an additional TOP ligand. The Ag–TOP precursor is a better choice to prevent the reduction of silver ions by oleylamine (OLA) into metallic silver.35 On the other hand, benzoyl chloride is well known for strong reactivity toward nucleophilic compounds (amines and carboxylic acids) in which the reactive CO–Cl bonds can interact with amines and/or carboxylic acids and release HCl. Moreover, TOP can facilitate the nucleophilic reaction with benzoyl chloride or HCl that results in the formation of phosphonium chloride intermediates which can serve as both halide sources and surface capping ligands.36 In a typical synthesis of Fe3+-doped Cs2NaInCl6 DP NCs, cesium carbonate, sodium acetate, indium(III) acetate, and iron(III) acetylacetonate along with oleic acid (OA), OLA and 1-octadecene (ODE) were mixed and heated at 110 °C under N2 protection for 20 min so that the precursors were completely dissolved (Scheme 1a). After that, 300 μL of TOP was injected into the hot reaction solution, and the solution was kept for another 10 min. Subsequently, the temperature of the reaction mixture was increased to 140 °C (Scheme 1b). Finally, 300 μL of neat benzoyl chloride was injected into the hot reaction solution and the color of precursor solution was rapidly changed from dark red to yellow indicating the prompt nucleation and growth of the DP NCs (Scheme 1c). After 5 s, the mixture was rapidly immersed in an ice-cooled water bath to quench the reaction. The Fe3+-doped Cs2Ag0.5Na0.5InCl6 and Cs2AgInCl6DP NCs were synthesized following the same approach, by simply replacing the sodium precursor with a silver precursor. The detailed description of the synthetic procedures and the purification method are provided in the Experimental section and Fig. S1 (ESI†). The following samples were prepared: Fe3+-doped Cs2NaInCl6, as well as Fe3+ and Ag+ co-doped DP denoted as Fe3+-doped Cs2Ag0.5Na0.5InCl6 and Fe3+-doped Cs2AgInCl6 DP NCs, respectively. They were labelled as Cs2NaInCl6: y% Fe, Cs2Ag0.5Na0.5InCl6: y% Fe, and Cs2AgInCl6: y% Fe, where y% (25% and 50%) is the molar ratio of the iron precursor relative to that of the indium precursor used during synthesis. It is important to note that the 25% and 50% are nominal amounts of Fe, which are very different from the actual amount of the incorporated Fe3+-dopant, as will be discussed later on.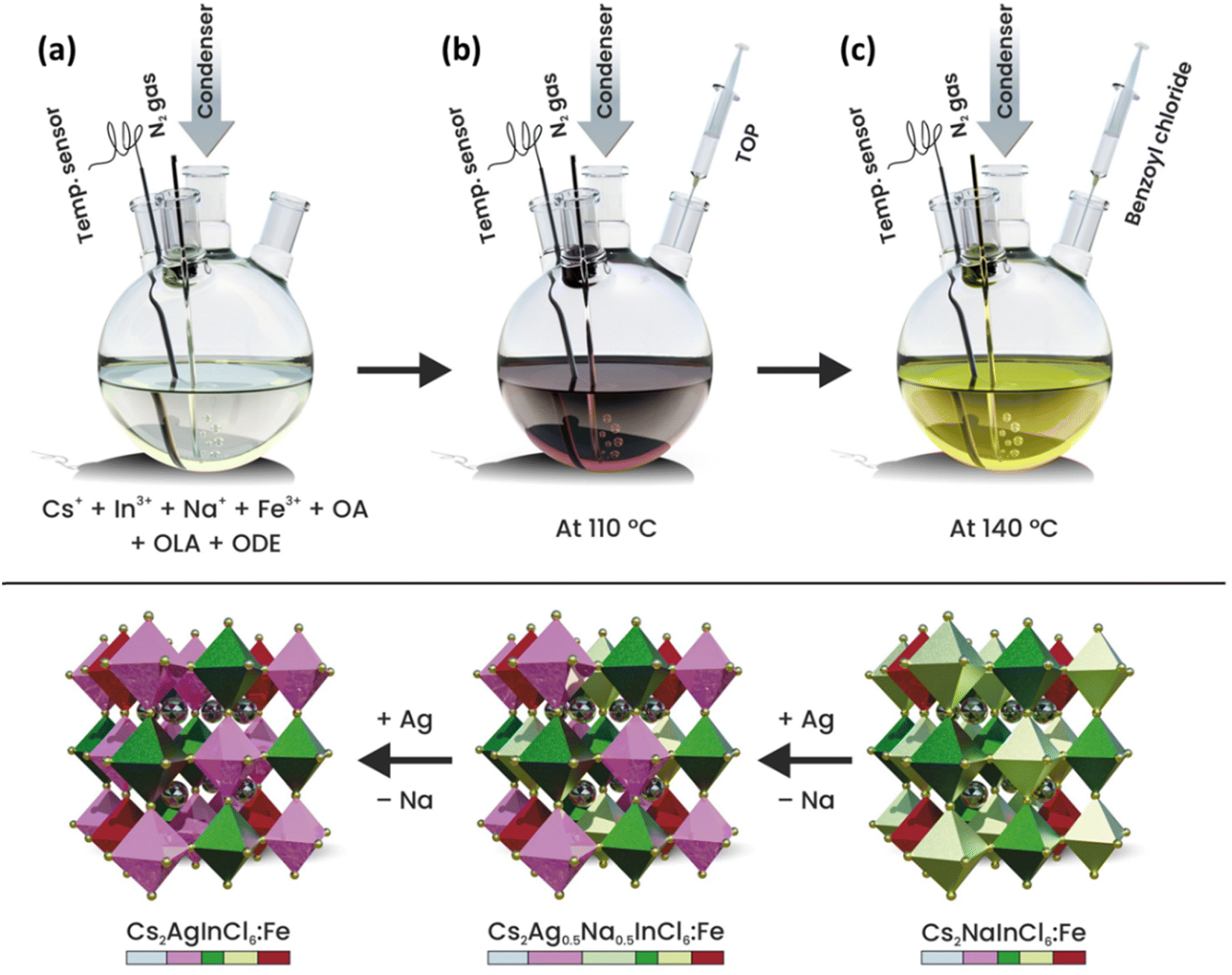 | ||
| Scheme 1 Schematic illustration of the colloidal synthesis of Fe3+-doped Cs2AgxNa1−xInCl6 (x = 0, 0.5 and 1) DP nanocrystals. | ||
Crystal structure, morphology, and elemental composition
The crystal structure and phase purity of undoped, Fe3+-doped, and Fe3+/Ag+ co-doped DP NCs were investigated by X-ray diffraction (XRD) measurements (Fig. 1a). All the observed diffraction peaks in both Fe3+-doped and undoped samples match well with the corresponding bulk reflections of Cs2NaInCl6 and do not contain any impurities. Upon closer examination of the (044) diffraction peak (right panel in Fig. 1a), a noticeable shift towards a higher diffraction angle is observed after the incorporation of Fe3+ ions due to the lattice shrinkage. The observed lattice shrinkage can be attributed to the smaller ionic radius of Fe3+ (0.64 Å) compared to In3+ (0.81 Å), providing clear evidence of the successful incorporation of Fe3+ ions into the host DP structure.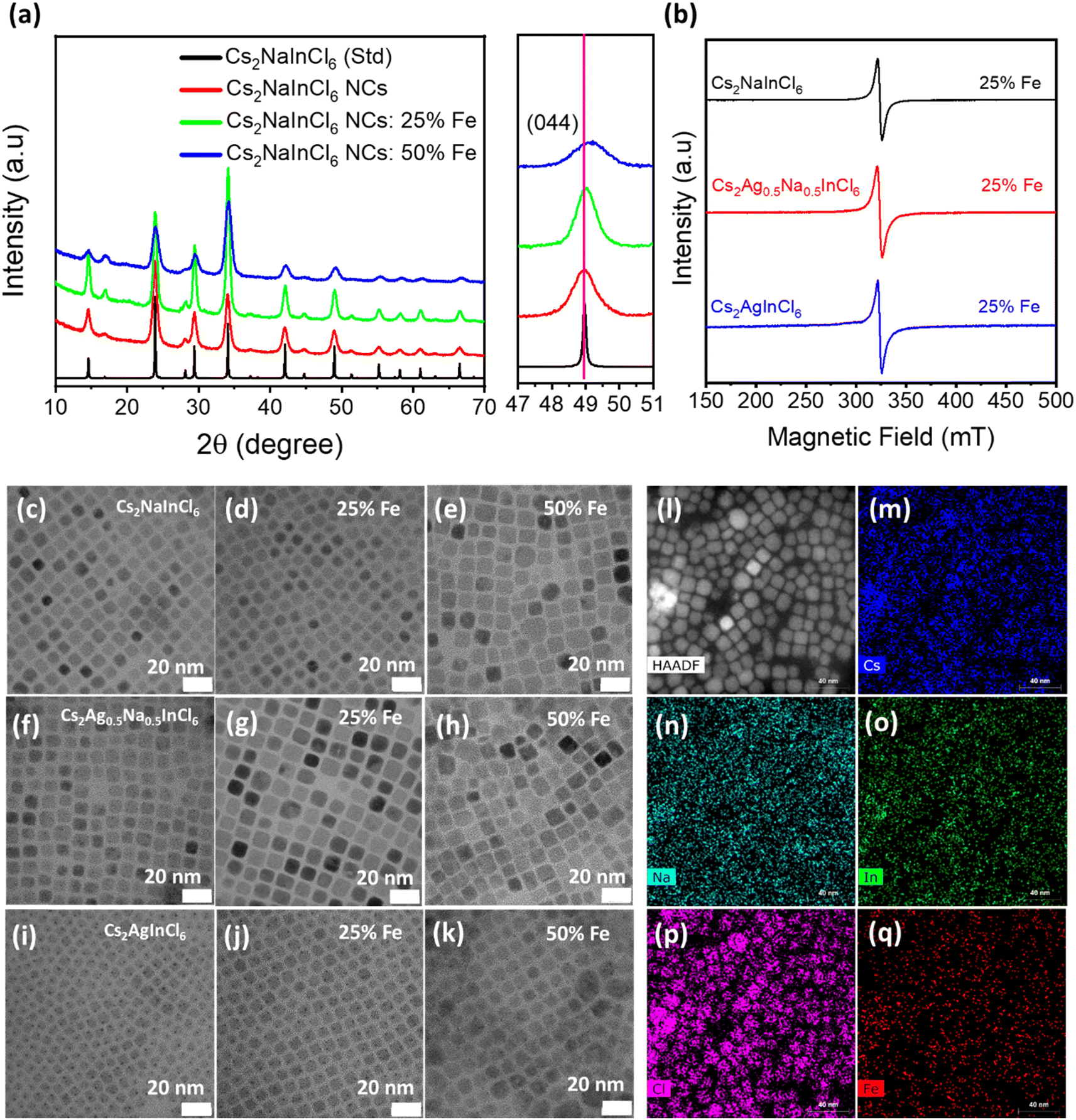 | ||
| Fig. 1 (a) XRD patterns of undoped and Fe3+-doped Cs2NaInCl6 NCs, with the corresponding reflections of bulk Cs2NaInCl6 (labeled as Std). The right panel shows an enlarged view of the (044) diffraction peaks of the DP samples. (b) EPR signals of Fe3+-doped Cs2AgxNa1−xInCl6 (x = 0, 0.5 and 1) NCs with a nominal 25% Fe amount, measured at room temperature. (c–k) TEM images of Fe3+-doped Cs2AgxNa1−xInCl6 (x = 0, 0.5 and 1) NCs. (l) High-angle annular dark-field (HAADF) image of Fe3+-doped Cs2NaInCl6 NCs with a nominal iron concentration of 50% and (m–q) STEM-EDS elemental mapping of Cs, Na, In, Cl, and Fe elements which show their homogeneous distribution within the cubic NCs. | ||
Regarding Fe3+/Ag+ co-doped samples, XRD patterns of Fe3+-doped Cs2Ag0.5Na0.5InCl6 (Fig. S2†) NCs show a similar trend and do not contain any impurity peaks, confirming the preservation of the cubic DP structure. The only exception is observed for 50% Fe-doped Cs2AgInCl6 NCs (Fig. S3†), where the synthesis does not yield pure phase NCs. This can be attributed to the distinct growth kinetics and some saturation limits in terms of the actual concentration of the iron ions doped into the host DP structure (as will also be discussed later).
The actual amount of the Fe3+-dopant in Cs2Ag1−xNaxInCl6 (x = 0, 0.5, 1) NCs was retrieved by inductively coupled plasma mass spectrometry (ICP-MS) measurements. The ICP-MS results (Tables S1–S3†) reveal that the Fe amount present in the host DP significantly varies for different DP systems. The Cs2NaInCl6 NCs accommodated a maximum concentration of Fe while the lowest amount of Fe can be incorporated into Cs2AgInCl6, under similar reaction conditions. In particular, for a nominal 25% Fe feeding during the synthesis, the actual atomic concentrations of the Fe dopant (related to In) in Cs2NaInCl6, Cs2Ag0.5Na0.5InCl6, and Cs2AgInCl6 are 8.1%, 3.4% and 0.8%, respectively. Notably, the actual contents of Na in the Cs2Ag0.5Na0.5InCl6 DP NCs (Table S2†) are in close agreement with the amount of Na precursor that was used in the reaction.
Next, EPR measurements were performed at room temperature to gain insight on the spin state of iron in the host NCs. EPR spectra (Fig. 1b) of Fe3+-doped (25% feeding concentration) Cs2AgxNa1−xInCl6 (x = 0, 0.5 and 1) NCs show a strong resonance signal from all samples located at g ∼ 2.03, ascribed to paramagnetic Fe3+ ions.39 In contrast, EPR spectra (Fig. S4†) of undoped Cs2NaInCl6 NCs do not show any resonance signal in the probed range of magnetic fields. Thus, the absence of any resonance signal in the EPR spectrum of undoped Cs2NaInCl6 NCs clearly validates that the strong resonance signal in all Fe3+-doped systems indeed originated from the presence of paramagnetic Fe3+ ions.
X-ray photoelectron spectroscopy (XPS) was performed to study the chemical composition of DP NCs. Full range survey and high-resolution XPS spectra for Cs 3d, Ag 3d, Na 1s, In 4d, Cl 3p and Fe 2p of iron doped (25% of Fe feeding with respect to In, which was hereafter used as the benchmark for all the following key measurements) Cs2Ag1−xNaxInCl6 (x = 0, 0.5, 1) NCs are shown in Fig. S5a–c and S6a–f,† respectively. The XPS spectra of Fe3+-doped Cs2NaInCl6 NCs confirm the presence of all constituent elements in the expected oxidation states. However, in the XPS spectrum of Fe3+-doped Cs2Ag0.5Na0.5InCl6 and Cs2AgInCl6, the presence of iron could not be verified, which may be due to the low concentration of Fe at the surface of the NC film which may be beyond the detection limit of XPS as previously reported by other research groups.27,30,39
The shape and size of the undoped and Fe3+-doped Cs2AgxNa1−xInCl6 (x = 0, 0.5 and 1) NCs were investigated by transmission electron microscopy (TEM). The TEM images of undoped (Fig. 1c, f and i) and 25% Fe3+ doped Cs2AgxNa1−xInCl6 (x = 0, 0.5 and 1) NCs are presented in Fig. 1d, g and j. These images confirm the formation of uniform size NCs with a well-defined cubic morphology, and an average edge size of 8–12 nm. Notably, the cubic-shape morphology of doped NCs is still retained even for 50% Fe3+ doping. However, the size distribution becomes broader and, in the case of Cs2AgInCl6 NCs, larger aggregates of the NCs are clearly visible (Fig. 1e, h and k). The morphology and elemental analysis of Cs2NaIn0.5Fe0.5Cl6 NCs were also investigated by high-angle annular dark-field (HAADF) scanning TEM (STEM) imaging and STEM-energy dispersive X-ray spectroscopy (EDS) elemental mapping. The HAADF-STEM image (Fig. 1l) further confirms the cubic shape of Cs2NaIn0.5Fe0.5Cl6 NCs, while STEM-EDS elemental mapping (Fig. 1m–q) demonstrates a homogeneous distribution of Cs, Na, In, Cl and Fe elements across the Cs2NaIn0.5Fe0.5Cl6 NCs. Additionally, the high-resolution TEM (HRTEM) image and the corresponding fast Fourier transform (FFT) (inset) of a single Cs2NaInCl6 NC (Fig. S7a†) reveal high crystallinity with a lattice spacing of 0.372 nm, corresponding to the (220) crystal plane. In the Fe3+-doped Cs2NaInCl6 NCs, the lattice spacing of the (220) plane slightly decreases to 0.370 nm and 0.367 nm for 25% and 50% Fe3+-doped Cs2NaInCl6 NCs (Fig. S7a–c†), respectively. This is ascribed to the substitution of larger In3+ by smaller Fe3+ ions. The decrease in the lattice spacing is in agreement with the shift of the diffraction peak towards a higher diffraction angle in the XRD patterns (Fig. 1a). The decrease in the lattice spacing of Fe3+-doped Cs2NaInCl6 NCs further corroborates the successful incorporation of Fe3+ ions. Similar trends were also observed in Fe3+-doped Cs2Ag0.5Na0.5InCl6 NCs (Fig. S7d–f†).
Optical properties of undoped and Fe3+-doped Cs2AgxNa1−xInCl6 (x = 0, 0.5 and 1) NCs
To explore the effect of iron and silver co-doping on the optical properties of Cs2NaInCl6 DP NCs, we performed UV-vis absorbance, steady-state PL, and time-resolved PL measurements on both undoped and Fe-doped Cs2AgxNa1−xInCl6 (x = 0, 0.5 and 1) NCs. As shown in Fig. 2a and e, both Cs2NaInCl6 and Cs2AgInCl6 NCs exhibit featureless absorption with the onset near 330 and 350 nm, respectively, in agreement with previous reports.35,36 In contrast to this, Cs2Ag0.5Na0.5InCl6 NCs show a clear excitonic absorption peak (Fig. 2c) at 330 nm which is attributed to the breaking of the parity-forbidden transition upon partial Na substitution with Ag.14 Interestingly, Fe3+-doped Cs2NaInCl6 NCs (Fig. 2a) display a dramatic shift of the optical band edge reaching towards 505 nm, and consequently the colour of their colloidal solution (inset of Fig. 2a) turns from colourless to yellow. Tauc plots (Fig. 2b) show that the optical band gap decreased from 4.29 eV for the undoped Cs2NaInCl6 NCs to 2.88 eV for the 25% Fe doped Cs2NaInCl6 NCs, in agreement with recently reported bulk and single crystal Fe3+-doped DP systems.27,30 On the other hand, Fe3+-doped Cs2AgInCl6 NCs (Fig. 2e) exhibit an obvious red-shift in the absorption edge, while in the case of Fe3+-doped Cs2Ag0.5Na0.5InCl6 NCs (Fig. 2c) the absorption edge lies in between that of Fe3+-doped Cs2NaInCl6 NCs and Fe3+-doped Cs2AgInCl6 NCs, respectively. According to Tauc plots of Cs2AgInCl6 NCs and Cs2Ag0.5Na0.5InCl6 NCs, the band gap is reduced from 4.1 to 2.7 eV and from 4.17 to 2.84 eV upon Fe3+-doping (Fig. 2f and d), respectively. We note that for all samples with nominal 50% Fe doping, the absorption intensity (Fig. 2a, c and e) increases, while the absorption onset and the band gap (Fig. S8a–c†) remain unchanged.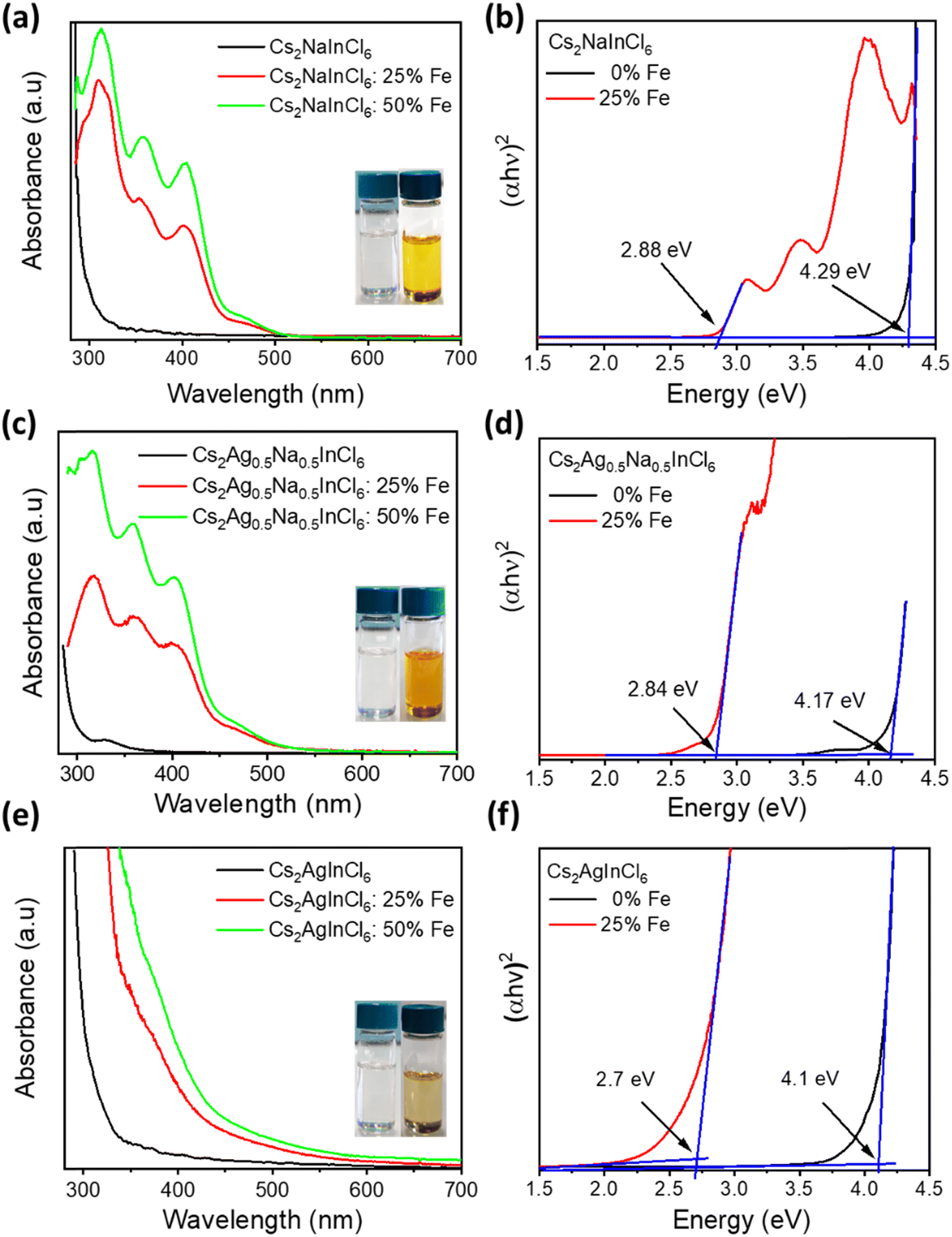 | ||
| Fig. 2 UV-vis absorption spectra of undoped and Fe3+-doped (a) Cs2NaInCl6, (c) Cs2Ag0.5Na0.5InCl6, and (e) Cs2AgInCl6 NCs, respectively. (Inset) Photographs of the undoped sample (left) and the Fe3+-doped sample (right) with a nominal iron concentration of 25%. Tauc plots of undoped and Fe3+-doped samples with a nominal iron concentration of 25%. (b) Cs2NaInCl6, (d) Cs2Ag0.5Na0.5InCl6, and (f) Cs2AgInCl6 NCs, respectively. | ||
Fig. S9a† shows steady state PL spectra of undoped Cs2AgxNa1−xInCl6 (x = 0, 0.5 and 1) NCs with different x values. The PL spectra of Cs2NaInCl6 NCs display a narrow emission band centered at 427 nm, while Cs2AgInCl6 NCs exhibit broadband emission with the PL maximum located at 580 nm. The PL signal of Cs2NaInCl6 NCs originates from defect related states, while the broadband emission from Cs2AgInCl6 NCs can be assigned to the emission via recombination of self-trapped excitons (STEs).35,36 Both NCs and bulk DP materials commonly display a very week PL signal due to the presence of parity-forbidden transitions.14 In the case of Cs2Ag0.5Na0.5InCl6 NCs, the intensity of the broadband emission is significantly enhanced compared to that of Cs2AgInCl6 NCs due to the breaking of parity-forbidden transitions upon partial Na alloying.14 Upon nominal 25% Fe3+ doping, the PL spectra of Cs2AgxNa1−xInCl6 (x = 0, 0.5 and 1) NCs exhibited distinct features compared to those of the respective undoped or exclusively Ag-alloyed counterparts. The PL spectra (Fig. S10a†) of Fe3+-doped Cs2NaInCl6, Cs2Ag0.5Na0.5InCl6, and Cs2AgInCl6 NC solutions exhibit PL maxima at 455, 468, and 503 nm, respectively. It can be seen that the broadband features of STE emission from undoped samples have disappeared upon Fe doping, and a relatively sharp PL peak emerged in all samples. These results indicate that the observed PL signals from the Fe-doped samples do not originate from STEs but rather may result from recombination of free-excitons via sub-band gap states.30
Next, time-resolved PL (TRPL) measurements were carried out in order to understand the charge recombination dynamics of Fe-doped Cs2AgxNa1−xInCl6 (x = 0, 0.5 and 1) NCs using the time correlated single photon counting technique. The TRPL spectra and their fitted kinetic parameters for all undoped and Fe-doped Cs2AgxNa1−xInCl6 (x = 0, 0.5 and 1) NCs are presented in Fig. S9b, c, S10b and Tables S4, S5,† respectively. The TRPL spectra of Cs2NaInCl6 NCs (Fig. S9c†) reveal an average PL lifetime of 6.4 ns. On the other hand, both Cs2AgInCl6 and Cs2Ag0.5Na0.5InCl6 NCs (Fig. S9b†) exhibit a much longer PL lifetime of about 2.9 μs and 4.2 μs, respectively, as expected for STE emission.14,35 The increase in PL intensity and PL lifetime of Cs2Ag0.5Na0.5InCl6 NCs can indeed be attributed to the improvement in radiative recombination of STEs enabled by breaking of parity-forbidden transitions upon Na-alloying into Cs2AgInCl6 NCs. Next, we analyzed the PL decay profiles of all Fe3+-doped NCs (see extracted parameters in Table S5†), and found out average PL lifetimes of 6.2, 6.8, and 4.7 ns for Fe3+-doped Cs2NaInCl6, Cs2Ag0.5Na0.5InCl6, and Cs2AgInCl6 NCs, respectively. All the Fe3+-doped NCs exhibit fast recombination carrier dynamics relative to undoped samples, with nanosecond PL lifetimes, most likely because the STEs can effectively dissociate to free electrons and holes. These results suggest that iron and silver co-doping is advantageous in order to manipulate the optoelectronic properties of DP NCs for use as photoactive materials in solar cells or photocatalysis where specific energies of band levels as well as charge separation are required.
Analysis of the optical properties of iron and silver co-doped DPs by DFT calculations
To explain the optical properties of Fe-doped DP NCs, we used DFT calculations with the implementation of the Heyd, Scuseria, and Ernzerhof (HSE03) hybrid functional,42,43 coupled with optical-transition calculations obtained by solving the Bethe–Salpeter equation (BSE)44–46 (see Table S8 in the ESI†). The calculated optical band gap of 4.43 eV for pristine Cs8Na4In4Cl24 agrees well with the experimental data (4.29 eV). Fe-doping reduces the band gap to approximately 2.85–3.12 eV depending on the Fe3+ electronic configuration, but it remains insensitive to the Fe3+ feeding amount in the range of 25–50%. The degree of band gap reduction caused by Fe doping is influenced by the geometry and spin state of Fe. Fe3+ ions in a 3d5 valence configuration in an octahedral ligand field have either a high spin or a low spin state, while the actual spin state depends on the relative strengths of crystal field splitting and electron–electron repulsion. The high-spin isomer of Fe3+ (t32ge2g) in the DP-NCs leads to better agreement with the experimental values (Table S8†). Fe3+ doping creates intra-gap states, which can occur in either both spin channels (in the low-spin configuration) or in the minority spin channel (in the high-spin configuration), as illustrated in Fig. S22.† The band structure shown in Fig. S23† reveals that these states are strongly localized on Fe atoms and remained nearly independent of the Fe doping amount. Experimental measurements have shown that substituting Ag for Na in pristine Cs8Na4In4Cl24 does not lead to a significant reduction of the band gap, which varies from 4.29 eV (undoped system) to 4.17 and 4.10 eV for Cs2Ag0.5Na0.5InCl6 and Cs2AgInCl6, respectively (Fig. 2b, d and f). A similar trend is observed in the DFT calculations, which suggests the strong impact of Ag alloying on the electronic structure of Cs8Na4In4Cl24 (Fig. S24 and S25†). As the degree of Ag incorporation increases, the electronic band gap narrows in a similar manner to the optical band gaps, resulting from the upshift/downshift of the valence/conduction band edges with each additional Ag+ ion. Ag+ ions, similar to Fe3+ ions, are situated within the octahedral ligand field of the six Cl atoms, which causes Ag+ 4d orbital splitting into two energy levels: the lower energy t2g and the higher-lying eg. However, the states introduced by the Ag+ dopant are unlikely to contribute to light absorption near the absorption edge, because the optical transition edge is located directly below the hybridized states of Ag and Cl. In contrast, the introduction of Fe3+ into Cs2AgInCl6 and Cs2Ag0.5Na0.5InCl6 significantly decreases the band gap to 2.93–3.28 eV, which is consistent with the experimental observations (Fig. 2). Furthermore, electronic structure calculations (Fig. 3) indicated that Ag doping results in an upshift of the valence band edge (VBE), primarily composed of Cl and Ag states (or Cl in the case of Cs2NaInCl6), as well as the conduction band edge (CBE), whose orbital composition depends on the spin channel. Specifically, for the spin-down channel, the CBE is formed by electronic states of Cl, In, and Ag, while for the spin-up channel, it is formed by Fe and less significantly by Cl. In Cs2NaInCl6, the VBE is contributed to by Cl and Fe in the spin-down channel, and by Cl in the spin-up channel, while the CBE is formed by Fe and Cl/Fe, respectively.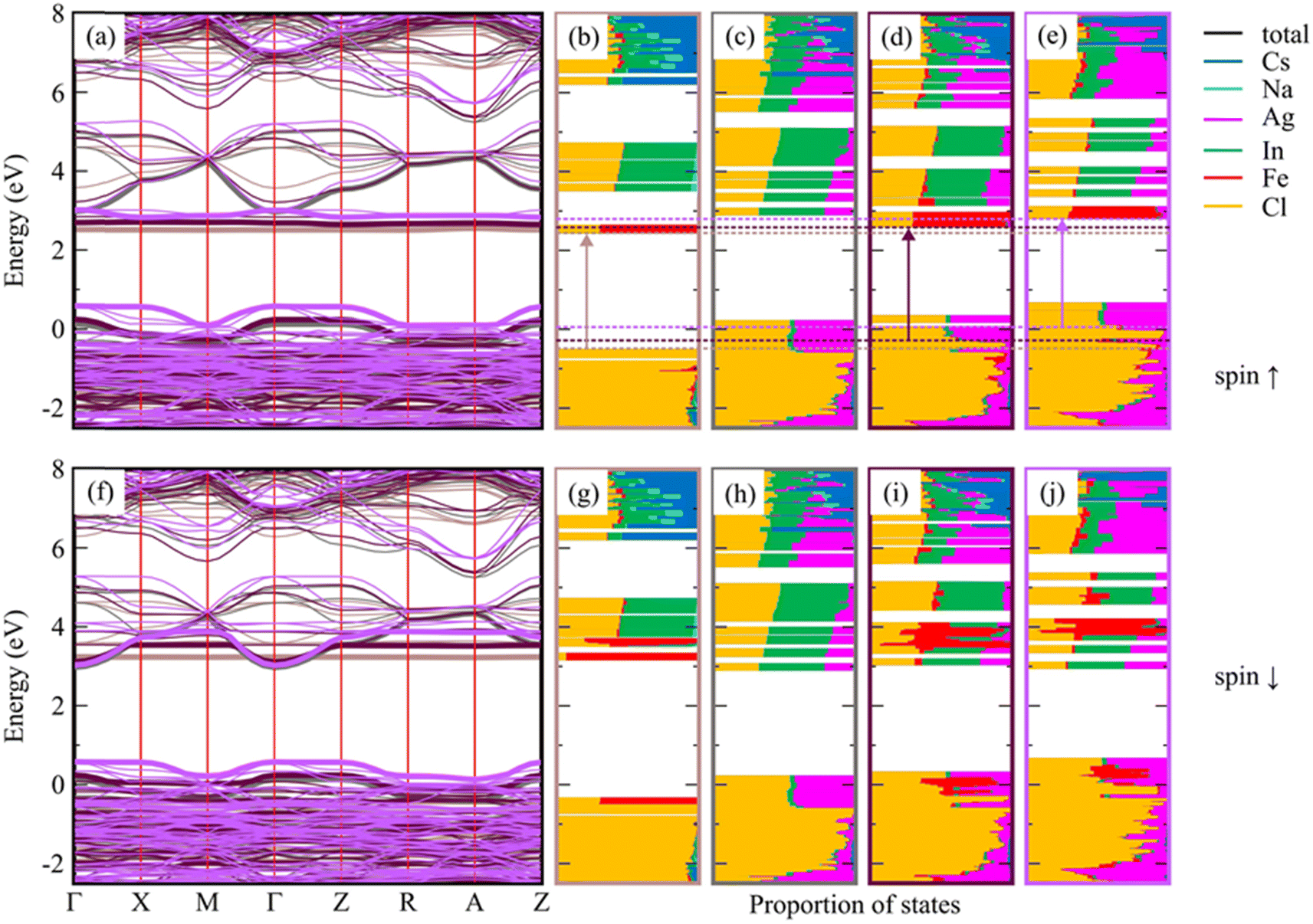 | ||
| Fig. 3 Spin-resolved electronic band structure of optimized low-spin Cs8Na4In3Fe1Cl24 with a constant In/Fe ratio, and the effect of Ag-doping on this system (a and f). The relaxed reference system without Fe, Cs8Na2Ag2In4Fe0Cl24, is also included for comparison. The colors in panels (a and f) conform to the bounding boxes of the corresponding DOS plots shown in (b–e and g–j), where the valence and conduction band edges are marked with bold lines. The DOS plots (see also Fig. S26†) of Cs8Na4Ag0In3Fe1Cl24 (b and g), Cs8Na2Ag2In4Fe0Cl24 (c and h), Cs8Na2Ag2In3Fe1Cl24 (d and i), and Cs8Na0Ag4In3Fe1Cl24 (e and j) are shown, and the transitions (corresponding to the optical band gap) are marked by arrows. The edges of these transitions are indicated by dashed horizontal lines. The electronic band structure and the DOS plots for the high-spin configuration of the same systems are presented in Fig. S27.† | ||
The analysis of the band gap and the atom-resolved density of states (DOS) shown in Fig. 3 leads to the conclusion that the optical transitions do not occur from the VBEs but instead from the Cl states located directly below the hybridized Ag and Cl VBE states. This same conclusion can be drawn for the high-spin configurations of the same system (Fig. S27†). The transition leads to the d-orbitals of Fe atoms, which is crucial for the photocatalysis of CO2 reduction as will be thoroughly discussed below.
Stability of Fe3+-doped Cs2AgxNa1−xInCl6 (x = 0, 0.5 and 1) NCs
Ensuring the long-term material stability of DP NCs is of utmost importance for their practical applications. We conducted a thorough examination of long-term colloidal, structural, and morphological stability of Fe3+-doped Cs2AgxNa1−xInCl6 (x = 0, 0.5 and 1) NCs. In our case, the extensively washed Fe3+ (with a nominal 25% Fe amount) doped Cs2AgxNa1−xInCl6 (x = 0, 0.5, and 1) NCs, displayed remarkable long-term colloidal, structural, and morphological stability when stored under ambient conditions. The photographs of both fresh and aged samples of Fe3+-doped (25% Fe) Cs2AgxNa1−xInCl6 (x = 0, 0.5, and 1) NCs, presented in the insets of Fig. S11a, S12a and S13a† show no signs of sedimentation; they appear identical even after 30 days of storage under ambient conditions. Furthermore, the absorption spectra (Fig. S11a, S12a and S13a†) of both fresh and aged Fe3+-doped Cs2AgxNa1−xInCl6 (x = 0, 0.5, and 1) NCs remain unchanged, indicating consistent optical properties over the storage period. The XRD patterns (Fig. S11b, S12b and S13b†) of the aged samples also exhibit minimal changes, attested to the sustained structural integrity of the NCs during storage under ambient conditions. Additionally, the TEM images (Fig. S11c, S12c and S13c†) of the aged Fe3+-doped Cs2AgxNa1−xInCl6 (x = 0, 0.5, and 1) NCs reveal their preserved cubic morphology, devoid of any signs of aggregation or degradation. This provides further evidence of their exceptional long-term structural stability. However, Fe3+-doped Cs2AgxNa1−xInCl6 (x = 0.5 and 1) NCs synthesized with a 50% Fe3+ loading exhibited relatively poor colloidal stability. Specifically, in the case of Fe3+-doped Cs2Ag0.5Na0.5InCl6 NCs, a small amount of yellow-orange precipitate was observed at the bottom of the vial after 30 days of storage under ambient conditions (inset of Fig. S12d†). Despite this sedimentation, the shape of the absorption spectra (Fig. S11d and S12d†), crystal structure (Fig. S11e and S12e†), and cubic morphology (Fig. S11f and S12f†) of both Fe3+-doped Cs2NaInCl6 and Cs2Ag0.5Na0.5InCl6 NCs, synthesized with a 50% Fe3+ loading, remained preserved. In contrast, Fe3+-doped Cs2AgInCl6 NCs synthesized with a 50% Fe3+ loading exhibited poor colloidal and structural stability, as evidenced by complete precipitation at the bottom of the vial within a few days of storage under ambient conditions (inset of Fig. S13d†). This instability in Fe3+-doped Cs2AgInCl6 NCs with a 50% nominal Fe3+ loading is also reflected in their irregular morphology (Fig. S13f†) along with the presence of impurity phases (Fig. S13e†), which likely resulted in poorly capped NCs. Overall, the colloidal and structural stability of Fe3+-doped Cs2AgxNa1−xInCl6 (x = 0.5 and 1) NCs varied depending on the Fe3+ loading, with the 50% nominal Fe3+ loading showing adverse effects on the stability of Cs2Ag0.5Na0.5InCl6 and Cs2AgInCl6 NCs, but not significantly affecting Cs2NaInCl6 NCs. These findings underscore the importance of carefully optimizing the doping concentration to achieve the desired stability and performance of these NCs in potential optoelectronic applications.Surface chemistry
Next, our investigation delved deeper into understanding the factors contributing to the remarkable stability observed in extensively washed Fe3+-doped DP NCs. It is widely recognized that the binding of capping ligands to the surface of colloidal NCs plays a pivotal role in achieving high-quality products with high colloidal and structural stability during the extraction and purification process. Given the uniform shape and size, as well as the colloidal and structural stability observed in both fresh and aged samples, we hypothesized that the superior quality and stability of the DP NCs could be attributed to efficient surface passivation through robust surface capping ligands. To research the surface chemistry and nature of ligands bound to the surface of iron-doped DP NCs, we conducted 1H and 31P nuclear magnetic resonance (NMR) measurements. Fig. S14a† displays the 1H NMR spectra of extensively washed Fe3+ (with a 25% nominal Fe3+ loading) doped Cs2Ag0.5Na0.5InCl6 NCs, showing several broad signals corresponding to bound ligands and characteristic sharp signals from the solvents used during the purification process and for dispersing the DP NCs. Importantly, the extensively washed NCs showed no sharp signals from unwanted organic residues, such as unbound ligands and octadecene. However, the intense and broad signals at 5.53 ppm from alkene protons of OA and/or OLA remained preserved, suggesting that either OLA or OA, or both are strongly bound to the surface of the NCs. Furthermore, the NMR spectra revealed the presence of characteristic broad peaks of oleylammonium ions at 7.5 and 4.1 ppm, corresponding to the α-proton (–NH3+) and β-proton nearest to the amine group (CH2–N), respectively, which verifies the binding of OLA to the NC surface. Additionally, the presence of a broad peak at 2.2 ppm corresponding to the proton (1) of OA, which is unique for OA, further supports the binding of OA to the NC surface. Hence, the presence of broad peaks from alkene protons along with the unique peaks of both OLA and OA in extensively washed samples clearly demonstrates that both oleylammonium and oleate ligands are bound together to the surface of the NCs. According to our previous reports on TOP-based synthesis of DP NCs, the in situ generated phosphonium species, which can be formed by a nucleophilic reaction between TOP and benzoyl chloride, serve as highly reactive halide sources and surface capping ligands.35,36 To further investigate the possible binding of phosphonium cations to the surface of iron-doped DP NCs, we performed 31P NMR spectroscopy measurements. The 31P NMR spectra (Fig. S14b†) of extensively washed NCs exhibited a broad signal at 35.2 ppm, accompanied by a small peak at 10.2 ppm, which correspond to benzoyl trioctylphosphonium and trioctylphosphonium cations, respectively.36 Moreover, the broadened peaks at 35.2 ppm and 10.2 ppm in the 31P NMR spectra (Fig. S14b†), along with the broadened peak of aromatic protons from the benzoyl group at 8.2 ppm in the 1H NMR spectra (Fig. S14a†), suggest the binding of benzoyl trioctylphosphonium and trioctylphosphonium cations at the Cl sites of the iron-doped DP NCs. Thus, NMR results confirm that the extensively washed iron-doped DP NCs are effectively passivated by robust cationic ligands, such as oleylammonium and phosphonium species, together with oleate anions. This combined surface passivation approach contributes to the high colloidal and structural stability observed in extensively washed Fe3+-doped DP NCs.Photocatalytic CO2 reduction
The combination of a suitable band gap and exceptional material stability in Fe-doped Cs2AgInCl6, Cs2NaInCl6, and Cs2Ag0.5Na0.5InCl6 DP NCs has sparked our interest in exploring their potential for photocatalytic CO2 reduction. The reactions were carried out in a CO2 saturated ethyl acetate/isopropanol solvent mixture under simulated solar light illumination with an intensity of 150 mW cm−2 (AM 1.5G). Ethyl acetate was chosen as the reaction medium due to high solubility of CO2 and excellent stability of DP NCs in this solvent, while isopropanol served as a hole scavenger and a proton source.47,48 We started from the comparison of the photocatalytic activity of nominal 25% Fe-doped DP NCs. Notably, although the detected photocatalytic CO2 reduction products were CH4 and CO for all the tested photocatalysts, we registered a marked difference in both the product yield and selectivity (Fig. 4a). In particular, Fe3+-doped Cs2Ag0.5Na0.5InCl6 DP NCs offer the highest activity with a CH4 yield of 18.3 μmol g−1 and CO yield of 1.1 μmol g−1, with a remarkable selectivity toward CH4 formation. In addition, as compared to the recently reported photocatalysts based on lead-free perovskites for CO2 reduction (Table S6, ESI†), Fe3+-doped Cs2Ag0.5Na0.5InCl6 NCs show comparable performance to most of them towards solar-light-driven CO2 reduction to CH4. To validate the role of Fe3+-doped Cs2Ag0.5Na0.5InCl6 NCs in photocatalytic CO2 reduction into CH4 and CO products, three control experiments (Fig. S15a†) were conducted under different experimental conditions. The first and second control experiments were performed without light (in the dark) and without a catalyst, respectively, and no hydrocarbon products were detected, indicating that the presence of both light irradiation and a photocatalyst is essential for CO2 reduction. The third control experiment was conducted similar to the standard photocatalytic CO2 reduction but under an argon atmosphere instead of CO2. The result shows (Fig. S15a†) that an insignificant amount of products was detected, which could be originated from the partial photooxidation of ethyl acetate and/or residual organic ligands bound to the surface of NCs, which is in agreement with the previous studies reported for perovskite NC based CO2 reduction by several groups.13,48–53 Additionally, the products of photocatalytic CO2 reduction were further verified by an isotope labelling experiment. As shown in Fig. S15b,† following the photocatalytic reaction in a 13CO2 atmosphere, the mass spectra revealed a major signal of 13CH4 (m/z = 17) and a trace of 13CO (m/z = 29). These results affirm that the CH4 indeed originated from the photocatalytic reduction of CO2 over the photocatalyst. On the other hand, Fe3+-doped Cs2AgInCl6 and Cs2NaInCl6 NCs demonstrate much lower catalytic performance, resulting in a CH4 yield of 2.52 and 1.28 μmol g−1, respectively. Undoped Cs2Ag0.5Na0.5InCl6 NCs (Fig. 4b) produce only a trace amount of CH4 (0.61 μmol g−1) and CO (2.02 μmol g−1) products. The poor photocatalytic performance of the undoped sample is attributed to the large band gap (4.17 eV) and thus poor light absorption capabilities. In contrast, 50% nominal Fe3+-doped Cs2Ag0.5Na0.5InCl6 NCs were significantly active but lower (Fig. 4b) in comparison with the 25% nominal Fe3+-doped sample, probably due to their poor quality and stability (see stability discussion in the ESI†). Thus, our results demonstrate that Fe3+ doping not only improves the light absorption capability of DP NCs but also regulates their photocatalytic performance towards selective reduction of CO2 into CH4. Moreover, to investigate the stability of the Fe3+-doped Cs2Ag0.5Na0.5InCl6 NCs, a cyclic experiment was carried out under similar reaction conditions. As shown in Fig. S16,† no obvious decay of the product yield was observed after three cycles of repeated photocatalytic reaction. Remarkably, both the double perovskite crystal structure (Fig. S17a†) and the cubic morphology (Fig. S17b†) of Fe3+-doped Cs2Ag0.5Na0.5InCl6 NCs are preserved after the photocatalytic reaction, demonstrating superior material stability.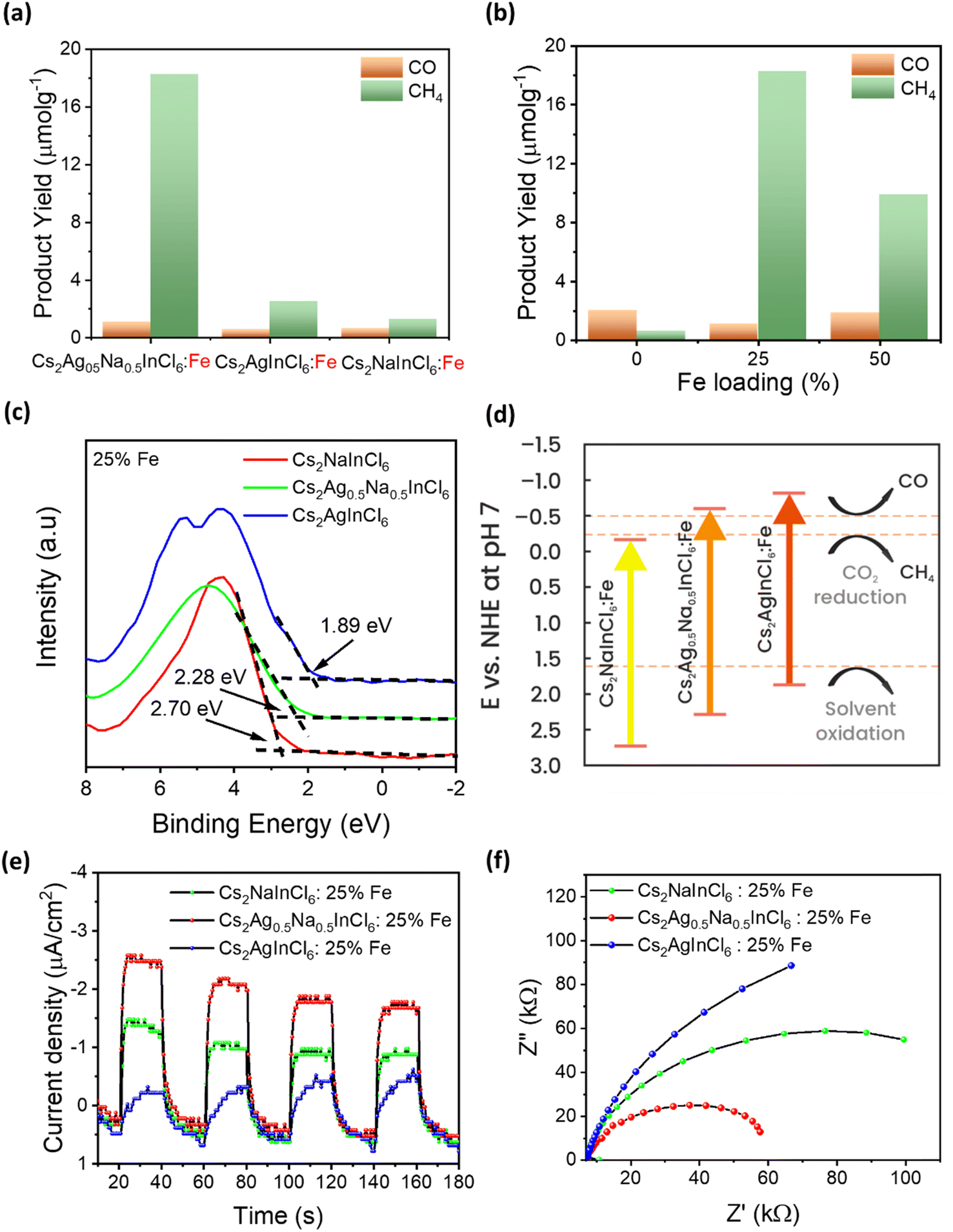 | ||
| Fig. 4 (a) Comparison of photocatalytic CO2 reduction performances after a 6 h reaction of Fe3+-doped Cs2Ag0.5Na0.5InCl6, Cs2AgInCl6, and Cs2NaInCl6 NCs with a nominal iron concentration of 25%. (b) Product yield of the Cs2Ag0.5Na0.5InCl6 NCs with various nominal amounts of Fe3+ (with respect to In) from photocatalytic CO2 reduction after 6 h of the reaction. (c) Valence band XPS spectrum and (d) schematic illustration of the energy band structures of Fe3+-doped Cs2NaInCl6, Cs2Ag0.5Na0.5InCl6, and Cs2AgInCl6 NCs with a nominal iron concentration of 25%, respectively. The energy band structures were constructed on the basis of UV-vis and VB XPS results. (e) Transient photocurrent response recorded at −0.4 VAg/AgCl under chopped AM 1.5G illumination. (f) Nyquist plots obtained under illumination with a light intensity of 150 mW cm−2 at a bias of −0.4 VAg/AgCl. | ||
To explore the reasons for the different performances of photocatalytic CO2 reduction, we analysed the energy band alignment and band gap of Fe-doped Cs2NaInCl6, Cs2Ag0.5Na0.5InCl6, and Cs2AgInCl6 NCs. The XPS valence band (VB) edges versus Fermi level of nominal 25% Fe3+-doped Cs2NaInCl6, Cs2Ag0.5Na0.5InCl6, and Cs2AgInCl6 NCs is located at 2.70, 2.28, and 1.89 eV, respectively (Fig. 4c). The optical band gap of the Fe3+-doped NCs calculated from the Tauc plots is 2.88, 2.84 and 2.70 eV, respectively. The corresponding conduction band (CB) positions are calculated using the equation ECB = EVB − Eg and on the basis of these values, their band energy alignment with respect to the CH4 and CO redox potentials is plotted in Fig. 4d. The CB position of Fe3-doped Cs2NaInCl6 NCs is located at −0.18 eV, which makes it energetically unfavourable for CO2 reduction, thus explaining the poor photocatalytic activity of this material. On the other hand, although Fe3+-doped Cs2AgInCl6 NCs show favourable CB positions for the formation of both CH4 and CO, the poor light absorption capabilities in the visible region could be the reason resulting in their poor performance. The strong light absorption in the visible region along with the suitable CB position of Fe3+-doped Cs2Ag0.5Na0.5InCl6 NCs make them efficient for selective photocatalytic CO2 reduction. DFT calculations (Fig. 3) show that Ag doping induces an upshift of the edges of optical transitions. The CB edges are primarily formed by the localized Fe states, whose positions are influenced by the level of Ag doping. The VB states formed by Ag and Cl states do not participate in the optical transition, and the edge is placed directly below these states. With increasing doping, the CB edge shifts up along with the optical transition edges, resulting in the band gap remaining nearly unchanged. Besides electronic arguments, therefore, the simultaneous presence of Na, Ag, and Fe, i.e. the NC chemical composition, is crucial to facilitate the activation of CO2 and its transformation into CH4.
In order to further probe the enhanced photocatalytic CO2 reduction ability of Fe3+-doped Cs2Ag0.5Na0.5InCl6 NCs, CO2 adsorption/desorption measurements were performed. The CO2 adsorption/desorption isotherms are shown in Fig. S18.† It can be seen that Fe3+-doped Cs2Ag0.5Na0.5InCl6 NCs exhibit higher CO2 adsorption capacity (5.1 cm3 g−1) than undoped Cs2Ag0.5Na0.5InCl6 NCs (2.9 cm3 g−1). The enhanced CO2 adsorption by Fe3+-doped Cs2Ag0.5Na0.5InCl6 NCs could be possibly assigned to the presence of extra adsorption sites induced by interstitial iron dopants. According to the previous reports, CO2 molecules generally adsorb on the metal centres of perovskite materials,54 and thus the introduction of iron dopants would manipulate the active sites for CO2 adsorption and iron could act as active adsorption sites along with other metal ions. Next, photoelectrochemical characterization studies were performed to investigate the photogenerated charge separation and transfer behaviour of the three photocatalysts. The transient photoresponse curve shown in Fig. 4e demonstrated that the photocurrent of the samples was repeatable. In comparison to the Fe3+-doped Cs2NaInCl6 and Cs2AgInCl6 NCs, the Fe3+-doped Cs2Ag0.5Na0.5InCl6 NCs show significantly higher photocurrent density, indicating efficient separation of photogenerated charge carriers. Furthermore, the interfacial charge transfer resistance of the three catalysts was investigated by using electrochemical impedance spectra. The Nyquist plot (Fig. 4f) of Fe3+-doped Cs2Ag0.5Na0.5InCl6 NCs has a smaller radius than that of Fe3+-doped Cs2NaInCl6 and Cs2AgInCl6 NCs, implying a lower interfacial charge transfer resistance in Fe3+-doped Cs2Ag0.5Na0.5InCl6 NCs. Thus electrochemical analysis clearly demonstrates the efficient separation and transfer of photogenerated charge carriers in Fe3+-doped Cs2Ag0.5Na0.5InCl6 NCs which further validates their efficient photocatalytic activity.
To shed more light on the mechanism behind the selective reduction of CO2 to CH4, a computational analysis of the possible reaction pathway on (Fe3+-doped) Cs8Na2Ag2In3Fe1Cl24 has been conducted and compared to that of the undoped system Cs8Na2Ag2In4Fe0Cl24. The energy profile, expressed in terms of the change in Gibbs energy, is illustrated in Fig. 5a. For the Fe3+-doped system, the reaction initiates with the adsorption of a CO2 molecule at the Fe3+ ion, followed by the successive addition of H atoms, and the removal of the resulting H2O by-product. The change in Gibbs free energy (ΔG) of *CO2 formation for an iron-doped system is lower than that of the undoped counterpart, implying that CO2 adsorption and activation on the surface of an Fe3+-doped system is more favourable compared to that of the undoped system (Fig. S28†). Moreover, the transformation of adsorbed CO2 (*CO2) into OCOH* on the Fe3+-doped surface is an easy step and requires a small energy barrier of 0.08 eV. This could be attributed to the stronger binding of OCOH* through an interaction with the surface Fe atom. In particular, the formation energy of OCOH* is 2.29 eV for the undoped surface, which is much higher than that of the Fe3+-doped surface (ΔG = −0.21 eV). Thus, our result indicates that the hydrogenation of *CO2 to the OCOH* intermediate is much easier on the Fe3+-doped surface. However, protonation of OCOH* and conversion into *CO is an endothermic process that requires an energy input to overcome the barrier of 1.19 eV. In contrast, downhill free energy profiles of the continuous protonation of OCOH* intermediates suggest that conversion of OCOH* to OCHOH* is an exothermic reaction which could further drive the continuous protonation of CO2. Nevertheless, the formation of HOCH* intermediates is the rate-limiting step for further protonation processes, but the hydrogenation of HOCH* to 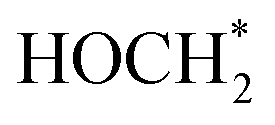 and finally to CH4 is thermodynamically spontaneous. According to the previous reports,55–59 the selective reduction of CO2 into CH4 generally proceeds on the catalyst surface through a series of proton-coupled photogenerated electron transfer processes via the formation of several key intermediates such as OCOH*, HCO* and
and finally to CH4 is thermodynamically spontaneous. According to the previous reports,55–59 the selective reduction of CO2 into CH4 generally proceeds on the catalyst surface through a series of proton-coupled photogenerated electron transfer processes via the formation of several key intermediates such as OCOH*, HCO* and 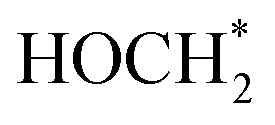 . The ΔG value of two key intermediates (OCOH*, and
. The ΔG value of two key intermediates (OCOH*, and 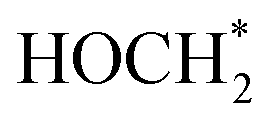 ) for selective conversion of CO2 to CH4 over an Fe3-doped surface is −0.21 eV for OCOH* and −0.93 eV for
) for selective conversion of CO2 to CH4 over an Fe3-doped surface is −0.21 eV for OCOH* and −0.93 eV for 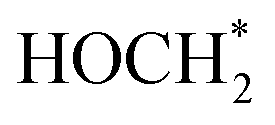 which is substantially lower than that over the undoped surface (2.29 eV for OCOH* and 1.43 eV for
which is substantially lower than that over the undoped surface (2.29 eV for OCOH* and 1.43 eV for 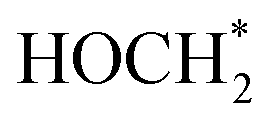 ). In general, lowering the free energy of key intermediates can concurrently reduce the activation energy barrier of the reaction, thereby enhancing the efficiency of CO2 reduction. Moreover, the calculated charge distribution at a CO2 adsorption distance of 3.9 Å above the surfaces (Fig. 5b and c) reveals only a slight difference in the vicinity of the Fe dopant, with a smaller gradient between Fe and Cl than that in the undoped surface. Consequently, CO2 tends to adsorb parallel to the surface on an Fe-doped system, favouring a slightly lower charged underlying atom for bonding. However, the change in Gibbs energy along the reaction profile shown in Fig. 5a indicates that the stabilization of the first intermediate in the subsequent steps is of greater significance. The results of DFT calculations clearly demonstrate that the Fe3-doped surface dramatically improves CO2 adsorption and activation, reducing the barrier for the formation of key intermediates for selective reduction of CO2 to CH4.
). In general, lowering the free energy of key intermediates can concurrently reduce the activation energy barrier of the reaction, thereby enhancing the efficiency of CO2 reduction. Moreover, the calculated charge distribution at a CO2 adsorption distance of 3.9 Å above the surfaces (Fig. 5b and c) reveals only a slight difference in the vicinity of the Fe dopant, with a smaller gradient between Fe and Cl than that in the undoped surface. Consequently, CO2 tends to adsorb parallel to the surface on an Fe-doped system, favouring a slightly lower charged underlying atom for bonding. However, the change in Gibbs energy along the reaction profile shown in Fig. 5a indicates that the stabilization of the first intermediate in the subsequent steps is of greater significance. The results of DFT calculations clearly demonstrate that the Fe3-doped surface dramatically improves CO2 adsorption and activation, reducing the barrier for the formation of key intermediates for selective reduction of CO2 to CH4.
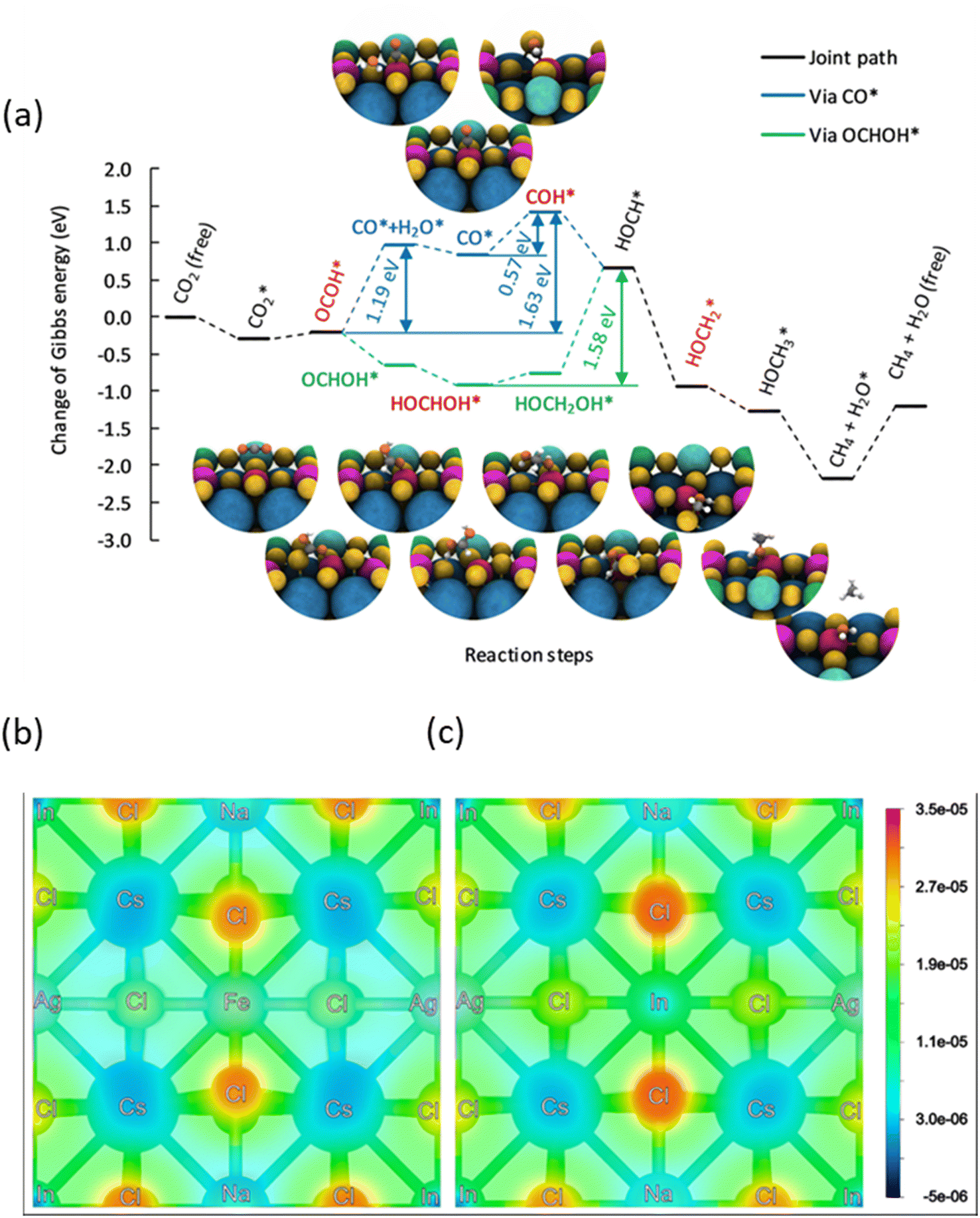 | ||
| Fig. 5 (a) Energy profile for the reduction of CO2 at a Fe3+ ion located on Cs8Na2Ag2In3Fe1Cl24, with each value calculated using a step-by-step procedure within the hybrid-DFT framework including the DFT-D3 vdW correction described in Computational methods (ESI).† The Gibbs energies are relative to the Gibbs energy of a free CO2 molecule and free surface. For comparison on the Cs8Na2Ag2In4Fe0Cl24 surface, see Fig. S28.† Charge density plot of (b) Fe-doped and (c) pristine Cs4NaAgIn2Cl6 surfaces corresponding to a CO2 adsorption distance of 3.9 Å above the surfaces. Slightly negative values are caused by integrations within the PAW spheres. | ||
Conclusions
In summary, we successfully synthesised a series of Fe3+-doped and Fe3+/Ag+ co-doped lead-free DP NCs. The incorporation of iron into Cs2NaInCl6 NCs results in extended visible light absorption, broadening the absorption edge from 330 nm to 505 nm, and reducing the band gap from 4.29 eV to 2.88 eV. The resulting absorption spectra are characterized by strong excitonic optical transitions. The presence of Fe and Ag, Na co-alloying enables a proper band energy alignment with the redox potential energy levels of CO and CH4 produced during photocatalytic CO2 reduction. This results in a stable and optimized photocatalyst (Fe-doped Cs2Ag0.5Na0.5InCl6) showing 94% methane selectivity for the reduction of CO2. Our findings open up a reliable pathway to enhance the light absorption properties of lead-free DP NCs and to tune their band energy alignment and surface chemistry for selective photocatalytic reactions. Furthermore, our synthetic methodology holds great potential for the development of stable and lead-free light absorber materials for solution-processed photovoltaic and optoelectronic applications.Data availability
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
RA and SK gratefully acknowledge support by the Czech Science Foundation, project GA CR 23-07971S. The work was further supported by the project Operational Programme Research, Development and Education – European Regional Development Fund, Project No. CZ.02.1.01/0.0/0.0/15_003/0000416 of the Ministry of Education, Youth and Sports of the Czech Republic, and by the Croucher Foundation of Hong Kong. RZ and SK acknowledge the funding from the Czech Science Foundation, project GA CR – EXPRO, 19-27454X. This work was also supported by the experimental and theoretical studies of near-infrared-emitting and chiral carbon dot luminophores project – from Moravian-Silesian Region, contract no. 00734/2023/RRC and European Union under the REFRESH – Research Excellence for Region Sustainability. The authors would also like to acknowledge High-tech Industries project number CZ.10.03.01/00/22_003/0000048 via the Operational Programme and ERDF/ESF project TECHSCALE (CZ.02.01.01/00/22_008/0004587). The IT4Innovations National Supercomputing Center is gratefully acknowledged for providing generous computational resources supported by the Ministry of Education, Youth and Sports of the Czech Republic through the e-INFRA CZ (No. 90254). JN acknowledges support by Palacký University Olomouc (project IGA_PrF_2024_017). LZ acknowledges the support from the Horizon Europe project HORIZON-WIDERA-2022-TALENTS “APPROACH” (No. 101120397).References
- Z. Xiao, Z. Song and Y. Yan, Adv. Mater., 2019, 31, 1803792 CrossRef CAS PubMed.
- H. Min, D. Y. Lee, J. Kim, G. Kim, K. S. Lee, J. Kim, M. J. Paik, Y. K. Kim, K. S. Kim, M. G. Kim, T. J. Shin and S. Il Seok, Nature, 2021, 598, 444–450 CrossRef CAS PubMed.
- Y.-H. Kim, S. Kim, A. Kakekhani, J. Park, J. Park, Y.-H. Lee, H. Xu, S. Nagane, R. B. Wexler, D.-H. Kim, S. H. Jo, L. Martínez-Sarti, P. Tan, A. Sadhanala, G.-S. Park, Y.-W. Kim, B. Hu, H. J. Bolink, S. Yoo, R. H. Friend, A. M. Rappe and T.-W. Lee, Nat. Photonics, 2021, 15, 148–155 CrossRef CAS.
- A. Swarnkar, V. K. Ravi and A. Nag, ACS Energy Lett., 2017, 2, 1089–1098 CrossRef CAS.
- T. C. Jellicoe, J. M. Richter, H. F. J. Glass, M. Tabachnyk, R. Brady, S. E. Dutton, A. Rao, R. H. Friend, D. Credgington, N. C. Greenham and M. L. Böhm, J. Am. Chem. Soc., 2016, 138, 2941–2944 CrossRef CAS PubMed.
- C. C. Stoumpos, L. Frazer, D. J. Clark, Y. S. Kim, S. H. Rhim, A. J. Freeman, J. B. Ketterson, J. I. Jang and M. G. Kanatzidis, J. Am. Chem. Soc., 2015, 137, 6804–6819 CrossRef CAS PubMed.
- B.-W. Park, B. Philippe, X. Zhang, H. Rensmo, G. Boschloo and E. M. J. Johansson, Adv. Mater., 2015, 27, 6806–6813 CrossRef CAS PubMed.
- B. Saparov, F. Hong, J.-P. Sun, H.-S. Duan, W. Meng, S. Cameron, I. G. Hill, Y. Yan and D. B. Mitzi, Chem. Mater., 2015, 27, 5622–5632 CrossRef CAS.
- A. H. Slavney, T. Hu, A. M. Lindenberg and H. I. Karunadasa, J. Am. Chem. Soc., 2016, 138, 2138–2141 CrossRef CAS PubMed.
- G. Volonakis, M. R. Filip, A. A. Haghighirad, N. Sakai, B. Wenger, H. J. Snaith and F. Giustino, J. Phys. Chem. Lett., 2016, 7, 1254–1259 CrossRef CAS PubMed.
- J. Zhou, Z. Xia, M. S. Molokeev, X. Zhang, D. Peng and Q. Liu, J. Mater. Chem. A, 2017, 5, 15031–15037 RSC.
- E. Greul, M. L. Petrus, A. Binek, P. Docampo and T. Bein, J. Mater. Chem. A, 2017, 5, 19972–19981 RSC.
- L. Zhou, Y.-F. Xu, B.-X. Chen, D.-B. Kuang and C.-Y. Su, Small, 2018, 14, 1703762 CrossRef PubMed.
- J. Luo, X. Wang, S. Li, J. Liu, Y. Guo, G. Niu, L. Yao, Y. Fu, L. Gao, Q. Dong, C. Zhao, M. Leng, F. Ma, W. Liang, L. Wang, S. Jin, J. Han, L. Zhang, J. Etheridge, J. Wang, Y. Yan, E. H. Sargent and J. Tang, Nature, 2018, 563, 541–545 CrossRef CAS PubMed.
- L. Zdražil, S. Kalytchuk, M. Langer, R. Ahmad, J. Pospíšil, O. Zmeškal, M. Altomare, A. Osvet, R. Zbořil, P. Schmuki, C. J. Brabec, M. Otyepka and Š. Kment, ACS Appl. Energy Mater., 2021, 4, 6445–6453 CrossRef.
- W. Zhu, W. Ma, Y. Su, Z. Chen, X. Chen, Y. Ma, L. Bai, W. Xiao, T. Liu, H. Zhu, X. Liu, H. Liu, X. Liu and Y. Yang, Light: Sci. Appl., 2020, 9, 112 CrossRef CAS PubMed.
- Z. Dong, S. Su, Z. Zhang, Y. Jiang and J. Xu, Inorg. Chem., 2023, 62, 1752–1761 CrossRef CAS PubMed.
- Z. Zhang, Q. Sun, Y. Lu, F. Lu, X. Mu, S.-H. Wei and M. Sui, Nat. Commun., 2022, 13, 3397 CrossRef CAS PubMed.
- R. Ahmad, G. V. Nutan, D. Singh, G. Gupta, U. Soni, S. Sapra and R. Srivastava, Nano Res., 2021, 14, 1126–1134 CrossRef CAS.
- Y. Jiang, K. Li, X. Wu, M. Zhu, H. Zhang, K. Zhang, Y. Wang, K. P. Loh, Y. Shi and Q.-H. Xu, ACS Appl. Mater. Interfaces, 2021, 13, 10037–10046 CrossRef CAS PubMed.
- S. Kumar, I. Hassan, M. Regue, S. Gonzalez-Carrero, E. Rattner, M. A. Isaacs and S. Eslava, J. Mater. Chem. A, 2021, 9, 12179–12187 RSC.
- M. Yu, N. Wang, K. Lin, D. Song, J. Chen, T. Liang, J. Sun, K. Pan and H. Fu, J. Mater. Chem. A, 2023, 11, 4302–4309 RSC.
- K. Du, W. Meng, X. Wang, Y. Yan and D. B. Mitzi, Angew. Chem., 2017, 129, 8270–8274 CrossRef.
- Y. Liu, P. Jain, I. J. Cleveland, M. Tran, S. Sarp, K. Sandrakumar, R. S. Rodriguez and E. S. Aydil, J. Mater. Chem. A, 2023, 11, 21099–21108 RSC.
- K. P. Lindquist, S. A. Mack, A. H. Slavney, L. Leppert, A. Gold-Parker, J. F. Stebbins, A. Salleo, M. F. Toney, J. B. Neaton and H. I. Karunadasa, Chem. Sci., 2019, 10, 10620–10628 RSC.
- F. Ji, Y. Huang, F. Wang, L. Kobera, F. Xie, J. Klarbring, S. Abbrent, J. Brus, C. Yin, S. I. Simak, I. A. Abrikosov, I. A. Buyanova, W. M. Chen and F. Gao, Adv. Funct. Mater., 2020, 30, 2005521 CrossRef CAS.
- G. Liu, Z. Zhang, C. Wu, Y. Zhang, X. Li, W. Yu, G. Yao, S. Liu, J. Shi, K. Liu, Z. Chen, L. Xiao and B. Qu, Adv. Funct. Mater., 2022, 32, 2109891 CrossRef CAS.
- Z. Li, S. R. Kavanagh, M. Napari, R. G. Palgrave, M. Abdi-Jalebi, Z. Andaji-Garmaroudi, D. W. Davies, M. Laitinen, J. Julin, M. A. Isaacs, R. H. Friend, D. O. Scanlon, A. Walsh and R. L. Z. Hoye, J. Mater. Chem. A, 2020, 8, 21780–21788 RSC.
- F. Ji, F. Wang, L. Kobera, S. Abbrent, J. Brus, W. Ning and F. Gao, Chem. Sci., 2021, 12, 1730–1735 RSC.
- H. Yin, Y. Xian, Y. Zhang, W. Chen, X. Wen, N. U. Rahman, Y. Long, B. Jia, J. Fan and W. Li, Adv. Funct. Mater., 2020, 30, 2002225 CrossRef CAS.
- Z. Liu, Y. Sun, T. Cai, H. Yang, J. Zhao, T. Yin, C. Hao, M. Chen, W. Shi, X. Li, L. Guan, X. Li, X. Wang, A. Tang and O. Chen, Adv. Mater., 2023, 35, 2211235 CrossRef CAS PubMed.
- F. Locardi, M. Cirignano, D. Baranov, Z. Dang, M. Prato, F. Drago, M. Ferretti, V. Pinchetti, M. Fanciulli, S. Brovelli, L. De Trizio and L. Manna, J. Am. Chem. Soc., 2018, 140, 12989–12995 CrossRef CAS PubMed.
- Y. Liu, Y. Jing, J. Zhao, Q. Liu and Z. Xia, Chem. Mater., 2019, 31, 3333–3339 CrossRef CAS.
- F. Locardi, E. Sartori, J. Buha, J. Zito, M. Prato, V. Pinchetti, M. L. Zaffalon, M. Ferretti, S. Brovelli, I. Infante, L. De Trizio and L. Manna, ACS Energy Lett., 2019, 4, 1976–1982 CrossRef CAS.
- R. Ahmad, L. Zdražil, S. Kalytchuk, A. Naldoni, E. Mohammadi, P. Schmuki, R. Zboril and Š. Kment, Appl. Mater. Today, 2022, 26, 101288 CrossRef.
- R. Ahmad, L. Zdražil, S. Kalytchuk, A. Naldoni, A. L. Rogach, P. Schmuki, R. Zboril and Š. Kment, ACS Appl. Mater. Interfaces, 2021, 13, 47845–47859 CrossRef CAS PubMed.
- H. Arfin, J. Kaur, T. Sheikh, S. Chakraborty and A. Nag, Angew. Chem., Int. Ed., 2020, 59, 11307–11311 CrossRef CAS PubMed.
- Y. Zhang, Z. Zhang, W. Yu, Y. He, Z. Chen, L. Xiao, J. Shi, X. Guo, S. Wang and B. Qu, Advanced Science, 2022, 9, 2102895 CrossRef CAS PubMed.
- W. Ning, J. Bao, Y. Puttisong, F. Moro, L. Kobera, S. Shimono, L. Wang, F. Ji, M. Cuartero, S. Kawaguchi, S. Abbrent, H. Ishibashi, R. De Marco, I. A. Bouianova, G. A. Crespo, Y. Kubota, J. Brus, D. Y. Chung, L. Sun, W. M. Chen, M. G. Kanatzidis and F. Gao, Sci. Adv., 2020, 6, eabb5381 CrossRef CAS PubMed.
- P. Han, C. Luo, W. Zhou, J. Hou, C. Li, D. Zheng and K. Han, J. Phys. Chem. C, 2021, 125, 11743–11749 CrossRef CAS.
- S. E. Creutz, E. N. Crites, M. C. De Siena and D. R. Gamelin, Nano Lett., 2018, 18, 1118–1123 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- J. Heyd and G. E. Scuseria, J. Chem. Phys., 2004, 121, 1187–1192 CrossRef CAS PubMed.
- E. E. Salpeter and H. A. Bethe, Phys. Rev., 1951, 84, 1232–1242 CrossRef.
- S. Albrecht, L. Reining, R. Del Sole and G. Onida, Phys. Rev. Lett., 1998, 80, 4510–4513 CrossRef CAS.
- M. Rohlfing and S. G. Louie, Phys. Rev. Lett., 1998, 81, 2312–2315 CrossRef CAS.
- S. Shyamal, S. K. Dutta and N. Pradhan, J. Phys. Chem. Lett., 2019, 10, 7965–7969 CrossRef CAS PubMed.
- Y. Jiang, H. Chen, J. Li, J. Liao, H. Zhang, X. Wang and D. Kuang, Adv. Funct. Mater., 2020, 30, 2004293 CrossRef CAS.
- F. Luo, M. Liu, M. Zheng, Q. Li, H. Wang, J. Zhou, Y. Jiang, Y. Yu and B. Jiang, J. Mater. Chem. A, 2023, 11, 241–250 RSC.
- Y.-F. Xu, M.-Z. Yang, B.-X. Chen, X.-D. Wang, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, J. Am. Chem. Soc., 2017, 139, 5660–5663 CrossRef CAS PubMed.
- J.-N. Huang, Y.-J. Dong, H.-B. Zhao, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, J. Mater. Chem. A, 2022, 10, 25212–25219 RSC.
- Z. Liu, H. Yang, J. Wang, Y. Yuan, K. Hills-Kimball, T. Cai, P. Wang, A. Tang and O. Chen, Nano Lett., 2021, 21, 1620–1627 CrossRef CAS PubMed.
- G. Yin, X. Qi, Y. Chen, Q. Peng, X. Jiang, Q. Wang, W. Zhang and X. Gong, J. Mater. Chem. A, 2022, 10, 22468–22476 RSC.
- S. S. Bhosale, A. K. Kharade, E. Jokar, A. Fathi, S. Chang and E. W.-G. Diau, J. Am. Chem. Soc., 2019, 141, 20434–20442 CrossRef CAS PubMed.
- X. Li, Y. Sun, J. Xu, Y. Shao, J. Wu, X. Xu, Y. Pan, H. Ju, J. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690–699 CrossRef CAS.
- Y. Chai, Y. Kong, M. Lin, W. Lin, J. Shen, J. Long, R. Yuan, W. Dai, X. Wang and Z. Zhang, Nat. Commun., 2023, 14, 6168 CrossRef CAS PubMed.
- Z. Zhang, D. Li, Z. Dong, Y. Jiang, X. Li, Y. Chu and J. Xu, Sol. RRL, 2023, 7, 2300038 CrossRef CAS.
- J. Qian, H. Hu, Y. Liang and Z. Zhang, Appl. Surf. Sci., 2024, 648, 159084 CrossRef CAS.
- Q. Sun, J. Xu, F. Tao, W. Ye, C. Zhou, J. He and J. Lu, Angew. Chem., Int. Ed., 2022, 61, e202200872 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta00676c |
| This journal is © The Royal Society of Chemistry 2024 |