
NiFeCo–OH/NiTe nanoarrays with amorphous/crystalline interfaces for highly efficient oxygen evolution reaction†
Jing
Liu
a,
Da
Liu
a,
Xiaoxiao
Yan
a,
Peifang
Guo
a,
Hongbin
Xu
b,
Peng
Chen
a and
Renbing
Wu
 *a
*a
aDepartment of Materials Science, Fudan University, Shanghai, 200438, P. R. China. E-mail: rbwu@fudan.edu.cn
bDepartment of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
First published on 11th April 2024
Abstract
Transition metal tellurides have drawn much interest as alternative electrocatalysts for the oxygen evolution reaction (OER) on account of the high electrical conductivity and variable phase and composition. However, they suffer from the low exposure of active sites and unfavourable adsorption energies of oxygenated intermediates, exhibiting a limited catalytic activity. Herein, one-dimensional hetero-structured NiFeCo–OH/NiTe nanorod arrays with amorphous/crystalline interfaces are constructed on a nickel foam scaffold (denoted as NiFeCo–OH/NiTe@NF) through a facile hydrothermal method followed by a rapid electrodeposition process. Both the experimental investigations and density functional theory (DFT) calculations reveal that the heterostructure with amorphous/crystalline interfaces not only provides abundant defects and disordered arrangement for high exposure of active sites but also optimizes the electronic structure for energetically favourable intermediates. Accordingly, the as-developed NiFeCo–OH/NiTe@NF exhibits an excellent electrocatalytic performance for the OER with a low overpotential of 276 mV at a current density of 100 mA cm−2 in 1.0 M KOH and a remarkable stability. This study may pave a new avenue for the optimal design and construction of highly efficient transition metal telluride-based electrocatalysts for the OER.
1. Introduction
On account of the carbon-free emission and high gravimetric energy density, hydrogen has shown significant dynamism and potential as a clean energy source.1–4 Electrochemical water splitting could provide a feasible and sustainable supply for large-scale hydrogen production, which involves the hydrogen evolution reaction (HER) at the cathode and the oxygen evolution reaction (OER) at the anode.5–8 In comparison with the HER, the OER process is more sluggish owing to multiple proton/electron-coupled transfer steps, and thus efficient OER catalysis has momentous impact on the overall efficiency of electrochemical water splitting.6,9–11 The noble IrO2 and RuO2 have been widely verified as the most active OER electrocatalysts so far, whereas their high economic cost and low abundance seriously hinder their practical applications on a large scale.12–15As an alternative, earth-abundant first-row (3d) transition metal-based electrocatalysts have been intensively investigated, including transition metal chalcogenides,1,16–18 oxides/(oxy)hydroxides,12,13 phosphides,2,19 nitrides20,21 and carbides.22,23 Among them, transition metal tellurides have received considerable attention owing to their high electrical conductivity, variable phase and composition, and unique physicochemical properties. Nevertheless, the catalytic performances of those tellurides without intentional modification are required to be further enhanced in order to satisfy the practical applications. In this regard, numerous strategies have been explored for enhancing the OER performance of telluride-based electrocatalysts, including heteroatom doping, morphological modulation and heterostructure engineering. For example, Chen et al. doped P anions into Te vacancies in CoTe2, inducing a phase transition from hexagonal structure to orthorhombic structure, and therefore achieving a significantly enhanced OER performance with an ultralow overpotential of 241 mV at a current density of 10 mA cm−2.16 Wang et al. developed the strategy of the integration of S doping and surface oxidation to activate CoTe nanoarrays, which has achieved a current density of 10 mA cm−2 with an overpotential of 246 mV.24 Hu et al. prepared NiFe-LDH/NiTe composite electrocatalysts and disclosed that the electronic interactions between the components could enhance the OER catalytic reaction kinetics.25 Xing et al. reported a NiTe/Ni2P heterostructure catalyst through a hydrothermal process and demonstrated that the strain engineering of the heterostructure via the lattice incompatibility not only boosted the OER activity but also stabilized the intrinsic structure of the catalyst.26 Despite the continuous progress that has been made so far, the rational design of telluride-based catalysts with better catalytic performance is still urgently desired.
Recently, it has been demonstrated that electrocatalysts with an amorphous phase show better electrocatalytic activity for the OER than the crystalline-based counterpart. On one hand, a large number of randomly oriented bonds are conducive to exposing more available active sites for catalytic reaction.27 On the other hand, the flexibility of the amorphous structure allows the self-regulation of the electronic configuration and geometry of metallic active centers, which may improve the adsorption/desorption process of oxygenated intermediates.28 Accordingly, the design of amorphous–crystalline interfaces in OER catalysts can be speculated as an effective strategy to further boost the electrocatalytic performance.
With these facts in mind, herein, we report a one-dimensional NiFeCo–OH/NiTe@NF with amorphous/crystalline interfaces by electrodepositing amorphous hydroxides on the crystalline NiTe nanorods supported on nickel foam (NF) within only a few minutes. The deposited amount of amorphous NiFeCo–OH can be precisely manipulated by finely tuning the electrodeposition duration and the electrolyte concentration. Conspicuously, NiFeCo–OH/NiTe@NF exhibits excellent electrocatalytic activities for the OER with a low overpotential of 276 mV at a large current density of 100 mA cm−2 in 1.0 M KOH.
2. Results and discussion
2.1 Structure and composition characterization
A facile strategy consisting of a hydrothermal reaction and a rapid electrodeposition process was carried out to synthesize the target product NiFeCo–OH/NiTe@NF as illustrated in Fig. 1a and Scheme S1.† As displayed in the field emission scanning electron microscopy (FESEM) images (Fig. S1†), well-aligned NiTe nanorod arrays on the NF surface are successfully prepared via a hydrothermal process, which can evidently provide an enlarged specific surface area, increased available active sites, and accelerated charge transfer.29–32 As displayed in Fig. S2,† transmission electron microscopy (TEM) was performed to observe the microstructure of NiTe, further confirming the nanorod morphology with a diameter of ∼100 nm. Subsequently, taking advantage of the increasing concentration of OH− ions near the working electrode, NiFeCo hydroxides (NiFeCo–OH) are expected to be epitaxially modified onto the NiTe nanorods (NiFeCo–OH/NiTe@NF). The FESEM image displayed in Fig. 1b indicates that the vertically distributed nanorod array morphology maintains well after the electrodeposition process. The rough surface of NiFeCo–OH/NiTe@NF can be clearly observed in the magnified FESEM image (Fig. 1c) and TEM image (Fig. 1d), demonstrating the formation of NiFeCo–OH layers. A similar morphology of NiFe–OH/NiTe@NF can be observed in Fig. S3,† indicating that the absence of Co does not affect the morphological structure. The high-resolution TEM (HRTEM) image (Fig. 1e) further discloses the heterostructure of the NiFeCo–OH/NiTe, where a clear demarcation between NiTe and NiFeCo–OH can be obviously identified. The uniform lattice fringes with a spacing of 0.17 nm present in the major structure correspond to the (200) plane of NiTe, while no distinct lattice fringes can be observed in the NiFeCo–OH, revealing the heterointerfaces between the crystalline NiTe and the amorphous NiFeCo–OH. The selected area electron diffraction (SAED) patterns of the individual NiFeCo–OH and NiFeCo–OH/NiTe (Fig. S4†) further confirm the amorphous/crystalline heterojunction structure. The elemental mapping images (Fig. 1f) demonstrate the uniform distribution of all the expected elements of O, Te, Ni, Fe and Co over the nanorod. Furthermore, the inductively coupled plasma atomic emission spectroscopy (ICP-AES) results reveal that the atomic ratio of Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Te:Fe:Co is 80:62:1.7:1 (Table S1†).
:Te:Fe:Co is 80:62:1.7:1 (Table S1†).
 | ||
| Fig. 1 (a) Schematic representation of the NiFeCo–OH/NiTe@NF catalyst for the OER. (b and c) FESEM images, (d) TEM image, (e) HRTEM image and (f) the corresponding EDS elemental mapping images of NiFeCo–OH/NiTe@NF. | ||
The X-ray diffraction (XRD) measurement was performed to investigate the crystal structure of all the as-synthesized samples. As displayed in Fig. 2a, apart from the peaks (44.5° and 51.8°) indexed to the cubic Ni (JCPDS no. 04-0850) derived from the nickel foam substrate, all the samples of NiTe@NF, NiFe–OH/NiTe@NF and NiFeCo–OH/NiTe@NF exhibit only one crystalline phase of NiTe (JCPDS no. 38-1393), further suggesting the amorphous structure of the corresponding hydroxides. To be specific, the peaks centered at 30.9°, 42.7°, 45.6°, 56.1° and 58.1° match well with the (101), (102), (110), (201) and (103) planes of the hexagonal structure of NiTe, respectively. Fourier transform infrared spectroscopy (FT-IR) was also employed to further confirm the formation of hydroxides (Fig. S5†). Compared with the spectra of NiTe@NF, two infrared bands around 3280 cm−1 and 1330 cm−1 of NiFeCo–OH/NiTe@NF and NiFeCo–OH@NF can be respectively attributed to the O–H stretching vibration and the NO3− vibration, evidently verifying the existence of hydroxides.33–35
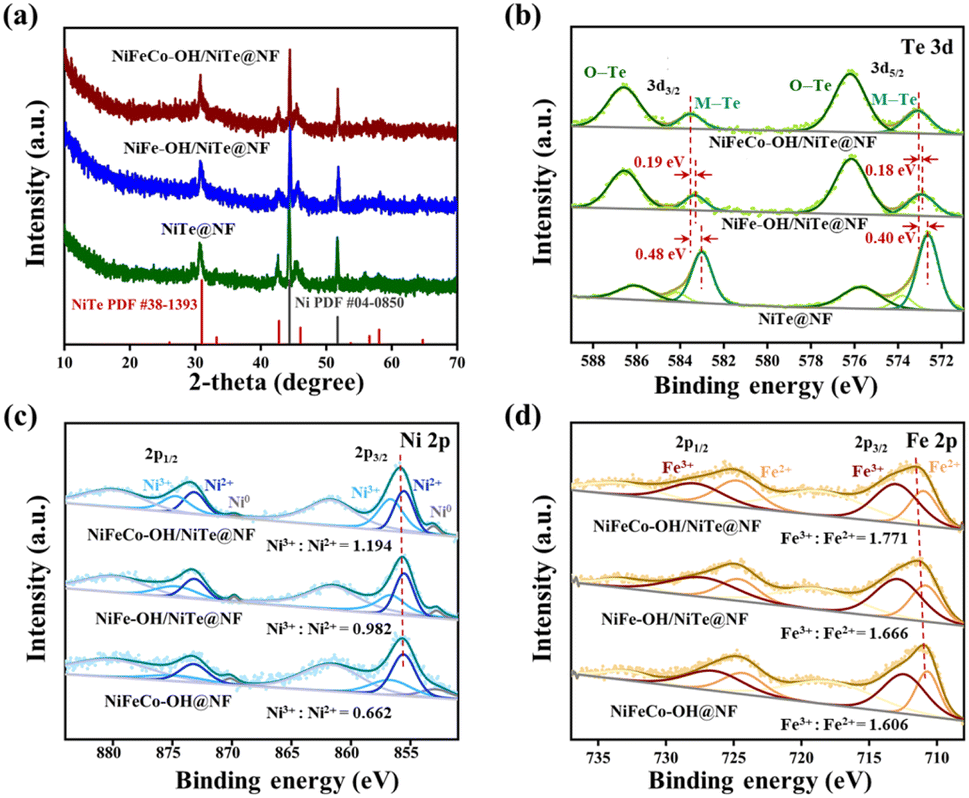 | ||
| Fig. 2 (a) XRD spectra and high-resolution (b) Te 3d XPS spectra of NiFeCo–OH/NiTe@NF, NiFe–OH/NiTe@NF and NiTe@NF. High-resolution (c) Ni 2p and (d) Fe 2p XPS spectra of NiFeCo–OH/NiTe@NF, NiFe–OH/NiTe@NF and NiFeCo–OH@NF. | ||
X-ray photoelectron spectroscopy (XPS) measurement was conducted to probe the surface chemical compositions and elemental valence states of NiTe@NF, NiFe–OH/NiTe@NF, NiFeCo–OH/NiTe@NF and NiFeCo–OH@NF. The XPS survey spectrum of NiFeCo–OH/NiTe@NF (Fig. S6c†) evidently demonstrates the existence of O, Te, Ni, Fe and Co elements. As displayed in Fig. 2b, after being decorated by NiFeCo–OH, the peaks attributed to Te2− 3d3/2 (583.51 eV) and Te2− 3d5/2 (573.10 eV) exhibit positive shifts with 0.48 and 0.40 eV compared with those of NiTe@NF, implying the built-in charge transfer pathways at the heterointerface. It is noteworthy that the positive shift of binding energy in NiFeCo–OH/NiTe@NF is more significant than that in NiFe–OH/NiTe@NF, suggesting a stronger electronic interaction at the heterointerface of the NiFeCo–OH/NiTe, which is more favourable to the electronic structure adjustment on the metallic active sites. Moreover, the peak strength of Te–O (576.20 and 586.60 eV) in NiFeCo–OH/NiTe@NF is significantly intensified in comparison with the weak ones in NiTe@NF resulting from the surface oxidation by air, further demonstrating the successful formation of the heterostructure in the NiFeCo–OH/NiTe@NF. As displayed in the high-resolution Ni 2p XPS spectra (Fig. 2c), the main peaks with binding energies of 855.89 and 873.53 eV can be indexed to the Ni 2p3/2 and Ni 2p1/2 orbitals of NiFeCo–OH/NiTe@NF, which can be fitted by two peaks assigned to Ni2+ and Ni3+. In comparison with the Ni characteristic peak locations of NiFeCo–OH@NF, NiFe–OH/NiTe@NF and NiTe@NF (Fig. 2c and S7†), the most electron deficient state of Ni in the NiFeCo–OH/NiTe@NF sample can be deduced. The specific ratio of Ni3+/Ni2+ for NiFeCo–OH/NiTe@NF is determined to be 1.194, higher than those of NiFeCo–OH@NF (0.662) and NiFe–OH/NiTe@NF (0.982), further revealing the electron depletion of Ni. Similar upshifts in binding energy and high-proportioned metal species with a high valence can also be observed in Co 2p3/2 (Fig. S8†) and Fe 2p XPS spectra (Fig. 3d). It is well known that electrophilic metal sites with a high valence usually exhibit excellent catalytic OER activities due to the promoted adsorption of hydroxyls and H2O molecules.36–38 In addition, the O 1s XPS spectrum of NiFeCo–OH/NiTe@NF (Fig. S9†) can be deconvoluted into three peaks centered at 532.5 (O1), 531.3 (O2) and 529.9 eV (O3), which can be attributed to the adsorbed water, hydroxyl groups and lattice oxygen bonding to the metal cations, respectively.
 | ||
| Fig. 3 Soft XAS spectra at the (a) Co L-edge, (b) Ni L-edge, (c) Fe L-edge, and (d) O K-edge (inset: magnified section of O peak) of NiTe, NiFe–OH/NiTe and NiFeCo–OH/NiTe. | ||
Moreover, the coordinatively unsaturated metal ions tend to accept the transferred electrons from the negatively charged hydroxyl ions, thus forming the built-in charge transfer channels with the assistance of the heterointerfaces. Similar deconvolution can be observed in the O 1s XPS spectra of NiFeCo–OH@NF and NiFe–OH/NiTe@NF, whereas two sub-peaks centered at 532.7 and 530.8 eV in the spectrum of NiTe@NF can be ascribed to the adsorbed water and surface oxygen bonding to Ni and Te, respectively.
X-ray absorption spectroscopy (XAS) in total electron yield (TEY) mode was further performed to gain an insight into the electronic interaction of the heterostructure. Above all, the Co L-edge spectrum can supplementally demonstrate the existence of Co atoms in the sample of NiFeCo–OH/NiTe (Fig. 3a). As shown in the Ni L-edge XAS spectra (Fig. 3b), the overall spectral line consists of the L3-edge region around 852 eV and the L2-edge region around 870 eV, resulting from the spin–orbital-coupling split.39 Taking advantage of the sensitivity of the spectral shape to the electronic configuration and oxidation state of the metal atoms, the difference in the charge density of Ni among the samples can be briefly deduced by comparing the intensity of peaks i and ii in the L2-edge region.40–42 The highest peak intensity ratio of ii/i can be observed in Fig. 3b, confirming the most electron deficient state of Ni in the NiFeCo–OH/NiTe electrocatalysts, in accordance with the results in Ni 2p XPS. NiFeCo–OH/NiTe also exhibits a similar higher peak intensity ratio of ii/i in the Fe L3-edge spectra (Fig. 3c), which corresponds to the lower electron density of Fe than that of NiFe–OH/NiTe.43,44 In the O K-edge XAS spectra (Fig. 3d), the peak around 532 eV in NiFeCo–OH/NiTe, which is assigned to the hybridization of O 2p with metal 3d states,45,46 is significantly intensified and shows negative shifts compared with that of NiFe–OH/NiTe and NiTe, in agreement with the afore-mentioned analysis of the decreased electron density and increased oxidation states in the Ni and Fe atoms in NiFeCo–OH/NiTe. Obviously, both the XPS and XAS analyses confirm the successful construction of the heterostructure and the electron depletion state of the metals in NiFeCo–OH/NiTe. It can be concluded that the electrodeposition of NiFeCo–OH onto the NiTe nanorods leads to strong electronic interaction between the NiFeCo–OH and NiTe.
2.2 Electrochemical performances for OER
All electrocatalytic tests for the OER were conducted in 1.0 M KOH electrolyte and all potentials were referenced to the reversible hydrogen electrode (RHE). To highlight the positive contribution of the introduction of NiTe nanorod arrays to the enhanced electrocatalytic OER activities, NiFeCo–OH@NF and NiFe–OH@NF samples were synthesized by directly electrodepositing hydroxides on the NF scaffold for comparison (see detailed morphology and structure characterization in Fig. S10 and S11†). It can be found that the surface of NF appears to be stacked by amorphous hydroxides, resulting in numerous electrochemically active sites severely buried, which is detrimental to the OER electrocatalysis. Fig. 4a displays the linear sweep voltammetry (LSV) curves of all the as-prepared samples with a scanning rate of 5 mV s−1. The target NiFeCo–OH/NiTe@NF electrode exhibits the best OER electrocatalytic performance with an ultralow overpotential of 276 mV at a large current density of 100 mA cm−2, which also outperforms recently reported transition metal telluride-based OER electrocatalysts (Table S2†). It is worth noting that both the NiFeCo–OH/NiTe@NF and the NiFe–OH/NiTe@NF display better electrocatalytic performance for the OER than the corresponding catalysts with hydroxides directly growing on the NF substrate. In addition to the synergy between the hydroxides and NiTe, the nanorod arrays provide an increased accessible surface area and continuous pathways for mass transfer, greatly accelerating the reaction kinetics and thus promoting the OER electrocatalytic activities. The same tendency can also be verified by the Tafel plots (Fig. 4b) derived from the corresponding LSV curves. To be specific, NiFeCo–OH/NiTe@NF delivers a Tafel slope of 105 mV dec−1, lower than those of NiFeCo–OH@NF (120 mV dec−1), NiFe–OH/NiTe@NF (117 mV dec−1), NiFe–OH@NF (125 mV dec−1), NiTe@NF (124 mV dec−1) and blank NF (217 mV dec−1). Moreover, electrochemical double-layer capacitance (Cdl) values were utilized to evaluate the electrochemical surface area (ECSA) of all the as-synthesized electrocatalysts, which were calculated from the cyclic voltammetry (CV) curves recorded within the non-Faraday region at different scan rates (Fig. S12†). As depicted in Fig. 4c, NiFeCo–OH/NiTe@NF possesses the largest Cdl value of 16.6 mF cm−2, higher than those of NiFeCo–OH@NF (13.5 mF cm−2), NiFe–OH/NiTe@NF (14.6 mF cm−2), NiFe–OH@NF (12.6 mF cm−2), NiTe@NF (10.6 mF cm−2) and blank NF (4.7 mF cm−2), further demonstrating that the enriched active sites originated from the vertically aligned architecture and amorphous/crystalline interfaces. The ECSA-normalized LSV curves show that NiFeCo–OH/NiTe@NF exhibits the best OER performance, further unclosing the high intrinsic activity (Fig. S13†). Electrochemical impedance spectroscopy (EIS) was subsequently conducted to assess the charge transfer efficiency at the electrolyte/electrode interfaces based on the simulated equivalent circuit models extracted from the Nyquist plots (Fig. 4d). As expected, NiFeCo–OH/NiTe@NF shows a charge transfer resistance (Rct) of 3.28 Ω, smaller than those of NiFeCo–OH@NF (13.74 Ω) and NiTe@NF (129.90 Ω), implying the rapid electron transfer rate (Table S3†). The excellent conductivity can be attributed to the diverse conductive pathways at the NiFeCo–OH/NiTe heterointerfaces along with the multi-dimensional charge transport pathways derived from the nanorod arrays. Furthermore, long-term durability is also an integral part to assess the potential value in commercial applications. As shown in Fig. 4e and S14,† NiFeCo–OH/NiTe@NF exhibits excellent stability with only 9% degradation for 45 h and 12% degradation for 140 h in current density around 100 mA cm−2. Additionally, the nanorod array morphology and the phase composition of NiFeCo–OH/NiTe@NF after the long-term stability test (inset in Fig. 4e, S15 and S16†) maintain well with negligible changes, indicating the outstanding electrochemical and structural stability.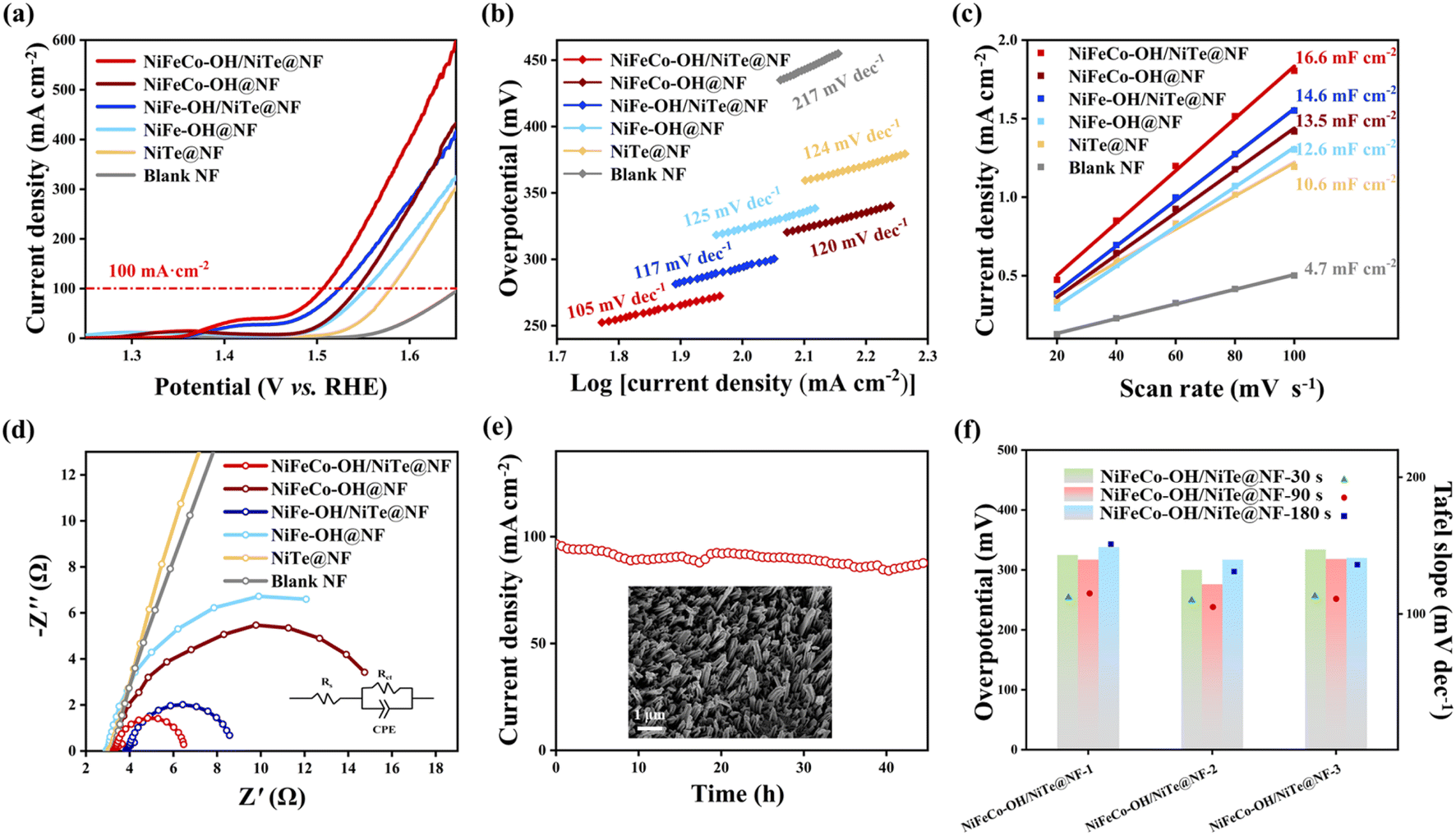 | ||
| Fig. 4 Electrocatalytic tests for the OER of NiFeCo–OH/NiTe@NF, NiFeCo–OH@NF, NiFe–OH/NiTe@NF, NiFe–OH@NF, NiTe@NF and blank NF. (a) LSV curves. (b) Tafel plots. (c) Variation of double-layer charging currents at 1.025 V as a function of different scan rate. (d) EIS plots. (e) Chronoamperometry curve of NiFeCo–OH/NiTe@NF at a constant potential of 1.52 V vs. RHE. Inset: FESEM image of the post-test catalysts. (f) The stacked bar plots of overpotential at 100 mA cm−2 and scatter plots of the corresponding Tafel slope of NiFeCo–OH/NiTe@NF electrodes synthesized under different conditions. | ||
To optimize the OER electrocatalytic performance, a series of control experiments with different electrodeposition durations (t = 30 s, 90 s, 180 s) and Co2+ concentrations (c = 1, 2, 3) were also conducted (denoted as NiFeCo–OH/NiTe@NF-c-t). Considering the best catalytic activity and highest conductivity of the samples (Fig. 4f, S17 and S18†) where the Co2+ concentration is fixed with 0.02 M, NiFeCo–OH/NiTe@NF-2 electrocatalysts with different electrodeposition durations were further characterized to probe the effects on the catalytic performances. As seen in the XRD patterns (Fig. S19†), only peaks assigned to crystalline NiTe and Ni can be found, implying negligible influence on the amorphous phase by the electrodeposition durations. As shown in Fig. 4f and Table S4,† NiFeCo–OH/NiTe@NF-2 synthesized under the electrodeposition duration of 90 s simultaneously exhibits the lowest overpotential at 100 mA cm−2, the lowest Tafel slope and the smallest charge transfer resistance among all the electrodes. Moreover, it can be evidently observed in Fig. S20† that the thickness of NiFeCo–OH nanosheets growing on the NiTe nanorods increases along with the prolonged electrodeposition durations. The best electrochemical performance of the NiFeCo–OH/NiTe@NF-2-90 s electrocatalyst suggests that the thickness of hydroxide should be appropriately regulated to ensure sufficiently exposed heterointerfaces for accelerated charge transfer channels.
2.3 Density functional theory (DFT) calculations
In order to gain an in-depth insight into the remarkable catalytic performance of NiFeCo–OH/NiTe and the electronic properties at the heterointerfaces, density functional theory (DFT) calculations were further conducted. It is well known that the hydroxides first transform to oxyhydroxides through reconstruction during the OER, which have been commonly considered as the real active phase.47–49 Hence, combined with the experimental results, the DFT calculations are based on the simulated heterostructure of Fe, Co-doped Ni–OOH/NiTe with the Ni atom as the active site. In consideration of the lattice match and the stability of the heterostructure, the junction of the (010) surface of NiTe and the (100) surface of FeCoNi–OOH was built with lattice mismatch less than 5% (see detailed calculations in the ESI†). Similarly, NiTe, NiFe–OOH/NiTe and NiFeCo–OOH models were also constructed for comparison. As shown in the free-energy diagrams at the potential of 1.23 V (Fig. 5a), the steepest uphill appears at the third step of the *OOH generation in the models of NiFe–OOH/NiTe, NiFeCo–OOH, and NiFeCo–OOH/NiTe, which can be recognized as the reaction rate-determining step (RDS) of the OER, while the second step of the dehydrogenation process is determined to be the RDS for NiTe. The reaction free energy of the RDS has been calculated to be 0.34 eV for NiFeCo–OOH/NiTe, lower than those of NiFe–OOH/NiTe (0.43 eV), NiFeCo–OOH (0.74 eV), and NiTe (0.80 eV). Obviously, the energy barrier is significantly reduced not only by the construction of the heterostructure between NiTe and oxyhydroxides, but also by the introduction of Co atoms in oxyhydroxides, which can account for the remarkable OER performance of NiFeCo–OH/NiTe. Besides, the projected density of states (PDOS) on the O 2p, Te 5p, and Ni 3d orbitals is displayed in Fig. 5d to further unveil the electronic interactions. Notably, in comparison with NiFe–OOH/NiTe, NiFeCo–OOH and NiTe, the shift of the Ni d-band center in NiFeCo–OOH/NiTe implies the less occupancy of the antibonding states of the oxygen-adsorbed intermediates and the corresponding strengthened interaction between adsorbates and the catalyst surface,50 which is beneficial for the generation of *OOH from *O during the OER process. Moreover, the charge transfer was further investigated through the analysis of Bader charge and differential charge density. It can be observed visually in Fig. 5c that Te, O and metal atoms around the interfacial region exhibit evident electron accumulation and depletion, indicating the significant electron transfer and electronic interaction between oxyhydroxides and NiTe. As displayed in Fig. S21,† the O, Te, Ni, Fe, and Co atoms are assigned the number from 1 to 6 in order to clearly diagrammatize the results of Bader charge analysis. Conspicuously, the Te atom simultaneously serves as the electron donor and acceptor, and therefore it can be interpreted as an electron transfer station of the heterostructure which is adaptive to the regulated interfacial interactions in oxyhydroxides/NiTe (Fig. 5e). In addition, NiFeCo–OOH/NiTe generally possesses the more negative valence charge for O atoms and more positive valence charge for Ni, Fe around the heterogeneous interface than NiFe–OOH/NiTe, which is consistent with the forementioned analysis of the modulated electronic structure in XPS and XAS. | ||
| Fig. 5 DFT theoretical models. (a) Calculated free-energy diagrams of the four-step elementary reaction for the OER on the active sites of NiFeCo–OOH/NiTe, NiFe–OOH/NiTe, NiFeCo–OOH, NiTe and the corresponding (b) schematic for the OER mechanism in the optimized model of NiFeCo–OOH/NiTe with different reaction intermediates. (c) Differential charge density plots for the heterostructure in the model of NiFeCo–OOH/NiTe with yellow and bright blue areas representing electron accumulation and depletion, respectively. (d) Projected DOS on the O 2p, Te 5p, Ni 3d orbitals in the model of NiFeCo–OOH/NiTe, NiFe–OOH/NiTe, and NiFeCo–OOH, NiTe. (e) Bar plots of calculated Bader charges on the Ni, Fe, Co, O, and Te atoms on the heterointerface of NiFe–OOH/NiTe and NiFeCo–OOH. | ||
3. Conclusions
In conclusion, we have developed a convenient method to synthesize hetero-structured NiFeCo–OH/NiTe nanorods with amorphous/crystalline interfaces for improving the OER performance, which involves the growth of NiTe nanorod arrays on nickel foam and the subsequent electrodeposition of NiFeCo–OH onto the surface of nanorods. The 1D heterostructure with amorphous/crystalline interfaces enables sufficiently exposed electrocatalytic active sites and facile charge transfer, and simultaneously enhances kinetics by reducing the adsorption/desorption energy barrier of the reaction rate-determining step during the OER. As a result, the NiFeCo–OH/NiTe@NF exhibited an excellent performance for the OER with an ultralow overpotential of 276 mV at a large current density of 100 mA cm−2 and a high operation stability in 1.0 M KOH. Our findings may be valuable to the optimal design and construction of robust telluride-based electrocatalysts for the OER.Author contributions
J. Liu, X. Yan and R. Wu conceived and designed the experiments and wrote the manuscript. R. Wu supervised the project. J. Liu and P. Chen conducted material synthesis. J. Liu, P. Guo and H. Xu performed characterization and performance measurements. D. Liu and J. Liu performed calculations and simulations. All authors discussed and analysed the experimental results and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (no. 52225104 and 52071084), “Shuguang Program” supported by the Shanghai Education Development Foundation and Shanghai Municipal Education Commission (no. 20SG03), and the Science and Technology Commission of Shanghai Municipality (no. 22520710600). The authors also thank BL10B in the National Synchrotron Radiation Laboratory (NSRL) for characterization by synchrotron radiation.Notes and references
- B. Fei, Z. Chen, J. Liu, H. Xu, X. Yan, H. Qing, M. Chen and R. Wu, Adv. Energy Mater., 2020, 10, 2001963 CrossRef CAS
.
- W. Li, J. Liu, P. Guo, H. Li, B. Fei, Y. Guo, H. Pan, D. Sun, F. Fang and R. Wu, Adv. Energy Mater., 2021, 11, 2102134 CrossRef CAS
- J. Xia, S. Li, H. Su, P. Liao, S. Wang, R. Xiang, Y. Zhang, Q. Liu and G. Li, Angew. Chem., Int. Ed., 2023, 62, e202306726 CrossRef PubMed
- Y. Zhong, P. Liao, J. Kang, Q. Liu, S. Wang, S. Li and G. Li, J. Am. Chem. Soc., 2023, 145, 4659–4666 CrossRef CAS PubMed
- P. Guo, R. Wu, B. Fei, J. Liu, D. Liu, X. Yan and H. Pan, J. Mater. Chem. A, 2021, 9, 21741–21749 RSC
- J. Yang, Y. Shen, Y. M. Sun, J. Xian, Y. Long and G. Li, Angew. Chem., Int. Ed., 2023, 62, e202302220 CrossRef CAS PubMed
- Y. J. Li, Y. J. Sun, Y. N. Qin, W. Y. Zhang, L. Wang, M. C. Luo, H. Yang and S. J. Guo, Adv. Energy Mater., 2020, 10, 1903120 CrossRef CAS
- Y. Hao, X. Cao, C. Lei, Z. Chen, Y. Yang and M. Gong, Mater. Today Catal., 2023, 2, 100012 CrossRef
- D. Zhong, T. Li, D. Wang, L. Li, J. Wang, G. Hao, G. Liu, Q. Zhao and J. Li, Nano Res., 2022, 15, 162–169 CrossRef CAS
- P. Guo, L. Shi, D. Liu, X. Wang, F. Gao, Y. Ha, J. Yin, M. Liu, H. Pan and R. Wu, Mater. Today Catal., 2023, 1, 100002 CrossRef
- H. M. Sun, Z. H. Yan, F. M. Liu, W. C. Xu, F. Y. Cheng and J. Chen, Adv. Mater., 2020, 32, 1806326 CrossRef CAS PubMed
- Y. Zhang, M. Du, Y. Ma, J. Shang and B. Qiu, Mater. Today Catal., 2023, 3, 100027 CrossRef
- D. Liu, X. Yan, P. Guo, Y. Yang, Y. He, J. Liu, J. Chen, H. Pan and R. Wu, ACS Catal., 2023, 13, 7698–7706 CrossRef CAS
- C. W. Liang, P. C. Zou, A. Nairan, Y. Q. Zhang, J. X. Liu, K. W. Liu, S. Y. Hu, F. Y. Kang, H. J. Fan and C. Yang, Energy Environ. Sci., 2020, 13, 86–95 RSC
- Z. P. Wu, X. F. Lu, S. Q. Zang and X. W. Lou, Adv. Funct. Mater., 2020, 30, 1910274 CrossRef CAS
- Z. Chen, M. Chen, X. Yan, H. Jia, B. Fei, Y. Ha, H. Qing, H. Yang, M. Liu and R. Wu, ACS Nano, 2020, 14, 6968–6979 CrossRef CAS PubMed
- T. Zhang, J. Sun and J. Guan, Nano Res., 2023, 16, 8684–8711 CrossRef CAS
- A. Hassan, L. Nisar, R. Iqbal, M. Sadaqat, F. Hussain, M. N. Ashiq, M. Najam-ul-Haq, A. Shah and K. S. Joya, J. Power Sources, 2021, 491, 229628 CrossRef CAS
- P. Ji, H. Jin, H. Xia, X. Luo, J. Zhu, Z. Pu and S. Mu, ACS Appl. Mater. Interfaces, 2020, 12, 727–733 CrossRef CAS PubMed
- A. Saad, H. Shen, Z. Cheng, R. Arbi, B. Guo, L. S. Hui, K. Liang, S. Liu, J. P. Attfield, A. Turak, J. Wang and M. Yang, Nano-Micro Lett., 2020, 12, 79 CrossRef CAS PubMed
- R. Q. Yao, H. Shi, W. B. Wan, Z. Wen, X. Y. Lang and Q. Jiang, Adv. Mater., 2020, 32, e1907214 CrossRef PubMed
- P. Wang, J. Zhu, Z. Pu, R. Qin, C. Zhang, D. Chen, Q. Liu, D. Wu, W. Li, S. Liu, J. Xiao and S. Mu, Appl. Catal., B, 2021, 296, 120334 CrossRef CAS
- L. Qiao, A. Zhu, W. Zeng, R. Dong, P. Tan, Z. Ding, P. Gao, S. Wang and J. Pan, J. Mater. Chem. A, 2020, 8, 2453–2462 RSC
- X. Wang, Z. Mao, X. Mao, X. Hu, F. Gao, M. Gao, Q. L. Wu, X. Lyu, A. Du, X. Xu, Y. Jia and L. Wang, Adv. Sci., 2023, 10, e2206204 CrossRef PubMed
- L. Y. Hu, X. Zeng, X. Q. Wei, H. J. Wang, Y. Wu, W. L. Gu, L. Shi and C. Z. Zhu, Appl. Catal., B, 2020, 273, 119014 CrossRef CAS
- M. Xing, Z. Qiao, Z. Niu, S. Wang, Z. Liu and D. Cao, ACS Appl. Mater. Interfaces, 2023, 15, 40428–40437 CrossRef CAS PubMed
- H. Xu, B. Fei, G. Cai, Y. Ha, J. Liu, H. Jia, J. Zhang, M. Liu and R. Wu, Adv. Energy Mater., 2019, 10, 1902714 CrossRef
- J. Kwon, H. Han, S. Jo, S. Choi, K. Y. Chung, G. Ali, K. Park, U. Paik and T. Song, Adv. Energy Mater., 2021, 11, 2100624 CrossRef CAS
- T. Zhou, Z. Cao, P. Zhang, H. Ma, Z. Gao, H. Wang, Y. Lu, J. He and Y. Zhao, Sci. Rep., 2017, 7, 46154 CrossRef CAS PubMed
- Y. Zhang and L. Qi, Nanoscale, 2022, 14, 12196–12218 RSC
- Z. Xue, X. Li, Q. Liu, M. Cai, K. Liu, M. Liu, Z. Ke, X. Liu and G. Li, Adv. Mater., 2019, 31, e1900430 CrossRef PubMed
- P. Zhai, Y. Zhang, Y. Wu, J. Gao, B. Zhang, S. Cao, Y. Zhang, Z. Li, L. Sun and J. Hou, Nat. Commun., 2020, 11, 5462 CrossRef CAS PubMed
- A. M. Cardinale, C. Carbone, S. Consani, M. Fortunato and N. Parodi, Crystals, 2020, 10, 443 CrossRef CAS
- G. Y. A. El-Reesh, A. A. Farghali, M. Taha and R. K. Mahmoud, Sci. Rep., 2020, 10, 587 CrossRef PubMed
- Y. Lin, H. Wang, C. K. Peng, L. Bu, C. L. Chiang, K. Tian, Y. Zhao, J. Zhao, Y. G. Lin, J. M. Lee and L. Gao, Small, 2020, 16, e2002426 CrossRef PubMed
- M. Han, N. Wang, B. Zhang, Y. Xia, J. Li, J. Han, K. Yao, C. Gao, C. He, Y. Liu, Z. Wang, A. Seifitokaldani, X. Sun and H. Liang, ACS Catal., 2020, 10, 9725–9734 CrossRef CAS
- G. H. Choi, N. C. S. Selvam, H. Kim, Y. S. Park, J. Jung, M. G. Nam, H. S. Jeon, A. S. Lee, W.-S. Yoon and P. J. Yoo, Appl. Catal., B, 2023, 333, 122816 CrossRef CAS
- H. Wang, T. Zhai, Y. Wu, T. Zhou, B. Zhou, C. Shang and Z. Guo, Adv. Sci., 2023, 10, e2301706 CrossRef PubMed
- D. Drevon, M. Gorlin, P. Chernev, L. Xi, H. Dau and K. M. Lange, Sci. Rep., 2019, 9, 1532 CrossRef PubMed
- Y. Tokura and N. Nagaosa, Science, 2000, 288, 462–468 CrossRef CAS PubMed
- H. X. Wang, D. S. Patil, W. W. Gu, L. Jacquamet, S. Friedrich, T. Funk and S. P. Cramer, J. Electron Spectrosc. Relat. Phenom., 2001, 114, 855–863 CrossRef
- E. J. Guo, Y. Liu, C. Sohn, R. D. Desautels, A. Herklotz, Z. Liao, J. Nichols, J. W. Freeland, M. R. Fitzsimmons and H. N. Lee, Adv. Mater., 2018, 30, e1705904 CrossRef PubMed
- S. Shen, J. Zhou, C. L. Dong, Y. Hu, E. N. Tseng, P. Guo, L. Guo and S. S. Mao, Sci. Rep., 2014, 4, 6627 CrossRef CAS PubMed
- H. Zhang, J. Liu, G. Zhao, Y. Gao, T. Tyliszczak, P. A. Glans, J. Guo, D. Ma, X. H. Sun and J. Zhong, ACS Appl. Mater. Interfaces, 2015, 7, 7863–7868 CrossRef CAS PubMed
- B. Zhang, X. L. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. García-Melchor, L. L. Han, J. X. Xu, M. Liu, L. R. Zheng, F. P. G. de Arquer, C. T. Dinh, F. J. Fan, M. J. Yuan, E. Yassitepe, N. Chen, T. Regier, P. F. Liu, Y. H. Li, P. De Luna, A. Janmohamed, H. L. L. Xin, H. G. Yang, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333–337 CrossRef CAS PubMed
- F. Frati, M. Hunault and F. M. F. de Groot, Chem. Rev., 2020, 120, 4056–4110 CrossRef CAS PubMed
- Q. Han, Y. Luo, J. Li, X. Du, S. Sun, Y. Wang, G. Liu and Z. Chen, Appl. Catal., B, 2022, 304, 120937 CrossRef CAS
- Y. Hao, Y. Li, J. Wu, L. Meng, J. Wang, C. Jia, T. Liu, X. Yang, Z. P. Liu and M. Gong, J. Am. Chem. Soc., 2021, 143, 1493–1502 CrossRef CAS PubMed
- J. Jiang, F. Sun, S. Zhou, W. Hu, H. Zhang, J. Dong, Z. Jiang, J. Zhao, J. Li, W. Yan and M. Wang, Nat. Commun., 2018, 9, 2885 CrossRef PubMed
- J. K. Norskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 937–943 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta00772g |
| This journal is © The Royal Society of Chemistry 2024 |