
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Water-triggered self-healing and reversible underwater adhesion in metalorganic polymers†
Elif
Kaymazlar
 ab,
Omer
Andac
a and
Santiago J.
Garcia
*b
ab,
Omer
Andac
a and
Santiago J.
Garcia
*b
aDepartment of Chemistry, Institute of Graduate Studies, Ondokuz Mayis University, Kurupelit, Samsun, 55200, Turkiye
bAerospace Structures and Materials Department, Faculty of Aerospace Engineering, Delft University of Technology, Kluyverweg 1, Delft, 2629 HS, The Netherlands. E-mail: S.J.GarciaEspallargas@tudelft.nl
First published on 19th June 2024
Abstract
The strategies used by organisms living in water to adhere to surfaces have been a major source of inspiration to develop synthetic underwater adhesives. Amongst the mechanisms explored, byssus-inspired metalorganic chemistry offers a broad range of possibilities due to the breath of coordination bonds, salts and polymer backbones available. This has led to a significant amount of research on bio-inspired synthetic glue-type (liquid) and tape-type (solid) adhesives. However, reversibility under water, durability and universality of adhesion remains elusive. We demonstrate that the combination of Ni-metalorganic chemistry with a flexible hydrophobic polymer allows developing fully healable and recyclable polymers able to reversibly adhere (under water) to substrates with surface energies as diverse as Teflon and glass. Other metal ions such as Fe3+ and Zn2+ did not provide the desired adhesion in water. The underlying mechanism is attributed to local water-induced chain re-orientation and the use of strong but dynamic metalorganic coordination (Ni2+-2,5-thiophenedicarboxaldehyde). The results unveil a versatile route to develop solid-state underwater adhesives and water-triggered healing polymers using a one-pot synthesis strategy (Schiff-base with metal coordination) with an underlying mechanism that can be extrapolated to different application domains such as biomedical, energy and underwater soft robotics.
1. Introduction
The adhesion of man-made materials to organic or inorganic surfaces in humid environments or under immersion is relevant in a broad range of applications such as artificial tissue engineering, drug delivery, biomedical devices, maritime and underwater soft robotics.1–5 However, developing synthetic (liquid or solid) polymers able to adhere to surfaces in water remains challenging due to the unique properties and conditions of highly humid environments. The challenge increases when dealing with solid adhesives (tape-type) that can adhere to hydrophobic and hydrophilic surfaces alike (universal adhesive) in water. On the one hand, polymers show a hydration layer at the surface leading to weak bonding between polymer and substrate.6–8 At long immersion times, water ingress further leads to plasticization, swelling, degradation and hydrolysis, ultimately leading to cohesive polymer failure.9 On the other hand, water is known to destabilize interfacial bonding between dissimilar materials (e.g., in adhesives and coatings) thereby leading to lower adhesive strengths and accelerated interfacial failure through hydrolysis and oxidation.10,11Attempts to develop underwater adhesives have been largely bio-inspired by the strategies that marine organisms such as mussels, octopus, barnacles, and clingfish use to adhere to ships and rocks. As a result, several (commercial) glue-type (liquid) and tape-type (solid) underwater adhesive polymers have been proposed.12–14 In general, bio-inspired glue-type adhesives are liquid, must be cured to solidify, and offer good adhesive strength to substrates.15 However, this type of adhesion is irreversible (i.e., once lost it cannot be recovered). In contrast, bio-inspired tape-type adhesives typically offer weaker but reversible adhesion16 in a similar way as Gecko-inspired solid polymer adhesives.17 Some of the most reported strategies to obtain solid adhesives include the use of hydrogen bonding,18 electrostatic interactions,19 π–π interactions20 and metal complexes.21 Due to their unique properties, metal complexes have been gradually gaining attention in this field. Besides their role in the underwater adhesion of biological species such as mussels, the dynamic nature of metal–ligand coordination is responsible for the damage repair or healing of the mussel byssus.22,23 Correspondingly, metal-coordination has been reported to be a relevant healing and recycling enabler of synthetic polymers using dopamine or histidine with Zn2+ or Fe2+/Fe3+ complexes amongst others.24–29 Moreover, polymer architecture control has been recognized, together with the reversible chemistry used, as a key fundamental factor to balance healing and mechanical properties.30 The implementation of a polymer based technology able to induce adhesion under water and to manage mechanical damage through local healing appears as a round up strategy to develop long-lasting underwater adhesives. Nevertheless, and even though there are several reported mussel-inspired hydrogels synthesized using DOPA-metal complexation, the practical implementation of the concept is complex due to, amongst others, the oxidation susceptibility of the two hydroxyl groups on DOPA's benzene ring leading to dopaquinone, an important natural cross-linker with negative impact on adhesion.31 Besides biological approaches, recent reports introduced the potential benefit of introducing hydrophilic groups in hydrophobic networks to increase adhesion under water.32,33
In this work we show that reversible solid underwater adhesives able to fully heal macroscopic damages (cuts) in the expected operating conditions (under water) and to be recycled multiple times can be developed using simple one-pot metalorganic chemistry. To the best of our knowledge, this work covers a research gap in solid adhesives (tape-type) with a new polymer strategy that brings us closer to durable and more environmentally friendly adhesives able to operate reversibly in humid environments. To this aim, we synthesized imine functional PDMS through the reaction of amine terminated PDMS monomers and 2,5-thiophenedicarboxaldehyde via Schiff base reaction and physically cross-linked the resulting polymer with different metal salts in a one-pot chemistry approach. Different metal salts (with Fe, Zn, and Ni ions) were used to study the role of the metal–ligand coordination bond strength on the polymer properties, healing in air and in water, recyclability, and underwater adhesion. We show that nickel complexes lead to the best polymers amongst the studied metalorganic in terms of mechanical strength, healing, recycling, and underwater adhesion. We demonstrate that the (underwater) healing and underwater adhesion mechanisms are both facilitated by the same water-induced physical phenomena. During exposure to humidity or water, the hydrophobic nature of PDMS minimizes the formation of a hydration layer between the polymer and the surface by repelling water. At the same time, polar groups such as imine, aldehyde, amine, and metal complexes within the polymer reorient and facilitate interfacial adhesion through surface polarity changes. The dynamic nature of the metal–ligand, and to a minor extent imine bonds, enhances adhesiveness, as well as self-healing and recyclability. Ni2+ coordination leads to a sweet spot between polymer strength and network dynamicity to allow full interfacial polymer healing and adhesion while Fe3+ and Zn2+ complexes are either too strong to allow healing and interfacial adhesion or too inefficient to lead to strong cohesive networks. The results introduce new insights on dynamic metallopolymers and present a novel approach to achieve reversible underwater adhesion and healing through humidity-triggered local chain orientation using a very versatile polymer chemistry platform.
2. Experimental section
2.1 Material synthesis
2,5-Thiophenedicarboxaldehyde (0.14 g, 1 mmol) was added to NH2-PDMS-NH2 (poly(dimethylsiloxane), bis(3-aminopropyl)terminated) (5 g, 1 mmol) in DCM (25 mL). The mixture solution was stirred at room temperature for one day until a solution of stable bright yellow colour confirming reaction completion was obtained. Subsequently, 5 mL of metal salt (ferric chloride, zinc acetate, and nickel nitrate) solution in MetOH (0.5 mmol metal salt) was added to the PDMS-ligand solution and was stirred at room temperature for 6 hours. The solution was concentrated to about 10 mL at 35 °C. The concentrated solution was poured into a Teflon mould and dried at room temperature for one day and further dried at 100 °C for two days. The synthesis route and chemical structure of samples are shown in Fig. S1.†2.2 Material characterization
The bond strengths of the metal complexes were analysed using the Gaussian program. The model structures were calculated using the Gaussian16 program, employing the density functional theory (DFT) with the B3LYP method. The calculations utilized the 6-311+G(2d-2p) basis set for non-metal atoms and LANL2DZ for the metal atoms (see the ESI† for details). The coordination bond energies were calculated using eqn (S1) and (S2).†GPC was conducted at room temperature in an Agilent LC GPC system (Agilent Technologies, Waldbronn, Germany) equipped with a refractive index detector and combination of mixed D and mixed E columns. THF (HPLC grade) was used as an eluent at a flow rate of 1.0 mL min−1.
The crosslinking density of polymers was calculated via swelling tests according to Flory–Rehner equation and as detailed in the ESI.†
Absorption spectra of polymers in DCM were obtained by UV-vis spectrometer (Thermo Scientific Evolution™ 201/220) in the spectral range from 200 nm to 800 nm at a resolution of 1 nm with a Quartz cuvette.
The ATR-FTIR analysis was performed using a PerkinElmer Spectrum Two™ by 32 scans from 4000 cm−1 to 400 cm−1, with a resolution of 4 cm−1 at room temperature. Background subtraction and baseline correction were applied prior to data analysis.
The mechanical properties of the elastomers were investigated using Zwick/Z010 universal testing machine according to ASTM D412 with crosshead speed of 100 mm min−1 at room temperature. Young's modulus was calculated from the initial slope (20% strain) of the stress–strain curve. At least four specimens per sample were tested for reproducibility.
Differential scanning calorimetry (DSC) experiments were performed using a PerkinElmer DSC8000 with temperature range from −100 °C to 100 °C at a heating and cooling speed of 10 °C min−1 under N2 flow.
Rheological behaviour of polymers was investigated using a Haake Mars III rheometer in parallel-plate geometry with a plate diameter of 8 mm. Frequency sweeps from 0.01 to 100 Hz were run at 0.1% strain at 25 °C. Temperatures sweeps were measured from 25 °C to 250 °C at 1 Hz fixed frequency.
Leaching of Ni ions into water was studied with leaching tests. To reproduce the reversible adhesion test conditions, 1.0 g of polymer was immersed in 150 mL deionized water (liquid/solid ratio of 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (L kg−1)). The extraction bottle was shaken with a shaker at room temperature for 1 week. Aliquots were taken after 1 hour, 1 day and 1 week extraction times and analysed with an ICP-MS for Ni2+.
:1 (L kg−1)). The extraction bottle was shaken with a shaker at room temperature for 1 week. Aliquots were taken after 1 hour, 1 day and 1 week extraction times and analysed with an ICP-MS for Ni2+.
2.3 Self-healing and recyclability tests
For the self-healing studies, the elastomers were cut into two pieces, then put together in air or in water and left to heal at 25 °C for 24 h. The healed interfaces were observed using optical microscopy. Stress–strain curves of the healed samples were obtained using the same procedure described above for the as prepared and uncut samples. The self-healing efficiencies for the different characteristic mechanical parameters were calculated as Xhealed/Xvirgin × 100%, where X is elastic modulus, yield stress, stress at break and strain a break obtained from the stress–strain curves.To investigate recyclability, the samples were cut into small pieces and re-moulded under 200 bar pressure for 15 min at room temperature. The re-moulded dog-bone shaped samples were then tested mechanically to obtain the stress–strain curves using the same procedure described above. The recycling protocol was repeated three times.
2.4 Underwater adhesion tests
Underwater adhesion strength was measured with a handmade setup using a Zwick/Z010 universal testing machine. The test setup consists of two parts: (1) a glass container with deionized water inside at the bottom; and (2) a steel bar connected to a load cell at the top. The polymers were prepared as disks of 12 mm diameter and 2 mm thickness. Prior to immersion, the polymer pieces were glued to the steel bar using super glue. The target substrates (Teflon, aluminium, steel, wood, and glass) were cut as squares of 20 mm × 20 mm and glued to the base of the glass container using super glue prior to immersion. During the test, the upper metallic stud approaches the target substrate and makes contact at a contact pressure of 3 N. The sample remains at this contact pressure for 1 min. Once passed this fixed time, the probe is retracted at a constant rate of 20 mm min−1 until the de-bonding is achieved. The force, displacement, and time were recorded during the process at an acquisition rate 100 data points per s. Representative adhesion test curves on a glass surface are shown in Fig. S11.† For each substrate-polymer couple the test was repeated three times for reproducibility.3. Results and discussion
3.1 Material design and characterizations
In order to achieve healable, recyclable, and underwater adhesive polymer networks, a polymer design strategy based on metal–ligand coordination as reversible crosslinks in hydrophobic polymer networks was selected (Fig. 1a and S1†). The material selection strategy envisioned the use of a flexible hydrophobic polymeric backbone for surface water repellence to prevent the formation of hydration layers at the polymer–substrate interface,34 and the presence of dynamic hydrophilic ligands (non-covalent ligands) able to reorient in the presence of humidity at the surface. This strategy allowed developing polymers with reversible underwater adhesion to hydrophobic and hydrophilic surfaces (Fig. 1b and c) and accelerated interfacial polymer–polymer healing in the presence of water (Fig. 1d).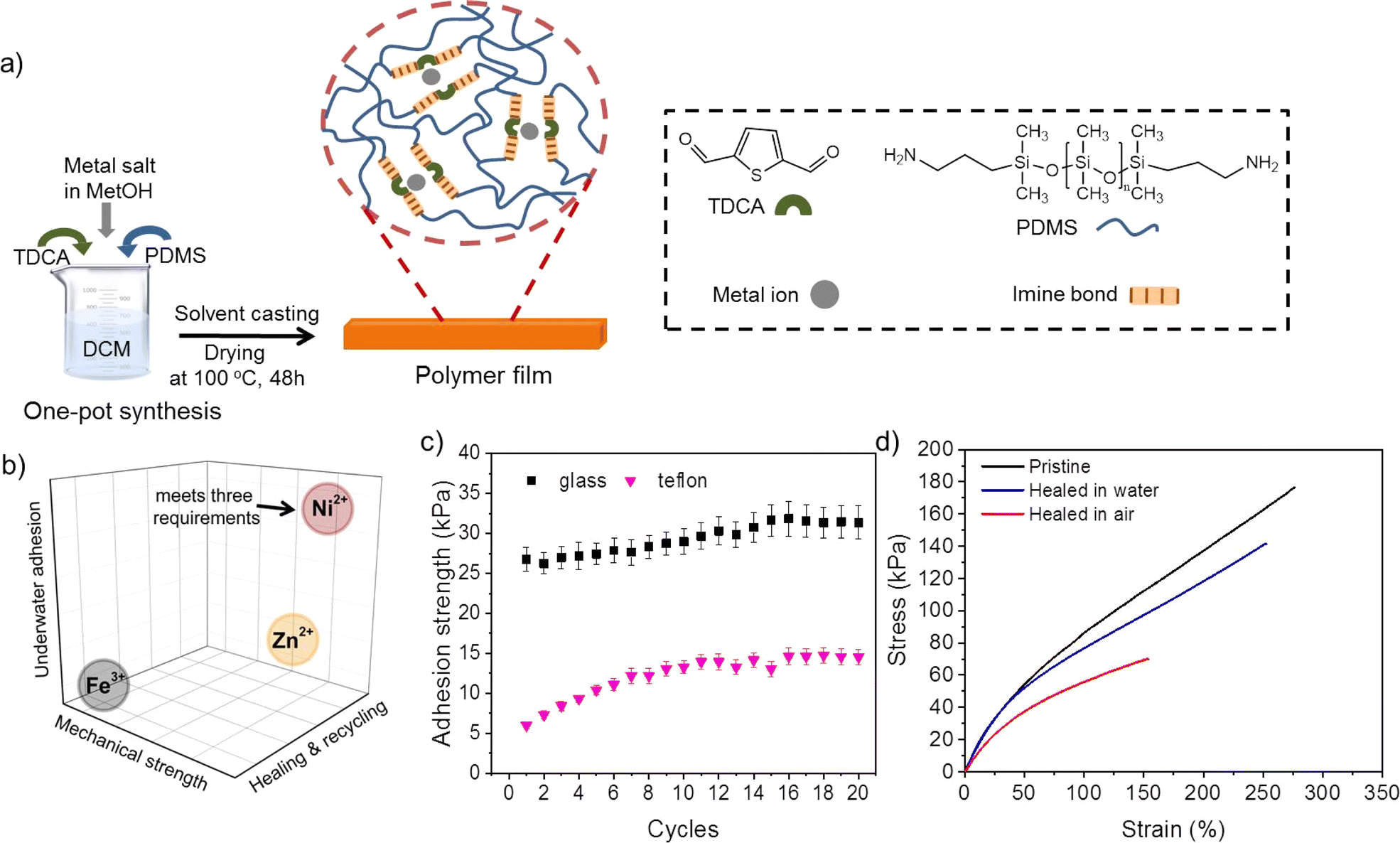 | ||
| Fig. 1 (a) Schematic illustration of the steps followed to synthesize PDMS-metalorganic water healable and reversible underwater adhesives; (b) Qualitative representation of the self-healing, recycling, and underwater adhesion properties of PMDS-imine metalorganics as function of the metal complexing cation. Plot shows Ni cation leads to the best property combination for a healable underwater reversible adhesive; (c) Underwater adhesive strength of 2,5-thiophenedicarboxaldehyde-PDMS-Ni (QTNi) to hydrophobic (Teflon) and hydrophilic (glass) surfaces as function of the attach–detach cycles; (d) Stress–strain curves of QTNi after cutting and subsequent healing in air and in water for 24 h at 25 °C. Plot shows how water accelerates interfacial healing of cuts. | ||
To demonstrate the idea, PDMS was selected as the polymer network due to high chain flexibility, low toxicity, chemical adaptability, and its hydrophobic nature. To include the dynamic ligands a 5 kDa amine-terminated PDMS was chain extended using one-pot synthesis via Schiff base formation between the amine in the PDMS and a di-aldehyde ligand (2,5-thiophenedicarboxaldehyde, TDCA). This leads to polymer chains of approximately 35 kDa (Fig. S2†) containing imine bonds (2,5-thiophenedicarboxaldehyde-PDMS polymer, QT). Imine bond formation between PDMS and TDCA was confirmed by UV-vis spectroscopy and FT-IR spectroscopy analyses (Fig. S3†): detection of an absorbance band at 317 nm in the UV-vis spectrum corresponding to π → π* transition of imine bond (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and detection of CN stretching peak located at 1634 cm−1 in the FT-IR spectra. In order to introduce sufficient mechanical properties beyond those given by entanglements and to increase the stability of the imine bonds yet implementing dynamic non-covalent crosslinks, a range of metallic salts were used. Fe3+, Zn2+ and Ni2+ were chosen as complexing metal ions due to their different affinities to the ligand used (thiophene) leading to complexes with different bond strengths. The complexation reaction was confirmed in UV-vis as a shift to higher wavelength of the imine-related absorbance band at 317 nm (Fig. S3†) due to metal ion coordination to nitrogen atom in the imine bond. QTNi (Ni complex of QT) shows a broader absorbance band at 440 nm as expected for nickel ligand coordination bonds.35 QTZn polymer (Zn complex of QT) has metal–ligand absorbance band at 364 nm slightly overlapping with the absorbance band of imine bond. Unlike Ni and Zn coordinated polymers, QTFe polymer (Fe complex of QT) shows two absorbance bands at 412 nm and 487 nm. This indicates QTFe has various complexation structures not further identified in this work yet attributed to the higher complexation diversity of Fe3+. The complexation in the polymers was further revealed in FTIR as the CN stretching peak signal related to imine at 1634 cm−1 shifted to higher wavelengths after coordination (Fig. S3†). UV and FTIR therefore confirmed the successful polymerization and metal complexation in octahedral geometry of the three showcased polymers based on thiophene ligand schematized in Fig. 1a. The midpoint Tg of all the metalorganic polymer networks was below room temperature (5 °C for Fe, −1 °C for Ni and −14 °C for Zn), Fig. S4.† This, together with the dynamic nature of the metalorganic interactions, provides a reliable basis for the rapid movement of molecular chains at ambient temperature needed for healing of macroscopic damages.
N) and detection of CN stretching peak located at 1634 cm−1 in the FT-IR spectra. In order to introduce sufficient mechanical properties beyond those given by entanglements and to increase the stability of the imine bonds yet implementing dynamic non-covalent crosslinks, a range of metallic salts were used. Fe3+, Zn2+ and Ni2+ were chosen as complexing metal ions due to their different affinities to the ligand used (thiophene) leading to complexes with different bond strengths. The complexation reaction was confirmed in UV-vis as a shift to higher wavelength of the imine-related absorbance band at 317 nm (Fig. S3†) due to metal ion coordination to nitrogen atom in the imine bond. QTNi (Ni complex of QT) shows a broader absorbance band at 440 nm as expected for nickel ligand coordination bonds.35 QTZn polymer (Zn complex of QT) has metal–ligand absorbance band at 364 nm slightly overlapping with the absorbance band of imine bond. Unlike Ni and Zn coordinated polymers, QTFe polymer (Fe complex of QT) shows two absorbance bands at 412 nm and 487 nm. This indicates QTFe has various complexation structures not further identified in this work yet attributed to the higher complexation diversity of Fe3+. The complexation in the polymers was further revealed in FTIR as the CN stretching peak signal related to imine at 1634 cm−1 shifted to higher wavelengths after coordination (Fig. S3†). UV and FTIR therefore confirmed the successful polymerization and metal complexation in octahedral geometry of the three showcased polymers based on thiophene ligand schematized in Fig. 1a. The midpoint Tg of all the metalorganic polymer networks was below room temperature (5 °C for Fe, −1 °C for Ni and −14 °C for Zn), Fig. S4.† This, together with the dynamic nature of the metalorganic interactions, provides a reliable basis for the rapid movement of molecular chains at ambient temperature needed for healing of macroscopic damages.
To understand the relation between coordination bond strength, mechanical properties and healing, DFT quantum calculations and mechanical testing in quasi-static loading were performed. For DFT, we designed a model structure with QT and different metal complexes formed by different cations. As the PDMS backbone is not expected to have any major contribution on the metal–ligand bond energy, the polymer backbone was represented as a –CH3 group as proposed elsewhere.28 The calculated bond energies of the complexes (ion-N and ion-S) shows that the overall coordination bond energy increases in the order Ni2+ > Zn2+ > Fe3+ (Fig. S5†).
In this kind of metalorganic dynamic polymers with the same backbone Mw (35 kDa), three factors govern the mechanical response: physical crosslinking density and strength provided by the metalorganic interactions, entanglements dominated by Mw and rate of the metalorganic dynamic interactions. Fig. S6,† shows representative stress–strain curves of the pristine polymers (as produced). Fe3+ complexes lead to brittle polymers with the highest elastic modulus (E), no yield strength (σy) and lowest strain at break (εb). Zn2+ and Ni2+ show clear elastic and plastic regions with more marked strain hardening for the Ni2+ sample. Table 1 summarizes the most relevant characteristic parameters: mechanical (E, σy, σb, εb), bond energy and crosslinking density (ν) calculated as shown in the ESI.†
| Samples | E (MPa) | σ y (MPa) | σb (MPa) | ε b (%) | Bond energy (kcal mol−1) | ν ×10−6 (mol g−1) |
|---|---|---|---|---|---|---|
| QTZn | 0.06 ± 0.009 | 0.010 ± 0.001 | 0.083 ± 0.003 | 303 ± 12 | 277 | 1.7 ± 0.12 |
| QTNi | 0.12 ± 0.016 | 0.023 ± 0.003 | 0.180 ± 0.007 | 272 ± 9 | 453 | 14.8 ± 0.08 |
| QTFe | 0.38 ± 0.018 | — | 0.085 ± 0.008 | 27 ± 5 | 134 | 34.9 ± 0.23 |
The mechanical behaviour of the Fe3+ samples can be explained by the fast complexation reaction observed in solution (immediate colour change) leading to relatively higher physical crosslinking density, as further confirmed by swelling tests (Table S1†) where Fe3+ polymers show the highest crosslinking density of the three metallopolymers (∼35 × 10−6 mol g−1) and highest Tg. It is hypothesized that the rapid crosslinking (complexation) reaction does not allow maximizing entanglements but leads to high metalorganic crosslinking density. This leads to higher elastic modulus and Tg, lower σb and brittle behaviour (Fig. S6†). Ni-metalorganic polymers, on the other hand, show half the physical crosslinking density of the Fe-complexes (∼15 × 10−6 mol g−1) in a slower reaction process (colour change happening later in time) that allows for more entanglements to take place in the reactive solution. This better balance in entanglements and physical crosslinking density with higher bond strength (453 kcal mol−1vs. 134 kcal mol−1 based on DFT) leads to higher (yield) strength and a significant strain hardening at ten times higher elongation (270% vs. 27%) yet with a lower elastic modulus (0.12 vs. 0.4 MPa). We argue that, for comparable crosslinking density and Mw, the relatively high complexation rate of stronger Ni-complexes positively contributes to the strain hardening in a process similar to stick-slip reported for other self-healing polymers relying on H-bonding and other reversible physical bonds.36,37 Zn-metalorganic polymers show the highest elongation and lowest strength and elastic modulus (Table 1). Following the same rationale, this is attributed to a relatively low cross-linking density (∼2 × 10−6 mol g−1), relatively low bond strength but comparable entanglement density as the Ni-based polymers.
Healing behaviour was tested by cutting the polymers in two parts, bringing them in contact in air and in water and letting the polymers heal at atmospheric pressure and 25 °C for a maximum of 24 h in air or in water (Movie S1†). After healing, the samples were taken out from the healing environment, dried and tested manually and with a universal testing machine in quasi-static tensile loading (see Experimental section and ESI†). Fig. 2a shows how all the samples were able to hold manual bending and stretching without visual failure. Fig. 2b–d, show representative stress–strain curves of the healed samples in air (red lines) and in water (blue lines) compared to the as-produced samples (black lines).
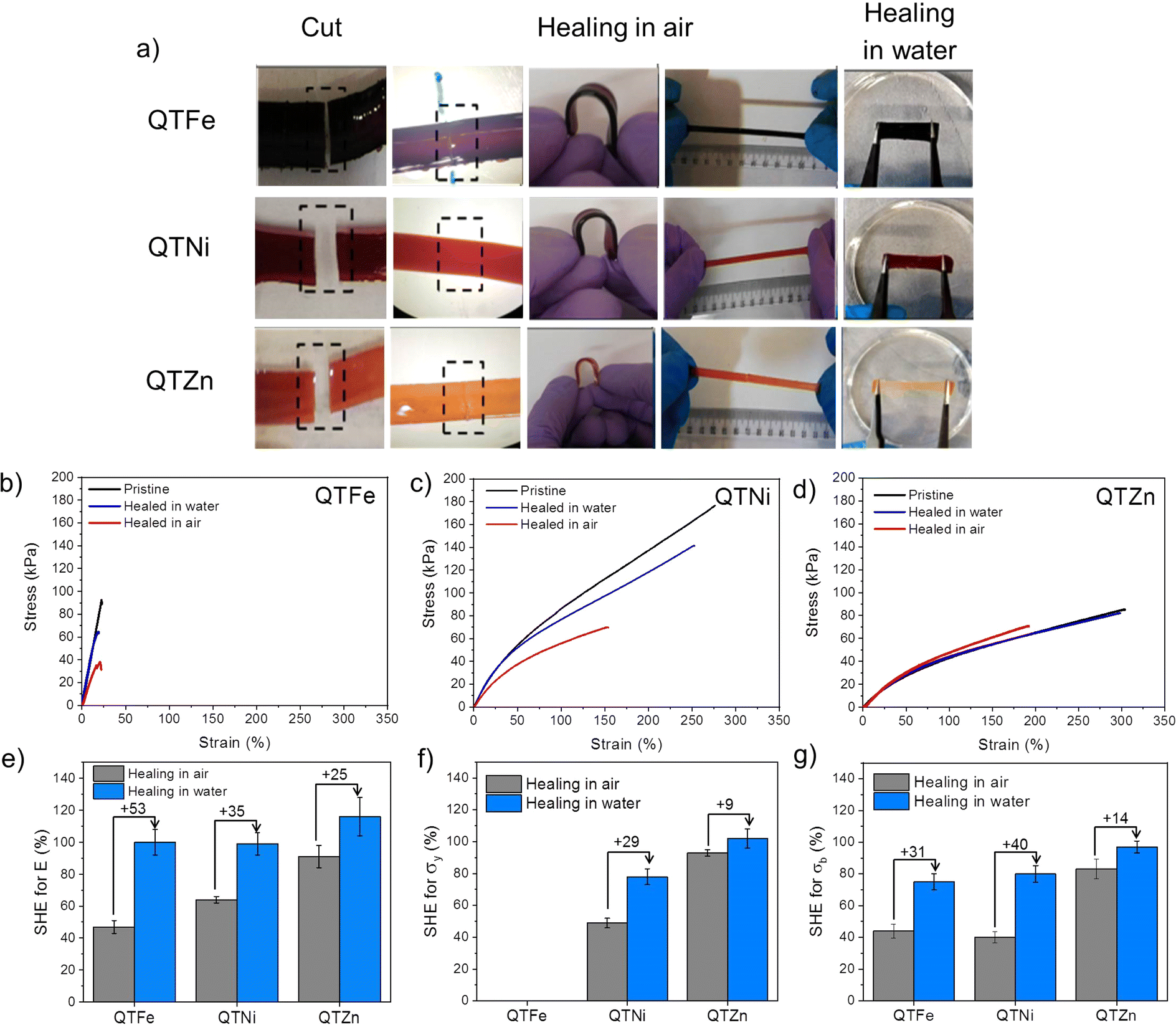 | ||
| Fig. 2 Comparation of self-healing properties of metallopolymers. (a) Images of damaged and healed polymers; (b)–(d) show representative stress–strain curves of original and healed in air and water of QTFe, QTNi and QTZn metallopolymers, respectively; (e)–(g) show self-healing efficiency values in air and in water of elastic modulus, yield strength and strength at break for the three metallopolymers. Figure shows the high level of healing achieved for all polymers in air and water and the increase of healing efficiency of all mechanical characteristic parameters when exposed to water. | ||
3.2 Self-healing and recycling properties
The self-healing efficiency (SHE) of the three metallopolymers for the characteristic mechanical parameters studied (E, σb, σy) was found to be significantly higher when healed in water than in air (Fig. 2e–g). These results confirm a water-accelerated healing mechanism for this kind of polymers. Overall, Zn polymers showed the highest healing efficiencies although differences between polymers decreased when healed in water, reaching 80–100% healing in all parameters for all samples. The samples with the highest cross-linking density (Fe and Ni) show comparable healing efficiencies in air and in water, independently of the bond strength and Tg differences. The results suggest a predominant role of the crosslinking density and chain mobility over the physical crosslink bond strength in the healing behaviour of these polymers.In order to test the effect of the cation on the recyclability of the metalorganic polymers a recycling protocol was implemented consisting in cutting the polymers in pieces and remoulding them with pressure (see Fig. 3 and ESI† for details). Since the polymers were able to heal at room temperature the recycling was planned at 25 °C. Temperature sweep rheology at 1 Hz was performed to identify possible G′/G′′ crossover points that could help selecting a different recycling or healing temperature (i.e. when G′′ > G′). Nevertheless, no T-dependent crossover was observed for any polymer before terminal flow at T > 200 °C (Fig. S7†). Unlike for most previously reported healable metalorganic polymers, the results indicate that healing and recycling is not temperature dependent at temperatures below terminal flow and is not dominated by macroscopic viscous flow. Fig. 3a and b show the stress strain curves of the recycled samples after three repeated recycling events. It should be noted that Fe-metalorganic could not be recycled under the same conditions (Fig. S8†). This is attributed to the high crosslink density and rapid complexation preventing sufficient chain reptation (interdiffusion). Ni and Zn show the same mechanical behaviour and colour after three recycling events. Nevertheless, and as for the healing experiments, the Ni samples showed lower mechanical properties after the first recycling event (close to 50%) while Zn-metallopolymer (QTZn) showed no significant mechanical loss after recycling.
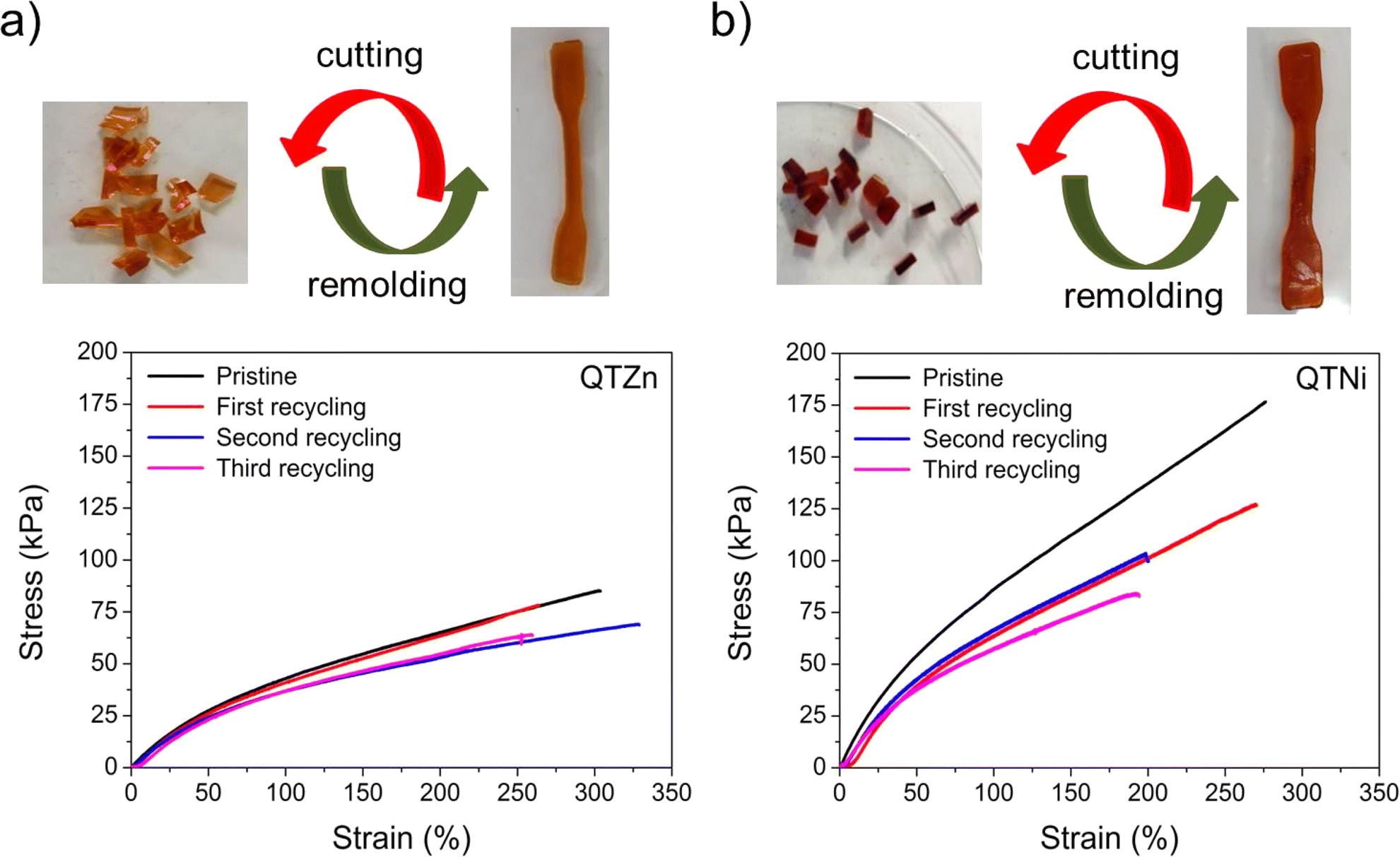 | ||
| Fig. 3 Recycling behavior of QTZn and QTNi (QTFe could not be recycled). (a) and (b) show the stress–strain behavior of the recycled samples (up to three recycling cycles) compared to the pristine samples for the QTZn and QTNi, respectively. The figure shows that both QTZn and QTNi elastomers can be recycled multiple times by pressure at room temperature. QTZn maintains most properties after recycling while QTNi loses mechanical properties in the first recycling event. | ||
3.3 Underwater adhesion properties
The adhesive properties of the developed healable and recyclable metallopolymers (QTNi and QTZn) to various surfaces in air and in water was initially observed by bringing two similar surfaces in contact and manually stretching the adhesive bond as shown in Fig. 4a and S9 of the ESI.† During this preliminary test, QTZn showed some tack to surfaces in air, but no adhesion to any tested surface when immersed in water. QTNi, on the other hand, showed adhesion in air and in water, to all the materials tested with varying surface energies (wood, glass, Teflon (PTFE)), aluminium, and steel) as shown in Fig. 4 and S9 of the ESI.† This remarkable result of adhesion in water of the nickel coordinated polymer has not been reported earlier, presumably because most works on underwater adhesion using mussel-bioinspired chemistries focus on the role of Fe3+/Fe2+ and Zn2+ coordination38,39 due to their major role on byssus adhesion.40 Given the potential toxicity of Ni ions in solution, the binding efficiency of Ni2+ in QTNi was evaluated by immersing the polymer in water for one week and taking aliquots at different immersion times (see Experimental section and Fig. S10 in the ESI†). The leaching experiment demonstrated that the maximum Ni leaching was achieved in the first day of immersion at a concentration of 1.5 μg L−1 in 150 mL water, much lower than maximum allowed nickel concentration (223.5 μg L−1 in drinking water according to World Health Organization (WHO).41 This result confirms the good fixation of the Ni to the polymer network and confirms the low toxicity of the developed Ni-metallopolymers in a potential working environment underwater.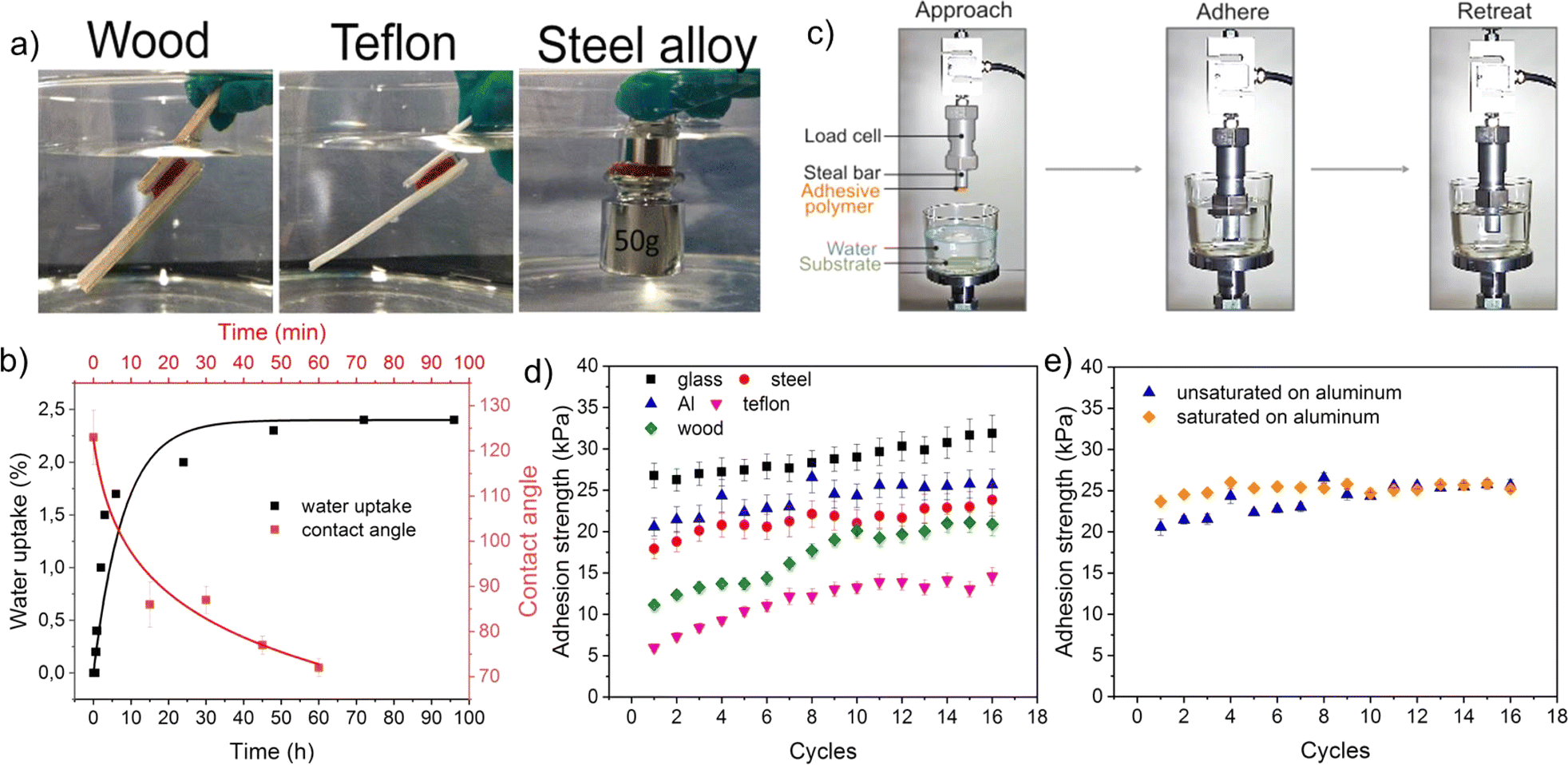 | ||
| Fig. 4 Reversible adhesion of polymers in water: (a) test setup used to measure cyclic adhesion under water; (b) visualization of the underwater adhesion of QTNi to different surfaces; (c) Underwater adhesion strength of QTNi to various substrates during adhesion–deadhesion cyclic testing. During the cyclic test the samples remain in water for 1.5 hours. The adhesion of polymer is reversible due to reversible dynamic bonds at the polymer surface. Adhesion strength is related to surface chemical structure of the substrate with higher adhesion to the more hydrophilic surfaces; (d) contact angle and water uptake values of QTNi over time; (e) underwater adhesion strength of unsaturated and water-saturated QTNi on an aluminium surface. Figure shows that cycles have no effect on the adhesion strength of the saturated polymer. Values of the saturated and unsaturated become equal and constant after 7 adhesion cycles. | ||
We argue that the polar groups such as imine bond, metal ion and amine and aldehyde end groups in the polymer are responsible for the interfacial adhesion with the surfaces through dipole–dipole interactions and hydrogen bonding. Additionally, the metal ion present in the polymer may form coordination bonds with the polar groups on the substrate surface. Water at the interface contributes to this interfacial adhesion process by inducing chain reorientation (hydrophobic chains reorient inwards) allowing more hydrophilic groups at the polymer surface as exemplified by the rapid water contact angle drop shown in Fig. 4b for QTNi. Despite containing the same polar groups and polymer backbone, QTNi and QTZn exhibit clear differences in underwater adhesion. As revealed by DFT calculations, the coordination bond formed with the thiophene ligand is stronger with nickel than with zinc. The weaker coordination bond in QTZn is responsible for lower cohesive strength while the stronger bonds in QTNi provide cohesive strength needed to facilitate water-induced chain reorientation of the PDMS and to allow the polymer to adhere more strongly to the substrates.
In order to test the reversible underwater adhesion of QTNi, an adhesion test was performed. As can be seen in Movie S2†, the QTNi polymer exhibits good adhesion to Teflon (and other surfaces) under water. After removing the Teflon plate from the water, a slight bending of the stud with the solid adhesives induces the detachment of the adhesive. This experiment was repeated up to 3 times (Movie S2†). To quantify the reversibility of the interfacial adhesion of QTNi in air and in water, a specific adhesion test set-up was designed as shown in Fig. 4c and detailed in the Experimental section. The mechanical adhesion–deadhesion tests were performed in air and in water and repeated up to 20 times (a total of 1.5 hours under immersion) as shown in Fig. S11 of the ESI†and exemplified in Movie S3.†Fig. 4d shows the adhesion strength of QTNi to all the substrates studied underwater during 16 cycles. Overall, QTNi is able to reversibly adhere under water to both hydrophilic and hydrophobic surfaces but adhesion strength is highest for the most hydrophilic surfaces. The lowest adhesion strength of QTNi to Teflon (∼15 kPa at 16 cycles) can be attributed to the weaker dipole–dipole interaction between the –CF3 groups on the substrate surface and the predominantly polar groups of the QTNi polymer in water (Fig. 4b shows how the contact angle of water in the polymer, initially hydrophobic, rapidly decreases when exposed to water). In line with this, the highest underwater adhesion strength is observed for the glass surface (∼32 kPa at 16 cycles). This is attributed to the lowest surface energy of glass with higher number of –OH polar groups able to form hydrogen bonds and coordination bonds with the metalorganic polymer. Interestingly, the reversible adhesion strength in air was the same as in water (Fig. S12†).
During the cyclic adhesion tests in water, it was observed that the adhesion strength increases over time for all surfaces (stabilizing at around 10 cycles, Fig. 4d). However, when the experiments were performed in air, no increase of the adhesion strength was observed with the cycles (Fig. S12†). The cyclic testing did not have any measurable effect on the viscoelastic properties of the polymer that could explain adhesion strength increase (see frequency sweep rheology of the polymer before and after cyclic adhesion testing in water in Fig. S13†). The low water uptake rate also indicates that capillarity does not play a role in adhesion and adhesion increase in this case (2.7 %wt. of water after 100 hours duration and close to 0 during the 1.5 h duration of the cyclic adhesion test as shown in Fig. 4b). Contact angle evolution with time revealed on the other hand a rapid transition of the polymer surface energy from hydrophobic (120°) to hydrophilic (70°) during the first hour of immersion (corresponding to around 13 cycles in the adhesion test). This points at a water-induced chain reorientation as the dominating phenomena for adhesion and adhesion strength increase. To further analyse the possible role of chain orientation on adhesion in water and to discard the role of water uptake on adhesion, a separate test was performed. For this, the QTNi polymer was soaked in water until it was fully saturated. This sample was then used to perform cyclic adhesion tests in water on an aluminium surface. As shown in Fig. 4e, the cyclic underwater adhesion strength of the fully water-saturated sample is comparable to that of the as-produced QTNi polymer. Moreover, the adhesion strength of the water-saturated polymer remains high and stable since the first cycle, in line with the idea of a completed surface chain reorientation in water.
In line with the above results, we propose that a chain reorientation at the polymer surface in the presence of water (Fig. 5) is behind the exceptional adhesion properties of the polymer in water to all substrates as well as behind the gradual increase of adhesion strength with the cycles. This hypothesis is in good agreement with a recent study reporting, for a different chemistry, a surface chain reorientation mechanism in the presence of water.42 However, previous studies did not demonstrate the effect of re-orientation on healing or on reversible adhesion in water. We argue that when the polar groups in the polymer (ionic coordination and imine bonds amongst others) get in contact with polar water molecules, they rearrange themselves towards the surface at the same time the more hydrophobic PDMS backbone moves inwards. This process increases the number of polar groups at the polymer surface, leading to an increase in underwater adhesion strength with time until the polymer is fully rearranged. We propose that this mechanism is also behind the higher and faster healing efficiency observed in water than in air (Fig. 2e–g). In healing, the surface re-organization of the polar and non-polar groups and a local crosslinking density decrease related to chain re-arrangement allows for an improved encounter probability of reactive groups and interfacial diffusion and therefore faster and improved mechanical strength restoration (i.e., mechanical healing). The low Tg values appears as a minimum requirement to allow sufficient molecular chain mobility needed for healing and adhesion. The role of the cation and the metal coordination strength seems to play a major in this process since, at an even lower Tg, QTZn did not show underwater adhesion but did show healing in water. We hypothesize that the weaker coordination bond in QTZn and its dynamic nature, allows the polymer chains to move more easily during reorientation in water, leading to a higher number of active sites on the surface, and to a drop in apparent crosslinking density at the surface. This allows the underwater self-healing efficiency of QTZn to be as high as for the QTNi. But at the same time, weak bonding ability of Zn causes weak polymer–surface interactions and provides weaker adhesion.43 A static contact angle test with olive oil (Fig. S14†) showed that the chain reorientation occurs rapidly (<4 min) leading to a transition to more oleophilic character and demonstrates the high surface mobility and polymer chain rearrangement character of the QTNi polymer.
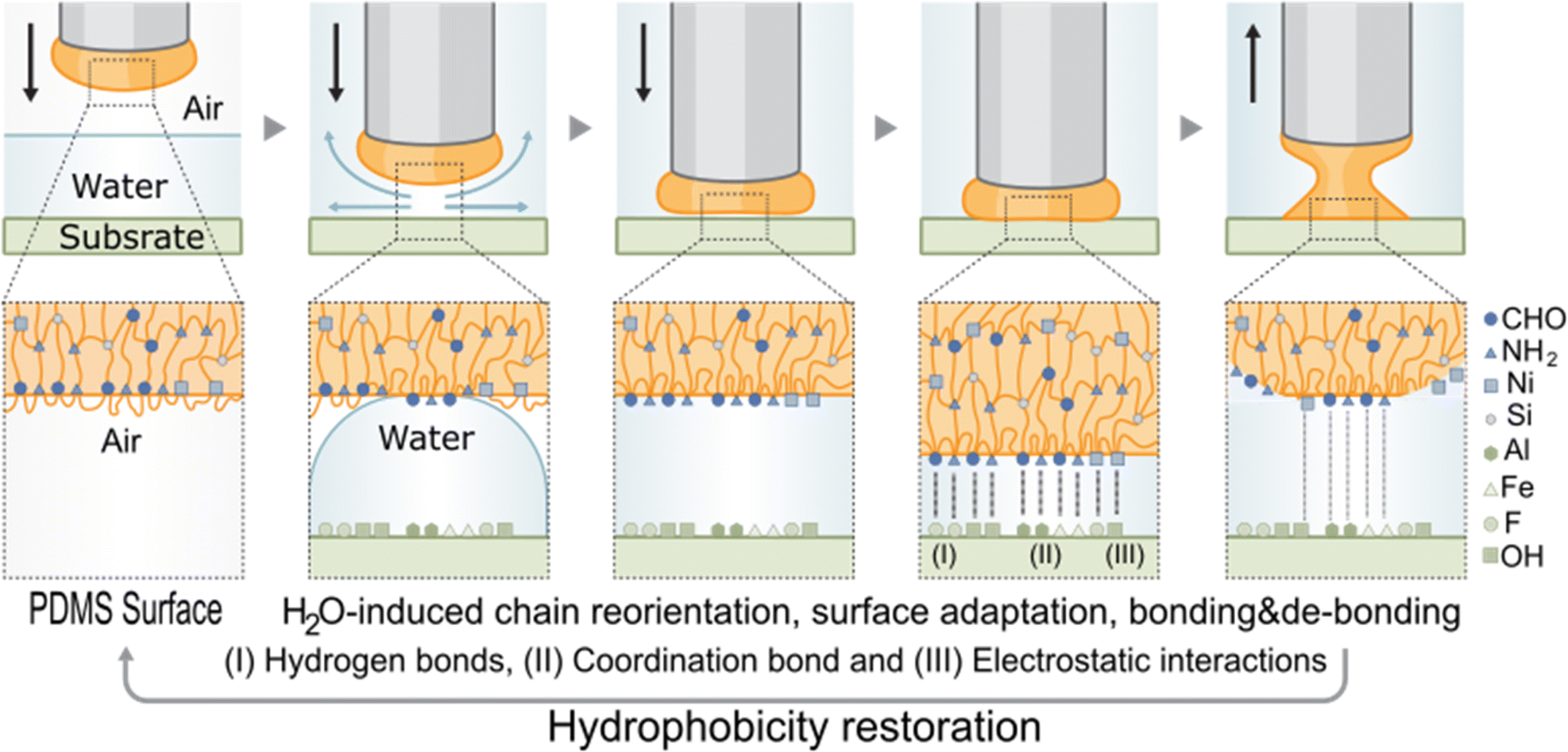 | ||
| Fig. 5 Underwater adhesion mechanism of QTNi showing reorientation of chains and repositioning of the hydrophilic groups in the polymer towards the surface. | ||
To further prove the idea of water-induced surface orientation, the adhesion of a commercial double-side tape in air and water to aluminium was compared to the reversible adhesion provided by QTNi (Fig. S12†). The adhesion of the commercial double-sided tape decreased over time with the cycles when tested in air to reach, after 4 cycles, values comparable to those obtained in water, probably due to a deactivation or removal of the active surface chemistry. The adhesion values of the double side tape to aluminium are furthermore significantly lower than those of QTNi to aluminium and comparable to QTNi adhesion to Teflon. We attribute this to the absence of a specific adapting technology in the case of the commercial double side tape to repel water and to stand numerous cyclic adhesion tests but confirms that not every polymer that exhibits adhesion in air will also exhibit adhesion underwater as presented here.
4. Conclusions
In this work, a new strategy to develop self-healable and recyclable tape-type adhesives able to reversibly adhere to hydrophobic and hydrophilic surfaces in air and in water is presented. To achieve this combination of properties a metallopolymer network was synthesized using a hydrophobic polymer backbone (amine terminated PDMS) extended with a thiophene ligand via imine bonds and crosslinked by metal complexation using one-pot synthesis. Three different metal ions (Zn2+, Ni2+ and Fe3+) with different Lewis acidity were used for complexation. This lead to polymers with a range of crosslinking densities and mechanical properties (from brittle to elastomeric). All metallopolymers showed good healing behaviour in air and accelerated and more efficient healing in water. Nevertheless, only the Ni-based metallopolymer showed reversible adhesion to all surfaces in air and in water. Both Ni and Zn based polymers showed good recyclability. Reversible adhesion of Ni2+-2,5-thiophenedicarboxaldehyde (QTNi) in air and in water to hydrophilic surfaces such as glass, and hydrophobic surfaces such as Teflon, was demonstrated for 20 cycles with no loss of the adhesion strength. We demonstrate that the underlying adhesion mechanism is due to an increase in polar groups at the surface in the presence of water due to a water-induced chain reorientation process of the hydrophobic backbone facilitated by the presence of relatively strong but dynamic Ni-based metal coordination. This work provides fundamental insights relevant for the development of surface adaptive polymers leading to adhesion and healing in the presence of water of interest in applications as diverse as artificial tissue engineering, underwater soft robotics, energy devices using water, strain sensors, and reversible adhesives for temporary repairs.Data availability
The data underlying this study are openly available via a link to 4TU.ReaserchData online repository (https://doi.org//10.4121/7ad0b47d-5dcf-4918-af1e-9fd7b082de07).Author contributions
Elif Kaymazlar: conceptualization, investigation, funding acquisition, writing – original draft preparation, methodology, visualization. Omer Andac: writing – review & editing, funding acquisition. Santiago J. Garcia: conceptualization, supervision, project administration, writing – original draft preparation, writing – review & editing, methodology, visualization, resources, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the economic support from the Scientific and Technological Research Council of Turkiye (TUBITAK) under Research Project 120Z846, TUBITAK BIDEB International Research Fellowship Programme awarded to E. K., the department of Aerospace Structures and Materials at the TU Delft for financial support and Dave Ruijtenbeek (TU Delft) for his support in the design of the testing device for underwater adhesion.References
- J. Chen, D. Wang, L. H. Wang, W. Liu, A. Chiu, K. Shariati, Q. Liu, X. Wang, Z. Zhong, J. Webb, R. E. Schwartz, N. Bouklas and M. Ma, Adv. Mater., 2020, 32, 2001628 CrossRef CAS PubMed.
- C. Christianson, C. Bayag, G. Li, S. Jadhav, A. Giri, C. Agba, T. Li and M. T. Tolley, Front. Robot. AI, 2019, 6, 126 CrossRef PubMed.
- J. Shin, J. S. Lee, C. Lee, H.-J. Park, K. Yang, Y. Jin, J. H. Ryu, K. S. Hong, S.-H. Moon, H.-M. Chung, H. S. Yang, S. H. Um, J.-W. Oh, D.-I. Kim, H. Lee and S.-W. Cho, Adv. Funct. Mater., 2015, 25, 3814–3824 CrossRef CAS.
- Y. Tang, Q. Zhang, G. Lin and J. Yin, Soft Robot., 2018, 5, 592–600 CrossRef PubMed.
- H. Zheng, J. Li, Y. Zhou, C. Zhang, W. Xu, Y. Deng, J. Li, S. Feng, Z. Yi, X. Zhou, X. Ji, P. Shi and Z. Wang, Nat. Commun., 2022, 13, 4584 CrossRef CAS PubMed.
- B. Cheng, J. Yu, T. Arisawa, K. Hayashi, J. J. Richardson, Y. Shibuta and H. Ejima, Nat. Commun., 2022, 13, 1892 CrossRef CAS PubMed.
- X. T. Ma, X. Zhou, J. J. Ding, B. Huang, P. Y. Wang, Y. Zhao, Q. Y. Mu, S. H. Zhang, C. G. Ren and W. L. Xu, J. Mater. Chem. A, 2022, 10, 11823–11853 RSC.
- Y. Wang, V. Kang, W. Federle, E. Arzt and R. Hensel, Adv. Mater. Interfaces, 2020, 7, 2001269 CrossRef.
- T. Nguyen, W. E. Byrd, D. Alshed, J. Chin, C. Clerici and J. Martin, J. Adhes., 2007, 83, 587–610 CrossRef CAS.
- M. Nijemeisland, Y. V. Meteleva-Fischer and S. J. Garcia, Int. J. Adhes. Adhes., 2022, 118, 103246 CrossRef CAS.
- W. Tian, L. Liu, F. Meng, Y. Liu, Y. Li and F. Wang, Corros. Sci., 2014, 86, 81–92 CrossRef CAS.
- C. Cui, C. Fan, Y. Wu, M. Xiao, T. Wu, D. Zhang, X. Chen, B. Liu, Z. Xu, B. Qu and W. Liu, Adv. Mater., 2019, 31, 1905761 CrossRef CAS PubMed.
- X. Su, W. Xie, P. Wang, Z. Tian, H. Wang, Z. Yuan, X. Liu and J. Huang, Mater. Horiz., 2021, 8, 2199–2207 RSC.
- Y. Wang, D. Liu, C. Wang, J. Wu, X. Xu, X. Yang, C. Sun, P. Jiang and X. Wang, Chem. Eng. J., 2023, 457, 141268 CrossRef CAS.
- H. Fan and J. P. Gong, Adv. Mater., 2021, 33, 2102983 CrossRef CAS PubMed.
- S. Huang, J. Sun, H. K. Bisoyi, Y. Tao, J. Wang, Y. Xu, Y. Huang, M. Wang, H. Yang and Q. Li, Chem. Mater., 2023, 35, 3705–3712 CrossRef CAS.
- S. Li, H. Tian, J. Shao, H. Liu, D. Wang and W. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 39745–39755 CrossRef CAS PubMed.
- P. Rao, T. L. Sun, L. Chen, R. Takahashi, G. Shinohara, H. Guo, D. R. King, T. Kurokawa and J. P. Gong, Adv. Mater., 2018, 30, 1801884 CrossRef PubMed.
- C. K. Roy, H. L. Guo, T. L. Sun, A. B. Ihsan, T. Kurokawa, M. Takahata, T. Nonoyama, T. Nakajima and J. P. Gong, Adv. Mater., 2015, 27, 7344–7348 CrossRef CAS PubMed.
- L. Xiang, J. Zhang, W. Wang, L. Gong, L. Zhang, B. Yan and H. Zeng, Acta Biomater., 2020, 117, 294–301 CrossRef CAS PubMed.
- B. J. Kim, S. Kim, D. X. Oh, A. Masic, H. J. Cha and D. S. Hwang, J. Mater. Chem. B, 2015, 3, 112–118 RSC.
- S. Mandal, A. Das, E. Euchler, S. Wiessner, G. Heinrich, J. Sawada, R. Matsui, T. Nagase and T. Tada, Rubber Chem. Technol., 2023, 96, 175–195 CrossRef CAS.
- T. Priemel, G. Palia, F. Förste, F. Jehle, S. Sviben, I. Mantouvalou, P. Zaslansky, L. Bertinetti and M. J. Harrington, Science, 2021, 374, 206–211 CrossRef CAS PubMed.
- A. Das, A. Sallat, F. Böhme, M. Suckow, D. Basu, S. Wießner, K. W. Stöckelhuber, B. Voit and G. Heinrich, ACS Appl. Mater. Interfaces, 2015, 7, 20623–20630 CrossRef CAS PubMed.
- K. V. Deriabin, N. A. Ignatova, S. O. Kirichenko, A. S. Novikov and R. M. Islamova, Polymer, 2021, 212 Search PubMed.
- H. Fu, B. Wang, J. Li, J. Xu, J. Li, J. Zeng, W. Gao and K. Chen, Mater. Horiz., 2022, 9, 1412–1421 RSC.
- L. He, D. Ye, S. Weng and X. Jiang, Eur. Polym. J., 2022, 181, 111650 CrossRef CAS.
- C.-H. Li, C. Wang, C. Keplinger, J.-L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X.-Z. You and Z. Bao, Nat. Chem., 2016, 8, 618–624 CrossRef CAS PubMed.
- J. Zhang, H. Wang, H. Feng, X. Li and N. Wu, Mater. Lett., 2023, 338, 134042 CrossRef CAS.
- S. J. Garcia, Eur. Polym. J., 2014, 53, 118–125 CrossRef CAS.
- B. K. Ahn, J. Am. Chem. Soc., 2017, 139, 10166–10171 CrossRef CAS PubMed.
- Z. Wang, J. Zhao, W. Tang, T. He, S. Wang, X. He, Y. Chen, D. Yang and S. Peng, ACS Appl. Mater. Interfaces, 2021, 13, 3435–3444 CrossRef CAS PubMed.
- H. Yang, W. Wang, C. Zhu, G. Chen, J. Zheng, S. Li, J. Ren and J. Fu, Chem. Eng. J., 2024, 480, 148064 CrossRef CAS.
- Y. Xu, Q. Liu, A. Narayanan, D. Jain, A. Dhinojwala and A. Joy, Adv. Mater. Interfaces, 2017, 4, 1700506 CrossRef.
- J. J. Rani, A. M. I. Jayaseeli, S. Rajagopal, S. Seenithurai, J.-D. Chai, J. D. Raja and R. Rajasekaran, J. Mol. Liq., 2021, 328, 115457 CrossRef CAS.
- A. Susa, R. K. Bose, A. M. Grande, S. van der Zwaag and S. J. Garcia, ACS Appl. Mater. Interfaces, 2016, 8, 34068–34079 CrossRef CAS PubMed.
- J. Verjans, A. André, E. Van Ruymbeke and R. Hoogenboom, Macromolecules, 2022, 55, 928–941 CrossRef CAS.
- S.-W. Maeng, T. Y. Park, Y. Park, T. Yoon, Y. M. Jung and H. J. Cha, Biomacromolecules, 2023, 25, 379–387 CrossRef PubMed.
- Z. Tang, M. Zhao, Y. Wang, W. Zhang, M. Zhang, H. Xiao, L. Huang, L. Chen, X. Ouyang, H. Zeng and H. Wu, Int. J. Biol. Macromol., 2020, 144, 127–134 CrossRef CAS PubMed.
- R. J. Stewart, T. C. Ransom and V. Hlady, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 757–771 CrossRef CAS PubMed.
- W. W. H. Organization, Nickel in Drinking-Water, 2021 Search PubMed.
- X. Zhang, R. Crisci, J. A. Finlay, H. Cai, A. S. Clare, Z. Chen and M. N. Silberstein, Adv. Mater. Interfaces, 2022, 9, 2200430 CrossRef CAS.
- D. S. Hwang, H. Zeng, A. Masic, M. J. Harrington, J. N. Israelachvili and J. H. Waite, J. Biol. Chem., 2010, 285, 25850–25858 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta01650e |
| This journal is © The Royal Society of Chemistry 2024 |