
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular interactions using β-cyclodextrin in controlling perovskite solar cell performance†
Parnian
Ferdowsi
a,
Sun-Ju
Kim
b,
Thanh-Danh
Nguyen
b,
Ji-Youn
Seo
b,
Jun-Ho
Yum
 *a and
Kevin
Sivula
a
*a and
Kevin
Sivula
a
aLaboratory for Molecular Engineering of Optoelectronic Nanomaterials, Institut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland. E-mail: junho.yum@epfl.ch
bDepartment of Nano Fusion Technology, Pusan National University, 46241 Busan, Republic of Korea
First published on 23rd May 2024
Abstract
In the context of perovskite solar cells (PSCs), enhancing device performance often involves adding a small excess of lead iodide (PbI2) to the precursor solution. However, the presence of unreacted PbI2 can lead to accelerated degradation compromising long-term stability. This study addresses this issue through supramolecular complex engineering by introducing beta-cyclodextrin (β-CD) into a triple cation perovskite to effectively prevent the crystallization of residual PbI2. This approach results in uniform crystal growth and the passivation of undercoordinated lead cation defects. The use of β-CD leads to a PSC with an improved power conversion efficiency (PCE) of 21.36%, surpassing the control, and enhanced stability against aggressive thermal stress and high humidity (85% RH). This is supported by optical and morphological investigations, underscoring the role of β-CD to maintain the desired perovskite phase. Notably, in comparison to the β-CD-free control, the β-CD-treated sample exhibited minimal bandgap shifts of 3 meV after 1170 hours of moisture exposure. Furthermore, this method not only passivates unreacted PbI2 but also provides valuable insights into the role of β-CD in hybrid perovskite solar cells. Additional tests with maltose as a non-cyclic control were conducted and confirm the superior ability of β-CD to enhance perovskite film stability under harsh conditions. The formation of a supramolecular system between β-CD and perovskite holds promise as a strategy to control perovskite precursor chemistry, material structure, and subsequent device performance and stability.
Introduction
In recent years, scientists have been fascinated by the remarkable properties of halide perovskite materials, which are leading the way in the development of advanced optoelectronic technologies, particularly in converting solar energy into electricity for photovoltaic systems. These materials have attracted widespread attention due to their remarkable photoelectric properties, featuring tunable band gaps, large absorption coefficients, long electron and hole diffusion lengths, low exciton binding energies, ease of fabrication, and low-cost processing.1–6 The perovskite material is structured with an ABX3 composition, consisting of an organic/inorganic monovalent cation, A (methylammonium (MA+), formamidinium (FA+), or cesium (Cs+)); a divalent metal, B (Pb2+ or Sn2+); and a halide anion, X (Cl−, Br−, or I−).7,8Perovskite solar cells (PSCs) have currently achieved impressive power conversion efficiencies (PCE) of up to 26.1%,9,10 solidifying their position as one of the frontrunners for next-generation thin-film photovoltaics. However, despite their improved efficiency, significant obstacles to their commercialization still persist.7,11 The primary obstacle hindering their practical deployment stems from their limited extrinsic stability, notably in the presence of oxygen and moisture, as well as intrinsic instability under typical device operating conditions, including voltage bias, exposure to light, and temperature fluctuations. These challenges are particularly associated with the migration of ions, weak binding and ion–electron properties of perovskite crystals, when subjected to an applied electric field.12 This can lead to a degradation of photoelectric properties, which, in turn, can adversely affect both the performance and long-term stability of PSCs.13,14
Inherent defects and grain boundaries are the main ion migration pathways within the perovskite layer. Notably, unreacted lead iodide (PbI2) at these sites, including surface of perovskite grains, and the interface between the perovskite layer and the charge transport layer,15–20 is a major cause of intrinsic instability under illumination. It can break down into iodine and metallic lead, acting as recombination centers and promoting ion migration.15,21 In stark contrast, some studies have shown that the intentional addition of a slight excess of PbI2 in the precursor for PSC fabrication can actually enhance efficiency.16,17,22–24 In this case several mechanisms that enhance the performance of these solar cells have been identified, including defect passivation in perovskite films, reduction in halide vacancy concentration, prevention of the formation of organic-rich surfaces, and enhanced charge transport.16,18–20,25–27 Moreover, the unreacted PbI2 formed at the interface between the perovskite layer and charge transport layer can effectively suppress charge recombination and improved carrier injection either due to surface passivation or band edge matching between charge transport layer, PbI2, and the perovskite.17,20,28,29 The conflicting observations that PbI2 can both enhance performance but degrade stability suggests a need for research efforts directed toward simultaneously mitigating crystallization of residual PbI2 while improving device stability, ensuring that efficiency gains do not compromise the long-term reliability of perovskite solar cells.
Numerous methods have emerged to mitigate the adverse impact of excess PbI2 in perovskite materials. For instance, a ligand modulation technique improved device performance by transforming excess PbI2 into vertically distributed PbI2 nanosheets, but it did not address PbI2 photolysis-induced instability.30 To eliminate residual PbI2 and enhance stability, a secondary growth method using organic cations and isopropyl alcohol (IPA) surface treatment was effective but requires additional effort.15,31–34 Hence, there remains a need to simultaneously eliminate residual PbI2 and enhance stability using a simple method.
In this respect there have been efforts to develop strategies to suppress ion migration, including passivation, controlling the crystal growth, and increasing ion-migration barriers through substitution.35–39 While significant efforts have been made to control ionic migration pathways, the ultimate solution depends on developing an effective method to preemptively inhibit ion mobility.
One promising approach involves supramolecular complex engineering. A particularly versatile strategy, which has recently proven highly effective in preventing these issues within perovskite compositions, relies on host–guest (HG) complexation. This method utilizes a variety of macrocyclic host (H) molecules, which can selectively bind and transport geometrically compatible guest (G) molecules through noncovalent interactions.40–43 By employing HG complexation, it becomes possible to regulate ion migration phenomena, as the host molecules effectively capture cations. This emerging approach has the potential to impact the crystal structure of perovskites and consequently influence the properties of materials and devices, leading to enhanced long-term stability.2,6,44–47 For instance, β(beta)-cyclodextrin (β-CD) is one the most well-known cyclic H molecules comprising seven glucose units connected by α-1,4 glycosidic bonds. The β-CD can capture hydrophobic substances in water and can be functionalized.11,44,48–50 This molecule is chosen for its remarkable capacity to interact with various chemical compounds effectively, aiming to establish a comprehensive supramolecular network involving all perovskite precursors in the solution. It also demonstrates non-toxicity, commercial availability, cost-effectiveness in comparison to alternative macrocyclic hosts, and the capability to form supramolecular HG complexes in aqueous solutions. Despite their potential, the application of CDs in HG complexes with metal halide perovskites in solar cells and LEDs has seen limited use.11,44,51–55 Previous studies highlight β-CD's pivotal role in regulating growth, improving device efficiency, and enhancing moisture resistance. Notable applications include stable MAPbBr3/β-CD nanodots for diverse use,44 and crucial role of β-CD in achieving standard blue emission and high photoluminescence quantum yield in CsPbBr3 nanorods, ensuring stability against moisture and oxygen.53 Investigations into CD's interaction with single cation perovskite materials showcase their dual advantage in enhancing crystallinity and moisture resistance by modulating solvation equilibria.51 Moreover, in an n–i–p configuration, β-CD has been utilized for moisture resistance and grain stabilization, leading to an improvements in stability and efficiency.56 Additional studies have explored the utility of β-CD under different operational challenges, employing it as an iodine-trapper in double cation PSCs to mitigate iodine-induced degradation, thereby enhancing stability and efficiency under light and heat.57 A moisture-dependent host–guest interaction between β-CD and adamantane enabled a self-healing system in flexible perovskite solar cells—promising prolonged device longevity in portable and wearable photovoltaics.54 Beyond LEDs and solar cells, β-CD proves valuable in applications like I2 trapping in Li–iodine batteries,55 highlighting its broad potential in advancing perovskite-based technologies.
Herein, we utilized the remarkable properties of β-CD for triple cation perovskite, drawing inspiration from its successful application in the aforementioned studies. We incorporated β-CD in the precursor solution of triple cation perovskite to remove the residual PbI2 in the perovskite composition through providing uniform crystal growth and passivating under coordinated metal/organic cation defects. A perovskite film modification using β-CD yields a PCE as high as 21.36% with competitive solar cell performance and enhanced device stability against aggressive thermal stress and high humidity (85% RH). Furthermore, it effectively prevented crystallization of unreacted PbI2 and provided valuable mechanistic insights into β-CD's role in hybrid perovskite solar cells.
Results and discussion
To evaluate the impact of β-CD in triple cation perovskites, we investigated Cs0.05(FA0.83MA0.17)0.95Pb(I0.83Br0.17)3 both with and without β-CD. Films were synthesized using a conventional solution processing method and deposited onto the hole transport layer (HTL) through a one-step procedure employing chlorobenzene (CB) as an anti-solvent (Scheme 1), following the detailed protocol outlined in the Experimental Section. In our study, we exclusively used β-CD due to its favorable interaction with PbI2 and MAI, supported by prior research.51 Alpha cyclodextrin (α-CD) was not examined due to its lower solubility and limited interaction.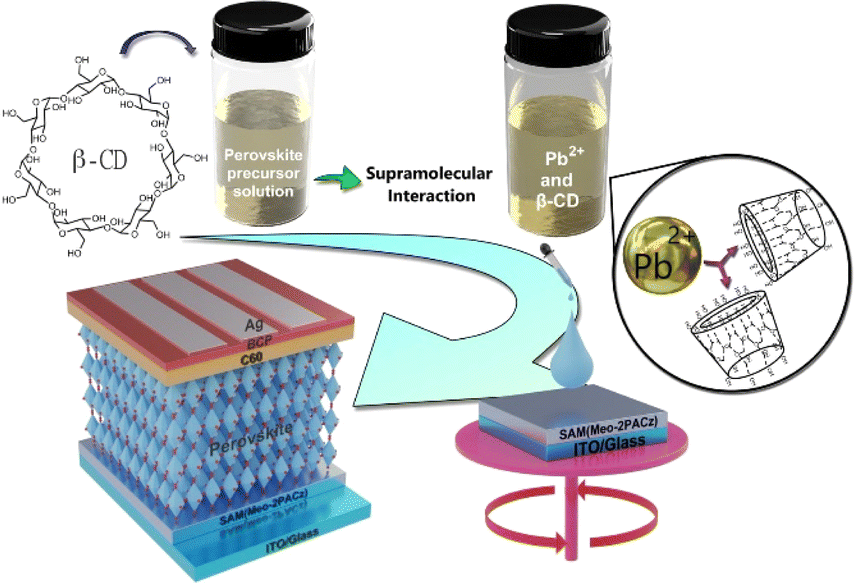 | ||
| Scheme 1 Representation of β-CD modulation mechanism and potential interaction sites in an inverted perovskite solar cell. | ||
Various concentrations of β-CD, specifically for thin film formation, were investigated, ranging from 0% to 5 weight/volume %, including intermediate values of 0.05%, 0.1%, 0.3%, 0.5%, 1%, 2%, and 5%. The outcomes of some of these experiments are presented in the ESI† (ESI, Fig S1–S3,† which present thin film XRD data (S1a†), steady-state photoluminescence (S1b†), UV-vis absorption (S1c†), top-down and cross-sectional SEM images (S2 and S3†), respectively). Results from these experiments will be discussed in detail below.
To assess the impact of β-CD on photovoltaic performance, we fabricated solar cells with varying β-CD concentrations (0%, 0.3%, 0.5%, 1%, 2%, and 5%) and compared their performance. Notably, the power conversion efficiency (PCE) of these solar cells under standard AM1.5G 1 sun conditions showed a notable improvement, increasing from an average of 15.55% to 19.88% with the addition of 0.5% β-CD. Furthermore, the optimized concentration of 0.5% β-CD led to enhancements in all parameters: the open-circuit voltage (VOC) increased from an average of 1.06 V to 1.12 V, the fill factor (FF) rose from 71.44% to 77.21%, and the short-circuit current density (JSC) increased from an average of 21.31 mA cm−2 to 23.08 mA cm−2 (Fig. 1).
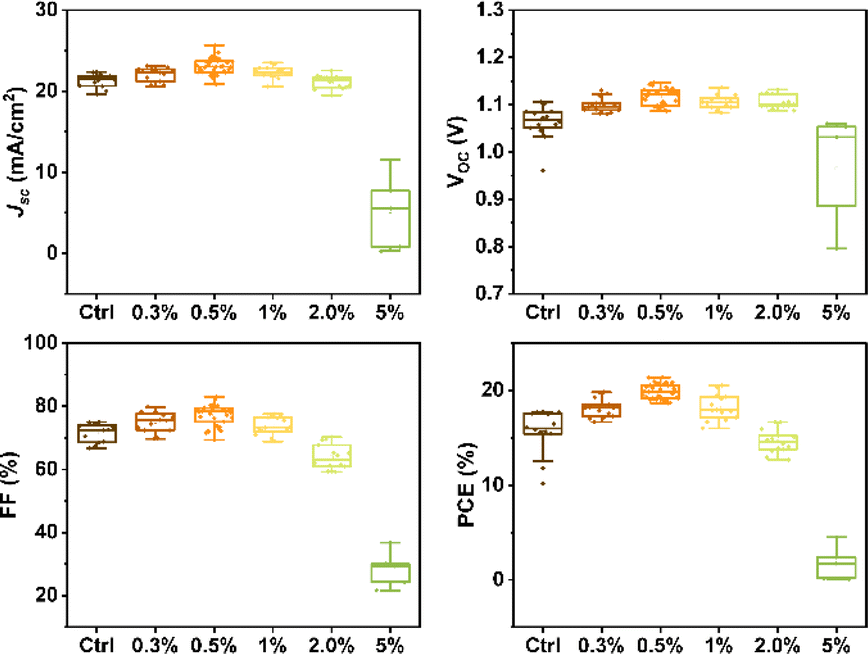 | ||
| Fig. 1 Statistical distribution of the photovoltaic parameters for control and β-CD-treated devices. | ||
Based on these results, a champion cell, attributed to the 0.5% β-CD treatment, exhibited an impressive PCE of 21.4%, high VOC of 1.14 V, a substantial JSC of 23.5 mA cm−2, and a notable FF of 80.2%. In contrast, the control champion cell without β-CD treatment achieved a PCE of 19.4%, VOC of 1.11 V, JSC of 23.3 mA cm−2, and FF of 74.8% (Fig. 2a) as detailed in Table 1. Notably, the characterization results conclusively indicated that the optimal concentration of β-CD was 0.5%. Consequently, this concentration was selected for further investigations and analysis in the subsequent stages of the study.
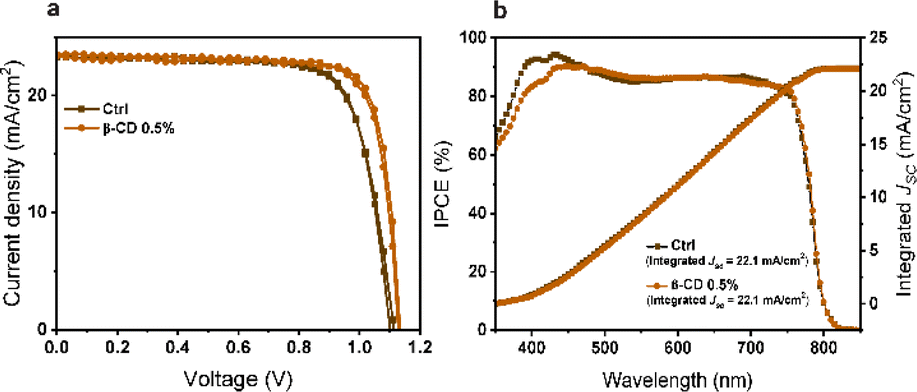 | ||
| Fig. 2 Photovoltaic characteristics. (a) Current–voltage scans for the best performing devices, and (b) corresponding IPCE spectra and photocurrent density calculated by integrating the IPCE with the AM1.5G solar spectrum. | ||
| J SC (mA cm−2) | V OC (V) | FF (%) | PCE (%) | ||
|---|---|---|---|---|---|
| Ctrl | Forward | 23.3 | 1.11 | 74.8 | 19.4 |
| Reverse | 23.3 | 1.10 | 76.3 | 19.6 | |
| β-CD 0.5% | Forward | 23.5 | 1.14 | 80.2 | 21.4 |
| Reverse | 23.4 | 1.13 | 79.4 | 20.9 | |
The expected JSC obtained by integrating the incident photon-to-current efficiency (IPCE) spectra for the treated device amounted to 22.1 mA cm−2, which closely matched the measured JSC value derived from the current–voltage characteristics (Fig. 2b).
X-ray diffraction (XRD) measurements were conducted to investigate the structural properties of perovskite films. The XRD spectra of the films exhibit the typical perovskite peaks at 2θ ≈14° and ≈31° for all concentrations, corresponding to the (100) and (200) perovskite lattice orientations, respectively. Notably, the presence of PbI2 crystals, evidenced by the peak around 12.5°, is observed in both control and the treated film with 0.05% β-CD. However, this PbI2 peak vanished with increasing concentrations of β-CD (0.1%, 0.3%, and 0.5%) (Fig. 3a and S1a†). The absence of this crystalline phase can likely prevent any PbI2 decomposition into iodine and metallic lead under light exposure, a process associated with recombination. Consequently, we anticipate a reduction in non-radiative recombination, leading to improved device efficiency.
 | ||
| Fig. 3 Characterization of the triple-cation perovskite without/with β-CD treatment. (a) XRD pattern on ITO substrates covered by a SAM layer (PbI2 reflection indicated by a star), (b) top-view SEM images, (c) steady-state PL spectra (ITO/MeO-2PACz/perovskite), and (d) absorption spectra (glass/perovskite) of control (ctrl) and treated sample by 0.5% of β-CD. | ||
The impact on the surface morphology was further examined using scanning electron microscopy (SEM). Fig. 3b clearly shows the absence of PbI2 crystals with the addition of β-CD at a concentration of 0.5%. Additionally, various concentrations of β-CD in treated films exhibit differences in surface coverage and morphology when compared to the control films (Fig. S2†). Cross section SEM images are shown in Fig. S3.† In the control sample, multiple grains and an internal grain boundary are observed. However, in the treated sample with 0.5% β-CD, the grains appear compact and nicely aligned alongside each other, and the excess PbI2 is notably absent. The SEM images provide additional evidence confirming the absence of a crystalline PbI2 phase in the films with the addition of β-CD, particularly from 0.3% and above. Furthermore, it is widely believed that ion migration often occurs across grain boundaries, where the macrocyclic host molecules are thought to assemble.14,35,39,58 Therefore, it is expected that the morphology changes, which have already shown their impact on photovoltaic characteristics, will also be reflected in the optoelectronic properties.
The optoelectronic properties were investigated by steady-state UV-vis absorption and photoluminescence (PL) spectroscopy of thin films with the glass/perovskite and ITO/MeO-2PACz/perovskite architectures, respectively. The PL spectra of the samples containing 0.5% of β-CD displays higher PL intensities compared to the others (Fig. 3c and S1b†). Typically, higher PL peak intensity points toward the suppression of non-radiative charge recombination. This observation aligns with the improvements in the crystallinity noted in the XRD analysis. However, the absorption spectra of both the control and treated films exhibit similar characteristics, suggesting that the presence of the β-CD does not significantly impact the film's thickness and composition (Fig. 3d and S1c†).
Furthermore, the space-charge-limited current (SCLC) method was employed to estimate the trap density as depicted in Fig. S4.† Electron-only devices (ITO/SnO2/Perovskite with and without β-CD/PCBM/Ag) were fabricated to quantify the trap-state density in the perovskite layer. The trap-state density (Ntrap) was determined using the specific equation equation,59 with calculation details provided in the experimental section. As illustrated by the SCLC curves (Fig. S4†), the trap-filled limit voltage (VTFL) for perovskite films without β-CD (Fig. S4a†) and those with 0.5% β-CD (Fig. S4b†) were found to be 0.487 V (average: 0.455 V) and 0.348 V (average: 0.353 V), respectively. Notably, the perovskite film incorporating 0.5% β-CD exhibited a lower VTFL compared to the control sample, suggesting a reduction in trap-state density. Based on the VTFL results, the average Ntrap for the control and β-CD-treated films were calculated to be 1.47 × 1016 cm−3 and 1.14 × 1016 cm−3, respectively (Table S1†). This outcome provides additional evidence that the introduction of β-CD can effectively mitigate the trap-state density of perovskite.
We further examined the charge recombination dynamics in the device by analysing the functional relationships between JSC (Fig. S5a†) and VOC (Fig. S5b†) with various incident light intensities. The ideality factor (α), indicating bimolecular recombination, was found to be 0.97 for the control and 1 for the 0.5% β-CD-treated sample. A higher α value closer to 1 suggests effective suppression of bimolecular recombination in perovskite solar cells with 0.5% β-CD. The light intensity dependence of VOC (Fig. S5b†) provides further insights into the recombination processes accompanying changes in light intensity. The ideality factor (ε) under open-circuit conditions, with a range of 1 < ε < 1.5, reflects the extent of charge recombination. A value of ε closer to 1 indicates lesser bimolecular recombination, while a value closer to 1.5 suggests more traps in the device, leading to increased bimolecular recombination, which is unfavourable for charge transmission and collection. Experimental results reveal that with the incorporation of 0.5% β-CD, the ε value is reduced from 1.40 kT/q to 1.09 kT/q, aligning well with the results obtained from SCLC measurements, indicating the significant role of β-CD in passivating defects through supramolecular interaction.
To explore deeper into the influence of β-CD treatment on the perovskite, X-ray photoelectron spectroscopy (XPS) analysis was conducted. Within the Pb 4f level range (Fig. 4a), the XPS spectra revealed two primary peaks corresponding to Pb 4f7/2 and Pb 4f5/2 at 138.8 and 143.6 eV, respectively, which are attributed to Pb–I species. Additionally, there were two smaller peaks observed at 136.9 and 141.9 eV, indicating the presence of metallic Pb. Notably, the metallic Pb peak disappeared in the treated film, suggesting that β-CD molecules could bind to the undercoordinated Pb2+ ions responsible for the formation of metallic Pb. This would greatly hold promising implications for enhancing the operational stability of the PSCs, which are adversely affected by the presence of elemental Pb. Furthermore, the peaks within the O 1s and C 1s level ranges at 533.5 eV and 287.1 eV, respectively, are associated with C–O binding energies and suggest that β-CD remains present on the surface of the film (Fig. 4b and c). In addition, as depicted in the normalized spectra (Fig. 4d and e), it is evident that these peaks (both in the O 1s and C 1s level ranges) exhibit a noticeable shift towards higher binding energies in the case of the β-CD 0.5% treated sample when compared to the sample containing only β-CD without perovskite. These results collectively suggest that β-CD is not merely adsorbed onto the surface but rather integrated into the perovskite layer.
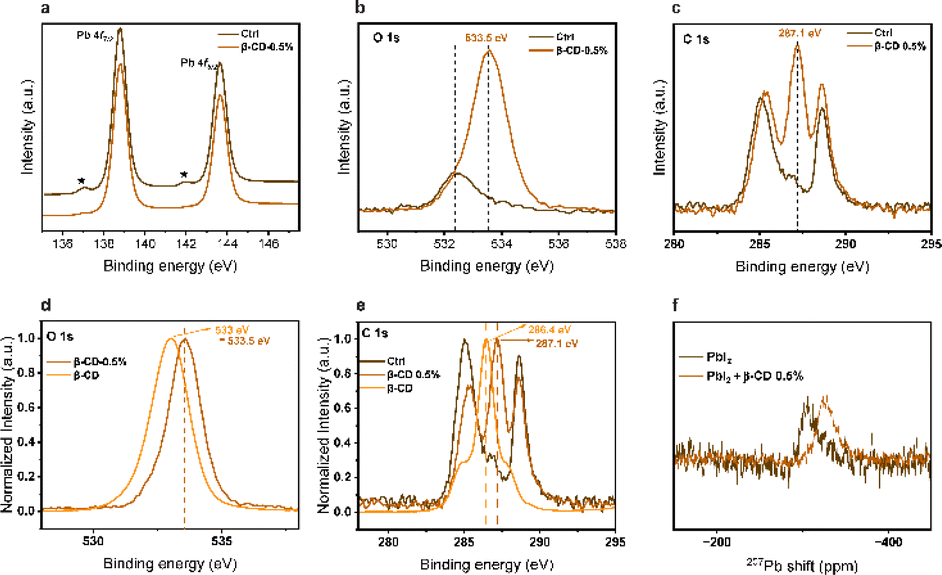 | ||
| Fig. 4 XPS core-level spectra of pristine perovskite film and β-CD-treated perovskite film for (a) Pb 4f, (b) O 1s, (c) C 1s, and normalized (d) O 1s, (e) C 1s, and (f) 207Pb NMR spectra of PbI2 and PbI2 + 0.5% β-CD precursor solutions in DMSO-d6. The asterisk (*) indicates metallic Pb species. | ||
To understand the mechanism responsible for the observed changes, nuclear magnetic resonance (NMR) spectroscopy was conducted. Initially, we performed solid-state NMR spectroscopy, the results of which are illustrated in Fig. S6a and b.† In this context, it is noteworthy that β-CD exhibited a broad peak, whereas the target substance displayed a heightened peak intensity when compared to the control within the same spectral region (Fig. S6a†). In the normalized spectra (Fig. S6b†), we were able to observe a slight distinction between the target and control samples, accompanied by the emergence of a minuscule new peak. However, it is important to note that we encountered difficulties drawing confident conclusions from these observations, primarily because the quantity of β-CD is exceedingly small, rendering it most likely undetectable through NMR techniques.
Subsequently, we opted to perform solution NMR analyses on the precursor solutions. Notably, a shift was observed in the 207Pb NMR spectra when comparing PbI2 at a concentration of 150 mM to PbI2 with the addition of 0.5% β-CD (Fig. 4f). Remarkably, no such shift was observed in the 1H NMR spectra for the other precursor solutions, implying that the shift (Pb) is unlikely to be influenced by environmental factors (Fig. S7†). The 1H NMR spectra revealed the presence of β-CD peaks in the mixture of precursor and β-CD, and these peaks did not exhibit any noticeable chemical shift. Moreover, a solution of control and 0.5% β-CD powder in DMSO-d6 was prepared, and 1H NMR analysis confirmed the presence of β-CD within the solution (Fig. S8†). The 207Pb NMR shift upon adding 0.5% β-CD indicates an interaction with PbI2, contrasting unchanged 1H NMR spectra, emphasizing the selective β-CD binding to PbI2.
Based on XPS and NMR observations, it is evident that β-CD effectively anchors uncoordinated Pb2+ ions. This interaction is pivotal in preventing the formation of metallic Pb, thereby enhancing the overall stability of the film under environmental stress.
Attenuated Total Reflectance-Fourier Transform Infrared (ATR-FTIR) spectroscopy was further employed to investigate the interactions between β-CD and the perovskite structure. Perovskite films with 0.5 and 5% β-CD were compared with the control film and β-CD powder. We opted to test 5% β-CD since exploring a higher β-CD concentration ensured the detectability of potential interactions. The resulting spectra depicted in Fig. S9.† The observed shifts in the C–O stretching vibrations from 1152 cm−1 and 1019 cm−1 in pure β-CD to higher wavenumbers in the treated sample with 5% β-CD (1158 and 1059 cm−1) suggest an interaction between β-CD and the perovskite. Additionally, the broader O–H stretching peaks in the 3200–3550 cm−1 range for the treated samples in contrast to the control, further support the hypothesis of likely supramolecular interaction involving the hydroxyl groups of β-CD and the perovskite structure.53,60 This implies the potential for oxygen from the ether bond and hydroxyl groups forming supramolecular interactions with Pb2+.
Drawing from literature on analogous systems where β-CD has demonstrated its ability to regulate crystallization behaviour and improve device efficiency and moisture resistance through host-guest interactions, it can be inferred that β-CD has a similar effect in this context. Previous studies have shown that β-CD can modulate nucleation rates by controlling the supersaturation levels of precursor solutions and equilibrating precursor solubility, leading to uniform nucleation. The supramolecular interactions established by β-CD, particularly at grain boundaries, result in a significant improvement in the crystalline-to-amorphous ratio, thereby altering the solution chemistry and influencing the final film properties. These findings highlight the important role of β-CD in modifying film characteristics and improving performance.51,53,56,57
We further investigated the electronic properties of perovskite film with and without β-CD treatment using kelvin probe force microscopy (KPFM). As illustrated in Fig. 5, the β-CD-treated perovskite film revealed a lower average surface potential compared to the control (−158 mV), which implies a change in the electronic property of the perovskite surface.61,62 More importantly, more homogeneous surface potential than that of control, which is associated with mitigation of residual PbI2 crystals at the surface of perovskite upon β-CD treatment was clearly observed. The similar root-mean-square roughness was obtained for the control samples and the β-CD treated samples (18.40 nm vs. 18.46 nm, respectively). The average surface contact potentials were determined as −333 mV (with a range of −286 to −380 mV) for the control samples and −441 mV (ranging from −444 to −438 mV) for the β-CD modified samples.
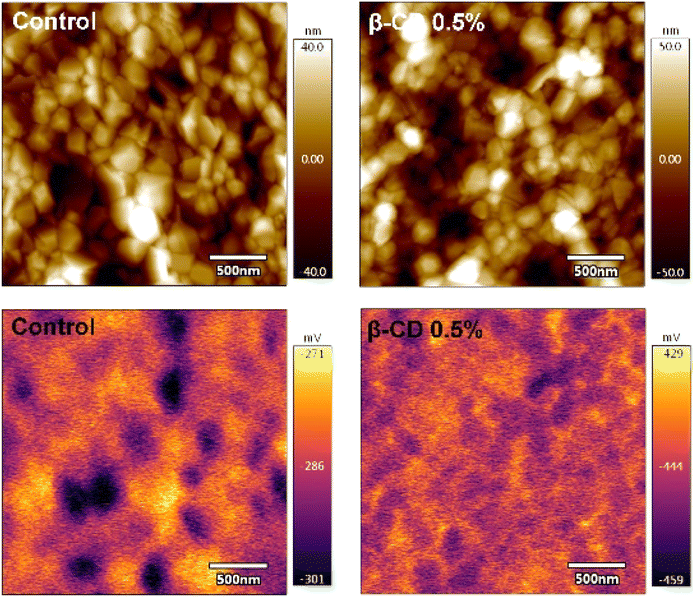 | ||
| Fig. 5 Topographical (top) and surface potential (bottom) images of control and β-CD-treated films by KPFM measurements in the dark condition. | ||
To further explore the impact of β-CD on the humidity resistance of perovskite films, we subjected both untreated control perovskite films and those treated with 0.5% β-CD to an 85% relative humidity chamber at 27 °C. We conducted assessments of the optical properties and morphology of the samples after exposure to humidity. The UV-Vis absorption spectra presented in Fig. 6a and b depict measurements taken at various time intervals, highlighting the beneficial influence of β-CD in enhancing the humidity stability of perovskite films. Analysis of the absorption spectra revealed a noteworthy decline in the control sample's absorption, particularly in the shorter wavelength region. Notably, the inset SEM images in Fig. 6a and b, revealed that the control samples exhibited degradation of the perovskite structure after 176 hours in the humid environment, while the presence of β-CD maintained the desired crystal phase. This observation implies that the reduction in absorption can be attributed to changes in the film morphological structure resulting from humidity stress.
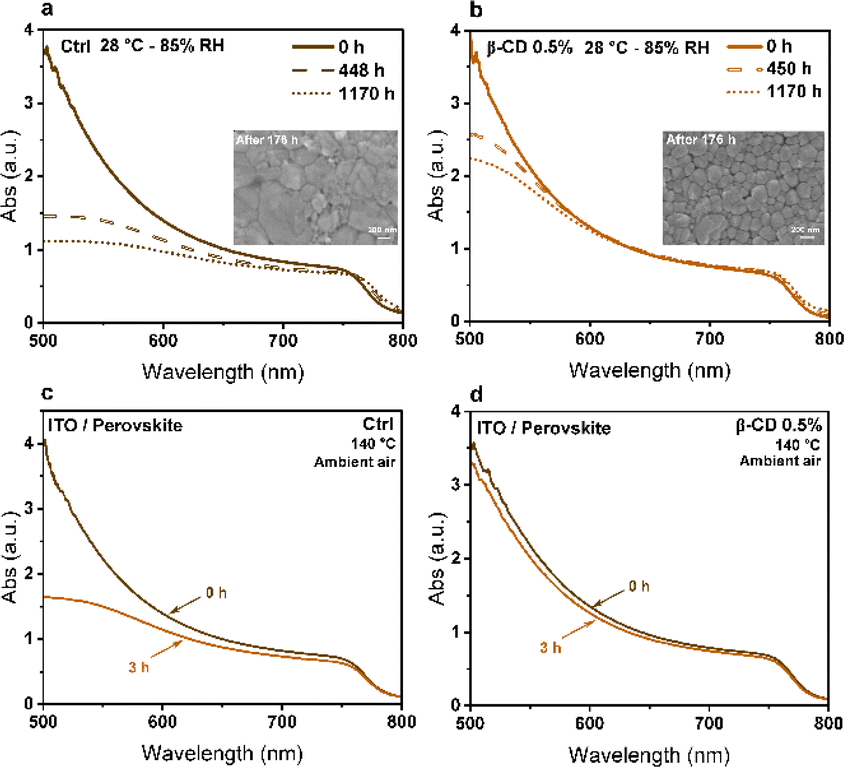 | ||
| Fig. 6 Absorption spectra of (a) control and (b) β-CD 0.5% treated films under humidity environment (85% RH) at 27 °C (insets show top-view SEM images of films after 176 hours humidity exposure). Absorption spectra of (c) control and (d) β-CD 0.5% treated films before and after exposure to aggressive thermal stress for 3 hours at 140 °C. | ||
Interestingly, exposure to moisture for 1170 hours caused a shift in the bandgap from 1.595 eV to 1.583 eV in the control sample (without β-CD), while only a minimal change of approximately 3 meV was observed in the treated sample with 0.5% β-CD (Fig. S10†), highlighting the moisture-stabilizing effect of β-CD. These findings reveal that prolonged exposure to moisture led to the decomposition of the control sample into PbI2, resulting in a redshift of the absorption edge and a reduction in the band gap. The smaller ionic radius of methylammonium (MA) compared to formamidinium (FA), along with the smaller size of bromine compared to iodine, results in a higher tolerance factor for FAPbI3 compared to MAPbBr3. This indicates greater stability for FAPbI3 over MAPbBr3. Consequently, MAPbBr3 perovskite is more prone to decomposition at high temperature. As the heating temperature increases, MAPbBr3 undergoes initial decomposition in triple-cation perovskite films, leading to an increased FA ratio, a redshift in absorption edge, and a decrease in bandgap energy.63–68 This decomposition also introduces defect states within the perovskite layer, impeding carrier transfer and exacerbating electron–hole recombination causing a decline in device performance.63,69 Importantly, our results show that the chemical and crystal phase deterioration of perovskite films due these environmental factors can be averted through supramolecular interaction using β-CD. Our findings align with previous research, which demonstrated that despite being hydrophilic, β-CD enhances the moisture stability of perovskite films by improving nucleation and crystallinity, thereby enhancing both optical properties and moisture resistance. The presence of β-CD leads to more compact and merged grains in perovskite films, slowing down moisture and oxygen permeation and increasing film stability.51
To better demonstrate this, both the control and treated sample (0.5% β-CD) films were subjected to an aggressive thermal stress test by exposing them to 140 °C for 3 hours in an ambient air environment. Before and after this thermal stress, an examination of both optical and structural characteristics of the perovskite films was conducted. Analysis of absorption spectra revealed a notable reduction in absorption for the control sample at shorter wavelengths (Fig. 6c and d). This mirrors the observed trend during humidity stress, indicating a potential correlation with alterations in the morphological structure of the film induced by thermal stress. The impact of harsh thermal stress on perovskite film morphology was investigated, with results shown in Fig. S11.† Following exposure to increased thermal stress, a notable increase in the intensity of the PbI2 peak was observed in the XRD data (Fig. S12†). This heightened intensity indicates that there are more PbI2 crystals in the perovskite film, which is supported by the SEM images. Thus, it is evident that thermal stress induces a significant amplification in the intensity of the PbI2 peak, reflecting substantial alterations in the structural of the perovskite film. A similar trend of a decreased bandgap was observed for thermal stability, mirroring the findings observed for moisture stability (Fig. S13†). This underscores the importance of β-CD in mitigating bandgap shifts and preserving the perovskite phase under both moisture and thermal stress.
Furthermore, the observed remarkable color change after storing the perovskite films for 5 months at a controlled environment with a temperature range of 18–20 °C and humidity between 40 to 45% provides additional compelling evidence of stability. The control film completely turned yellow, indicating the absence of the perovskite phase, while the treated film with β-CD remained black (Fig. S14†). This result confirms again the crucial role of β-CD in preserving the perovskite phase.
Long-term stability tests were conducted on both control and treated devices (0.5% β-CD), under constant 1 sun illumination and maximum power point tracking (MPPT) up to 320 hours, without encapsulation, in an air light soaking box, maintaining temperatures between 50 to 60 °C. The results are depicted in Fig. S15.† As shown, the control device experienced a significant performance decline, with over a 45% relative loss in PCE after 89 hours of MPPT ageing. Moreover, the control device ceased to function after 90 hours. In contrast, device treated with 0.5% β-CD demonstrated enhanced stability, retaining around 73% of its initial PCE after 320 hours of testing. These findings underscore the effectiveness of β-CD treatment in improving the long-term stability of our devices under harsh environmental conditions.
To further highlight the significance of β-CD's cavity in ion capture, a series of experiments involving maltose, which consists of two glucose units joined by an α-1,4-glycosidic bond (Fig. S16a†), was conducted to compare its ion-capturing capabilities against β-CD. Considering that maltose is a short linear submit of β-CD, this exploration aimed to delineate the distinctive attributes of β-CD's cavity structure. The film treated with 0.5% maltose underwent testing in an 85% relative humidity chamber at 27 °C alongside control and β-CD treated films. Fig. S16b† shows the UV-Vis absorption spectra of a 0.5% maltose treated film. A noteworthy decline in absorption, particularly in the shorter wavelength region is observed. An inset SEM image revealed that after 176 hours in the humid environment, the 0.5% maltose film showed degradation of the perovskite structure, whereas the presence of β-CD maintained the desired crystal phase, as previously mentioned (Fig. 6b). This result confirms that the cavity structure of β-CD's is key in bolstering the humidity stability of perovskite films. Fig. S16c† illustrates that exposure to moisture for 1170 hours led to a bandgap shift from 1.598 eV to 1.586 eV in the 0.5% maltose film, mirroring the behavior of the control sample (Fig. S10a†). However, the film treated with 0.5% β-CD exhibited only a nominal change (Fig. S10b†).
To examine the influence of maltose on thermal stability, film treated with 0.5% maltose underwent an aggressive thermal stress test at 140 °C for 3 hours in an ambient air environment. This test revealed a significant decrease in absorption, particularly noticeable in the shorter wavelength range, as depicted in Fig. S17a.† Moreover, Fig. S17b† illustrates that the aggressive thermal stress caused a shift in the bandgap of the maltose 0.5% film, aligning it more closely with the behavior of the control sample shown in Fig. S13a.† Additionally, an evident enhancement in the intensity of the PbI2 peak (2θ ≈ 12.5°) in the XRD data of the maltose 0.5% film is observed when compared to β-CD 0.5%, as displayed in Fig. S17c.†
Moreover, an investigation into the impact of thermal stress on the morphology of the perovskite film treated with maltose 0.5% revealed degradation, as illustrated in Fig. S17d.† Thermal stress was found to induce degradation in the morphology of both the control films and those treated with maltose 0.5% (Fig. S11 and S17d†).
The results indicate that β-CD demonstrates superior performance in enhancing the stability of perovskite films under harsh conditions compared to maltose. This outcome can be attributed to the inherent architecture of β-CD, which likely provides better protection and support against the deleterious effects of harsh environmental conditions on the perovskite films through formation of supramolecular interaction.
Conclusions
In conclusion, our study demonstrates a promising implementation of perovskite film modification through supramolecular interaction with β-CD in triple cation perovskite devices. This approach effectively eliminated unreacted PbI2, as confirmed by XRD and SEM analyses, leading to enhanced device performance, likely attributed to defect passivation as evidenced by PL, SCLC and the dependence of VOC on the incident light intensity studies. Moreover, we observed improved device stability against aggressive thermal stress and high humidity conditions (85% RH), as supported by morphological investigations, underlining ability of β-CD to maintain the desired perovskite phase. In addition, the devices treated with a 0.5% of β-CD showcased improved stability, maintaining over 73% of their initial PCE even after undergoing 320 hours of testing at 50–60 °C.Further characterizations were conducted to gain more insight into the impact of the supramolecular complexation. XPS analysis revealed that β-CD treatment led to the disappearance of metallic Pb peaks, suggesting binding of β-CD to undercoordinated Pb2+ ions. This finding holds promise for enhancing the operational stability of PSCs by mitigating the presence of elemental Pb. Furthermore, the observed shifts in O 1s and C 1s binding energies imply integration of β-CD into the perovskite, supporting the absence of phase segregation at the surface.
To understand the mechanism responsible for these effects, the interaction between the β-CD and perovskite was studied by NMR spectroscopy. The distinct shift observed in the 207Pb NMR spectra with the addition of 0.5% β-CD suggests an interaction between β-CD and PbI2, while the absence of similar shifts in 1H NMR spectra can imply minimal environmental influence, reinforcing the selective binding of β-CD with PbI2. These NMR findings align with the observations from XPS, collectively highlighting the potential of supramolecular interaction with β-CD as a valuable strategy in optimizing perovskite device performance and stability. Additional tests, employing maltose as a non-cyclic control, confirmed the superior efficacy of β-CD in enhancing perovskite film stability under harsh conditions, attributed to its unique cavity structure. Overall, the formation of a supramolecular system between β-CD and perovskite presents a promising strategy for controlling perovskite precursor chemistry, material structure, and subsequent device performance and stability. Indeed, we believe that our research can open a novel pathway toward more efficient and environmentally friendly perovskite solar cells.
Experimental methods
Materials
The materials used were commercially available. All solvents and chemicals were used without further purification.Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMSO 4:1 (v/v) according to a formula of triple cation perovskite as Cs0.05(FA0.83MA0.17)0.95Pb(I0.83Br0.17)3. The β-CD was added into the precursor solution at different ratios (0, 0.05, 0.1, 0.3, 0.5, 1, 2 and 5 w/v%).
:DMSO 4:1 (v/v) according to a formula of triple cation perovskite as Cs0.05(FA0.83MA0.17)0.95Pb(I0.83Br0.17)3. The β-CD was added into the precursor solution at different ratios (0, 0.05, 0.1, 0.3, 0.5, 1, 2 and 5 w/v%).
Inverted p–i–n architecture PSCs were fabricated (Scheme 1). In a typical procedure, a hole transport material of (2-(3,6-dimethoxy-9H-carbazol-9-yl)ethyl)phosphonic acid (MeO-2PACz) (100 μL) with a concentration of 0.5 mg mL−1 dissolved in absolute ethanol was spin-coated on the ITO substrates with a speed of 3000 rpm for 30 s, followed by annealing at 100 °C for 10 min. Subsequently, 80 μL perovskite precursor was spin-coated on ITO/MeO-2PACz layer at 6000 rpm for 30 s with a ramp of 2000 rpm s−1. About 15 s prior to the end of the program, 200 μL CB as anti-solvent was dripped onto the pre-perovskite film. Then the substrates were quickly transferred to a hot plate for 30 min at 100 °C. After those, 15 nm C60, 6 nm bathocuproine (BCP) and 100 nm Ag electrode via the controllable power were thermally deposited on the ITO/MeO-2PACz/perovskite substrate.
Using SCLC measurement, the trap-state density (Ntrap) was determined through the following equation:
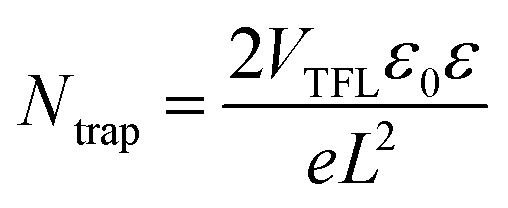
ATR-FTIR spectra were recorded with a PerkinElmer 1600 Series FTIR spectrophotometer.
UV-vis measurements were performed with a UV-3600 (Shimadzu) using a 2D integrating sphere. The samples were measured using ITO or glass as a baseline correction according to the substrates.
Photoluminescence measurements on perovskite films were performed using a Fluorolog-3 modular spectrofluorometer (Horiba) with an excitation light of 600 nm and 8 nm bandpass.
Scanning electron microscopy images were carried out with SEM (Zeiss Merlin In-Lens detectors). Optical microscopy was done using a Nikon Eclipse LV100ND.
X-ray diffraction measurements of thin films were performed on a by a D8 Discover Vario diffractometer (Bruker) equipped with a Johansson Kα1 monochromator.
X-ray photoelectron spectroscopy measurements were carried out on an Axis Supra (Kratos Analytical) using the monochromated Kα X-ray line of an aluminium anode. The pass energy was set to 20 eV with a step size of 0.1 eV. The samples were electrically grounded to limit charging effects.
Nuclear magnetic resonance spectra were acquired on a Bruker 400 MHz spectrometer (9.4 T) equipped with an AV-IV console and a 5 mm liquid-state BBO Z-gradient field three channel (1H/2H/BB) probe head. Experiments were performed at room temperature while locking to the deuterium signal of the d6-DMSO. 1H chemical shifts were referenced to Si(CH3)4 (δ(1H) = 0 ppm) using the signals of the residual protons of the deuterated solvent (δ1H = 2.50 ppm) as secondary reference. 207Pb chemical shifts were indirectly referenced with the 1H signals of the residual protons of the deuterated solvent using the Ξ-scale (Ξ = 20.920599 MHz) with Me4Pb + 5% C6D6 (δ(207Pb) = 0 ppm) as secondary references.
Solid-state nuclear magnetic resonance-1H MAS NMR spectra were recorded on a 900 MHz Bruker spectrometer (21.1T) equipped with an Avance NEO console and 1.3 mm HCND quadruple-channel CPMAS probe. Samples were packed into 1.3 mm zirconia rotors under ambient atmosphere spun to 60 kHz spinning speed using nitrogen gas at room temperature. 1H MAS NMR spectra were recorded using Hahn echo sequences. τ90 pulse length was 2.6 μs. 16 scans were cumulated for every spectrum. Recycle delays were chosen to be at 1.3 × T1. 1H chemical shifts were referenced to the secondary reference adamantane at 1.82 ppm.
Author contributions
The manuscript was written by P. F. with the support of all authors. The study was conceptualized by P. F. and J.-H. Y., who directed the project. P. F. performed material, device fabrication and characterization with the supervision of K. S. and J.-H. Y., who were actively involved in discussions. Furthermore, device fabrication and IPCE analysis were carried out by S.-J. K. and T.-D. N., under the supervision of J.-Y. S. All authors were involved in the discussions and approved the final manuscript version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the EPFL and the research agreement between EPFL and the Korea Electric Power Corporation (KEPCO) for financial support of this work. Additionally, the authors appreciate the contributions of Dr Laura Piveteau and Dr Aurélien Bornet from EPFL for their assistance with NMR spectroscopy, and acknowledge Dr Mounir Mensi for conducting XPS measurements.References
- R. Chen, Y. Hui, B. Wu, Y. Wang, X. Huang, Z. Xu, P. Ruan, W. Zhang, F. Cheng, W. Zhang, J. Yin, J. Li and N. Zheng, J. Mater. Chem. A, 2020, 8, 9597–9606 RSC.
- M. A. Ruiz-Preciado, D. J. Kubicki, A. Hofstetter, L. McGovern, M. H. Futscher, A. Ummadisingu, R. Gershoni-Poranne, S. M. Zakeeruddin, B. Ehrler, L. Emsley, J. V. Milić and M. Grätzel, J. Am. Chem. Soc., 2020, 142, 1645–1654 CrossRef CAS PubMed.
- X. Hu, X.-F. Jiang, X. Xing, L. Nian, X. Liu, R. Huang, K. Wang, H.-L. Yip and G. Zhou, Sol. RRL, 2018, 2, 1800083 CrossRef.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- P. Ferdowsi, E. Ochoa-Martinez, U. Steiner and M. Saliba, Chem. Mater., 2021, 33, 3971–3979 CrossRef CAS.
- H. Zhang, F. T. Eickemeyer, Z. Zhou, M. Mladenović, F. Jahanbakhshi, L. Merten, A. Hinderhofer, M. A. Hope, O. Ouellette, A. Mishra, P. Ahlawat, D. Ren, T.-S. Su, A. Krishna, Z. Wang, Z. Dong, J. Guo, S. M. Zakeeruddin, F. Schreiber, A. Hagfeldt, L. Emsley, U. Rothlisberger, J. V. Milić and M. Grätzel, Nat. Commun., 2021, 12, 3383 CrossRef CAS PubMed.
- H. Zai, Y. Ma, Q. Chen and H. Zhou, J. Energy Chem., 2021, 63, 528–549 CrossRef CAS.
- J. Peng, Y. Wu, W. Ye, D. A. Jacobs, H. Shen, X. Fu, Y. Wan, T. Duong, N. Wu, C. Barugkin, H. T. Nguyen, D. Zhong, J. Li, T. Lu, Y. Liu, M. N. Lockrey, K. J. Weber, K. R. Catchpole and T. P. White, Energy Environ. Sci., 2017, 10, 1792–1800 RSC.
- G. Szabó, N.-G. Park, F. De Angelis and P. V. Kamat, ACS Energy Lett., 2023, 8, 3829–3831 CrossRef.
- https://www.nrel.gov/pv/cell-efficiency.html .
- P. Ferdowsi, U. Steiner and J. V. Milić, JPhys Mater., 2021, 4, 042011 CrossRef CAS.
- B. Yu, Z. Xu, H. Liu, Y. Liu, K. Ye, Z. Ke, J. Zhang and H. Yu, ACS Appl. Mater. Interfaces, 2024, 16, 2408–2416 CrossRef CAS PubMed.
- L. Meng, J. You and Y. Yang, Nat. Commun., 2018, 9, 1–4 CrossRef CAS PubMed.
- T.-S. Su, F. T. Eickemeyer, M. A. Hope, F. Jahanbakhshi, M. Mladenović, J. Li, Z. Zhou, A. Mishra, J.-H. Yum, D. Ren, A. Krishna, O. Ouellette, T.-C. Wei, H. Zhou, H.-H. Huang, M. D. Mensi, K. Sivula, S. M. Zakeeruddin, J. V. Milić, A. Hagfeldt, U. Rothlisberger, L. Emsley, H. Zhang and M. Grätzel, J. Am. Chem. Soc., 2020, 142, 19980–19991 CrossRef CAS PubMed.
- H. Zhang, W. Yu, J. Guo, C. Xu, Z. Ren, K. Liu, G. Yang, M. Qin, J. Huang, Z. Chen, Q. Liang, D. Shen, Z. Wu, Y. Zhang, H. T. Chandran, J. Hao, Y. Zhu, C. Lee, X. Lu, Z. Zheng, J. Huang and G. Li, Adv. Energy Mater., 2022, 12, 2201663 CrossRef CAS.
- Y. C. Kim, N. J. Jeon, J. H. Noh, W. S. Yang, J. Seo, J. S. Yun, A. Ho-Baillie, S. Huang, M. A. Green, J. Seidel, T. K. Ahn and S. I. Seok, Adv. Energy Mater., 2016, 6, 1502104 CrossRef.
- T. J. Jacobsson, J.-P. Correa-Baena, E. Halvani Anaraki, B. Philippe, S. D. Stranks, M. E. F. Bouduban, W. Tress, K. Schenk, J. Teuscher, J.-E. Moser, H. Rensmo and A. Hagfeldt, J. Am. Chem. Soc., 2016, 138, 10331–10343 CrossRef CAS PubMed.
- B. Park, N. Kedem, M. Kulbak, D. Y. Lee, W. S. Yang, N. J. Jeon, J. Seo, G. Kim, K. J. Kim, T. J. Shin, G. Hodes, D. Cahen and S. I. Seok, Nat. Commun., 2018, 9, 3301 CrossRef PubMed.
- S. P. Dunfield, L. Bliss, F. Zhang, J. M. Luther, K. Zhu, M. F. A. M. van Hest, M. O. Reese and J. J. Berry, Adv. Energy Mater., 2020, 10, 1904054 CrossRef CAS.
- Q. Chen, H. Zhou, T.-B. Song, S. Luo, Z. Hong, H.-S. Duan, L. Dou, Y. Liu and Y. Yang, Nano Lett., 2014, 14, 4158–4163 CrossRef CAS PubMed.
- L. Zhao, Q. Li, C.-H. Hou, S. Li, X. Yang, J. Wu, S. Zhang, Q. Hu, Y. Wang, Y. Zhang, Y. Jiang, S. Jia, J.-J. Shyue, T. P. Russell, Q. Gong, X. Hu and R. Zhu, J. Am. Chem. Soc., 2022, 144, 1700–1708 CrossRef CAS PubMed.
- L. Chen, J. Chen, C. Wang, H. Ren, Yu-X. Luo, K.-C. Shen, Y. Li, F. Song, X. Gao and J.-X. Tang, ACS Appl. Mater. Interfaces, 2021, 13, 24692–24701 CrossRef CAS PubMed.
- B. Roose, K. Dey, Y.-H. Chiang, R. H. Friend and S. D. Stranks, J. Phys. Chem. Lett., 2020, 11, 6505–6512 CrossRef CAS PubMed.
- Y. Li, K. Cui, X. Xu, J. Chen, Y. Liu, J. Wu, S. Lu, W. Qin and X. Wu, J. Phys. Chem. C, 2020, 124, 15107–15114 CrossRef CAS.
- D. Bi, W. Tress, M. I. Dar, P. Gao, J. Luo, C. Renevier, K. Schenk, A. Abate, F. Giordano, J.-P. Correa Baena, J.-D. Decoppet, S. M. Zakeeruddin, M. K. Nazeeruddin, M. Grätzel and A. Hagfeldt, Sci. Adv., 2016, 2, e1501170 CrossRef PubMed.
- D. H. Cao, C. C. Stoumpos, C. D. Malliakas, M. J. Katz, O. K. Farha, J. T. Hupp and M. G. Kanatzidis, APL Mater., 2014, 2, 091101 CrossRef.
- F. Liu, Q. Dong, M. K. Wong, A. B. Djurišić, A. Ng, Z. Ren, Q. Shen, C. Surya, W. K. Chan, J. Wang, A. M. C. Ng, C. Liao, H. Li, K. Shih, C. Wei, H. Su and J. Dai, Adv. Energy Mater., 2016, 6, 1502206 CrossRef.
- C. Roldán-Carmona, P. Gratia, I. Zimmermann, G. Grancini, P. Gao, M. Graetzel and M. K. Nazeeruddin, Energy Environ. Sci., 2015, 8, 3550–3556 RSC.
- S. Nakayashiki, H. Daisuke, Y. Ogomi and S. Hayase, J. Photonics Energy, 2015, 5, 057410 CrossRef.
- H. Wang, Z. Wang, Z. Yang, Y. Xu, Y. Ding, L. Tan, C. Yi, Z. Zhang, K. Meng, G. Chen, Y. Zhao, Y. Luo, X. Zhang, A. Hagfeldt and J. Luo, Adv. Mater., 2020, 32, 2000865 CrossRef CAS PubMed.
- D. Luo, W. Yang, Z. Wang, A. Sadhanala, Q. Hu, R. Su, R. Shivanna, G. F. Trindade, J. F. Watts, Z. Xu, T. Liu, K. Chen, F. Ye, P. Wu, L. Zhao, J. Wu, Y. Tu, Y. Zhang, X. Yang, W. Zhang, R. H. Friend, Q. Gong, H. J. Snaith and R. Zhu, Science, 2018, 360, 1442–1446 CrossRef CAS PubMed.
- A. Kogo, T. Miyadera and M. Chikamatsu, ACS Appl. Mater. Interfaces, 2019, 11, 38683–38688 CrossRef CAS PubMed.
- Z. Hu, Q. An, H. Xiang, L. Aigouy, B. Sun, Y. Vaynzof and Z. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 54824–54832 CrossRef CAS PubMed.
- M. Qin, H. Xue, H. Zhang, H. Hu, K. Liu, Y. Li, Z. Qin, J. Ma, H. Zhu, K. Yan, G. Fang, G. Li, U.-S. Jeng, G. Brocks, S. Tao and X. Lu, Adv. Mater., 2020, 32, 2004630 CrossRef CAS PubMed.
- D. Wei, F. Ma, R. Wang, S. Dou, P. Cui, H. Huang, J. Ji, E. Jia, X. Jia, S. Sajid, A. M. Elseman, L. Chu, Y. Li, B. Jiang, J. Qiao, Y. Yuan and M. Li, Adv. Mater., 2018, 30, 1707583 CrossRef PubMed.
- Y. Shao, Z. Xiao, C. Bi, Y. Yuan and J. Huang, Nat. Commun., 2014, 5, 5784 CrossRef CAS PubMed.
- D. Wei, J. Ji, D. Song, M. Li, P. Cui, Y. Li, J. M. Mbengue, W. Zhou, Z. Ning and N.-G. Park, J. Mater. Chem. A, 2017, 5, 1406–1414 RSC.
- W. Ke, C. Xiao, C. Wang, B. Saparov, H.-S. Duan, D. Zhao, Z. Xiao, P. Schulz, S. P. Harvey, W. Liao, W. Meng, Y. Yu, A. J. Cimaroli, C.-S. Jiang, K. Zhu, M. Al-Jassim, G. Fang, D. B. Mitzi and Y. Yan, Adv. Mater., 2016, 28, 5214–5221 CrossRef CAS PubMed.
- E. Bi, Z. Song, C. Li, Z. Wu and Y. Yan, Trends Chem., 2021, 3, 575–588 CrossRef CAS.
- G. Yu and X. Chen, Theranostics, 2019, 9, 3041–3074 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- J.-M. Lehn, Chem. Soc. Rev., 2017, 46, 2378–2379 RSC.
- L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS PubMed.
- S. Huang, T. Zhang, C. Jiang, R. Qi, C. Luo, Y. Chen, H. Lin, J. Travas-sejdic and H. Peng, ChemNanoMat, 2019, 5, 1311–1316 CrossRef CAS.
- J. Li, T. Jiu, S. Chen, L. Liu, Q. Yao, F. Bi, C. Zhao, Z. Wang, M. Zhao, G. Zhang, Y. Xue, F. Lu and Y. Li, Nano Lett., 2018, 18, 6941–6947 CrossRef CAS PubMed.
- Y. Takashima and A. Harada, J. Inclusion Phenom. Macrocyclic Chem., 2017, 87, 313–330 CrossRef CAS.
- E. S. Parrott, J. B. Patel, A.-A. Haghighirad, H. J. Snaith, M. B. Johnston and L. M. Herz, Nanoscale, 2019, 11, 14276–14284 RSC.
- K. M. Cheah, J. V. Jun, K. D. Wittrup and R. T. Raines, Mol. Pharm., 2022, 19, 3869–3876 CrossRef CAS PubMed.
- A. R. Khan, P. Forgo, K. J. Stine and V. T. D'Souza, Chem. Rev., 1998, 98, 1977–1996 CrossRef CAS PubMed.
- E. Engeldinger, D. Armspach and D. Matt, Chem. Rev., 2003, 103, 4147–4174 CrossRef CAS PubMed.
- S. Masi, F. Aiello, A. Listorti, F. Balzano, D. Altamura, C. Giannini, R. Caliandro, G. Uccello-Barretta, A. Rizzo and S. Colella, Chem. Sci., 2018, 9, 3200–3208 RSC.
- X.-Y. Du, K. Ma, R. Cheng, X.-J. She, Y.-W. Zhang, C.-F. Wang, S. Chen and C. Xu, J. Hazard. Mater., 2019, 361, 329–337 CrossRef CAS PubMed.
- C. Ye, Y. Wang and Y. Xiao, Chem. Commun., 2023, 59, 916–919 RSC.
- Z. Yang, Y. Jiang, D. Xu, Z. Wang, X. Gao, X. Lu, G. Zhou, J.-M. Liu and J. Gao, J. Mater. Chem. A, 2022, 10, 22445–22452 RSC.
- F. Cai, Y. Duan and Z. Yuan, J. Mater. Sci.: Mater. Electron., 2018, 29, 11540–11545 CrossRef CAS.
- D. Wei, H. Huang, P. Cui, J. Ji, S. Dou, E. Jia, S. Sajid, M. Cui, L. Chu, Y. Li, B. Jiang and M. Li, Nanoscale, 2019, 11, 1228–1235 RSC.
- X. Li, H. Yang, A. Liu, C. Lu, H. Yuan, W. Zhang and J. Fang, Energy Environ. Sci., 2023, 16, 6071–6077 RSC.
- A.-F. Castro-Méndez, J. Hidalgo and J.-P. Correa-Baena, Adv. Energy Mater., 2019, 9, 1901489 CrossRef.
- J. Liang, Z. Chen, G. Yang, H. Wang, F. Ye, C. Tao and G. Fang, ACS Appl. Mater. Interfaces, 2019, 11, 23152–23159 CrossRef CAS PubMed.
- Z.-K. Zhou, Z.-H. Mo and X.-L. Wei, Chem. Commun., 2019, 55, 11916–11919 RSC.
- M. Cai, N. Ishida, X. Li, X. Yang, T. Noda, Y. Wu, F. Xie, H. Naito, D. Fujita and L. Han, Joule, 2018, 2, 296–306 CrossRef CAS.
- Q. Wang, Y. Shao, H. Xie, L. Lyu, X. Liu, Y. Gao and J. Huang, Appl. Phys. Lett., 2014, 105, 163508 CrossRef.
- Q. Meng, Y. Chen, Y. Y. Xiao, J. Sun, X. Zhang, C. B. Han, H. Gao, Y. hang and H. Yan, J. Mater. Sci.: Mater. Electron., 2021, 32, 12784–12792 CrossRef CAS.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Nature, 2015, 517, 476–480 CrossRef CAS PubMed.
- T. Leijtens, G. E. Eperon, N. K. Noel, S. N. Habisreutinger, A. Petrozza and H. J. Snaith, Adv. Energy Mater., 2015, 5, 1500963 CrossRef.
- J. A. Christians, P. A. Miranda Herrera and P. V. Kamat, J. Am. Chem. Soc., 2015, 137, 1530–1538 CrossRef CAS PubMed.
- K. Jung, J.-H. Lee, K. Oh, C. Im, J. Do, J. Kim, W.-S. Chae and M.-J. Lee, Nano Energy, 2018, 54, 251–263 CrossRef CAS.
- L.-Q. Xie, L. Chen, Z.-A. Nan, H.-X. Lin, T. Wang, D.-P. Zhan, J.-W. Yan, B.-W. Mao and Z.-Q. Tian, J. Am. Chem. Soc., 2017, 139, 3320–3323 CrossRef CAS PubMed.
- Enhancing Thermal Stability of Perovskite Solar Cells with a Polymer Through Grain Boundary Passivation, Electrical & Computer Engineering Dissertation, PhD thesis, Old Dominion University, 2021 DOI:10.25777/x3e3-5x18.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta01741b |
| This journal is © The Royal Society of Chemistry 2024 |