
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-oxidative calcination enhances the methane dry reforming performance of Ni/CeO2−x catalysts under thermal and photo-thermal conditions†
Kristijan
Lorber
a,
Vasyl
Shvalya
c,
Janez
Zavašnik
 c,
Damjan
Vengust
c,
Iztok
Arčon
bc,
Matej
Huš
ade,
Andraž
Pavlišič
a,
Janvit
Teržan
a,
Uros
Cvelbar
c,
Blaž
Likozar
a and
Petar
Djinović
*ab
c,
Damjan
Vengust
c,
Iztok
Arčon
bc,
Matej
Huš
ade,
Andraž
Pavlišič
a,
Janvit
Teržan
a,
Uros
Cvelbar
c,
Blaž
Likozar
a and
Petar
Djinović
*ab
aNational Institute of Chemistry, Hajdrihova 19, 1000 Ljubljana, Slovenia. E-mail: petar.djinovic@ki.si
bUniversity of Nova Gorica, Vipavska 13, SI-5000 Nova Gorica, Slovenia
cJožef Stefan Institute, Jamova Cesta 39, SI-1000 Ljubljana, Slovenia
dAssociation for Technical Culture (ZOTKS), Zaloška 65, 1000 Ljubljana, Slovenia
eInstitute for the Protection of Cultural Heritage (ZVKDS), Poljanska 40, 1000 Ljubljana, Slovenia
First published on 1st July 2024
Abstract
We analyzed the effect of the calcination atmosphere and visible-light contribution to an accelerated reaction rate and improved H2 selectivity over 2 wt% Ni/CeO2−x nanorod catalysts. Spectroscopic and structural characterization was performed by operando DRIFTS, in situ Raman, UV-vis and XAS techniques, which were complemented by DFT calculations. Calcination in an argon or H2 atmosphere yields 15% more active catalysts in the thermally driven reaction, which are also more susceptible to light-induced rate acceleration compared to the catalyst calcined in air. The most active 2Ni/CeO2 catalyst calcined in hydrogen converts methane with a rate of 7.5 mmol (gcat min)−1 and produces a H2/CO ratio of 0.6 at 460 °C when stimulated by a combination of visible light and thermal energy. In the absence of visible light illumination and at an identical catalyst temperature, the achieved methane rate was 4.2 mmol (gcat min)−1 and the H2/CO ratio was 0.49. The non-oxidative calcination improves nickel dispersion and the formation of subnanometer sized Ni clusters, together with a higher abundance of surface and bulk oxygen vacancies in ceria nanorods. The Ni–Ov–Ce3+components constitute the catalytically active sites under visible light illumination, which enable the DRM reaction to proceed with an Ea value of 20 kJ mol−1. Visible light also induces the following changes in the 2Ni/CeO2−x catalyst during the DRM reaction: (1) decomposition and desorption of carbonates from the nickel–ceria interface sites, (2) reduced population of nickel surface with carbonyl species and (3) promoted adsorption and dissociation of methane.
1. Introduction
A steady increase of CO2 concentration in Earth's atmosphere is closely connected to the growth of population, industrialization, transport and agricultural sectors. These segments rely heavily on increasing demands for energy as their driving force.1,2 The combustion of fossil fuels is a dominant source for energy production and presents the main contribution to the elevated CO2 emissions.3The methane dry reforming reaction (DRM, eqn (1)) has gained a lot of attention in the last few decades because it converts two greenhouse gases into syngas.4 The CO-rich syngas produced during DRM can be used for the synthesis of various chemicals and synthetic fuels through Fischer–Tropsch chemistry.5,6 The DRM reaction still faces some challenges that need to be resolved, such as: (a) high catalytic activity at low reaction temperatures through active site optimization and (b) high H2 selectivity due to the deceleration of H2O causing side reactions.5
| CO2 + CH4 ↔ 2H2 + 2CO | (1) |
The complexity of the DRM reaction is signified by the co-occurrence of side reactions, such as methane cracking (CH4 → C + 2H2), the Boudouard reaction (2CO ↔ CO2 + C) and reverse water-gas shift (RWGS, eqn (2)), which produce carbon or lead to lower H2 selectivity.
| H2 + CO2 ↔ H2O + CO | (2) |
Nickel is an abundant and affordable metal with high intrinsic DRM activity,7–10 which can be further improved by dispersing it over ceria (CeO2) or other refractory supports that help maintain nano-sized metallic particles during the reaction.11 The strong metal–support interactions help maintain highly dispersed nickel particles and the high oxygen mobility of ceria. This enables carbon gasification through the spillover of oxygen species, thus providing a highly carbon resistant catalyst. Carbon accumulation on the catalyst is among the main drawbacks of the DRM reaction, as it causes an increased pressure drop across the catalytic bed and reactor blocking, as well as catalyst deactivation due to active site coverage.12–14
The implementation of visible light into the thermally driven DRM reaction is an attractive method for breaking the thermodynamic equilibrium limitations and achieving high catalytic activity and selectivity at low temperatures.15–17 In photo-thermal catalysis over semiconductor based catalysts, three categories of performance enhancement are possible, based on the distinct utilization of light energy: (i) photo-excited charge carrier transfer (herein denoted as the PT mechanism) into the conduction band and valence band of the semiconductors, (ii) strong electromagnetic near field enhancement (NF mechanism) due to the illumination by photons having insufficient energy for electron promotion across the optical bandgap and (iii) the conversion of the photon energy into thermal energy, classified as the light promoted thermal effect.15,18,19
Hu et al.20 studied the synergy between thermal and photon energy over a Pt/TiO2−x catalyst in the DRM reaction. The Pt/black TiO2−x photocatalyst exploits oxygen deficiency and the occupation of mid-bandgap energy states activated by visible light, resulting in 3 orders of magnitude higher H2 and CO yields compared to the thermal DRM reaction.
Zhao et al.21 demonstrated an efficient solar light driven thermo-catalytic DRM process via Ni nanocrystals embedded within silica clusters. They reported exceptionally high rates for H2 and CO (17.1 and 19.9 mmol (g min)−1), achieved by means of elevated catalyst surface temperature through strong surface plasmon absorption of Ni and the IR heating effect.
Our previous studies analyzed the effect of the ceria shape on DRM activity, and the nanorod morphology emerged as the most active and most carbon resistant.22 Further, by changing the nickel content on the nanorod shaped ceria, we identified 2 wt% nickel as an optimal catalyst for thermal and combined photo-thermal DRM processes.23 Enhancing nickel dispersion and the interface with ceria is highly beneficial for thermally driven catalytic DRM activity,24 but for the photo-thermal reaction, the structure–activity relationship still remains unknown. Also, the following important questions regarding the photo-thermal DRM reaction still need to be answered: what are the relative contributions of PT and NF on the observed photocatalytic gain, how is the surface reaction chemistry impacted by light and how this influences CH4 and CO2 activation, as well as H2 selectivity? It is also important to understand the role of Ni size in synergy with the CeO2−x support in generating and utilizing the PT and NF effects optimally.
In this work, catalysts containing 2 wt% Ni on CeO2 nanorods were calcined in inert, oxidative and reducing atmospheres during their synthesis. The calcination atmosphere was identified as an efficient tool to increase oxygen defect density in ceria and to increase nickel dispersion through variation of surface defect sites in ceria, which act as anchoring sites for nickel. Thermocatalytic and the combined photo-thermal catalytic tests were complemented by DFT calculations and in situ structural, chemical, optical and operando spectroscopic characterization to analyze the origins of the light-induced promotion of the DRM reaction performance.
2. Experimental section
2.1. Synthesis of catalysts
The CeO2 nanorods were synthesized and decorated with 2 wt% nickel. Such a low loading produces very small nickel particles, which prevent carbon accumulation during the catalytic reaction.22 For the CeO2 nanorod synthesis, 58.8 g of NaOH (purity 99%) and Ce(NO3)3·6H2O (purity 99%) were dissolved in ultrapure water and stirred for 0.5 h. The suspension was transferred into Teflon®-clad stainless steel autoclaves and heated to 100 °C for 24 h. After hydrothermal treatment, the product was washed with ultrapure water and freeze dried. Calcination was performed in one of the following atmospheres (air, argon or 5% H2/N2) at 500 °C for 4 h, using a heating ramp of 5 °C min−1. Nickel was deposited by dissolving Ni(NO3)2·6H2O (purity 99%) in ultrapure water, followed by the addition of the CeO2 nanorod powder during mixing and increasing the pH to 7.5 by adding aqueous ammonia solution. After adsorption and precipitation, the precursor was filtered and dried. The calcination of the as-synthesized Ni/CeO2 samples was repeated in an air, argon, or 5% H2/N2 flow.The synthesized catalysts, calcined in air, argon and 5% H2/N2, are denoted as NC-Air, NC-Ar and NC-H2, respectively. The actual Ni content was analyzed by the ICP-OES method and equals 1.80 wt% for NC-Air and 1.78 wt% for both NC-Ar and NC-H2.
2.2. Characterization and catalytic testing
The UV-vis analysis was performed on a Lambda 650 apparatus (PerkinElmer) equipped with a Praying Mantis accessory and reaction chamber from Harrick. After reduction at 500 °C for 0.5 h in 5% H2/N2 (15 ml min−1), the samples were cooled to 400 °C, followed by the substitution of reducing gas with an equimolar CO2/CH4 gas mixture and a 20 ml min−1 total flow.
The photoluminescence of the 2Ni/CeO2 catalysts was analyzed using a PerkinElmer LS 55 spectrometer. 25 mg of sample was excited with a wavelength of 330 nm and the emission spectrum was recorded between 350 and 650 nm.
In situ DRIFTS DRM pulse analysis under thermal and photo-thermal conditions was performed to analyze the behavior of formate species, which are masked by methane vibrations in a continuous flow. The experimental setup is described above and the protocol was injecting equimolar CO2/CH4 gas pulses (0.5 ml) into the argon flow passing through the catalyst bed between 500 °C and 100 °C.
A 0.5 mm thin layer of finely powdered sample (2 mg) was placed over inert powdered SiC (Sicat, 30–150 μm). Before the reaction, the catalysts were reduced in the dark at 500 °C for 0.5 h using a 15 ml min−1 flow of 5% H2/N2. After reduction, an equimolar flow of CH4 and CO2 (10 ml min−1 each) was introduced into the reaction chamber. Thermocatalytic experiments were performed between 460 and 380 °C, whereas photo-thermal experiments were performed between 460 and 140 °C. The reaction products were analyzed by GC (Agilent 490, equipped with MS5A and Poraplot U columns). Conversion, reaction rates and H2 selectivity were calculated using eqn (1)–(3) in the ESI.†
The structures of bulk Ni and CeO2 were calculated with a 16 × 16 × 16 K point sampling of the Monkhorst–Pack mesh. For large p(4 × 4) slabs of CeO2(111), single-point gamma sampling was used. For gaseous molecules, a box of 20 × 21 × 22 Å sufficed to minimize inter-image interactions and eliminate any spurious symmetry effects.
All the relaxations were performed until the forces dropped below 0.01 eV Å−1. The geometries were confirmed with vibrational analysis to contain zero (stable structures) or exactly one (saddle points) imaginary frequency. The latter were determined using the dimer method with a threshold of 0.01 eV Å−1.37–39
A CeO2 slab consisted of 12 layers with the bottom six fixed layers in their bulk positions of a cubic unit cell (space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (225)) with a lattice constant of 5.53 Å.22 The nickel cluster was made from bulk Ni (unit cell of 3.48 Å), which was cut into one half of an icosahedron containing 55 Ni atoms, yielding a Ni34/CeO2 structure. It was positioned atop CeO2 in the most stable position and allowed to relax. Note that this is a computationally tractable model structure, while on a real catalyst several shapes, geometries and sizes of nickel clusters are present.
m (225)) with a lattice constant of 5.53 Å.22 The nickel cluster was made from bulk Ni (unit cell of 3.48 Å), which was cut into one half of an icosahedron containing 55 Ni atoms, yielding a Ni34/CeO2 structure. It was positioned atop CeO2 in the most stable position and allowed to relax. Note that this is a computationally tractable model structure, while on a real catalyst several shapes, geometries and sizes of nickel clusters are present.
The adsorption energies were calculated as Eads = Eadsorbed − Eslab − Egaseous. Eadsorbed corresponds to the energy of the adsorbate on the slab, Eslab is the energy of an empty slab and Egaseous is the energy of an empty molecule. Activation barriers were computed as EA = ETS − Einitial, where ETS is the energy of the transition state and Einitial is the energy of the initial state. The zero-point energy corrections were included. The entropic effects, required to obtain the free energies, were computed within the harmonic approximation (accounting only for the vibrational contributions) for adsorbed species and full ideal gas approximation for gaseous species, as implemented in ASE 3.22.1 software.40
3. Results and discussion
3.1. Catalytic activity
Catalytic activity and H2 selectivity (expressed as the H2/CO ratio) are presented in Fig. 1a–c. The CH4 and CO2 rates were under thermo-catalytic conditions (full bars) at 460 °C about 15% higher over the catalysts calcined in H2 and argon, compared to the catalyst calcined in air. The CH4 rates for NC-H2, NC-Ar and NC-Air at 460 °C were 4.2, 4.1 and 3.6 mmol (gcat min)−1, respectively. The H2/CO ratio produced by the most active NC-H2 sample increased from 0.27 to 0.51 when the temperature increased from 380 to 460 °C. No catalytic activity was observed in the dark below 360 °C. Since all catalysts went through identical activation protocols directly prior to the catalytic experiment (0.5 h reduction in 5% H2/N2 at 500 °C), the observed differences in performance can strictly be correlated to the calcination history in different atmospheres (Fig. S3 and ESI†).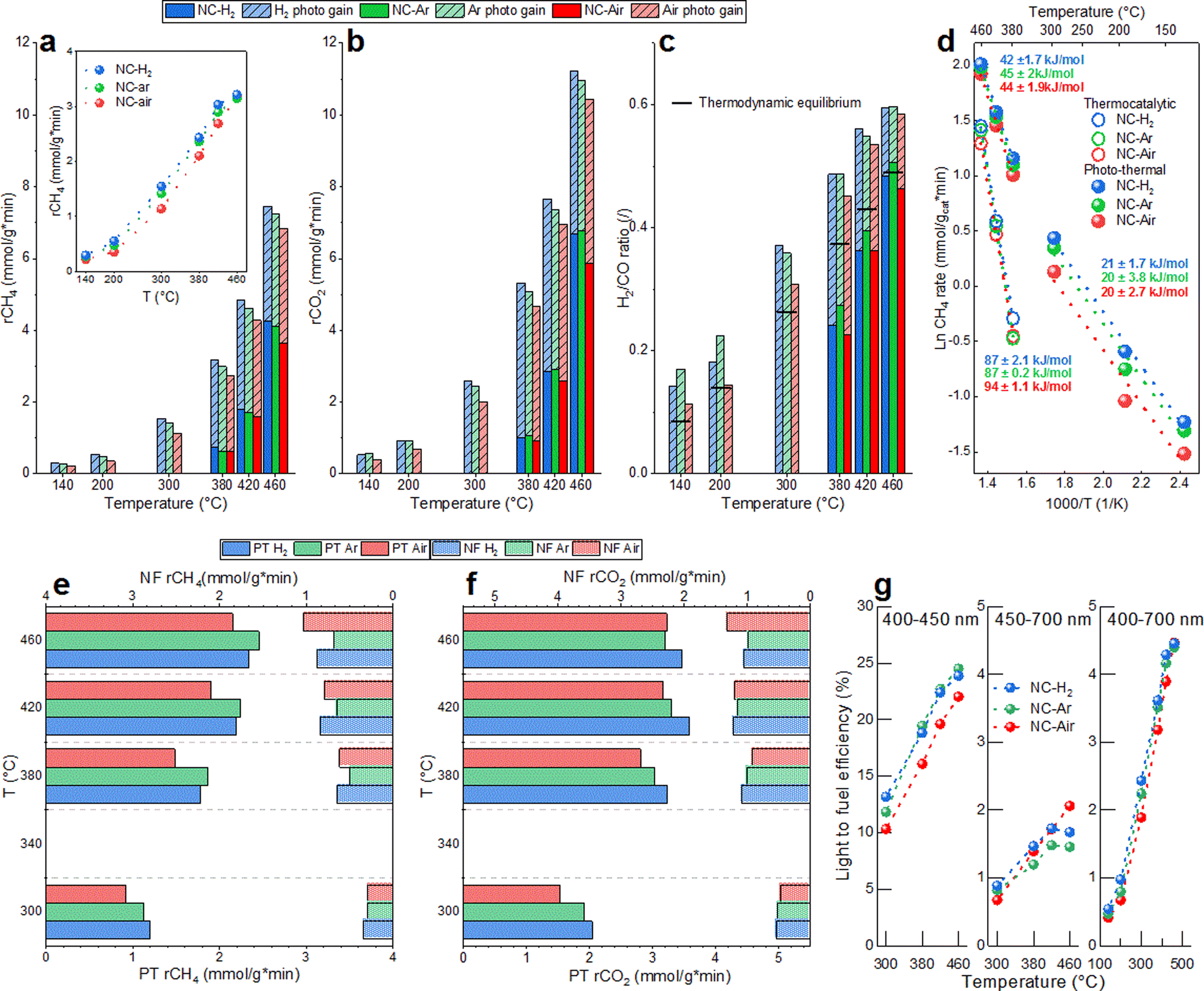 | ||
| Fig. 1 The reaction rate for (a) CH4 and (b) CO2 and (c) the H2/CO ratio under thermal and photo-thermal conditions for NC-Air, NC-Ar and NC-H2 catalysts. Horizontal lines in (c) represent H2/CO values predicted by thermodynamic equilibrium. (d) Comparison of methane apparent activation energies in thermo-catalytic and photo-thermal DRM reactions. Photocatalytic gain of CH4 and CO2 (inset in (a)) achieved by 780 mW cm−2 of visible light illumination. The NF and PT contributions to the photocatalytic gain of (e) CH4 and (f) CO2 rates. (g) Wavelength dependent light-to-fuel efficiency of 2Ni/CeO2 catalysts. | ||
In photo-thermal mode (dashed bars in Fig. 1a–c), a constant visible light irradiance of 780 mW cm−2 was used and the electric heating power was adjusted to control the catalyst temperature. The lowest catalyst temperature where the reaction occurs photo-thermally without electric heating was 140 °C, yielding a CH4 rate of about 0.27 mmol (gcat min)−1 and a H2/CO ratio of 0.14. It has to be noted that illumination causes the sample temperature to reach 140 °C. At a catalyst temperature of 460 °C, the photocatalytic gain (difference between the reaction rate in photo-thermal and thermal modes for the most active NC-H2 catalyst) was 3 and 4.5 mmol (gcat min)−1 for CH4 and CO2, which corresponds to 71 and 68%, respectively. Catalysts calcined in H2 and Ar enable around 15–25% higher photocatalytic gains in the temperature range 420–300 °C (inset in Fig. 1a). In photo-thermal mode, the H2/CO ratio increased from 0.19 to 0.6 as the catalyst temperature increased from 140 to 460 °C, and even surpassed the values predicted by thermodynamic equilibrium (Fig. S4†). The impact of visible light on the DRM reaction mechanism was confirmed through analysis of apparent activation energies (Fig. 1d). The Ea values calculated in thermo-catalytic mode (∼90 kJ mol−1) decreased to ∼40 kJ mol−1 in photo-thermal mode. At temperatures below 300 °C where no thermally driven DRM reaction occurs, the light effect becomes even more substantial and results in a further decrease of Ea values to about ∼20 kJ mol−1. The substantial lowering of the Ea values upon illumination reveals a change of the reaction mechanism and a great alleviation of the activation barriers related to the rate determining step of the DRM reaction.
Wavelength dependent experiments were used to quantify the contribution of the phase transfer (PT) and the near field enhancement (NF) mechanisms to the acceleration of the DRM rate and more precisely identify the optimal wavelength to drive the DRM reaction. In the PT mechanism, electron promotion across the bandgap of CeO2−x occurs, followed by migration to Ni sites, where it assists in methane activation and C–H bond cleavage. This process is driven by photons having higher energy than the bandgap of CeO2−x.
The NF mechanism is driven by photons which have insufficient energy to promote the electrons across the bandgap of CeO2−x, yet result in substantial electromagnetic near field enhancement at the Ni/CeO2−x interface.23 The resulting vibrational and rotational energy is transferred to the surface species and reaction intermediates, thus influencing the reaction rates.41
The optical bandgap energies of 2Ni/CeO2−x materials (2.85–2.89 eV, Fig. S5†) were analyzed in situ under DRM conditions at 400 °C, which correspond to the optical absorption edge of ∼430 nm. As a result, if only the PT phenomenon contributes to the DRM rate, there should be no photocatalytic response with longer wavelengths.
The PT contribution to the CH4 rates is about 2–3 fold larger compared to the NF contribution (Fig. 1e). The largest PT effect on the reaction rate was observed on the NC-Ar catalyst (closely followed by NC-H2), while the lowest was observed on the NC-Air catalyst (Fig. 1e). With smaller nickel size, the energy of the Fermi level decreases, making the charge transfer from CeO2−x to Ni faster and e−/h+ pair recombination slower.42 This is exactly what was observed when probing the photoluminescence of the catalysts. The highest photoluminescence, indicative of the most extensive emissive hot carrier recombination, was observed over the NC-Air catalyst (Fig. S6†).
When we probed the NF contribution to the CH4 rate acceleration, the smallest effect was observed on the NC-Ar sample. It can be expected that a reduction of the mean free path of electrons due to nickel particle size reduction increases the losses in the material and quenches the near field enhancement.43 It appears that the structural properties of the nickel/ceria catalysts, required to optimally utilize the PT and NF pathways, are very different: small Ni particles boost the PT effect while larger ones improve the NF contribution.
The wavelength dependent CO2 rate acceleration is shown in Fig. 1f. The highest PT effect is recorded on NC-H2, followed by the NC-Ar and NC-Air samples. The NF effect on CO2 conversion is much less dependent on the calcination of the samples, as no clear and consistent trend is observable. This is because light cannot directly dissociate CO2 in the absence of hydrogen and the hydrogen-assisted CO2 dissociation via the RWGS reaction is negligibly accelerated by visible light illumination (Fig. 4 and S7†) at temperatures above 300 °C.
The light-to-fuel efficiencies (Fig. 1g) for photons in the wavelength range between 400 and 450 nm reached values up to 25% on NC-Ar and NC-H2, followed by the NC-Air catalyst. The light to fuel efficiency of less energetic photons (450–700 nm) is much lower and reaches values below 2%. With the entire visible spectrum (400–700 nm), the light-to-fuel efficiencies reach up to 4.6% and are highest for the NC-Ar and NC-H2 catalysts.
3.2. Structural characterization
In situ Ni K-edge EXAFS analysis was used to determine the local structure around Ni (Fig. S10, ESI and Tables S2–S5†). In all as-synthesized catalysts, the coordination numbers of Ni and oxygen shells at larger distances are significantly lower than that in bulk NiO (Table S3†), indicating that the majority of nickel exists as NiO clusters smaller than 1 nm.44–47 In addition, Ce neighbors are identified at about 3.6 Å, which clearly indicates that some of the Ni cations are directly attached to the CeO2 support by Ni–O–Ce bridges. Small differences in the average Ni local structure exist between the catalysts calcined in different atmospheres: the smallest NiO clusters and the largest number of Ni–O–Ce bridges are present in the sample calcined in H2.
During reduction in a 10% H2/He flow at 500 °C, small metallic Ni clusters are formed (Table S5†). The distribution of Ni neighbors is similar, but not identical to that in the Ni metal with a fcc crystal structure. The coordination numbers of Ni neighbors in all coordination shells are significantly lower than in the case of bulk nickel, indicating that the majority of nickel is present as small nanoclusters and their average size is below 1 nm. A small fraction of nickel remains attached to the CeO2 support, forming Ni–O–Ce bridges which are most abundant in NC-Ar and NC-H2 catalysts, suggesting their higher nickel dispersion and metal–support interface compared to the NC-Air catalyst. A structural change in the nickel–ceria interface is observed upon visible light illumination at 500 °C: in the NC-H2 and NC-Ar catalysts, the number of Ni–O–Ce bridges is reduced, while in the NC-Air sample, their number is increased. This is in line with the Raman analysis of the surface oxygen (vide infra) and is related to the thermal history of the samples and varying oxygen abundance in ceria.
m) (Fig. 2 and S11†). The individual crystallites have a rod-like shape of ∼10–20 nm in diameter and ∼200 nm in length. The CeO2 crystals show well-developed crystal facets, having chisel-like morphology, terminated predominantly by {111} crystal planes with minor {100} facets (Fig. 2c).
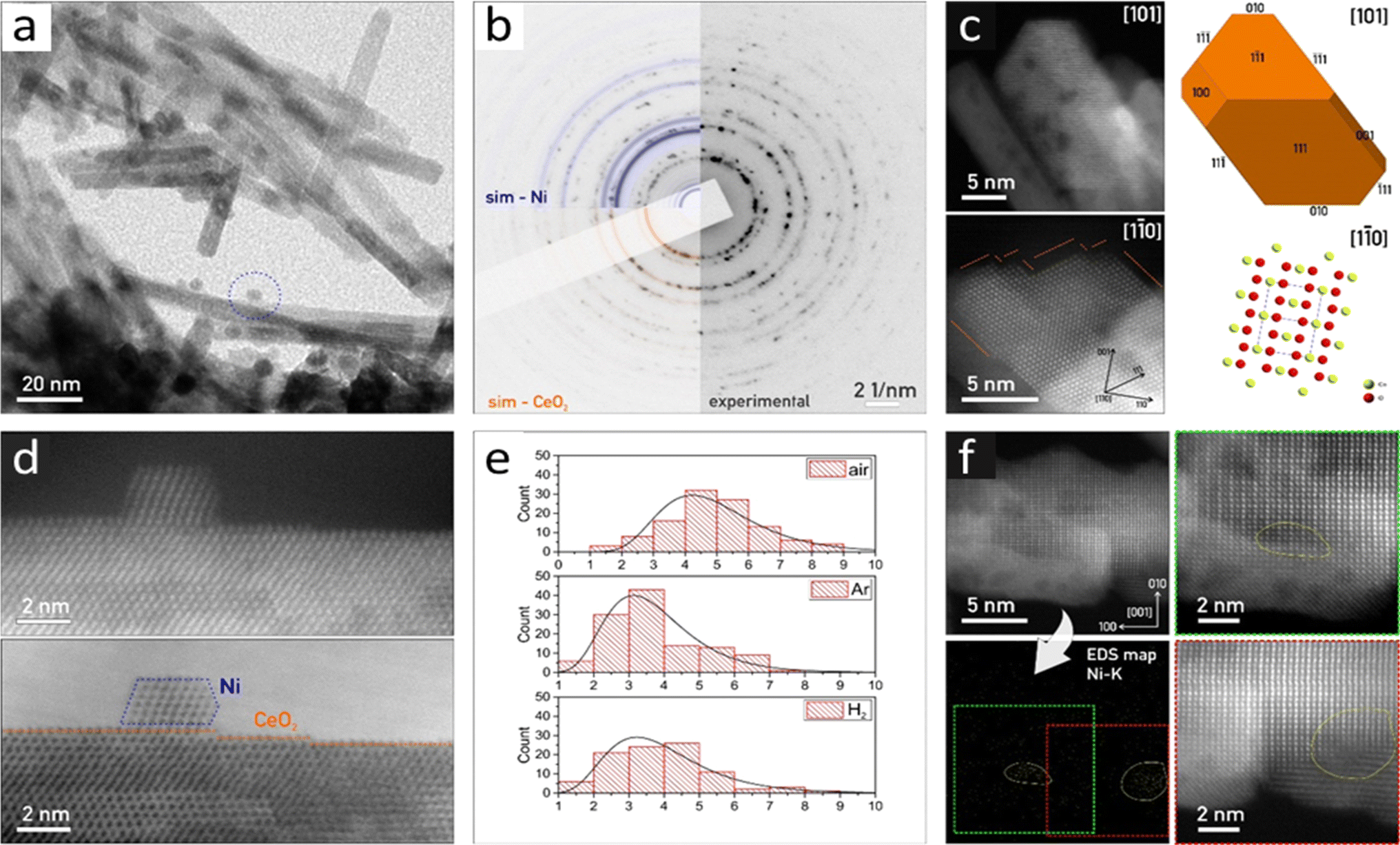 | ||
| Fig. 2 (a) TEM overview of rod-like CeO2 crystals with scattered Ni nanoparticles (one marked with a blue dashed circle) of the NC-Ar sample. (b) Experimental SAED pattern with over-imposed Ni (blue) and CeO2 (red) simulations. (c) HAADF-STEM micrograph for the morphology reconstruction of the CeO2 crystals corresponding to the NC-H2 sample, with the correlated 3D model and crystal structure in identical orientation. The surface shows multiple steps in {111}. (d) HAADF-STEM and BF-STEM micrographs of Ni (outlined blue) on CeO2 in the NC-Ar sample. CeO2 crystal shows a stepped {111} surface in the [110] zone axis. (e) Nickel particle size distribution histogram for samples reduced in the air, Ar, and H2 atmospheres. (f) HAADF-STEM of CeO2 with the corresponding Ni–K EDS map and magnified regions where Ni clusters were detected (outlined in yellow). | ||
The visualized Ni nanoparticles are <10 nm in size, forming cuboctahedrons (Fig. 2a and d). The Ni particle size distribution histograms considering the Feret diameter48 show a clear trend: NC-Air: 4.9 nm, NC-Ar: 3.7 nm and NC-H2: 3.9 nm (Fig. 2e). Besides Ni nanocrystals, we identified many regions where EDS confirmed the presence of Ni on the CeO2 nanorods, although we could not visualize the Ni crystallites (Fig. S11†). Due to the absence of HAADF-STEM and SAED signals originating in the periodic ordering of nickel atoms (Fig. 2f), we conclude that these regions are covered by nanometer sized Ni clusters (Fig. S11†).49
| Catalyst | Peak position (cm−1) | Fitted peak area at different conditions (a.u.) | |||||
|---|---|---|---|---|---|---|---|
| Air | Argon | Hydrogen | |||||
| RT | 500 °C | RT | 500 °C | RT | 500 °C | ||
| CeO2 | 240 | 3.2 | 2.8 | 2.2 | 2.4 | 2.2 | 3.3 |
| 2Ni/CeO2 | 1.7 | 1.8 | 0.6 | 0.1 | 0.3 | 0.1 | |
| CeO2 | 550 and 600 | 4.2 | 12.6 | 3.2 | 12.9 | 4.1 | 13.5 |
| 2Ni/CeO2 | 6.4 | 13.6 | 14.3 | 23.4 | 11.7 | 29.1 | |
In the as-synthesized NC-Ar and NC-H2 materials at ambient temperature, the subsurface/bulk oxygen vacancy sites are present in about 2-fold greater abundance compared to the NC-Air sample (Fig. 3a–c, S13† and Table 1).
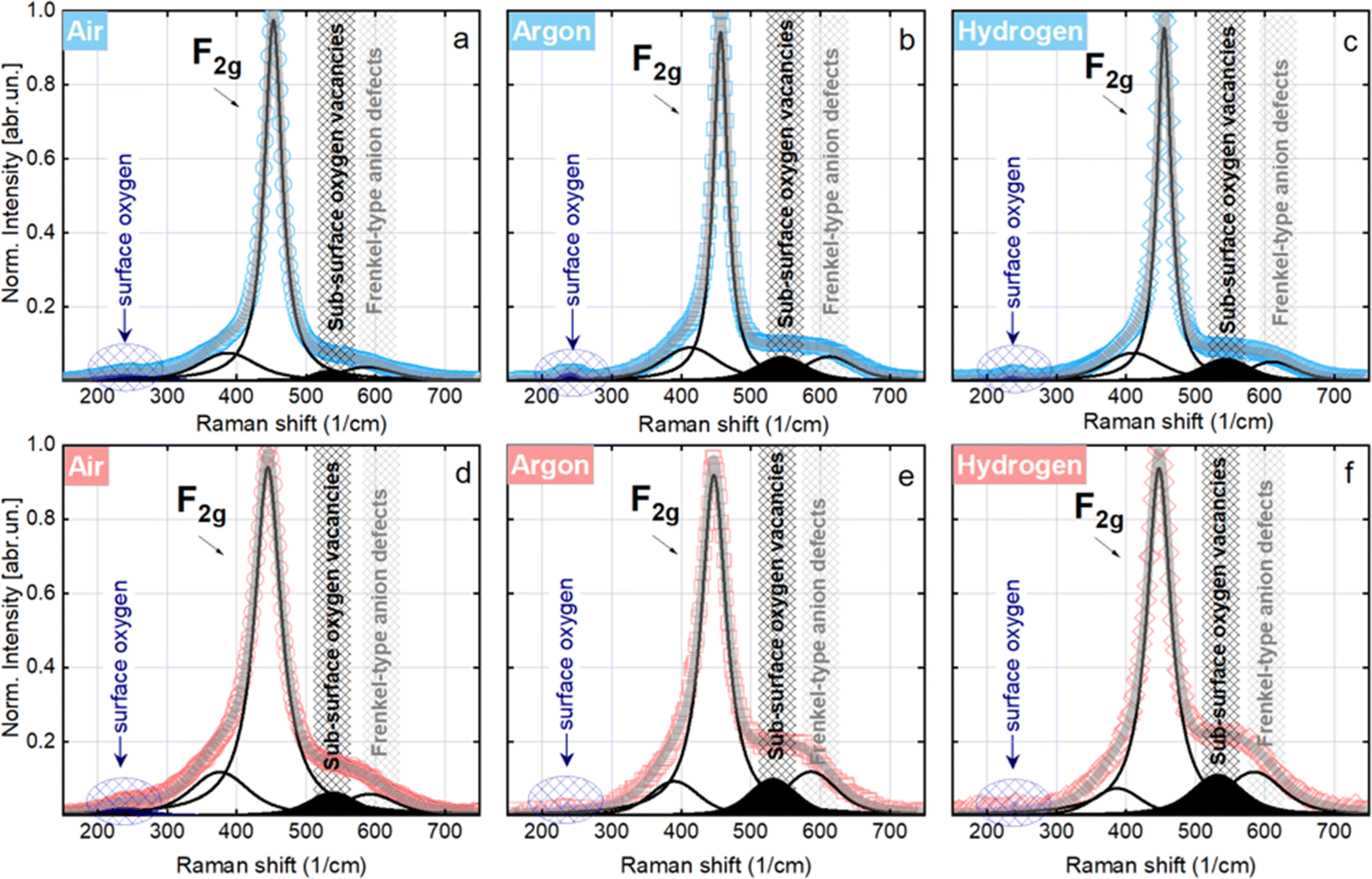 | ||
| Fig. 3 Raman spectra of Ni/CeO2−x catalysts calcined in different atmospheres at room temperature (blue data) and 500 °C (red data) in an equimolar CH4/CO2 flow, accompanied by Gaussian peak deconvolution. | ||
At 500 °C, an evident reduction of surface oxygen is observed especially in the NC-Ar and NC-H2 samples, together with an increased contribution of subsurface/bulk oxygen defects (Fig. 3d–f and Table 1). The surface oxygen removal is greatly improved by the presence of nickel, since the peak area under 240 cm−1 mode is 2 to 10 times lower for Ni/CeO2 catalysts compared to bare CeO2 (Table 1).
The combination of in situ XAS and TEM results revealed that calcination in argon or hydrogen enables higher dispersion of nickel, which is present mainly as subnanometer sized metallic clusters. Furthermore, in situ Raman analysis showed that calcination in an Ar or H2 atmosphere lowers the surface oxygen population by about 18 times compared to the sample calcined in air, and the abundance of bulk oxygen defects is about two times higher (Table 1). To summarize, higher dispersion of nickel and a higher abundance of surface as well as bulk oxygen defects in ceria are achieved by calcination of Ni/CeO2 samples in argon or hydrogen.
The XRD results (Fig. S15†) confirmed the presence of CeO2 in the fcc crystal structure in all fresh 2Ni/CeO2−x catalysts. The differences in average CeO2 crystal size among samples are negligible (Table S6†). The absence of diffraction lines belonging to nickel containing phases (inset Fig. S15†) is due to its low content and high dispersion.
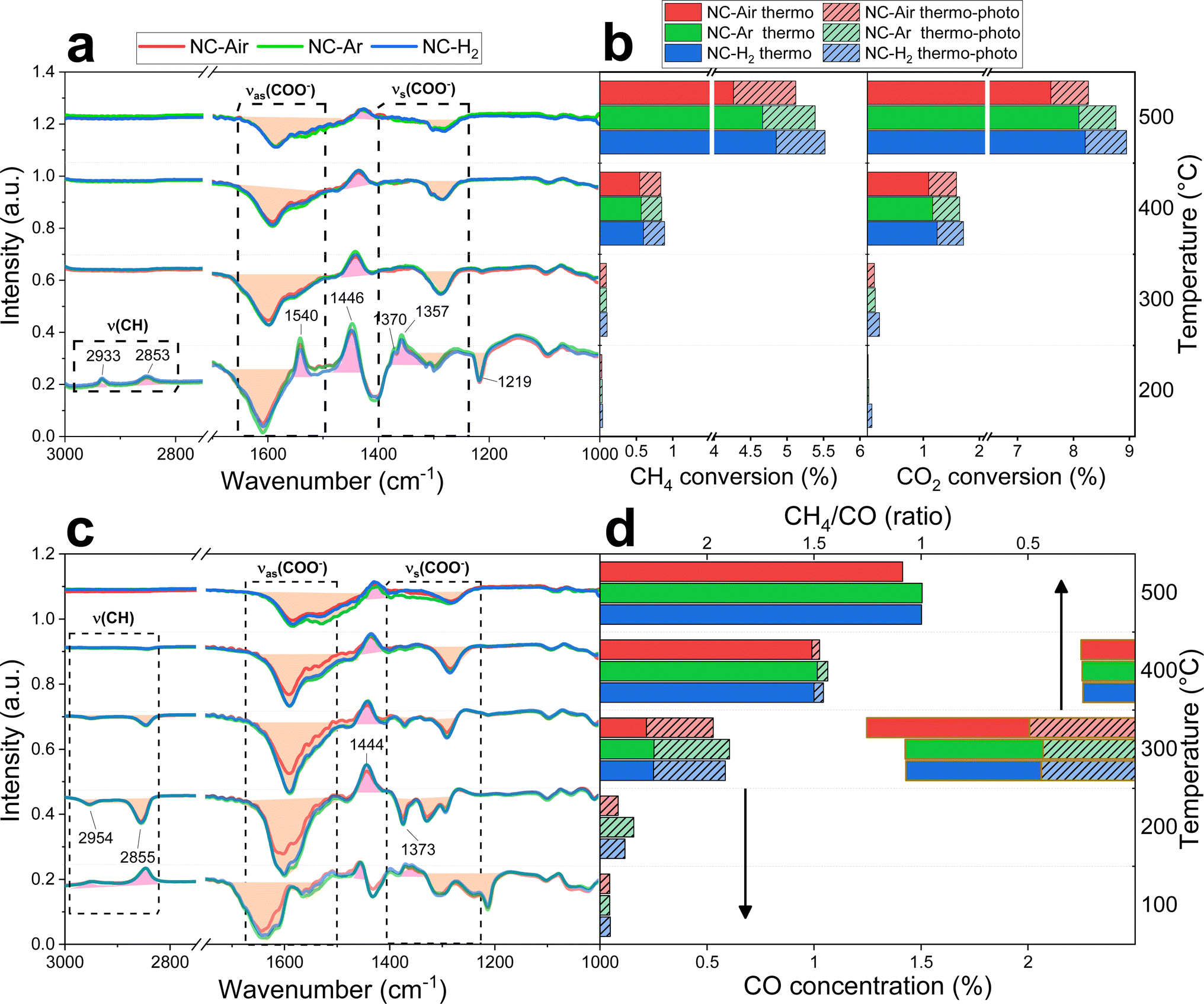 | ||
| Fig. 4 Difference spectra between thermal and photo-thermal conditions during operando DRM (a) and RWGS (c), combined with the measured CH4 and CO2 conversions for DRM reactions (b), and (d) CO concentration and the CH4/CO ratio during the RWGS reaction. | ||
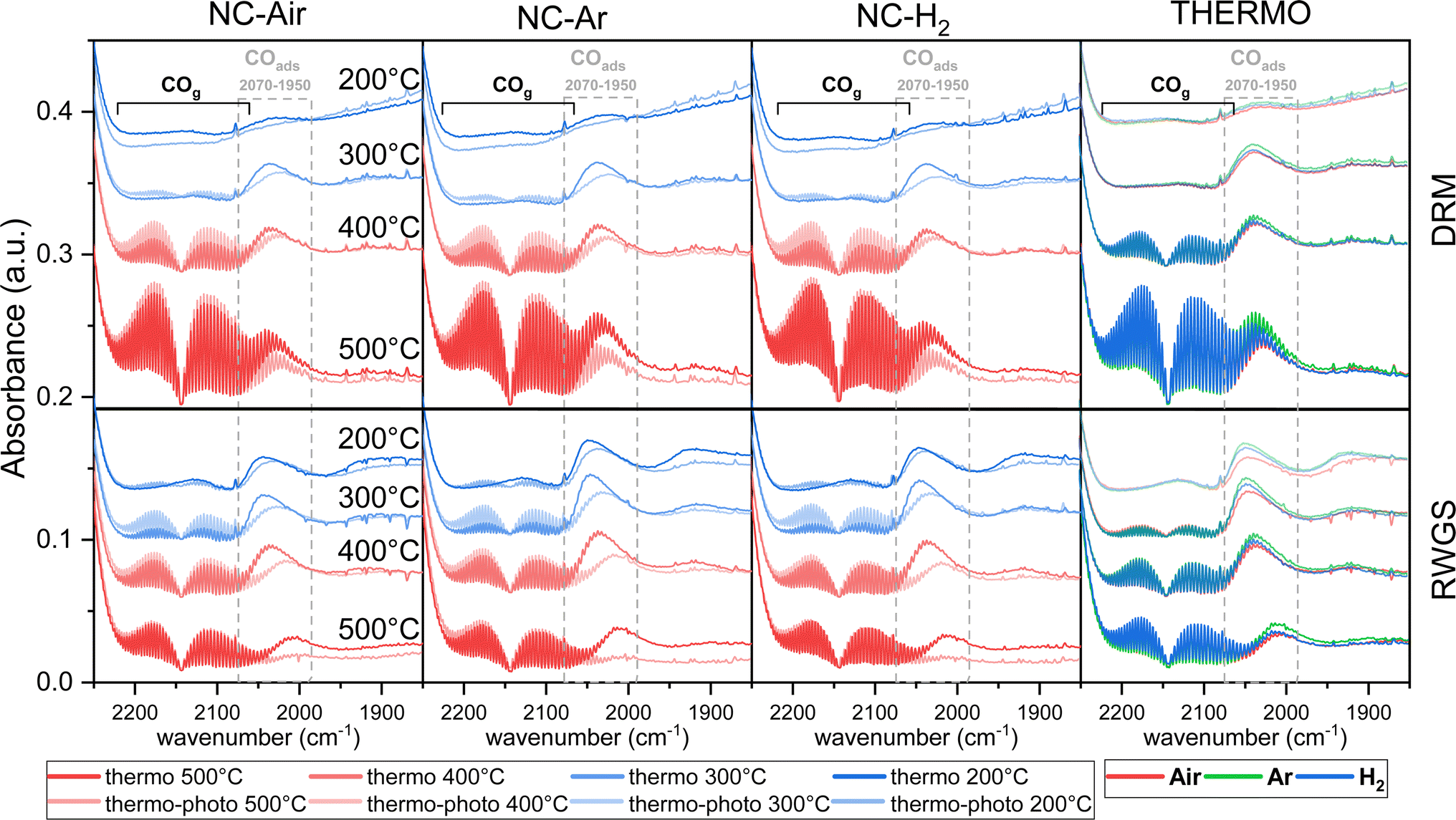 | ||
| Fig. 5 Operando DRIFT spectra of the carbonyl region during DRM and RWGS reactions at different temperatures in thermocatalytic and photo-thermal modes for NC-Air, NC-Ar and NC-H2 catalysts. | ||
Under thermal DRM reaction conditions, νasCOO− (1650–1540 cm−1) and νsCOO− (1450–1360 cm−1) vibrations were identified between 200 and 500 °C. Both νasCOO− and νsCOO− vibrations can be assigned to bidentate (1570–1550 cm−1) and monodentate (1610–1590 cm−1) carbonate groups, while the bands between 1490 and 1385 cm−1 are assigned to the stable polydentate carbonate species (see Fig. S16 and detailed explanation in the ESI†). Carbonyl groups on nickel are also observed in the steady state (2070–1950 cm−1, Fig. 5). No significant differences in the DRIFT spectra were observed in the carbonate region (1700–1200 cm−1) among samples calcined in different atmospheres (Fig. 4a and S16†).
When comparing the DRIFT spectra under thermal and photo-thermal conditions, a decrease of the νasCOO− and νsCOO− vibrational bands (orange area in Fig. 4a) and an increase of the band at 1446 cm−1 (pink area in Fig. 4a) were observed regardless of the temperature (Fig. 4a and S16†). The band at 1219 cm−1 decreases synchronously with the bands between 1650 and 1540 cm−1 at 200 °C, suggesting that the band at 1219 cm−1 originates from mono- or bidentate carbonates.54 Apparently, mono- and bidentate carbonates partially desorb as CO2, decompose to CO or convert to the most stable polydentate upon illumination. It has to be mentioned that carbonates populate the ceria surface.22 Compared with thermal mode, new peaks appeared upon illumination at 200 °C (Fig. 4a), denoted as split CH3 umbrella bending vibrational modes (1370 and 1357 cm−1),55 C–H symmetric deformation vibrational mode of adsorbed CH4 (1540 cm−1),56 and asymmetric and symmetric C–H vibrational modes for CH2 and CH3 groups (2933 and 2853 cm−1). The assignation of the bands at 2933 and 2853 cm−1 to CH3 functional groups (and not formate HCOO species) is substantiated by the fact that they do not follow the intensity trends of C–O formate vibrations in the range of 1650–1520 cm−1. At 200 °C when the methane related peaks appear, the photo-thermal DRM activity is very low, which allows for the accumulation of weakly bound methane reaction intermediates (Fig. 4b).
The RWGS reaction (eqn (2)) negatively influences H2 selectivity during the DRM reaction and operando DRIFTS analysis was applied to study the effect of light on this side reaction. A mixture of 2.5% H2, 50% CO2 and 47.5% N2 (Fig. 4c, d and S16†) was used because it mimics the gas composition during the DRM reaction (excluding methane).
CO formation becomes observable at 100 °C only in photo-thermal mode and CO is the only reaction product. The thermal decomposition of formate on ceria surfaces becomes notable around 200 °C.22 In line with this, in photo-thermal mode at 200 °C and 300 °C, the selectivity for CH4 is lower and the asymmetric and symmetric C–H vibrations (2954 and 2855 cm−1) together with the 1373 cm−1 band of bidentate formate55 decrease (Fig. 4c, d and S16†). The observed phenomena can be related to the oxidation of the formate species on the valence band of reduced ceria, thus preventing its further hydrogenation to CH4. Formate was previously identified as a crucial intermediate governing methane formation.57 At 400 °C, the steady state formate population is negligible and the leveraging power of light to increase the CO selectivity via the RWGS pathway is greatly diminished. Illumination causes a decrease of the mono- and bidentate C–O vibrations in the regions 1650–1540 and 1450–1360 cm−1 (Fig. 4c and S16†) and an increase of the polydentate carbonates (band at 1444 cm−1). The carbonate signal difference is smaller on NC-Air compared to the other two samples (red trace in Fig. 4c), whereas the differences in the CO formation do not follow the same trend (Fig. 4d). As a result, carbonates likely do not influence the kinetically relevant steps of the RWGS reaction.58 On the other hand, the CH4 selectivity at 300 °C is highest on the NC-Air catalyst (Fig. 4d), which can be explained by the presence of larger Ni clusters on this sample.59 Also, the carbonyl bands characteristic of CO chemisorbed on nickel are larger on NC-Ar and NC-H2 samples compared to NC-Air (Fig. 5), suggesting higher nickel dispersion in the former.
Light also induces the desorption of the carbonyl groups from the nickel surface (Fig. 5). As a result, a larger number of Ni and Ni/CeO2−x interfacial sites are available and populated with methane under visible light, thus accelerating the CH4 rate (Fig. 4b). The light effect on the formate species was evaluated only at 140, 200 and 300 °C (Fig. 4a and S17†) because at 400 and 500 °C their surface population is negligible. The bands at 2934 and 2846 cm−1 belonging to the asymmetric and symmetric C–H vibrations are less intense during illumination, which reveals their lower surface coverage as a result of their increased reactivity.
In stoichiometric CeO2(111), the average Bader charge is calculated to be +2.38 for Ce and −1.19 for O. All atoms are equivalent as there is negligible charge transfer towards the surface. In the partially reduced CeO2−x where a fraction of surface oxygen atoms are removed, a migration of the subsurface oxygen to the surface is observed, together with the formation of subsurface vacancies.22 The oxygen atoms adjacent to the vacancies are more negative (−1.30), while the adjacent Ce is less charged (+2.25).
As the interface with a nickel cluster is formed, there is a considerable charge transfer of 2.69 e0 from nickel to the cerium atoms of CeO2. This is limited mostly to the first layer of nickel atoms, which have a charge of +0.20. The Ce atoms in contact with them are slightly less charged at +2.23 per atom. However, when the ceria surface was partially reduced (two oxygen atoms were removed in the model), the overall charge transfer from the Ni cluster was only 1.35 e0 and the cerium atoms adjacent to the oxygen vacancy in contact with nickel were further reduced to +2.13. The nickel atoms near the oxygen vacancy were even slightly negative (−0.15), and the rest of nickel also exhibited lower charge transfer (+0.18 per atom).
It has been previously shown that the first C–H dissociation in CH4 is the rate-determining step.60–63 Thus, we limit our DFT investigation to adsorption 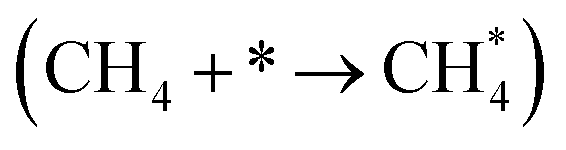 and activation
and activation 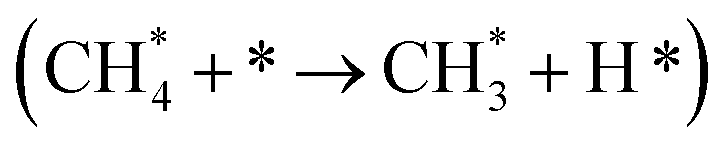 . We study four cases: an extended Ni(111) surface, an icosahedral Ni55 nanoparticle, the interface between Ni and CeO2, modelled as Ni34/(CeO2)n, and a reduced interface, where two oxygen atoms adjacent to the reaction site were removed, Ni34/(CeO2−x)n.
. We study four cases: an extended Ni(111) surface, an icosahedral Ni55 nanoparticle, the interface between Ni and CeO2, modelled as Ni34/(CeO2)n, and a reduced interface, where two oxygen atoms adjacent to the reaction site were removed, Ni34/(CeO2−x)n.
In all instances, CH4 physisorbs with a weak interaction of 0.25–0.29 eV. The calculated Gibbs free energies are positive above 210 K, meaning that the surface coverage with CH4 will be negligible. More importantly, the activation barrier of the C–H bond cleavage is substantially lowered at the interface from 0.71 eV on Ni(111) to 0.50 eV on the stoichiometric Ni34/(CeO2). Upon partial reduction of the ceria support, the barrier is further decreased to 0.45 eV. The unsupported nanoparticle (Ni55) is similarly active as the extended surface (see Table 2). When considering the entropic effects, the trend is even more pronounced. The barriers are increased at higher temperatures, which is consistent with the experimental data (Fig. 1d), and the difference in barriers between the stoichiometric Ni34/(CeO2)n and oxygen-vacancy Ni34/(CeO2−x)n grows larger in favor of the latter. See Fig. S18† for structures and free energies.
| Ni(111) | Ni55 | Ni34/(CeO2)n | Ni34/(CeO2−x)n | ||
|---|---|---|---|---|---|
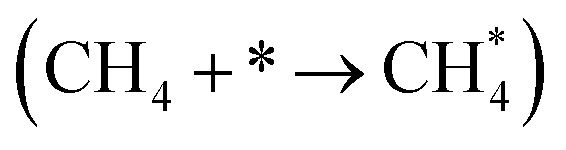
|
ΔE | −0.25 | −0.25 | −0.25 | −0.29 |
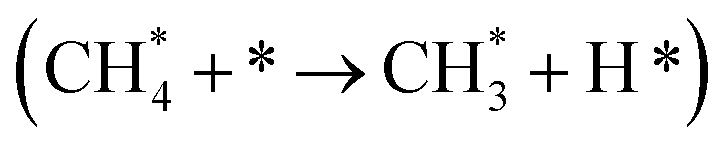
|
E a | 0.71 | 0.70 | 0.50 | 0.45 |
| ΔE | −0.20 | −0.21 | −0.82 | −0.30 |
We show that the calcination of Ni/CeO2 catalysts in air, argon or hydrogen can be used as a simple and versatile technique to manipulate nickel dispersion and the abundance of surface, as well as bulk oxygen vacancies in ceria. The possibility to influence nickel dispersion through thermal aging is associated with the formation of oxygen defects in ceria during calcination, which act as anchoring sites for nickel.64–67 Higher nickel dispersion as a result of calcination in Ar or H2 was identified via in situ EXAFS through a lower nickel coordination number in all shells, and lower abundance of the Ni–O–Ce bridges, which suggests extensive surface reduction of ceria. In addition to the subnanometer nickel clusters which represent the majority of the nickel phase, a minor fraction of Ni crystals was observed by TEM. Their average size increased from 3.7 (NC-H2) to 3.9 (NC-Ar) and 4.9 nm (NC-Air). Again, inert and reductive calcination atmospheres favored smaller nickel crystals.
Thermal history has a pronounced effect on the number of oxygen vacancies in ceria: bulk oxygen defects are 1.7 times more abundant after calcination in argon (23.4/13.6, Table 2) and 2.1 times more abundant after calcination in hydrogen (29.1/13.6, Table 2) compared to calcination in air. The chemistry of the Ni/ceria interface and its catalytic activity depend strongly on the presence of oxygen. The oxygen deficient interface, as identified by in situ Raman and in situ EXAFS techniques, is achieved during photo-thermal reaction over samples with higher inherent oxygen defects (calcined in Ar and H2).
The DRM is a structure sensitive reaction where the C–H bond cleavage (rate determining step) occurs with an energy barrier (Ea = 0.71 eV) on extended Ni terraces and (Ea = ∼0.6 eV) on coordinatively unsaturated Ni step sites. The Ea is further decreased to 0.50 and 0.45 eV at the Ni/CeO2 and Ni/CeO2−x interfaces, respectively. This shows the following: (i) the interface is more active than the extended Ni surface, (ii) the increase in activity is not due to the rugged surface or nanoparticle geometry but the electronic effects, (iii) partially reducing the Ni/Ceria interface further increases the activity, although to a smaller extent, and (iv) at higher temperatures, the reaction barrier is higher, but this is overcome by the faster kinetics.
The calcination atmosphere induced differences among Ni/CeO2−x catalysts cause notable consequences in the DRM reaction activity: samples calcined in argon and H2 exhibit about a 15% higher rate under thermo-catalytic conditions and between 15 and 25% higher photocatalytic gains under photo-thermal conditions, compared to the catalyst calcined in air.
The Ea values in thermo-catalytic mode are 87–94 kJ mol−1, which is in good agreement with previous studies, as well as barriers estimated for extended Ni(111) facets.68,69 As a result, Ni crystallites visualized by TEM likely act as the main methane dissociation sites when the DRM reaction is performed in the dark.
Visible light illumination of the catalysts causes several changes in the surface chemistry of the investigated materials: (i) carbonyl species are desorbed from the nickel sites, (ii) carbonates on ceria are destabilized and desorbed/decomposed and (iii) adsorbed/activated methane is more abundant.
In addition to freeing up the highly active nickel clusters through desorption of CO and decomposition of carbonates, the hot electron injection into the methane molecule can accelerate its dissociation into C and H2, further alleviating methane dissociation. In the reaction temperature range between 360 and 460 °C, the Ea values for methane dropped to 42–45 kJ mol−1, which resulted in the acceleration of CH4 and CO2 rates and the H2/CO ratio in the produced syngas exceeding the values predicted by thermodynamic equilibrium. The origins of the improved H2 selectivity were analyzed by operando DRIFTS analysis, which revealed no direct CO2 dissociation under thermal and photo-thermal conditions. In the presence of both CO2 and H2, the RWGS rate (and consequently CO formation) is accelerated only at temperatures below 400 °C, where formate species, crucial RWGS intermediates, are present in notable amounts on ceria and are oxidized to CO using photo-catalytically generated hot holes in ceria.
The wavelength dependent DRM studies showed that the PT effect, induced by high energy photons, enabled up to 25% light-to-fuel efficiency, whereas the highest light-to-fuel efficiency reached 2% when illuminated by wavelengths longer than 450 nm. The PT reaction channel is clearly dominant for accelerating the DRM reaction rate under photo-thermal conditions over the Ni/CeO2−x catalysts. From the aspect of photon utilization, the nanometer sized nickel clusters would shorten the required distance for excited carrier migration from bulk CeO2−x nanorods to their surface and nickel–ceria interface, resulting in higher light-to-fuel efficiency. The PT driven reaction channel is favored by the presence of highly dispersed nickel clusters, whose formation is favored by calcination in Ar and H2 atmospheres. The hot holes which remain on the ceria surface participate in the oxidation of formate to OH and CO.
The methane dissociation mechanism is crucial in the DRM reaction and is discussed next. From a mechanistic point of view, the elementary steps of CH4 dissociation in DRM involve stepwise CH4 dissociation to 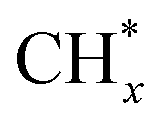 (x = 0–3) and H*, the interaction of
(x = 0–3) and H*, the interaction of 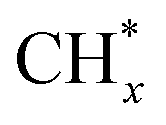 with O* or lattice oxygen of the support to CHxO*, and further oxidation of CHxO* to CO. Our DRIFTS analysis at different temperatures in the dark and under illumination shows that the C–H symmetric vibration mode of adsorbed CH4 (1540 cm−1) and bands at 1370 and 1357 cm−1 from CH3 umbrella bending vibrational modes were observed only at the lowest temperature (200 °C), and CHxO* species (1120 cm−1) were not observed at all. This suggests enhanced reactivity and higher surface coverage of
with O* or lattice oxygen of the support to CHxO*, and further oxidation of CHxO* to CO. Our DRIFTS analysis at different temperatures in the dark and under illumination shows that the C–H symmetric vibration mode of adsorbed CH4 (1540 cm−1) and bands at 1370 and 1357 cm−1 from CH3 umbrella bending vibrational modes were observed only at the lowest temperature (200 °C), and CHxO* species (1120 cm−1) were not observed at all. This suggests enhanced reactivity and higher surface coverage of 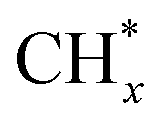 intermediates upon illumination. In the DRM reaction, methane represents the hydrogen source needed for CO2 dissociation, meaning that the activation of methane dictates the conversion of carbonate species on the catalyst surface to the CO product. Despite a similar decrease of the carbonate species intensity under photo-thermal DRM conditions (Fig. 4a and b), the lowest methane conversion over the NC-Air catalyst provides evidence of a lower amount of hydrogen, enabling the lowest CO2 conversion compared to the catalyst calcined in argon and H2. Consequently, carbonates on the NC-Air catalyst are more prone to desorption as CO2 compared to the NC-Ar and NC-H2 catalysts.
intermediates upon illumination. In the DRM reaction, methane represents the hydrogen source needed for CO2 dissociation, meaning that the activation of methane dictates the conversion of carbonate species on the catalyst surface to the CO product. Despite a similar decrease of the carbonate species intensity under photo-thermal DRM conditions (Fig. 4a and b), the lowest methane conversion over the NC-Air catalyst provides evidence of a lower amount of hydrogen, enabling the lowest CO2 conversion compared to the catalyst calcined in argon and H2. Consequently, carbonates on the NC-Air catalyst are more prone to desorption as CO2 compared to the NC-Ar and NC-H2 catalysts.
Based on the spectroscopic, DFT and catalytic data, we propose that the sequential C–H dissociation at the oxygen deficient Ni/ceria interface without oxygen involvement is likely the most active reaction channel. Upon the C–H bond cleavage at the nickel–oxygen deficient ceria interface, the hydrogen–oxygen interaction is less preferable. As a result, easier CH4 dissociation and higher H2 selectivity are achieved by visible light, especially on catalysts with a higher oxygen deficiency. Based on the above, we postulate that highly active sub-nanometer nickel CeO2−x interface sites are enabled by visible light, which can dissociate methane with an Ea value of about 42–45 kJ mol−1. These sites are unable to participate in the thermally driven DRM reaction at temperatures below 460 °C due to relatively low oxygen vacancy density at the nickel–ceria interface, but even more importantly, due to the high coverage of the nickel surface with CO and ceria surface coverage with carbonate spectator species.
With further decrease of the catalyst temperature below 350 °C, the Ea values further decrease to 20 kJ mol−1, which is well below the DFT predicted values. This indicates a clear alteration in the DRM reaction mechanism and enables a methane rate of 0.22 mmol (gcat min)−1 at a catalyst temperature of 140 °C without electric heating of the reactor. The activation energies are very similar on all catalysts, which suggests that the working mechanism is the same, and they are differentiated mainly by the number of active sites, which is determined by the initial calcination.
4. Conclusion
The calcination of Ni/CeO2 catalysts in reductive, inert or oxidative atmospheres was used to tailor nickel dispersion and oxygen vacancy density in ceria nanorods. The formation of subnanometer sized nickel clusters was favored and surface and bulk oxygen density in ceria could be improved by about 18 and 2 times, after calcination in argon or hydrogen. Calcination induced active site promotion enabled ∼15% methane rate acceleration under thermo-catalytic conditions compared to calcination in air. The photo-thermal DRM rate is higher by about 70% at 460 °C and 780 mW cm−2 of visible light illumination, compared to the rate in the dark at an identical catalyst temperature. The photo-thermal rate is mainly driven by the PT mechanism, with higher nickel dispersion and a larger number of subsurface oxygen defects retarding hot carrier recombination. In addition, hot carrier mediated oxidation of formate species on ceria prevents methane formation, causing improved H2 selectivity at temperatures below 400 °C. Visible light illumination also stimulates the desorption of CO and promotes methane adsorption and activation on nickel. Over the ceria surface, illumination causes destabilization and desorption of the carbonates as CO and CO2. As a result of light-induced nickel and ceria surface chemistry modification, outstandingly active surface sites can be unlocked at temperatures below 400 °C for the DRM reaction, which otherwise remain covered by strongly adsorbed species.This work outlines a simple calcination approach for maximizing nickel dispersion and oxygen vacancy abundance in ceria, which in photo-thermal mode enables running the DRM reaction under mild conditions with exceptional rates and selectivity.
Author contributions
Kristijan Lorber: writing – original draft, conceptualization, investigation, and writing – review & editing; Vasyl Shvalya: writing – review & editing, investigation, and visualization; Janez Zavašnik: investigation and visualization; Damjan Vengust: investigation; Iztok Arčon: investigation and writing – original draft; Matej Huš: formal analysis and methodology; Andraž Pavlišič: formal analysis and visualization; Janvit Teržan: investigation; Uros Cvelbar: supervision and funding acquisition; Blaž Likozar: supervision and funding acquisition; Petar Djinović: writing – review & editing, supervision, and funding acquisition.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The Slovenian Research and Innovation Agency (ARIS) financed this research through projects and programs: P2-0152, J1-3020, N1-0303, I0-0039, J2-4424, P1-0417, P1-0112, J2-1726, N2-0265 and P1-0418. The HPC RIVR consortium and EuroHPC JU provided computing resources of the HPC system Vega at the Institute of Information Science. The access to the SR facilities of ESRF (beamline BM23, experiment number MA 5036) is acknowledged. Dr Kirill A. Lomachenko, Dr Cesare Atzori and Prof. Dr Katarina Vogel Mikuš advised and assisted during XAS experiments. Dr Matjaž Mazaj helped with the analysis of XRD data.References
- D. J. Hofmann, J. H. Butler and P. P. Tans, Atmos. Environ., 2009, 43, 2084–2086 CrossRef CAS.
- X. Li, Energy Policy, 2005, 33, 2237–2243 CrossRef.
- S. S. Ali, S. S. Ali and N. Tabassum, J. Environ. Chem. Eng., 2022, 10, 106962 CrossRef CAS.
- M. Zhang, J. Zhang, Y. Wu, J. Pan, Q. Zhang, Y. Tan and Y. Han, Appl. Catal., B, 2019, 244, 427–437 CrossRef CAS.
- A. Abdulrasheed, A. A. Jalil, Y. Gambo, M. Ibrahim, H. U. Hambali and M. Y. Shahul Hamid, Renewable Sustainable Energy Rev., 2019, 108, 175–193 CrossRef CAS.
- H. J. Venvik and J. Yang, Catal. Today, 2017, 285, 135–146 CrossRef CAS.
- Z. Bian, S. Das, M. H. Wai, P. Hongmanorom and S. Kawi, ChemPhysChem, 2017, 18, 3117–3134 CrossRef CAS PubMed.
- D. Pakhare and J. Spivey, Chem. Soc. Rev., 2014, 43, 7813–7837 RSC.
- Y. Wang, L. Yao, S. Wang, D. Mao and C. Hu, Fuel Process. Technol., 2018, 169, 199–206 CrossRef CAS.
- J. Khatri, A. S. Al-Fatesh, A. H. Fakeeha, A. A. Ibrahim, A. E. Abasaeed, S. O. Kasim, A. I. Osman, R. Patel and R. Kumar, Mol. Catal., 2021, 504, 111498 CrossRef CAS.
- A. S. Al-Fatesh, N. Patel, A. H. Fakeeha, M. F. Alotibi, S. B. Alreshaidan and R. Kumar, Catal. Rev., 2023, 1–99 CrossRef.
- A. Trovarelli, Catal. Rev., 1996, 38, 439–520 CrossRef CAS.
- P. Djinović and A. Pintar, Appl. Catal., B, 2017, 206, 675–682 CrossRef.
- M. Boaro, C. de Leitenburg, G. Dolcetti and A. Trovarelli, J. Catal., 2000, 193, 338–347 CrossRef CAS.
- K. Lorber and P. Djinović, iScience, 2022, 25(4), 104107 CrossRef CAS PubMed.
- S. Shoji, X. Peng, A. Yamaguchi, R. Watanabe, C. Fukuhara, Y. Cho, T. Yamamoto, S. Matsumura, M.-W. Yu, S. Ishii, T. Fujita, H. Abe and M. Miyauchi, Nat. Catal., 2020, 3, 148–153 CrossRef CAS.
- Q. Zhang, M. Mao, Y. Li, Y. Yang, H. Huang, Z. Jiang, Q. Hu, S. Wu and X. Zhao, Appl. Catal., B, 2018, 239, 555–564 CrossRef CAS.
- M. Li, Z. Sun and Y. H. Hu, Chem. Eng. J., 2022, 428, 131222 CrossRef CAS.
- Z. Wang, Z. Yang, R. Fang, Y. Yan, J. Ran and L. Zhang, Chem. Eng. J., 2022, 429, 132322 CrossRef CAS.
- B. Han, W. Wei, L. Chang, P. Cheng and Y. H. Hu, ACS Catal., 2016, 6, 494–497 CrossRef CAS.
- H. Huang, M. Mao, Q. Zhang, Y. Li, J. Bai, Y. Yang, M. Zeng and X. Zhao, Adv. Energy Mater., 2018, 8, 1702472 CrossRef.
- K. Lorber, J. Zavašnik, I. Arčon, M. Huš, J. Teržan, B. Likozar and P. Djinović, ACS Appl. Mater. Interfaces, 2022, 14, 31862–31878 CrossRef CAS PubMed.
- K. Lorber, J. Zavašnik, J. Sancho-Parramon, M. Bubaš, M. Mazaj and P. Djinović, Appl. Catal., B, 2022, 301, 120745 CrossRef CAS.
- C. Vogt, J. Kranenborg, M. Monai and B. M. Weckhuysen, ACS Catal., 2020, 10, 1428–1438 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- C. Penschke and J. Paier, Phys. Chem. Chem. Phys., 2017, 19, 12546–12558 RSC.
- S. Fabris, G. Vicario, G. Balducci, S. de Gironcoli and S. Baroni, J. Phys. Chem. B, 2005, 109, 22860–22867 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. Neugebauer and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 16067–16080 CrossRef CAS PubMed.
- G. Makov and M. C. Payne, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 4014–4022 CrossRef CAS PubMed.
- P. Xiao, D. Sheppard, J. Rogal and G. Henkelman, J. Chem. Phys., 2014, 140, 174104 CrossRef PubMed.
- J. Kästner and P. Sherwood, J. Chem. Phys., 2008, 128, 014106 CrossRef PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 1999, 111, 7010–7022 CrossRef CAS.
- C. J. Cramer, Essentials of Computational Chemistry: Theories and Models, Wiley, 2nd edn, 2004 Search PubMed.
- R. Asapu, N. Claes, R.-G. Ciocarlan, M. Minjauw, C. Detavernier, P. Cool, S. Bals and S. W. Verbruggen, ACS Appl. Nano Mater., 2019, 2, 4067–4074 CrossRef CAS.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Am. Chem. Soc., 2004, 126, 4943–4950 CrossRef CAS PubMed.
- H. Amekura, Y. Takeda and N. Kishimoto, Nucl. Instrum. Methods Phys. Res., Sect. B, 2004, 222, 96–104 CrossRef CAS.
- M. Zabilskiy, I. Arčon, P. Djinović, E. Tchernychova and A. Pintar, ChemCatChem, 2021, 13, 1814–1823 CrossRef CAS.
- N. N. Tušar, D. Maučec, M. Rangus, I. Arčon, M. Mazaj, M. Cotman, A. Pintar and V. Kaučič, Adv. Funct. Mater., 2012, 22, 820–826 CrossRef.
- I. Arčon, J. Kolar, A. Kodre, D. Hanžel and M. Strlič, X-Ray Spectrom., 2007, 36, 199–205 CrossRef.
- R. Dominko, C. Sirisopanaporn, C. Masquelier, D. Hanzel, I. Arcon and M. Gaberscek, J. Electrochem. Soc., 2010, 157, A1309 CrossRef CAS.
- J. Zavašnik, A. Šestan and V. Shvalya, in Comprehensive Analytical Chemistry, 2021, vol. 93, pp. 241–284 Search PubMed.
- S. Manna, Y. Wang, A. Hernandez, P. Lile, S. Liu and T. Mueller, Sci. Data, 2023, 10, 308 CrossRef CAS PubMed.
- P. G. Lustemberg, P. J. Ramírez, Z. Liu, R. A. Gutiérrez, D. G. Grinter, J. Carrasco, S. D. Senanayake, J. A. Rodriguez and M. V. Ganduglia-Pirovano, ACS Catal., 2016, 6, 8184–8191 CrossRef CAS.
- C. Schilling, A. Hofmann, C. Hess and M. V. Ganduglia-Pirovano, J. Phys. Chem. C, 2017, 121, 20834–20849 CrossRef CAS.
- G. Ferré, M. Aouine, F. Bosselet, L. Burel, F. J. Cadete Santos Aires, C. Geantet, S. Ntais, F. Maurer, M. Casapu, J.-D. Grunwaldt, T. Epicier, S. Loridant and P. Vernoux, Catal. Sci. Technol., 2020, 10, 3904–3917 RSC.
- Z. Wu, M. Li, J. Howe, H. M. Meyer and S. H. Overbury, Langmuir, 2010, 26, 16595–16606 CrossRef CAS PubMed.
- C. Li, K. Domen, K.-I. Maruya and T. Onishi, J. Catal., 1990, 125, 445–455 CrossRef CAS.
- B. Smith, Infrared Spectral Interpretation A Systematic Approach, CRC Press, 1st edn, 1999 Search PubMed.
- W. Jiang, J. Low, K. Mao, D. Duan, S. Chen, W. Liu, C.-W. Pao, J. Ma, S. Sang, C. Shu, X. Zhan, Z. Qi, H. Zhang, Z. Liu, X. Wu, R. Long, L. Song and Y. Xiong, J. Am. Chem. Soc., 2021, 143, 269–278 CrossRef CAS PubMed.
- T. H. Tan, B. Xie, Y. H. Ng, S. F. B. Abdullah, H. Y. M. Tang, N. Bedford, R. A. Taylor, K.-F. Aguey-Zinsou, R. Amal and J. Scott, Nat. Catal., 2020, 3, 1034–1043 CrossRef CAS.
- N. C. Nelson, M. T. Nguyen, V. A. Glezakou, R. Rousseau and J. Szanyi, Nat. Catal., 2019, 2, 916–924 CrossRef CAS.
- H. Zheng, W. Liao, J. Ding, F. Xu, A. Jia, W. Huang and Z. Zhang, ACS Catal., 2022, 12, 15451–15462 CrossRef CAS.
- Y. Guo, J. Feng and W. Li, Chin. J. Chem. Eng., 2017, 25, 1442–1448 CrossRef CAS.
- L. Xu, H. Wen, X. Jin, Q. Bing and J. Liu, Appl. Surf. Sci., 2018, 443, 515–524 CrossRef CAS.
- Y.-A. Zhu, D. Chen, X.-G. Zhou and W.-K. Yuan, Catal. Today, 2009, 148, 260–267 CrossRef CAS.
- S.-G. Wang, D.-B. Cao, Y.-W. Li, J. Wang and H. Jiao, J. Phys. Chem. B, 2006, 110, 9976–9983 CrossRef CAS PubMed.
- M. Bugnet, S. H. Overbury, Z. L. Wu and T. Epicier, Nano Lett., 2017, 17, 7652–7658 CrossRef CAS PubMed.
- P. A. Crozier, R. Wang and R. Sharma, Ultramicroscopy, 2008, 108, 1432–1440 CrossRef CAS PubMed.
- C. Yang, X. Yu, S. Heißler, A. Nefedov, S. Colussi, J. Llorca, A. Trovarelli, Y. Wang and C. Wöll, Angew. Chem., Int. Ed., 2017, 56, 375–379 CrossRef CAS PubMed.
- P. G. Lustemberg, Z. Mao, A. Salcedo, B. Irigoyen, M. V. Ganduglia-Pirovano and C. T. Campbell, ACS Catal., 2021, 11, 10604–10613 CrossRef CAS PubMed.
- Z. Liu, D. C. Grinter, P. G. Lustemberg, T.-D. Nguyen-Phan, Y. Zhou, S. Luo, I. Waluyo, E. J. Crumlin, D. J. Stacchiola, J. Zhou, J. Carrasco, H. F. Busnengo, M. V. Ganduglia-Pirovano, S. D. Senanayake and J. A. Rodriguez, Angew. Chem., Int. Ed., 2016, 55, 7455–7459 CrossRef CAS PubMed.
- F. Abild-Pedersen, O. Lytken, J. Engbæk, G. Nielsen, I. Chorkendorff and J. K. Nørskov, Surf. Sci., 2005, 590, 127–137 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta01823k |
| This journal is © The Royal Society of Chemistry 2024 |