
A dynamically stable self-assembled CoFe (oxy)hydroxide-based nanocatalyst with boosted electrocatalytic performance for the oxygen-evolution reaction†
Ming
Zhu‡
ab,
Hengyue
Xu‡
c,
Jie
Dai
 d,
Daqin
Guan
*c,
Zhiwei
Hu
e,
Sixuan
She
f,
Chien-Te
Chen
g,
Ran
Ran
*a,
Wei
Zhou
a and
Zongping
Shao
*c
d,
Daqin
Guan
*c,
Zhiwei
Hu
e,
Sixuan
She
f,
Chien-Te
Chen
g,
Ran
Ran
*a,
Wei
Zhou
a and
Zongping
Shao
*c
aState Key Laboratory of Materials-Oriented Chemical Engineering, College of Chemical Engineering, Nanjing Tech University, Nanjing, 211800, China. E-mail: ranr@njtech.edu.cn
bInstitute for Smart City of Chongqing University in Liyang, Chongqing University, Jiangsu 213300, China
cWA School of Mines: Minerals, Energy and Chemical Engineering, Curtin University, Perth, Western Australia 6845, Australia. E-mail: daqin.guan@curtin.edu.au; zongping.shao@curtin.edu.au
dSchool of Environmental Science and Engineering, Shanghai Jiao Tong University, Shanghai, 200240, China
eMax-Planck-Institute for Chemical Physics of Solids, Nöthnitzer Str. 40, Dresden 01187, Germany
fDepartment of Applied Physics and Materials Research Center, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong
gNational Synchrotron Radiation Research Center, 101 Hsin-Ann Road, Hsinchu 30076, Taiwan
First published on 9th August 2024
Abstract
Surface reconstruction or elemental leaching is generally involved in the oxygen evolution reaction (OER) process on transition metal-based oxides during alkaline water electrolysis, which gives rise to both opportunities and challenges for the development of OER electrocatalysts. Reaction-derived metal (oxy)hydroxides have been proven to be the actual active species for many metal oxides but have suffered from the dissolution of active elements. Here, the construction of a dynamically stable CoFe (oxy)hydroxide OER nanocatalyst is proposed, which delivers a low overpotential of 253 mV at 10 mA cm−2 and high mass activity (3.78 A mg−1 at an overpotential of 300 mV), among the highest of all the hydroxides reported previously. The in situ synthesized nanocatalyst acts as a stable CoOxHy host for Fe adsorption and facilitates active Co–O–Fe motif formation. The weak metal–O coordination environment and hydrophilic surface morphology lead to superficial mass transfer. The drawbacks of Fe depletion for conventional CoFe (oxy)hydroxides are overcome, attributed to tardy lattice oxygen ion diffusion and dynamic Fe-saturated dual metal active sites, which lead to a stable performance for over 100 h. This study provides a design strategy for OER electrocatalysts with both high mass activity and durability, which shows application prospects for large-scale electrochemical water splitting.
Introduction
Electrochemical energy generation and storage will play an important role in future sustainable green energy systems, while the oxygen-evolution reaction (OER) was found to govern the overall efficiency of many energy conversion processes.1–4 During the past few decades, considerable research activities have been directed towards the development of efficient electrocatalysts to overcome the sluggish kinetics of the OER.5–7 To realize the commercial applications of the related electrochemical energy technologies, such as metal–air batteries and water electrolyzers, OER electrocatalysts should be cost effective, highly catalytically active, and durable. Therefore, precious metal-free transition metal compounds, such as oxides and hydroxides, have been extensively exploited as alternative OER catalysts for operation in alkaline solution, attributing to their abundance, rich properties, and compositional flexibility.8–10 With the in-depth investigation, it was found that many transition metal oxides, nitrides and sulfides actually experienced surface reconstruction during the OER process, leading to the formation of surface metal (oxy)hydroxide layers, which were believed to be the actual active sites.11–13 Therefore, reaction-derived transition metal (oxy)hydroxides are of particular interest in OER electrocatalysis.Considering that reaction-derived transition metal (oxy)hydroxides often exhibit high OER activity, the origin of their boosted intrinsic electrocatalytic activity and strategies to promote the local electrochemical restructuration process have been widely studied. With the development of operando characterization techniques, extensive research studies have revealed that the self-reconstruction process was often accompanied by the modification of local electronic and geometric structures of the electrocatalysts, such as, phase transformation,14,15 increased structural disorder,16 or more exposed surface area.17 Even though these pioneering studies have elaborated the dynamic changes of surface catalytically active sites, the reconstruction process is spontaneous and challenging to control. In order to promote the electrochemical transition process, researchers have tried to tailor the surface properties of pre-catalysts, for example, by establishing atomic scale heterointerfaces,18 introducing cation and anion defects, 19,20 or loading Pt single-atoms,15 which have successfully optimized the intrinsic activity of OER catalysts. However, these mainstream methods to boost the electrochemical restructuration reaction require complex steps and additives. Moreover, a key factor that has been largely overlooked in previous research studies is that the reaction-derived transition metal (oxy)hydroxide layer is very thin, which limits the mass activity of the entire catalyst. Therefore, a facile strategy to construct OER electrodes with a high proportion of reaction-derived transition metal hydroxides holds the potential to enhance their mass activity.
Another intrinsic weakness of reaction-derived transition metal hydroxides is that electrochemical restructuration processes can change integrity of surface crystal configurations,21–23 which can lead to subsequent activity decay. Among all the transition metal (oxy)hydroxides OER catalysts, (oxy)hydroxides containing double metals, especially Fe containing (oxy)hydroxides (Fe–Ni and Fe–Co), have exhibited extraordinary activity derived from the synergistic effect between two metallic sites.24–28 However, Fe containing (oxy)hydroxides experience severe Fe active site depletion29 and structure dissolution30 during long term usage, which limit their practical application. Previous studies have revealed that Fen+ in electrolyte contributed to preserving the high activity levels of Fe-containing (oxy)hydroxides through Fe dynamic exchange taking place at the interface.31 Other research studies involving oxide-based catalysts have also achieved similar results. For example, Guan et al. introduced pre-leaching soluble compounds into perovskites to lower the difference in interfacial ion concentrations and thus endow the host phase Ba0.35Sr0.65Co0.8Fe0.2O3−δ with a stable surface structure.32 Hence, considering that the ionic exchange between the electrode and electrolyte plays a key role in the oxygen evolution process, adjusting the characteristics of electrolyte is a promising strategy to realize sustained OER performance.
Herein, we report a self-assembled CoFe (oxy)hydroxide electrode (CoFe-in situ) via an in situ electrochemical method under anodic bias and analyzed its superiority in comparison with CoFe (oxy)hydroxide prepared by a conventional cathodic electrodeposition method. The CoFe-in situ nanocatalyst exhibited a remarkable OER performance (253 mV overpotential to reach 10 mA cm−2) with ultrahigh mass activity (3.78 A mg−1 at η = 300 mV), 5 times higher than that of the conventionally prepared CoFe (oxy)hydroxide. X-ray absorption spectroscopy (XAS) and X-ray photoelectron spectroscopy (XPS) spectra revealed that the weak coordination bond of Co–O in CoFe-in situ facilitated the Co–O–Fe active site formation, and its low Fe–O coordination number contributed to a hydrophilic surface. The tardy lattice oxygen ion diffusion together with suppressed metal dissolution during the OER activation process contributed to the structure integrity of the CoFe-in situ nanocatalyst and overcame the instability of metal (oxy)hydroxides. The dynamically stable surface of the CoFe-in situ nanocatalyst guaranteed the Fe saturation coverage and a high number of Co–O–Fe active sites, leading to excellent durability for running over 100 h. Our study provides new insights into the regulating strategy of (oxy)hydroxides and deepens the understanding of designing efficient electrocatalysts for large-scale industrial manufacturing.
Experimental methods
Synthesis
Basic characterization
The crystal X-ray diffraction (XRD) patterns of the CoFe LDH catalysts were collected using an X-ray powder diffractometer (λ = 1.5418 Å, Bruker D8 Advance). The morphology of the catalysts was investigated using a scanning electron microscope (SEM, Hitachi S-4800). The high-resolution transmission electron microscopy (HRTEM) images were obtained using an FEI Tecnai G2T20 electron microscope operating at 200 kV. The amounts of Co and Fe contained in samples were recorded with inductively coupled plasma mass spectrometry (ICP-MS, Optima 7000 DV). The X-ray absorption spectroscopy (XAS) data of Co-L3 and Fe-L3 were obtained using the total electron yield (TEY) mode at the BL 11A of the National Synchrotron Radiation Research Center (NSRRC) in Taiwan. The contact angles were measured using a KRUSS DSA100 system. The Co 2p and O 1s spectra were recorded using an X-ray photoelectron spectrometer (XPS, PHI5000 Versa Probe). Raman spectroscopy was conducted using a Horiba LabRAM HR evolution with a 473 nm blue laser.Electrochemical characterization
The electrochemical measurements were controlled using a CHI 760E bipotentiostat with a three-electrode configuration. The as-prepared samples deposited on carbon papers were used as working electrodes. The counter electrode was a graphite rod and reference electrode was Hg/HgO. All OER measurements were performed using O2-saturated 1 M KOH solution as electrolyte at room temperature. The calibration of the reference electrode was performed in H2-saturated electrolyte before the tests. Electrochemically active surface area (ECSA) was analyzed by obtaining the electrochemical double layer capacitance (Cdl). The cycle voltammetry (CV) method was used to measure the Cdl, with a potential range from 1.10 to 1.20 V vs. RHE in a non-faradic current region. The scan rates of CV were 20, 40, 60, 80 and 100 mV s−1. The halves of the positive and negative current density differences at the center of the scanning potential range were plotted versus scan rates, and the slopes obtained were Cdl.Oxygen intercalation and diffusion rate measurements
The oxygen intercalation measurements were performed in an Ar saturated 1 M KOH electrolyte with the same three-electrode configuration as that in the electrocatalytic activity test. CV was run at 20 mV s−1 to determine the E1/2, which is the potential half way between the redox peak pair. Then a chronoamperometry test was conducted at a potential 50 mV more anodic than the E1/2 with a vigorous rotation to get rid of mass-transfer effects. The current was plotted versus the inverse square root of time (i vs. t−1/2), and the linear section of the curve was fit to obtain the intercept on the t−1/2 axis. The oxygen ion diffusion coefficient was calculated according to the equation λ = α(DOt)−1/2, where λ is a dimensionless shape factor of 2.01, α is the radius of the particle (in this case 50 nm was used for CoFe-in situ and 150 nm for CoFe LDH), t−1/2 is the intersection with the t−1/2 axis, and DO is the oxygen ion diffusion coefficient.DFT calculation parameters
Spin-polarized DFT calculations for periodic material systems were performed with the Vienna ab initio simulation package34 with the projector-augmented wave method.35 The exchange–correlation function was handled using the generalized gradient approximation formulated using the Perdew–Burke–Ernzerhof functional. The effective U values for Fe and Co were determined to be 4.0 eV and 3.3 eV, respectively, using the Hubbard U model (DFT+U) in the metal oxide system.36 The van der Waals interactions are described with the DFT-D3 method in Grimme's scheme.37 The interaction between the atomic core and electrons was described by the projector augmented wave method. The plane-wave basis set energy cutoff was set to 500 eV.38 The Brillouin zone was sampled with 3 × 3 × 1 gamma (Γ) centered Monkhorst–Pack mesh sampling for geometry relaxation. All the slabbed models possessed a sampled vacuum spacing of ≈15 Å, ensuring negligible lateral interaction of adsorbates.39 The bottom layers about half of the structure were kept frozen in the lattice position. All structures with a dynamic magnetic moment were fully relaxed to optimize without any restriction until their total energies were converged to <1 × 10−6 eV,40 and the average residual forces were <0.02 eV Å−1.41Results and discussion
For the in situ synthesis of the CoFe-in situ sample in the potential range of the OER, a three-electrode system was applied, in which carbon paper, a graphite rod and Hg/HgO were used as the working electrode, counter electrode and reference electrode, respectively. Co and Fe chlorides were added into the 1 M KOH electrolyte simultaneously while the linear sweep voltammetry (LSV) test was performed. The metal ions and hydroxyl ions in the solution adsorbed on the surface of the working electrode driven by the anode potential. The amount of depositions gradually increased along with the continuous application of anodic bias, which contributed to a prominent decrease in overpotential (η) at the early stage of the formation of the active substance. However, the LSV curves of samples showed no more obvious change after about 20 loops and stabilized at their optimal performances (Fig. S1†). It is worth noting that the effect of chlorine ions in electrolyte on OER can be eliminated, because the operating potential is lower than the theoretical overpotential of the chlorine redox reaction (η = 480 mV).42 The samples with an optimized loading amount on carbon papers, in only Co-containing electrolyte, only Fe-containing electrolyte, and both Co and Fe containing electrolyte, are named Co-in situ, Fe-in situ and CoFe-in situ, respectively. The CoFe layered double hydroxide (LDH) was first prepared via a traditional electrosynthesis method at cathodic potential and then served as an OER pre-catalyst to induce surface CoFe (oxy)hydroxide formation driven by the anodic potential. The deposition amount of the CoFe LDH pre-catalyst was tuned to make sure that it could achieve the same OER performance as that of CoFe-in situ.The electrocatalytic activities of these samples for the OER are shown by the iR-corrected polarization curves in Fig. 1a. The in situ synthesized bimetallic catalyst (namely CoFe-in situ) exhibited much smaller overpotential (253 mV) than the single metal catalysts (359 mV for Co-in situ and 395 mV for Fe-in situ) to reach a current density of 10 mA cm−2. It is not surprising that CoFe-in situ presents better activity than Co-in situ and Fe-in situ because of the increased active sites and synergistic effects between Co and Fe as reported before.43–45
 | ||
| Fig. 1 (a) The OER polarization curves of the catalysts in 1 M KOH, (b) mass activities of the catalysts at an overpotential (η) of 300 mV, (c) TEM figure of CoFe-in situ and (d) Raman spectra of CoFe-in situ and CoFe LDH after the OER (CoFe LDH AR) and CoFe-in situ and CoFe LDH post stability test. | ||
To reveal the mass activity of catalysts, which is vital for the practical application, the electrocatalytic activity was normalized to the mass loading measured by inductively coupled plasma mass spectrometry (ICP-MS). The contents of Co and Fe are 0.0106 mg cm−2 and 0.004 mg cm−2 for CoFe-in situ and 0.0526 mg cm−2 and 0.0292 mg cm−2 for CoFe LDH, respectively. As shown in Fig. 1b, CoFe-in situ achieved a remarkable high mass activity of 3.78 A mg−1 at η = 300 mV, which is 5 times higher than that of CoFe LDH (0.65 A mg−1). The mass activities of CoFe-in situ and CoFe LDH normalized to the same mass loading show similar results (Fig. S2†). Electrochemically active surface areas (ECSAs) of CoFe-in situ and CoFe LDH are 4.17 mF cm−2 and 3.24 mF cm−2, respectively (Fig. S3†). Specific activity normalized to ECSA of CoFe-in situ (0.91 A cm−2 ECSA) is still ∼5 times higher than that of CoFe LDH catalysts (0.2 A cm−2 ECSA), which further proves the superiority of CoFe-in situ in intrinsic activity. Notably, to the best of our knowledge, this value is higher than that of most transition metal-based catalysts reported previously (Table S1†).
To define the active structure of catalysts, the samples were analyzed using the X-ray diffraction (XRD) spectrum and high-resolution transmission electron microscopy (HRTEM). The XRD pattern of CoFe LDH (Fig. S4†) exhibited characteristic diffraction peaks of the CoFe LDH nanosheets (PDF#50-0235) at 11.6°, 23.4°, 34.1°, and 59.1°.46 The HRTEM image (Fig. S5†) indicates interplanar distances of 0.269 nm, which is also consistent with the (101) plane characteristic of CoFe LDH nanosheets (PDF#50-0235), agreeing well with the XRD result. These characterization results confirm that CoFe LDHs were successfully synthesized. As for CoFe-in situ, the HRTEM image (Fig. 1c) displays the crystalline phase of the material, in which crystalline and amorphous domains coexisted. Lattice fringe with a d-spacing of 0.236 nm corresponds to the (011) plane of CoOOH (PDF 96-101-0268), which signifies that the in situ synthesized substance can be a metal oxyhydroxide. Considering the trace amount of CoFe-in situ on carbon paper and its poor crystallinity, XRD peak intensity is too weak to be detected even in the synchrotron X-ray powder diffraction (SXRPD) spectrum (Fig. S6†). To further explore the crystallographic phase of catalysts, we obtained the Raman spectra of CoFe-in situ and CoFe LDH catalysts pre and post stability test (10 h at 10 mA cm−2) (Fig. 1d). For the as-prepared CoFe-in situ, the Raman spectra consist of main peaks assigned to Co(OH)2, CoO2 and Co3O4.47,48 After long-term running, the spectral features of Co(OH)2 at 290 cm−1, 386 cm−1 and 732 cm−1 are well maintained, and two broad signals centered at around 510 cm−1 and 608 cm−1 appear, corresponding to Eg and A1g vibrational modes of CoOOH.49–51 The presence of the signal of CoOOH and the decreased intensity of Co–O vibration peaks are attributed to the partial conversion of Co–O species into oxyhydroxide electroactive phases.
The dynamic formation process of the active phase in CoFe-in situ was further studied. To figure out the doping form of Fe ions in CoFe-in situ, some contrast experiments were conducted via adjusting the metal ion constituents in electrolyte. Considering that CoFe-in situ was one-step synthesized in the alkaline solution containing both Co and Fe ions, we test the performance of catalysts prepared in two steps in single-metal-containing electrolyte respectively. Co-in situ was first prepared in Co-containing electrolyte, and then the electrode was taken out and immediately put into another three-electrode system with pure KOH solution as electrolyte. Fe ions were subsequently added into the electrolyte during the LSV tests at anodic potential. The obtained catalyst is named Co-in situ-Fe, and the sample prepared with the ions added in reverse order is named Fe-in situ-Co. As shown in Fig. S7,† Co-in situ-Fe exhibits high activity which is similar to CoFe-in situ (η = 253 mV), but Fe-in situ-Co shows a relatively larger overpotential (η = 414 mV) which is similar to that of Fe-in situ. These results suggest that the homogeneous Co ion fails to form an active Co–O–Fe interaction at the surface of the Fe oxyhydroxide host and the Fe ion in electrolyte is indispensable to the formation of the CoFe-in situ sample. Therefore, we speculate that the dynamically formed Co–O–Fe motif is the key active site for CoFe-in situ. To further investigate the Fe-incorporation processes in our sample, experiments of CoFe-in situ synthesis at different pH (1 M and 6 M KOH) and temperatures (25 °C and 60 °C) were conducted. As shown in Fig. S8b,† the increase in temperature brought about obviously increased mass loading of the metal compound on the electrodes, which illustrates that the formation process of CoFe-in situ can be accelerated via the thermodynamic process for electroadsorption of metal ions from electrolyte to the working electrode. The increase in pH and temperature for preparation conditions also speed up the Fe-incorporation process, where the molar ratios of Fe ions in the CoFe (oxy)hydroxides are 0.28, 0.42 and 0.41 for electrodes prepared at 1 M KOH 25 °C, 6 M KOH 25 °C, and 1 M KOH 60 °C, respectively. As the value of pH or temperature of synthesis conditions increases, the reaction activity of CoFe-in situ improves obviously (Fig. S8a†). It has been reported that Fe ions in the electrolyte exhibit a higher incorporation rate at OER potentials than those in the OER dormant state and favour locating on electrochemically more reactive edge sites.44,52,53 So the increased Fe ratios in CoFe-in situ (6 M KOH 25 °C and 1 M KOH 60 °C) samples were caused by the increased OER rate in high concentration electrolyte and at high temperature, which makes it easier for Fe ions to incorporate into the as-deposited CoFe (oxy)hydroxides on the working electrode. According to the above analysis, the dynamic formation process of CoFe-in situ can be described as follows: the metal ions and hydroxyl ions in the alkaline electrolyte tend to adsorb on the surface of the working electrode driven by the anode potential. Fe ions in electrolyte incorporate onto the surface of the Co oxyhydroxide host to form the highly OER active Co–O–Fe motifs.54 Some of the Fe ions in CoFe (oxy)hydroxide may leach during the OER and Fe ions in electrolyte redeposit on those vacancies to reform the Co–O–Fe active structure. The redeposited Fe undergoes exchange with the Fe species in the hosts to establish dynamically stable active sites.31 It is worth noting that, during the electrochemical depositing process, the increasing loading amount of CoFe-in situ has little effect on the catalytic activity after reaching its optimal conditions. As a result, it is reasonable to speculate that most of the outer surface layer of the sample actually acts as the active phase and provides remarkable electrocatalytic activity.
Our previous experiments have confirmed that the dynamic active site for CoFe-in situ is the O-bridged Fe–Co reaction center, which is similar to the previously reported active site for CoFe LDH.14 To further explore the origin of the significant improvement in mass activity of CoFe-in situ, the surface properties of these two CoFe-based catalysts after the OER were detected using Co-L3, O-K and Fe-L3 X-ray absorption spectroscopy (XAS) spectra using the total electron yield (TEY) mode, which is highly sensitive to the information of electronic structures and local coordination environments on near-surface catalysts.55 As shown in Fig. 2a, the peak of the Co-L3 edge for CoFe LDH AR appears at ∼779.5 eV, which is between the energy positions of the standard reference for Co3+ (∼779.1 eV) and Co4+ (∼779.7 eV). In addition, no characteristic peak appears at the energy position for the Co2+ standard reference (∼777.5 eV), implying that CoFe LDH presents a mixture Co valence state of Co3+ and Co4+. For CoFe-in situ, the main peak of the Co-L3 edge appears at energy positions between those of Co3+ and Co4+ with a new peak at ∼777.5 eV, which represents that the Co valence state of CoFe-in situ is a mixture of Co2+, Co3+ and Co4+. The main peak of the Co-L3 edge for CoFe-in situ appears at lower energy positions than that for CoFe LDH AR, suggesting that CoFe-in situ presents a lower Co valence state than CoFe LDH AR. This result is further proved via the O-K XAS results. Fig. 2b (bottom) shows that the peaks of standard reference samples shift to lower energies and gain spectral weight with increasing Co valence, reflecting the increased covalent mixing between the Co 3d orbitals and O 2p states. Hence, compared with the O-K XAS spectra of CoFe LDH AR, the shift of the peak to higher energy indicates a lower Co valence and a weaker Co–O bond for CoFe-in situ (Fig. 2b top). It has been reported that for the process of dynamic Fe incorporation into 3d transition-metal (oxy)hydroxides, the Fe adsorption energy on the MOxHy host materials shows an inverse correlation with the M–O bond energy.31 Therefore, the weak coordination environment of Co–O for the CoFe-in situ surface provides stable sites for dynamic Fe doping. Since the number of Co–O–Fe active sites on the surface of the electrode is the key to OER activity, the high mass activity of CoFe-in situ can be explained.
 | ||
| Fig. 2 (a) The Co-L3, (b) O-K, and (c) Fe-L3 XAS spectra and (d) XPS spectra of O 1s for CoFe-in situ and CoFe LDH after the OER (CoFe LDH AR). | ||
The electronic structures of Fe ions in these two (oxy)hydroxides were tested, where the charge states of Fe are both 3+ as shown in Fe-L3 XAS spectra (Fig. 2c). The Fe–O coordination structure can be identified from the resolved degree of the t2g and eg related peaks. As the Fe-L3 edge of standard reference samples shows, the tetrahedral coordination FeO4 exhibits much lower peak intensity at ∼707 eV than that of octahedral coordination FeO6, indicating that the weaker the Fe-L3 peak at ∼707 eV, the lower the coordination number of Fe–O.56,57 The spectral feature t2g peak for CoFe-in situ was poorly resolved as compared with that of CoFe LDH AR, indicating a decrease in the coordination number of Fe ions. It can be justified that oxygen vacancies tend to form during the in situ electrosynthesis process. The oxygen defects and relatively low coordination environment of metal ions in CoFe-in situ are conducive to facilitating electron exchange and interaction with oxygen intermediates on the catalyst surface.58–61 These results were further testified by XPS spectra. The O 1s spectra consisted of three kinds of oxygen species (Fig. 2d). From the lowest energy to the highest one respectively correspond to the metallic oxides (denoted as M–O), the absorbed hydroxyl groups (denoted as OHad), and the absorbed H2O (denoted as H2Oad).62 The content of M–O in CoFe-in situ is far lower than that of CoFe LDH AR, indicating a weaker bond between metal and oxygen. These oxygen vacancies as active sites could reduce the initial adsorption energy of water.63 A higher ratio of surface OH− species was observed on CoFe-in situ (76.91%) than that on CoFe LDH AR (66.75%), which was attributed to the increased strength of OH− adsorption. Considering that most of the reported OER processes on hydroxides involve adsorption and desorption processes of oxygenated intermediates on unsaturated coordinated metal active sites,64 it can be deduced that the relatively weak coordination environments of metal ions in CoFe-in situ are one of the reasons for its improved intrinsic activity. Apart from the reactant OH− adsorption, the hydrophilicity of the catalyst is another important factor that affects mass transfer of the OER. Fig. S9† shows the results of contact-angle measurement for our samples. The contact angles of water droplets towards blank carbon paper dropped obviously after electrochemical deposition. For the CoFe LDH before and after the OER, the contact angle increased from 56.32° to 78.49°, implying that the surface reconstruction process of CoFe LDH during the OER led to a more hydrophobic surface. In contrast, CoFe-in situ (18.56°) demonstrated better surface wettability than CoFe LDH, allowing greater accessibility of liquid electrolyte. These results may be attributed to the surface morphology of catalysts, where CoFe-in situ exhibited nanosheet arrays with smaller lateral length (Fig. 3b, ∼100 nm) than that of CoFe LDH (Fig. 3a, ∼300 nm). Since the exfoliated ultrathin LDHs have been previously reported to show significantly increased hydrophilicity than those with thicker nanosheets,63 it is reasonable to speculate that the decrease in the size of nanoarrays for (oxy)hydroxides contributed to the enhanced solid–liquid contact and faster mass transfer. In conclusion, the weak coordination environment of M–O and the hydrophilic morphology of CoFe-in situ facilitate the adsorption of OH− at the catalyst/electrolyte interface.
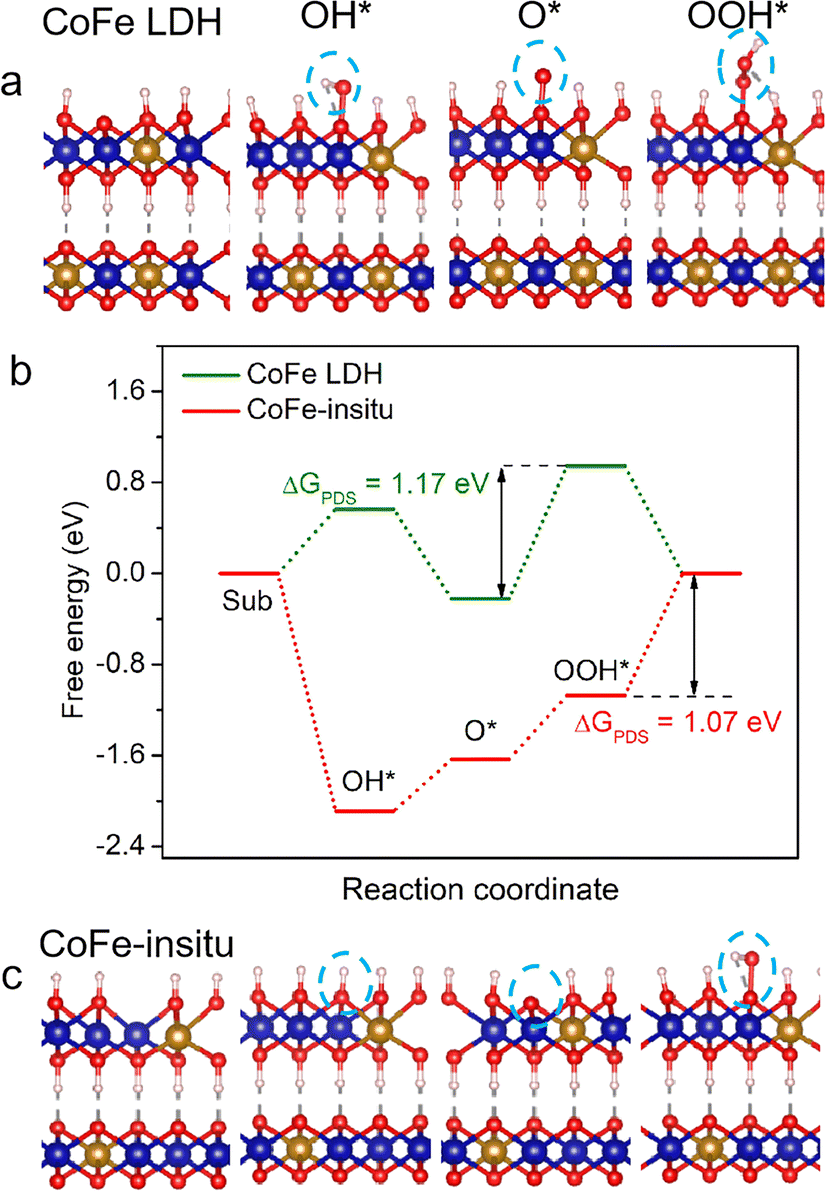 | ||
| Fig. 3 Structural configurations of (a) CoFe LDH and (c) CoFe-in situ. (Note: the blue ball represents the Co ion, yellow ball represents the Fe ion, red ball represents the O ion, white ball represents the H ion, and OER intermediates are highlighted by blue circles). (b) Reaction free-energy diagrams for CoFe LDH and CoFe-in situ. | ||
The effects of structural properties of the catalysts on their OER activity were further elaborated using density functional theory (DFT) calculations. The calculation models of catalysts were set up according to the stoichiometric ratio of Co/Fe and coordination number of metal–oxygen, where CoFe LDH adopted the structure of perfect Co0.6Fe0.4OOH (Fig. 3a) and CoFe-in situ adopted the structure of Co0.75Fe0.25O0.875OH with oxygen vacancies (Fig. 3c). The Gibbs free energy of a four-step associative mechanism was calculated to evaluate the OER performance. As demonstrated in Fig. 3b, the Gibbs free energy profile for CoFe LDH shows that the OH adsorption step suffers from a high energy barrier, while this reaction step becomes spontaneous with negative Gibbs free energy at the surface of CoFe-in situ, validating that the oxygen unsaturated metal site acts as the active site to facilitate the formation of OH* intermediates. As we mentioned above, it is more feasible for CoFe-in situ to adsorb surface OH−/H2O, which makes it easier to generate OOH* intermediates and lower the free energy barrier of the rate-determining step (RDS) for perfect CoFe LDH. Moreover, the Gibbs free energy of the RDS for CoFe-in situ is 1.07 eV, lower than that for CoFe LDH (1.17 eV), leading to a boosted OER catalytic performance.
Stability is another important criterion to estimate the value of a catalyst for industrial scale water splitting, while structural inconstancy and deactivation for long-term usage have always been the bottlenecks hindering the application of hydroxide-type catalysts. The plot from the chronopotentiometry test shows that the CoFe-in situ shows excellent stability with potential remaining at about 1.47 V at 10 mA cm−2 for 100 h, while the OER activity of CoFe LDH decays obviously with increased overpotential only after 10 h of operation (Fig. 4c). The stability of CoFe-in situ at 1 A cm−2 was also measured to evaluate its potential to be used in anion exchange membrane water electrolysers (Fig. 4i). Given that the carbon paper is brittle at high currents, we used Ti foam as a substrate. The contribution of Ti foam to OER activity has been excluded, due to its poor electrochemical performance shown in the LSV results (Fig. S10†). The chronopotentiometry test verified that CoFe-in situ can be operated stably at 1 A cm−2 for 80 h without an obvious increase in the operating voltage. The variations in microstructures for both the catalysts after the stability test were examined by SEM. Obvious dilation of nanoplatelets is observed in the used CoFe LDH sample (Fig. 4a and d), which might lead to poor electrical connectivity and retarded mass transformation from the outer layer of the catalytic material into the conductive substrate, resulting in increased electrical resistance and receded electrocatalytic performances. In contrast, the nanosheet morphology of CoFe-in situ is well preserved after 10 h of testing with nearly unchanged diameter size, but a more densely packed structure composed of larger nodules (Fig. 4b and e). The denser coating film with thin nanoplates retains a porous structure for electrolyte accessibility and provided better electrical conductivity.65 The variations in electrical transfer abilities for both catalysts after 10 h of operation have been confirmed by electrochemical impedance spectroscopy (EIS) (Fig. 4f and g). The impedance arc of CoFe LDH after the reaction turns larger, suggesting its increased charge-transfer resistance. In comparison, the used CoFe-in situ shows a smaller semicircle in the EIS spectrum than the fresh one, which indicates that the continuous dynamic reconstruction process of CoFe-in situ improves its conductivity for long-term running and overcomes the drawbacks of poor durability of traditional CoFe (oxy)hydroxides.
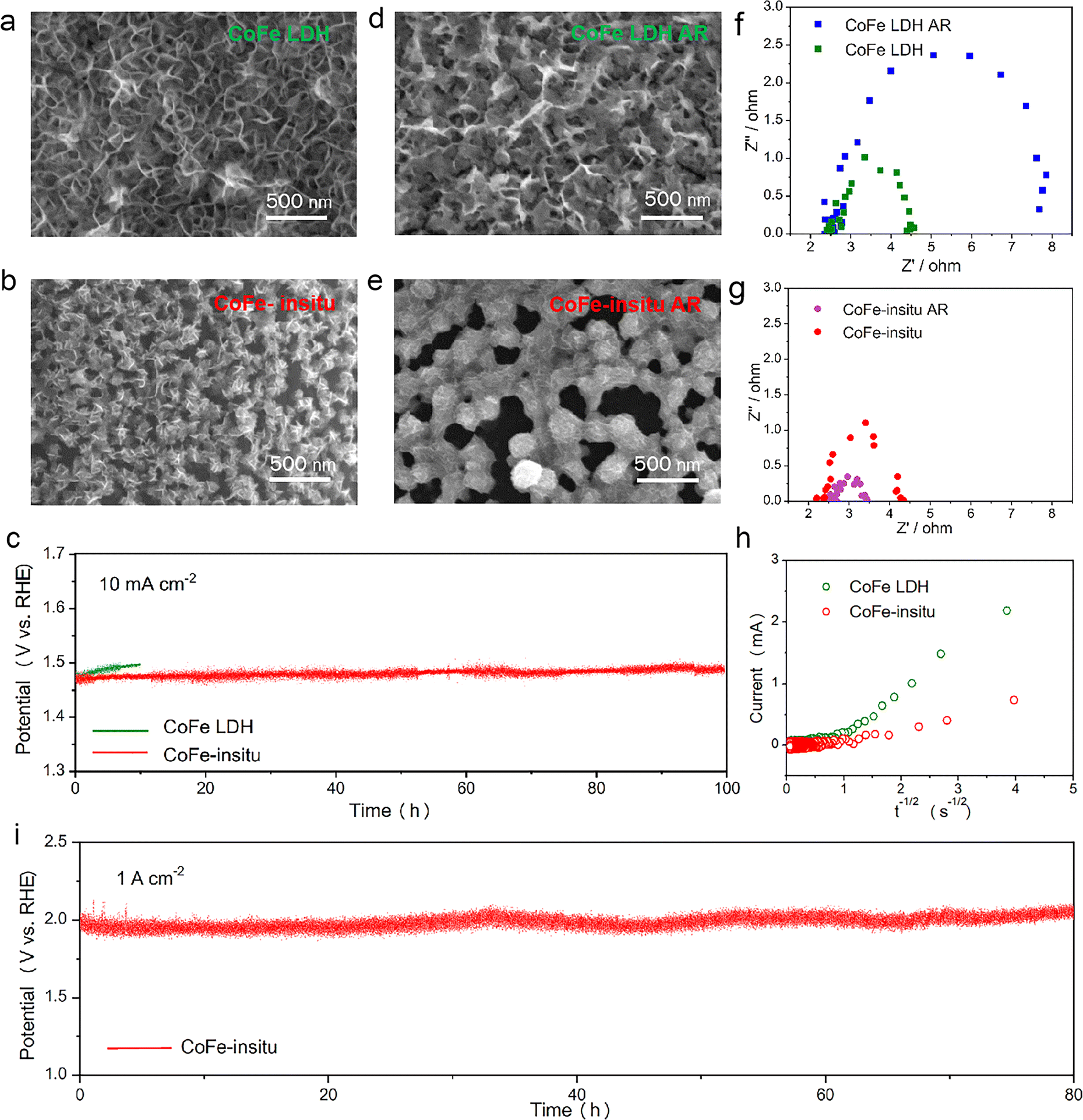 | ||
| Fig. 4 SEM figures of (a) CoFe LDH, (b) CoFe-in situ, (d) CoFe LDH after the 10-h OER (CoFe LDH AR) and (e) CoFe-in situ after the 10-h OER (CoFe-in situ AR). (c) Stability tests for CoFe LDH and CoFe-in situ at 10 mA cm−2. EIS plots of (f) CoFe LDH, (g) CoFe-in situ before and after the 10-h OER. (h) Chronoamperometry data (i vs. t−1/2) for CoFe LDH and CoFe-in situ. (i) Chronopotentiometry test for CoFe-in situ on a Ti foam substrate at 1 A cm−2. | ||
In addition to the beneficial effects of morphology on the durability of CoFe-in situ, the participation of lattice oxygen affects the stability of the catalyst as well. As it has been reported previously, the interlayer basal plane in bulk LDHs is involved in the oxygen evolution process, while the shrinkage of interlayer spacing during the OER would slow down the diffusion of proton acceptors (e.g., OH−) into LDH interlayers, leading to the dissolution of LDHs.29 Hence, the degrees of lattice oxygen involvement in CoFe LDH and CoFe-in situ nanocatalysts for the OER were estimated using the oxygen ion diffusion coefficients (DO). As depicted in Fig. 4h, currents measured by chronoamperometry are plotted as a function of the inverse square root of time. DO is calculated according to the bounded three dimensional diffusion model,66,67 where the CoFe LDH (DO = 20.42 × 10−11 cm2 s−1) displays faster oxygen ion diffusion than CoFe-in situ (DO = 0.22 × 10−11 cm2 s−1). These results represent that oxygen ions in CoFe LDH had a greater propensity to migrate from the bulk than those in CoFe-in situ,68 which might cause structural instability and the absence of proton acceptors.
As mentioned in the introduction part, iron content in Fe-based (oxy)hydroxides is a key factor that affects the activity of the OER in alkaline media for long-term running. Thus, inductively coupled plasma mass spectrometry (ICP-MS) was employed to reveal the mechanism of the excellent stability of the CoFe-in situ nanocatalyst. The mass of Co and Fe in catalysts, as well as the calculated molar ratio of Fe/Co + Fe before and after the 10-h OER are shown in Fig. 5a and b. The ICP results indicate that both Co and Fe contents in CoFe LDH underwent an obvious decrease after the 10-h OER (16.7% decrease for Co, and 24.6% decrease for Fe) with a decline in the Fe/Co + Fe molar ratio (0.36 to 0.33) (Fig. 5b). The dissolved Co and Fe ions resulted from the unstable adsorption between the catalyst and substrate of the self-standing electrodes prepared by the traditional electrodeposition method under cathodic bias. The Fe loss in CoFe LDH during the OER activation process agrees with the previously reported results, where Fe depletion in Fe–M (oxy)hydroxide happened because of the very unstable Fe active sites and the failure of Fe redeposition when the OER was conducted in Fe-free electrolyte.23 The results of Co–Fe active site loss in CoFe LDH further explain its decayed activity after the 10-h OER. However, the poor activity retention of Fe-containing (oxy)hydroxides could be solved by introducing Fe ions in electrolyte and facilitating Fe dynamic exchange at the electrode/electrolyte interface. The ICP results of the mass loading variation on the electrode show that the Fe/Co + Fe molar ratio in CoFe-in situ was well maintained after 10 h of running (Fig. 5a). The concentrations of Fe and Co ions on the surface of CoFe-in situ before and after the stability test were further detected, using the semiquantitative analysis of XPS measurement. As shown in Table S2,† the molar ratios of Fe/Fe + Co in the catalysts pre- and post-10 h running are 0.456 and 0.472, respectively. The Fe content both in the bulk and on the surface of CoFe-in situ exhibits a relatively stable value after long-term running, which can lead to the stability mechanism that, even though a portion of Fe ions dissolve from the crystalline matrix of CoFe-in situ during the OER, dissociative Fe ions in electrolyte redeposit on the electrode surface to reconstruct the Co–O–Fe active motifs. Furthermore, there is an Fe saturation coverage for each surface (with Fen+(aq.) content above 0.1 ppm).31 For CoFe-in situ, neither the OER activity nor the ratio of Fe incorporated into Co (oxy)hydroxide present an increase along with the deposition time. These results show that our in situ synthesis method successfully constructed a dynamically stable electrode for electrochemical OER activation by keeping the Fe saturated surface. To conclude, by controlling the reconstruction process of the solid/liquid interface, we successfully attained a high number of stable Co–O–Fe active sites and maintained their long durability, which is favorable for the industrial application of the OER.
 | ||
| Fig. 5 Mass of Co and Fe, and the molar ratio of Fe/Co + Fe in (a) CoFe-in situ and (b) CoFe LDH electrodes before and after the 10-h OER. Schemes of the (c) ‘dynamically stable Fe’ for CoFe-in situ and (d) ‘Fe leaching’ for CoFe LDH at the electrode/electrolyte interface, highlighting the role of CoOxHy with a weak Co–O bond as a suitable host for Fe adsorption and Fe-containing electrolyte ensuring Fe redeposition for Co–O–Fe active site formation. | ||
Conclusions
In summary, the in situ synthesized CoFe (oxy)hydroxide nanocatalyst successfully broke the bottleneck of low mass activity and instability of traditional heterogeneous catalysts for the OER. Compared to the CoFe (oxy)hydroxides based on LDH precatalysts, the as-prepared CoFe-in situ nanocatalyst exhibited 5 times higher mass activity, attributed to its excellent intrinsic activity and beneficial morphology. We elucidated that the self-assembling synthesis process of CoFe-in situ forms Co–O–Fe active sites with a weak metal–O coordination environment, leading to strong adsorption of hydroxyl species. The ultrasmall nanosheet structure of CoFe-in situ showed outstanding hydrophilicity, which further increased the exposure of active sites for the OER. The degree of lattice oxygen participation in oxygen evolution and metal dissolution was suppressed for CoFe-in situ, which ensures structure integrity and overcomes the instability of metal (oxy)hydroxides. Owing to the dynamic stable surface with a saturated amount of active sites, the CoFe-in situ nanocatalyst retained its performance for over 100 h of running. Our study provides a new strategy for the development of alternative cost-effective and durable OER catalysts for industrial application.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Writing – original draft: M. Z.; writing – review & editing: J. D., D. G., Z. H., and S. S.; resources: C. C. and H. X.; supervision: Z. S., R. R., and W. Z.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support from the Max Planck-POSTECH-Hsinchu Center for Complex Phase Materials, and Jiangsu Funding Program for Excellent Postdoctoral Talent.Notes and references
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- J. Dai, Y. Zhu, Y. Chen, X. Wen, M. Long, X. Wu, Z. Hu, D. Guan, X. Wang, C. Zhou, Q. Lin, Y. Sun, S.-C. Weng, H. Wang, W. Zhou and Z. Shao, Nat. Commun., 2022, 13, 1189 CrossRef CAS PubMed.
- B. M. Hunter, H. B. Gray and A. M. Müller, Chem. Rev., 2016, 116, 14120–14136 CrossRef CAS PubMed.
- D. Guan, B. Wang, J. Zhang, R. Shi, K. Jiao, L. Li, Y. Wang, B. Xie, Q. Zhang, J. Yu, Y. Zhu, Z. Shao and M. Ni, Energy Environ. Sci., 2023, 16, 4926–4943 RSC.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- J. Song, C. Wei, Z.-F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- V. Tripkovic, H. A. Hansen, J. M. Garcia-Lastra and T. Vegge, J. Phys. Chem. C, 2018, 122, 1135–1147 CrossRef CAS.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- J. A. Carrasco, A. Harvey, D. Hanlon, V. Lloret, D. McAteer, R. Sanchis-Gual, A. Hirsch, F. Hauke, G. Abellán, J. N. Coleman and E. Coronado, Chem. Commun., 2019, 55, 3315–3318 RSC.
- J. Bauer, D. H. Buss, H. J. Harms and O. Glemser, J. Electrochem. Soc., 1990, 137, 173 CrossRef CAS.
- L. Gao, X. Cui, C. D. Sewell, J. Li and Z. Lin, Chem. Soc. Rev., 2021, 50, 8428–8469 RSC.
- N. C. S. Selvam, L. Du, B. Y. Xia, P. J. Yoo and B. You, Adv. Funct. Mater., 2021, 31, 2008190 CrossRef CAS.
- S. Zuo, Z.-P. Wu, H. Zhang and X. W. Lou, Adv. Energy Mater., 2022, 12, 2103383 CrossRef CAS.
- F. Dionigi, Z. Zeng, I. Sinev, T. Merzdorf and P. Strasser, Nat. Commun., 2020, 11, 2522 CrossRef CAS PubMed.
- W. Chen, B. Wu, Y. Wang, W. Zhou, Y. Li, T. Liu, C. Xie, L. Xu, S. Du, M. Song, D. Wang, Y. liu, Y. Li, J. Liu, Y. Zou, R. Chen, C. Chen, J. Zheng, Y. Li, J. Chen and S. Wang, Energy Environ. Sci., 2021, 14, 6428–6440 RSC.
- S. Zuo, Z.-P. Wu, G. Zhang, C. Chen, Y. Ren, Q. Liu, L. Zheng, J. Zhang, Y. Han and H. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202316762 CrossRef CAS PubMed.
- O. O. Mabayoje, A. Shoola, B. R. Wygant and C. B. Mullins, ACS Energy Lett., 2016, 1, 195–201 CrossRef CAS.
- N. Clament Sagaya Selvam, S. J. Kwak, G. H. Choi, M. J. Oh, H. Kim, W.-S. Yoon, W. B. Lee and P. J. Yoo, ACS Energy Lett., 2021, 6, 4345–4354 CrossRef CAS.
- Y.-j. Wu, J. Yang, T.-x. Tu, W.-q. Li, P.-f. Zhang, Y. Zhou, J.-f. Li, J.-t. Li and S.-G. Sun, Angew. Chem., Int. Ed., 2021, 60, 26829–26836 CrossRef CAS PubMed.
- B. Zhang, K. Jiang, H. Wang and S. Hu, Nano Lett., 2019, 19, 530–537 CrossRef CAS PubMed.
- E. Fabbri, M. Nachtegaal, T. Binninger, X. Cheng, B.-J. Kim, J. Durst, F. Bozza, T. Graule, R. Schäublin, L. Wiles, M. Pertoso, N. Danilovic, K. E. Ayers and T. J. Schmidt, Nat. Mater., 2017, 16, 925–931 CrossRef CAS PubMed.
- M. Risch, A. Grimaud, K. May, K. Stoerzinger, T. Chen, A. Mansour and Y. Shao-Horn, J. Phys. Chem. C, 2013, 117, 8628–8635 CrossRef CAS.
- Y. Lin, H. Ren, S. Zhang, S. Liu, T. Zhao, W.-J. Jiang, W. Zhou, J.-S. Hu and Z. Li, Adv. Energy Mater., 2024, 14, 2302403 CrossRef CAS.
- D. Wang, S. Ruan, P. Ma, R. Wang, X. Ding, M. Zuo, L. Zhang, Z. Zhang, J. Zeng and J. Bao, Nano Res., 2023, 16, 8793–8799 CrossRef CAS.
- D. Zhou, P. Li, X. Lin, A. Mckinley and X. Duan, Chem. Soc. Rev., 2021, 50, 8790–8817 RSC.
- P. Babar, A. Lokhande, H. H. Shin, B. Pawar, M. G. Gang, S. Pawar and J. H. Kim, Small, 2018, 14, 1702568 CrossRef PubMed.
- W. Liu, H. Liu, L. Dang, H. Zhang, X. Wu, B. Yang, Z. Li, X. Zhang, L. Lei and S. Jin, Adv. Funct. Mater., 2017, 27, 1603904 CrossRef.
- G. Abellán, J. A. Carrasco, E. Coronado, J. Romero and M. Varela, J. Mater. Chem. C, 2014, 2, 3723–3731 RSC.
- R. Chen, S. Hung, D. Zhou, J. Gao and B. Liu, Adv. Mater., 2019, 31, 1903909 CrossRef CAS PubMed.
- C. Kuai, Z. Xu, C. Xi, A. Hu, Z. Yang, Y. Zhang, C.-J. Sun, L. Li, D. Sokaras, C. Dong, S.-Z. Qiao, X.-W. Du and F. Lin, Nat. Catal., 2020, 3, 743–753 CrossRef CAS.
- Y. C. Dong, P. P. Lopes, P. Martins, H. He, T. Kawaguchi, P. Zapol, H. You, D. Tripkovic, D. Strmcnik and Y. Zhu, Nat. Energy, 2020, 5, 222–230 CrossRef.
- D. Guan, G. Ryu, Z. Hu, J. Zhou, C.-L. Dong, Y.-C. Huang, K. Zhang, Y. Zhong, A. C. Komarek, M. Zhu, X. Wu, C.-W. Pao, C.-K. Chang, H.-J. Lin, C.-T. Chen, W. Zhou and Z. Shao, Nat. Commun., 2020, 11, 3376 CrossRef CAS PubMed.
- R. A. Márquez, K. Kawashima, Y. J. Son, G. Castelino, N. Miller, L. A. Smith, C. E. Chukwuneke and C. B. Mullins, ACS Energy Lett., 2023, 8, 1141–1146 CrossRef.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- D. Guan, H. Xu, Q. Zhang, Y. C. Huang, C. Shi, Y. C. Chang, X. Xu, J. Tang, Y. Gu, C. W. Pao, S. C. Haw, J. M. Chen, Z. Hu, M. Ni and Z. Shao, Adv. Mater., 2023, 35, 2305074 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- Q. Zheng, H. Xu, Y. Yao, J. Dai, J. Wang, W. Hou, L. Zhao, X. Zou, G. Zhan, R. Wang, K. Wang and L. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202401386 CrossRef CAS PubMed.
- C. Cheng, H. Xu, M. Ni, C. Guo, Y. Zhao and Y. Hu, Appl. Catal., B, 2024, 345, 123705 CrossRef CAS.
- H. Xu, J. Z. Zhu, C. Zou, F. Zhang, D. Ming, D. Guan and L. Ma, Energy Fuels, 2023, 37, 16781–16789 CrossRef CAS.
- W. Xiao, K. Yoo, J. H. Kim and H. Xu, Adv. Sci., 2023, 10, 2303916 CrossRef CAS PubMed.
- F. Dionigi, T. Reier, Z. Pawolek, M. Gliech and P. Strasser, ChemSusChem, 2016, 9, 962–972 CrossRef CAS PubMed.
- F. Dionigi, J. Zhu, Z. Zeng, T. Merzdorf, H. Sarodnik, M. Gliech, L. Pan, W.-X. Li, J. Greeley and P. Strasser, Angew. Chem., Int. Ed., 2021, 60, 14446–14457 CrossRef CAS PubMed.
- Z. Sun, A. Curto, J. Rodríguez-Fernández, Z. Wang, A. Parikh, J. Fester, M. Dong, A. Vojvodic and J. V. Lauritsen, ACS Nano, 2021, 15, 18226–18236 CrossRef CAS PubMed.
- R. D. L. Smith, C. Pasquini, S. Loos, P. Chernev, K. Klingan, P. Kubella, M. R. Mohammadi, D. Gonzalez-Flores and H. Dau, Nat. Commun., 2017, 8, 2022 CrossRef PubMed.
- Z. Li, H. Duan, M. Shao, J. Li, D. O'Hare, M. Wei and Z. L. Wang, Chem, 2018, 4, 2168–2179 CAS.
- A. Moysiadou, S. Lee, C.-S. Hsu, H. M. Chen and X. Hu, J. Am. Chem. Soc., 2020, 142, 11901–11914 CrossRef CAS PubMed.
- M. Ludvigsson, J. Lindgren and J. Tegenfeldt, J. Mater. Chem., 2001, 11, 1269–1276 RSC.
- Y.-C. Liu, J. A. Koza and J. A. Switzer, Electrochim. Acta, 2014, 140, 359–365 CrossRef CAS.
- J. A. Koza, C. M. Hull, Y.-C. Liu and J. A. Switzer, Chem. Mater., 2013, 25, 1922–1926 CrossRef CAS.
- R. Sanchis-Gual, D. Hunt, C. Jaramillo-Hernández, A. Seijas-Da Silva, M. Mizrahi, C. Marini, V. Oestreicher and G. Abellán, ACS Catal., 2023, 13, 10351–10363 CrossRef CAS PubMed.
- C. Kuai, C. Xi, A. Hu, Y. Zhang, Z. Xu, D. Nordlund, C.-J. Sun, C. A. Cadigan, R. M. Richards, L. Li, C.-K. Dong, X.-W. Du and F. Lin, J. Am. Chem. Soc., 2021, 143, 18519–18526 CrossRef CAS PubMed.
- M. B. Stevens, C. D. M. Trang, L. J. Enman, J. Deng and S. W. Boettcher, J. Am. Chem. Soc., 2017, 139, 11361–11364 CrossRef CAS PubMed.
- S. Anantharaj, S. Kundu and S. Noda, Nano Energy, 2020, 80, 105514 CrossRef.
- Y. Y. Chin, Z. Hu, H. J. Lin, S. Agrestini, J. Weinen, C. Martin, S. Hébert, A. Maignan, A. Tanaka, J. C. Cezar, N. B. Brookes, Y. F. Liao, K. D. Tsuei, C. T. Chen, D. I. Khomskii and L. H. Tjeng, Phys. Rev. B, 2019, 100, 205139 CrossRef CAS.
- D. Guan, J. Zhong, H. Xu, Y.-C. Huang, Z. Hu, B. Chen, Y. Zhang, M. Ni, X. Xu, W. Zhou and Z. Shao, Appl. Phys. Rev., 2022, 9, 011422 CAS.
- N. Hollmann, Z. Hu, M. Valldor, A. Maignan, A. Tanaka, H. H. Hsieh, H. J. Lin, C. T. Chen and L. H. Tjeng, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 085111 CrossRef.
- Y. Dou, D. Yuan, L. Yu, W. Zhang, L. Zhang, K. Fan, M. Al-Mamun, P. Liu, C.-T. He and H. Zhao, Adv. Mater., 2021, 34, 2104667 CrossRef PubMed.
- H. Sun, W. Zhang, J.-G. Li, Z. Li, X. Ao, K.-H. Xue, K. K. Ostrikov, J. Tang and C. Wang, Appl. Catal., B, 2021, 284, 119740 CrossRef CAS.
- Y. Wang, Y. Zhang, Z. Liu, C. Xie, S. Feng, D. Liu, M. Shao and S. Wang, Angew. Chem., Int. Ed., 2017, 56, 5867–5871 CrossRef CAS PubMed.
- Q. Xie, Z. Cai, P. Li, D. Zhou, Y. Bi, X. Xiong, E. Hu, Y. Li, Y. Kuang and X. Sun, Nano Res., 2018, 11, 4524–4534 CrossRef CAS.
- S. She, Y. Zhu, H. A. Tahini, Z. Hu, S.-C. Weng, X. Wu, Y. Chen, D. Guan, Y. Song, J. Dai, S. C. Smith, H. Wang, W. Zhou and Z. Shao, Appl. Phys. Rev., 2021, 8, 11407 CAS.
- R. Liu, Y. Wang, D. Liu, Y. Zou and S. Wang, Adv. Mater., 2017, 29, 1701546 CrossRef PubMed.
- C. Hu, L. Zhang and J. Gong, Energy Environ. Sci., 2019, 12, 2620–2645 RSC.
- A. S. Batchellor and S. W. Boettcher, ACS Catal., 2015, 5, 6680–6689 CrossRef CAS.
- F. R. Van Buren, G. H. J. Broers, A. J. Bouman and C. Boesveld, J. Electroanal. Chem. Interfacial Electrochem., 1978, 87, 389–394 CrossRef CAS.
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Nat. Commun., 2016, 7, 11053 CrossRef CAS PubMed.
- Y. Pan, X. Xu, Y. Zhong, L. Ge, Y. Chen, J.-P. M. Veder, D. Guan, R. O'Hayre, M. Li, G. Wang, H. Wang, W. Zhou and Z. Shao, Nat. Commun., 2020, 11, 2002 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta01848f |
| ‡ These authors contributed equally: Ming Zhu and Hengyue Xu. |
| This journal is © The Royal Society of Chemistry 2024 |