
Engineering semiconductor quantum dots for co-upcycling of CO2 and biomass-derived alcohol†
Lin-Xing
Zhang
a,
Zi-Rong
Tang
 a,
Ming-Yu
Qi
*ab and
Yi-Jun
Xu
*a
a,
Ming-Yu
Qi
*ab and
Yi-Jun
Xu
*a
aCollege of Chemistry, State Key Laboratory of Photocatalysis on Energy and Environment, Fuzhou University, Fuzhou, 350116, China. E-mail: yjxu@fzu.edu.cn
bInstitute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu, 611731, China. E-mail: myqi@uestc.edu.cn
First published on 28th June 2024
Abstract
Utilizing semiconductor quantum dots (QDs) to construct a bifunctional reaction system of coupling CO2 reduction with biomass valorization represents an appealing approach for the production of useable fuels and value-added chemicals. Herein, we present an efficient cooperative photocatalytic process for simultaneously achieving the reduction of CO2 to syngas and the oxidation of biomass-derived furfuryl alcohol to furfural and hydrofuroin over SiO2-supported CdSe/CdS QDs (CdSe/CdS–SiO2). The type-II band alignment in CdSe/CdS core/shell heterostructures enables effective charge separation and interfacial charge migration concurrently. By further assembly onto a spherical SiO2 support, the optimized CdSe/CdS–SiO2 composite exhibits remarkably enhanced activities for syngas and furfural/hydrofuroin production, which are 2.3 and 3.5 times higher than those of binary CdSe/CdS core/shell QDs, and 90.4 and 18.5 times higher than those of bare CdSe QDs, along with good stability. In particular, by altering the thickness of the CdS shell, the syngas CO/H2 ratio can be precisely modulated within a wide range (1.6 to 7.1), which serves as crucial feedstock for the production of liquid fuels. This work is expected to develop core/shell QD-based photocatalysts for versatile and available photoredox-catalyzed reaction systems that integrate CO2 valorization with biomass upgrading.
 Ming-Yu Qi | Ming-Yu Qi obtained his PhD degree from Fuzhou University in May 2023 and now works as a postdoctoral researcher at the University of Electronic Science and Technology of China. In the recent five years, he has published over 50 papers in many prestigious journals, such as Chem. Rev., Chem. Soc. Rev., Nat. Commun., Angew. Chem., Energy Environ. Sci., ACS Nano, and ACS Catal., among which 15 papers are selected as ESI highly cited papers. His publications have generated more than 4500 citations, and he has been recognized in the “World's Top 2% Scientists 2023” (identified by Stanford University). In addition, he was sponsored by the 2024 National “Postdoctoral Innovative Talent Support Program” and awarded the “2023 Nano Research Energy Young Star Researcher Gold Award” and “2024 IUPAC International Award for Young Chemists”. |
Introduction
Leveraging artificial photosynthesis for the transformation of CO2 into valuable fuels and chemicals represents a highly attractive approach to tackle the escalating energy crisis and environmental challenges.1–4 Unfortunately, the sluggish reaction kinetics of the O2-producing half-reaction pose significant obstacles to the efficiency and selectivity of overall CO2 reduction (CO2 + 2H2O = CH4 + 2O2).5 Furthermore, the use of hole sacrificial reagents, such as triethanolamine (TEOA), sodium sulfite (Na2SO3), ascorbic acid, and isopropyl alcohol, to facilitate CO2 conversion results in the wasteful consumption of hole energy and generates undesired oxidation products.6 Biomass, as the Earth's most abundant renewable chemical resource, holds considerable value in the fragrance and polymeric industries.7,8 Upon pretreatment of raw biomass materials, there are a great number of biomass-derived intermediates that require further upgrading to become value-added products.9,10 In this scenario, exploiting biomass derivatives (for example, alcohols, sugars, lignin and bio-oils) to consume holes for high-value fine chemical production, while leaving electrons and protons for CO2 reduction to form a redox cycle, opens a fascinating avenue for valorizing two waste streams (CO2 and biomass) synergistically into value-added fuels and chemicals.11,12Recently, the transformation of biomass-derived alcohols, such as furfuryl alcohol to value-added furfural or C–C coupled products, has been investigated; nevertheless, these mushrooming demonstrations have focused on O2-mediated furfuryl alcohol oxidation, instead of cooperative coupling photoredox catalysis by simultaneous use of photogenerated electrons and holes for co-upcycling of CO2 and furfuryl alcohol.13–15 Notably, such coupling reactions crucially rely on the utilization of a semiconductor with an appropriate band gap position for both oxidative and reductive reactions.16,17 Among various materials, colloidal CdSe quantum dots (QDs) stand out due to their tunable band-edge positions, excellent light harvesting and abundant surface-active sites, making them ideal for multi-electron photoredox reactions.18,19 Recent years have witnessed the application of CdSe QDs with matched redox potentials as photocatalysts toward various oxidative organic syntheses or the CO2 reduction reaction.20,21 However, owing to the severe charge recombination faced by CdSe QDs, their photocatalytic performance is severely suppressed. To address these challenges, the epitaxial growth of a CdS shell around the CdSe QD core has been verified as an effective method to construct a staggered type-II band alignment, which leads to the spatial separation of the photogenerated electrons and holes within the core and shell materials, respectively, therefore improving their activity.22–25
Herein, we for the first time have reported the immobilization of core/shell CdSe/CdS QD heterostructures onto spherical SiO2, enabling the efficient co-upcycling of CO2 and biomass-derived furfuryl alcohol in one reaction system. Consequently, compared to bare CdSe QDs, the optimized CdSe/CdS–SiO2 composite shows a 90.4-fold and 18.5-fold enhancement in activity for the production of syngas and furfural/hydrofuroin, respectively. Notably, the syngas CO/H2 ratio can be precisely adjusted within a broad range, from 1.6 to 7.1, encompassing the optimal syngas composition required for both methanol production and Fischer–Tropsch hydrocarbon generation. Mechanism studies have demonstrated that the core/shell QD heterostructure, combined with the near-field scattering resonances of spherical SiO2, promotes light absorption, charge separation, and CO2 adsorption/activation, thereby boosting the efficiency of valorizing CO2 and furfuryl alcohol. It is hoped that this work could provide new insights for developing innovative QD-based photocatalysts toward cooperative photoredox catalysis of CO2 reduction and biomass upgrading.
Results and discussion
The process for fabricating the CdSe/CdS–SiO2 composite is illustrated in Fig. 1a. First, CdSe/CdS quantum dots (QDs) are acquired by a seeded-growth approach, where a CdS shell is epitaxially grown on the surface of CdSe QDs.26 According to their high-resolution transmission electron microscopy (HRTEM) observations, CdSe QDs exhibit a diameter of approximately 2.8 nm and a lattice spacing of 0.215 nm, corresponding to the (220) facet, while the CdSe/CdS QDs have a larger size distribution than bare CdSe QDs (Fig. 1b and c). As the CdS shell thickness increases, the size of the CdSe/CdS QDs increases, and the lattice fringes gradually converge toward those of CdS, confirming the successful synthesis of core/shell structured QDs (Fig. S1 and S2†).21,27,28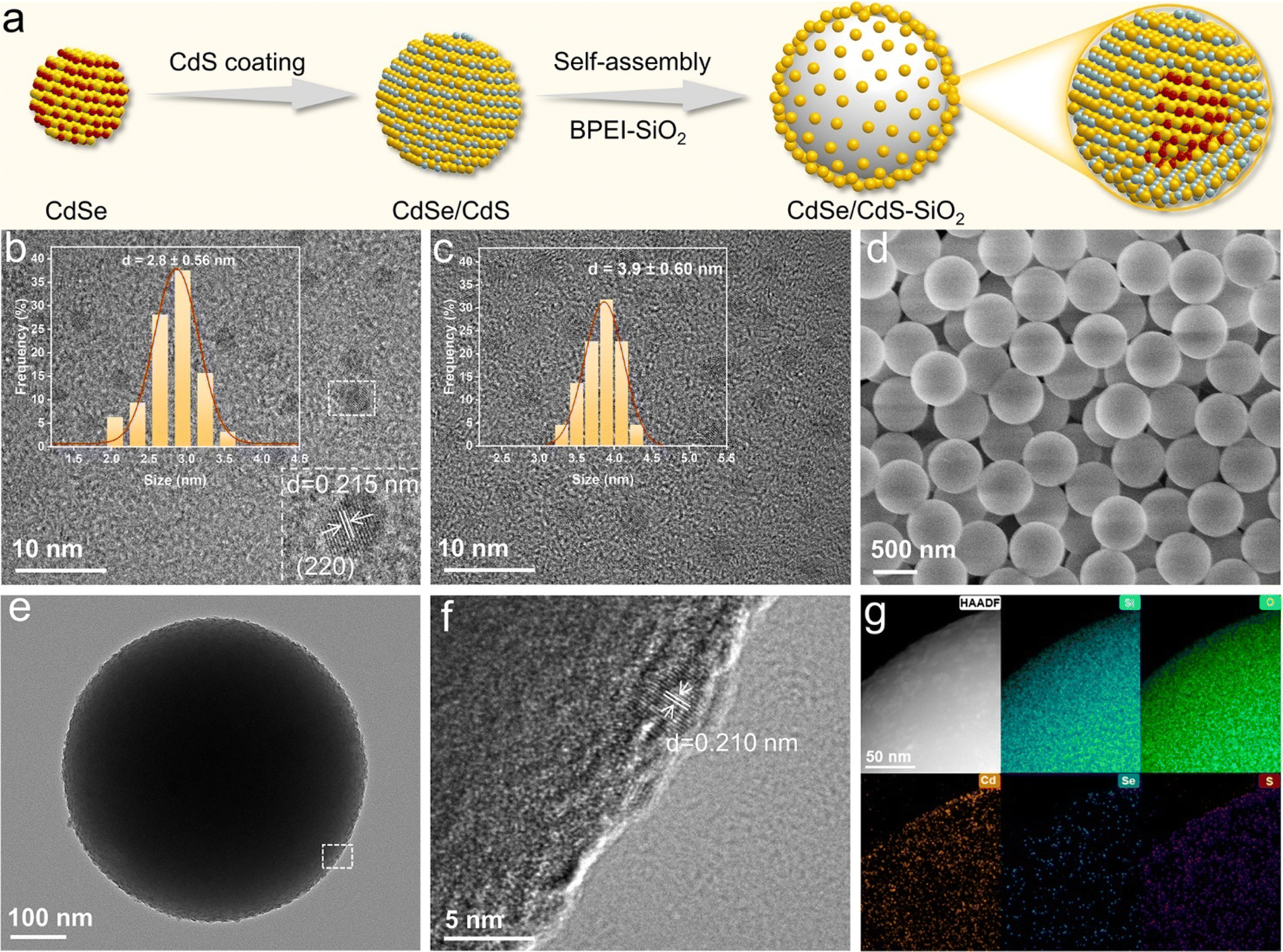 | ||
| Fig. 1 (a) Schematic representation of the CdSe/CdS–SiO2 composite synthesis procedure. HRTEM images of (b) CdSe QDs and (c) CdSe/3CdS QDs (where 3 signifies approximately 3 atomic layers of the CdS shell). Inset: size distribution histograms of the corresponding QDs. (d) Scanning electron microscopy image of BPEI-SiO2 spheres. (e) TEM image, (f) HRTEM image of the selected region in (e) and (g) high-angle annular dark field and corresponding elemental mapping images of the CdSe/CdS–SiO2 composite. | ||
To enhance the light-harvesting efficiency of core/shell QDs, we then fabricated a SiO2-supported CdSe/CdS QD (CdSe/CdS–SiO2) composite through an electrostatic self-assembly method.12,29–31 Fig. S3† depicts the robust electrostatic attraction that arises from the opposing surface charge of CdSe/CdS QDs (−34 mV) and the branched poly-ethylenimine functionalized SiO2 (denoted as BPEI-SiO2) (+38 mV). After self-assembly, a dense arrangement of QDs can be clearly seen on the surface of BPEI-SiO2, whereas the surface of BPEI-SiO2 is smooth (Fig. 1d–f). Elemental mapping analysis further demonstrates the even distribution of Cd, Se and S elements, manifesting the dispersal of CdSe/CdS QDs onto the SiO2 sphere surface (Fig. 1g).
The surface-capped groups on BPEI-SiO2 and CdSe/CdS QDs are then verified via Fourier transform infrared (FTIR) spectroscopy. As displayed in Fig. 2a, the appearance of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration peaks at 1620 and 1400 cm−1, along with the O–H stretching vibration peak at 3310 cm−1, suggests the presence of a –COOH group in CdSe/CdS QDs.32 The characteristic peaks, located at 1620 and 1140 cm−1, are attributed to N–H and C–N vibrations in BPEI-SiO2, indicating the successful attachment of BPEI across the SiO2 sphere surface. The X-ray diffraction (XRD) pattern is employed to ensure the successful synthesis of CdSe/CdS QDs. As shown in Fig. 2b, the CdSe QDs show a cubic structure (JCPDS No. 19-0191). In contrast, the feature diffraction peaks of CdSe/CdS QDs progressively shift to larger angles with the increase of CdS shell thickness, which agrees with the published literature on the formation of a CdS shell (JCPDS No. 75-0581) on CdSe QDs.28,33 Besides, no discernible characteristic peaks associated with CdSe/3CdS QDs are observed in the XRD pattern of the CdSe/CdS–SiO2 composite, presumably due to their low content and uniform dispersion throughout the composite (Fig. S4†).34
O vibration peaks at 1620 and 1400 cm−1, along with the O–H stretching vibration peak at 3310 cm−1, suggests the presence of a –COOH group in CdSe/CdS QDs.32 The characteristic peaks, located at 1620 and 1140 cm−1, are attributed to N–H and C–N vibrations in BPEI-SiO2, indicating the successful attachment of BPEI across the SiO2 sphere surface. The X-ray diffraction (XRD) pattern is employed to ensure the successful synthesis of CdSe/CdS QDs. As shown in Fig. 2b, the CdSe QDs show a cubic structure (JCPDS No. 19-0191). In contrast, the feature diffraction peaks of CdSe/CdS QDs progressively shift to larger angles with the increase of CdS shell thickness, which agrees with the published literature on the formation of a CdS shell (JCPDS No. 75-0581) on CdSe QDs.28,33 Besides, no discernible characteristic peaks associated with CdSe/3CdS QDs are observed in the XRD pattern of the CdSe/CdS–SiO2 composite, presumably due to their low content and uniform dispersion throughout the composite (Fig. S4†).34
 | ||
| Fig. 2 (a) FTIR spectra of CdSe QDs, BPEI-SiO2 and CdSe/CdS–SiO2 composites. (b) XRD patterns of CdSe QDs, CdS QDs and CdSe/CdS QDs with different atomic thicknesses of the CdS shell. (c) DRS spectra of CdSe QDs, CdS QDs, BPEI-SiO2, CdSe/CdS QDs and CdSe/CdS–SiO2 composites (to ascertain the quantitative comparativeness of DRS spectra, an equal amount of QDs (2.5% weight content) was mixed with BaSO4). XPS spectra for (d) Cd 3d, (e) Se 3d and (f) S 2p of CdSe QDs, CdS QDs and CdSe/CdS QDs. | ||
Ultraviolet-visible diffuse reflectance spectroscopy (DRS) shows that the absorption edges of CdSe and CdS QDs lie at 530 and 557 nm, respectively. The CdSe/CdS–SiO2 composite shows six distinct and well-defined absorption peaks at 235, 259, 288, 310, 360 and 425 nm.12 Nevertheless, these characteristic absorption peaks are absent in both BPEI-SiO2 and CdSe/CdS QDs, indicating that their presence in the CdSe/CdS–SiO2 composite is ascribed to the enhanced light–matter interaction facilitated by near-field scattering between the CdSe/CdS QDs and SiO2 spheres.12,29–31 Furthermore, the surface chemical state of CdSe/CdS QDs has been examined by X-ray photoelectron spectroscopy (XPS) to explore the heterointerface interaction. The XPS survey spectra presented in Fig. S5† clearly reveal the existence of Cd, Se and S within the CdSe/CdS QDs, corroborating the findings obtained from element mapping. The Cd 3d spectra (Fig. 2d) of CdSe/CdS QDs exhibit two narrow peaks at 404.80 and 411.55 eV, attributed to the binding energies of Cd 3d5/2 and Cd 3d3/2, respectively. These characteristic peaks are negatively shifted by 0.16 eV compared to CdSe QDs. Similarly, the Se 3d spectra (Fig. 2e) of CdSe/CdS QDs also display a negative shift (0.18 eV) with the binding energies of Se 3d5/2 and Se 3d3/2 emerging at 53.45 and 54.30 eV, respectively, while those peaks in CdSe QDs are located at higher values (53.63 and 54.48 eV, respectively). In comparison to CdS QDs, the Cd 3d and S 2p spectra of CdSe/CdS QDs exhibit an opposing trend (positive shifted by 0.10 eV), indicating electron migration from CdS to CdSe.34 Besides, the S 2p spectra can be resolved into four peaks, with binding energies of 162.24 eV (S 2p3/2) and 163.17 eV (S 2p1/2) attributed to the capped ligand of MPA, while the S2− in the CdS lattice is located at 160.98 eV (S 2p3/2) and 161.33 eV (S 2p1/2).35,36 These findings suggest the existence of robust chemical interactions between CdSe and CdS components within the core/shell QD heterostructure.37
Subsequently, the photocatalytic performance of CO2 reduction coupled with the oxidation of furfuryl alcohol is assessed in a CO2-saturated acetonitrile solution at room temperature (Fig. 3a). Gas chromatography-mass spectrometry (GC-MS) has been used to detect primary oxidation products, namely furfural and hydrofuroin (Fig. S6†). As depicted in Fig. 3b and c, CdSe/CdS QDs exhibit superior catalytic performance compared to bare CdSe and CdS QDs, with the maximum performance observed for CdSe/3CdS QDs, where 3 signifies approximately 3 atomic layers of the CdS shell. Specifically, upon 4 h of continuous illumination, CdSe/3CdS QDs show a syngas production rate of 3.10 mmol gQDs−1 h−1, and the production rate for furfural/hydrofuroin stands at 0.89 mmol gQDs−1 h−1. These rates represent a 38.8-fold and 5.2-fold increase, respectively, compared to the corresponding bare CdSe QDs (0.08 mmol gQDs−1 h−1 for syngas and 0.17 mmol gQDs−1 h−1 for furfural/hydrofuroin). The significantly improved catalytic performance of core/shell CdSe/CdS QDs can be primarily attributed to the efficient photogenerated charge separation. However, an excessively thick CdS shell would create a large hindrance to charge transfer (Fig. 3b).24 In addition, the syngas CO/H2 ratio can be flexibly adjusted within a broad range by altering the CdS shell thickness, ranging from 1.6 to 7.1, which encompasses the optimal syngas composition required for both methanol production and Fischer–Tropsch hydrocarbon generation.38 To further enhance the light-harvesting properties of CdSe/CdS QDs, they are attached to spherical SiO2 to recover scattered light in the near-field of SiO2 supports.12,29–31 Notably, the CdSe/CdS–SiO2 composite exhibits greatly improved catalytic performance, achieving syngas and furfural/hydrofuroin production rates of up to 7.23 and 3.15 mmol gQDs−1 h−1, respectively, which are approximately 2.3 and 3.5 times higher than those of CdSe/CdS QDs. As expected, the CdSe–SiO2 and CdS–SiO2 composites exhibit superior catalytic performance compared to bare CdSe QDs and CdS QDs, respectively.
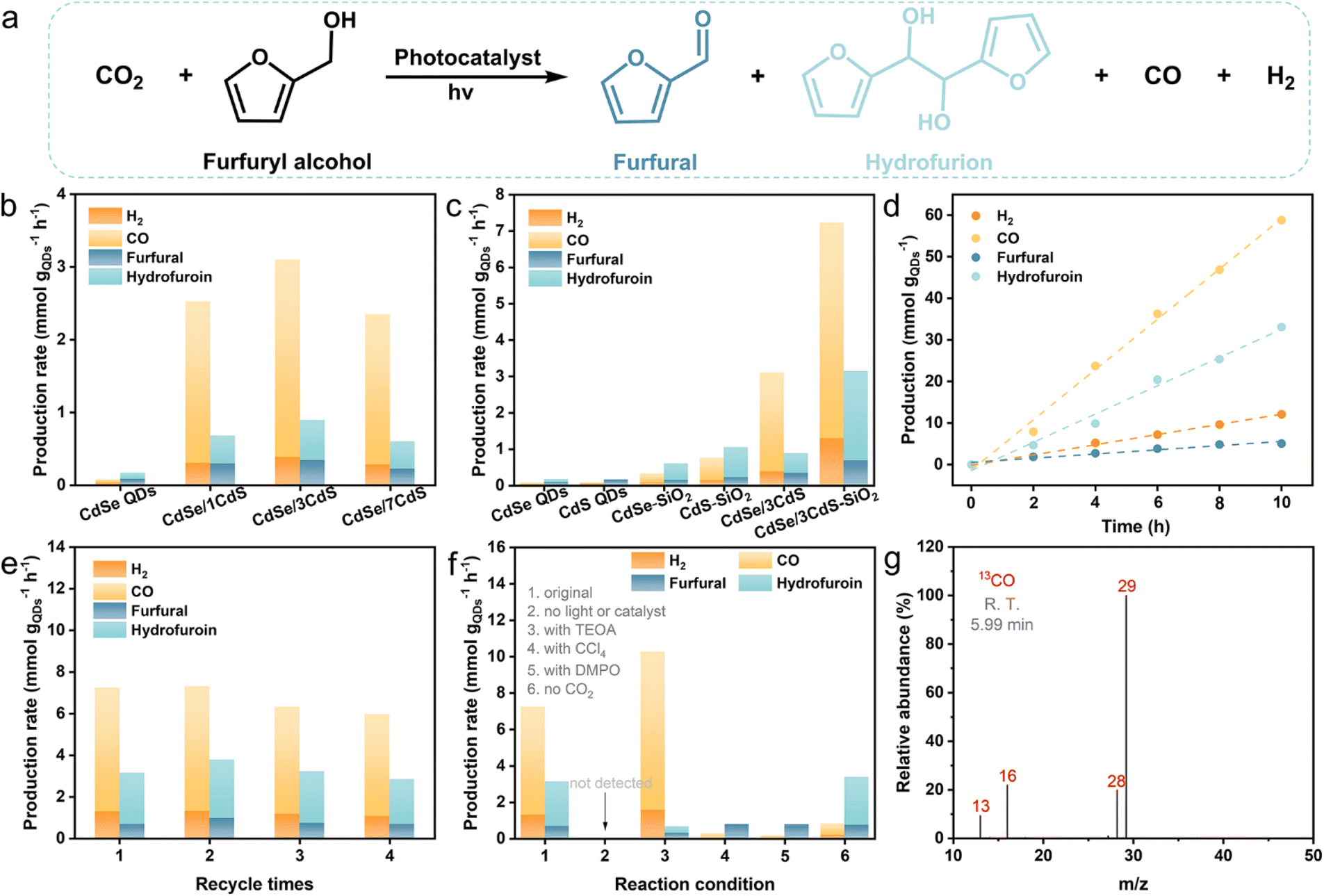 | ||
| Fig. 3 (a) The formula for the bifunctional reaction of photocatalytic CO2 reduction combined with the oxidation of furfuryl alcohol. (b) and (c) Photocatalytic performances of CdSe QDs, CdS QDs, and CdSe/CdS QDs with varying CdS shell thickness, as well as CdSe–SiO2, CdS–SiO2 and CdSe/CdS–SiO2 composites. (d) Prolonged photocatalytic performance and (e) four-cycle recycling photocatalytic performance of the CdSe/CdS–SiO2 composite. (f) Comparative experiments on CdSe/CdS–SiO2 catalysis under different conditions. (g) Mass spectrum of 13CO produced by the photoreduction of 13CO2 over the CdSe/CdS–SiO2 composite. | ||
When the catalytic stability of the photocatalyst is taken into account, it is found that the CdSe/CdS–SiO2 photocatalyst displays good long-term stability and recyclability. As depicted in Fig. 3d, after a duration of 10 h, the production of syngas and furfural/hydrofuroin is 70.90 and 38.13 mmol gQDs−1, respectively, and these production levels maintain a linear correlation with the duration of the reaction. In addition, the cycling test verified that the photocatalytic activity remains stable, with no significant decrease observed after four cycles (Fig. 3e). Notably, there is no obvious change in the crystalline configuration and light-capturing characteristics of fresh and used CdSe/CdS–SiO2 composites (Fig. S7†), which proves its good stability in the dual-functional reaction process.
To gain a deeper understanding of the photocatalytic conversion of CO2 paired with the furfuryl alcohol oxidation reaction, controlled experiments have been performed, as shown in Fig. 3f. Without light or catalysts, the reaction does not occur at all, indicating that the redox reaction is driven entirely by light. The introduction of triethanolamine (TEOA) as a hole sacrificial agent significantly inhibits the formation of furfural and hydrofuroin, meaning that the oxidation of furfuryl alcohol relies on photogenerated holes.39 Simultaneously, almost no H2 and CO are observed when carbon tetrachloride (CCl4), an electron sacrificial agent, is incorporated into the reaction system, thus highlighting the crucial role of electrons as the main reductive species for the evolution of CO and H2. In addition, the photocatalytic activity is significantly reduced, and the hydrofuroin is undetectable when 5,5-dimethyl-1-pyrroline N-oxide (DMPO) is used as a free radical scavenger, illustrating that the bifunctional reaction is mediated by radical intermediates.34 The decrease in furfural/hydrofuroin production correspondingly results in a reduced output of CO and H2, underscoring the close interdependence between reduction and oxidation reactions.40 Substituting Ar with CO2 results in the detection of only minimal quantities of CO, presumably originating from the decomposition of carbon residues.41,42 Furthermore, a 13CO2 isotope tracing experiment (Fig. 3g and S8†) exhibits that the primary mass spectrum signal with a m/z value of 29 (13CO) is distinctly visible, demonstrating that the majority of the produced CO molecules come from the photocatalytic conversion of CO2.43,44 Additionally, the low peak with a m/z value of 28 derives from the decomposition of carbon residues on the catalyst surface during the isotopic experiment.
To explore the charge transfer dynamics, photoluminescence (PL) spectra, electrochemical and photoelectrochemical measurements have been performed for typical samples. Fig. S9a† presents the steady-state PL spectra of CdSe QDs, CdSe/CdS QDs and CdSe/CdS–SiO2 composites, in which CdSe QDs show a prominent fluorescence peak at around 624 nm (345 nm excitation). Clearly, the greatly quenched PL intensity observed in CdSe/CdS QDs suggests that the formation of the core/shell heterojunction effectively blocks the radiative recombination of electron–hole pairs in CdSe QDs. The immobilization of CdSe/CdS QDs onto spherical SiO2 leads to further quenching of the PL intensity due to an enhanced light–matter interaction promoted by the near-field scattering of the spherical SiO2.29–31 These conclusions are also in accordance with the time-resolved PL (TRPL) decay plots (Fig. 4a), where the CdSe/CdS–SiO2 composite (τa = 30.99 ns) displays a decreased fluorescence lifetime with reference to CdSe QDs (τa = 53.54 ns) (Table S1†). As illustrated in Fig. 4b, the CdSe/CdS–SiO2 composite presents a significantly smaller semicircle radius in electrochemical impedance spectroscopy (EIS) compared to CdSe/CdS QDs and CdSe QDs, suggesting a lower charge transfer resistance.45 Furthermore, the CdSe/CdS–SiO2 composite shows an enhanced photocurrent intensity when illuminated, confirming its superior charge transfer capability (Fig. 4c). The enhancement of current density in the cyclic voltammetry (CV) curve of the CdSe/CdS–SiO2 composite suggests more efficient interfacial charge transfer across the contact surface (Fig. S9b†). All these results imply the presence of effective interfacial charge transfer in the core/shell CdSe/CdS QD heterostructure, further diminishing electron–hole recombination by the attachment of CdSe/CdS QDs onto SiO2 spheres. Cathodic linear sweep voltammetry (LSV) tests have been performed in either Ar-saturated or CO2-saturated Na2SO4 solution without light. As depicted in Fig. 4d, there is no distinct difference in the onset overpotentials for H2 production among CdSe QDs, CdSe/CdS QDs and CdSe/CdS–SiO2 electrodes when operated in an Ar atmosphere. After introducing CO2 into the electrolyte solution, notable increases in reduction current are observed, indicating the occurrence of electrocatalytic CO2 reduction on the electrode surface. Notably, the CdSe/CdS–SiO2 electrode demonstrates a more positive overpotential and an enhanced cathodic current compared to CdSe/CdS QDs and CdSe QDs. This observation means that the formation of a core/shell QD heterostructure, followed by the decoration of CdSe/CdS QDs onto SiO2 spheres, gradually enhances the activation of CO2.41,46
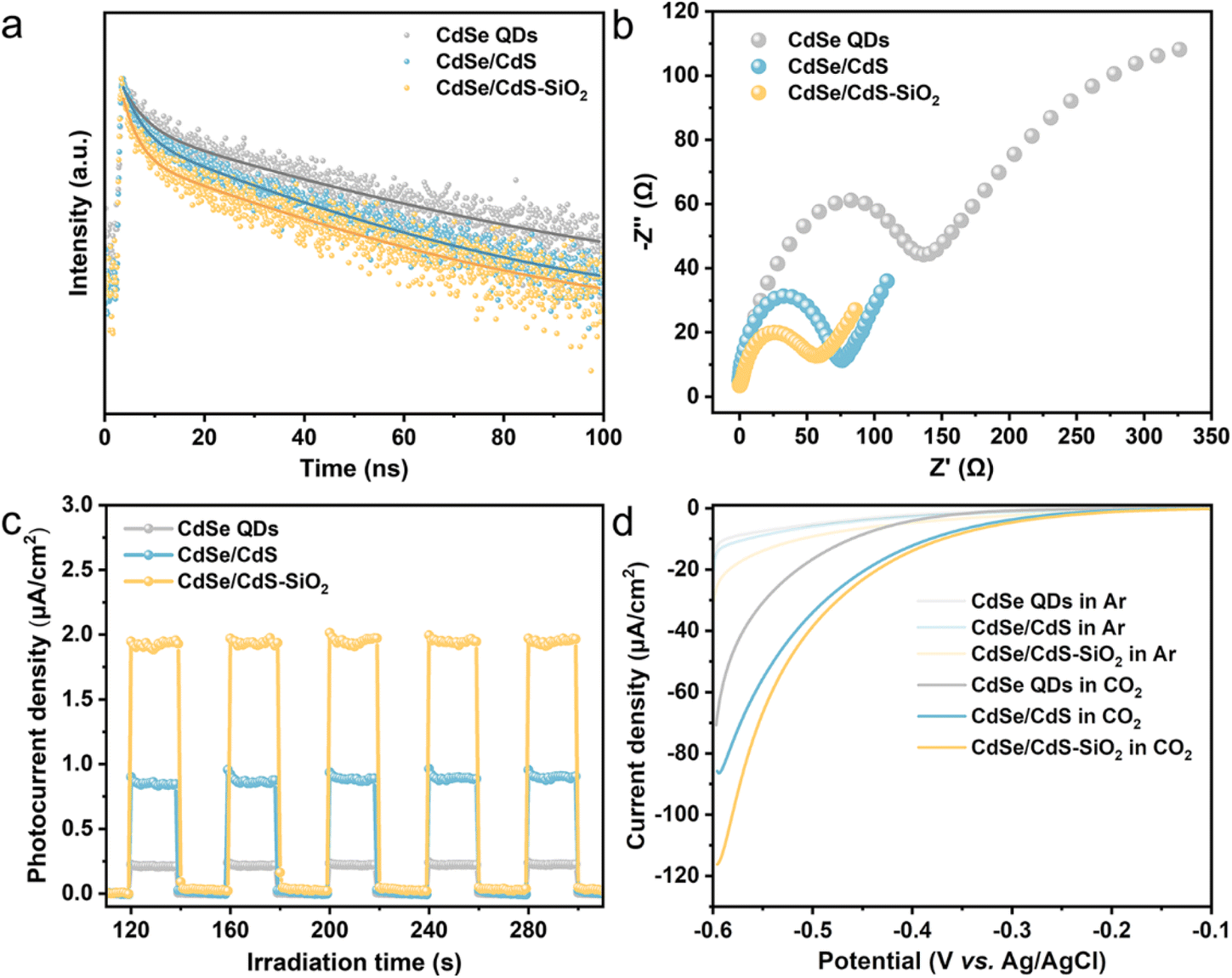 | ||
| Fig. 4 (a) TRPL decay plots, (b) EIS plots and (c) transient photocurrent spectra of CdSe QDs, CdSe/CdS QDs and CdSe/CdS–SiO2 composites. (d) LSV curves of CdSe QDs, CdSe/CdS QDs and CdSe/CdS–SiO2 composites under Ar or CO2 atmospheres. | ||
The CO2 adsorption/CO desorption performance has been evaluated by temperature-programmed-desorption (TPD). Two primary CO2 desorption peaks appeared at approximately 130 and 440 °C for both the CdSe–SiO2 composite and the CdSe/CdS–SiO2 composite (Fig. 5a and S10†). This illustrates that the QDs-SiO2 composite possesses a strong CO2 adsorption capacity, which is crucial for CO2 photoreduction.47 Besides, the CdSe/CdS–SiO2 composite manifests a decreased CO adsorption capacity compared to the CdSe–SiO2 composite, signifying an easier desorption of CO on the surface of the CdSe/CdS–SiO2 composite (Fig. 5b).48 Furthermore, electron paramagnetic resonance (EPR) spectroscopy has been performed to pinpoint the free radical intermediates that are involved in the furfuryl alcohol oxidation half-reaction. As displayed in Fig. 5c, six signal peaks with equal intensities have been detected under light irradiation. These signals, corresponding to nitrogen hyperfine splitting αN = 15.7 G and hydrogen hyperfine splitting αH = 22.0 G, can be associated with DMPO–CH(OH)C4H3O.49 Remarkably, the CdSe/CdS–SiO2 composite displays higher signal intensities, indicating that the CdSe/CdS–SiO2-catalyzed system is conducive to the generation of ˙CH(OH)C4H3O radicals in higher concentrations, subsequently driving the reaction process.
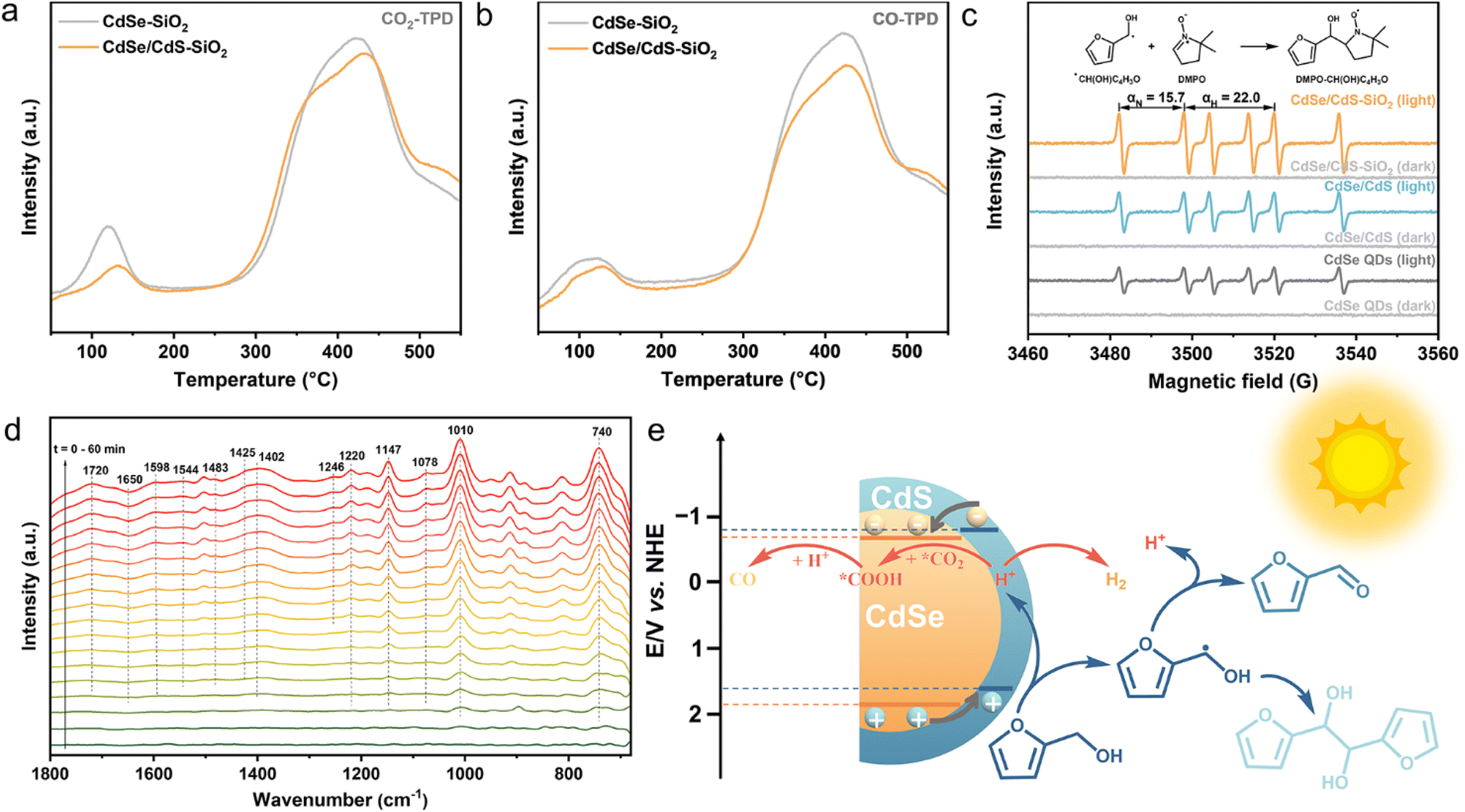 | ||
| Fig. 5 (a) CO2-TPD and (b) CO-TPD measurements of the CdSe–SiO2 composite and CdSe/CdS–SiO2 composite. (c) EPR spectra of CdSe QDs, CdSe/CdS QDs and CdSe/CdS–SiO2 suspensions in Ar-saturated furfuryl alcohol solution under light or dark conditions. (d) In situ FTIR spectra for CO2 photoreduction coupled with the oxidation of furfuryl alcohol over the CdSe/CdS–SiO2 composite. (e) Proposed mechanism for the cooperative photoredox reaction catalysed by CdSe/CdS–SiO2. | ||
In situ FTIR spectroscopy has been applied to characterize the adsorbed species and their intermediate products on the surface during such a dual-functional reaction. As shown in Fig. 5d, bidentate bicarbonates (b-HCO3− at 1650, 1425 and 1220 cm−1),48 bidentate carbonates (b-CO32− at 1598 and 1483 cm−1) and a monodentate carbonate (m-CO32− at 1402 cm−1) have been generated on the surface of the CdSe/CdS–SiO2 composite.50 CO2− (at 1246 cm−1) and COOH* (at 1544 cm−1) are also seen in this photocatalyzed-redox reaction process.51,52 These findings highlight the critical role played by the intermediate species CO2−, COOH*, HCO3− and CO32− in facilitating the conversion of CO2 to CO, following the sequential pathway of *CO2 → *COOH → *CO → CO.41,47 Moreover, the C–OH peaks of furfuryl alcohol/hydrofuroin appeared at 1078 and 1010 cm−1, and the characteristic β-ring bending vibration of furan rings exhibits a sharp intense peak at 740 cm−1. The adsorption peak observed at 1147 cm−1 is associated with the C–O–C bond of the furan ring. Meanwhile, a prominent peak at 1720 cm−1, which intensifies as the reaction time progresses, is attributed to furfural, indicating a dehydrogenation process of furfuryl alcohol to furfural.53,54
In light of the aforementioned results, a possible photocatalytic reaction mechanism for the transformation of CO2 combined with furfuryl alcohol oxidation over the CdSe/CdS–SiO2 composite can be described, as shown in Fig. 5e and S11.† The conduction band (CB) values of CdSe and CdS QDs are calculated to be −0.67 and −0.76 V vs. the normal hydrogen electrode (NHE), respectively. The valence band (VB) values are 1.87 and 1.66 V vs. NHE for CdSe and CdS QDs, respectively, resulting in a staggered band alignment. When exposed to light, the core/shell CdSe/CdS QDs exhibit a dual capability of absorbing incident light and recycling scattered light in the near field of SiO2 spheres, thus facilitating the generation of more electron–hole pairs.12,29–31 Holes located in the VB of CdSe are transferred to CdS following the alignment in the type-II band structure.21,55–57 Furfuryl alcohol reacts with holes, leading to the formation of carbon radicals (˙CH(OH)C4H3O), which are subsequently oxidized to furfural or undergo a C–C coupling reaction to produce hydrofuroin.7,18,58 On the other hand, electrons are transferred from the CB of CdS to CdSe QDs to participate in the CO2 reduction reaction.59,60 Specifically, the adsorbed CO2 molecules are hydrogenated with the assistance of protons extracted from the C–H bond of furfuryl alcohol to form COOH*, ultimately resulting in the generation of H2 and CO.
Conclusions
In summary, we have developed a photoredox reaction system for simultaneously converting CO2 to syngas and oxidizing biomass-derived furfuryl alcohol to furfural and hydrofuroin over CdSe/CdS–SiO2. In contrast to bare CdSe QDs, core/shell CdSe/CdS QDs with a type-II structure enable effective spatial separation of electrons and holes. By further assembly onto spherical SiO2 supports, the CdSe/CdS–SiO2 composite exhibits greatly enhanced dual-functional activity in the production of syngas and furfural/hydrofuroin, which is 90.4 and 18.5 times higher than those of bare CdSe QDs, respectively. It is noteworthy that the syngas CO/H2 ratio that can be precisely adjusted within a wide range of 1.6 to 7.1 by altering the CdS shell thickness is crucial for the production of liquid fuels from non-petroleum carbon resources. In situ Fourier transform infrared (FTIR) spectroscopy reveals crucial intermediates such as CO2−, COOH*, HCO3− and CO32− involved in promoting photoreduction of CO2. We believe that this work will present an approach that utilizes core/shell QD-based heterostructured photocatalysts to rationally design cooperative reaction systems, enabling efficient CO2 valorization and biomass transformation.Experimental
Materials
All chemicals were of analytical quality and could be used without additional treatment. Sodium sulfite anhydrous (Na2SO3), cadmium chloride hemi(pentahydrate) (CdCl2·2.5H2O), absolute ethanol (C2H5OH), sodium hydroxide (NaOH), methanol anhydrous (CH3OH), acetonitrile (CH3CN), hydrochloric acid (HCl), ethyl acetate (C4H8O2), carbon tetrachloride (CCl4), ammonium hydroxide (NH3·H2O), triethanolamine (C6H15NO3, TEOA) and N,N-dimethylformamide (C3H7NO, DMF) were obtained from Sinopharm Chemical Reagent Co., Ltd. Cadmium perchlorate hexahydrate (Cd(ClO4)2·6H2O), 3-mercaptopropionic acid (C3H6O2S, MPA), isopropyl alcohol (C3H8O), sodium sulfide nonahydrate (Na2S·9H2O), tetramethylammonium hydroxide pentahydrate (C4H13NO, TMAOH), 5, 5-dimethyl-1-pyrroline N-oxide (C6H11NO, DMPO), branched poly-ethylenimine (Mw = 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, BPEI), furfuryl alcohol (C5H6O2), selenium powder (Se, 200 mesh) and tetraethyl orthosilicate (C8H20O4Si, TEOS) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. The deionized (DI) water used in our experiments was sourced locally.
000, BPEI), furfuryl alcohol (C5H6O2), selenium powder (Se, 200 mesh) and tetraethyl orthosilicate (C8H20O4Si, TEOS) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. The deionized (DI) water used in our experiments was sourced locally.
Synthesis of CdSe quantum dots (QDs)
Typically, 400 mg of Na2SO3 and 79 mg of Se powder were introduced in 200 mL of DI water. Next, the mixture was refluxed at a temperature of 120 °C until Se powder completely dissolved, resulting in a clear solution of Na2SeSO3. Subsequently, CdCl2·2.5H2O aqueous solution (0.01 mol L−1, 40 mL) was mixed with 60 μL of MPA, followed by the adjustment of the pH to about 11 with NaOH solution. The resulting solution was deaerated with argon (Ar) bubbling for 30 min. Afterwards, 20 mL of the previously prepared Na2SeSO3 solution was added to this mixture solution and refluxed at 120 °C for 1 h. In the end, the as-synthesized CdSe QDs were separated by precipitation with ethanol and stored in DI water.Synthesis of CdS QDs
Briefly, a solution of CdCl2·2.5H2O (0.05 mol L−1) in 20 mL of DI water was mixed with 148 μL of MPA. After adjusting the pH to around 11 with a NaOH solution, the resulting solution was purged with Ar for 30 min. Following this, 5 mL of Na2S·9H2O solution (0.2 mol L−1) was quickly injected into the mixture solution. The mixture was then heated to a temperature of 100 °C and stirred for 0.5 h to boost the growth of QDs. After the solution cooled down, ethanol was introduced to isolate the product, which was subsequently stored in DI water.Synthesis and ligand exchange of CdSe/xCdS QDs
A protocol was followed to synthesize CdSe/xCdS QDs, where x represents approximately x atomic layers of the CdS shell.26,61 Initially, 50 mg of CdSe QDs were dispersed in 40 mL DI water degassed with Ar for 0.5 h, and then the temperature was raised to 50 °C with stirring. Subsequently, Na2S·9H2O (5 mL × x, 5 × 10−3 mol L−1) and Cd(ClO4)2·6H2O (4 mL × (x − 1), 5 × 10−3 mol L−1) aqueous solutions were pumped into the flask at rates of 5 mL/20 min and 4 mL/15 min, respectively. After adding these precursors, the mixture was allowed to react for another 1 h to boost the growth of CdSe/xCdS QDs. The sulfide ions were believed to stabilize the CdSe/CdS QDs in the absence of additional ligands. To acquire MPA-capped QDs, the sulfide stabilized CdSe/CdS QDs were precipitated by the addition of HCl (0.1 mol L−1) and washed three times with DI water. MPA (100 μL) was dissolved in 20 mL of methanol, and the pH of the solution was maintained at around 11 with TMAOH. Then, 20 mg of CdSe/CdS precipitate was added to the solution and refluxed at 65 °C overnight. Finally, the resulting QDs were washed three times with ethyl acetate and redispersed in DI water for future use.Synthesis and functionalization of SiO2 spheres
Typically, a mixture consisting of 200 mL of isopropyl alcohol, 5 mL of DI water and 10 mL of NH3·H2O was prepared. Subsequently, 10 mL of TEOS was added to the above solution, and the reaction progressed for a duration of 4 h. After isolation, the colloidal SiO2 was then rinsed repeatedly with ethanol and DI water, followed by drying at 60 °C. To introduce positive charges on the surface of SiO2 spheres, a dispersion of 400 mg of these spheres in 200 mL of ethanol was achieved through sonication treatment. Afterwards, 344 mg of BPEI was added, and the reaction mixture was heated under reflux at 60 °C for 4 h to ensure complete modification of the SiO2 surfaces. The BPEI-functionalized SiO2 spheres were then rinsed with ethanol and DI water before being dried overnight at 60 °C.Synthesis of the CdSe/CdS–SiO2 composite
200 mL of DI water was utilized to disperse 0.2 g of BPEI-SiO2 spheres, followed by the addition of a certain concentration of CdSe/CdS QD solution to the dispersion. The resulting mixture was refluxed at 60 °C for 4 h and then washed with DI water three times and dried at 60 °C. The weight content of CdSe/CdS QDs in the obtained CdSe/CdS–SiO2 composite was 2.5%, which was calculated based on the total weight of the sample. The synthesis method of CdSe–SiO2 and CdS–SiO2 composites was similar to that of the CdSe/CdS–SiO2 composite, except that CdSe QDs or CdS QDs were used instead of CdSe/CdS QDs.Photocatalytic performance testing
Photocatalytic CO2 reduction integrated with the oxidation of furfuryl alcohol reaction was executed in a 40 mL sealed double-walled quartz reactor. A typical experiment involved the addition of 20 mg of the catalyst (2.5% CdSe/CdS–SiO2) to 10 mL of CH3CN containing 100 μmol of furfuryl alcohol. Prior to illumination, the solution was bubbled with CO2 for 20 min. Utilizing a 300 W Xe arc lamp (PLS-SME300E H1, Beijing Perfectlight Technology Co., Ltd), the reaction was then initiated by exposing the solution to UV-vis light (300 nm ≤ λ ≤ 800 nm) for 4 h at ambient temperature. Employing a PL-MW2000 photoradiometer from Beijing Perfectlight Technology Co., Ltd, the incident light intensity was determined to be 500 mW cm−2. The production of H2 and CO was quantitatively analyzed via a gas chromatograph (Shimadzu GC-2014C, equipped with a 5A column and an Ar carrier gas). Oxidized liquid products were characterized by gas chromatography-mass spectroscopy (GC-MS; Shimadzu GC-MS QP 2020, interfaced with a Q-Exactive mass spectrometer).Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Lin-Xing Zhang: investigation, validation, writing-original draft, formal analysis, and writing-review & editing. Zi-Rong Tang: funding acquisition, formal analysis, supervision, writing-review & editing, and resources. Ming-Yu Qi: funding acquisition, conceptualization, resources, project administration, writing-review & editing, and supervision. Yi-Jun Xu: funding acquisition, conceptualization, resources, project administration, writing-review & editing, and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22172030, 22072023, 21872029, U1463204, and 21173045), the Program for Leading Talents of Fujian Universities, the Program for National Science and Technology Innovation Leading Talents (00387072), the First Program of Fujian Province for Top Creative Young Talents, the China Postdoctoral Science Foundation (2023M740513), the China National Postdoctoral Program for Innovative Talents (BX20240055), the Jiangxi Province “Double Thousand Plan” (No. jxsq2023102143) and the Natural Science Foundation of Fujian Province (2017J07002 and 2019J01631).References
- B. Zhao, M. Sun, F. Chen, Y. Shi, Y. Yu, X. Li and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 4496–4500 CrossRef CAS PubMed
.
- Y. Li, D. Hui, Y. Sun, Y. Wang, Z. Wu, C. Wang and J. Zhao, Nat. Commun., 2021, 12, 123 CrossRef CAS PubMed
- J. Li, W. Pan, Q. Liu, Z. Chen, Z. Chen, X. Feng and H. Chen, J. Am. Chem. Soc., 2021, 143, 6551–6559 CrossRef CAS PubMed
- G. Chen, R. Gao, Y. Zhao, Z. Li, G. I. N. Waterhouse, R. Shi, J. Zhao, M. Zhang, L. Shang, G. Sheng, X. Zhang, X. Wen, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2018, 30, 1704663 CrossRef PubMed
- X. Zhang, Y. Liu, M. Zhang, T. Yu, B. Chen, Y. Xu, M. Crocker, X. Zhu, Y. Zhu, R. Wang, D. Xiao, M. Bi, D. Ma and C. Shi, Chem, 2020, 6, 3312–3328 CAS
- C. Han, Y.-H. Li, J.-Y. Li, M.-Y. Qi, Z.-R. Tang and Y.-J. Xu, Angew. Chem., Int. Ed., 2021, 60, 7962–7970 CrossRef CAS PubMed
- X. Wu, J. Li, S. Xie, P. Duan, H. Zhang, J. Feng, Q. Zhang, J. Cheng and Y. Wang, Chem, 2020, 6, 3038–3053 CAS
- W. J. Liu, W. W. Li, H. Jiang and H. Q. Yu, Chem. Rev., 2017, 117, 6367–6398 CrossRef CAS PubMed
- Q. Lin, Y.-H. Li, Z.-R. Tang and Y.-J. Xu, Trans. Tianjin Univ., 2020, 26, 325–340 CrossRef CAS
- S. Pang, Biotechnol. Adv., 2019, 37, 589–597 CrossRef CAS PubMed
- L. Yuan, M.-Y. Qi, Z.-R. Tang and Y.-J. Xu, Angew. Chem., Int. Ed., 2021, 60, 21150–21172 CrossRef CAS PubMed
- M.-Y. Qi, Y.-H. Li, M. Anpo, Z.-R. Tang and Y.-J. Xu, ACS Catal., 2020, 10, 14327–14335 CrossRef CAS
- G. Huang, L. Lu, H. Jiang and B. Yin, Chem. Commun., 2017, 53, 12217–12220 RSC
- Y. Guo, B. Liu, J. Zhang, G. Wang, C. Pan, H. Zhao, C. Wang, F. Yu, Y. Dong and Y. Zhu, Appl. Catal., B, 2024, 340, 123217 CrossRef CAS
- Z. Yang, X. Xia, M. Fang, L. Wang and Y. Liu, Chem. Eng. J., 2023, 476, 146544 CrossRef CAS
- M.-Y. Qi, M. Conte, M. Anpo, Z.-R. Tang and Y.-J. Xu, Chem. Rev., 2021, 121, 13051–13085 CrossRef CAS PubMed
- H.-L. Wu, M.-Y. Qi, Z.-R. Tang and Y.-J. Xu, J. Mater. Chem. A, 2023, 11, 3262–3280 RSC
- Z.-K. Xin, M.-Y. Huang, Y. Wang, Y.-J. Gao, Q. Guo, X.-B. Li, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2022, 61, e202207222 CrossRef CAS PubMed
- Z. W. Xi, L. Yang, D. Y. Wang, C. D. Pu, Y. M. Shen, C. D. Wu and X. G. Peng, J. Org. Chem., 2018, 83, 11886–11895 CrossRef CAS PubMed
- X.-B. Li, Z.-J. Li, Y.-J. Gao, Q.-Y. Meng, S. Yu, R. G. Weiss, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2014, 53, 2085–2089 CrossRef CAS PubMed
- Q. Guo, F. Liang, X.-B. Li, Y.-J. Gao, M.-Y. Huang, Y. Wang, S.-G. Xia, X.-Y. Gao, Q.-C. Gan, Z.-S. Lin, C.-H. Tung and L.-Z. Wu, Chem, 2019, 5, 2605–2616 CAS
- H.-L. Wu, X.-B. Li, C.-H. Tung and L.-Z. Wu, Adv. Mater., 2019, 31, 1900709 CrossRef PubMed
- N. Jin, Y. Sun, W. Shi, P. Wang, Y. Nagaoka, T. Cai, R. Wu, L. Dube, H. N. Nyiera, Y. Liu, T. Mani, X. Wang, J. Zhao and O. Chen, J. Am. Chem. Soc., 2023, 145, 21886–21896 CrossRef CAS PubMed
- X.-B. Li, C.-H. Tung and L.-Z. Wu, Nat. Rev. Chem, 2018, 2, 160–173 CrossRef CAS
- X. B. Fan, S. Yu, B. Hou and J. M. Kim, Isr. J. Chem., 2019, 59, 762–773 CrossRef CAS
- X.-B. Li, Y.-J. Gao, Y. Wang, F. Zhan, X.-Y. Zhang, Q.-Y. Kong, N.-J. Zhao, Q. Guo, H.-L. Wu, Z.-J. Li, Y. Tao, J.-P. Zhang, B. Chen, C.-H. Tung and L.-Z. Wu, J. Am. Chem. Soc., 2017, 139, 4789–4796 CrossRef CAS PubMed
- G. Liu, W. Liang, X. Xue, F. Rosei and Y. Wang, Adv. Sci., 2021, 8, 2102784 CrossRef PubMed
- A. C. A. Silva, E. S. F. Neto, S. W. da Silva, P. C. Morais and N. O. Dantas, J. Phys. Chem. C, 2013, 117, 1904–1914 CrossRef CAS
- N. Zhang, C. Han, Y.-J. Xu, J. J. Foley Iv, D. Zhang, J. Codrington, S. K. Gray and Y. Sun, Nat. Photonics, 2016, 10, 473–482 CrossRef CAS
- C. Han, S.-H. Li, Z.-R. Tang and Y.-J. Xu, Chem. Sci., 2018, 9, 8914–8922 RSC
- M.-Y. Qi, Z.-R. Tang and Y.-J. Xu, ACS Catal., 2023, 13, 3971–3982 CrossRef CAS
- M. Yang, Y. Wang, Y. Ren, E. Liu, J. Fan and X. Hu, J. Alloys Compd., 2018, 752, 260–266 CrossRef CAS
- A. P. Rangappa, D. Praveen Kumar, Y. Hong, S. Jeong, D. A. Reddy, J. K. Song and T. K. Kim, ACS Appl. Energy Mater., 2020, 3, 10533–10540 CrossRef CAS
- L.-X. Zhang, M.-Y. Qi, Z.-R. Tang and Y.-J. Xu, Research, 2023, 6, 0073 CrossRef CAS PubMed
- J. Wang, T. Xia, L. Wang, X. Zheng, Z. Qi, C. Gao, J. Zhu, Z. Li, H. Xu and Y. Xiong, Angew. Chem., Int. Ed., 2018, 57, 16447–16451 CrossRef CAS PubMed
- K. H. Do, D. P. Kumar, A. P. Rangappa, Y. Hong, D. A. Reddy and T. K. Kim, ChemCatChem, 2020, 12, 4550–4557 CrossRef CAS
- Y. Jiang, H. Y. Chen, J. Y. Li, J. F. Liao, H. H. Zhang, X. D. Wang and D. B. Kuang, Adv. Funct. Mater., 2020, 30, 2004293 CrossRef CAS
- J. Wu, T. Wang, L. Zhang and P. Du, ACS Sustainable Chem. Eng., 2024, 12, 1625–1631 CrossRef CAS
- M.-Y. Qi, Q. Lin, Z.-R. Tang and Y.-J. Xu, Appl. Catal., B, 2022, 307, 121158 CrossRef CAS
- J.-H. Zheng, M.-Y. Qi, Z.-R. Tang and Y.-J. Xu, J. Mater. Chem. A, 2023, 11, 4013–4019 RSC
- M.-Y. Qi and Y.-J. Xu, Angew. Chem., Int. Ed., 2023, 62, e202311731 CrossRef CAS PubMed
- L. Yuan, Y.-H. Li, M.-Y. Qi, Z.-R. Tang and Y.-J. Xu, J. Catal., 2020, 390, 244–250 CrossRef CAS
- L. Wang, X. Zhao, D. Lv, C. Liu, W. Lai, C. Sun, Z. Su, X. Xu, W. Hao, S. X. Dou and Y. Du, Adv. Mater., 2020, 32, e2004311 CrossRef PubMed
- S. Wang, B. Jiang, J. Henzie, F. Xu, C. Liu, X. Meng, S. Zou, H. Song, Y. Pan, H. Li, J. Yu, H. Chen and J. Ye, Nat. Commun., 2023, 14, 2534 CrossRef CAS PubMed
- N. Zhang, S. Xie, B. Weng and Y.-J. Xu, J. Mater. Chem. A, 2016, 4, 18804–18814 RSC
- S. Cao, B. Shen, T. Tong, J. Fu and J. Yu, Adv. Funct. Mater., 2018, 28, 1800136 CrossRef
- C. Feng, T. Bo, P. Maity, S. Zuo, W. Zhou, K. W. Huang, O. F. Mohammed and H. Zhang, Adv. Funct. Mater., 2024, 34, 2309761 CrossRef CAS
- X. Xiong, C. Mao, Z. Yang, Q. Zhang, G. I. N. Waterhouse, L. Gu and T. Zhang, Adv. Energy Mater., 2020, 10, 2002928 CrossRef CAS
- C.-L. Tan, M.-Y. Qi, Z.-R. Tang and Y.-J. Xu, Appl. Catal., B, 2021, 298, 120541 CrossRef CAS
- Y. Liu, D. Shen, Q. Zhang, Y. Lin and F. Peng, Appl. Catal., B, 2021, 283, 119630 CrossRef CAS
- Y. Yu, X. a. Dong, P. Chen, Q. Geng, H. Wang, J. Li, Y. Zhou and F. Dong, ACS Nano, 2021, 15, 14453–14464 CrossRef CAS PubMed
- Z.-K. Xin, Y.-J. Gao, Y. Gao, H.-W. Song, J. Zhao, F. Fan, A.-D. Xia, X.-B. Li, C.-H. Tung and L.-Z. Wu, Adv. Mater., 2022, 34, 2106662 CrossRef CAS PubMed
- S. Li, Y. Fan, C. Wu, C. Zhuang, Y. Wang, X. Li, J. Zhao and Z. Zheng, ACS Appl. Mater. Interfaces, 2021, 13, 8507–8517 CrossRef CAS PubMed
- B. He, Z. Wang, P. Xiao, T. Chen, J. Yu and L. Zhang, Adv. Mater., 2022, 34, 2203225 CrossRef CAS PubMed
- S. Shenoy and K. Tarafder, J. Phys.: Condens. Matter, 2020, 32, 275501 CrossRef CAS PubMed
- J. Low, J. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Adv. Mater., 2017, 29, 1601694 CrossRef PubMed
- Y. Nandan and M. S. Mehata, Sci. Rep., 2019, 9, 2 CrossRef PubMed
- L. Wang, Z. Huang, S. Xie, Q. Zhang, H. Wang and Y. Wang, Catal. Commun., 2021, 153, 106300 CrossRef CAS
- H. Cai, B. Wang, L. Xiong, J. Bi, L. Yuan, G. Yang and S. Yang, Appl. Catal., B, 2019, 256, 117853 CrossRef CAS
- M. Ou, W. Tu, S. Yin, W. Xing, S. Wu, H. Wang, S. Wan, Q. Zhong and R. Xu, Angew. Chem., Int. Ed., 2018, 57, 13570–13574 CrossRef CAS PubMed
- Z.-J. Li, X.-B. Fan, X.-B. Li, J.-X. Li, F. Zhan, Y. Tao, X. Zhang, Q.-Y. Kong, N.-J. Zhao, J.-P. Zhang, C. Ye, Y.-J. Gao, X.-Z. Wang, Q.-Y. Meng, K. Feng, B. Chen, C.-H. Tung and L.-Z. Wu, J. Mater. Chem. A, 2017, 5, 10365–10373 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta01996b |
| This journal is © The Royal Society of Chemistry 2024 |