
Low-coordinated Co–Mn diatomic sites derived from metal–organic framework nanorods promote electrocatalytic CO2 reduction†
Jiajing
Pei‡
ab,
Guikai
Zhang‡
ac,
Jiangwen
Liao
ac,
Shufang
Ji
d,
Huan
Huang
a,
Ping
Wang
b,
Pengfei
An
*a,
Shengqi
Chu
*a and
Juncai
Dong
 *ac
*ac
aBeijing Synchrotron Radiation Facility, Institute of High Energy Physics, Chinese Academy of Science, Beijing 100049, China. E-mail: anpf@ihep.ac.cn; chusq@ihep.ac.cn; dongjc@ihep.ac.cn
bSpallation Neutron Source Science Center, Dongguan 523803, China
cUniversity of Chinese Academy of Sciences, Beijing 100049, People's Republic of China
dDepartment of Chemistry, University of Toronto, Toronto, Ontario M5S3H6, Canada
First published on 20th May 2024
Abstract
The manipulation of the geometric coordination structure of diatomic site (DS) catalysts to promote the CO2-to-CO conversion process has garnered significant attention. However, the challenge remains in how to rationally design the catalytic microenvironment of DSs to enhance the kinetics of CO product formation. Herein, we present a post-synthetic co-substitution (PSCR) method for producing Co–Mn DS catalysts with low N coordination numbers (referred to as L-Co1Mn1-NC) on pre-designed N-doped carbon derived from metal–organic framework nanorods. When utilized in the process of CO2 electroreduction, the L-Co1Mn1-NC catalyst demonstrates a CO faradaic efficiency (FE) of up to 97.6% at −0.47 V, which is significantly higher compared to those of Co1Mn1-NC, Co1-NC, and Mn1-NC catalysts. In situ ATR-SEIRAS and theoretical simulations demonstrate that the creation of Co–Mn DSs with a lower N coordination number can notably facilitate the desorption of CO*, thus expediting the kinetics of the CO2-to-CO conversion process. This study introduces a novel approach to fine-tune the catalytic microenvironment of multi-atomic sites in order to facilitate the conversion and utilization of CO2.
 Juncai Dong | Juncai Dong serves as a senior scientist at the Beijing Synchrotron Radiation Facility (BSRF) in China. He earned his PhD in condensed matter physics in 2014 from the University of Chinese Academy of Sciences. Afterwards, Dong underwent specialized training as a beamline scientist at BSRF, where he honed his expertise in utilizing X-ray spectroscopy techniques to delve into the intricate relationship between structure and functionality in the fields of condensed matter physics and materials science. Dong's current research focuses on applying in situ/operando synchrotron-radiation-based X-ray spectroscopies to uncover the dynamic processes and fundamental physicochemical mechanisms of various electrocatalytic systems under real-time operational conditions. |
Introduction
The electrochemical reduction of CO2 to produce high-value fuels and fine chemicals (CO2RR), powered by electricity generated from renewable clean energy sources, holds significant importance for mitigating environmental issues and achieving global carbon neutrality.1,2 However, challenges persist in the CO2RR process, such as the intrinsic CO2 inertness, complex reaction environments, difficulties in controlling the surface interface, and competitive hydrogen evolution reaction (HER).3,4 The key to addressing these challenges lies in the development of highly efficient electrocatalysts with superior catalytic activity, stability, and targeted control over product selectivity.5,6 In recent years, single-atom catalysts (SACs), typically atomically dispersed metal–nitrogen–carbon (M–N–C) materials, have sparked a research boom in the CO2RR process.7–10 This is due to their unique chemical environment properties, and they are anticipated to facilitate the large-scale development of electrochemical CO2-to-CO conversion.11–13 Generally, in M–N–C materials, the coordination-saturated M–N4 moiety is a common structure that determines the activity of CO2 reduction.14 Although considerable progress has been made in the CO2RR application of SACs with the M–N4 moiety,15,16 the highly symmetrical M–N4 configuration actually restrains the modulation of the electronic structure of the metal center, thereby limiting its potential applications.Inspired by the fact that single-atom metal centers are stabilized on the carrier by coordination interactions with anchoring nodes based on coordination chemistry, the catalytic activity of SACs is closely related to their catalytic coordination microenvironment.17,18 The coordination number of the first shell is a significant indicator that influences the local electronic properties of the catalytic microenvironment in SACs, and can be considered a key factor determining catalytic activity and selectivity.19,20 Specifically, altering the coordination number of neighboring N atoms around the metal center can lead to more defects and accumulation of more negative charges in the support.21,22 This, in turn, induces differentiation in the local electron density of active sites, enhancing the adsorption mode and capacity of intermediates, ultimately influencing the CO2RR performance. For instance, a series of Co SACs featuring Co-Nx (where x = 4, 3, 2) active sites synthesized at temperatures of 800, 900, and 1000 °C, respectively, were employed to explore the correlation between the Co-Nx sites and the catalytic performance in the CO2-to-CO conversion process.23 The findings indicated that a modulation of the Co–N coordination number results in an augmentation of the unoccupied 3d orbitals of the Co center, and such an alteration is conducive to the adsorption of the CO2− intermediate, thus enhancing the CO2RR performance. Likewise, Ni SACs known as NiSA-Nx-C derived from metal–organic frameworks with alterable N coordination numbers were developed through precise regulation of the pyrolysis temperature.24 Interestingly, it had been observed that NiSA-N2-C catalysts with the lowest N coordination number exhibited the highest selectivity and activity for CO generation. Consequently, there is growing interest in modifying the coordination environment to enable the directed synthesis of SACs with varying coordination numbers, in order to enhance the conversion of CO2 into CO.25,26 In addition, the approach of incorporating a homo/hetero-metal atom to form a diatomic site (DS) has been demonstrated to adjust the electronic configuration of individual metal catalytic sites dispersed within the carbon matrix.27–29 The DS catalyst not only retains the same characteristics as SACs, but also exhibits synergistic interactions, altering the chemical surroundings to match the complex reaction mechanism in the CO2 conversion process, thereby enhancing catalytic efficiency.30,31 An atom-pair catalyst composed of stable Cu10–Cu1x+ pair structures was created, and the Cu10 site bound one CO2 molecule, while the Cu1x+ site absorbed one H2O molecule to boost the activation of CO2.32 Despite some research progress in the construction of DSs, to the best of our knowledge, there has been limited attention given to the precise regulation of their low-coordination microenvironment at the atomic level. Furthermore, manipulating the catalytic microenvironment through the low-coordination effect has the potential to enhance the intrinsic activity of DSs, thereby improving the kinetics of CO production, but it remains challenging.
In this study, an approach of post-synthetic co-substitution (PSCR) has been proposed to achieve precise control of Co–Mn DS catalysts in a low-coordination microenvironment on a N-doped carbon host (L-Co1Mn1-NC) derived from nanorod-shaped zeolitic imidazolate framework-8 (ZIF-8). In the first stage of pyrolysis, the Zn nodes and ligand within ZIF-8 undergo gradual degradation, resulting in the formation of vacancy defects. Subsequently, the pre-evaporated Zn species are rapidly removed at elevated temperatures, leading to the creation of numerous Zn vacancies surrounded by varying N atoms for the co-infiltration of the Co/Mn DS. At a temperature of 1000 °C, the Co/Mn DS exhibits simultaneous occupation of the Zn vacancy with a 3 N atom coordination, resulting in the creation of a low-coordinated Co/Mn DS. Conversely, at 900 °C, the Co–Mn DS is filled with Zn vacancies with a 4 N atom coordination. The L-Co1Mn1-NC DS catalyst exhibits the highest CO faradaic efficiency (FECO) reaching 97.6%, along with a turnover frequency (TOF) of 1067 h−1 at −0.47 V during the CO2RR, and its CO selectivity and activity exceed the performance of Co1Mn1-NC, Co1-NC, and Mn1-NC catalysts. In situ ATR-SEIRAS reveals that the low coordination microenvironment endows the Co/Mn DS with the ability to produce CO at a low potential. Moreover, theoretical calculations demonstrate that the presence of low-coordinated Co–Mn active sites in the L-Co1Mn1-NC catalyst significantly decreases the adsorption energy of the CO* intermediate, leading to an expedited CO2 reduction process.
Results and discussion
The low-coordinated structure of the L-Co1Mn1-NC DS catalyst was rationally modulated by a PSCR strategy, as illustrated in Fig. 1a. Typically, the ZIF-8 substance was chosen as the supporting structure for the metallic active site.33 The as-prepared ZIF-8 has a uniform nanorod-like morphology, which is different from the conventional dodecahedron morphology, as demonstrated by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images (Fig. S1†). By encapsulating with a SiO2 layer, the initial calcination step not only promotes the distortion and disruption of the Zn-Nx structure to form pre-evaporated Zn species, but also effectively preserves the nanorod-like structure of ZIF-8 for subsequent stages. During secondary pyrolysis conducted at 1000 °C, the pre-evaporated Zn species are rapidly eliminated, while simultaneously, a partial loss of N element occurs within the imidazole ligands, resulting in the formation of Zn vacancies and N defects for the co-substitution and insertion of Co–Mn metal sites. When the pyrolysis is performed at 900 °C, both saturated coordination Co–Mn bimetallic (Co1Mn1-NC) and individual Co and Mn monometallic (Co1-NC and Mn1-NC) catalysts can be synthesized. The L-Co1Mn1-NC DS catalyst featuring a low-coordinated N structure can be synthesized. Both TEM and SEM as well as high-angle annular dark-field scanning TEM (HAADF-STEM) images show that the L-Co1Mn1-NC DS catalyst inherits the nanorod-like morphology of ZIF-8, but the surface is rougher and the spatial structure distribution is more open (Fig. 1b and c and S2†). This facilitates the desorption separation of CO intermediates and gas products. Significantly, the absence of metallic Co, Mn, or CoMn is evident in these findings of L-Co1Mn1-NC, indicating the potential atomic dispersion of Co and Mn components. Fig. 1d displays the energy-dispersive X-ray spectroscopy (EDS) mapping of the L-Co1Mn1-NC DS catalyst, illustrating a uniform dispersion of Co, Mn, N, and C atoms within the nanorod-shaped carbon host. To further confirm the distribution of metal Co/Mn species, aberration-corrected HAADF-STEM (AC HAADF-STEM) was adopted. As exhibited in Fig. 1e, the area observed in the AC HAADF-STEM image chosen at random indicate the presence of either Co or Mn species, confirming their dispersion at the atomic level. And the selected area electron diffraction (SAED) pattern analysis of the L-Co1Mn1-NC DS catalyst, as presented in Fig. S3,† reveals the presence of amorphous carbon structure rings exclusively, with no observable diffraction spots corresponding to metal nanoparticles. Following this, a 3D topographic atom imaging analysis was utilized to examine the current distribution of the Co/Mn DS pair based on the blue ellipse in Fig. 1f. The observed hump pair, observed by a minor strength variation, is likely assigned to the Co/Mn DS pair, as indicated by the discrepancy in atomic number. The spacing between the Co/Mn DS pair is measured at 0.25 nm, slightly exceeding the theoretical spacing for a direct Co–Mn connection. In order to minimize the uncertainty attributed to human activities, more than 100 distinct locations (specifically 150 groups of metal sites) were chosen for statistical analyses based on different AC HAADF-STEM images. These statistical analyses include measurements of atomic site density and distance distribution (Fig. 1g and S4†). The findings indicate that the L-Co1Mn1-NC catalyst primarily exists in the form of Co/Mn DS pairs, with a frequency above 67%, consistent with previous literature on dual-atom structures,34,35 and the atomic spacing between Co/Mn pairs is concentrated in the range of 0.24 to 0.26 nm. The metal quantification analysis of the L-Co1Mn1-NC is specifically presented in Table S1† using the inductively coupled plasma optical emission spectroscopy (ICP-OES) method. In short, the L-Co1Mn1-NC catalyst featuring rich Co/Mn pairs undergoes oriented synthesis by the PSCR method. | ||
| Fig. 1 The morphology and structure characterizations of the L-Co1Mn1-NC catalyst. (a) Illustration depicting the PSCR approach utilized in the production of L-Co1Mn1-NC catalysts for the CO2RR. (b) TEM image. (c) HAADF-STEM image. (d) EDS mapping (C in red, N in green, Co in blue, Mn in yellow). (e) AC HAADF-STEM image. (f) 3D map of Co/Mn DSs and intensity spacing diagram. (g) The statistical results of atomic site density and distance distribution. | ||
The X-ray diffraction (XRD) patterns of L-Co1Mn1-NC, Co1Mn1-NC, Co1-NC, and Mn1-NC catalysts exhibit two broad peaks attributed to the (002) and (101) planes of the carbon matrix, with no discernible crystalline phases containing any metal observed (Fig. S5†). This observation aligns with the findings from the aforementioned electron microscopy.
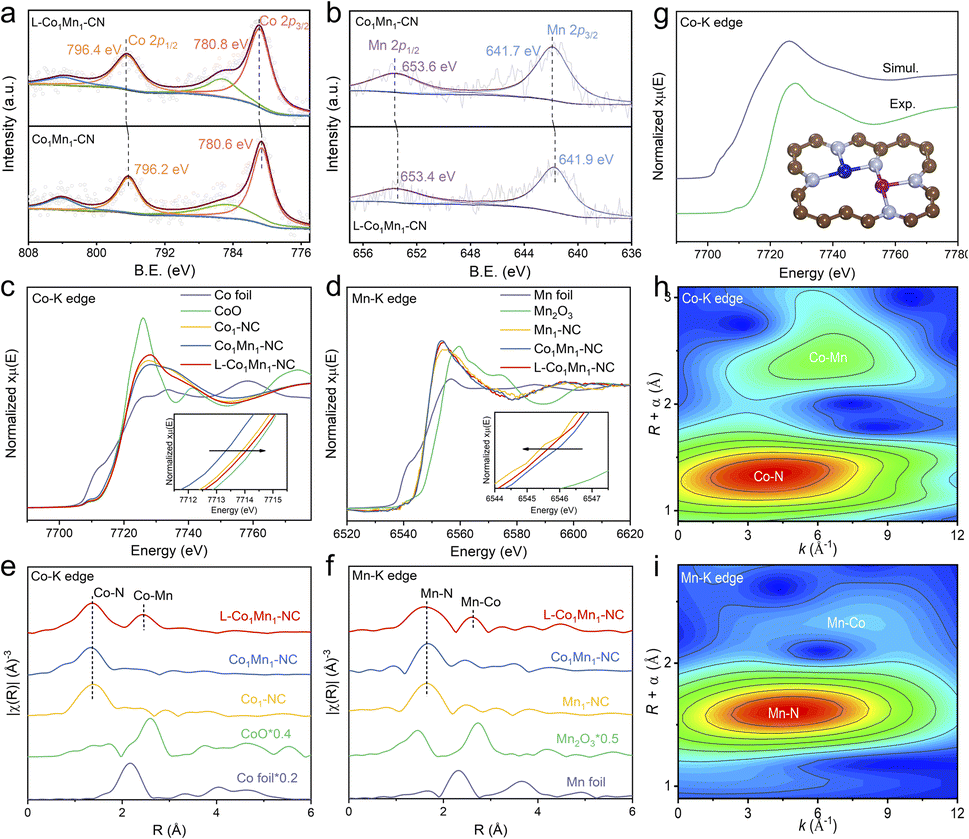
The Raman spectra analysis of the four atomic catalysts reveals that the L-Co1Mn1-NC DS catalyst exhibits a greater intensity ratio between the D band and G band, suggesting a higher propensity for vacancy defect formation (Fig. S6†). To further explain the influence of the low coordination environment, an examination of the chemical composition and electronic state of L-Co1Mn1-NC and Co1Mn1-NC was conducted through X-ray photoelectron spectroscopy (XPS). In the N 1s XPS spectra (Fig. S7†), there is a noticeable shift in the distribution of metallic N (399.5 eV) and graphitic N (401.2 eV) in L-Co1Mn1-NC compared to Co1Mn1-NC. Specifically, there is a decrease in metallic N and an increase in graphitic N in L-Co1Mn1-NC. This suggests a lower N coordination number of Co/Mn DSs in the former, along with a higher level of graphitization overall, likely induced by elevated temperatures. Furthermore, the high-resolution Co 2p and Mn 2p XPS analyses reveal that Co in L-Co1Mn1-NC shifts to higher binding energy relative to Co1Mn1-NC (Fig. 2a), while Mn migrates toward lower binding energy (Fig. 2b), which may be due to the fact that Mn has fewer d electrons and easily receives external electrons.
 | ||
| Fig. 2 Geometric and electronic structure characterization of atomic catalysts. XPS spectra of (a) Co 2p and (b) Mn 2p. XANES spectra of (c) the Co K-edge and (d) Mn K-edge. Fourier-transform EXAFS fitting curves of (e) the Co K-edge and (f) Mn K-edge. (g) Comparison between the experimental XANES spectrum of L-Co1Mn1-NC at the Co K-edge and the theoretical spectrum based on the structure in the inset. WTs for the EXAFS signals of (h) the Co K-edge and (i) Mn K-edge for L-Co1Mn1-NC. | ||
To delve deeply into the variances in the chemical environments of L-Co1Mn1-NC and Co1Mn1-NC, as well as individual SACs, X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) measurements were carried out. XANES spectra at the Co K-edge for Co1-NC, Co1Mn1-NC and L-Co1Mn1-NC exhibit a comparable absorption edge to that of the CoO reference, suggesting a valence state close to +2 (Fig. 2c). In contrast to Co1Mn1-NC, the valence state of the low-coordination L-Co1Mn1-NC exhibits a slight increase, while the valence state of the Mn atom in L-Co1Mn1-NC experiences a decrease (Fig. 2d), thus indicating the electron transfer from Co to Mn. The coordination environments surrounding Co and Mn were additionally investigated using EXAFS analysis. The coordination peaks at 1.4 Å and 2.5 Å in L-Co1Mn1-NC can be assigned to the scattering path of Co–N and Co–Mn, respectively (Fig. 2e). Likewise, the presence of Mn–N and Mn–Co coordination can be detected in the vicinity of the Mn atom (Fig. 2f). Furthermore, the theoretical XANES spectrum is calculated based on the structural model in Fig. 2g, and the oscillation characteristics of the experimental spectrum are reproduced well.
The wavelet transform (WT) analysis, along with the quantitative fitting outcomes of EXAFS, provides additional confirmation regarding the presence of Co/Mn–N coordination in the compound. In the WT plots (Fig. 2h and i), the Co–Mn coordination observed at the Co K-edge is marginally shifted to the left relative to the Mn–Co coordination observed at the Mn K-edge. This discrepancy can be rationalized by considering that the scattering ability of Mn, functioning as a coordinating atom, is slightly lower than that of Co. Subsequently, the WT analyses were conducted on the EXAFS spectra obtained at the Co K-edge for samples including Co foil, Co1-NC, and L-Co1Mn1-NC, as well as at the Mn K-edge for samples comprising Mn foil, Mn1-NC, and L-Co1Mn1-NC (Fig. S8†). These findings illustrate that in L-Co1Mn1-NC, the Co–Mn coordination at the Co K-edge lies between the Co–Co coordination in Co foil and Co–N coordination in Co1-NC. This tendency is also observed at the Mn K-edge. Consequently, it can be inferred that the coordination arrangement of the nearest neighboring metal atom in L-Co1Mn1-NC predominantly features Co–Mn pairs rather than self-pairing of identical metals. Moreover, the extracted EXAFS fitting parameters indicate a relatively low N coordination in the L-Co1Mn1-NC sample (Fig. S9 and S10, and Tables S2 and S3†). The findings presented suggest that L-Co1Mn1-NC possesses a low-coordination structure of Co/Mn dual sites, with evidence of charge transfer occurring between Co and Mn, resulting in a more pronounced interaction.
Given the variances in the coordination microenvironments, electrocatalytic CO2 reduction over Co–Mn DSs and their corresponding single sites has been investigated. The CO2RR assessments were conducted on L-Co1Mn1-NC, Co1Mn1-NC, Co1-NC, and Mn1-NC catalysts in an H-type cell utilizing a CO2-saturated 0.5 M KHCO3 solution (Fig. S11†). The L-Co1Mn1-NC sample was initially examined using linear sweep voltammetry (LSV) profiles in KHCO3 electrolyte saturated with Ar and CO2, respectively. As revealed in Fig. S12,† the L-Co1Mn1-NC sample exhibits elevated total current densities and a decreased onset potential in the electrolyte saturated with CO2 compared to that of the Ar saturated environment, indicating that the structure of low-coordinated DSs may facilitate the CO2RR activity. All electrochemical data were corrected by iR compensation through the test impedance spectra (Fig. S13†), from which it can be found that the L-Co1Mn1-NC shows a lower value of solution impedance than those of Co1Mn1-NC, Co1-NC and Mn1-NC catalysts. Significantly, the L-Co1Mn1-NC catalyst also exhibits a notably higher current response and smaller onset potential for CO2 conversion compared to the Co1Mn1-NC, Co1-NC, and Mn1-NC catalysts (Fig. 3a). This underscores the enhanced activity associated with the low coordination effect of Co–Mn dual sites. Furthermore, based on the results of online gas chromatography, the products on the four atomic catalysts consist of CO and H2, with no liquid-phase products generated, as confirmed by 1H nuclear magnetic resonance spectroscopy (Fig. S14†). The enhancement of CO faradaic efficiency (FECO) on the four atomic catalysts can be divided into two stages: firstly, a significant increase can be achieved by utilizing DSs; secondly, further improvement can be attained by reducing their coordination number. Specifically, the L-Co1Mn1-NC catalyst shows a maximum FECO of 97.6% obtained at −0.47 V, which can be sustained at levels exceeding 90% across a broad 400 mV potential window (Fig. 3b), which exceeds the performance of Co1Mn1-NC (84.7% at −0.57 V), Co1-NC (64.6% at −0.57 V), and Mn1-NC (52.8% at −0.67 V) catalysts. Following the creation of a low-coordinated DS microenvironment, there is a notable enhancement in CO selectivity across all operational potential windows, particularly at low overpotential conditions. The carbon substrates produced at varying pyrolysis temperatures may also play a role in influencing the activity of the CO2RR (Fig. S15†). The polarization curves demonstrate a notable disparity in current density between the carbon carrier and the loading Co–Mn diatomic site, with the CO product exhibiting significantly lower efficiency. These findings suggest that the enhanced catalytic performance of the CO2RR on Co1Mn1-NC and L-Co1Mn1-NC catalysts primarily stems from the specific microenvironment surrounding the Co–Mn diatomic sites. Besides, a 13C isotope labeling experiment was conducted to investigate the precise source of the CO product. By utilizing 13CO2 as the feedstock for CO2 conversion, it is confirmed that the formation of CO originates solely from the consumption of the CO2 molecule, rather than from alternative carbon sources within the catalyst (Fig. S16†).36 The CO partial current density (JCO) is significantly enhanced after the construction of low-coordinated DSs, as shown in Fig. 3c, achieving a value of 34.7 mA cm−2 at −0.97 V on the L-Co1Mn1-NC catalyst, which demonstrates that the low-coordinated microenvironment promotes the intrinsic activity of the reaction Co/Mn site.
 | ||
| Fig. 3 The CO2RR performance of L-Co1Mn1-NC, Co1Mn1-NC, Co1-NC, and Mn1-NC catalysts. (a) LSV profiles. (b) FECO curves. The error bars correspond to the standard deviation of three independent measurements. (c) JCO curves. (d) TOF comparison image. (e) The stability test curve of the L-Co1Mn1-NC catalyst at −0.8 V. | ||
Afterwards, we proceed to quantitatively assess and compare the intrinsic activity of each metal site through the turnover frequency (TOF). As shown in Fig. 3d, the TOF of L-Co1Mn1-NC reaches 1067 h−1 at −0.47 V, significantly surpassing the TOFs of Co1Mn1-NC (255 h−1), Co1-NC (104 h−1), and Mn1-NC (51 h−1) catalysts when subjected to identical operating conditions. In comparison with the reported atomic catalysts,37–39 the performance of L-Co1Mn1-NC also ranks at the forefront among similar systems reported in the literature (Table S4†). Among these, L-Co1Mn1-NC nearly fills the radar chart under equivalent conditions. The construction of Co/Mn DSs and modulation of the coordination environment significantly enhance the activity and selectivity of the CO2RR. The low-coordinated M–N moieties play a crucial role in promoting this process. In addition, the L-Co1Mn1-NC catalyst exhibits acceptable stability, maintaining the current density at approximately 86% at −0.8 V for a duration of 30 hours, while the FECO level remains consistently above 85% throughout the stability assessment, as illustrated in Fig. 3e. Following the stability assessment, several examinations were conducted to analyze the morphology and structural integrity of the L-Co1Mn1-NC DS catalyst. The SEM and TEM images still show that the L-Co1Mn1-NC sample maintains a rough and uneven nanorod-like structure after CO2RR operations (Fig. S17†). The EDS mappings also demonstrate the even dispersion of Co, Mn, N, and C on the carbon host (Fig. S18†). The XRD (Fig. S19†) analysis of L-Co1Mn1-NC reveals the presence of only two peaks corresponding to the C (002) and C (101) phases, indicating that the Co/Mn metallic species remain stable during the CO2RR process without aggregating to form bulk crystalline structures. Furthermore, the micro-distribution state of the Co/Mn active sites was confirmed by AC HAADF-STEM results, still remaining atomically dispersed (Fig. S20†). In short, these findings unequivocally indicate that the Co/Mn DS, characterized by a lower N coordination number, displays superior catalytic activity, selectivity, and stability for the CO2RR, underscoring the crucial significance of regulating the microenvironment coordination of atoms in catalysis.
To assess the practical application of the L-Co1Mn1-NC catalyst under large current density, a custom-designed CO2 flow cell was utilized, incorporating gas diffusion electrodes (Fig. S21†). Fig. S22a† displays the polarization curve obtained from flow cell measurements, showing significantly higher current densities due to improved mass transfer compared to those obtained using an H-cell at a specific applied potential. At −0.84 V, the flow cell achieved a remarkable current density of −330 mA cm−2. Additionally, it exhibited excellent CO selectivity, with FECO exceeding 90% across a wide current density range from −25 to −275 mA cm−2 (Fig. R22b†). The endurance of the L-Co1Mn1-NC catalyst was evaluated by chronoamperometry at −0.55 V versus RHE in the flow cell setup (Fig. R22c†). The L-Co1Mn1-NC catalyst demonstrates good durability, with a gradual decrease in current over time from an initial density of −120 mA cm−2 to −107.7 mA cm−2 after 80 hours of electrolysis, retaining 89% of its initial value post stability assessment. FECO levels were monitored every 10 hours, consistently remaining high at 87% throughout the 80 hour stability evaluation. These findings highlight the potential application of the L-Co1Mn1-NC catalyst at high current density conditions.
In order to track the catalytic behavior of the reaction interface, we utilized in situ attenuated total reflectance surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) to observe the key intermediates.40 The ATR-SEIRAS spectra were acquired by incrementally reducing the potential from 0 to −1.0 V, as illustrated in Fig. S23.† In an electrolyte saturated with CO2, the IR spectrum recorded at 0 V is regarded as the baseline reference (R0), with no discernible reactive group signal initially observed. Two significant peaks corresponding to the adsorbed CO2 (2343 cm−1) and COOH* (1425 cm−1) signals are observed starting from a potential of −0.4 V. With a progressive rise in potentials, the CO2 present at the active interface is continuously depleted within the potential range of −0.4 to −1.0 V. The formation of the COOH* intermediate primarily occurs through the rapid protonation of the adsorbed *CO2− species, followed by further protonation to generate CO*. Importantly, the emergence of CO* is also identified on the low-coordinated Co/Mn DS at −0.3 V, which is consistent with the experimental results of CO production at a low potential. As the potential increases, the accumulation of adsorbed CO* gradually occurs. Combined with the application potential of CO*, it can be inferred that the CO* desorption is the step that limits the potential of the CO2RR. Furthermore, across all potential ranges, the strength of H2O adsorption at the low-coordinated Co/Mn DS remains relatively constant, serving as a potential stable proton donor.41 Additionally, due to the consumption of H+ during the reaction process, a small amount of CO32− will form in the localized buffer equilibrium saturated with CO2.42 Furthermore, we also recorded the in situ ATR-SEIRAS spectra for the Co1Mn1-NC catalyst (Fig. S24†). The adsorption peaks of CO* and COOH* species begin to appear at a higher applied potential compared to the L-Co1Mn1-NC catalyst, indicating the effect of low coordination structure has significantly improved the catalytic performance. Therefore, we have provided experimental evidence through in situ ATR-SEIRAS to unveil the reduction process of CO2 on the low-coordination Co/Mn DS.
In view of the excellent catalytic performance of the low-coordinated Co/Mn DS microenvironment toward the CO2RR, density functional theory (DFT) calculations have been further performed to explore its essential influence mechanism. Based on structural characterization analysis, we establish the structural models of L-Co1Mn1-NC and Co1Mn1-NC catalysts and optimize the adsorption models of intermediates at different sites (Fig. S25–S28†). The differential charge density diagrams (Fig. 4a–d) illustrate the charge redistribution within Co1Mn1-NC and L-Co1Mn1-NC. Such redistribution has the potential to alter the electronic configuration of the metal active sites via charge interactions, thereby modulating their reactivity.43,44 Subsequently, as depicted in Fig. 4e and f, the free energy diagrams for the CO2RR and HER at U = 0 V elucidate the impact of the low coordination effect of the Co/Mn DS structure on performance. Except for Co site in Co1Mn1-NC, the potential-limiting step of other sites within Co1Mn1-NC and L-Co1Mn1-NC is CO desorption (*CO → *+CO), and the Co site at L-Co1Mn1-NC exhibits a relatively small energy barrier (0.92 eV). Previous studies have indicated that the slow desorption of CO is a significant factor contributing to the poor selectivity and activity of CO2-to-CO conversion.45,46 The calculation results demonstrate that the adsorption energy of CO can be decreased and the desorption of CO can be enhanced by utilizing a low coordination bimetallic center (CoN3–MnN3) in comparison to Co1Mn1-NC. The competitive response of the CO2RR is primarily represented by the HER, thereby necessitating the calculation of the limiting potential difference between the CO2RR and HER (UL(CO2)–UL(H2)) for both catalysts to assess reaction selectivity. The more positive difference value signifies enhanced selectivity, and thus L-Co1Mn1-NC shows higher CO selectivity (Fig. 4g).47 Therefore, the low-coordinated microenvironment of L-Co1Mn1-NC facilitates the attainment of enhanced performance in the CO2RR, aligning with the outcomes observed in experimental results.
 | ||
| Fig. 4 Charge density difference of (a) Co1-NC, (b) Mn1-NC, (c) Co1Mn1-NC and (d) L-Co1Mn1-NC, where electron depletion and electron accumulation are shown in yellow and cyan, respectively. (e) Gibbs free energy diagrams for the CO2-to-CO conversion process. (f) Gibbs free energy diagrams for the HER. (g) Calculated limiting potential difference of the CO2RR and HER. ELF analysis for (h) Co1Mn1-NC and (i) L-Co1Mn1-NC. (j) PDOS of the 3d-orbital. | ||
Besides, we examined the L-Co2-NC and L-Mn2-NC models through DFT calculations, possessing a structure akin to that of the L-Co1Mn1-NC model (Fig. S29 and S30†). Subsequently, we evaluated their performance in the CO2RR and HER. As exhibited in Fig. S31,† the results show that the overpotentials of the CO2RR of L-Co2-NC and L-Mn2-NC are 1.08 V and 1.54 V, respectively, both of which are significantly higher than that of L-Co1Mn1-NC. In addition, their HER overpotential is lower, resulting in a lower limiting potential difference. Consequently, the CO selectivity on L-Co2-NC and L-Mn2-NC models is compromised, marking a notable deviation from the observed performance in sample testing.
Moreover, an analysis of the electronic structure was conducted to elucidate the inherent origin of low-coordination L-Co1Mn1-NC. The electron localization function (ELF) analysis indicates a higher concentration of electrons shared between the Co and Mn sites within the L-Co1Mn1-NC (Fig. 4h and i). This phenomenon suggests an enhanced charge interaction and a direct electron transfer between the Co and Mn sites. Quantitatively, the Bader charge analysis (Fig. S32†) reveals a rise in the valence state of Co and a decline in Mn when compared to Co1Mn1-NC, which substantiates the charge transfer occurring between Co and Mn sites, aligning with the findings from XPS and XANES analyses. Subsequently, the electronic configuration of Co and Mn 3d orbitals was probed by the projected density of states (PDOS). It is observed that there is a rise in the d-band center of Co and Mn in the L-Co1Mn1-NC, with a more pronounced alteration noted in the case of Co (Fig. 4j). This leads to a transfer of electrons from the Co 3d orbital to lower energy states, specifically the Mn 3d orbital. In addition, a strong d–d orbital coupling and delocalization of electrons promote the desorption of CO, thus reducing the reaction energy barrier of the CO2RR.48 Additionally, the total DOS near the Fermi energy within L-Co1Mn1-NC surpasses that of Co1Mn1-NC, as illustrated in Fig. S33.† This indicates that the enhanced conductivity and the facilitation of charge transfer at the reaction interface are attributed to the lower coordination structure that emerged in the L-Co1Mn1-NC catalyst.
Conclusions
In brief, a direct and rational PSCR approach was employed to fabricate Co–Mn DS catalysts dominated by a low-coordination microenvironment via simultaneously substituting Co and Mn atoms for the atomically dispersed Zn sites in the Zn-Nx-C material derived from ZIF-8 nanorods. When altering the pyrolysis temperature and substituting metal in the PSCR strategy, a relatively high N coordination of Co–Mn DSs and the common 4-coordination of Co/Mn–N4 structures can be obtained, respectively. The development of this PSCR strategy has simplified the cumbersome steps involved in synthesizing DS catalysts derived from MOFs, and has achieved the regulation of DS coordination environments through simple temperature control. Accordingly, the optimal L-Co1Mn1-NC catalyst exhibits superior efficiency in CO2 electroreduction, achieving a high FECO of 97.6% and a TOF value of 1067 h−1 at −0.47 V, which significantly surpass those of Co1Mn1-NC and individual Co/Mn single sites. A combination of in situ investigation and theoretical analysis unveils that the lower Co/Mn coordination number in the L-Co1Mn1-NC catalyst efficiently boosts the desorption of the critical CO* species, thereby enhancing the CO2 reduction kinetics. This study presents a convenient and rational control approach for manipulating the catalytic microenvironment of atomic catalysts, laying a solid experimental foundation for improving the reaction kinetics of energy conversion processes.Author contributions
J. D. conceived the idea and designed the research. J. P., S. J., P. W. and H. H. performed the synthesis, structural characterisations, and electrochemical measurements. P. A. and J. L. performed the XAFS measurements and analysed the data. G. Z. and S. C. performed DFT simulations. J. P. and G. Z. analysed and discussed the experimental results and wrote the manuscript. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. D. acknowledges financial support from the Youth Innovation Promotion Association of the Chinese Academy of Sciences (Y2022006) and the Innovation project of Institute of High Energy Physics, Chinese Academy of Sciences. J. P. acknowledges financial support from the China Postdoctoral Science Foundation (grant no. 2023M742047) and the Beijing Natural Science Foundation (no. 1244064).Notes and references
- C. Chen, J. F. Khosrowabadi Kotyk and S. W. Sheehan, Chem, 2018, 4, 2571–2586 CAS
.
- B. S. Crandall, S. Overa, H. Shin and F. Jiao, Acc. Chem. Res., 2023, 56, 1505–1516 CrossRef CAS PubMed
- A. D. Handoko, F. Wei, Jenndy, B. S. Yeo and Z. W. Seh, Nat. Catal., 2018, 1, 922–934 CrossRef CAS
- L. Fan, C. Xia, F. Yang, J. Wang, H. Wang and Y. Lu, Sci. Adv., 2020, 6, eaay3111 CrossRef CAS PubMed
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed
- M. B. Ross, P. De Luna, Y. Li, C.-T. Dinh, D. Kim, P. Yang and E. H. Sargent, Nat. Catal., 2019, 2, 648–658 CrossRef CAS
- T. N. Nguyen, M. Salehi, Q. V. Le, A. Seifitokaldani and C. T. Dinh, ACS Catal., 2020, 10, 10068–10095 CrossRef
- D. Yang, B. Ni and X. Wang, Adv. Energy Mater., 2020, 10, 2001142 CrossRef CAS
- C. Zhu, J. Yang, J. Zhang, X. Wang, Y. Gao, D. Wang and H. Pan, Interdiscip. Mater., 2024, 3, 74–86 CrossRef CAS
- Y. Wang, Y. Zhang, W. Yu, F. Chen, T. Ma and H. Huang, J. Mater. Chem. A, 2023, 11, 2568–2594 RSC
- S. Zhu, E. P. Delmo, T. Li, X. Qin, J. Tian, L. Zhang and M. Shao, Adv. Mater., 2021, 33, 2005484 CrossRef CAS PubMed
- J. Zhuang and D. Wang, Mater. Today Catal., 2023, 2, 100009 CrossRef
- J. Dong, Y. Liu, J. Pei, H. Li, S. Ji, L. Shi, Y. Zhang, C. Li, C. Tang, J. Liao, S. Xu, H. Zhang, Q. Li and S. Zhao, Nat. Commun., 2023, 14, 6849 CrossRef CAS PubMed
- T. Tang, Z. Wang and J. Guan, Adv. Funct. Mater., 2022, 32, 2111504 CrossRef CAS
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, Adv. Mater., 2020, 32, 2001848 CrossRef CAS PubMed
- Y. Wang, H. Su, Y. He, L. Li, S. Zhu, H. Shen, P. Xie, X. Fu, G. Zhou, C. Feng, D. Zhao, F. Xiao, X. Zhu, Y. Zeng, M. Shao, S. Chen, G. Wu, J. Zeng and C. Wang, Chem. Rev., 2020, 120, 12217–12314 CrossRef CAS PubMed
- R. Qin, K. Liu, Q. Wu and N. Zheng, Chem. Rev., 2020, 120, 11810–11899 CrossRef CAS PubMed
- X. Li, L. Liu, X. Ren, J. Gao, Y. Huang and B. Liu, Sci. Adv., 2020, 6, eabb6833 CrossRef CAS PubMed
- C. Jia, X. Tan, Y. Zhao, W. Ren, Y. Li, Z. Su, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2021, 60, 23342–23348 CrossRef CAS PubMed
- S. Li, X. Lu, S. Zhao, M. Ceccato, X.-M. Hu, A. Roldan, M. Liu and K. Daasbjerg, ACS Catal., 2022, 12, 7386–7395 CrossRef CAS
- L. Yuan, S. Zeng, G. Li, Y. Wang, K. Peng, J. Feng, X. Zhang and S. Zhang, Adv. Funct. Mater., 2023, 33, 2306994 CrossRef CAS
- D. Xi, J. Li, J. Low, K. Mao, R. Long, J. Li, Z. Dai, T. Shao, Y. Zhong, Y. Li, Z. Li, X. J. Loh, L. Song, E. Ye and Y. Xiong, Adv. Mater., 2021, 34, 2104090 CrossRef PubMed
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS PubMed
- Y. N. Gong, L. Jiao, Y. Qian, C. Y. Pan, L. Zheng, X. Cai, B. Liu, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2020, 59, 2705–2709 CrossRef CAS PubMed
- M. Wang, Z. Zhang, S. Zhang, W. Liu, W. Shang, X. Su, Y. Liang, F. Wang, X. Ma, Y. Li and Y. Liu, Angew. Chem., Int. Ed., 2023, 62, e202300826 CrossRef CAS PubMed
- Y. Deng, J. Zhao, S. Wang, R. Chen, J. Ding, H.-J. Tsai, W.-J. Zeng, S.-F. Hung, W. Xu, J. Wang, F. Jaouen, X. Li, Y. Huang and B. Liu, J. Am. Chem. Soc., 2023, 145, 7242–7251 CrossRef CAS PubMed
- D.-C. Zhong, Y.-N. Gong, C. Zhang and T.-B. Lu, Chem. Soc. Rev., 2023, 52, 3170–3214 RSC
- Y.-X. Zhang, S. Zhang, H. Huang, X. Liu, B. Li, Y. Lee, X. Wang, Y. Bai, M. Sun, Y. Wu, S. Gong, X. Liu, Z. Zhuang, T. Tan and Z. Niu, J. Am. Chem. Soc., 2023, 145, 4819–4827 CrossRef CAS PubMed
- Z.-H. Zhao, J.-R. Huang, P.-Q. Liao and X.-M. Chen, J. Am. Chem. Soc., 2023, 145, 26783–26790 CrossRef CAS PubMed
- Y. Chen, J. Lin, Q. Pan, X. Liu, T. Ma and X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202306469 CrossRef CAS PubMed
- Y. N. Gong, C. Y. Cao, W. J. Shi, J. H. Zhang, J. H. Deng, T. B. Lu and D. C. Zhong, Angew. Chem., Int. Ed., 2022, 61, e202215187 CrossRef CAS PubMed
- J. Jiao, R. Lin, S. Liu, W.-C. Cheong, C. Zhang, Z. Chen, Y. Pan, J. Tang, K. Wu, S.-F. Hung, H. M. Chen, L. Zheng, Q. Lu, X. Yang, B. Xu, H. Xiao, J. Li, D. Wang, Q. Peng, C. Chen and Y. Li, Nat. Chem., 2019, 11, 222–228 CrossRef CAS PubMed
- S. Ma, W. Han, W. Han, F. Dong and Z. Tang, J. Mater. Chem. A, 2023, 11, 3315–3363 RSC
- L. Zhang, R. Si, H. Liu, N. Chen, Q. Wang, K. Adair, Z. Wang, J. Chen, Z. Song, J. Li, M. N. Banis, R. Li, T.-K. Sham, M. Gu, L.-M. Liu, G. A. Botton and X. Sun, Nat. Commun., 2019, 10, 4936 CrossRef PubMed
- Q. Hao, H.-X. Zhong, J.-Z. Wang, K.-H. Liu, J.-M. Yan, Z.-H. Ren, N. Zhou, X. Zhao, H. Zhang, D.-X. Liu, X. Liu, L.-W. Chen, J. Luo and X.-B. Zhang, Nat. Synth., 2022, 1, 719–728 CrossRef
- J. Pei, H. Shang, J. Mao, Z. Chen, R. Sui, X. Zhang, D. Zhou, Y. Wang, F. Zhang, W. Zhu, T. Wang, W. Chen and Z. Zhuang, Nat. Commun., 2024, 15, 416 CrossRef CAS PubMed
- T. Wang, Q. Zhang, K. Lian, G. Qi, Q. Liu, L. Feng, G. Hu, J. Luo and X. Liu, J. Colloid Interface Sci., 2023, 655, 176–186 CrossRef PubMed
- S. Liu, M. Jin, J. Sun, Y. Qin, S. Gao, Y. Chen, S. Zhang, J. Luo and X. Liu, Chem. Eng. J., 2022, 437, 135294 CrossRef CAS
- S. Gao, S. Chen, Q. Liu, S. Zhang, G. Qi, J. Luo and X. Liu, ACS Appl. Nano Mater., 2022, 5, 12387–12394 CrossRef CAS
- W. Cheng, H. Su and Q. Liu, Acc. Chem. Res., 2022, 55, 1949–1959 CrossRef CAS PubMed
- H. Xiao, W. A. Goddard, T. Cheng and Y. Liu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6685–6688 CrossRef CAS PubMed
- C. Liu, Y. Wu, K. Sun, J. Fang, A. Huang, Y. Pan, W.-C. Cheong, Z. Zhuang, Z. Zhuang, Q. Yuan, H. L. Xin, C. Zhang, J. Zhang, H. Xiao, C. Chen and Y. Li, Chem, 2021, 7, 1297–1307 CAS
- Z. Chen, X. Zhang, W. Liu, M. Jiao, K. Mou, X. Zhang and L. Liu, Energy Environ. Sci., 2021, 14, 2349–2356 RSC
- J. Zhu, M. Xiao, D. Ren, R. Gao, X. Liu, Z. Zhang, D. Luo, W. Xing, D. Su, A. Yu and Z. Chen, J. Am. Chem. Soc., 2022, 144, 9661–9671 CrossRef CAS PubMed
- Y. Li, W. Shan, M. J. Zachman, M. Wang, S. Hwang, H. Tabassum, J. Yang, X. Yang, S. Karakalos, Z. Feng, G. Wang and G. Wu, Angew. Chem., Int. Ed., 2022, 61, e202205632 CrossRef CAS PubMed
- X. Qin, T. Vegge and H. A. Hansen, J. Am. Chem. Soc., 2023, 145, 1897–1905 CrossRef CAS PubMed
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed
- Z. Zeng, L. Y. Gan, H. Bin Yang, X. Su, J. Gao, W. Liu, H. Matsumoto, J. Gong, J. Zhang, W. Cai, Z. Zhang, Y. Yan, B. Liu and P. Chen, Nat. Commun., 2021, 12, 4088 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta02261k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |