
2D crumpled nitrogen-doped carbon nanosheets anode with capacitive-dominated behavior for ultrafast-charging and high-energy-density Li-ion capacitors†
Fangyan
Liu‡
a,
Tong
Yu‡
b,
Jieqiong
Qin
*c,
Liangzhu
Zhang
a,
Feng
Zhou
a,
Xiong
Zhang
 def,
Yanwei
Ma
def,
Feng
Li
*b and
Zhong-Shuai
Wu
*a
def,
Yanwei
Ma
def,
Feng
Li
*b and
Zhong-Shuai
Wu
*a
aState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: wuzs@dicp.ac.cn
bShenyang National Laboratory for Materials Science, Institute of Metal Research, Chinese Academy of Science, Shenyang, 110016, China. E-mail: fli@imr.ac.cn
cCollege of Science, Henan Agricultural University, 63 Agricultural Road, Zhengzhou 450002, China. E-mail: qinjieqiong@henau.edu.cn
dInstitute of Electrical Engineering, Chinese Academy of Sciences, Beijing 100190, China
eSchool of Engineering Sciences, University of Chinese Academy of Sciences, Beijing 100049, China
fInstitute of Electrical Engineering and Advanced Electromagnetic Drive Technology, Qilu Zhongke, Jinan 250013, China
First published on 4th June 2024
Abstract
Li-ion capacitors (LICs) are promising to simultaneously achieve battery-level energy density and supercapacitor-level power density, but the slow kinetics of diffusion-controlled battery anodes lead to unmatched two-electrode kinetics at the device level. Herein, we report a capacitive-dominated anode of two-dimensional (2D) crumpled nitrogen-doped carbon nanosheets (N-CNS) with tailored nitrogen incorporation and abundant mesopore distribution, exhibiting large capacities and superior rate performance. The optimized N-CNS delivers large reversible capacities of 620 and 121 mA h g−1 at 0.1 and 100 A g−1, respectively. The introduced nitrogen is found to contribute to providing additional pseudocapacity and high Li+ diffusion coefficients in the medium–high voltage region and enhancing the capacitive-dominated charge storage process. The structural reversibility and “adsorption-intercalation” mechanism are supported by in situ and ex situ measurements. Furthermore, it is theoretically revealed that N-CNS with superior electrochemical properties benefits from the increase in Li+ adsorption energy and the decline in the Li+ diffusion barrier. A LIC coupling the N-CNS anode with a porous carbon cathode outputs a high energy density of 75 W h kg−1 at an ultrahigh power density of 65 kW kg−1. This study provides a novel and effective approach to developing high-performance carbon-based anodes for constructing advanced LICs featuring high energy and power density.
Introduction
With the urgent requirement for new energy storage devices, Li-ion capacitors (LICs) merge a high-energy battery-type electrode and a fast-kinetic capacitive electrode into one cell, burdened with the high expectation of delivering battery-grade energy density with a power delivery rate close to that of electrical double-layer capacitors (EDLCs).1,2 However, LICs currently remain in an embarrassing situation where they can boost the specific energy of the device but sacrifice the power density. The major obstruction lies in the fact that the slow diffusion-controlled kinetics in battery-type anodes lead to poor rate capability and fail to accept the large capacity from the capacitive electrode at high rates. Therefore, it is desired to develop a high-capacity anode with excellent rate performance to assemble LICs that retain high energy density even at ultrahigh power output, which is highly required for wider application in the current market.Hard carbons (HCs) are disordered non-graphitic carbons that are composed of randomly stacked, rumpled graphene sheets that are in a “house of cards” arrangement.3–5 They are generally considered to be a promising anode choice for realizing Li-ion fast charging. In general, Li ions can be stored in HCs via intercalation between graphite-like layers, adsorption on the surface, defects, and nanopores.6,7 Accordingly, the galvanostatic discharge/charge voltage profiles of HCs usually present sloping lines followed by a plateau representing a diffusion-controlled process with slow kinetics that restrict the rate capability.8–10 Hence, some HCs still face the dilemma of being unable to meet the high demands from LICs that simultaneously achieve high capacity and superior rate capability.
Surface-controlled charge storage behavior with a high capacitive contribution demonstrates excellent rate performance and reversibility.11,12 Increasing the surface defects by techniques such as nitrogen doping has been demonstrated to be effective in promoting the capacitive contribution.13–16 The doped-N acting as active sites can adsorb ions and produce reversible extra pseudocapacity to enhance the total capacity of carbon materials to satisfy the requirement of high energy density of devices.16,17 Being different from the sluggish ion diffusion within a crystalline structure, the doped-N reacts quickly on the surface or in the near-surface region while well maintaining the carbon structure, enabling great rate performance and cycling stability.18 It has been experimentally confirmed that the Li-ion storage performance has a strong correlation with the N-doping species.13,14 The adsorption ability of N-doping species for ions in carbon materials has been extensively studied from the perspective of theoretical calculations;19,20 however, the effects on Li-ion diffusion still need further investigation.
Although capacitive-dominated behavior exhibits faster kinetics than a diffusion-controlled Li-ion storage process, it is still not enough to achieve supercapacitor-level rate performance just via surface modification. To overcome this limitation, the adjustment of the pore structure especially mesopores is indispensable since abundant mesopores could shorten ion transport paths and largely buffer the ion diffusivity constraints at high rates,21 playing a critical role in determining the rate capability of carbon materials. The generation of mesopores usually comes from etching, template methods and self-arrangement of structural units.22–26 The first two methods are usually accompanied by some complicated processes, and the acquired porous carbons possess huge specific surface areas (SSAs), leading to limited initial coulombic efficiency.27,28 The last one could produce abundant mesopore size distribution with a limited specific surface area by taking advantage of the structural features of structural units, such as the folded pore structure of crumpled graphene.25,29,30 As inspired by the wrinkled structure, 2D crumpled carbon materials could be proposed to get abundant mesopores and a limited SSA. Taking all these considerations into account, it is imperatively demanded to take advantage of the synergistic effect between N-doping and 2D crumpled structures to realize large Li-storage at fast charging rates.
Herein, we report a capacitive-dominant graphene oxide@polypyrrole (GO@PPy)-derived anode of 2D crumpled N-doped carbon nanosheets (N-CNS) prepared by a structure-designed and nitrogen-tailoring strategy. The unique 2D crumpled structure featuring abundant mesopore distribution guarantees fast ion diffusion, while N-doping offers lots of active sites for Li-ion storage. Benefiting from the synergistic effect of the 2D wrinkled structure and N-doping, the as-synthesized N-CNS delivers a large specific capacity of 620 mA h g−1 at 0.1 A g−1 and incredible rate capability with 19.5% retention after a 1000-fold current density increase from 0.1 to 100 A g−1. The surface-controlled capacity contribution dominates in the charge storage process and can be significantly improved with increasing N-5 and N-6 contents, which is mainly attributed to the N-doping producing additional pseudocapacity and high Li+ diffusion coefficients, both of which occur in the medium–high voltage region. The charge storage mechanism is also investigated by in situ Raman and ex situ XRD. Furthermore, it is theoretically revealed that the N-5 and N-6 doped at the nanopore-defect sites can improve the Li+ adsorption energy and lower the Li+ diffusion barrier. The as-prepared N-CNS-Ar-20 could function as an outstanding anode for a LIC full cell. The resulting device could offer a large energy density of 75 W h kg−1 even at an ultrahigh power density of 65 kW kg−1 within an extremely short time of only 4.2 s, along with stable cycling ability with 91.5% capacity retention after 5000 cycles at 5 A g−1.
Results and discussion
The designed synthesis of 2D crumpled N-CNS is schematically illustrated in Fig. 1a. First, graphene oxide (GO) was used as the hard template of the 2D structure to uniformly adsorb the pyrrole monomers. By initiating the polymerization of the monomers using ammonium persulfate as the oxidizing agent, polypyrrole (PPy) was generated on the surface of GO, resulting in the formation of 2D GO@PPy nanosheets. Finally, the GO@PPy nanosheets were carbonized at 800 °C under different atmospheres, Ar and Ar/H2, to achieve the crumpled N-CNS products with different nitrogen contents, labeled N-CNS-Ar and N-CNS-Ar/H2, respectively. To further tailor the nitrogen content, GO@PPy and urea with varying mass ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 and 1:20 were mixed and then carbonized at 800 °C under an Ar atmosphere, and correspondingly, the as-obtained crumpled N-CNS samples were labeled N-CNS-Ar-5 and N-CNS-Ar-20, respectively. Such rationally designed N-CNS samples with 2D crumpled structures and tailored nitrogen contents are expected to achieve not only high capacity by storing more Li+ on active sites, but also excellent rate capability by allowing unhindered Li-flow (Fig. 1b).
:5 and 1:20 were mixed and then carbonized at 800 °C under an Ar atmosphere, and correspondingly, the as-obtained crumpled N-CNS samples were labeled N-CNS-Ar-5 and N-CNS-Ar-20, respectively. Such rationally designed N-CNS samples with 2D crumpled structures and tailored nitrogen contents are expected to achieve not only high capacity by storing more Li+ on active sites, but also excellent rate capability by allowing unhindered Li-flow (Fig. 1b).
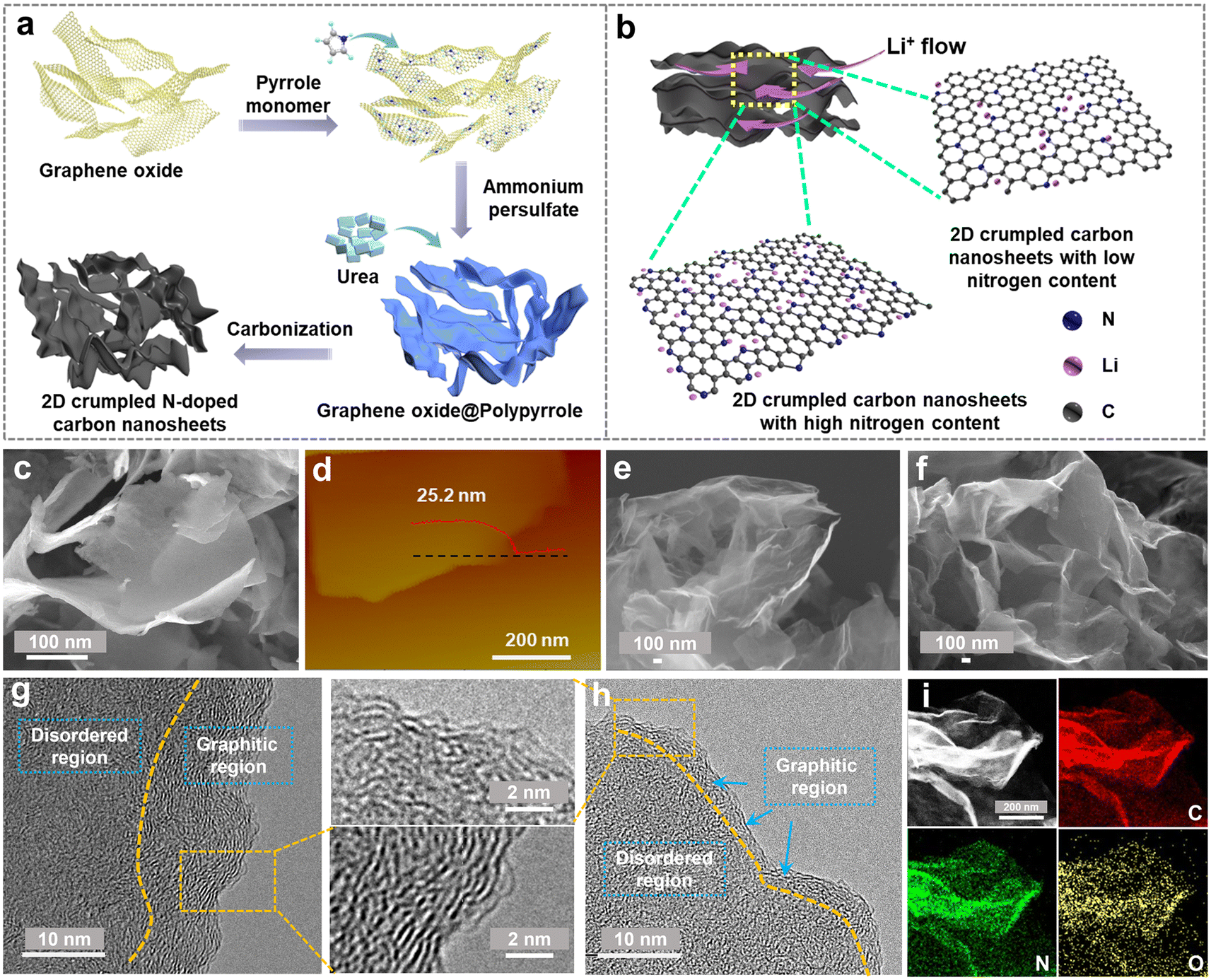 | ||
| Fig. 1 (a) Schematic diagram of the preparation process of 2D crumpled N-CNS. (b) Charge storage of 2D crumpled N-CNS with different N contents. (c) SEM image and (d) atomic force microscopy (AFM) image and corresponding height profile of 2D GO@PPy. SEM images of (e) N-CNS-Ar and (f) N-CNS-Ar-20. HRTEM images of (g) N-CNS-Ar and (h) N-CNS-Ar-20. (i) HADDF-STEM image of N-CNS-Ar-20 and its corresponding mapping images (C, N, and O). | ||
The morphologies and microstructures of 2D crumpled GO@PPy and N-CNS materials were examined by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). GO@PPy has a 2D sheet-like wrinkled morphology with a thickness of about 25 nm (Fig. 1c and d). The sharp wrinkles are formed owing to the in-plane partial restacking of nanosheets, which plays a vital role in maintaining the structural stability of GO@PPy.31 After carbonization, the obtained N-CNS materials show similar morphologies to those of GO@PPy, maintaining the 2D crumpled structure (Fig. 1e, f and S1, ESI†). These 2D nanosheets are randomly arranged accompanied by graphene shrinkage, resulting in a paper-ball-like shape with opened channels outward and wide voids inside the crumpled nanosheets (Fig. S1, ESI†). Fig. S2, ESI† displays the microstructure of the N-CNS sample, which mainly consists of many wrinkles and relatively flat parts. High-resolution TEM (HRTEM) images further present that the relatively flat part consists of a dominantly disordered amorphous region and some distorted carbon lattices (Fig. 1g, h, S3 and S4, ESI†). Especially, the microstructure of N-CNS-Ar-20 tends to be more disordered than that of N-CNS-Ar (enlarged sections of Fig. 1g and h). The wrinkled parts present apparent lattice fringes and the corresponding average d-spacings of N-CNS-Ar/H2, N-CNS-Ar, N-CNS-Ar-5, and N-CNS-Ar-20 are measured to be 0.362, 0.354, 0.358, and 0.359 nm (Fig. S4, ESI†), respectively. These average d-spacings are higher than 0.335 nm of graphite, which highly favors Li-ion diffusion and storage. Moreover, the high-angle annular dark-field scanning transmission electron microscopy (HADDF-STEM) elemental mapping images suggest that carbon, nitrogen, and oxygen are uniformly distributed in the N-CNS materials (Fig. 1i and S5, ESI†).
The structural properties of 2D crumpled N-CNS-Ar, N-CNS-Ar/H2, N-CNS-Ar-5, and N-CNS-Ar-20 were characterized using X-ray diffraction (XRD) patterns. The broad diffraction peak (002) in Fig. 2a is attributed to the amorphous structure in the partially graphitized samples and the imperfect alignment of graphitic carbon.32,33 Compared with N-CNS-Ar, the (002) peaks of N-CNS-Ar/H2, N-CNS-Ar-5, and N-CNS-Ar-20 show no obvious shift, indicating unchanged average interlayer distances for (002) planes. The interlayer distances of N-CNS samples are calculated to be around 0.352 nm, which is almost consistent with HRTEM results (Fig. S4, ESI†). The similar interlayer spacings of N-CNS materials suggest the structural stability of sharp wrinkles against the attack of hydrogen and urea during the pyrolysis process. All crumpled N-CNS materials present I/IV isotherms, showing meso/macroporous structures (Fig. 2b). The specific surface areas (SSAs) of N-CNS-Ar/H2, N-CNS-Ar-5, and N-CNS-Ar-20 are measured to be 115, 123, and 164 m2 g−1, respectively, larger than that of N-CNS-Ar (102 m2 g−1). It can be explained that the reactions of H2 and urea with the precursor increase the SSA of N-CNS, especially the micropore areas (Table S1, ESI†). This is because H2 can react with the nitrogen-containing functional groups in polypyrrole to produce NH3, which will decompose to free radicals such as NH2, NH, and atomic hydrogen and nitrogen.34,35 When carbon materials are treated with NH3 at elevated temperatures, these free radicals attack the carbon, forming nitrogen-containing functional groups and etching the carbon fragments, resulting in an increase in porosity.36 As for urea, it will decompose into carbon dioxide and ammonia at high temperatures, both of which can work as pore-forming agents and react with carbon materials to form porosity to increase the specific surface area.36,37 These samples exhibit abundant and similar mesopore/macropore distributions as well as similar external surface areas, which suggests that these mesopores are mainly the interior pores formed from the original arrangement of these crumpled carbon nanosheets and are not significantly affected by the additional treatments (Fig. 2b). It is worth noting that the micropores could store Li ions to produce sloping capacity,6,7 while the mesopores could provide wide channels for fast ion adsorption/diffusion, contributing to high specific capacity and superior rate capability.21,38 Raman spectroscopy was used to further investigate the microstructure of the resulting carbon samples. As shown in Fig. 2c, the two typical peaks of the D band at 1352 cm−1 and the G band at 1588 cm−1 fitted by using the Gaussian function are assigned to the disordered or defective graphite (sp3) and graphitic lattice structures (sp2), respectively.39 The integral area ratios of the fitted D- and G-peaks ID/IG continuously increase from 1.83 of N-CNS-Ar/H2 to 1.90 of N-CNS-Ar, 1.98 of N-CNS-Ar-5, and 2.08 of N-CNS-Ar-20, indicating that more defects are created in these carbon materials with increasing nitrogen doping (Fig. 2e). X-ray photoelectron spectroscopy (XPS) was conducted to analyze the surface composition of N-CNS materials (Fig. 2d, f, S6 and Table S2, ESI†). Compared with a N content of 6.3 at% in N-CNS-Ar, the treatment in the Ar/H2 atmosphere leads to a lower nitrogen content, while the N content increases markedly up to 13.2 at% when GO@PPy is carbonized with urea (Fig. 2e). As the N content of N-CNS samples increases, the pyridinic N (N-6) located at around 398 eV gradually dominates (up to 45.5% for N-CNS-Ar-20) (Fig. 2f and Table S2, ESI†), which together with N-5 was formed predominantly by substituting a carbon atom on defect sites or edges in the plane. The N-5 and N-6 act as highly active chemical sites and reversibly react with Li+ to enhance the surface-controlled pseudocapacity and improve fast Li+ diffusion.14,40,41 The quaternary-N (N-Q) proportion of N-CNS materials shows a downtrend with N content increasing. Although the N-Q is conducive to the electrical conductivity of carbon materials owing to the electron donor characteristic,42 it is more prone to breakage after lithiation than the N-6 and N-5.43 Additionally, oxygenated species such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O can also react with Li+ (Fig. S6, ESI†), resulting in enhanced capacity.44
O can also react with Li+ (Fig. S6, ESI†), resulting in enhanced capacity.44
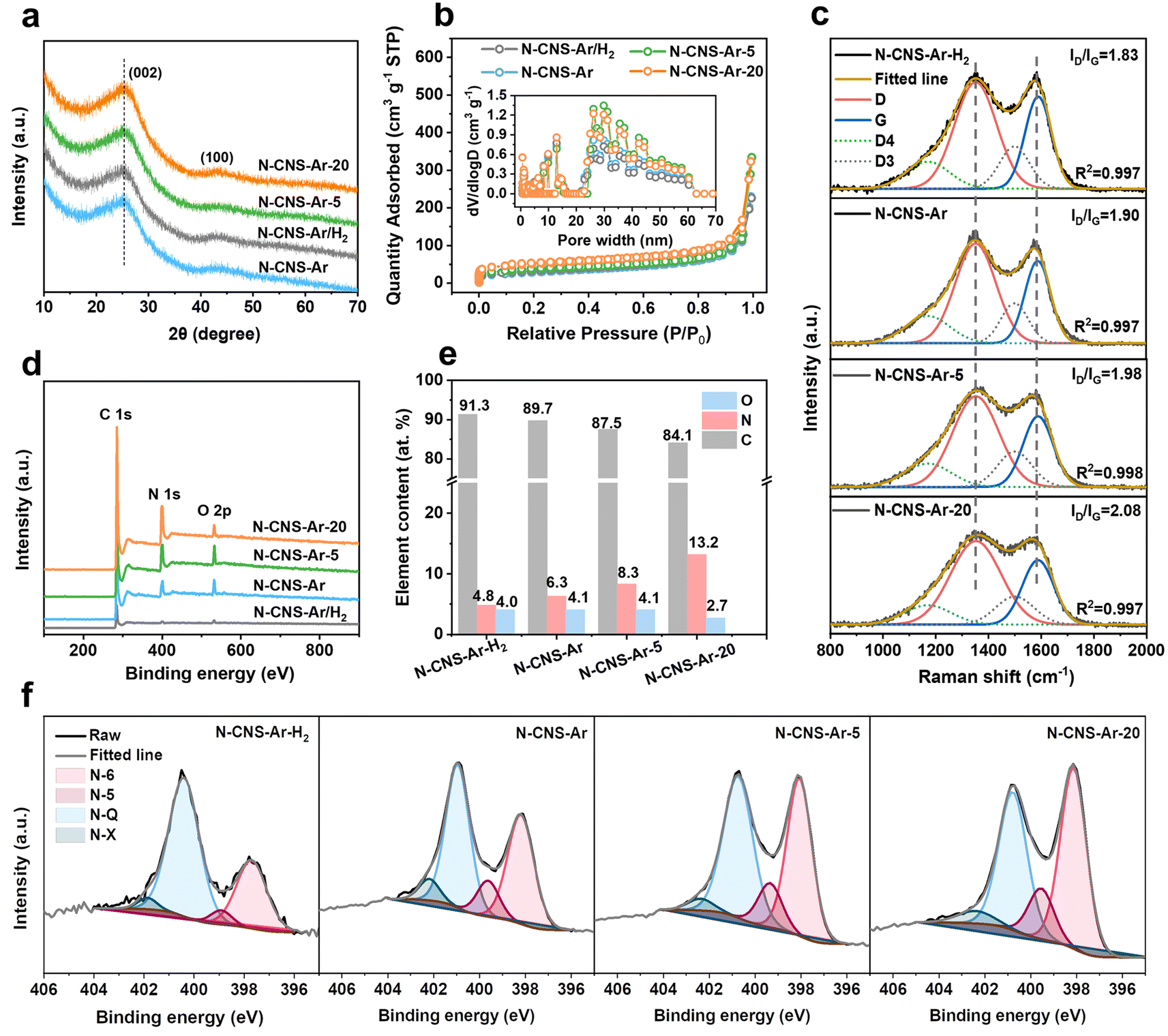 | ||
| Fig. 2 Morphological and structural characterization of 2D crumpled N-CNS-Ar, N-CNS-Ar/H2, N-CNS-Ar-5, and N-CNS-Ar-20. (a) XRD patterns, (b) nitrogen adsorption–desorption isotherms (inset presents the pore size distributions obtained by the DFT method), (c) fitted Raman spectra curves, (d) XPS spectra, (e) element content of C, N and O, and (f) high-resolution N 1s XPS spectra. | ||
The electrochemical performance of 2D crumpled N-CNS-Ar, N-CNS-Ar/H2, N-CNS-Ar-5, and N-CNS-Ar-20 was measured by assembling half cells with lithium foil as the counter electrode and reference electrode. Fig. S7, ESI† presents the first four cyclic voltammetry (CV) curves of 2D crumpled N-CNS-Ar, N-CNS-Ar/H2, N-CNS-Ar-5, and N-CNS-Ar-20 within 0.02–3 V vs. Li/Li+ at a scan rate of 0.1 mV s−1. There are two cathodic peaks observed during the first lithiation process, corresponding to the irreversible reaction between Li+ and surface functional groups (first peak)45,46 and the formation of a solid electrolyte interphase (SEI) film (second peak),47 which together with the trapped Li+ in the pores can well explain the first-cycle capacity loss. As depicted in Fig. 3a, the first galvanostatic charge/discharge (GCD) profiles of N-CNS-Ar-20 deliver the maximum initial reversible charge capacity of 765 mA h g−1 at 0.1 A g−1, which is much higher than the 554 mA h g−1 of N-CNS-Ar/H2 with the lowest N content and also twice the theoretical capacity (372 mA h g−1) of graphite. The initial coulombic efficiency (ICE) of N-CNS-Ar-5 is 68%, much higher than those of N-CNS-Ar/H2 (58%), N-CNS-Ar (60%), and N-CNS-Ar-20 (59%), respectively. To further increase the coulombic efficiency, the prelithiation strategy provided in the ESI† has been adopted for N-CNS samples before utilization. Notably, in the first charging curves of N-CNS samples (Fig. 3a), a significantly increased capacity can be observed in the voltage range above around 1.0 V with increasing nitrogen content. Correspondingly, their CV shapes gradually tend to be more rectangular in the medium–high voltage range above 1.0 V with the increase in nitrogen content and a rising tendency of the integrated area of CV curves in the medium–high voltage region (highlighted by a black dotted box) is visually presented (Fig. 3b). It is suggested that the increased surface-controlled pseudocapacity offered by N-doping mainly occurs in the medium–high voltage region. Since the binding of ions at defective sites in graphene is energetically favorable,48 the increased capacity in the medium–high potential region is ascribed to the ion adsorption on active sites, especially N-doping locations. It should be pointed out that the nitrogen content is not the only factor that affects the sloping capacity, while other factors, such as SSA, also influence the electrochemical performance.
 | ||
| Fig. 3 Electrochemical performance of 2D crumpled N-CNS-Ar, N-CNS-Ar/H2, N-CNS-Ar-5, and N-CNS-Ar-20 as anodes for LICs. (a) The first-cycle GCD profiles tested at a current density of 0.1 A g−1. (b) The 4th CV curves measured at 0.1 mV s−1. GCD profiles of (c) N-CNS-Ar-20 and (d) N-CNS-Ar obtained from 0.1 to 100 A g−1. (e) Rate capability from 0.1 to 100 A g−1. (f) Comparison of the rate capability of N-CNS-Ar-20 with the typical carbon anodes reported previously. (g) Cycling performance of N-CNS-Ar-20 tested at 2 and 10 A g−1. | ||
Significantly, the 2D crumpled N-CNS material as an anode exhibits an excellent ability to store and release capacity. At a current density of 0.1 A g−1, N-CNS-Ar-20 shows a large reversible capacity of 620 mA h g−1 in the 10th cycle. With further increasing current densities from 0.2, 0.5, 1, 2, 5, 10, 20, 40 to 60 A g−1, the corresponding specific capacities of N-CNS-Ar-20 decrease from 552, 484, 445, 410, 365, 327, 277, 219 to 173 mA h g−1, respectively, higher than those of N-CNS-Ar, N-CNS-Ar/H2, and N-CNS-Ar-5 at the same current densities (Fig. 3c–e and S8, ESI†). Impressively, even at an ultrahigh current density of 100 A g−1, the reversible capacity of N-CNS-Ar-20 can still reach a high value of 121 mA h g−1 within a charge time of 4.4 s, showing an ultrahigh rate capability with a capacity retention of 19.5% after a 1000-fold increase in current density from 0.1 to 100 A g−1. The most important merit of 2D crumpled N-CNS-Ar-20 is that it simultaneously achieves supercapacitor-like power output rate and battery-like specific capacity. This remarkable rate capability is among the best reported for carbon anodes (Fig. 3f and Table S3†).19,22,23,27,32,46,49–56 When comparing the charging profiles of N-CNS-Ar-20 to those of N-CNS-Ar, N-CNS-Ar/H2, and N-CNS-Ar-5 (Fig. 3c, d and S8, ESI†), it is observed that the slope of their outlines in the medium–high voltage range gradually becomes smaller due to the increase in specific capacity. Such slope-dominated curves could avoid the possibility of Li metal plating and dendrite formation for safety that concerns high-power LICs.57 The linear-capacity-dominated electrochemical behavior enables N-CNS-Ar-20 to be well-suited for high-capacity and fast-charging LICs. In brief, the high-rate capability and large capacity of 2D crumpled N-CNS-Ar-20 can be attributed to the synergistic effect of (i) the 2D structure with abundant mesopore size distribution that favors Li-ion diffusion, guaranteeing superior rate capability, and (ii) the introduced more defects especially the N-5 and N-6, which can contribute to extra capacitive capacity and still store Li ions at high rates. Furthermore, N-CNS-Ar-20 presents excellent cyclability with a capacity retention of 90% and 95% after 3000 cycles at 2 and 10 A g−1, respectively (Fig. 3g). Besides, N-CNS-Ar, N-CNS-Ar/H2, and N-CNS-Ar-5 also exhibit outstanding cyclability at a high rate of 10 A g−1 (Fig. S9, ESI†).
The structural evolution of N-CNS-Ar-20 during the first discharge and charge process is investigated by in situ Raman spectroscopy. As shown in Fig. 4a, there is no obvious position shift of the D band without peak fitting during the discharge process, but its intensity gradually weakens, which is attributed to the lithium adsorption on the defective sites and nanopores of N-CNS-Ar-20.32 As for the G band, its position shifts downward and its intensity decreases with the drop in discharging potential, which is attributed to the insertion of lithium between the graphene layers, and the increase in electrical conductivity caused by lithium insertion.58 As the voltage decreases further from 0.2 to 0.02 V in the lithiation process, lithium continues to intercalate between graphene layers, and therefore the variation of the G band is continuous, and no change suggesting staged graphite intercalation compound (GIC) formation was observed.58 It can be seen that the position and intensity of the D and G bands of N-CNS-Ar-20 can return to their initial state after full charging, suggesting its reversible structural stability, which is also proven by the ex situ XRD patterns. As presented in Fig. 4b, the (002) peak shifts to a lower position upon discharging and recovers back to the pristine angle after delithiation, suggesting an enlarged interlayer spacing caused by lithium-ion intercalation in the graphitic lattices and also its reversible structure. In situ Raman spectra and ex situ XRD patterns support the adsorption-intercalation mechanism for lithium ion storage in N-CNS-Ar-20.
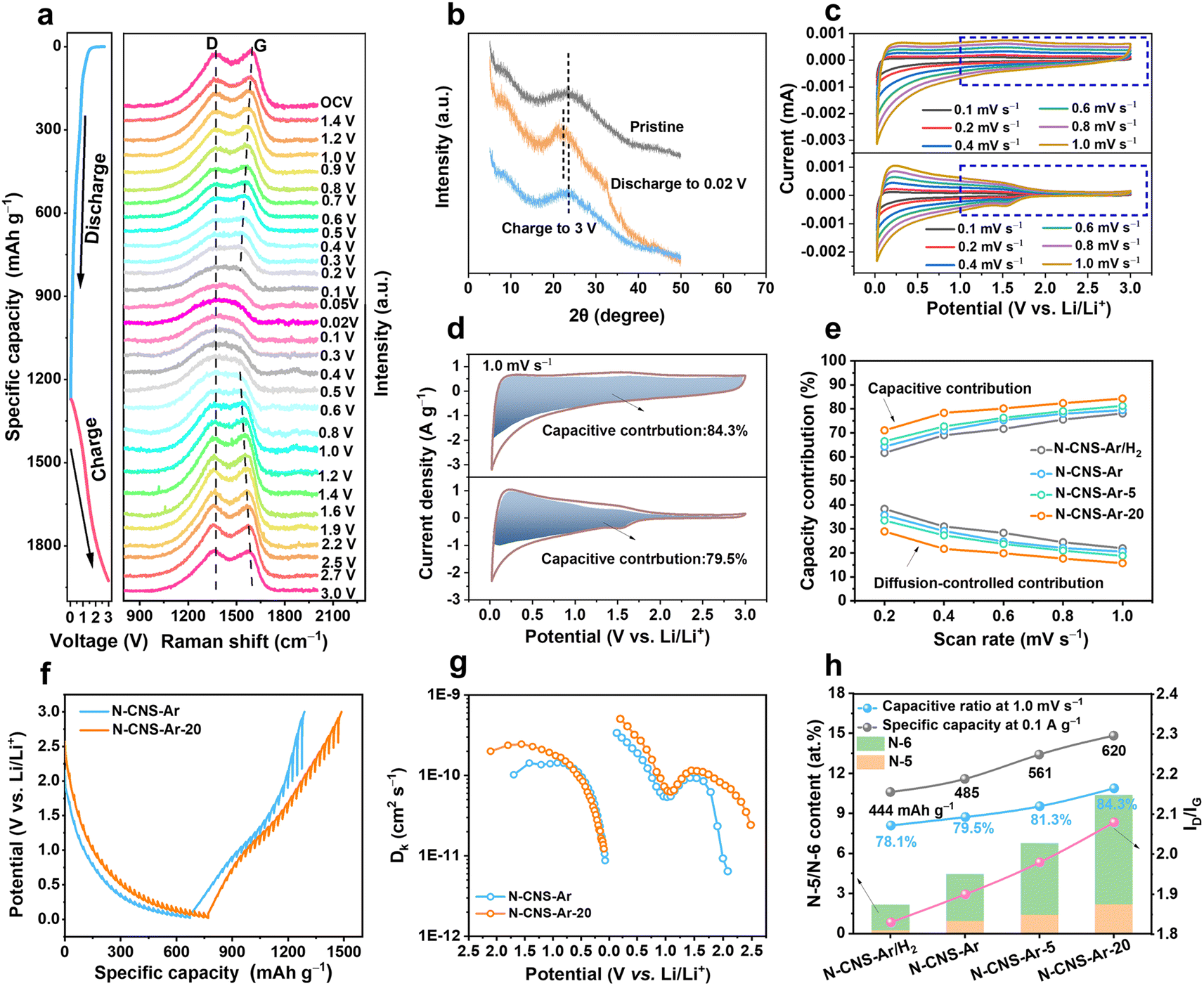 | ||
| Fig. 4 (a) In situ Raman spectra and (b) ex situ XRD patterns obtained during the first discharge/charge process of N-CNS-Ar-20 at 0.1 A g−1. (c) The CV curves at different scan rates and (d) the capacitive contributions at a scan rate of 1.0 mV s−1 of N-CNS-Ar and N-CNS-Ar-20. (e) Normalized capacitive contribution ratios of N-CNS-Ar, N-CNS-Ar/H2, N-CNS-Ar-5, and N-CNS-Ar-20 at different scan rates. (f) GITT profiles and (g) corresponding Li+ diffusion coefficients of N-CNS-Ar and N-CNS-Ar-20 after the 5th cycle at 0.05 A g−1. (h) The relationship among the N-5/N-6 content, ID/IG, capacitive ratio, and specific capacity. | ||
Electrochemical kinetics analyses were performed based on the CV curves and galvanostatic intermittent titration technique (GITT). As shown in Fig. 4c and S10, ESI†, these samples exhibit different CV shapes at the same scan rate, and the CV curves tend to be more rectangular especially in the voltage region above 1.0 V, which is attributed to the increased pseudocapacity caused by nitrogen element doping. To quantitatively distinguish the contribution from different mechanisms, the current at a fixed potential (V) can be regarded as a combination of capacitive (k1v) and diffusion-controlled (k2v1/2) parts.59,60 The calculated results show that the capacitive charge storage process dominates in the N-CNS materials owing to their high capacitive contributions, greater than 60% at different scan rates (Fig. 4e). In particular, N-CNS-Ar-20 has the highest capacitive contributions ranging from 71.0% to 84.3% at scan rates from 0.2 to 1.0 mV s−1. For example, when comparing the capacitive fractions of N-CNS samples at 1.0 mV s−1, N-CNS-Ar-20 possesses the highest capacitive proportion of 84.3% (Fig. 4d, S10b and d, ESI†), indicating that N-CNS-Ar-20 has fast Li+ transport kinetics and can achieve excellent rate capability.
Furthermore, Li+ diffusion was evaluated through the GITT (Fig. 4f). The Li+ diffusion coefficient (DLi+) of N-CNS-Ar-20 is significantly greater than that of N-CNS-Ar before voltage discharging to 0.7 V and after charging to 1.2 V (Fig. 4g), due to the higher capacitive capacity of N-CNS-Ar-20 in the medium–high voltage region, which is mainly contributed by N-doping. The substantial enhancement of DLi+ for N-CNS-Ar-20 is extraordinarily favorable for high-rate capability. However, there is no obvious difference in DLi+ in the low voltage region, revealing that N-doping has no significant contribution to Li+ diffusion in the low potential region.
To understand the higher capacitive contribution of 2D crumpled N-CNS-Ar-20, the correlation between the electrochemical performance and physicochemical parameters (nitrogen configuration and content, d002, ID/IG, micropore/mesopore area, etc.) is further explored. Generally, the defects, heteroatoms, and SSA are regarded as the critical factors that can influence the capacitive capacity.57 It is disclosed that the specific capacity and capacitive contribution display positive correlations with the N-5 and N-6 content, ID/IG, and micropore area, while no apparent relationship is observed with the mesopore area and d002 (Fig. 4h and S11, ESI†). Compared with N-CNS-Ar, N-CNS-Ar/H2 possesses a slightly larger SSA but lower N-5 and N-6 contents, presenting a lower specific capacity and capacitive contribution, demonstrating the importance of N-5 and N-6 in achieving capacitive capacity. Based on the 2D crumpled structure with the abundant mesopore distribution, N-CNS-Ar-20 displays higher N-5 and N-6 contents, ID/IG, and micropore area, and these combined features result in a larger capacitive contribution together with higher specific capacity.
It is clearly demonstrated that Li-storage occurs in the highly N-doped CNS with both large specific capacity and excellent rate capability endowed by a surface-dominated charge storage process. To further unveil the experimental results, first-principles calculations based on density functional theory (DFT) were performed to illustrate the Li+ adsorption and diffusion abilities of the N-doped carbon structure. The optimized graphene models and their Li+ adsorption energies (Eads) are provided in Fig. S12 and Table S4, ESI†, and their corresponding charge density differences are calculated (Fig. 5a–c, S13 and 14, ESI†). The lower the adsorption energy value, the more favorable it is for Li+ adsorption. Compared with the Eads of −1.61 eV for pristine graphene (P-G) (Fig. S13a, ESI†), the Eads of nanopore-defect graphene (ND-G) is calculated to be −4.05 eV and the transferred charge gathersaround the defect, implying that the presence of nanopore defects contributes to Li+ adsorption (Fig. 5a). After incorporating N-Q into P-G and N-6 and N-5 into ND-G, the Eads values are calculated to be −1.33, −4.33, and −4.94 eV (Fig. S12b, d and e, ESI†), respectively. As a result, the charge density tends to accumulate more around the N-5 and N-6 sites, and a less favorable electron transfer for N-Q occurs (Fig. 5b, c and S13b, ESI†). It is indicated that the N-5 and N-6 located at nanopore defects can provide stronger chemical Li+ adsorption ability and are more favorable for improving charge transfer kinetics, while the N-Q exhibits an adverse effect on Li+ adsorption. When the doping number of N-6 is increased to two and four, the Eads values are −5.38 and −6.20 eV, respectively (Fig. S12f, g and S14, ESI†), implying that the defective N-CNS with more N-6 has stronger adsorption ability to grab Li+ at high rates and thus to produce large capacitive capacity. Therefore, it is confirmed that the higher capacity of N-CNS-Ar-20 could benefit from the enhancement of Li+ adsorption energy through the introduction of more defects such as N-6.
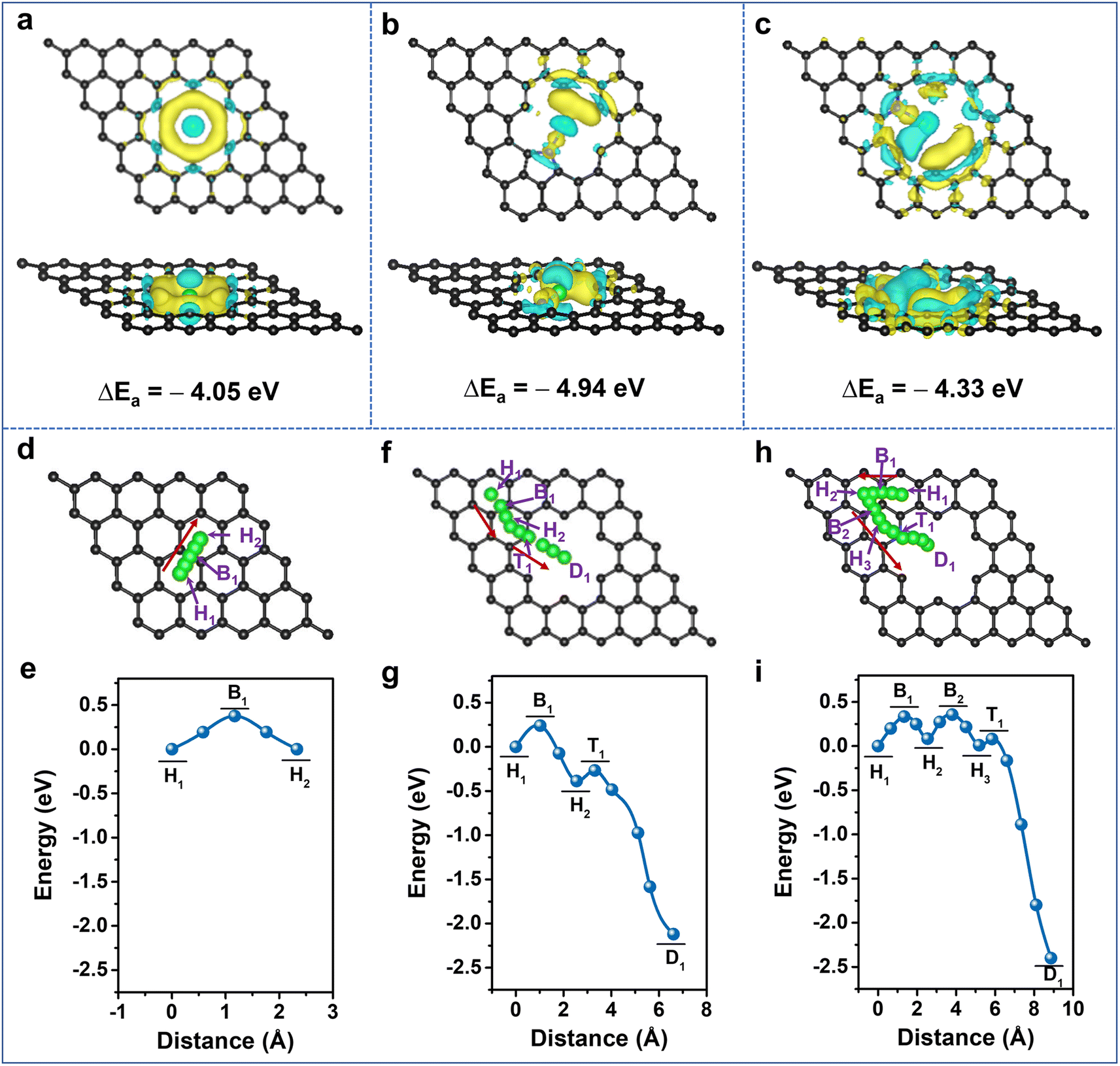 | ||
| Fig. 5 Theoretical calculations of Li+ adsorption and diffusion in different graphene configurations. Top and side views of charge density differences of Li+ adsorbed on (a) ND-G, (b) N-5, and (c) N-6 doped graphene. Yellow and light blue areas represent electron accumulation and depletion, respectively. The isosurfaces are 0.0015 electron bohr−3. Schematic representations and potential–energy curves of Li+ diffusion on graphene following the arrow paths for (d and e) P-G, (f and g) ND-G, and (h and i) N-6 doped graphene. Black, blue, and green balls represent carbon, nitrogen, and lithium, respectively. The Li ion is designed to cross carbon hexatomic rings during the diffusion process to investigate the effect of N-doping and nanopore defects on the diffusion barrier of Li+ crossing nearby carbon hexatomic rings. | ||
Given the importance of heteroatoms and nanopore defects in Li+ diffusion energetics, the diffusion of Li+ near active sites is explored. As presented in Fig. 5d–i and S15, ESI†, the designed diffusion pathways of different models along with the arrows are analyzed, and the corresponding diffusion energy barriers (Ed) are listed in Table S5, ESI†. For P-G, when the distance between Li+ and the hollow site H1 is 1.17 Å, the system energy reaches the maximum value (Fig. 5e), which suggests that Li+ diffuses between adjacent H sites by crossing the C–C bridge site B1 and the Ed is 0.38 eV, which is close to the reported value.61,62 The Ed values between H1 and H2 sites are 0.24, 0.20, 0.33, and 0.42 eV for nanopore-defect, N-5, N-6, and N-Q doped graphene, respectively. It is suggested that nanopore-defect, N-5, and N-6 are good for nearby Li+ diffusion along with carbon hexatomic rings, while the higher Ed value of N-Q manifests its adverse effect on Li+ diffusion (Fig. 5f–i, S15 and Table S5, ESI†). Comparing the Ed values from the H2 (H3) sites to the destination (D) (Fig. 5f–i, S15c, d and Table S5, ESI†), it can be found that Li+ diffusion can be very fast with negligible barriers as small as 0.04 and 0.07 eV for N-5 and N-6 models, respectively,63 indicating the ability of N-5 and N-6 to further reduce the diffusion barrier of ND-G. Besides the investigation of Li+ diffusion in planes, the diffusion energy barriers of Li+ along the direction perpendicular to the graphene were also explored (Fig. S16, ESI†). In comparison with the virtual impossibility of Li+ diffusion in the direction perpendicular to P-G due to a large Ed of 7.71 eV (Fig. S16a and Table S6, ESI†), the defects with nanopores and N-6 allow Li+ to diffuse easily towards themselves and go through the pore to the opposite surface or adjacent graphene's surface (Fig. S16b, c and Table S6, ESI†). Therefore, the N-doping, together with the nanopore defects, is validated to promote Li+ diffusion and contribute to improving the rate capability of highly N-doped carbon nanosheets.
To verify the practical applicability of 2D crumpled N-CNS-Ar-20, a N-CNS-Ar-20//porous carbon LIC (Fig. 6a) was fabricated with pre-lithiated N-CNS-Ar-20 as the anode and porous carbon as the cathode (Fig. S17, ESI†) and operated in the voltage range of 0–4.5 V. Considering the double match of capacity and kinetics between the two electrodes especially at high rates, the mass ratio between the anode and the cathode is selected as 1:2. As displayed in Fig. 6b–c, the reversible capacities of the N-CNS-Ar-20//porous carbon LIC are measured to be 99, 93, 88, 80, 72, 67, 62, 54, and 46 mA h g−1 at the current densities of 0.5, 1, 2, 5, 10, 15, 20, 30, and 40 A g−1, respectively, demonstrating its excellent rate capability. It is noted that the capacity retention of 46.5% of the N-CNS-Ar-20//porous carbon LIC is much higher than the 29.6% of the N-CNS-Ar//porous carbon LIC, which delivers the specific capacity of 98 mA h g−1 at 0.5 A g−1 and 29 mA h g−1 at 40 A g−1. Furthermore, the N-CNS-Ar-20//porous carbon LIC offers highly stable cycling performance with 91.5% capacity retention at 5 A g−1 after 5000 cycles (Fig. 6d). Significantly, our N-CNS-Ar-20//porous carbon LIC provides a high energy density of 223 W h kg−1 at a power density of 1.13 kW kg−1 and still maintains an impressive energy density of 75 W h kg−1 even at an ultrahigh power density of 65 kW kg−1, outputting in a very short time of 4.2 s (Fig. 6e). Such high energy density and power density of the N-CNS-Ar-20//porous carbon LIC outperform those of most previously reported LICs (Fig. 6e and Table S7, ESI†), including high defect mesopore-dominant porous carbon (HDMPC)//HDMPC,28 nitrogen atoms into carbon nanospheres (ANCS)//ANCS,54 Fe2N//activated carbon (Fe2N//AC),64 cyclohexanehexanone//activated carbon (C6O6//AC),65 NbN@C//activated carbon (NbN@C//AC),66 hierarchical pore structural graphene//edge-carbonylated graphene nanosheets (MP-G//G-COOH),27 Co4Fe-BDC//self-made activated carbon (Co4Fe-BDC//SMAC),67 FeSe2@carbon nanofibers//carbon nanofibers@activated carbon (FeSe2@CNF//CNF@AC),68 and nitrogen-doped carbon nanosheets (NCNs-2)//NCNs-2.22
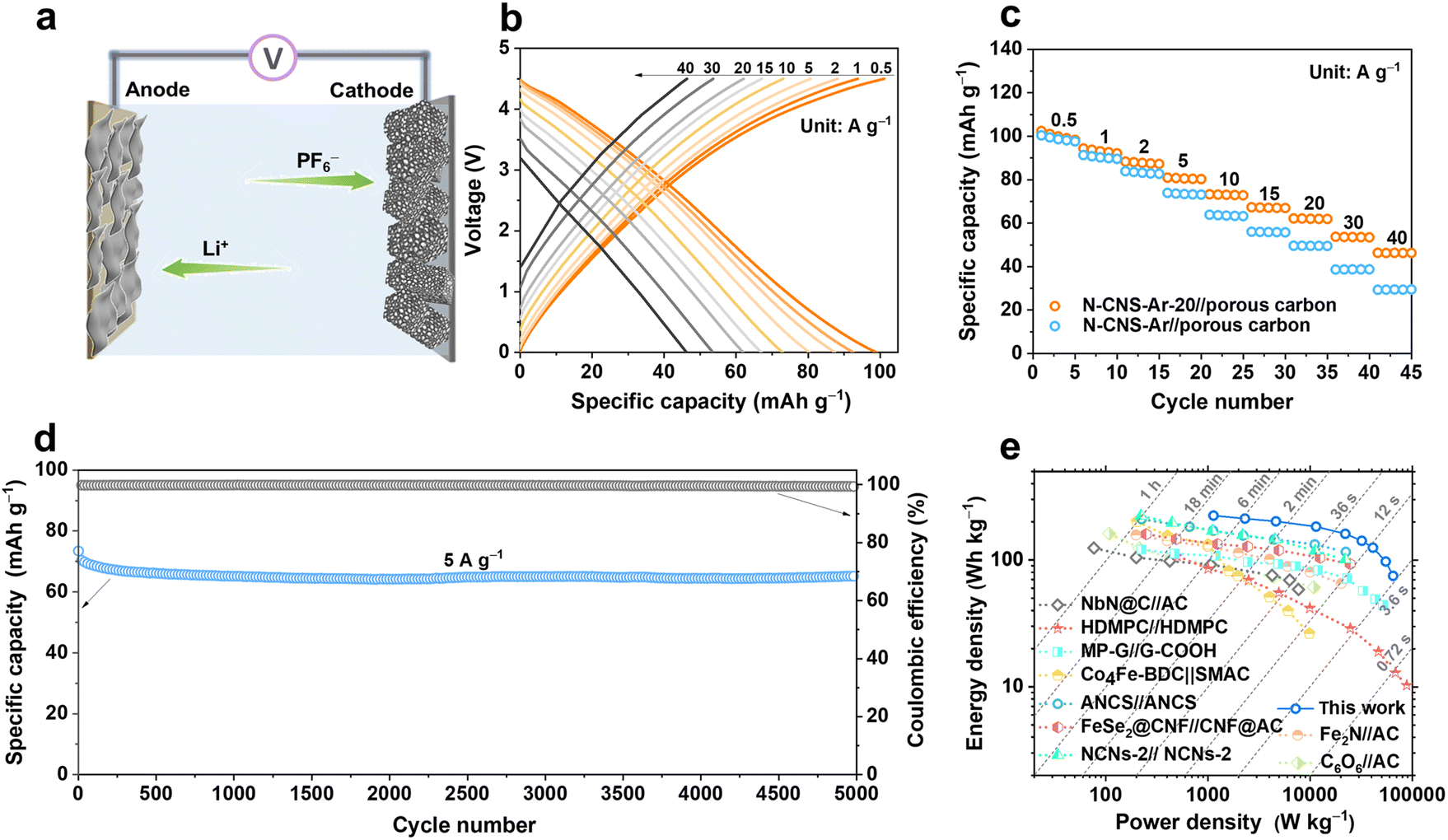 | ||
| Fig. 6 Electrochemical performance of the N-CNS//porous carbon LICs. (a) Schematic illustration of the N-CNS//porous carbon LICs. (b) GCD profiles at different current densities from 0.5 to 40 A g−1 of N-CNS-Ar-20//porous carbon LIC. (c) The rate capability of N-CNS-Ar-20//porous carbon and N-CNS-Ar//porous carbon LICs. (d) Cycling performance at 5 A g−1 of N-CNS-Ar-20//porous carbon LIC. (e) Ragone plot of N-CNS-Ar-20//porous carbon LIC in comparison with recently reported LICs. | ||
Conclusions
In summary, we report a new capacitive-dominated charge storage anode of 2D GO@PPy-derived crumpled N-CNS synthesized by a structure-designed and nitrogen-tailoring strategy, achieving high-energy and ultrafast-charging LICs. The synergistic effect of the unique 2D crumpled structure and abundant nitrogen doping enables the N-CNS material to possess excellent electrochemical performance with large sloping capacity, ultrahigh rate capability and superior cyclability. Especially, the surface-controlled capacity dominates the charge storage process and can be enhanced with increasing N-5 and N-6 content. This is ascribed to the N-doping producing additional pseudocapacity and high Li+ diffusion coefficients, both of which emerged in the medium–high voltage region. The analysis of in situ Raman and ex situ XRD reveals the “adsorption-intercalation” mechanism for lithium storage of N-CNS-Ar-20. Importantly, it is theoretically confirmed that the nitrogen-doping and nanopore defects could increase the Li+ adsorption energy and decrease the Li+ diffusion barrier, which is favorable for improving capacitive capacity and rate performance. The LIC device based on N-CNS-Ar-20//porous carbon delivers a high energy density of 223 W h kg−1 and offers an ultrahigh power density of 65 kW kg−1 at a large energy density of 75 W h kg−1. The pragmatic design that takes advantage of the unique structure and surface modification opens a new avenue to develop fast-charging anodes for achieving advanced electrochemical energy storage with high energy and power density.Author contributions
Z.-S. Wu, F. Li and J. Q. Qin conceived the experiments and supervised this project. F. Y. Liu synthesized the materials and conducted the characterization and performance measurement of electrodes and coin cells. T. Yu performed the theoretical calculation. L. Z. Zhang conducted the AFM characterization. F. Y. Liu, T. Yu, J. Q. Qin, F. Zhou, X. Zhang, Y. W. Ma, F. Li and Z.-S. Wu wrote the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 22125903 and 22005298), the National Key R&D Program of China (Grant 2022YFA1504100), the “Transformational Technologies for Clean Energy and Demonstration” Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA21000000), the Dalian National Laboratory For Clean Energy (DNL), CAS, DNL Cooperation Fund, CAS (DNL202016 and DNL202019), DICP (DICP I2020032 and DICP I202222), the Joint Fund of the Yulin University and the Dalian National Laboratory for Clean Energy (YLU-DNL Fund 2021002 and 2021009).Notes and references
- P. Han, G. Xu, X. Han, J. Zhao, X. Zhou and G. Cui, Adv. Energy Mater., 2018, 8, 1801243 CrossRef.
- B. Li, J. Zheng, H. Zhang, L. Jin, D. Yang, H. Lv, C. Shen, A. Shellikeri, Y. Zheng, R. Gong, J. P. Zheng and C. Zhang, Adv. Mater., 2018, 30, 1705670 CrossRef PubMed.
- L. Xie, C. Tang, Z. Bi, M. Song, Y. Fan, C. Yan, X. Li, F. Su, Q. Zhang and C. Chen, Adv. Energy Mater., 2021, 11, 2101650 CrossRef CAS.
- K. H. Chen, V. Goel, M. J. Namkoong, M. Wied, S. Müller, V. Wood, J. Sakamoto, K. Thornton and N. P. Dasgupta, Adv. Energy Mater., 2021, 11, 2003336 CrossRef CAS.
- J. R. Dahn, T. Zheng, Y. Liu and J. S. Xue, Science, 1995, 270, 590–593 CrossRef CAS.
- G. Yang, X. Li, Z. Guan, Y. Tong, B. Xu, X. Wang, Z. Wang and L. Chen, Nano Lett., 2020, 20, 3836–3843 CrossRef CAS PubMed.
- S. Alvin, H. S. Cahyadi, J. Hwang, W. Chang, S. K. Kwak and J. Kim, Adv. Energy Mater., 2020, 10, 2000283 CrossRef CAS.
- H. Kim, J. C. Hyun, D. H. Kim, J. H. Kwak, J. B. Lee, J. H. Moon, J. Choi, H. D. Lim, S. J. Yang, H. M. Jin, D. J. Ahn, K. Kang, H. J. Jin, H. K. Lim and Y. S. Yun, Adv. Mater., 2023, 35, 2209128 CrossRef CAS PubMed.
- S. Gautier, F. Leroux, E. Frackowiak, A. M. Faugère, J.-N. Rouzaud and F. Béguin, J. Phys. Chem. A, 2001, 105, 5794–5800 CrossRef CAS.
- W. Li, M. Chen and C. Wang, Mater. Lett., 2011, 65, 3368–3370 CrossRef CAS.
- J. Wu, X. Zhang, Z. Li, C. Yang, W. Zhong, W. Li, C. Zhang, N. Yang, Q. Zhang and X. Li, Adv. Funct. Mater., 2020, 30, 2004348 CrossRef CAS.
- G. Wang, M. Yu, J. Wang, D. Li, D. Tan, M. Loffler, X. Zhuang, K. Mullen and X. Feng, Adv. Mater., 2018, 30, 1800533 CrossRef PubMed.
- X. Hu, X. Sun, S. J. Yoo, B. Evanko, F. Fan, S. Cai, C. Zheng, W. Hu and G. D. Stucky, Nano Energy, 2019, 56, 828–839 CrossRef CAS.
- F. Su, C. K. Poh, J. S. Chen, G. Xu, D. Wang, Q. Li, J. Lin and X. W. Lou, Energy Environ. Sci., 2011, 4, 717–724 RSC.
- Q. Sun, D. Li, J. Cheng, L. Dai, J. Guo, Z. Liang and L. Ci, Carbon, 2019, 155, 601–610 CrossRef CAS.
- S. Huang, D. Yang, X. Qiu, W. Zhang, Y. Qin, C. Wang and C. Yi, Adv. Funct. Mater., 2022, 32, 2203279 CrossRef CAS.
- S. N. Lauro, J. N. Burrow, B. G. Broekhuis, P. E. Papa and C. B. Mullins, J. Mater. Chem. A, 2024, 12, 7923–7931 RSC.
- C. Chen, Z. Wang, B. Zhang, L. Miao, J. Cai, L. Peng, Y. Huang, J. Jiang, Y. Huang, L. Zhang and J. Xie, Energy Storage Mater., 2017, 8, 161–168 CrossRef.
- J. Jiang, J. Yuan, P. Nie, Q. Zhu, C. Chen, W. He, T. Zhang, H. Dou and X. Zhang, J. Mater. Chem. A, 2020, 8, 3956–3966 RSC.
- F. Zheng, Y. Yang and Q. Chen, Nat. Commun., 2014, 5, 5261 CrossRef CAS PubMed.
- C. Li, X. Zhang, K. Wang, X. Sun and Y. Ma, Carbon, 2018, 140, 237–248 CrossRef CAS.
- Z. Li, L. Cao, W. Chen, Z. Huang and H. Liu, Small, 2019, 15, 1805173 CrossRef PubMed.
- C. Li, X. Zhang, K. Wang, X. Sun and Y. Ma, J. Power Sources, 2018, 400, 468–477 CrossRef CAS.
- L. Yao, Q. Wu, P. Zhang, J. Zhang, D. Wang, Y. Li, X. Ren, H. Mi, L. Deng and Z. Zheng, Adv. Mater., 2018, 30, 1706054 CrossRef PubMed.
- B. Lee, M. Kim, S. Kim, J. Nanda, S. J. Kwon, H. D. Jang, D. Mitlin and S. W. Lee, Adv. Energy Mater., 2020, 10, 1903280 CrossRef CAS.
- J. Zhu, T. Huang, M. Lu, X. Qiu and W. Zhang, Green Chem., 2024, 26, 5441–5451 RSC.
- L. Jin, X. Guo, R. Gong, J. Zheng, Z. Xiang, C. Zhang and J. P. Zheng, Energy Storage Mater., 2019, 23, 409–417 CrossRef.
- J. Niu, R. Shao, M. Liu, J. Liang, Z. Zhang, M. Dou, Y. Huang and F. Wang, Energy Storage Mater., 2018, 12, 145–152 CrossRef.
- J. Zhang, W. Lv, D. Zheng, Q. Liang, D.-W. Wang, F. Kang and Q.-H. Yang, Adv. Energy Mater., 2018, 8, 1702395 CrossRef.
- J. Zhao, Y.-Z. Zhang, F. Zhang, H. Liang, F. Ming, H. N. Alshareef and Z. Gao, Adv. Energy Mater., 2019, 9, 1803215 CrossRef.
- M. J. Lee, K. Lee, J. Lim, M. Li, S. Noda, S. J. Kwon, B. DeMattia, B. Lee and S. W. Lee, Adv. Funct. Mater., 2021, 31, 2009397 CrossRef CAS.
- S. Huang, Z. Li, B. Wang, J. Zhang, Z. Peng, R. Qi, J. Wang and Y. Zhao, Adv. Funct. Mater., 2018, 28, 1706294 CrossRef.
- X. Yao, Y. Ke, W. Ren, X. Wang, F. Xiong, W. Yang, M. Qin, Q. Li and L. Mai, Adv. Energy Mater., 2018, 9, 1803260 CrossRef.
- J. F. Espinal, T. N. Truong and F. Mondragón, Carbon, 2007, 45, 2273–2279 CrossRef CAS.
- W. Shen and W. Fan, J. Mater. Chem. A, 2013, 1, 999–1013 RSC.
- B. Stöhr, H. P. Boehm and R. Schlögl, Carbon, 1991, 29, 707–720 CrossRef.
- T. Tsuchiya, T. Mori, S. Iwamura, I. Ogino and S. R. Mukai, Carbon, 2014, 76, 240–249 CrossRef CAS.
- Y. Xu, C. Zhang, M. Zhou, Q. Fu, C. Zhao, M. Wu and Y. Lei, Nat. Commun., 2018, 9, 1720 CrossRef PubMed.
- X. Yin, Z. Lu, J. Wang, X. Feng, S. Roy, X. Liu, Y. Yang, Y. Zhao and J. Zhang, Adv. Mater., 2022, 34, 2109282 CrossRef CAS PubMed.
- D. Qiu, J. Guan, M. Li, C. Kang, J. Wei, Y. Li, Z. Xie, F. Wang and R. Yang, Adv. Funct. Mater., 2019, 29, 1903496 CrossRef.
- R. Shi, C. Han, H. Li, L. Xu, T. Zhang, J. Li, Z. Lin, C.-P. Wong, F. Kang and B. Li, J. Mater. Chem. A, 2018, 6, 17057–17066 RSC.
- J. Yang, Z. Ju, Y. Jiang, Z. Xing, B. Xi, J. Feng and S. Xiong, Adv. Mater., 2018, 30, 1700104 CrossRef PubMed.
- J. Chen, Z. Mao, L. Zhang, D. Wang, R. Xu, L. Bie and B. D. Fahlman, ACS Nano, 2017, 11, 12650–12657 CrossRef CAS PubMed.
- S. W. Lee, N. Yabuuchi, P. T. Hammond, Y. Shao-Horn, B. M. Gallant, S. Chen, B.-S. Kim, P. T. Hammond and Y. Shao-Horn, Nat. Nanotechnol., 2010, 5, 531–537 CrossRef CAS PubMed.
- Y. Matsumura, S. Wang and J. Mondori, J. Electrochem. Soc., 1995, 142, 2914–2918 CrossRef CAS.
- Z.-L. Wang, D. Xu, H.-G. Wang, Z. Wu and X.-B. Zhang, ACS Nano, 2013, 7, 2422–2430 CrossRef CAS PubMed.
- K. Tang, R. J. White, X. Mu, M. M. Titirici, P. A. van Aken and J. Maier, ChemSusChem, 2012, 5, 400–403 CrossRef CAS PubMed.
- C. Bommier, T. W. Surta, M. Dolgos and X. Ji, Nano Lett., 2015, 15, 5888–5892 CrossRef CAS PubMed.
- Z.-S. Wu, W. Ren, L. Xu, F. Li and H.-M. Cheng, ACS Nano, 2011, 5, 5463–5471 CrossRef CAS PubMed.
- H. Zhang, X. Xu, H. Wang, Y. Lyu, X. Liu, Y. Zhao, J. Shi, W. Liu, E. Paek and D. Mitlin, ACS Sustainable Chem. Eng., 2018, 7, 2867–2877 CrossRef.
- L. Qie, W.-M. Chen, Z.-H. Wang, Q.-G. Shao, X. Li, L.-X. Yuan, X.-L. Hu, W.-X. Zhang and Y.-H. Huang, Adv. Mater., 2012, 24, 2047–2050 CrossRef PubMed.
- Q. Xia, H. Yang, M. Wang, M. Yang, Q. Guo, L. Wan, H. Xia and Y. Yu, Adv. Energy Mater., 2017, 7, 1701336 CrossRef.
- Y. Yang, Q. Lin, B. Ding, J. Wang, V. Malgras, J. Jiang, Z. Li, S. Chen, H. Dou, S. M. Alshehri, T. Ahamad, J. Na, X. Zhang and Y. Yamauchi, Carbon, 2020, 167, 627–633 CrossRef CAS.
- F. Sun, X. Liu, H. B. Wu, L. Wang, J. Gao, H. L and Y. Lu, Nano Lett., 2018, 18, 3368–3376 CrossRef CAS PubMed.
- R. Mo, F. Li, X. Tan, P. Xu, R. Tao, G. Shen, X. Lu, F. Liu, L. Shen, B. Xu, Q. Xiao, X. Wang, C. Wang, J. Li, G. Wang and Y. Lu, Nat. Commun., 2019, 10, 1474 CrossRef PubMed.
- Y. Qian, S. Jiang, Y. Li, Z. Yi, J. Zhou, T. Li, Y. Han, Y. Wang, J. Tian, N. Lin and Y. Qian, Adv. Energy Mater., 2019, 9, 1901676 CrossRef.
- F. Xie, Y. Niu, Q. Zhang, Z. Guo, Z. Hu, Q. Zhou, Z. Xu, Y. Li, R. Yan, Y. Lu, M. M. Titirici and Y. S. Hu, Angew. Chem., Int. Ed., 2022, 61, e202116394 CrossRef CAS PubMed.
- M. Inaba, H. Yoshida and Z. Ogum, J. Electrochem. Soc., 1996, 143, 2572–2578 CrossRef CAS.
- T.-C. Liu, W. G. Pell, B. E. Conway and S. L. Roberson, J. Electrochem. Soc., 1998, 145, 1882–1888 CrossRef CAS.
- T. Brezesinski, J. Wang, S. H. Tolbert and B. Dunn, Nat. Mater., 2010, 9, 146–151 CrossRef CAS PubMed.
- X. Fan, W. T. Zheng and J.-L. Kuo, ACS Appl. Mater. Interfaces, 2012, 4, 2432–2438 CrossRef CAS PubMed.
- K. Toyoura, Y. Koyama, A. Kuwabara, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 214303 CrossRef.
- J. Hu, B. Xu, S. A. Yang, S. Guan, C. Ouyang and Y. Yao, ACS Appl. Mater. Interfaces, 2015, 7, 24016–24022 CrossRef CAS PubMed.
- F. Li, Y. Li, L. Zhao, J. Liu, F. Zuo, F. Gu, H. Liu, R. Liu, Y. Li, J. Zhan, Q. Li and H. Li, Adv. Sci., 2022, 9, 2203895 CrossRef CAS PubMed.
- S. Li, J. Lin, Y. Zhang, S. Zhang, T. Jiang, Z. Hu, J. Liu, D. Y. Wu, L. Zhang and Z. Tian, Adv. Energy Mater., 2022, 12, 2201347 CrossRef CAS.
- S. Zhou, C. L. Chiang, J. Zhao, G. Cheng, T. Bashir, W. Yin, J. Yao, S. Yang, W. Li, J. Wang, X. Wang, Y. G. Lin and L. Gao, Adv. Funct. Mater., 2022, 32, 2112592 CrossRef CAS.
- X. Xiao, X. Deng, Y. Tian, S. Tao, Z. Song, W. Deng, H. Hou, G. Zou and X. Ji, Nano Energy, 2022, 103, 107797 CrossRef CAS.
- T. Liang, Z. Mao, L. Li, R. Wang, B. He, Y. Gong, J. Jin, C. Yan and H. Wang, Small, 2022, 18, 2201792 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta02681k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |