
A nanoarchitectured 2D–2D heterointerface of Pt@Ti3C2Tx–rGO aerogels via in situ γ-radiolysis induced self-assembly: interplay between strain and ligand effects in electrocatalytic interfaces†
Linsha
Vazhayal‡
,
Sharon Benny
Alex‡
and
Santosh K.
Haram
 *
*
Department of Chemistry, Savitribai Phule Pune University, Ganeshkhind, Pune, 411007, India. E-mail: santosh.haram@unipune.ac.in; Tel: +91 8888840630
First published on 19th September 2024
Abstract
Achieving high-performance and cost-effective Pt-based catalysts with low Pt content and thereby boosting Pt utilization remains a significant challenge in the field of oxygen and hydrogen electrocatalysis. The authentic performance of Pt is often hindered by the occupancy and poisoning of active sites, weak Pt–support interaction, and the degradation of catalysts. To address these issues, we demonstrate a rational design of a low Pt loaded 3D porous aerogel support through self-assembly and reduction of a 2D–2D heterostructure comprising MXene (Ti3C2Tx) and reduced graphene oxide (rGO) via a γ-radiolytic synthesis process. The aerogel heterointerface effectively prevents Ti3C2Tx restacking and aggregation, thereby enhancing the interaction of the electrocatalyst with the electrolyte. Through precise regulation of the heterojunction interface with a strong metal–support interaction (SMSI), the Pt@Ti3C2Tx–rGO catalyst demonstrates excellent electrocatalytic performance for the HER, OER, and ORR. The Pt@Ti3C2Tx–rGO catalyst exhibits efficient ORR activity, with a high onset potential of 0.957 V, and low overpotentials for the HER (43 mV) and OER (490 mV) at a current density of 10 mA cm−2, as well as excellent stability against degradation under acidic conditions. Furthermore, we studied the role of the electronic effects (ligand and strain) induced by SMSI. Spectroscopic analysis confirms that the observed downward shift in the Pt d-band center, attributed to both charge transfer from the support to Pt and compressive strain exerted on the Pt lattice, is responsible for the enhanced electrocatalytic activity. This work successfully offers strategic guidance for charge transfer and strain equilibration in heterointerfaces toward the rational design of advanced electrocatalysts.
1. Introduction
Electrocatalysis plays a significant role within the domain of materials science due to widespread engagement in critical energy related processes. These processes include the oxygen reduction reaction (ORR) for fuel cells, the hydrogen evolution reaction (HER) for green hydrogen production, and the oxygen evolution reaction (OER) for bi-polar metal–air batteries.1 Further progress in these and related technologies hinges on attaining a deeper fundamental understanding of new catalyst interfaces. Pt-based catalyst materials are most widely utilized and extensively studied for oxygen and hydrogen electrocatalysis in both fundamental and applied research. This is primarily due to their inherent low overpotential for these reactions and chemical stability working conditions.1,2 Within this domain, prominent research objectives include: (i) minimizing Pt loading, (ii) optimizing Pt dispersion with smaller particle size; (iii) enhancing the exposure of individual Pt active sites; and (iv) developing a stable support material with a large surface area, chemical stability and excellent electrical conductivity.2,3In pursuit of the aforementioned goals, one feasible approach involves utilizing advanced support materials capable of substantially decreasing the Pt loading without compromising its activity with effective dispersion.4,5 Consequently, this can result in an improved Pt utilization efficiency. Recently, there has been a significant increase in research interest in 2D MXene-based support materials. This is due to their adjustable and uniformly exposed lattice planes, which provide additional catalytic sites, as well as their unique physicochemical properties and electronic structure. Early studies suggest that the integration of different 2D MXenes, such as Ti3C2Tx, Mo2CTx, Mo2TiC2Tx, and V2CTx, into Pt catalytic systems effectively controls the electronic configuration and enhances tolerance to poisoning, resulting in better catalytic performance.5–8
Xie et al. were the first to introduce Pt nanoparticles supported on Ti3C2Tx MXene, serving as a substitute for the conventional carbon support to enhance the ORR activity.6 Subsequent studies have further validated this work, demonstrating that Pt undergoes minimal agglomeration when supported on Ti3C2Tx MXene. Additionally, it retains its conductivity and improves the ability to adsorb oxygen intermediates during the ORR in both acidic and alkaline environments.7 Furthermore, it has been demonstrated that both the electronic structure and terminating groups play a vital role in enabling Ti3C2Tx MXene supports to function effectively in the HER and OER.5 Recent spectroscopic and theoretical investigations have emphasized the significance of the metal d-band in MXene-supported systems, influencing active sites via electronic equilibration. This outcome is attributed to strong metal–support interactions (SMSIs).8,9
However, similar to many 2D materials, MXene demonstrates a pronounced tendency for inter-sheet aggregation (restacking) via hydrogen bonding and van der Waals interaction among its layers.4,5,10,11 This proclivity may degrade its electrocatalytic performance through decreasing the active surface area and thus leading to mass transport limitations. Coupling MXene with diverse active components, viz. 0D, 1D, and 2D nanomaterials, through a heterostructure approach can overcome these limitations.5,12,13 Within the realm of 2D materials, the heterostructure formed by MXene and graphene oxide (GO) or reduced GO (rGO) has garnered significant attention in the field of electrocatalysis. Wang et al. demonstrated that a Mo2CTx MXene–rGO heterostructure, modified with Pt, exhibited a 1.5 times larger electrochemically active surface area, 1.9 times higher mass activity, and 33.6% improved durability for the methanol oxidation reaction (MOR).14 A Ti3C2Tx–rGO heterostructure, synthesized through layer-by-layer assembly and dip coating techniques, exhibited enhanced electrocatalytic efficiency as illustrated by Reveendran et al., for the OER, HER, and MOR.15
Moving beyond conventional heterostructure assembly, the integration of MXene into three-dimensional (3D) aerogel frameworks with the inclusion of Pt opens up an intriguing prospect for improving catalytic performance.16 The intended outcome of this MXene 3D aerogel heterostructure is to increase the internal specific surface area (enabling more active sites) and induce high porosity (conducive to an improved mass transfer rate). To the best of our knowledge, there is only one report illustrating the enhanced performance of 3D rGO–Ti3C2Tx aerogel supports for Pt towards the MOR compared to conventional Pt/C catalysts.12 Nevertheless, further in-depth analysis correlating geometric and electronic properties through the SMSI effect would be a value addition towards enhanced performance of this heterostructure. Furthermore, apart from the MOR, it would be interesting to understand the performance of this catalyst towards the ORR, HER, and OER, through an integrated approach of electro-analysis.
With this motivation, we introduce a distinctive one-pot synthesis method involving γ-ray-mediated technology to form a 3D porous aerogel heterostructure, integrating Pt into the Ti3C2Tx–rGO framework. Previous studies from our group have already established that the γ-radiolysis method serves as a cleaner and effective reduction method of metal salts on catalyst supports.9,17,18 Moreover, this method does not require the addition of any other reducing chemicals and avoids the generation of undesirable by-products on the catalyst surface. In the present work, γ-radiolysis not only reduces the metal salt, as mentioned previously, but also induces the co-reduction of GO to rGO, accompanied by the self-assembly to form the 3D Ti3C2Tx–rGO framework. This approach additionally ensures the uniform distribution of Pt within the interwoven 3D network.
In this study, the ORR, OER, and HER were chosen as proof-of-concept reactions to assess the electrocatalytic characteristics of the resulting Pt@Ti3C2Tx–rGO aerogel catalyst. By virtue of its unique structural advantages as well as strong synergetic coupling effects, the prepared Pt@Ti3C2Tx–rGO aerogel catalyst exhibited enhanced electrocatalytic performance in oxygen and hydrogen electrocatalysis, which are apparently superior to those of conventional Pt/C catalysts. Through systematic spectroscopy analyses, we have unveiled that the surface properties and reactivity of the Pt@Ti3C2Tx–rGO aerogel are intricately influenced by the geometric and electronic structure modifications. These alterations result from the “ligand effect” initiated by SMSI and the “strain effect”, achieved by Pt lattice mismatch, in conjunction with d-band theory.
It is a well-documented fact in the literature that the electrochemical activity of a Pt-based catalyst can be altered by tuning the d-band center through various approaches, such as alloying, changing particle size, introducing strain through single atom doping, substitution, and intercalation processes.3,19 This study highlights that the heterostructure approach causes a downshift in the Pt d-band center, driven by lattice strain and ligand effects, contributing to an enhancement in electrocatalytic performance. Therefore, this work represents the first report of its kind, which unveils the strategic development of heterostructured Pt@Ti3C2Tx–rGO aerogel electrocatalysts, showcasing the interplay of ligand and strain effects that lead to improved activity and durability in both oxygen and hydrogen electrocatalysis.
2. Experimental section
2.1 Materials
All the chemicals and solvents of analytical grade were purchased commercially and utilized without any further purification. Titanium aluminum carbide MAX phase powder (Ti3AlC2), potassium hexachloroplatinate(IV) (K2PtCl6), Nafion 117 (5 wt%) and Pt/C (10 wt%) were procured from Sigma Aldrich. The etchant hydrofluoric acid (HF) (40.0%) and graphite powder were obtained from Loba Chemie Pvt. Ltd. Solvents including ethanol (>99.9%) and dimethyl sulfoxide (DMSO) (>99.7%) were purchased from SD-Fine chemicals, while isopropanol was procured from High Purity Laboratory Chemicals (HPLC). Milli-Q water was utilized for the preparation of all aqueous solutions.2.2 Methodology
Scheme 1 illustrates the formation of the 3D Pt@Ti3C2Tx–rGO aerogel framework, involving the following steps. (a) Synthesis of 2D Ti3C2Tx nanosheets from commercial Ti3AlC2 powder by a wet etching method; (b) synthesis of GO nanosheets from commercial graphite through a modified Hummers' process;20 and (c) synthesis of 3D Pt@Ti3C2Tx–rGO frameworks via a γ-radiolysis process. Detailed procedural steps for each stage are given below.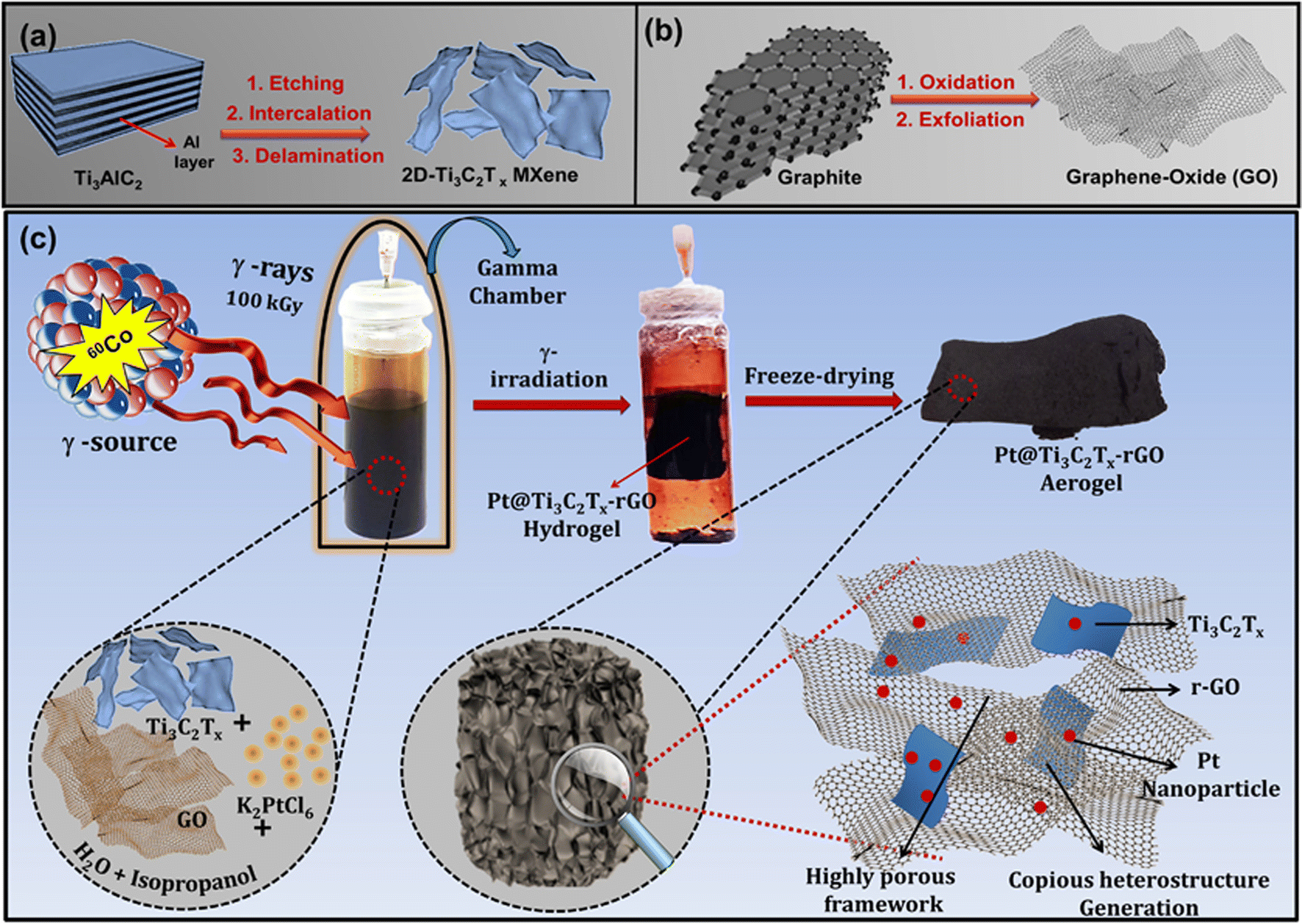 | ||
| Scheme 1 Graphical illustration outlining: (a) synthesis of 2D Ti3C2Tx nanosheets from Ti3AlC2 using wet etching method; (b) synthesis of GO nanosheets from natural graphite through a modified Hummers' process and (c) synthesis of 3D Pt@Ti3C2Tx-rGO aerogel framework via γ-radiolysis process. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :rGO feeding ratios (1:3, 1:1, and 3:1), was accomplished through a straightforward one-step γ-radiolysis method. During the radiolytic synthesis, approved Standard Operating Procedures (SOPs) were ensured in handling radiation facilities with a 60Co source. The typical synthetic pathway, with a Ti3C2Tx:rGO feeding ratio of 1:1, proceeds as follows: 5 mM stock solution of K2PtCl6 was mixed with Ti3C2Tx (2.5 mg mL−1) and GO (2.5 mg mL−1) nanosheets in a solution containing 18.0 mL water and 2.0 mL isopropanol using ultrasonication for 15 min. The reaction vessel was then sealed with a silicone lid and degassed in an inert environment. Subsequently, it was exposed to a 100 kGy dose from a 60Co source at a dose rate of 15 Gy min−1 to obtain a Pt@Ti3C2Tx–rGO hydrogel. (The details of the dose rate calibration with Fricke dosimetry are provided in ESI S2†). The obtained hydrogel underwent successive washes with water and ethanol, followed by freeze-drying at −60 °C (with pressure <10 Pa), resulting in the formation of a 3D Pt@Ti3C2Tx–rGO aerogel. This resultant dried product served as a catalyst in electrochemical reactions.
:rGO feeding ratios (1:3, 1:1, and 3:1), was accomplished through a straightforward one-step γ-radiolysis method. During the radiolytic synthesis, approved Standard Operating Procedures (SOPs) were ensured in handling radiation facilities with a 60Co source. The typical synthetic pathway, with a Ti3C2Tx:rGO feeding ratio of 1:1, proceeds as follows: 5 mM stock solution of K2PtCl6 was mixed with Ti3C2Tx (2.5 mg mL−1) and GO (2.5 mg mL−1) nanosheets in a solution containing 18.0 mL water and 2.0 mL isopropanol using ultrasonication for 15 min. The reaction vessel was then sealed with a silicone lid and degassed in an inert environment. Subsequently, it was exposed to a 100 kGy dose from a 60Co source at a dose rate of 15 Gy min−1 to obtain a Pt@Ti3C2Tx–rGO hydrogel. (The details of the dose rate calibration with Fricke dosimetry are provided in ESI S2†). The obtained hydrogel underwent successive washes with water and ethanol, followed by freeze-drying at −60 °C (with pressure <10 Pa), resulting in the formation of a 3D Pt@Ti3C2Tx–rGO aerogel. This resultant dried product served as a catalyst in electrochemical reactions.
2.3 Characterization
Elaborate descriptions of the electrochemical characterization methods are provided in the ESI (S4–S11†).
3. Results and discussion
3.1 Chemical and physical characterization of the Pt@Ti3C2Tx–rGO aerogel
Fig. 1a–f reveal a panoramic view illustrating the efficient exfoliation and delamination of ultrathin Ti3C2Tx MXene from the solid, dense 3D Ti3AlC2 structure. Initially, an accordion-shaped Ti3C2Tx MXene was obtained by selectively etching the Al layer using HF from the block-like laminated structure of Ti3AlC2.6 Further delamination resulted in the formation of several Ti3C2Tx layers, each measuring 1–2 μm in size (see Fig. 1g and S1†). The AFM images of the Ti3C2Tx MXene nanosheets (Fig. S1†) show an average thickness of around 2.7 nm, indicating that the nanosheets are composed of either single or double layers, which confirms the effectiveness of the exfoliation and delamination process. XRD patterns in Fig. 1h and i further confirm the successful transformation of Ti3AlC2 to Ti3C2Tx MXene nanosheets by the disappearance of the strongest diffraction peak at 38.7° corresponding to the (104) plane of the Al layer from the Ti3AlC2 MAX phase.12,15 Additionally, the downshift from 9.6 to 6.9° and the broadening of the (002) basal plane peaks of Ti3C2Tx indicate an increase in the interlayer distance (from 9.3 to 13.4 nm) and a reduction in the thickness of Ti3C2Tx MXene layers. Furthermore, the average crystallite size of the Ti3AlC2 MAX phase and Ti3C2Tx MXene reduced from 32.7 to 6.6 nm after etching, intercalation and delamination processes (the summary of the d(200) XRD peaks is provided in Table S1†). These changes indicate the successful transformation of solid dense Ti3AlC2 to ultrathin Ti3C2Tx nanosheets. Additionally, XPS and FTIR analyses offered a detailed understanding of the surface composition and functional groups, such as Ti–Tx, –OH, and –F, shedding light on the chemical environment and potential reactivity of delaminated Ti3C2Tx (detailed analysis is presented in Fig. S2 and S3†). | ||
| Fig. 1 SEM images of (a) Ti3AlC2 MAX phases, (b) exfoliated Ti3C2Tx and (c) delaminated Ti3C2Tx MXenes and (d)–(f) respective magnified images. (g) TEM image of a few layers of Ti3C2Tx MXene. XRD pattern of (h) bulk Ti3AlC2 in comparison with exfoliated and delaminated Ti3C2Tx MXenes, indicating the peak shift, and (i) corresponding magnified image showing the peak shift after Ti3C2Tx MXene formation. | ||
GO was selected as the spacer precursor due to its layered configuration similar to Ti3C2Tx, featuring a large number of hydrophilic functional groups and good solution processability. The successful chemical oxidation and exfoliation of graphite into GO were confirmed by the shift of the (002) peak in XRD (Fig. S4†) towards a smaller angle of 11.8°, indicating an increase in d-spacing from 0.342 to 0.881 nm (Table S2†).20 Furthermore, the FTIR spectrum of GO (Fig. S5†) confirms the successful oxidation of graphite, revealing various oxygen-containing functional groups. These groups enhance its hydrophilic nature and chemical reactivity. The SEM and AFM images (Fig. S6†) of the GO nanosheets reveal a lateral dimension of 3–5 μm and a thickness ∼2 nm for monolayers to a few layers. Both Ti3C2Tx MXene and GO sheets are negatively charged when dispersed in water with zeta potentials of −40.46 and −37.95 mV, respectively. Therefore, they can form a stable and uniformly mixed colloidal solution after ultrasonic treatment and the subsequent radiolysis process.
During radiolysis, γ-radiation cleaves water molecules to produce both reductive (eaq.−, H2, and H˙) and oxidative (˙OH, H3O+, and H2O2) species (eqn (1)).17,21
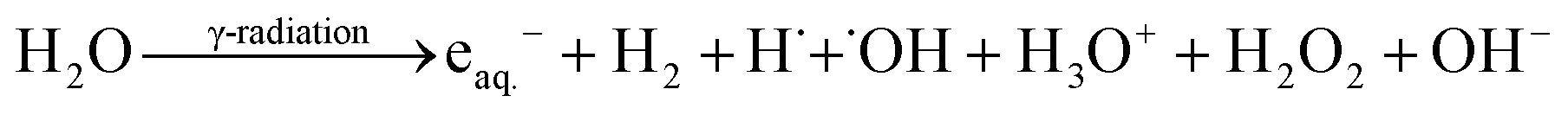 | (1) |
˙OH is the prominent oxidizing species with redox potential E0 (˙OH/H2O) = +2.8 VNHE, while eaq.− and H˙ are the active and strong reducing agents with redox potentials of E0 (H2O/eaq.−) = −2.87 VNHE and E0 (H+/H˙) = −2.3 VNHE, respectively.17 Additionally, ˙OH can be scavenged efficiently by (CH3)2CHOH to be converted into reductive radicals [(CH3)2C˙OH] (E0 ((CH3)2CHO/(CH3)2C˙OH) = −1.8 VNHE), generating an exclusively reducing environment in the reaction mixture.
| (CH3)2CHOH + ˙OH → (CH3)2C˙OH + H2O | (2) |
These reductive species play a dual role of reducing GO to rGO22 and converting [PtCl6]2− ions into Pt[0] nanoparticles (eqn (3) and (4)). It also efficiently triggers the self-assembly and gelation of Ti3C2Tx and rGO nanosheets through van der Waals interactions and hydrogen bonding.
| [PtCl6]2− + 2eaq.− → [PtCl4]2− + 2Cl− (E0 = 0.68 V) | (3) |
| [PtCl4]2− + 2eaq.− → Pt + 4Cl− (E0 = 0.75 V) | (4) |
Moreover, in situ generated Pt nuclei get readily integrated with unsaturation in the 3D framework of the Ti3C2Tx–rGO surface to form a Pt@Ti3C2Tx–rGO hydrogel. The subsequent freeze drying process yielded the Pt@Ti3C2Tx–rGO aerogel architecture without disrupting the porous architecture.
The crystalline and chemical structures of the Pt@Ti3C2Tx–rGO aerogel catalyst were characterized by XRD and Raman spectroscopy. Fig. 2a shows the XRD pattern of the Pt@Ti3C2Tx–rGO aerogel, along with control samples Ti3C2Tx, rGO, Pt@rGO and Pt@Ti3C2Tx, obtained after radiolysis. The diffraction peaks at 7.2, 18.1, 27.4, and 60.5° were assigned to the crystal planes (002), (004), (006), and (110) of Ti3C2Tx, respectively. A weak peak at 25° is attributed to the formation of TiO2, resulting from the mild oxidation of Ti3C2Tx by oxidative species (˙OH, H3O+, and H2O2) (eqn (1)) formed during radiolysis.21 The conversion of GO to rGO during the radiolysis process is evident from a broad peak at 23.1° corresponding to the (002) plane of rGO.20 Primary diffraction peaks corresponding to both Ti3C2Tx MXene and rGO were identified in the Pt@Ti3C2Tx–rGO aerogel, supporting the prospective self-assembly of Ti3C2Tx and rGO. Additionally, in the Pt@Ti3C2Tx–rGO, Pt@rGO, and Pt@Ti3C2Tx samples, three characteristic peaks were evident at 2θ = 39.8, 46.3, and 67.5°, corresponding to the (111), (200), and (220) planes of the cubic Pt nanocrystals, respectively (JCPDS 87-0646).7,23
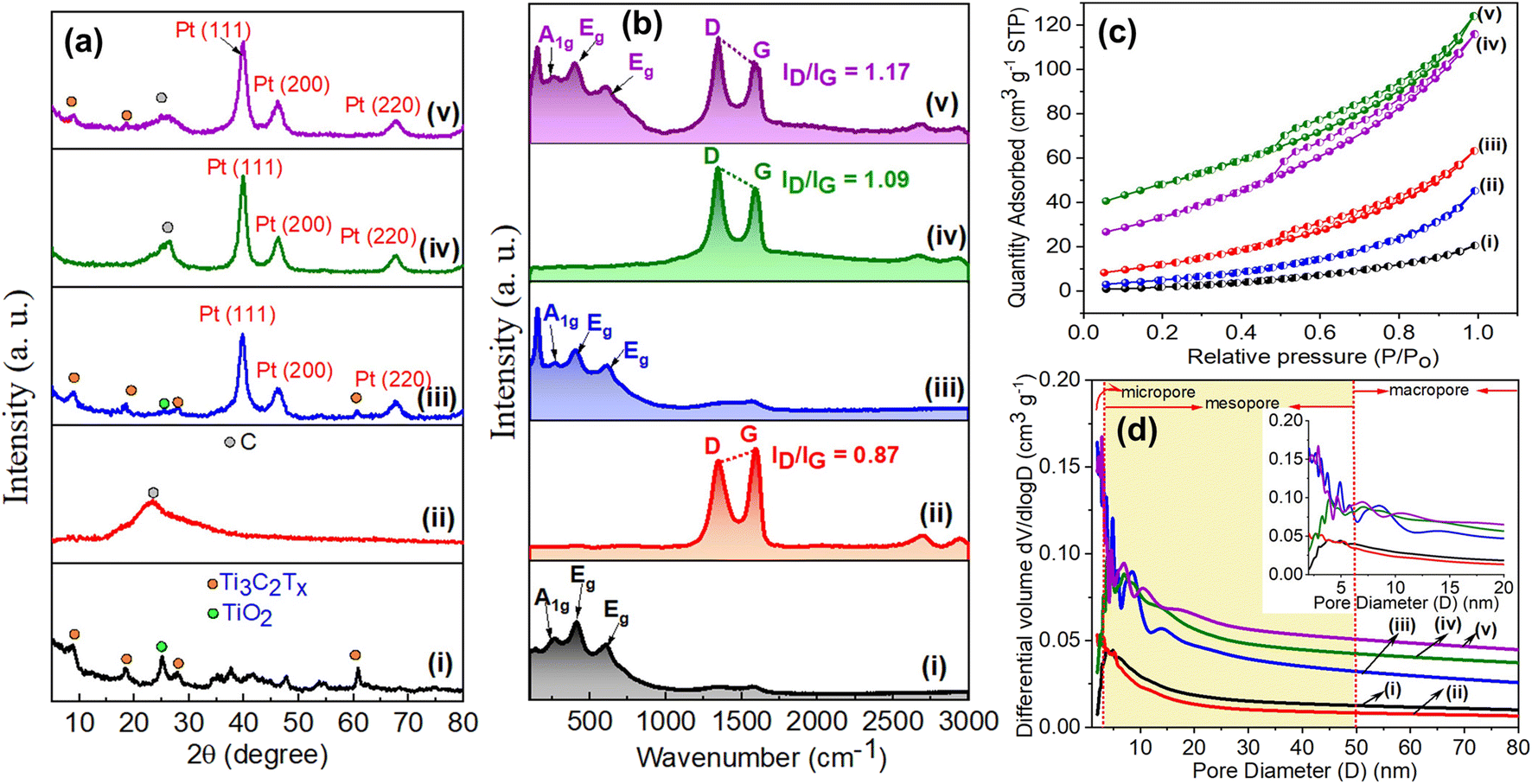 | ||
| Fig. 2 (a) X-ray diffraction pattern, (b) Raman spectra, (c) N2 adsorption–desorption isotherms, and (d) pore size distribution curve of (i) Ti3C2Tx, (ii) rGO, (iii) Pt@Ti3C2Tx, (iv) Pt@rGO and (v) Pt@Ti3C2Tx–rGO. | ||
Fig. 2b shows the Raman spectrum of the Pt@Ti3C2Tx–rGO aerogel and the control samples as described in XRD analysis. The Raman spectrum of the Pt@Ti3C2Tx–rGO aerogel displays a combined feature of the fingerprint signals of Ti3C2Tx MXene and rGO in a range of 120–750 and 1300–1600 cm−1, respectively.24 The peak at 270 cm−1 is attributed to the A1g symmetry out-of-plane vibrations of Ti and C atoms. The signals at 414 and 622 cm−1 were assigned to the Eg group vibrations resulting from the in-plane (shear) modes of Ti, C, and surface functional groups of Ti3C2Tx.12,24 Two strong peaks at 1346 and 1594 cm−1 are assigned to the D and G bands of graphitic carbon from rGO, respectively.12 No discernible shift in the Raman peaks of the Pt@Ti3C2Tx–rGO aerogel is observed relative to pristine samples, suggesting that the self-assembly process does not change the chemical structure of Ti3C2Tx and rGO. The intensity ratio of D and G bands (ID/IG) is used to evaluate the extent of defects in samples. Apparently, the ID/IG value for Pt@Ti3C2Tx–rGO (1.17) is higher than that for Pt@rGO (1.09) and rGO (0.84). This observation underlines a higher defect density in Pt@Ti3C2Tx–rGO interactive layers.12 Therefore, Raman analysis of these samples suggests the formation of a heterostructure through self-assembly of Ti3C2Tx MXene and rGO nanosheets.
Furthermore, XPS measurements of the Pt@Ti3C2Tx–rGO architecture reveal the presence of Pt, Ti, C, O, and F (Fig. S7a†). The Ti 2p spectrum (Fig. S7b†) shows six peaks: Ti–C 2p1/2 and Ti–C 2p3/2 at 461.5 eV and 455.3 eV, respectively; Ti–X 2p1/2 and Ti–X 2p3/2 at 459.8 eV and 456.1 eV, indicating substoichiometric titanium carbide or oxycarbides; and Ti–O 2p1/2 and Ti–O 2p3/2 at 463.68 eV and 457.8 eV.7,12 The slight upward shift in Ti 2p3/2 binding energy suggests strong electronic coupling between Ti3C2Tx and rGO. The C 1s spectrum (Fig. S7c†) shows peaks for C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (284.5 eV), C–O (286.5 eV), CO (287.5 eV), and O–CO (288.8 eV), indicating functional groups such as epoxides, hydroxyls, and carboxyls, with observed shifts due to interactions between rGO and Ti3C2Tx.25 Fig. S7d† presents the Pt 4f spectrum of Pt@Ti3C2Tx–rGO, showing two doublet pairs. Peaks at 71.0 and 74.4 eV correspond to metallic Pt0, while peaks at 72.0 and 76.9 eV indicate Pt2+. The lower binding energies of these peaks in Pt@Ti3C2Tx–rGO compared to Pt@rGO and Pt@Ti3C2Tx highlight a strong electronic interaction between Pt and Ti3C2Tx–rGO.6 This interaction enhances the stability of Pt and facilitates electron transfer from Ti3C2Tx–rGO to Pt nanoparticles, reflecting changes in the electronic structure and bonding due to the heterostructure formation.7
C (284.5 eV), C–O (286.5 eV), CO (287.5 eV), and O–CO (288.8 eV), indicating functional groups such as epoxides, hydroxyls, and carboxyls, with observed shifts due to interactions between rGO and Ti3C2Tx.25 Fig. S7d† presents the Pt 4f spectrum of Pt@Ti3C2Tx–rGO, showing two doublet pairs. Peaks at 71.0 and 74.4 eV correspond to metallic Pt0, while peaks at 72.0 and 76.9 eV indicate Pt2+. The lower binding energies of these peaks in Pt@Ti3C2Tx–rGO compared to Pt@rGO and Pt@Ti3C2Tx highlight a strong electronic interaction between Pt and Ti3C2Tx–rGO.6 This interaction enhances the stability of Pt and facilitates electron transfer from Ti3C2Tx–rGO to Pt nanoparticles, reflecting changes in the electronic structure and bonding due to the heterostructure formation.7
The N2 adsorption–desorption analysis was carried out to further investigate the porous features and specific surface area of the prepared samples. All adsorption–desorption isotherms (Fig. 2c) correspond to the combination of typical type II and type III behaviour according to IUPAC classification, indicating the presence of meso- and macro-pores in the structure.12,20 The isotherms of Pt@Ti3C2Tx–rGO, Pt@rGO, and rGO exhibit a distinct hysteresis loop of type H3, validating the existence of slit-like pores between nanosheets.20 The pore size distribution of the samples was calculated from the desorption branch using the Barrett–Joyner–Halenda (BJH) method, as shown in Fig. 2d. Pt@Ti3C2Tx–rGO displays a hierarchical porous structure, with meso- and macro-pores. This porous architecture plausibly originated from the interspaces between Ti3C2Tx and rGO nanosheets formed in the self-assembly process during radiolysis. The Brunauer–Emmett–Teller (BET) surface area of the Pt@Ti3C2Tx–rGO aerogel was calculated to be in the range of 125.32–101.21 m2 g−1, which is significantly larger than that of bare Ti3C2Tx (13.23 m2 g−1) and rGO (96.56 m2 g−1). The BET surface area, pore size and volume of all the samples are summarized in Table S3.† The existence of such a hierarchical porous structure and substantial surface area allows the catalyst to facilitate effective mass transfer at the active sites, thereby lowering diffusion resistance and expectedly boosting catalytic activity.
Fig. 3a illustrates the fluffiness of the aerogel, able to rest effortlessly on a dandelion. The calculated density of the prepared samples ranged between 23 and 30 mg cm−3. It should be noted that after radiolysis and freeze-drying, the sample surface remained intact. The morphology and nanostructures of the 3D Pt@Ti3C2Tx–rGO architecture were examined by FE-SEM, TEM, and HAADF-STEM (all the following results are shown for the sample Pt@Ti3C2Tx–rGO 3:1). As can be seen from SEM images in Fig. 3b–d, the Pt@Ti3C2Tx–rGO aerogel displays a 3D crosslinked framework of numerous nanosheets, with Ti3C2Tx MXene wrapped in the large nanosheets of rGO. This unique configuration not only prevents the restacking or re-aggregation of Ti3C2Tx and rGO but is also expected to enhance the permeation of electrolyte into the interior active metal sites during electrocatalysis. TEM images in Fig. 3e–g clearly reveal that Pt nanoparticles have a rough spheroid-like morphology with size ranging from 2 to 6 nm, which are well dispersed within the Ti3C2Tx and rGO matrix with no much aggregation. The Gaussian fitting of the size histogram for approximately 30 different Pt particles resulted in an average particle size of 4.90 nm (see the inset of Fig. 3f and g). The HR-TEM image of Pt reveals well-resolved lattice fringes with an interplanar distance of 0.227 nm indexed to the (111) plane (Fig. S8a†). The selected area electron diffraction (SAED) patterns (Fig. S8b and c†) displayed clear diffraction rings of the (006), (103), (105), and (110) planes of Ti3C2Tx, along with the (111), (200), and (220) planes of fcc crystals of Pt. These observations align with the results obtained from XRD analysis. These findings collectively imply a robust connection between Pt and the Ti3C2Tx–rGO support, while also revealing the polycrystalline structure of the catalyst.
 | ||
| Fig. 3 (a) Photograph of the Pt@Ti3C2Tx–rGO 3:1 aerogel placed on a dandelion, suggesting the featherweight density of the sample. (b)–(d) SEM images and (e)–(g) HR-TEM images (inset shows the particle size distribution histogram of Pt over Pt@Ti3C2Tx–rGO 3:1) and (h)–(i) HAADF-STEM images and the corresponding elemental maps of Ti, C, Pt, O and F for Pt@Ti3C2Tx–rGO 3:1. | ||
The HAADF-STEM image, along with the corresponding EDS elemental mapping, provides additional insights into the heterostructure of the Pt@Ti3C2Tx–rGO aerogel. The larger rGO nanosheets enveloping the smaller Ti3C2Tx nanosheets are well confirmed by the HAADF-STEM image in Fig. 3h. The EDS elemental mapping (Fig. 3i) of Pt@Ti3C2Tx–rGO indicates an even distribution of the characteristic elements Ti, F, and O from Ti3C2Tx, along with the C element from rGO and Ti3C2Tx. Furthermore, the Pt nanoparticles exhibit excellent dispersion across these Ti3C2Tx–rGO nanosheets, highlighting their effective support. This suggests that the binding of Pt to the porous Ti3C2Tx–rGO heterostructure can enhance the conductive pathways between the 2D/2D layers, which is anticipated to increase catalytic activity by promoting efficient charge transfer.
3.2 Electrocatalytic performance of the Pt@Ti3C2Tx–rGO aerogel catalyst
Motivated by the intriguing structural features, we investigated the ORR activities of the Pt@Ti3C2Tx–rGO aerogel catalyst featuring different ratios of Ti3C2Tx–rGO support (1:3, 1:1, and 3:1) using the Rotating Disk Electrode (RDE) and Rotating Ring Disk Electrode (RRDE). The Pt content of all the samples was calculated by ICP-MS measurement and kept constant within the range of ∼7–9 wt% (Table S4†). Accordingly, Pt/C with low Pt content (E-TEK 10 wt% Pt) was chosen as the benchmark for comparison. Fig. 4a shows CV (Cyclic Voltammetry) curves for Pt@Ti3C2Tx–rGO, Pt@Ti3C2Tx, Pt@rGO and Pt/C in an O2-saturated 0.1 M HClO4 solution. All the samples displayed the essential features of the Pt surface viz. hydrogen adsorption/desorption (0.05–0.35 V) and formation/reduction of Pt oxide (0.6–1.2 V), resulting in the commonly recognized “butterfly” shape.26
 | ||
| Fig. 4 (a) Cyclic voltammetry curves in O2-saturated 0.1 M HClO4 at a scan rate of 100 mV s−1, (b) ORR polarization curves of 1600 rpm at 10 mV s−1, (c) Tafel curves obtained from polarization curves in (b) for Pt@Ti3C2Tx–rGO 1:3, Pt@Ti3C2Tx–rGO 1:1, Pt@Ti3C2Tx–rGO 3:1, Pt@Ti3C2Tx, Pt@rGO, and Pt/C catalysts. (d) RRDE polarization curve of Pt@Ti3C2Tx–rGO 3:1 at different rotation rates; (e) number of electrons transferred by Pt@Ti3C2Tx–rGO 3:1 and Pt/C determined using the K–L plot and RRDE method; (f) accelerated durability test (ADT) for Pt@Ti3C2Tx–rGO 3:1 and Pt/C before and after 10k cycles. | ||
Previous investigations have confirmed that hydrogen adsorption/desorption behaviours are influenced by the surface atomic arrangement of Pt. In fact, the CV curve for clean Pt (111) exhibits broad symmetrical features, while a well-defined redox peak is observed for the Pt (100) surface.27,28 Here, all catalysts exhibit similar features in the hydrogen adsorption/desorption region to Pt/C, except the Pt@Ti3C2Tx–rGO 3:1 catalyst. Notably, the Pt@Ti3C2Tx–rGO 3:1 catalyst presents a small redox peak at 0.31 V in the “hydrogen desorption region,” characteristic of the Pt (100) surface. The volume fraction of Pt crystallites with (100) orientation was computed by integrating the intensity ratio between the (111) and (220) peaks (details are provided in ESI Table S5†).27 From the table, it is inferred that Pt@Ti3C2Tx–rGO 3:1 has a higher fraction of (100) sites compared to other compositions. Therefore, the Pt within Pt@Ti3C2Tx–rGO 3:1 is enclosed by the (100) surface in addition to the (111) plane, resulting in the observed additional peak in the CV.27,28 These results suggest that Pt anchored to an optimized combination of Ti3C2Tx and rGO heterostructures could expose more abundant (111) and (100) surfaces that are active towards the ORR.
The electrochemically active surface areas (ECSAs) of all the catalysts derived from hydrogen under the potential desorption (HUPD) area (details given in S5†) are summarized in Table S6.† Among them, Pt@Ti3C2Tx–rGO 3:1 demonstrated the highest ECSA (80.12 m2 g−1), suggesting effective dispersion of Pt on it, thereby providing the largest number of active sites for the reaction. A discernible positive shift in cathodic peak potential is observed in the CV curve during the reduction of oxygen species adsorbed on the Pt@Ti3C2Tx–rGO 3:1 catalyst, relative to Pt/C. This suggests that Pt@Ti3C2Tx–rGO 3:1 reduces the desorption free energy for Pt–O, Pt–OH, or Pt–O2 species, probably due to the participation of the Ti3C2Tx–rGO heterostructure support. Consequently, this enhances their availability during the ORR.
Fig. 4b displays the ORR polarization curves of all the catalysts recorded at 1600 rpm. The onset (Eonset) and half-wave (E1/2) potentials, derived from Fig. 4b, follow the order: Pt@Ti3C2Tx–rGO 3:1 > Pt@Ti3C2Tx > Pt@Ti3C2Tx–rGO 1:1 > Pt@Ti3C2Tx–rGO 1:3 > Pt/C > Pt@rGO. This result suggests that Pt@Ti3C2Tx–rGO 3:1 is the best among them with the highest Eonset and E1/2 of 72 and 47 mV respectively, higher than those of commercial Pt/C. Moreover, Pt@Ti3C2Tx–rGO 3:1 displayed a higher mass transport limiting diffusion current density of −6.98 mA cm−2, signifying efficient diffusion and transport of reactants. Additionally, the higher intrinsic electrocatalytic activity of the Pt@Ti3C2Tx–rGO 3:1 catalyst was confirmed by its higher mass and specific activities (details are given in S6†) of 0.132 mA μgPt−1 and 0.146 mA cm−2 compared to Pt/C, 0.021 mA μgPt−1 and 0.097 mA cm−2, respectively. All the parameters that quantify the catalytic efficiency of the investigated catalysts in the ORR have been evaluated and included in Table S6.† The optimal catalytic performance of Pt@Ti3C2Tx–rGO 3:1 arises from the well-suited ratio of Ti3C2Tx–rGO (3:1). This ratio achieves a harmonious combination of the 3D porous heterostructure and optimized Pt electronic structure, leading to the observed increment of activity.
To quantify the ORR efficiency of all catalysts, Linear Sweep Voltammetry (LSV) curves were studied at different rotation rates (Fig. S9†). Based on the Koutecky–Levich (K–L) equation (details are given in S7†) for the diffusion controlled region, a series of plots were obtained showing a linear relationship between j−1 and ω−1/2 over the potential range of 0.2–0.6 V (inset of Fig. S9†). The K–L plot of all catalysts indicated first-order reaction kinetics, demonstrating that the rate of the ORR is predominantly governed by mass transport.29 The electron transfer number (n) was calculated to be in the range of 4.1–4.6 from the slopes of K–L plots, indicating that the ORR proceeds via a four-electron pathway. Herein, the ‘n’ value obtained is slightly larger than that of Pt/C (3.98–4.0), the theoretical value (∼4) and other reported values.29–31 This is probably attributed to the accumulation of O2 within the macropores of the aerogel framework, which can influence the kinetics of the ORR process and potentially lead to deviations from the expected electron-transfer number.32Fig. 4c shows a plot of mass transfer corrected current density (jk) from LSV experiments vs. overpotential for all the samples. From all plots, the Tafel slope values were calculated to be ∼120 mV dec−1. This slope value suggests the presence of an oxide-free Pt surface with single electron transfer as the rate determining step.31,33
To obtain a more comprehensive understanding of the ORR pathway, the best performing Pt@Ti3C2Tx–rGO 3:1 catalyst was further characterized using the RRDE (Fig. 4d), by monitoring the formation of intermediate peroxide species (HO2−) (details are given in S8 and S9†). On the basis of the disk and ring currents, the n values for Pt@Ti3C2Tx–rGO 3:1 fell within the range of 3.92 to 4.12 across the potential range of 0.2–0.6 V, similar to that of Pt/C (3.80–3.91) (Fig. 4e). This confirms that the reaction proceeds predominantly via a four-electron mechanism, in line with the results determined from K–L plots. The calculated yields of H2O2 for the Pt@Ti3C2Tx–rGO 3:1 and Pt/C electrodes are less than ∼6% and ∼4% respectively, in the potential range of 0.2–0.6 V (Fig. S10†). The higher electron transfer number and lower H2O2 production yield observed for Pt@Ti3C2Tx–rGO 3:1 indicate a greater contribution of the parallel reactions compared to Pt/C.34
To assess the electrochemical stability of Pt@Ti3C2Tx–rGO 3:1, accelerated durability tests (ADTs) were conducted. Following 10000 consecutive potential cycles, Pt@Ti3C2Tx–rGO 3:1 exhibited a minimal negative shift of 18 mV in E1/2 (Fig. 4f). This remarkable durability surpasses that of Pt/C, which experienced a shift of 30 mV under similar testing conditions. These results again highlight the superior stability of Pt@Ti3C2Tx–rGO 3:1.
Moreover, the methanol tolerance of a cathode material is a critical factor in alcohol fuel cells. Fig. S11† compares the methanol tolerance of Pt@Ti3C2Tx–rGO 3:1 and Pt/C using chronoamperometry for the ORR in O2-saturated 0.1 M HClO4. When methanol was introduced at 1500 s, the Pt@Ti3C2Tx–rGO 3:1 catalyst showed only a slight current density fluctuation before stabilizing. In contrast, the commercial Pt/C catalyst showed a significant current density increase and took longer to return to its original level. This indicates that Pt@Ti3C2Tx–rGO 3:1 has better methanol tolerance compared to Pt/C.
In rechargeable batteries, a single electrode has to play a dual role in both the OER and the ORR.35 In such situations, only Pt can deliver optimum performance towards these reactions. With this motivation, we have investigated the electrocatalytic OER performance of Pt@Ti3C2Tx–rGO catalysts in N2-saturated 0.1 M HClO4 using the RDE. From the polarization curve in Fig. 5a, it can be noted that Pt@Ti3C2Tx–rGO 3:1 showed the lowest onset potential (∼1.58 V vs. RHE) among the other tested counterparts. As expected, Pt@Ti3C2Tx–rGO 3:1 outperformed by achieving the lowest overpotential of 490 mV (at a current density of 10 mA cm−2), which is 42, 90, 150, 151 and 185 mV less than that achieved by Pt@Ti3C2Tx–rGO 1:1, Pt@Ti3C2Tx–rGO 1:3, Pt@Ti3C2Tx, Pt@ rGO and Pt/C, respectively (Fig. S12†). For further analysis, LSV data were fitted into the Tafel equation and are plotted as shown in Fig. 5b. The relatively low Tafel slope value for Pt@Ti3C2Tx–rGO 3:1 (165.3 mV dec−1) reflected its intrinsic facile kinetics towards the OER.
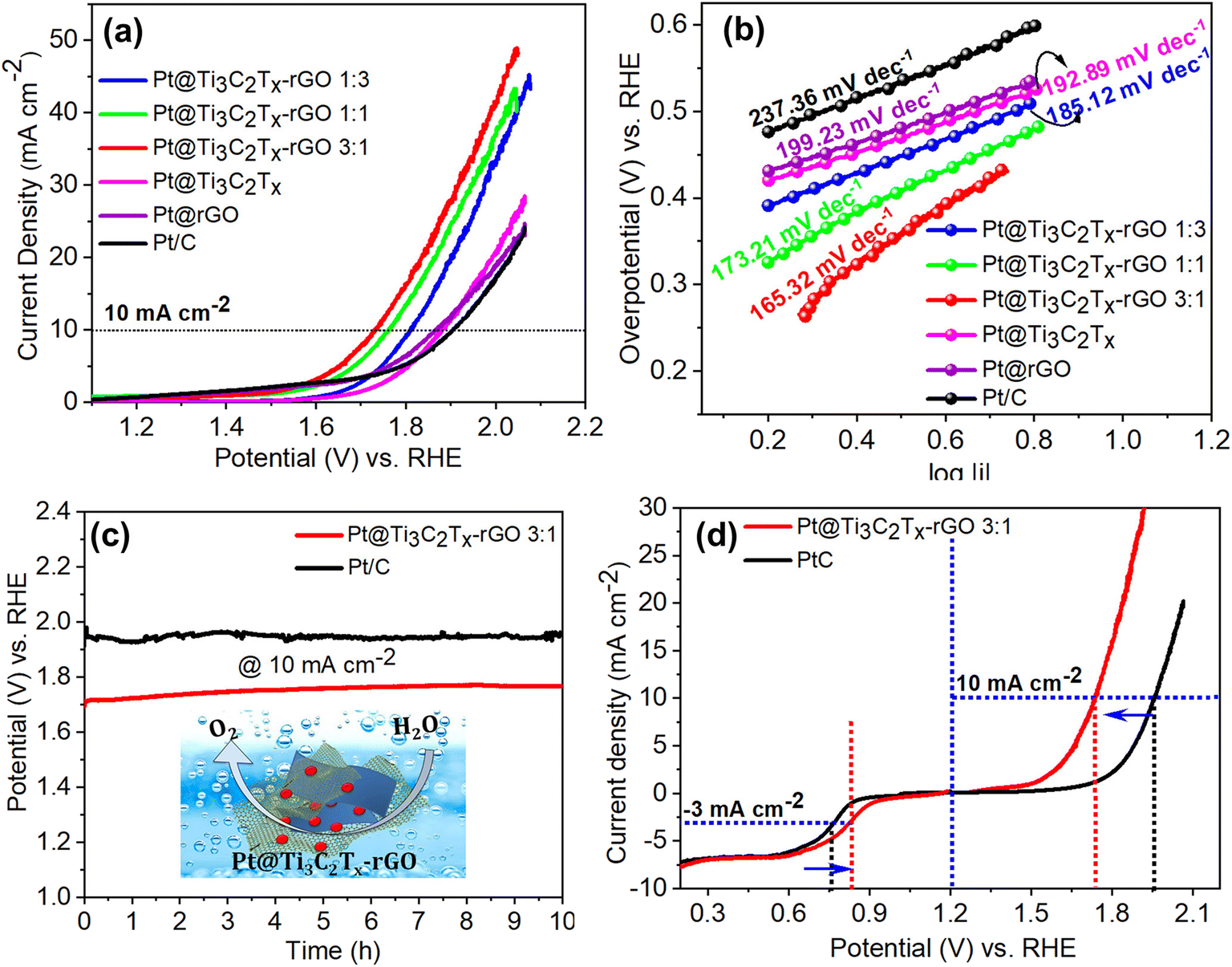 | ||
| Fig. 5 (a) OER polarization curves in N2-saturated 0.1 M HClO4 at 1600 rpm and (b) corresponding Tafel plots of Pt@Ti3C2Tx–rGO 1:3, Pt@Ti3C2Tx–rGO 1:1, Pt@Ti3C2Tx–rGO 3:1, Pt@Ti3C2Tx, Pt@rGO, and Pt/C catalysts derived from (a). (c) Chronopotentiometric response of Pt@Ti3C2Tx–rGO 3:1 and Pt/C at 10 mA cm−2. (d) Overall LSV curves of Pt@Ti3C2Tx–rGO 3:1 and Pt/C showing their bifunctional ORR/OER activities. | ||
The electrochemical stability of Pt@Ti3C2Tx–rGO 3:1 and its comparison with Pt/C towards redox degradation was assessed by chronopotentiometry for several hours. The stability curve shown in Fig. 5c for Pt@Ti3C2Tx–rGO 3:1 and Pt/C reveals their ability to endure uninterrupted electrolysis, ensuring long-term stability for over 10 hours. The experimental results manifest that the Pt@Ti3C2Tx–rGO 3:1 catalyst exhibits a better OER activity and stability in acidic medium. This enhanced performance can be attributed to the catalyst's efficient charge transfer capabilities, which are facilitated by the heterointerface between the 3D-interconnected network structures of the aerogel framework.
The overall bifunctionality of the oxygen electrode is estimated from the potential difference (ΔE) between the ORR at a current density of −3 mA cm−2 and the OER at a current density of 10 mA cm−2.9,36 The acidic ORR/OER bifunctionality of Pt@Ti3C2Tx–rGO 3:1 and Pt/C is shown in Fig. 5d. Pt@Ti3C2Tx–rGO 3:1 exhibited smaller ΔE (0.910 V) compared to Pt/C (1.192). The excellent ORR/OER bifunctional catalytic performance of Pt@Ti3C2Tx–rGO 3:1 may be ascribed to the combination of its structural advantages, including highly dispersed Pt nanoparticles, which allow efficient adsorption and activation of H2O or oxygen species and a 3D porous framework for enhanced electron and mass transport.
In addition to the ORR and OER, the activity of the Pt@Ti3C2Tx–rGO catalyst was investigated towards the HER in N2-saturated 0.1 M HClO4. Fig. 6a displays the polarization curves of all tested catalysts. It is observed that Pt@Ti3C2Tx–rGO catalysts exhibited significantly superior HER activity compared to their counterparts and commercial Pt/C catalysts. The Pt@Ti3C2Tx–rGO 3:1 catalyst exhibited a lower onset potential (−1.4 mV) and lower overpotential of 43 mV at 10 mA cm−2 (this current density was chosen as a performance metric for the HER) (Fig. 6b).
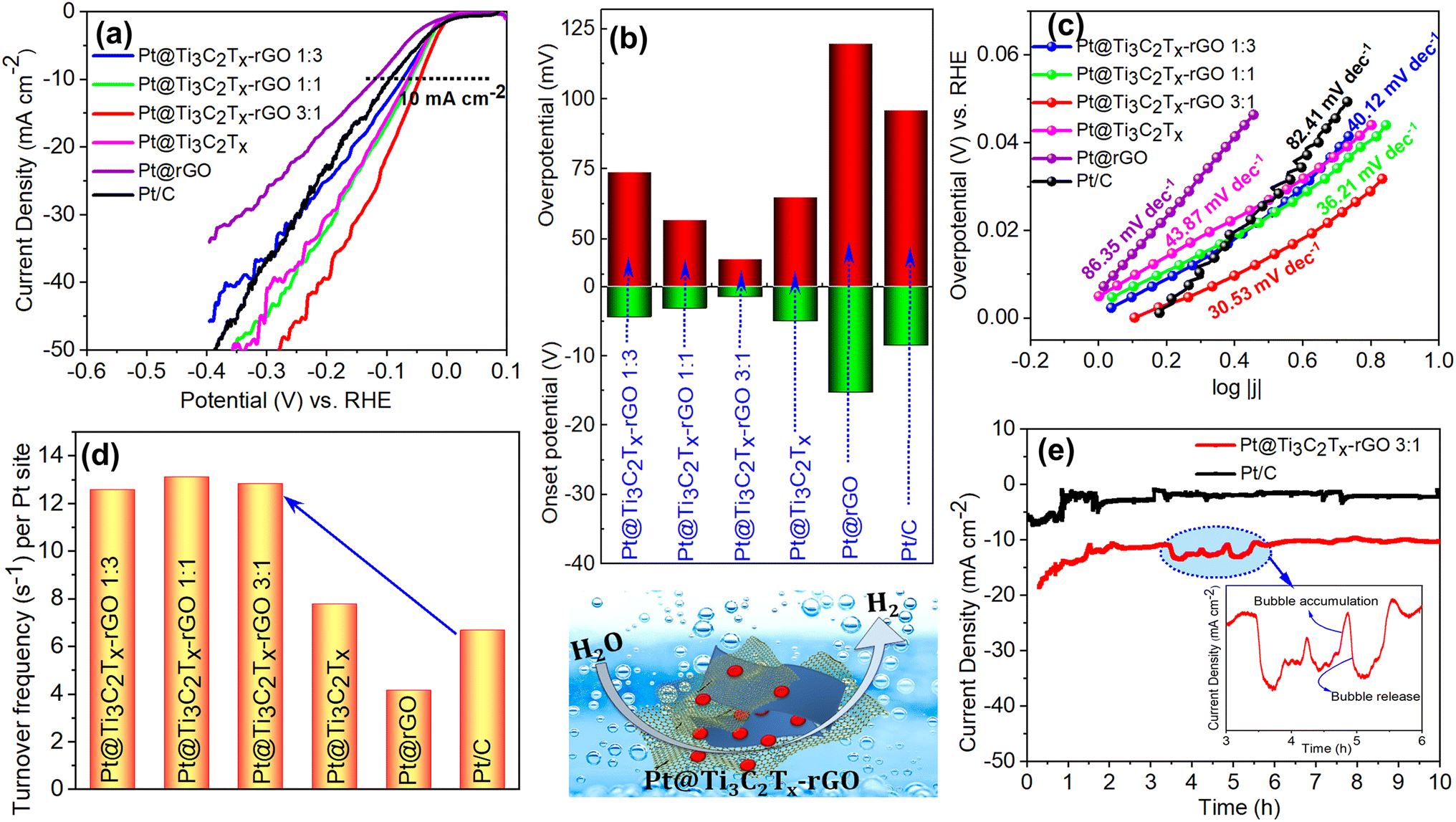 | ||
| Fig. 6 (a) HER polarization curves, (b) histograms of onset potential and HER overpotential at a current density of 10 mA cm−2, (c) Tafel fitting plots derived from data in (a), and (d) turnover frequency (TOF) per Pt site towards the HER at an overpotential of 20 mV for all the developed catalysts. (e) Chronoamperometry plots of Pt@Ti3C2Tx–rGO 3:1 and Pt/C recorded under a constant applied potential of −0.1 V vs. RHE for 10 h (current variation due to bubble formation and release has been depicted in the inset). | ||
The linear regions of polarization curves in Fig. 6c are fitted with the Tafel equation, and the Tafel slope provides insights into the reaction mechanism and kinetics of the HER. In acidic solution, the HER proceeds through three possible elementary steps: (i) H3O+ + e− → Hads + H2O (Volmer reaction, b = 120 mV dec−1), (ii) H3O+ + Hads + e− → H2 + H2O (Heyrovsky reaction, b = 40 mV dec−1) and (iii) Hads + Hads → H2 (Tafel reaction, b = 30 mV dec−1). The HER reported in an acidic solution proceeds via either the combination of Volmer–Heyrovsky or Volmer–Tafel mechanisms. As the amount of rGO content in the Pt@Ti3C2Tx–rGO catalyst increases, the Tafel slope shows a gradual increase from 30 to 40 mV dec−1, which signifies a transition in the rate-limiting step from the Volmer–Tafel to the Volmer–Heyrovsky mechanism. These results suggest that the rate-limiting step is associated with an increase in proton discharge when a small amount of Ti3C2Tx is present in the aerogel heterostructure (Pt@Ti3C2Tx–rGO 1:3), whereas in Pt@Ti3C2Tx–rGO 3:1, the HER proceeds through the recombination of two adsorbed hydrogen atoms. Therefore, the lower Tafel slope of Pt@Ti3C2Tx–rGO 3:1 (30.6 mV dec−1) in comparison with Pt/C (82.4 mV dec−1) signifies faster reaction kinetics and improved H2 generation efficiency under the given experimental conditions.
The property enhancement of Pt@Ti3C2Tx–rGO over the other tested catalysts was further highlighted by the comparisons of turnover frequency (TOF) as shown in Fig. 6d and Table S7.† TOF represents the number of hydrogen molecules evolved per active site per second37 (details are given in S11†). The TOF values for the Pt@Ti3C2Tx–rGO catalysts at an overpotential of 20 mV were ∼12 s−1 per Pt site, representing a significant increase compared to the TOF values of Pt/C, which was estimated to be 6.5 s−1 per Pt site. This demonstrates that the Ti3C2Tx–rGO heterostructure support facilitates full participation of Pt with a high adsorption/desorption rate in the HER.37
To assess the catalytic stability of Pt@Ti3C2Tx–rGO 3:1, chronoamperometry was performed at a potential of −0.1 V and compared with that of Pt/C (Fig. 6e). At the onset, a transient response was observed for both catalysts, characterized by the rapid increase in current, followed by the attainment of a steady state. This may be attributed to the initial activation of the catalyst surface, followed by a steady and sustained catalytic activity. The high signal to noise ratio observed (inset Fig. 6e) in the obtained data suggests rapid H2 gas bubble formation and release at the electrode–electrolyte interface, resulting in a rise and fall of current. Moreover, the higher current density of Pt@Ti3C2Tx–rGO 3:1 suggests that it is a more effective and stable catalyst for the HER compared to Pt/C. These results demonstrate that the Ti3C2Tx–rGO aerogel architecture plays a crucial role in facilitating the exposure of catalytically active sites in Pt nanoparticles. A comparison of the catalyst with others from the literature was also performed, revealing that Pt@Ti3C2Tx–rGO 3:1 exhibited comparable or better performance. Details are provided in Tables S8 and S9.†
3.3 Origin of the enhanced electrochemical performance of the Pt@Ti3C2Tx–rGO catalyst
To comprehend the contributions of the Ti3C2Tx–rGO supported Pt catalyst to oxygen and hydrogen electrocatalysis in acidic medium, we correlated the electrochemical activity with the surface electronic structure using XPS and XAS measurements. Fig. 7a shows the Pt 4f core level spectra of Pt@Ti3C2Tx–rGO (1:3, 1:1 and 3:1) and Pt/C. The Pt 4f spectrum of all the catalysts consists of multiple chemical valences of Pt0 and Pt2+ in the corresponding spin orbital coupling states of Pt 4f7/2 and Pt 4f5/2.23 Comparing the relative intensities of Pt0 and Pt2+ of all the catalysts, it is evident that the peaks associated with Pt0 are considerably stronger in Pt@Ti3C2Tx–rGO than in Pt/C (values are summarized in Table S10†). This indicates the beneficial role of Pt0 in catalytic performance, as observed during the ORR/OER/HER. Furthermore, the Pt 4f peaks in Pt@Ti3C2Tx–rGO samples were downshifted by ∼0.3–0.5 eV relative to those of Pt/C. This suggests a lowered affinity of 4f electrons for Pt in the Pt@Ti3C2Tx–rGO catalysts, possibly resulting from partial electron transfer from the support to Pt.37,38 The extent of shift increases with increasing Ti3C2Tx content in the order Pt@Ti3C2Tx–rGO 1:3 < Pt@Ti3C2Tx–rGO 1:1 < Pt@Ti3C2Tx–rGO 3:1. These results again underline the electronic interaction between Pt and the support, significantly affected by the Ti3C2Tx:rGO ratio. Therefore, the notable electron transfer evident in Pt@Ti3C2Tx–rGO 3:1 due to the profusion of Ti3C2Tx relative to rGO is duly attributed to the strong metal–support interaction (SMSI) effect.38,39
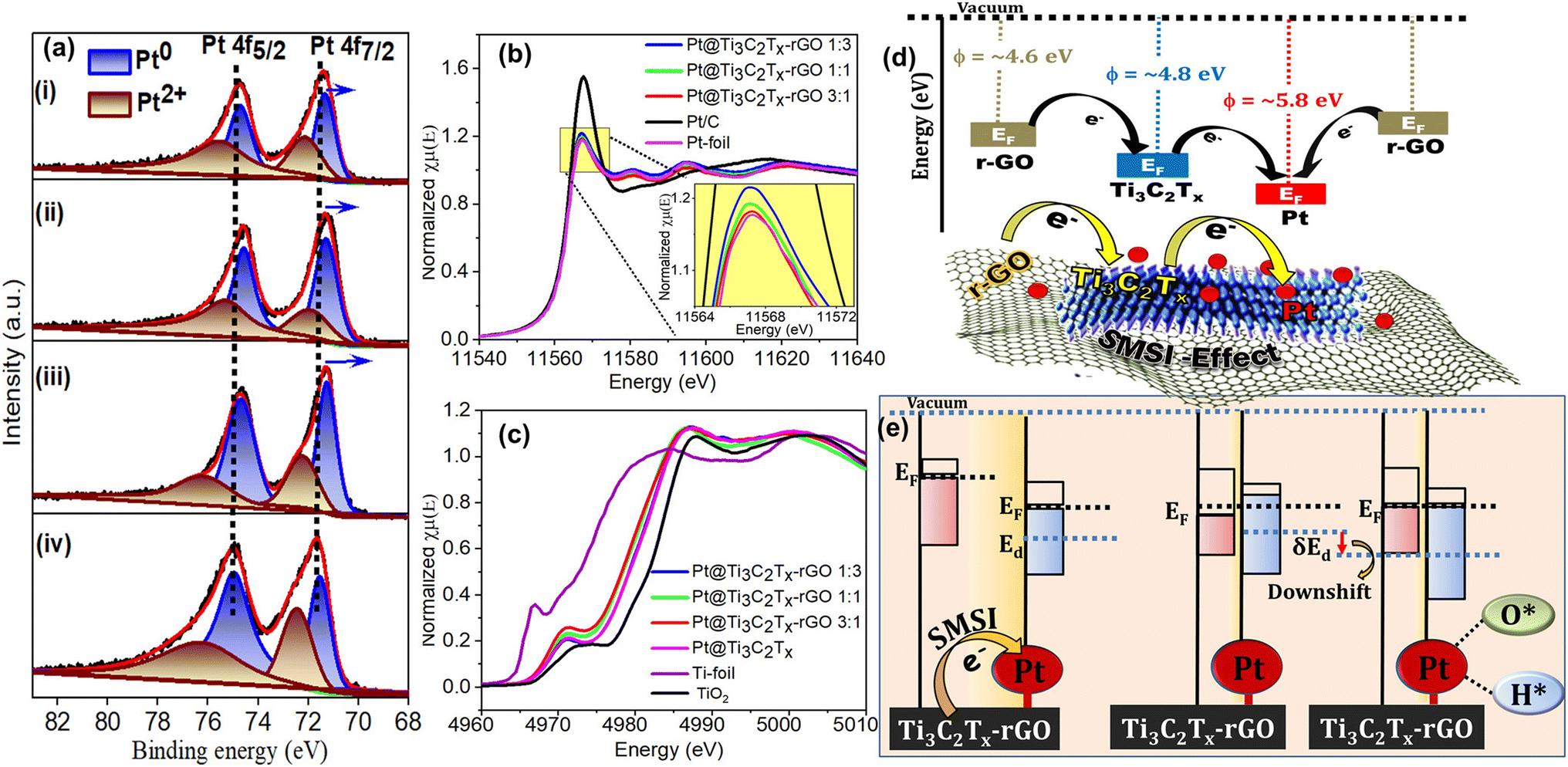 | ||
| Fig. 7 (a) XPS of the Pt 4f region of (i) Pt@Ti3C2Tx–rGO 1:3, (ii) Pt@Ti3C2Tx–rGO 1:1, (iii) Pt@Ti3C2Tx–rGO 3:1 and (iv) Pt/C. Normalized XANES at (b) the Pt L3 edge and (c) the Ti K-edge for all the developed catalysts along with the control samples (Pt-foil, Pt/C, Ti-foil and TiO2). (d) Schematic illustration of the charge transfer mechanism facilitated by the difference in work function (ϕ) between Pt and the Ti3C2Tx–rGO support. (e) Illustration of charge transfer induced downshift in the Pt d-band of the Pt@Ti3C2Tx–rGO catalyst. | ||
To further unveil the electronic properties and reconfirm the SMSI of Pt@Ti3C2Tx–rGO 3:1, Pt L3-edge XANES measurements were performed. The spectrum was plotted and compared with those of Pt foil and Pt/C as the control samples (Fig. 7b). The spectrum of Pt@Ti3C2Tx–rGO is closer to that ofPt-foil than to Pt/C, indicating a higher metallic character of Pt species. This result aligns with the XPS analysis shown in Fig. 7a. The rising edge of the absorption spectra in the Pt L3 energy level often known as the “white line” (WL) is associated with an electron transition from 2p3/2 to a vacant 5d orbital.40 Generally, alteration in WL intensity is influenced by the Pt electronic structure (d-band vacancy).41 Upon careful analysis, the WL intensity shows a decline in the order: Pt/C > Pt@Ti3C2Tx–rGO 1:3 > Pt@Ti3C2Tx–rGO 1:1 > Pt@Ti3C2Tx–rGO 3:1 > Pt-foil. This confirms relatively more electron density at Pt in the Pt@Ti3C2Tx–rGO 3:1 catalyst, due to electron transfer from Ti3C2Tx–rGO to Pt, compared to the transfer from C to Pt in Pt/C. This result is attributed to the SMSI of the Pt@Ti3C2Tx–rGO 3:1 catalyst, thereby leading to higher activity and stability observed during electrochemical reactions.
Further evidence supporting this charge transfer can be discerned through the Ti K-edge of the XANES spectra (Fig. 7c). The dipole-forbidden weak pre-edge (∼4960–4980 eV) indicates 1s → 3d quadrupole transitions associated with the centrosymmetry of the Ti species.42,43 The lower intense peak of the Pt@Ti3C2Tx–rGO catalyst indicates the formation of distorted Ti species. The post-edge peak (∼4980–5020 eV) consisting of a WL arises from the 1s → 4p transition, offering insights into the average oxidation state since the outer p-orbitals are highly sensitive to electronic structural changes. As the oxidation state increases, the adsorption peak shifts to higher energy values.38,44 Here, the observed energy shifts are noted to be in the sequence” TiO2 > Pt@Ti3C2Tx > Pt@Ti3C2Tx–rGO 1:3 > Pt@Ti3C2Tx–rGO 1:1 > Pt@Ti3C2Tx–rGO 3:1 > Ti foil, which suggests that the valence state of Ti in the Pt@Ti3C2Tx–rGO catalyst ranges between zero and positive tetravalency. The observed lower oxidation states in the Ti3C2Tx–rGO supported Pt catalyst, compared to Pt@Ti3C2Tx, can be attributed to the electron transfer characteristics of rGO, demonstrating the evident synergy between the heterojunctions of rGO and Ti3C2Tx.
The inferences drawn from XPS and XANES results were corroborated by the electron transfer mechanism facilitated by the difference in work function (ϕ) between Pt and Ti3C2Tx–rGO. From the literature, Pt has a ϕ of 5.84 eV,45 while the ϕ values of Ti3C2Tx and rGO were reported to be ∼4.8–5.0 (ref. 46) and ∼4.5–4.6 eV, respectively.47 If the Fermi level (EF) of the electrons in the support is larger than that of the metal, then the electrons tend to transfer from the support to the metal.48 The measured ϕ for the Ti3C2Tx–rGO support is 4.9 eV (see the details of the measurement technique in S12 and Fig. S13†). Consequently, significant electron transfer is anticipated to occur from the Ti3C2Tx–rGO support (with lower ϕ and higher EF) to Pt (with higher ϕ and lower EF), in order to equilibrate their respective EF, as shown in Fig. 7d and e.
The aforementioned charge transfer from the support to Pt, confirmed by XPS and XANES measurements, and its consequential impact on the observed electrocatalytic performance can be elucidated through d-band theory proposed by Hammer and Norskov.49 The transfer of charge results in an increased electron density around the Pt atoms, leading to a subsequent downshift of the Pt d-band center (εd) with respect to the Fermi level (EF) (as shown in Fig. 7e). Hence, any such perturbation in the electronic configuration due to charge transfer leads to an electronic effect, viz. “ligand effect”.50 This effect can directly influence the adsorption affinity of oxygen/hydrogen intermediates [(O*/*OH/*OOH) for the ORR/OER and (H* for the HER)] on the surface of an electrocatalyst during oxygen and hydrogen electrocatalysis.51 The aforementioned analysis also indicates that the Pt εd, and thereby the electrocatalysis pertaining to oxygen and hydrogen (ORR/OER/HER) on Pt anchored onto the Ti3C2Tx–rGO support, can be deliberately controlled by suitably choosing the nature of the composition of Ti3C2Tx and rGO.
In addition to charge transfer, the modulation in the electronic structure of electrocatalysts also results from lattice strain, a phenomenon known as the strain effect.48,50 Such strain induced by hybrid support systems featuring heterointerfaces has received limited attention within the realm of electrocatalysis. Nonetheless, this phenomenon is a pivotal factor influencing catalytic activity. To scrutinize the influence of lattice strain in Pt@Ti3C2Tx–rGO, EXAFS analysis has been used as a promising tool for comprehending the intricate structure within a lattice.
The raw background-subtracted k2-weighted Pt L3-edge EXAFS spectra (shown in Fig. S14†) exhibited well-resolved data quality and a high signal-to-noise ratio for the Pt@Ti3C2Tx–rGO catalyst with distinct EXAFS oscillations extending up to 16 Å−1. The k3-weighted Pt L3-edge Fourier-transformed-EXAFS (FT-EXAFS) R-space (radial-space) spectra are shown in Fig. 8a. From the reference spectrum (Pt foil and Pt/C), the peaks sited at around (2.57, 3.65, 4.54, and 5.09 Å) and ∼1.59 Å represent the contribution of Pt–Pt bonds (first, second, third and fourth coordination shells, respectively) and Pt–X (X = C/O) to the EXAFS oscillations, respectively.52 The FT characteristics of the Pt@Ti3C2Tx–rGO (1:3, 1:1 and 3:1) catalyst closely resemble those of Pt-foil, albeit with a comparatively lower intensity. This suggests that the samples are predominantly characterized by a metallic Pt phase, identified by the fcc structure with small Pt crystallite size uniformly distributed within the heterostructure.39,52 Moreover, in Pt@Ti3C2Tx–rGO samples, the scattering peak at ∼2.41 Å and ∼1.6 Å may be assigned to Pt–Pt/Ti and Pt–C/O bonds, respectively. This again underlines the unambiguous interaction of Pt with Ti3C2Tx and rGO, respectively. Interestingly, the EXAFS oscillations of Pt@Ti3C2Tx–rGO showed a shift towards the lower R direction, which may be attributed to the lattice distortion of Pt caused by the formation of metal–support interfaces due to the SMSI effect. Consequently, this suggests the possible compressive strain of Pt in the Ti3C2Tx–rGO heterostructure.
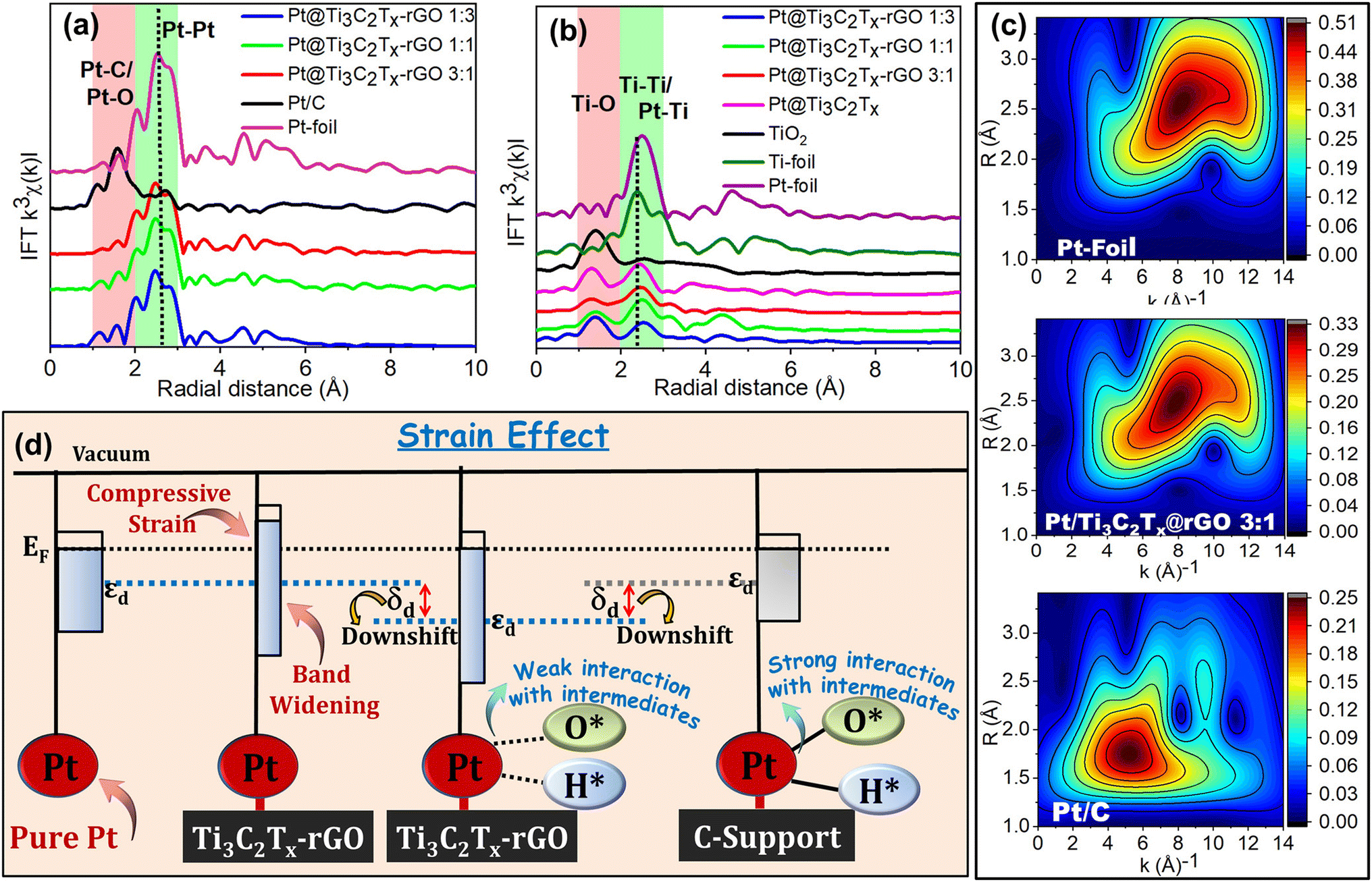 | ||
| Fig. 8 Fourier transform-EXAFS (FT-EXAFS) oscillations of (a) the k3-weighted Pt L3 edge and (b) the Ti K-edge. (c) Wavelet transforms (WT) of Pt@Ti3C2Tx–rGO, Pt-foil and Pt/C for reference. (d) Schematic illustration of the effect of compressive strain on the bandwidth and the center of the d-band of Pt@Ti3C2Tx–rGO with reference to Pt/C. | ||
Additionally, this was corroborated by the Ti coordination environment determined by Ti K edge FT-EXAFS (Fig. 8b). As evidenced, the broad peak at ∼1.3–1.6 Å depicts the Ti–O bond, while the 2.55 Å in Ti foil is attributed to direct Ti–Ti bonding.38,43 Apparently, Pt@Ti3C2Tx–rGO displays coordination peaks at ∼2.48–2.55 Å, which are ascribed to Ti–Pt and Ti–Ti scattering.38 Moreover, a slight positive shift in these peaks corresponds to an increase in the bond length, attributed to strong interactions between Pt and the Ti sites of the support. This again underlines the SMSI effect between Pt and the Ti3C2Tx–rGO support through Ti sites.
Furthermore, Fig. S15† shows the best fitting of the Pt L3-edge FT-EXAFS data for the catalyst, and the associated fit parameters are detailed in Table S11.† The coordination number (N) of Pt–Pt bonds in Pt@Ti3C2Tx–rGO catalysts (N ≈ 8–9) is smaller compared to that of the standard Pt-foil (N = 12). This is attributed to the higher proportion of Pt sites in the heterointerfaces of the Ti3C2Tx–rGO support.39 The Pt strain is intricately linked to the Pt–Pt bond length (RPt–Pt) derived from the EXAFS analysis, providing a means to monitor intraparticle strains within the sample.53 The Pt–Pt bond length (RPt–Pt) in Pt@Ti3C2Tx–rGO 3:1 (RPt–Pt = 2.731 Å) is ∼1.5% and ∼1.2% shorter than that in Pt/C (RPt–Pt = 2.771 Å) and Pt foil (RPt–Pt = 2.765 Å). This inference suggests a more robust electronic interaction between Pt and the Ti3C2Tx–rGO support.39 It clearly reveals the generation of minute compressive stress on the Pt lattice within the Pt@Ti3C2Tx–rGO catalyst as a result of SMSI. A closer examination of Pt (111) facets in the XRD pattern further validates this, as the Pt@Ti3C2Tx–rGO catalyst exhibited a positive shift to the higher angle side (lower d-spacing) (Fig. S16†). This shift points to a lattice contraction, which is attributed to the generation of a compressive strain within the heterostructure. Based on the changes in the lattice constant (shown in Table S12†), the calculated magnitudes of overall compressive strains are ∼1.7% for Pt@Ti3C2Tx–rGO with respect to Pt/C, aligning well with the EXAFS data.
This was further corroborated by the analysis of Wavelet-Transformed EXAFS (WT-EXAFS), enabling simultaneous resolution in both k-space (wavevector-space) and R-space. This approach furnishes details regarding atomic dispersions and bonding conditions.53 The WT-EXAFS contour plot in Fig. 8c illustrates intensity maxima for Pt foil around (R: 2.6 Å; k: 8.3 Å−1), indicating the presence of Pt–Pt contributions. In contrast, the WT signal derived from the Pt–Pt contribution of Pt@Ti3C2Tx–rGO 3:1 shifted to lower R and k-space (R = 2.4 Å; k = 7.8 Å−1), indicating lattice distortion induced by the closely interacting metal–support interfaces. In the case of Pt/C, an intense signal is observed at (R = 5.1 Å; k = 1.5 Å−1), corresponding to Pt–C/O bonding attributed to O backscattering. This observation further substantiates the claim of its high oxidized state (Fig. 7b).
Hence, the combination of strain or lattice mismatch introduced on the surface of Pt@Ti3C2Tx–rGO is also identified as a contributing factor in enhancing the observed electrocatalytic activity. This can also be effectively elucidated using d-band center theory, particularly its correlation with the adsorption energies of oxygen-containing intermediates.49,54 As depicted in Fig. 8d, the compressive strain causes an increase in the Pt d-band width, leading to a shift of εd away from EF in order to preserve the d-band filling level. It is well documented that the compressive strain-induced downward shift of εd reduces the adsorption of surface poisoning species, thereby weakening the interaction with surface adsorbates.3,19 Accordingly, with a downward shift of εd, Pt@Ti3C2Tx–rGO exhibits a greater ability to remove generated reaction intermediate species [(O*/*OH/*OOH) for the ORR/OER and (H* for HER)] compared to Pt/C (Fig. 8d), thereby contributing to the improved ORR/OER/HER performance noted in this study.
Therefore, the synergy between electronic interaction induced by both ligand and strain effects contributes to the downshift of εd at the heterointerface within the porous aerogel framework. This collectively boosts the electrocatalytic activity and durability of Pt@Ti3C2Tx–rGO for hydrogen and oxygen electrocatalysis. However, it should be noted that there are also studies suggesting that an upshift in εd (increase in d-band vacancies) can also contribute to improved catalytic activity.55,56 Hence, there is still room for discussion regarding the characterization of the electronic state of Pt to optimize electrocatalytic activity. Research in this direction is in progress in our laboratory.
4. Conclusions
In summary, our study introduces a highly active and low Pt content 3D Pt@Ti3C2Tx–rGO aerogel catalyst synthesized via a one-step γ-radiolytically induced reduction and self-assembly process. The Pt@Ti3C2Tx–rGO catalyst showcases enhanced electrocatalytic performance towards the ORR, OER, and HER. outperforming the benchmark Pt/C in terms of lower overpotentials, improved stability and better reaction kinetics. The improved electrocatalytic efficiency can be attributed to the combined beneficial effects of the porous aerogel framework, which aids in ion transport and diffusion, and the interconnected heterointerface, which provides numerous pathways for charge transport. This study underlines that the strong metal–support interaction (SMSI) at the interface of the Pt@Ti3C2Tx–rGO heterostructure significantly improves the electronic properties of Pt active sites, making them thermodynamically favorable for both oxygen and hydrogen electrocatalysis. Through spectroscopic analysis, we established the combined interplay of ligand and strain effects, which effectively fine-tune the energy of the d-band center of Pt sites, thereby enhancing the electrocatalytic activities. In conclusion, we offer herewith a novel approach for designing high-performance support structures in the realm of electrocatalysis.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
L. V. and S. B. A. contributed equally to this work. The whole work was supervised by S. K. H. The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the funding from UGC-DSK PDF (File No. F.4-2/2006(BSR)/CH/20-21/0057) and CSIR (File No. 09/0137(15346)2022-EMR-I). The authors express gratitude to Dr Biplab Ghosh and Dr Rajashri R. Urkude, Beamline Development and Application Section, Raja Ramanna Centre for Advanced Technology (RRCAT), for their valuable assistance and support in conducting XAS measurements using the scanning EXAFS beamline (BL-09) at Indus-2, RRCAT, Indore, India. The authors are grateful for the support received from the TEM facility at the Centre for Nano and Soft Matter Sciences (CENS), Bangalore, India. The authors thank S. Debalina for her assistance during the synthesis and electrochemical characterization of the catalyst. The authors are thankful to Dr J. Sarika and Dr V. L. Mathe for helping with XPS and retarding field method for work function measurements.References
- L. Huang, S. Zaman, X. Tian, Z. Wang, W. Fang and B. Y. Xia, Acc. Chem. Res., 2021, 54, 311–322 CrossRef CAS PubMed.
- G. A. Kamat, J. A. Zamora Zeledón, G. T. K. K. Gunasooriya, S. M. Dull, J. T. Perryman, J. K. Nørskov, M. B. Stevens and T. F. Jaramillo, Commun. Chem., 2022, 5, 20 CrossRef CAS.
- R. Chattot, T. Asset, P. Bordet, J. Drnec, L. Dubau and F. Maillard, ACS Catal., 2017, 7, 398–408 CrossRef CAS.
- N. H. Ahmad Junaidi, W. Y. Wong, K. S. Loh, S. Rahman and W. R. W. Daud, Int. J. Energy Res., 2021, 45, 15760–15782 CrossRef CAS.
- M. Zubair, M. M. Ul Hassan, M. T. Mehran, M. M. Baig, S. Hussain and F. Shahzad, Int. J. Hydrogen Energy, 2022, 47, 2794–2818 CrossRef CAS.
- X. Xie, S. Chen, W. Ding, Y. Nie and Z. Wei, Chem. Commun., 2013, 49, 10112–10114 RSC.
- C. Zhang, B. Ma, Y. Zhou and C. Wang, J. Electroanal. Chem., 2020, 865, 114142 CrossRef CAS.
- J. Zhu, L. Xia, R. Yu, R. Lu, J. Li, R. He, Y. Wu, W. Zhang, X. Hong, W. Chen and Y. Zhao, J. Am. Chem. Soc., 2022, 144, 15529–15538 CrossRef CAS.
- S. B. Alex, L. Vazhayal, P. P. Waghmaitar, R. R. Urkude, B. B. Chandashive, D. Khushalani and S. K. Haram, ACS Appl. Energy Mater., 2024, 7, 1890–1901 CrossRef CAS.
- L. Yang, M. Li, M. Li, Y.-C. Zhu, S. Seunghyun, L. Shi-Neng, A. Jolyon, T. Long-Cheng and B. Joonho, Composites, Part B, 2023, 251, 110465 CrossRef.
- B. F. Guo, Y. J. Wang, C. F. Cao, Z. H. Qu, J. Song, S. N. Li, J. F. Gao, P. Song, G. D. Zhang, Y. Q. Shi and L. C. Tang, Adv. Sci., 2024, 11, 2309392 CrossRef CAS PubMed.
- C. Yang, Q. Jiang, W. Li, H. He, L. Yang, Z. Lu and H. Huang, Chem. Mater., 2019, 31, 9277–9287 CrossRef CAS.
- H. Y. Chen, Z. Y. Chen, M. Mao, Y. Y. Wu, F. Yang, L. X. Gong, L. Zhao, C. F. Cao, P. Song, J. F. Gao and G. D. Zhang, Adv. Funct. Mater., 2023, 33, 2304927 CrossRef CAS.
- R. Wang, X. Zhang, S. Chang, S. Jin, J. Wang, C. Wang, Y. Chang, Z. Yu, Q. Hu and A. Zhou, Fuel, 2023, 360, 130507 CrossRef.
- A. Raveendran, M. Chandran, M. R. Siddiqui, S. M. Wabaidur, M. Eswaran and R. Dhanusuraman, ACS Omega, 2023, 8, 34768–34786 CrossRef CAS PubMed.
- T. C. Yang Li, Z. Yong-Cheng, V. Sowjanya, L. Man, S. Seunghyun, L.-C. Tang and B. Joonho, J. Energy Chem., 2024, 89, 313–323 CrossRef.
- K. M. Samant, V. S. Joshi, G. Sharma, S. Kapoor and S. K. Haram, Electrochim. Acta, 2011, 56, 2081–2086 CrossRef.
- D. C. Poudyal, R. Dugani, B. S. Dash, M. Dhavale, A. K. Satpati and S. K. Haram, ACS Omega, 2021, 6, 13579–13587 CrossRef PubMed.
- X. Yang, Y. Wang, X. Tong and N. Yang, Adv. Energy Mater., 2022, 12, 2102261 CrossRef.
- A. Ramesh, M. Jeyavelan and M. S. Leo Hudson, Dalton Trans., 2018, 47, 5406–5414 RSC.
- S. Li, X. Que, X. Chen, T. Lin, L. Sheng, J. Peng, J. Li and M. Zhai, ACS Appl. Energy Mater., 2020, 3, 10882–10891 CrossRef CAS.
- J. Shi, W. Jiang, L. Liu, M. Jing, F. Li, Z. Xu and X. Zhang, Curr. Appl. Phys., 2019, 19, 780–786 CrossRef.
- D. Dang, H. Zou, Z. A. Xiong, S. Hou, T. Shu, H. Nan, X. Zeng, J. Zeng and S. Liao, ACS Catal., 2015, 5, 4318–4324 Search PubMed.
- A. Sarycheva and Y. Gogotsi, Chem. Mater., 2020, 32, 3480–3488 CrossRef CAS.
- Q. Chen, K. Zhang, L. Huang, C. Zhang, Y. Yuan and Y. Li, Adv. Eng. Mater., 2022, 24, 2200098 Search PubMed.
- Y. Garsany, O. A. Baturina, K. E. Swider-Lyons and S. S. Kocha, Anal. Chem., 2010, 82, 6321–6328 Search PubMed.
- N. Sacré, G. Hufnagel, J. Galipaud, E. Bertin, S. A. Hassan, M. Duca, L. Roué, A. Ruediger, S. Garbarino and D. Guay, J. Phys. Chem. C, 2017, 121, 12188–12198 Search PubMed.
- J. Zhang, H. Yang, K. Yang, J. Fang, S. Zou, Z. Luo, H. Wang, I. T. Bae and D. Y. Jung, Adv. Funct. Mater., 2010, 20, 3727–3733 CrossRef CAS.
- K. Mohanraju and L. Cindrella, RSC Adv., 2014, 4, 11939–11947 Search PubMed.
- R. Zhou, Y. Zheng, M. Jaroniec and S. Z. Qiao, ACS Catal., 2016, 6, 4720–4728 CrossRef CAS.
- Y. Liu, S. Shrestha and W. E. Mustain, ACS Catal., 2012, 2, 456–463 CrossRef CAS.
- L. Lin, Z. K. Yang, Y. F. Jiang and A. W. Xu, ACS Catal., 2016, 6, 4449–4454 Search PubMed.
- G. F. Wei, Y. H. Fang and Z. P. Liu, J. Phys. Chem. C, 2012, 116, 12696–12705 Search PubMed.
- J. T. Ren and Z. Y. Yuan, ACS Sustain. Chem. Eng., 2019, 7, 10121–10131 CrossRef CAS.
- A. Mathura and A. Halder, Catal. Sci. Technol., 2019, 9, 1245–1254 RSC.
- T. C. Nagaiah, D. Gupta, S. D. Adhikary, A. Kafle and D. Mandal, J. Mater. Chem. A, 2021, 4, 9228–9237 RSC.
- T. Kim, S. B. Roy, S. Moon, S. Yoo, H. Choi, V. G. Parale, Y. Kim, J. Lee, S. C. Jun, K. Kang and S. H. Chun, ACS Nano, 2022, 16, 1625–1638 CrossRef CAS PubMed.
- R. Li, Z. Liu, Q. T. Trinh, Z. Miao, S. Chen, K. Qian, R. J. Wong, S. Xi, Y. Yan, A. Borgna and S. Liang, Adv. Mater., 2021, 33, 2101536 Search PubMed.
- C. He and J. Tao, J. Power Sources, 2016, 324, 317–324 CrossRef CAS.
- R. Wang, Y. Xie, K. Shi, J. Wang, C. Tian, P. Shen and H. Fu, Chem.–Eur. J., 2012, 18, 7443–7451 CrossRef CAS.
- X. Han, X. Wu, Y. Deng, J. Liu, J. Lu, C. Zhong and W. Hu, Adv. Energy Mater., 2018, 8, 1800935 CrossRef.
- L. Zhang, D. Sun, J. Kang, H. T. Wang, S. H. Hsieh, W. F. Pong, H. A. Bechtel, J. Feng, L. W. Wang, E. J. Cairns and J. Guo, Nano Lett., 2018, 18, 4506–4515 CrossRef CAS.
- W. Xu, T. Zhang, R. Bai, P. Zhang and J. Yu, J. Mater. Chem. A, 2020, 8, 9677–9683 Search PubMed.
- C. Xu, F. Xiong, Y. Wang, J. Nai and W. Zhang, Nanotechnology, 2023, 34, 255402 Search PubMed.
- C. Jackson, G. T. Smith, D. W. Inwood, A. S. Leach, P. S. Whalley, M. Callisti, T. Polcar, A. E. Russell, P. Levecque and D. Kramer, Nat. Commun., 2017, 8, 15802 CrossRef CAS.
- H. Kim and H. N. Alshareef, ACS Mater. Lett., 2020, 2, 55–70 CrossRef CAS.
- J. Lin, P. Hu, Y. Zhang, M. Fan, Z. He, C. K. Ngaw, J. S. C. Loo, D. Liao and T. T. Y. Tan, RSC Adv., 2013, 3, 9330–9336 RSC.
- J. H. Kim, S. Chang and Y. T. Kim, Appl. Catal., B, 2014, 158, 112–118 Search PubMed.
- B. Hammer and J. K. Nørskov, Surf. Sci., 1995, 343, 211–220 Search PubMed.
- C. Lim, A. R. Fairhurst, B. J. Ransom, D. Haering and V. R. Stamenkovic, ACS Catal., 2023, 13, 14874–14893 Search PubMed.
- F. Ando, T. Gunji, T. Tanabe, I. Fukano, H. D. Abruna, J. Wu, T. Ohsaka and F. Matsumoto, ACS Catal., 2021, 11, 9317–9332 CrossRef CAS.
- Y. Jeon, Y. Ji, Y. I. Cho, C. Lee, D. Park and Y. Shul, ACS Nano, 2018, 12, 6819–6829 Search PubMed.
- M. Lao, K. Rui, G. Zhao, P. Cui, X. Zheng, S. X. Dou and W. Sun, Angew. Chem., 2019, 131, 5486–5491 CrossRef.
- H. Chen, Q. Wu, Y. Wang, Q. Zhao, X. Ai, Y. Shen and X. Zou, Chem. Commun., 2022, 58, 7730–7740 RSC.
- D. Abrun, N. Dimitrov, J. Fang, Y. Xie, Y. Yang and D. A. Muller, ACS Catal., 2020, 10, 9967–9976 CrossRef.
- X. Wang, Y. Tang, J. Lee and G. Fu, Chem Catal., 2022, 2, 967–1008 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and physicochemical and electrochemical characterization of the Pt@Ti3C2Tx–rGO aerogel catalyst. Preparation of GO. Evaluation of the dosage rate through Fricke dosimetry, Electrochemical Surface Area (ECSA), Mass Activity (MA) and Specific Activity (SA) of the ORR, n and p values obtained using the RRDE, Tafel plot, collection efficiency, n-value obtained using the KL plot, Turnover Frequency (TOF) of the HER, XRD of GO before and after radiolysis, SEM and HR-TEM images and SAED pattern of the Pt@Ti3C2Tx–rGO aerogel, ORR polarization curves and corresponding K–L plots, peroxide yield, comparison of OER performance, and quantitative fitting of Fourier-transformed EXAFS in R-space and k-space. Tabulation of various materials and electrochemical properties/parameters. See DOI: https://doi.org/10.1039/d4ta02688h |
| ‡ L. V. and S. B. A. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |